

Abstract
Am 18. Gestationstag, bei einer Länge des Embryos von etwa 2,5 mm, verdickt sich das distale Ende des Vorderdarms und beginnt, sich als Leberknospe vorzuwölben (Leberdivertikel; Tab. 13.1).
Zu Beginn des Lebens ist der Dottersack für Ernährung und alle Stoffwechselfunktionen verantwortlich. Mit dem Beginn der Entwicklung der einzelnen Organe übernimmt die Leber immer mehr der wesentlichsten Funktionen. Im Gegensatz zur Situation bei der Leberregeneration im späteren Leben ist über die Steuerungsmechanismen in der Frühphase der menschlichen Entwicklung bis zum Sichtbarwerden der Leberknospe am distalen Ende des Vorderdarms nichts bekannt.
Struktur und Morphologie
Am 18. Gestationstag, bei einer Länge des Embryos von etwa 2,5 mm, verdickt sich das distale Ende des Vorderdarms und beginnt, sich als Leberknospe vorzuwölben (Leberdivertikel; ◘ Tab. 13.1).
| Zeitraum | Entwicklungsschritte |
|---|---|
| 18. Gestationstag | Entwicklung der Leberknospe aus dem verdickten Endoderm des Vorderdarms |
| 22. Gestationstag | Kontakt des Leberdivertikulums mit dem Mesoderm des Septum transversum |
| 23. Gestationstag | Beginn der Invasion von Hepatoblasten in das Mesenchym |
| 23.–24. Gestationstag | Expression von α-Fetoprotein und Albumin in den Hepatoblasten |
| 3.–8. Woche | Expression von Zytokeratin 19, Öffnung des Ductus hepaticus |
| 5. Woche | Verbindung der Vv. umbilicales mit dem sinusoidalen Plexus der Leber |
| 5.–9. Woche | Beginn der Gallensäurensynthese |
| 6.–16. Woche | Bildung der Duktalplatte vom Hilus aus, hämatopoetische Zellen zwischen den Hepatozyten erkennbar, Expression von α1-Antitrypsin und γ-GT |
| 7. Woche | Bildung der V. portae aus den Vitellin-Venen und des Ductus venosus aus dem sinusoidalen Plexus |
| 8. Woche | Entstehung von Glykogengranula in den fetalen Hepatozyten |
| 8.–9. Woche | Expression der Zytochrom-P450-Enzyme |
| 9. Woche | Beginn der Glykogensynthese |
| 8.–12. Woche | Bildung der 2-lagigen Duktalplatte |
| 10.–12. Woche | Sinusendothel-, Kupffer- und Ito-Zellen nachweisbar |
| 12. Woche | Beginn der Remodellierung der Duktalplatte in tubuläre Strukturen, Entstehung von Mesenchym, Verbindung der offenen intrahepatischen Gallenwege mit dem Darmlumen, Entstehung der Ductus hepatici (erste Generation der Gallengänge), Beginn der Gallesekretion |
| 12.–14. Woche | Beginn der Glykogenese und der Fettakkumulation |
| 15. Woche | Entstehung der 2. Generation der Gallengänge, Bilirubin in Galle und Mekonium nachweisbar |
| 17.–25. Woche | Entstehung der 3. Generation der Gallengänge |
| 25. Woche | Zusammenhängende Duktalplatte nicht mehr erkennbar |
| 25.–28. Woche | Beginn der Bildung von hämatopoetischen Inseln aus bisher diffus verteilter Hämatopoese |
| 35. Woche | Gallengänge in den meisten Portalfeldern nachweisbar |
| Geburt | Diskontinuierlich Duktalplatten in peripheren Portalfeldern erkennbar; Lebergewicht: 75–125 g |
| Geburt bis 11. Lebenstag | Verschluss des Ductus venosus |
| Geburt bis Alter von 4 Wochen | Ausreifung der Gallenwege bis über die 7. intrahepatische Generation hinaus |
| 3 Monate | Ausreifung der Phase-1-Enzyme |
| 1 Jahr | Lebergewicht: 250–300 g |
| 2 Jahre | Lebergewicht: 430 g |
| 5 Jahre | Lebergewicht: 530 g |
| Erwachsene | Lebergewicht: 1400 g; 400.000–500.000 terminale Gallengänge für jeweils 2–3 mm³ Lebergewebe |
γ-GT γ-Glutamyltranspeptidase.
In den folgenden Tagen wächst diese endodermale Sprosse in kranioventraler Richtung gegen eine Mesenchymplatte vor (Septum transversum). Danach durchdringen die endodermalen Zellen gleichzeitig mit dem sinusoidalen Netzwerk der Blutgefäße mit immer weiteren Aufzweigungen das Mesoderm des Septum transversum und bilden die Leber. Innerhalb der folgenden 3 Gestationswochen füllt die Leber den größten Teil der Bauchhöhle aus.
Durch den Kontakt mit den mesenchymalen Elementen des Septum transversum differenziert sich die Lebersprosse weiter in proliferierende Stränge von Hepatoblasten (ab der 4. Woche; Länge des Embryos: 5–7 mm), die sich sowohl zu Hepatozyten als auch zu intrahepatischen Gallenwegen weiterentwickeln. Sie bleiben dabei in engem Kontakt mit den sich weiter verzweigenden Ästen der Vitellin-Venen und bilden ein System miteinander in Verbindung stehender Leberzellplatten. Gleichzeitig entwickelt sich aus den größeren Venen (V. mesenterica, V. umbilica, Ductus venosus) ein Netzwerk kleiner venöser Verzweigungen zwischen den Strängen von Hepatozyten.
Vom Leberhilus ausgehend (etwa 6. Gestationswoche) flachen die Hepatoblasten an der Grenze zwischen Leberparenchym und Mesenchym des Portaltrakts ab und bilden eine kontinuierliche Lage gallengangähnlicher, kubischer Zellen ( Duktalplatte). Die Zellen zeigen eine gallengangspezifische Immunreaktivität für Zytokeratin 19, während weiter entfernt vom Portaltrakt befindliche Hepatoblasten diese Immunreaktivität verlieren. Dieser Prozess setzt sich in die Peripherie fort, so dass jede Region der Leber (bis etwa zur 16. Gestationswoche) eine Duktalplatte erhält.
Danach bildet sich eine zweite Lage gallengangähnlichen Epithels durch Verdopplung der Duktalplatte. Daraus entsteht ein Lumen, das einen Kranz von plattenähnlichen, luminalen Strukturen rund um die Portalvene bildet. Die Remodellierung der Duktalplatte in tubuläre Gallengangstrukturen beginnt in der gleichen Weise an der Porta hepatis zwischen der 11. und 13. Gestationswoche. Die tubulären Strukturen zeigen zunächst ein elliptisches, später ein kreisrundes Lumen und liegen schließlich zentral im Portaltrakt – Mesenchym. Verbleibende periphere Duktalplattenstrukturen verschwinden. Die Remodellierung der Duktalplatten in das duktuläre Gallengangsystem ist etwa 4 Wochen nach der Geburt abgeschlossen. Das reife Gallenwegsystem wird durch einen vaskulären Plexus versorgt, der von der Leberarterie gespeist wird. Die arteriellen Gefäße und der peribiliäre Plexus beginnen ebenfalls, vom Leberhilus aus in die Peripherie vorzuwachsen. Sowohl das Gallenwegsystem als auch das hepatische arterielle Blutgefäßsystem reifen bis zur Adoleszenz weiter aus.
Die Lumina des Ductus choledochus, der Gallenblase und des Ductus hepaticus communis sind immer offen und stehen mit dem Lumen des Verdauungstrakts in Verbindung. Auf der anderen Seite stehen sie mit den plattenähnlichen Duktalstrukturen der Leber in Verbindung, d. h. dass (von der 12. Gestationswoche an) die intrahepatischen Gallengangstrukturen immer offen mit dem Darmlumen in Verbindung stehen.
Derzeit ist noch unklar, wie das Gallenwegsystem an das Leberparenchym Anschluss gewinnt. Intralobuläre Duktuli entspringen von terminalen Gallenwegen und erstrecken sich bis etwa ein Drittel der Distanz zur terminalen Lebervene in das Parenchym. Die Duktuli gehen in den Hering-Kanal über und bilden dadurch die Basis für die Drainage der Galle aus dem Leberparenchym.
Die extrahepatischen und größeren intrahepatischen Gallenwege besitzen peribiliäre Drüsen, die unmittelbar angrenzend an das Gallenganglumen in das Mesenchym eingebettet sind. Diese extrahepatischen Drüsen entwickeln sich parallel zur Drüsenbildung des gesamten Verdauungstrakts als mikroskopisch kleine, divertikuläre Ausbuchtungen entlang der extrahepatischen Gallenwege. Intrahepatische peribiliäre Drüsen entwickeln sich auch vom Duktalplattenepithel aus. In der 40. Gestationswoche werden diese Drüsen auch am Leberhilus, entlang der großen Gallenwege sichtbar. Sie formen azinäre Strukturen, die sich im weiteren Verlauf zahlenmäßig vermehren und organisieren, bis sie ungefähr im Alter von 15 Jahren ausgereift sind.
Blutfluss
Fetale Situation
Beim Fetus erfolgt die Oxygenierung des Blutes in der Plazenta. Sauerstoffarmes Blut erreicht die Plazenta über die Nabelarterien aus der Aorta descendens, oxygeniertes Blut strömt durch die Nabelvene zurück, die sich mit der V. portae vereinigt und das Blut zunächst zur Leber transportiert. Fünfzig Prozent des Nabelvenenblutes fließt durch den Ductus venosus an der Leber vorbei, direkt durch die V. cava inferior oder die linke Lebervene zum Herz. Die fetale Leber erhält Blut aus der V. umbilicalis, der A. hepatica und der V. portae. Die V. umbilicalis versorgt mit Ästen den linken Leberlappen, gibt den Ductus venosus ab, wendet sich nach rechts zum Portalsinus und vereinigt sich mit der V. portae. Aus dem Ductus venosus zweigen keine Gefäße zum Leberparenchym ab. Die A. hepatica gibt symmetrisch zu beiden Leberlappen Äste ab; zahlreiche anatomische Varianten wurden registriert. Das Blut wird in 3 Lebervenen (rechte, mittlere und linke) gesammelt. Der Ductus venosus mündet entweder direkt in die V. cava inferior oder vereinigt sich zuerst mit der linken und der mittleren Lebervene.
Aus der V. umbilicalis stammen 75 % des Blutvolumens der fetalen Leber, 15–20 % aus der V. portae und 5–10 % aus der A. hepatica. Das Blut der V. portae fließt fast ausschließlich in den rechten Leberlappen. Der Blutfluss aus A. hepatica und V. umbilicalis teilt sich etwa gleich auf beide Leberlappen auf. Beim Fetus ist der Leberblutfluss pro Gramm Lebergewebe ungefähr 4-mal so hoch wie beim Erwachsenen.
Der linke Leberlappen erhält 95 % seiner Blutversorgung aus der V. umbilicalis und 5 % aus der A. hepatica, der rechte 60 % aus der V. umbilicalis, 30 % aus der V. portae und 10 % aus der A. hepatica.
Daher ist die Sauerstoffversorgung der beiden Leberlappen unterschiedlich. Das Blut des linken Leberlappens ist zu etwa 80 % mit Sauerstoff gesättigt, das Blut des rechten Leberlappens zu etwa 70 %.
Dem entspricht das Ausmaß der Hämatopoese, welche im rechten Leberlappen deutlich stärker ausgeprägt ist als im linken. Analog ist die Sauerstoffsättigung in der linken Lebervene höher als in der rechten (73 % vs. 63 %).
Es ist unklar, wie die Verteilung des Blutflusses aus der V. umbilicalis in Richtung Leber oder in Richtung Ductus venosus beeinflusst wird. Ein Sphinkter nahe dem Abgang des Ductus venosus aus der V. umbilicalis mit autonomer Innervation wurde beschrieben.
Änderungen bei und nach der Geburt
Bei der Geburt wird die Plazenta eliminiert, und es fließt kein Blut mehr durch die V. umbilicalis. Der Gasaustausch erfolgt nun über die Lungen. Die fetalen Shunts (Ductus arteriosus, Foramen ovale und Ductus venosus) sind nicht mehr notwendig; sie schließen sich im Verlauf von Stunden bis Tagen (der Ductus venosus schließt sich innerhalb von 11 Tagen nach der Geburt).
Damit sinkt der Leberblutfluss auf etwa ein Drittel, und der Blutfluss durch die V. portae steigt innerhalb von 2–10 h um das 3- bis 6-Fache an. In diesem Alter werden noch 50 % des portalvenösen Blutflusses durch den Ductus venosus an der Leber vorbeigeführt. Das restliche portalvenöse Blut wird gleichmäßig auf den rechten und den linken Leberlappen verteilt.
Daraus resultiert ein 50%iger Abfall des Sauerstoffverbrauchs der Leber bei der Geburt.
In den folgenden Tagen nimmt der Blutfluss durch den Ductus venosus kontinuierlich ab. Wahrscheinlich verstärkt ein offener Ductus venosus beim Neugeborenen Leberschäden oder die Wirkung von Stress auf die Leber.
Der gesamte Leberblutfluss ist beim Neugeborenen 2,5-mal höher als beim Erwachsenen, wobei 25–33 % aus der A. hepatica stammen. Im Gegensatz zum Erwachsenen kann der Blutfluss durch die Leber bei Fetus und Neugeborenem jedoch nicht konstant gehalten werden.
Beim Erwachsenen fungiert die Leber als Blutreserve für die systemische Zirkulation.
Etwa 25–30 % des Lebervolumens bestehen aus Blut, entsprechend etwa 10–15 % des gesamten Blutvolumens des Körpers. Etwa 50–60 % des Blutvolumens der Leber können innerhalb von 90 s durch Stimulierung des Sympathikus aus der Leber ausgestoßen werden, ohne die Leberfunktion zu verschlechtern. Ein Anstieg des zentralen Venendrucks führt bis zur Verdopplung des Leberblutvolumens. Beim Erwachsenen wird der Leberblutfluss durch den Gefäßwiderstand der A. hepatica und der intrahepatischen Äste der V. portae sowie durch den Portalvenenfluss kontrolliert.
Die Entwicklung der in ▶ Abschn. 13.1 geschilderten dreidimensionalen Leberstruktur durch die gleichzeitig stattfindende Differenzierung von Hepatoblasten zu reifen Hepatozyten, die Verbindung mit dem wachsenden Gefäßsystem sowie die Bildung des intrahepatischen Gallenwegsystems und dessen Beziehung zum umgebenden Mesenchym werden durch ein genetisch festgelegtes Entwicklungsprogramm gesteuert, das erst in Grundzügen aus Tierversuchen bekannt ist.
Energiestoffwechsel
Die Leber benötigt für ihre zahlreichen Stoffwechselprozesse eine ständige Energieproduktion. Zudem ist sie vor allem durch die Bereitstellung von Glukose aus Glykogenolyse und Glukoneogenese sowie durch die Regulierung des Aminosäuren- und Fettstoffwechsels für den Energiestoffwechsel des Organismus verantwortlich.
Die Entwicklung der Leberfunktion sowie die Erlangung der vollen Kapazität für Stoffwechsel, Biotransformation und Transportprozesse folgen verschiedenen Entwicklungsmustern:
hohe Aktivität beim Fetus und Abnahme der Aktivität nach der Geburt (z. B. Thymidinkinase, Ornithindecarboxylase),
Expression während der frühen fetalen Entwicklung und weiterer Anstieg der Aktivität nach der Geburt (z. B. Fruktose-1,6-Diphosphatase, Aspartataminotransferase),
perinatale Expression und weiterer Aktivitätsanstieg postnatal (z. B. Phosphoenolpyruvatcarboxykinase, Uridin-5-Diphosphat-Glukuronyltransferase),
Expression erst nach der Geburt und maximale Aktivität während des Abstillens (z. B. Alaninaminotransferase, Alkoholdehydrogenase).
Besonderheiten des Leberstoffwechsels in der sich entwickelnden Leber
Glykogenakkumulation in der fetalen Leber (bis zum 3-fachen Wert des Erwachsenen zum Zeitpunkt der Geburt)
Geringe Glukoneogenese der fetalen Leber
Geringer Glukoseverbrauch der fetalen Leber
Aminosäuren als bedeutende Energiequelle der fetalen Leber
Hohe Fettsäuresynthesekapazität der fetalen Leber
Fettsäureoxidation als Quelle für hepatische Glukoneogenese in der fetalen Leber
Anstieg der Aktivitäten von Thyroxin und Trijodthyronin in der 9.–10. Schwangerschaftswoche
Anstieg der Plasmakonzentration von Glukagon bei der Geburt
Rascher Anstieg der hepatischen Ketogenese nach der Geburt
Rasche Induktion der Fettsäureoxidation während der ersten Lebenstage
-
Verringerte Fähigkeit der neonatalen Leber für Metabolisierung, Entgiftung und Ausscheidung vieler Medikamente:
- Frühzeitige Expression vieler Zytochrom-P450-Enzyme bei Embryo und Fetus, z. B. CYP3A7 (Steroidstoffwechsel)
- Verzögerte Expression anderer Zytochrom-P450-Enzyme, z. B. CYP1A2 (Medikamentenstoffwechsel, z. B. Koffeinstoffwechsel)
- Geringe Aktivität vieler Phase-2-Enzyme bei Fetus und Neugeborenem, z. B. Uridin-5-Diphosphat-Glukuronyltransferase
Anstieg der Konzentrationen von Kortison und Thyroxin während des Abstillens
Die Veränderungen der Enzymkonzentrationen während der Entwicklung werden wahrscheinlich von der sequenziellen Änderung der Aktivität zirkulierender Hormone beeinflusst.
Die strukturelle und funktionelle Entwicklung der Leber beeinflusst auch Absorption, Verteilung, Ausscheidung und Metabolisierung von Medikamenten und anderen Xenobiotika, sowohl der Phase-1- (Oxidation–Reduktion, Hydrolyse) als auch der Phase-2-Reaktionen (Konjugation mit Sulfat, Acetat, Glukuronsäure, Glycin und Glutathion).
Kohlenhydratstoffwechsel
Überschüssige Kohlenhydrate werden in der Leber als Glykogen gespeichert. Von dort werden sie in Phasen des Hungers rasch wieder durch Hydrolyse freigesetzt. Unmittelbar nach der Geburt ist das Neugeborene von der hepatischen Glykogenolyse abhängig, jedoch bereits zur Glukoneogenese fähig. Bevor das Neugeborene Muttermilch bekommt, entsteht ein Mangel an Glukose. Ketonkörper stehen aufgrund einer verzögerten Ketogenese nicht zur Verfügung, daher wird das Neugeborene durch andere Stoffwechselprodukte wie Laktat als Energiequelle versorgt. Nicht nur die Leber, sondern auch Lunge, Herz und Gehirn verwenden Laktat für Energieproduktion und Lipogenese.
Die Glykogensynthese beginnt beim Fetus um die 9. Schwangerschaftswoche. Die Glykogenspeicher nehmen unmittelbar vor der Geburt stark zu, sie sind 2- bis 3-mal höher als in der Erwachsenenleber (40–60 mg/g Leber). Der überwiegende Anteil dieser Glykogenspeicher wird unmittelbar postnatal vom Neugeborenen wieder verbraucht. Um die 2. postnatale Woche kommt es wieder zu einer Zunahme der Glykogenspeicher, so dass sie bei normalen, reifgeborenen Säuglingen um die 3. Lebenswoche die Konzentration Erwachsener erreichen. In dieser Phase kann der Blutzuckerspiegel des Neugeborenen etwa 10–12 h lang durch Glykogenolyse konstant gehalten werden; der Leberglykogengehalt wird dabei bis auf 12 mg/g Leber reduziert. Der Beginn der Glykogenolyse wird wahrscheinlich durch einen Anstieg des Glukagon- und einen Abfall des Insulinspiegels im Plasma nach der Geburt stimuliert.
Glukoneogenese (Synthese von Glukose aus Laktat, Aminosäuren und anderen kleinen Molekülen) ist in der fetalen Leber kaum nachweisbar.
Erst nach der Geburt kommt es zu einem signifikanten Anstieg des für die Glukoneogenese limitierenden Enzyms (Phosphoenolpyruvatcarboxykinase).
Aminosäurenstoffwechsel
Aminosäuren sind eine wichtige Energiequelle für den Fetus, entsprechend der Größenordnung von Glukose. Ungefähr 40 % des fetalen Energiebedarfs und etwa ein Drittel des fetalen Kohlenstoffbedarfs stammen aus Aminosäuren. Die hohe Konzentration freier Aminosäuren in der fetalen Leber ist für die Regulation des Leberwachstums von Bedeutung. Die Harnstoffsynthese ist wahrscheinlich bereits in der Mitte der Schwangerschaft voll ausgebildet.
Die Leber ist der Hauptort für den Abbau von Ammoniak (aus peripheren Geweben wie Muskulatur). Zum Zeitpunkt der Geburt sind (mit 2 Ausnahmen) die meisten Enzyme vorhanden, welche den Aminosäurenstoffwechsel regeln.
Der relative Mangel an p-OH-Phenylpyruvatoxidase könnte die Ursache der transienten neonatalen Hypertyrosinämie sein, die bei Frühgeborenen häufiger vorkommt. Ebenso könnte Cystein für den Fetus und das Neugeborene eine essenzielle Aminosäure darstellen, da es erst nach der Geburt zu einem raschen Anstieg der Cystathionaseaktivität in der Leber kommt.
Die Glutaminsynthese ist von der Mitte der Schwangerschaft bis zur Geburt in allen fetalen Hepatozyten möglich. Nach der Geburt kommt es zur vorwiegenden Expression der Glutaminsynthetase in den perivenösen Hepatozyten. Diese zonale Verteilung persisiert bis in das Erwachsenenalter und ändert sich auch bei Leberzellschaden oder -regeneration nicht.
Fettstoffwechsel
Die Fettsäureoxidation ist eine größere Energiequelle, die sowohl die Glykogenolyse als auch die Glukoneogenese verstärkt.
Der Fetus erhält Fettsäuren durch De-novo-Synthese, passive Diffusion unveresterter Fettsäuren und einen selektiven maternofetalen Transport durch die Plazenta, besonders der physiologisch wichtigen langkettigen, mehrfach ungesättigten Fettsäuren. Die geringen durch die Plazenta aufgenommenen Mengen freier Fettsäuren werden in der Leber und im Fettgewebe gespeichert. Die fetale Fettsäuresynthese in der Leber zeigt einen Gipfel in der Mitte der Schwangerschaft. Mütterliche Ketone und Glukose können Vorstufen für die Fettsäuresynthese in der fetalen Leber sein. Das in der fetalen Leber gespeicherte Fett wird nach der Geburt mobilisiert und lokal verbraucht. Die Oxidation des Fetts führt zur Produktion von Adenosintriphosphat (ATP), aus dem Energie und Ketonkörper für die peripheren Gewebe freigesetzt werden. Während der ersten Lebenstage kommt es zu einer raschen Ausreifung der Fettsäureoxidation. Die Leber ist die Hauptquelle für die Synthese von Ketonkörpern, die von anderen Geweben verwendet werden. Während der ersten 24 h nach der Geburt steigen die Konzentrationen der Ketonkörper (z. B. 3-OH-Butyrat und Aceton) im Blut an. Die postnatale Entwicklung der Oxidation langkettiger Fettsäuren und der Ketogenese wird durch Pankreashormone reguliert.
Der rasche Anstieg der hepatischen Fettsäureoxidation und die Aufnahme von Milch (fettreich, kohlenhydratarm) unterstützen die aktive Glukoneogenese, um den Blutzuckerspiegel konstant zu halten. Nach der Geburt kommt es zu einem deutlichen Konzentrationsanstieg der freien Fettsäuren im Plasma. Kurzkettige Fettsäuren stellen lokale Wachstumsfaktoren für den Darm dar, mittelkettige und gesättigte langkettige Fettsäuren bedeutende Energiequellen. Überlangkettige Fettsäuren dienen dem Aufbau von Membranstrukturen. Bei Neugeborenen stellen freie Fettsäuren pro Tag 42 kJ/kg KG an Energie zur Verfügung.
Ontogenese und Stoffwechsel der Gallensäuren
Physiologie der Gallebildung
Galle wird in den Gallekanalikuli gebildet, in den Gallengängen modifiziert, in der Gallenblase konzentriert und schließlich im Dünndarm mit Nahrung vermischt. Dort werden organische Säuren, vor allem Gallensäuren, rückresorbiert und gelangen über die Zirkulation zur Leber, wo sie sehr effizient in die Hepatozyten aufgenommen und im weiteren Verlauf wieder im intrahepatischen Kreislauf verwendet werden. Gallensäuren und Gallensalze sind die bestimmende Kraft bei der Gallebildung.
Galle ist eine komplexe Lösung von Cholesterin (0,9–3,2 g/l), Phospholipiden (vor allem Phosphatidylcholin, 1,4–8 g/l), konjugierten Gallensäuren bzw. -salzen (3–45 mmol/l), Eiweiß (0,3–3 g/l) und Elektrolyten.
Die Gallensäuren regen die Sekretion von Lipiden an und stabilisieren sie in der Galle. Die Osmolarität der Galle ähnelt derjenigen des Plasmas, da Cholesterin, Phospholipide und Gallensäuren zu gemischten Mizellen aggregieren. Das Eiweiß enthält auch Immunglobulin A (IgA) in einer Konzentration, die 10-fach über der des Plasmas liegt. Die Elektrolytkonzentrationen entsprechen den Plasmakonzentrationen, mit Ausnahme einer wesentlich höheren Konzentration von Bicarbonat.
Die Gallebildung umfasst 4 Hauptschritte:
Aufnahme oder Synthese von Bestandteilen durch die Leber- bzw. Gallengangzellen,
Änderung dieser Substanzen auf dem Weg zur Galle durch das endoplasmatische Retikulum und den Golgi-Apparat,
vesikulärer Transport von Gallebestandteilen (die Vesikel werden an der basolateralen Membran innerhalb der Hepatozyten gebildet und gelangen schließlich in die Gallekanalikuli)
Ausscheidung von Wasser sowie organischen und anorganischen Verbindungen in die Gallekanalikuli.
Zusätzlich erfolgt eine parazelluläre Sekretion von Elektrolyten, Wasser und unlöslichen Bestandteilen über den „junction complex“.
Die Zusammensetzung der Galle, die bei Menschen in das Lumen der Kanalikuli sezerniert wird, ist nicht bekannt. Die Menge der Galle, die innerhalb von 24 h (gesammelt durch T-Tuben) produziert wird, liegt bei 500–600 ml. Bei Säuglingen und Kindern ist diese Menge wahrscheinlich proportional geringer. Die Elektrolytkonzentration der Galle ist bei Tieren und Menschen ähnlich (wahrscheinlich auch beim sich entwickelnden menschlichen Organismus). Es existieren deutliche Speziesunterschiede bezüglich anderer Gallebestandteile und des Galleflusses. Die Konzentration der Gallensalze ist während der fetalen und neonatalen Periode niedrig und steigt mit der Reifung der Gallensäurenbiosynthese, der steigenden Transportkapazität innerhalb des Dünndarms und des hepatischen Anteils der enterohepatischen Zirkulation an, und zwar von <0,05 mM bei menschlicher fetaler Galle vor der 17. Schwangerschaftswoche bis zur 20-fachen Konzentration zwischen der 16. und 18. Schwangerschaftswoche (0,1 mM). Bei der Geburt eines reifen Säuglings sind die Gallensäurenkonzentrationen verglichen mit älteren Kindern und Erwachsenen (3–45 mM) noch immer gering (1–2 mM). Auch die Gallenblase des Fetus und des Neugeborenen konzentriert Gallensalze geringer als bei Erwachsenen. Bei menschlichen Neugeborenen beträgt die Gallensäurenkonzentration im Dünndarm nach Stimulation durch Mahlzeiten 1–2 mmol und zeigt keine Änderung während des Tages. Beim reifen Neugeborenen beträgt die Größe des Cholsäurepools nur 50 % des Pools bei Erwachsenen. Die Gesamtgröße des Gallensäurenpools ist bei Frühgeborenen noch niedriger und korreliert direkt mit den extrem niedrigen postprandialen intraluminalen Gallensäurenkonzentrationen im Duodenum.
Gallensäurenproduktion, Gallesekretion und Gallensäurenausscheidung erfolgen in den frühen Entwicklungsstadien des Lebens nahe einem Maximum.
Während des ersten Lebensjahres kommt es zu einem langsamen Anstieg der Gallensäurensynthese, der Gallensäurepoolgröße, der intraluminalen Gallensäurenkonzentration und auch der Gallesekretion.
Die Entwicklung der Serumgallensäurenkonzentrationen ist ein Maß für die Effizienz des hepatischen Transports: Die Aufnahme von Gallensäuren aus dem Blut in die Leber ist extrem hoch (90 % bei einmaliger Passage). Der Gallensäurenspiegel im Serum wird daher beim gesunden Menschen durch die geringe restliche Menge bestimmt.
Beim menschlichen Fetus sind die Gallensäurenkonzentrationen in der Nabelarterie niedriger als in der Nabelvene. Nach der Geburt steigen die Konzentrationen der Konjugate der primären Gallensäuren ( Cholsäure und Chenodesoxycholsäure) stetig an und erreichen während der ersten Lebenswoche höhere Konzentrationen als bei gesunden älteren Kindern und Erwachsenen, ähnlich wie bei Patienten mit cholestatischen Lebererkrankungen. Dies dauert bis zum Alter von 6–8 Wochen an; erst ab dem 6. Lebensmonat fällt der Gallensäurenspiegel allmählich auf Erwachsenenwerte ab. Die Konzentration der Gallensäuren wird auch durch passive und aktive Rückresorption aus dem Darm beeinflusst. Auch die Extraktion von Gallensäuren aus dem Blut nach einer Testmahlzeit ist geringer als später; als Folge erreichen höhere Gallensäurenkonzentrationen die systemische Zirkulation.
Die Erhöhung der Serumgallensäurenkonzentration während des ersten Lebensjahres wird als „physiologische Cholestase des Säuglings“ bezeichnet.
Die klinische Bedeutung einer unreifen Ausscheidungsfunktion der Leber ist bekannt (z. B. bei Sepsis, parenteraler Ernährung und angeborenen Stoffwechselerkrankungen).
Die Gallebildung im Bereich der Kanalikuli ist ein Resultat des aktiven Transports gelöster Substanzen in die Kanalikuli, gefolgt vom passiven Transport von Wasser. Diese Transportprozesse sind energieabhängig.
Die kanalikuläre Galle kann in 2 Komponenten unterteilt werden, eine gallensäurenabhängige und eine gallensäurenunabhängige Fraktion (abhängig vom aktiven Transport anorganischer Elektrolyte und anderer gelöster Substanzen in die Galle).
Die gallensäurenabhängige Fraktion beginnt mit der carrier-abhängigen Aufnahme von Gallensäuren aus dem Portalblut durch die sinusoidale Membran der Leberzellen. Die Exkretion über die Kanalikulusmembran ist der limitierende Faktor für den hepatischen Transport der Gallensäuren; sie korreliert bei den meisten Spezies mit der kanalikulären Gallebildung. Der choleretische Effekt ist von der chemischen Zusammensetzung abhängig; er verringert sich durch Konjugation (mit Glycin oder Taurin). Auch die negative Ladung der Seitenketten der Gallensäuren bestimmt das Ausmaß des Gallensäurentransports. Monohydroxygallensäuren wie Lithocholsäure sind cholestatisch wirksam, während Ursodesoxycholsäure durch die Stimulation der Bicarbonatsekretion in die Galle choleretisch wirkt. Die Gallensäuren beeinflussen auch direkt die Aktivität der membrangebundenen Transporter (wie die Na+-K+-ATPase) und können die Membranfunktion wie auch die Permeabilität verändern.
Die Bestimmung der gallensäurenunabhängigen Fraktion des Galleflusses ist schwierig, da ein kleiner Prozentsatz von Gallensäuren in der Leberzelle neu gebildet wird. Die Menge des gallensäurenunabhängigen Galleflusses dürfte mit der Menge des gallensäurenabhängigen Flusses korrelieren.
Gallensäuren sind der wichtigste Faktor für die Lipidsekretion (Phospholipide und Cholesterol).
Die Lipide werden in der Galle durch die Bildung von gemischten Mizellen mit Gallensäuren in Lösung gehalten. In diese Mizellen können auch andere Bestandteile der Galle wie Bilirubin inkorporiert sein.
Im Bereich der Kanalikuli werden Volumen und Zusammensetzung der Galle durch Rückresorption und Sekretion von Wasser und Elektrolyten geändert. Hier bestehen große Speziesunterschiede. Hormone wie Sekretin und vasoaktives intestinales Peptid (VIP) führen zu einer bicarbonatreichen Cholerese. Im Gegensatz dazu hemmt Somatostatin den Gallefluss, entweder durch eine gesteigerte duktuläre Rückresorption oder durch Behinderung der duktalen Sekretion einer bicarbonatreichen Elektrolytlösung. Das Gallengangepithel besitzt Mechanismen zum Transport von Nahrungsbestandteilen und zur Regulierung des intrazellulären pH-Wertes, zudem kann es Wasser und Elektrolyte rückresorbieren. Durch die enge Nachbarschaft zur A. hepatica und zum peribiliären Plexus ist ein Austausch von Substanzen zwischen Blut und Galle möglich.
Schließlich wird die Galle durch die Speicherfunktion der Gallenblase modifiziert, welche die Galle während Fastenperioden bis zur 10-fachen Konzentration eindicken kann. Durch Cholezystokinin kann die Gallenblase bei Bedarf rasch entleert werden. Die Gallenblasengalle ist isoton zum Plasma; sie enthält höhere Konzentrationen von Kalium, Natrium, Kalzium, Gallensäuren und niedrigere Konzentrationen von Chlorid und Bicarbonat als die Lebergalle. Die Gallebildung wird auch durch Proteinkinase C, zyklisches Adenosinmonophosphat (cAMP) sowie Hormone wie Glukagon, Sekretin und Histamin beeinflusst.
Physiologische Funktionen der Gallensäuren
-
Leber und Gallenwege:
- Cholerese (gallensäurenabhängiger Flow)
- Regulierung der Phospholipid- und Cholesterinsynthese
- Regulierung des Transports und der Sekretion von Phospholipiden und Cholesterin in die Kanalikuli
- Lösung und Transport von Cholesterin in der Galle
- Verminderung der Sättigung der Galle
- Präzipitation von Kalzium
-
Darm:
- Mizellenbildung: Fettverdauung, Resorption von Fett und fettlöslichen Substanzen
- Regulierung der Resorption von Wasser und Elektrolyten aus dem Dickdarm
- Regulierung der gastrointestinalen Motilität: reinigende Wirkung, Effekt auf Neurotransmitter, Steigerung der intrazellulären Kalziumkonzentration
- Beeinflussung der Sekretinfreisetzung
Ontogenese der Gallebildung
Die Galleproduktion beginnt am Anfang des 4. Schwangerschaftsmonats. Von diesem Zeitpunkt an enthält das Gallenwegsystem ständig Galle, die in den Darm ausgeschieden wird und seinem Inhalt eine dunkelgrüne Färbung verleiht (Mekonium). Aus Tierstudien ist bekannt, dass Gallebildung und -ausscheidung beim Fetus unreif sind: Bis zu 40 % werden über die Plazenta entfernt. Bei neugeborenen Tieren ist die Gallesekretion maximal stimuliert. Auch die Kontraktion des Sphinkter des Ductus choledochus kann durch Mahlzeiten erst im Alter von 4 Wochen (ähnlich wie bei Erwachsenen) stimuliert werden. Man nimmt daher an, dass diese verminderte Funktion zur physiologischen Cholestase und zu den verringerten intraduodenalen Gallensäurenkonzentrationen des Neugeborenen beiträgt. Es gibt wenige Informationen über die Gallenblasenfunktion während der Entwicklung.
Ontogenese der zellulären Mechanismen der Gallebildung
Ähnlich wie bei erwachsenen Tieren ist die Gallebildung während der Entwicklung von der funktionellen Kapazität der basolateralen Na+-K+-ATPase sowie von der Entwicklung spezifischer Carrier für Gallensäuren und andere Ionen an der Plasmamembran abhängig (◘ Abb. 13.1). Die Transportsysteme für die unterschiedlichen Substrate an der basolateralen und kanalikulären Membran werden unabhängig voneinander reguliert. Die Zunahme der Gallensäurensynthese in den Zellen erfolgt parallel mit der Reifung anderer Teile der enterohepatischen Zirkulation. Auch die Größe des Gallensäurenpools kann die Produktion der Gallensäure-Carrier während der Entwicklung beeinflussen. Dies kann durch orale Zufuhr exogener Gallensäuren oder Hormone erfolgen.


Gallensäuren werden wahrscheinlich für die morphologische Differenzierung der Kanalikulusmembran benötigt.
Bildung der Gallensäuren
Die Gallensäuren werden in der Leber aus Cholesterin synthetisiert; dabei werden sie durch verschiedene steroidbindende Proteine in die einzelnen Teile der Zelle weitergeleitet. Chemisch (◘ Abb. 13.2) bestehen sie aus einem Zyklopentanperhydrophenanthrenring und besitzen eine Seitenkette aus 5 Kohlenstoffatomen mit einer terminalen Carboxylsäuregruppe.


Die beiden primären Gallensäuren Cholsäure und Chenodesoxycholsäure werden aus Cholesterin zunächst durch Modifikationen des ABCD-Ringkerns und danach durch Oxidation der Seitenkette modifiziert (◘ Abb. 13.3). Unter normalen Bedingungen erfolgen die chemischen Reaktionen nicht immer in dieser Reihenfolge. Abweichungen finden sich besonders während der Entwicklung (physiologische Cholestase) und bei Erkrankungen, welche die Funktion des enterohepatischen Kreislaufs beeinträchtigen.
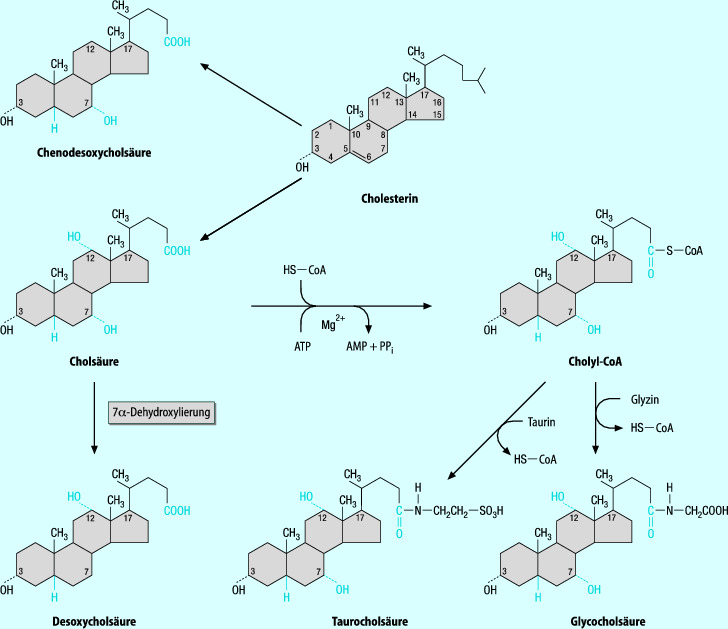

Als erster Schritt wird eine Hydroxylgruppe an Position C7 des Cholesterinkerns eingeführt, katalysiert durch eine mikrosomale Cholesterol-7α-Hydroxylase (ein leberspezifisches Zytochrom-P450-Enzym). Die Synthese dieses Schrittes wird durch primäre Gallensäuren herabreguliert. Gallensäuren mit einer 7β-Hydroxylgruppe wie Ursodesoxycholsäure können diese Synthese steigern. Dies ist für die Behandlung von Erkrankungen wie angeborene Stoffwechselstörungen der Gallensäurensynthese von Bedeutung. Cholesterol-7α-Hydroxylase zeigt einen Tagesrhythmus, synchron mit der Aktivität der HMG-Koenzym-A-Reduktase.
Danach wird der Steroidkern durch Oxidoreduktion und Hydroxylierung modifiziert, woraus entweder die Cholsäure oder die Chenodesoxycholsäure entsteht. Durch 12α-Hydroxylierung (leberspezifische mikrosomale Zytochrom-P450-12α-Hydroxlase) wird die Umwandlung in Cholsäure eingeleitet. Die Aktivität der 12α-Hydroxylase bestimmt das Ausmaß der Synthese von Cholsäure und Chenodesoxycholsäure. Danach kommt es zur Reduktion in 5β-Stellung und Hydrogenierung in 3α-Stellung, bevor die Seitenketten oxidiert werden, woraus Cholsäure und Chenodesoxycholsäure entstehen. Der erste Schritt der Seitenkettenoxidierung ist die Hydroxylierung des C27-Atoms (durch ein mitochondriales Zytochrom-P450-Enzym).
Alternative Stoffwechselwege
Alternative Stoffwechselwege haben geringere Bedeutung:
25-Hydroxylierung zu Cholsäure: Synthese von 5 % des Gesamtgallensäurenspiegels bei gesunden Erwachsenen, möglicherweise bei angeborenen Stoffwechselerkrankungen wie der zerebrotendinösen Xanthomatose wichtig;
Bildung von 27-Hydroxycholesterol (durch die Oxidierung der Seitenkette), danach Oxidierung zu Lithochol- und Chenodesoxycholsäure; unter Normalbedingungen von geringer Bedeutung, unter cholestatischen Bedingungen und während der Entwicklung für die Produktion von Lithocholsäure von größerer Bedeutung;
27-Hydroxylierung von Cholesterol und weitere Umwandlung durch eine mitochondriale 27-Hydroxycholesterol-7α-Hydroxylase in Cholsäure und Chenodesoxycholsäure; möglicherweise werden bis zu 50 % der Chenodesoxycholsäure über saure Zwischenprodukte ohne die Cholesterol-7α-Hydroxylase synthetisiert;
27-Hydroxylierung der 3-Oxo-Δ-4-Sterol-Zwischenprodukte (z. B. bei der zerebrotendinösen Xanthomatose).
Konjugation der Gallensäuren
Cholsäure und Chenodesoxycholsäure werden mit den Aminosäuren Glycin und Taurin konjugiert (in Peroxisomen und Zytosol), anschließend in die kanalikuläre Galle ausgeschieden und in der Gallenblasengalle gespeichert. Bei Menschen überwiegt bei Erwachsenen die Konjugierung mit Glycin diejenige mit Taurin (Verhältnis von 3,1 : 1), bei Neugeborenen und Säuglingen sind >80 % der Gallensäuren an Taurin konjugiert, abhängig von der hepatischen Speicherung von Taurin. Zusätzlich werden andere Konjugate gebildet: Sulfate, Glukuronidester und -äther, Glukoside, N-Acetylglukosaminide und Konjugate mit Medikamenten. Diese Konjugate sind im Harn nachweisbar. Durch die Konjugierung werden die physikochemischen Eigenschaften der Gallensäuren deutlich geändert (z. B. die Polarität des Moleküls), die Ausscheidung durch die Nieren erleichtert und das Potenzial der hydrophoben unkonjugierten Gallensäuren zur Membranschädigung reduziert.
Bei cholestatischen Lebererkrankungen steigen die Konzentrationen der Konjugate in biologischen Flüssigkeiten an.
Gallensäurenkonjugierende Enzyme wurden auch in den Nieren nachgewiesen: 80 % der Gallensäuren im Harn werden als Sulfate ausgeschieden; bei Cholestase ist die Ausscheidung erhöht. Die Konjugation von Gallensäuren mit Schwefelsäure erfolgt meist an C3-Position (manchmal auch an Position C7) und wird durch eine Gallensäuresulfotransferase katalysiert. In der fetalen Galle findet sich nur ein kleiner Prozentsatz von Gallensäuresulfaten, ebenso beim Neugeborenen.
Die Glukuronierung der verschiedenen Gallensäuren erfolgt durch Isoenzyme, die für die einzelnen Gallensäuren spezifisch sind. Glukoside und N-Acetylglukosaminide der Gallensäuren werden in quantitativ ähnlichen Mengen mit dem Harn ausgeschieden wie Glukuronidkonjugate.
Sekundäre Gallensäuren
Die in den Darm ausgeschiedenen primären Gallensäuren werden durch bakterielle Enzyme metabolisiert und die dabei entstehenden sekundären Gallensäuren in der Hauptsache mit dem Stuhl ausgeschieden. Die wichtigsten Reaktionen sind eine Dekonjugierung und eine 7α-Dehydroxylierung der Gallensäuren. Es finden aber auch eine Oxidoreduktion und eine Epimerisierung an verschiedenen Stellen des Gallensäurekerns statt.
Im proximalen Jejunum und im mittleren Dünndarm finden sich hohe Prozentsätze sekundärer Gallensäuren. Durch die 7α-Dehydroxylierung von Cholsäure und Chenodesoxycholsäure entstehen Desoxycholsäure und Lithocholsäure, die den größten Anteil an der Gesamtmenge der Gallensäuren im Stuhl darstellen. Beide sind relativ wenig löslich und werden daher nur geringfügig rückresorbiert, können aber durch einen Feedback-Mechanismus die Gallensäurensynthese in der Leber hemmen.
Die Serumkonzentrationen der Desoxycholsäure dienen daher als Maß für die Hemmung der enterohepatischen Zirkulation bei cholestatischen Lebererkrankungen.
Gallensäurensynthese und -stoffwechsel während der Fetalzeit und beim Neugeborenen
Unser Wissen über die Ontogenese der Gallensäurensynthese und des Gallensäurenstoffwechsels stammt vorwiegend von Tieren. Die Gallensäurenproduktion beginnt sehr früh im Laufe der Entwicklung und steigt bei Ratten bis zum 5. Lebenstag um das 40-Fache an. Beim Menschen können die primären Gallensäuren bereits in der 10. Schwangerschaftswoche nachgewiesen werden, allerdings überwiegen beim Fetus die Chenodesoxycholsäure und die Konjugierung mit Taurin. Im ersten Schwangerschaftsdrittel sind die Gallensäurenkonzentrationen niedrig, korrelieren mit der geringen Größe des Gallensäurenpools und steigen im Laufe der Schwangerschaft laufend an. Ein Überwiegen von Chenodesoxycholsäure (im Verhältnis 3 : 1 zu Cholsäure) und niedrige Konzentrationen von Gallensäuren zeigen sich auch in der Amnionflüssigkeit. Bei Frühgeburten in der 32. Gestationswoche beträgt der Gesamtgallensäurenpool nur ein Sechstel des Pools von Erwachsenen. Der Pool der primären Gallensäuren vergrößert sich bis zum 40. Lebenstag auf das Doppelte. Bei reif geborenen Kindern kommt es während des gesamten ersten Lebensjahres zu einem langsamen, stetigen Anstieg der Größe des Gallensäurenpools. Die diese Poolgröße steuernden Faktoren sind noch nicht ausreichend bekannt.
Auffällig ist der große Anteil „atypischer“ Gallensäuren (die nicht in der Erwachsenengalle gefunden werden). Diese sind für die Entwicklungsphase des Leberstoffwechsels typisch. Die Zusammensetzung des Gallensäurenpools spiegelt in dieser Phase sehr genau die Verhältnisse bei Erwachsenen mit schweren cholestatischen Lebererkrankungen wider, d. h. in der erkrankten Leber erfolgt ein Rückschritt auf diese ersten Stoffwechselwege.
Der wichtigste Unterschied ist die Fähigkeit zur Hydroxylierung (zu Trihydroxygallensäuren), die im Laufe des ersten Lebensjahres abnimmt. Durch die Hydroxylierung werden die Polarität der Gallensäuren vergrößert, die Ausscheidung über die Nieren erleichtert und das membranschädigende Potenzial der Gallensäuren verringert (hepatoprotektiver Mechanismus).
Beim Fetus werden 85 % der Gallensäuren mit Taurin konjugiert. Dieses wird von der Plazenta selektiv transportiert und steht damit der Leber in größerem Ausmaß zur Verfügung. Die hepatische Sulfatierung ist von geringer Bedeutung. Sulfate der Monohydroxygallensäuren finden sich im Mekonium; ob diese aus Leber, Niere oder dem Darm stammen, ist unbekannt.
Es gibt nur wenige Daten über die Entwicklung der Gallensäurezusammensetzung bei Neugeborenen, Säuglingen und älteren Kindern. Parallel mit der Kolonisierung des Darms durch die bakterielle Mikroflora kommt es zum Anstieg der Konzentration sekundärer Gallensäuren. Als Antwort auf die Notwendigkeit der Fettresorption vergrößert sich der Gallensäurenpool. Die Syntheseraten primärer Gallensäuren steigen parallel mit der Ausreifung der Transportsysteme für organische Anionen der basolateralen kanalikulären und intestinalen Membran.
Gallensäurensynthese und -stoffwechsel während der Entwicklung
Serumgallensäurenspiegel bei Frühgeborenen und während des ersten Lebensjahres erhöht
Geringe Größe des Gallensäurenpools bis zur 7. Lebenswoche
Gallensäurensynthese vermindert (bis zur Geburt)
Überwiegen von Chenodesoxycholsäure beim Fetus
Hohe Cholsäure-Chenodesoxycholsäure-Ratio beim Neugeborenen
Geringere Konjugation der Gallensäuren bei Fetus und Neugeborenem, vorwiegend mit Taurin
Produktion „atypischer“ Gallensäuren (Fetus und Neugeborenes)
Gallensäurenbiotransformation (Amidierung, Sulfierung) beim Fetus, beim Neugeborenen und beim Säugling reduziert
Geänderte Verteilung des Gallensäurenpools beim Fetus (vorwiegend enterohepatisch)
Geringe Gallensäurenausscheidung aus der Leber und geringer Gallefluss beim Fetus
Geringe intraluminale Gallensäurenkonzentration beim Fetus
Geringe Bildung sekundärer Gallensäuren im Stuhl während des ersten Lebensjahres
Ineffizienter aktiver ilealer Gallensäurentransport (verminderte Funktion der Gallensäuren-Carrier) bis zum Ende der Stillzeit
Verminderte Aufnahme von Gallensäuren aus dem portalvenösen Blut in die Leberzellen (Fetus, Neugeborenes und Säugling)
Fehlender portal-zentrilobulärer Gradient für die Gallensäurenaufnahme beim Fetus
Galle
Die Zusammensetzung der Gallensäuren bei Neugeborenen unterscheidet sich deutlich von der Zusammensetzung bei Erwachsenen, besonders die relativen Anteile der Gallensäuren und ihrer Konjugate. So fehlt z. B. die Desoxycholsäure, und die Gallensäuren werden hauptsächlich mit Taurin konjugiert. Der Anteil von Cholsäure überwiegt denjenigen von Chenodesoxycholsäure (2,5-fach); die Cholsäurekonzentrationen sind bei Neugeborenen höher als bei Erwachsenen (1,6-fach). Während des ersten Lebensjahres verringert sich dieses Verhältnis, ebenso die Konjugation mit Taurin. Die Taurinkonjugation nimmt besonders stark während der ersten 7 Lebenstage ab. Es finden sich signifikant erhöhte Anteile von Metaboliten mit C25-Hydroxylierung, C3- und C7-Oxidation sowie 6α- bzw. 4β-Hydroxylierung.
Serum
Während der ersten Lebenstage steigen die Konzentrationen von Cholsäure und Chenodesoxycholsäure deutlich an und fallen ab einem Alter von 4–8 Wochen langsam zu den Erwachsenenspiegeln ab, die am Ende des ersten Lebensjahres erreicht werden (physiologische Cholestase; ▶ Abschn. 13.4.1). Die Erhöhung der Serumgallensäurenkonzentrationen ist bei Frühgeborenen noch stärker ausgeprägt. Analog zur fetalen Galle finden sich beim Fetus und beim reifen Neugeborenen 1β- und 6α-hydroxylierte Metabolite. Etwa 10 % der Gesamtgallensäuren im Serum sind in den ersten 3 Lebensmonaten nicht konjugiert (bei Erwachsenen 50 %). Die Konjugation erfolgt hauptsächlich mit Glycin und Taurin (Ratio von 1 bei der Geburt, Anstieg auf 3 innerhalb der ersten Lebenswochen und Anstieg auf 6–7 am Ende des ersten Lebensjahres).
Die Konjugierung mit Sulfaten ist im Serum des Fetus und des reifen Neugeborenen gering, nimmt aber bei neonatalen Cholestasesyndromen deutlich zu.
Harn
Unter physiologischen Bedingungen werden nur geringe Mengen von Gallensäuren mit dem Harn ausgeschieden (<20 μmol/l). Bei Cholestase steigen die Gallensäurenkonzentrationen deutlich an. Bei Neugeborenen wurden mehr als 20 unterschiedliche Gallensäuren identifiziert. Nach der Neugeborenenzeit ist der größte Anteil der Gallensäuren mit Schwefelsäure konjugiert. Zusätzlich finden sich beim Neugeborenen tetrahydroxylierte Isomere, die für cholestatische Erkrankungen typisch sind.
Amnionflüssigkeit
Mehr als 50 verschiedene Gallensäuren wurden identifiziert und quantifiziert. Viele finden sich auch in der fetalen Galle und sind repräsentativ für die hepatische Synthese in utero. Amnionflüssigkeit enthält hohe Anteile polarer tetrahydroxylierter Gallensäuren.
Mekonium
Die Bestandteile des Mekoniums stammen von Fetus, Plazenta und Amnionflüssigkeit. Es enthält etwa 225 μg Gallensäuren pro Gramm Feuchtgewicht, vorwiegend als Taurin- und Sulfatkonjugate; unkonjugierte Gallensäuren und Glycinkonjugate kommen nur in geringen Konzentrationen vor. Die hauptsächlichen Gallensäuren sind Hyocholsäure und Hyodesoxycholsäure (6α-Hydroxylierung), zusätzlich finden sich tetrahydroxylierte (kern- bzw. seitenkettenhydroxylierte) Gallensäuren, daneben auch Oxo-, Allo- und Norgallensäuren sowie kurzkettige Gallensäuren.
Stuhl
Während der ersten 7 Lebenstage gesunder Neugeborener überwiegt Cholsäure. In den ersten beiden Tagen werden Gallensäuren fast ausschließlich in konjugierter Form ausgeschieden. Ab dem 3. Lebenstag (Kolonisierung des Darms mit Bakterien) werden primäre Gallensäuren in unkonjugierter Form ausgeschieden. Die sekundären Gallensäuren, die Desoxycholsäure und die Lithocholsäure werden während des gesamten ersten Lebensjahres nur in geringen Mengen ausgeschieden. Die Gesamtausscheidung von Gallensäuren beträgt 3–6 mg/kg KG/Tag und ist von der Nahrung abhängig.
Literatur
Literatur zu Abschn. 13.1
- Amenta PS, Harrison D. Expression and potential role of the extracellular matrix in hepatic ontogenesis: a review. Microscop Res Techn. 1997;39:372–386. doi: 10.1002/(SICI)1097-0029(19971115)39:4<372::AID-JEMT7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Colon AR. Textbook of pediatric hepatology. Chicago: Year Book Med Pub; 1990. [Google Scholar]
- Crawford JM. Development of the intrahepatic biliary tree. Semin Liver Dis. 2002;22:213–226. doi: 10.1055/s-2002-34508. [DOI] [PubMed] [Google Scholar]
- Karpen SJ, Suchy FJ. Structural and functional development of the liver. In: Suchy FJ, Sokol RJ, Balistreri WF, editors. Liver disease in children. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 3–21. [Google Scholar]
- Kaufman SS. Organogenesis and histologic development of the liver. In: Polin RA, Fox WW, editors. Fetal and neonatal physiology. Philadelphia: Saunders; 1998. pp. 1433–1441. [Google Scholar]
- MacSween RNM, Desmet VJ, Roskams T, Scothorne RJ, et al. Developmental anatomy and normal structure. In: MacSween RNM, Ishak KG, Burt AD, et al., editors. Pathology of the liver. London: Churchill Livingstone; 2002. pp. 1–66. [Google Scholar]
- Malarkey DE, Johnson K, Ryan L, Boorman G, Maronpot RR. New insights into functional aspects of liver morphology. Toxicol Pathol. 2005;33:27–34. doi: 10.1080/01926230590881826. [DOI] [PubMed] [Google Scholar]
- McLin VA, Yazigi N. Developmental anatomy and physiology of the liver and bile ducts. In: Wyllie R, Hyams JS, editors. Pediatric gastrointestinal and liver disease. 3. Philadelphia: Saunders Elsevier; 2006. pp. 841–849. [Google Scholar]
- Nakanuma Y, Hoso M, Sanzen T, Sasaki M. Microstructure and development of the normal and pathologic biliary tract in humans, including blood supply. Microsc Res Tech. 1997;38:552–570. doi: 10.1002/(SICI)1097-0029(19970915)38:6<552::AID-JEMT2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Peters RL. Early development of the liver: a review. In: Fisher MM, Roy CC, editors. Pediatric liver disease. New York: Plenum Press; 1983. pp. 1–15. [Google Scholar]
- Sergi C, Adam S, Kahl P, Otto HF. The remodelling of the primitive human biliary system. Early Hum Dev. 2000;58:167–178. doi: 10.1016/S0378-3782(00)00065-7. [DOI] [PubMed] [Google Scholar]
- Shankle WR, Landing BH, Gregg J. Normal organ weights of infants and children: graphs of values by age, with confidence intervals. Pediatr Pathol. 1983;1:399–408. doi: 10.3109/15513818309025871. [DOI] [PubMed] [Google Scholar]
- Shiojiri N. The origin of the intrahepatic bile duct cells in the mouse. J Embryol Exp Morphol. 1984;79:25–39. [PubMed] [Google Scholar]
- Wuestefeld T, Zaret KS. Liver development: from endoderm to hepatocyte. In: Suchy J, Sokol RJ, Balistreri WF, editors. Liver disease in children. 3. Cambridge: Cambridge University Press; 2007. pp. 3–13. [Google Scholar]
- Zakim D, Boyer TD. Hepatology. A textbook of liver disease. Philadelphia: Saunders; 2003. [Google Scholar]
Literatur zu Abschn. 13.2
- Arias IM, Popper H, Schachter D, Shafritz DA. The liver, biology and pathobiology. New York: Raven Press; 1982. [Google Scholar]
- Fahey JT. Developmental aspects of hepatic blood flow. In: Suchy FJ, editor. Liver disease in children. St Louis: Mosby; 1994. pp. 31–38. [Google Scholar]
- Laut WW, Greenway Conceptual review of the hepatic vascular bed. Hepatology. 1987;7:952–963. doi: 10.1002/hep.1840070527. [DOI] [PubMed] [Google Scholar]
- Rudolph CD, Rudolph AM. Fetal and postnatal hepatic vasculature and blood flow. In: Polin RA, Fox WW, editors. Fetal and neonatal physiology. Philadelphia: Saunders; 1998. pp. 1442–1449. [Google Scholar]
- Taylor IM. Developmental aspects of the hepatic circulation. In: Fisher MM, Roy CC, editors. Pediatric liver disease. New York: Plenum Press; 1983. pp. 17–42. [Google Scholar]
Literatur zu Abschn. 13.3
- Battaglia FC, Thureen PJ. Nutrition of the fetus and premature infant. Nutrition. 1997;13:903–906. doi: 10.1016/S0899-9007(97)00264-5. [DOI] [PubMed] [Google Scholar]
- Battaglia FC, Thureen PJ. Nutrition of the fetus and the premature infant. Diabetes Care. 1998;21:B70–B74. [PubMed] [Google Scholar]
- Berghaus TM, Demmelmair H, Koletzko B. Fatty acid composition of lipid classes in maternal and cord plasma at birth. Eur J Pediatr. 1998;157:763–768. doi: 10.1007/s004310050931. [DOI] [PubMed] [Google Scholar]
- Greengard O. Enzymatic differentiation of human liver: comparison with the rat model. Pediatr Res. 1977;11:669–676. doi: 10.1203/00006450-197705000-00009. [DOI] [PubMed] [Google Scholar]
- Hay WW., Jr Nutrient and metabolic needs of the fetus and very small infant: a comparative approach. Biochem Soc Trans. 1998;26:75–78. doi: 10.1042/bst0260075. [DOI] [PubMed] [Google Scholar]
- Koletzko B, Demmelmair H, Socha P. Nutritional support of infants and children: supply and metabolism of lipids. Clin Gastroenterol. 1998;12:671–696. doi: 10.1016/s0950-3528(98)90003-3. [DOI] [PubMed] [Google Scholar]
- MacSween RNM, Ishak KG, Burt AD, et al. Pathology of the liver. London: Churchill Livingstone; 2002. [Google Scholar]
- Narkewicz MR. Hepatic energy metabolism in the fetus and neonate. In: Suchy FJ, editor. Liver disease in children. St Louis: Mosby; 1994. pp. 39–56. [Google Scholar]
- Suchy FJ. Functional development of the liver. In: Suchy J, Sokol RJ, Balistreri WF, editors. Liver disease in children. 3. Cambridge: Cambridge University Press; 2007. pp. 4–27. [Google Scholar]
- Uauy R, Treen M, Hoffman DR. Essential fatty acid metabolism and requirements during development. Semin Perinatol. 1989;13:118–130. [PubMed] [Google Scholar]
- Zlotkin SH, Anderson GH. The development of cystathionase activity during the first year of life. Pediatr Res. 1982;16:65–68. doi: 10.1203/00006450-198201001-00013. [DOI] [PubMed] [Google Scholar]
Literatur zu Abschn. 13.4
- Argao EA, Balistreri WF. Bile acid physiology and alterations in the enterohepatic circulation. In: Willie R, Hyams JS, editors. Pediatric gastrointestinal disease. Philadelphia: Saunders; 1993. pp. 31–45. [Google Scholar]
- Björkhem I, Boberg KM. Inborn errors in bile acid biosynthesis and storage of sterols other than cholesterol. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 1995. pp. 2073–2099. [Google Scholar]
- Bucuvalas JC. Bile acid metabolism during development. In: Polin RA, Fox WW, editors. Fetal and neonatal physiology. Philadelphia: Saunders; 1998. pp. 1450–1456. [Google Scholar]
- Burt AD, Day CP, et al. Pathophysiology of the liver. In: MacSween RNM, Ishak KG, Burt AD, et al., editors. Pathology of the liver. London: Churchill Livingstone; 2002. pp. 67–105. [Google Scholar]
- Chen HL, Liu YJ, et al. Developmental expression of canalicular transporter genes in human liver. J Hepatol. 2005;43:472–477. doi: 10.1016/j.jhep.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Chuang E, Haber BA. Bile secretion and its control in the mature and immature organism. In: Polin RA, Fox WW, editors. Fetal and neonatal physiology. Philadelphia: Saunders; 1998. pp. 1457–1471. [Google Scholar]
- Colombo C, Zuliani G, Ronchi M, Breidenstein J, Setchell KD. Biliary bile acid composition of the human fetus in early gestation. Pediatr Res. 1987;21:197–200. doi: 10.1203/00006450-198702000-00017. [DOI] [PubMed] [Google Scholar]
- Erlinger S. Physiology of bile secretion and enterohepatic circulation. In: Johnson LR, editor. Physiology of the gastrointestinal tract. New York: Raven Press; 1987. pp. 1557–1580. [Google Scholar]
- Gerok W. Physiology of bile formation. In: Lentze MJ, Reichen J, editors. Paediatric cholestasis. Novel approaches to treatment. Dordrecht: Kluwer; 1992. pp. 3–26. [Google Scholar]
- Karpen SJ. Mechanisms of bile formation and cholestasis. In: Suchy J, Sokol RJ, Balistreri WF, editors. Liver disease in children. 3. Cambridge: Cambridge University Press; 2007. pp. 28–34. [Google Scholar]
- Lecureur V, Courtois A, Payen L, et al. Expression and regulation of hepatic drug and bile acid transporters. Toxicology. 2000;153:203–219. doi: 10.1016/S0300-483X(00)00315-2. [DOI] [PubMed] [Google Scholar]
- Löffler G, Petrides PE. Biochemie und Pathobiochemie. Berlin Heidelberg New York: Springer; 1997. [Google Scholar]
- Setchell KDR, O’Connell NC. Disorders of bile acid synthesis and metabolism: a metabolic basis for liver disease. In: Suchy FJ, Sokol RJ, Balisteri WF, editors. Liver disease in children. 3. Cambridge: Cambridge University Press; 2007. p. 736. [Google Scholar]
- Suchy FJ. Functional development of the liver. In: Suchy J, Sokol RJ, Balistreri WF, editors. Liver disease in children. 3. Cambridge: Cambridge University Press; 2007. pp. 4–27. [Google Scholar]
- Trauner M, Wagner M, Fickert P, Zollner G. Molecular regulation of hepatobiliary transport systems: clinical implications for understanding and treating cholestasis. J Clin Gastroemterol. 2005;39(2):111–124. doi: 10.1097/01.mcg.0000155551.37266.26. [DOI] [PubMed] [Google Scholar]
- Vonk RJ, Kuipers F, Smit MJ, et al. Bile acid metabolism in children. In: Lentze MJ, Reichen J, et al., editors. Paediatric cholestasis. Novel approaches to treatment. Dordrecht: Kluwer; 1992. pp. 27–37. [Google Scholar]
- Wagner M, Trauner M. Transcriptional regulation of hepatobiliary transport systems in health and disease: implications for a rationale approach to the treatment of intrahepatic cholestasis. Ann Hepatol. 2005;4:77–99. [PubMed] [Google Scholar]
- Watkins JB. Developmental aspects of bile acid metabolism and hepatic function. In: Fisher MM, Roy CC, editors. Pediatric liver disease. New York: Plenum Press; 1983. pp. 43–53. [Google Scholar]
- Zakim D, Boyer TD. Hepatology. A textbook of liver disease. Philadelphia: Saunders; 1982/2003. [Google Scholar]