

Abstract
The control of viral outbreaks requires nucleic acid diagnostic tests that are sensitive, simple and fast. Here, we report a highly sensitive and specific one-pot assay for the fluorescence-based detection of RNA from pathogens. The assay, which can be performed within 30–50 min of incubation time and can reach a limit of detection of 0.1-attomolar RNA concentration, relies on a sustained isothermal reaction cascade producing an RNA aptamer that binds to a fluorogenic dye. The RNA aptamer is transcribed by the T7 RNA polymerase from the ligation product of a promoter DNA probe and a reporter DNA probe that hybridize with the target single-stranded RNA sequence via the SplintR ligase (a Chlorella virus DNA ligase). In 40 nasopharyngeal SARS-CoV-2 samples, the assay reached positive and negative predictive values of 95 and 100%, respectively. We also show that the assay can rapidly detect a range of viral and bacterial RNAs.
Subject terms: Synthetic biology, DNA probes
A one-pot enzymatic assay for the fluorescence detection of RNA accurately and rapidly detects specific viral and bacterial pathogens, as shown for SARS-CoV-2 RNA in clinical samples.
Main
Increasing global trade and travel are considered the cause of frequent emergence and rapid dissemination of infectious diseases around the world. Some life-threatening infectious diseases often have signs and symptoms similar to cold or flu-like syndromes. Early diagnosis is therefore essential to identify the diseases and provide the correct treatment. Immediate and onsite diagnostic decisions also help to prevent the spread of epidemic and pandemic infectious diseases1–3. To rapidly diagnose infectious diseases, nucleic acid-based diagnosis has emerged as an alternative to the conventional culture-based or immunoassay-based approaches due to its rapidity and specificity4–6.
To increase their sensitivity, current nucleic acid detection methods generally involve a target amplification step before the detection step. The conventional amplification method is based on PCR, which requires a thermocycler for delicate temperature modulation. As an alternative to the thermal cycling-based amplification, isothermal amplification methods are available, which rely primarily on a strand-displacing polymerase or T7 RNA polymerase at a constant temperature7. However, the complex composition of the isothermal amplification mixtures often renders these approaches incompatible with detection methods and whole diagnosis generally becomes a multi-step process8–11. The diagnostic regimen with multi-step procedures requires additional time, instruments and reagents, as well as skilled personnel to perform the diagnostic procedure. This aspect limits the broad applicability of nucleic acid diagnostics, especially in situations where rapid and simple detection is required.
Ligation-dependent nucleic acid detection is a sequence-specific method that primarily depends on the ligation of two separate probes that hybridize to adjacent sites of the target sequence12. Because of their specificity, the ligation-dependent methods are used for the detection of markers of genetic disorders13,14 and pathogens15,16, typically combined with subsequent amplification and signal generation methods. In particular, the SplintR ligase can efficiently ligate two DNA probes using a target single-stranded RNA as a splint, enabling the sequence-specific detection of RNA molecules17,18. Because the reaction components of the ligation-dependent methods are relatively simple, we hypothesized that the ligation-dependent method could be exploited to establish a one-pot RNA detection method when combined with compatible amplification and signal generation methods in a single reaction mixture.
In this study, we developed a one-pot, ligation-dependent isothermal reaction cascade that enables the rapid detection of RNAs with high sensitivity, termed sensitive splint-based one-pot isothermal RNA detection (SENSR). SENSR consists of two simple enzymatic reactions: a ligation reaction by SplintR ligase and subsequent transcription by T7 RNA polymerase. The resulting transcript forms an RNA aptamer that binds to a fluorogenic dye and produces fluorescence only when target RNA exists in a sample. SENSR was able to detect target RNA of methicillin-resistant Staphylococcus aureus (MRSA) in 30 min with a limit of detection of 0.1 aM. We further applied this method to detect various pathogens, including Vibrio vulnificus, Escherichia coli O157:H7, Middle East respiratory syndrome-related coronavirus (MERS-CoV) and influenza A viruses, by merely redesigning the hybridization regions of the probes. Finally, we demonstrated the fast development of the SENSR assay for the latest epidemic pathogen, severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2), using minimal, publicly available information. Rapid SENSR assay workflows were validated using clinical samples acquired from patients with coronavirus disease 2019 (COVID-19).
Results
Design of the one-pot isothermal reaction cascade
We designed a reaction cascade that allows the one-pot diagnostic test, in which all reaction steps for nucleic acid detection occur simultaneously in a single tube (Fig. 1). The cascade consists of four core components, including only two enzymes: a set of oligonucleotide probes, SplintR ligase, T7 RNA polymerase and a fluorogenic dye. The components were mixed in a buffer solution with ribonucleotides. Upon addition of the pathogen-derived RNA sample, the reaction steps of ligation, transcription and dye–aptamer binding enabled detection, amplification and signal production, respectively.
Fig. 1. Schematic of SENSR, a one-pot isothermal reaction cascade for the rapid detection of RNA.

The reaction is composed of four main components: a set of probes, SplintR ligase, T7 RNA polymerase and a fluorogenic dye. In the presence of target RNA, hybridization, ligation, transcription and aptamer–dye binding reactions occur sequentially in a single reaction tube at a constant temperature.
Two single-stranded DNA probes were designed to include several functional parts involved in amplification, detection and signal generation, thereby eliminating the need for human intervention during the entire diagnostic process (Fig. 1). First, the promoter probe consists of an upstream hybridization sequence (UHS) and a stem–loop T7 promoter. The UHS hybridizes to the 5′ half of a target RNA region. The stem–loop T7 was adopted from the literature19 (Supplementary Table 1) to form an active, double-stranded T7 promoter using a single-stranded oligonucleotide. The sequence of the UHS was designed by Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi) to ensure specific binding to the target RNA. Among candidate UHS sequences, we chose the one with minimal secondary structure at 37 °C predicted by NUPACK20 to maximize the hybridization between the UHS and its target region (Supplementary Table 2). The 5′ end of the promoter probe was then phosphorylated for ligation. Next, a reporter probe consists of a downstream hybridization sequence (DHS) and a template sequence for a dye-binding RNA aptamer. The DHS contains the complementary sequence to the other half of the target RNA region. Similar to the UHS, the DHS was selected to have minimal predicted secondary structure (Supplementary Table 2).
Once both UHS and DHS probes hybridize correctly to the target RNA, SplintR ligase can initiate the cascade by connecting the probes that have all features built for the one-pot diagnostic test. Subsequently, T7 RNA polymerase can synthesize the RNA aptamer using the full-length, ligated probe as a DNA template, which can be bound with the fluorogenic dye (Fig. 1). When the fluorogenic dye binds to the RNA aptamer, the planar structure of the fluorogenic dye is stabilized, emitting fluorescence as an output. In contrast, the unbound fluorogenic dye dissipates its energy in the form of molecular vibration and heat, preventing an increase in fluorescence21. Notably, the reaction scheme of SENSR inherently supports two mechanisms that could amplify the signal: (1) multiple transcription events from the full-length, ligated probe by T7 RNA polymerase; and (2) the presence of target RNA sequence on the full-length transcript, which could be utilized as an additional splint for unligated probes in the reaction mixture. Accordingly, SENSR could enable sensitive RNA detection without any pre-amplification steps.
Construction of each component reaction in SENSR
In this study, we used MRSA as a model case to validate each reaction step that constitutes SENSR. Rapid detection of MRSA is of particular interest because it offers the potential to reduce the morbidity caused by healthcare-related infections22.
First, we designed a pair of probes that target the mecA transcript of MRSA, following the probe design process described in the previous section (Supplementary Note 1 and Supplementary Tables 1 and 3), and the RNA-splinted ligation between the two probes was tested. The probes were ligated using SplintR ligase with or without the target RNA, and the reaction products were further amplified with a pair of PCR primers and analysed (see Methods). The correct size of the PCR product was obtained only when the two probes and target RNA were added together to the ligation mixture (Fig. 2a). This result indicates that our probes were successfully ligated only in the presence of the target RNA.
Fig. 2. Construction of the three component reactions of SENSR.

a, The ligation reaction. The ligation products were amplified with a pair of PCR primers (LigChk_F and LigChk_R in Supplementary Table 7) and analysed using Bioanalyzer. The ligation reaction occurred only when the promoter probe, reporter probe, SplintR ligase and target RNA were all present. A full-length probe combining the promoter and reporter probes was amplified with the same set of PCR primers and used as a size control, as indicated by the red arrow. The full scan of the Bioanalyzer image is provided in Supplementary Fig. 10a. bp, base pairs. b, The transcription reaction. The ligated mixtures were used as a DNA template to validate transcription. The transcript was obtained only in the presence of target RNA and all other components, demonstrating both target-dependent ligation and subsequent transcription. The red arrow points to the correct size of the transcript. The full scan of the Bioanalyzer image is provided in Supplementary Fig. 10b. nt, nucleotides. c, The aptamer–fluorogenic dye binding reaction. After sequential ligation and transcription reactions, the reaction mixture with the correct size of the transcript produced higher fluorescence compared with other conditions that lacked one of the necessary components. Fluorescence tests were performed as four biological replicates (two-tailed Student’s t-test; ****P < 0.0001; bars represent means ± s.d). RFU, relative fluorescence units.
We then used the ligated probe as a DNA template to test whether transcription could occur. The ligation mixture was added at a 1/10 ratio to the in vitro transcription reaction mixture with T7 RNA polymerase. Only when the target RNA was present in the ligation reaction was the full-length transcript (92 nucleotides) observed from transcription, thereby confirming both target-dependent ligation and subsequent transcription (Fig. 2b).
Finally, we confirmed that the transcript from the full-length ligated probe could produce fluorescence upon binding to the fluorogenic dye. The reaction mixture of sequential ligation and transcription reactions were purified, and an equal amount of RNA (500 ng) from each combination was incubated with the fluorogenic dye (Fig. 2c). The RNA product from the reaction mixture with the two probes and target RNA produced higher fluorescence than that of the other combinations. Therefore, we confirmed that the target RNA could be detected using a set of probes by performing each component reaction in SENSR.
Development of the one-pot isothermal reaction cascade
Since all component reactions in SENSR were validated in their respective buffers, we then sought to develop a one-pot reaction condition with a single reaction buffer at a single temperature, where all reaction steps, including probe annealing, ligation, transcription and the aptamer fluorescence reaction occur simultaneously. To accomplish this, we first investigated a wide range of temperatures (25–95 °C) for hybridization of the probes and target. Then, each mixture was subjected to the sequential ligation, transcription and fluorescence reaction described in the previous section. Remarkably, fluorescence was observed at all hybridization temperatures, including 35 and 40 °C (Supplementary Fig. 1)—the optimal temperature ranges for enzyme activities in SENSR—thereby suggesting that the entire reaction can be built up as an isothermal reaction.
Additionally, a single reaction buffer composition suitable for all reaction steps was configured to establish all reactions in one pot. Since T7 RNA polymerase reaction buffer has the most inclusive composition of the four reaction buffers (probe annealing, ligation, transcription and aptamer fluorescence reaction buffers), we used T7 RNA polymerase buffer as a basis for the optimization. Various reaction conditions were optimized, including the reaction temperature, concentrations of the various enzymes and components to enhance the fluorescence signal (Supplementary Note 2 and Supplementary Figs. 2 and 3). The optimized SENSR condition enabled the detection of target RNA in a one-pot isothermal reaction at 37 °C (Supplementary Fig. 4).
Rapid and sensitive RNA detection by SENSR
Once the one-step and one-pot isothermal reaction condition was established, we then assessed the sensitivity and turnaround time of SENSR. We evaluated the sensitivity by measuring fluorescence from one-pot reactions containing the mecA probe pair and synthetic mecA RNA in the range of 0.1 aM to 220 nM (Fig. 3a). Notably, the detection limit was as low as 0.1 aM (corresponding to six molecules per 100 µl reaction), indicating the high sensitivity of SENSR. Moreover, the linearity of the fluorescence intensity over a wide range of concentrations (coefficient of determination (R2) = 0.9932) suggests that SENSR can be used for target RNA quantification. The real-time PCR with reverse transcription (rRT–PCR) result correlated well with the SENSR result (R2 = 0.9937), corroborating the validity of SENSR (Supplementary Fig. 5a). Additionally, independent replicate experiments confirmed the high reproducibility of SENSR (Supplementary Fig. 5b and Supplementary Table 4). There are two potential explanations for the high analytical sensitivity of SENSR. First, concentrations of some components increased in the optimized SENSR condition compared with other studies that utilized SplintR ligase and fluorogenic aptamer for RNA detection17,18, improving the efficiencies of individual reaction steps (Supplementary Table 5). Second, the one-pot reaction scheme generated cooperation between ligation and transcription. The full-length transcript contains the target RNA sequence, thus providing an additional splint for unligated DNA probes. Therefore, ligation and transcription can feed each other, creating a signal amplification cycle (Fig. 1). Indeed, a coupled one-pot reaction produced higher fluorescence intensity than an uncoupled two-step reaction (Supplementary Fig. 6).
Fig. 3. Sensitivity and turnaround time of SENSR.

a, Sensitivity of SENSR. The target RNA from 220 nM to 0.1 aM was tested. The detection limit is 0.1 aM (approximately six RNA copies in a 100-μl reaction). High linearity suggests that SENSR can be used for the quantification of target RNA. b, Left: turnaround time of SENSR. To check the time required for the SENSR reaction, the incubation time of SENSR was varied. The target RNA of 0.1 aM was detected as early as 30 min. Right: fluorescence of the 30-min SENSR reaction using target RNA at varying concentrations from 0–10 aM. All of the tests were performed with four biological replicates (two-tailed Student’s t-test; ****P < 0.0001; horizontal lines represent means ± s.d).
We then measured the minimal turnaround time required to confirm the presence of the target RNA in a sample. The target RNA ranging from 0.1–10 aM was added to the SENSR reaction and the fluorescence was measured every 30 min. The fluorescence with 0.1 aM was discernible against the negative control in just 30 min (Fig. 3b). Further incubation of the reaction better distinguished the target RNA-containing reaction from the negative control reaction. Notably, the fluorescence intensity slightly increased from 30 min, even in the absence of the target RNA. A possible source of this background fluorescence might be the DNA probes (Supplementary Fig. 7). Collectively, the SENSR reaction was able to specifically detect the target RNA within 30 min with a detection limit of 0.1 aM.
Broad adaptability of SENSR for pathogen detection
With the fast and sensitive RNA detection using SENSR, we next attempted to reconfigure this method for the detection of RNA markers from various pathogens. Target RNA sequences for SENSR are specified by only two hybridization regions (UHS and DHS) of probes, making the probe design process fast and straightforward without many computational steps. This design feature, requiring only nucleotide sequences to build molecular diagnostics, allows for the easy construction of SENSR probes for any infectious diseases (Fig. 4a).
Fig. 4. Broad adaptability of SENSR.

Two pathogenic microbes and three viruses were targeted by redesigning the probe sequences. a, Schematic of SENSR with easy reconfiguration and rapid development. b,c, Detection of bacterial RNA markers for V. vulnificus (b) and E. coli O157:H7 (c). d–f, Detection of viral RNA markers for MERS-CoV (d), influenza A (e) and SARS-CoV-2 (f). All probe pairs tested showed high sensitivity and linearity to detect RNA markers. The detection limit of 0.1 aM approximately corresponds to six RNA copies in a 100-μl reaction. All tests were performed with four biological replicates (two-tailed Student’s t-test; *P < 0.05; ***P < 0.001; ****P < 0.0001; bars represent means ± s.d).
To demonstrate SENSR for various pathogens, we first targeted two pathogenic microorganisms: V. vulnificus and E. coli O157:H7. V. vulnificus is known to cause gastroenteritis, wound infection and sepsis in humans. We designed a probe pair targeting vvhA (Supplementary Table 3), a V. vulnificus-specific target encoding extracellular haemolysin with haemolytic activity and cytotoxic effect. The sensitivity of the SENSR reaction using the probe pair and in vitro-transcribed vvhA RNA was as low as 0.1 aM (Fig. 4b), and a linear correlation was observed between the concentration of RNA and the fluorescence intensity (R2 = 0.9566).
Next, we used SENSR to detect E. coli O157:H7, which causes foodborne illness. Similar to the process for V. vulnificus, we designed a probe pair for an E. coli O157:H7-specific target gene, tir (Supplementary Table 3), for SENSR reaction. Similarly, RNA concentrations as low as 0.1 aM were detected by SENSR and a high linear correlation between the concentration of RNA and the fluorescence intensity was observed (Fig. 4c; R2 = 0.9684).
The target was expanded to human-infective RNA viruses that cause fatal diseases23. First, we aimed at MERS-CoV. The mortality rate of MERS-CoV infection has been reported to be 35%24, and it can be transmitted from human to human25, which raises the need for a fast and sensitive onsite diagnostic test. The probe pairs for the MERS-CoV-specific gene upE (Supplementary Table 3) exhibited similar sensitivity and linearity to the bacterial cases (Fig. 4d). Likewise, we designed a probe pair for the influenza A virus-specific target gene HA (Supplementary Table 3). SENSR was able to detect the influenza A RNA target with similar sensitivity and linearity (Fig. 4e). Finally, we designed a probe pair for a recently emerging pathogen, SARS-CoV-2. The target sequence was selected based on the standard rRT–PCR assay for SARS-CoV-2 (ref. 26), which aimed at the RNA-dependent RNA polymerase (RdRp) gene (SARS-CoV-2-MG1; Supplementary Table 3). Again, SENSR successfully detected its target RNA at a concentration as low as 0.1 aM, which corroborates the high adaptability of this method to various RNA markers (Fig. 4f). Notably, the discrepancy in background fluorescence between SENSR assays in the absence of the target RNAs could be derived from the reporter probe sequence unique to each SENSR assay (Supplementary Fig. 7).
Taken together, we demonstrated that SENSR could be easily reconfigured to detect various RNA markers of pathogens by redesigning the probes. The probe design process is simple and requires a small amount of computation using open web-based software. All probe pairs tested showed high sensitivity and linearity for detecting RNA markers, reinforcing the robustness of the probe design process and the wide expandability of SENSR.
Direct detection of a pathogen using SENSR
Next, we employed SENSR for the detection of RNA samples derived from the live cells of a pathogen. We targeted MRSA, whose marker RNA was detected by SENSR. Methicillin-sensitive S. aureus (MSSA) containing no target messenger RNA was used as a negative control. MRSA and MSSA cells were heated to 95 °C to lyse the cells and to release RNA. The samples were then diluted and added to the SENSR reaction to investigate the specificity and sensitivity (Fig. 5a). We observed a notable difference in fluorescence intensity between MRSA and MSSA (Fig. 5b). The RNA sample from only two colony-forming units (c.f.u.) per 100 μl reaction of MRSA was clearly detected by SENSR, thereby indicating its high sensitivity and specificity, even with samples of the living pathogen. Finally, the performance of SENSR was further validated using samples prepared in human serum (Fig. 5c). The sensitivity and specificity of SENSR were unaffected by the presence of human serum (Fig. 5d), indicating the suitability of SENSR in practical applications.
Fig. 5. Live cell and proxy clinical sample detection by SENSR.

a, Direct detection of bacterial cells. Thermal cell lysates of MRSA and MSSA were subjected to the SENSR reaction. b, Clear distinction in the fluorescence intensity between MRSA and MSSA samples. The detection limit of SENSR was as low as 2 c.f.u. per 100-µl reaction. c, Detection of bacterial cells diluted in human serum as a proxy clinical sample. Human serum containing bacteria was thermally lysed and subjected to the SENSR reaction. d, An obvious distinction in the fluorescence intensity between human serum containing MRSA and that containing MSSA was observed. The detection limit of SENSR was as low as 2 c.f.u. per 100-µl reaction. All SENSR reactions were run for 2 h. All tests were performed as four biological replicates (two-tailed Student’s t-test; NS, not significant (P > 0.5); **P < 0.01; ****P < 0.0001; bars represent means ± s.d).
Dual-target detection using orthogonal SENSR probes
We further expanded the capability of SENSR to enable the simultaneous detection of two target RNAs in a single reaction by leveraging the simple probe design. The detection of multiple biomarkers is frequently needed to make a better decision by reducing false positive and false negative results. Based on the high specificity of SENSR probes and the availability of light-up RNA aptamers with distinct spectral properties27, we hypothesized that we could design two sets of SENSR probes that operate orthogonally in a single reaction to detect their respective target RNAs.
First, we developed an orthogonal reporter probe for influenza A virus. Since the MRSA infection causes flu-like symptoms, discrimination of this pathogen from the common influenza A virus is required. Furthermore, patients with influenza A infection are more susceptible to MRSA infection28. Collectively, simultaneous detection and discrimination of both pathogens can help inform the diagnosis and follow-up actions. An orthogonal reporter probe for influenza A virus was designed by replacing its aptamer template region with the template for the Broccoli aptamer29–32, which binds to (5Z)-5-[(3,5-difluoro-4-hydroxyphenyl)methylene]-3,5-dihydro-2-methyl-3-(2,2,2-trifluoroethyl)-4H-imidazol-4-one (DFHBI-1T) and exhibits spectral properties distinct from those of the malachite green aptamer. Secondary structures of the new reporter probe and corresponding full-length RNA transcript were simulated using NUPACK and satisfied the probe design criteria without further optimization (Supplementary Table 2). Dual detection of MRSA and influenza A virus was tested in SENSR reactions in which the two probe pairs, their cognate fluorogenic dyes and various concentrations of the target RNAs were added (Fig. 6a). When the probe pairs were hybridized to their respective target RNAs, and successful transcription followed, the RNA aptamers would bind their cognate dyes and emit distinguishable fluorescence. The presence of each target RNA could be determined by the fluorescence patterns from the SENSR reaction: malachite green aptamer fluorescence for MRSA; and Broccoli aptamer fluorescence for influenza A virus. Indeed, the presence of either target RNA (1 nM) was easily detected by the fluorescence pattern (Fig. 6b). Across various concentrations of each target RNA, the SENSR probes specifically produced fluorescence that responded only to respective targets, thereby enabling orthogonal dual detection of two pathogens (Fig. 6c).
Fig. 6. One-pot dual detection of RNAs by SENSR.

a, One-pot dual detection of MRSA and influenza A. The dual-SENSR mixture contains two orthogonal pairs of probes and fluorogenic dyes with other components. Each probe pair hybridizes to the corresponding target RNA and allows the SENSR reaction to take place, emitting fluorescences that are distinguishable from each other. MG, malachite green. b, Validation of the orthogonal dual-SENSR reaction. The presence of each target RNA (1 nM) was determined by the intensities of non-overlapping fluorescence. c, One-pot dual-SENSR detection of MRSA and influenza A with various concentration combinations. All tests were performed with four biological replicates.
Lastly, we applied the orthogonal dual detection to SARS-CoV-2, which has many related viruses with high sequence homology. Simultaneous detection of multiple target sites along its genome would enable specific discrimination of this emerging pathogen from others. In addition to the previously demonstrated SARS-CoV-2 probe pair (Fig. 4f), we designed three additional probe pairs for other regions in the RdRp gene with either the malachite green aptamer or the Broccoli aptamer (Fig. 7a). Each probe pair contained a discriminatory base at either the 5′ end of the promoter probe or the 3′ end of the reporter probe, which are unique to SARS-CoV-2 compared with other similar viruses. Mismatches between the probes and non-target RNAs would inhibit ligation and subsequent SENSR reaction and could enable more specific detection of SARS-CoV-2 (Supplementary Fig. 8). Indeed, all four probe pairs were able to detect 1 aM SARS-CoV-2 RNA, thereby exhibiting a higher fluorescence intensity compared with that of the related viral RNA sequences (Fig. 7b). We then tested the orthogonal dual detection of two target regions using the SARS-CoV-2-MG1 and SARS-CoV-2-BR1 probe pairs. The dual-SENSR assay effectively detected the target RNA and maintained the specificity of each probe pair (Fig. 7c). Therefore, the dual-SENSR assay could be used to assist diagnostic decision making by providing two detection results that can complement each other.
Fig. 7. Dual detection of SARS-CoV-2 by SENSR.
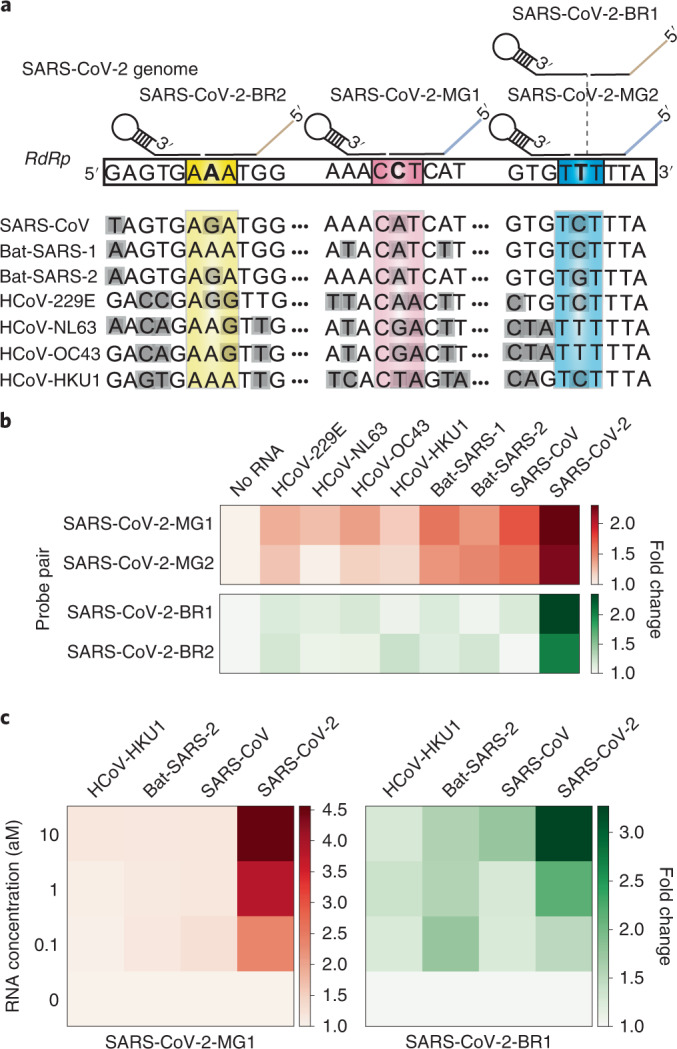
a, Probe design for dual-SENSR detection. Three regions in the RNA-dependent RNA polymerase (RdRp) gene of SARS-CoV-2 were targeted. Discriminatory bases that enable the specific detection of SARS-CoV-2 against viruses with highly similar sequences are marked by bold letters. Grey shading indicates mismatches between the sequences of SARS-CoV-2 and other viruses. Only several bases around the ligation junction are displayed here. Entire probe-binding sites on the SARS-CoV-2 genome and sequence alignments are shown in Supplementary Fig. 8. b, Singleplex detection of 1 aM SARS-CoV-2 RNA by SENSR. c, One-pot dual detection of SARS-CoV-2 by the orthogonal probe pairs SARS-CoV-2-MG1 and SARS-CoV-2-BR1. All tests were performed with two biological replicates. Fold changes were calculated by dividing the normalized fluorescence values by the value with no target RNA. Bat-SARS-1, bat SARS-related coronavirus 1 (accession code: MG772933); Bat-SARS-2, bat SARS-related coronavirus 2 (accession code: NC_014470); HCoV-229E, human coronavirus 229E; HCoV-HKU1, human coronavirus HKU1; HCoV-NL63, human coronavirus NL63; HCoV-OC43, human coronavirus OC43.
Detection of SARS-CoV-2 from clinical samples using SENSR
Finally, we validated the feasibility of SENSR for the detection of pathogens from clinical samples. Here, we sought to further improve the SENSR workflow by streamlining the sample preparation stage to make the whole procedure more suitable for time- and resource-limited settings. An RNA extraction is often required to remove complex components from a clinical sample that may inhibit reactions in molecular diagnostics. However, the RNA extraction consists of multiple steps that necessitate additional reagents, instruments and time. Given the successful RNA detection directly from the cell lysate of MRSA, we speculated that SENSR could be compatible with the clinical sample components. Therefore, we set up two SENSR workflows that employed thermal or chemical lysis to obtain crude viral lysates directly from clinical samples before SENSR assays (Fig. 8a,b). For the chemical lysis workflow, we tested several non-ionic detergents for viruses and selected Nonidet P-40 owing to its low background fluorescence (Supplementary Note 3 and Supplementary Fig. 9).
Fig. 8. SARS-CoV-2 detection from clinical samples by SENSR.

a,b, Schematics of SARS-CoV-2 detection from clinical samples by SENSR. Clinical samples in UTM were treated by either thermal (a, c and e) or chemical lysis (b, d and f) and mixed directly with the SENSR mixture. c,d, SARS-CoV-2 detection from clinical samples. A total of 40 samples (20 positives and 20 negatives; confirmed by rRT–PCR) were tested with the thermal lysis SENSR workflow (c, left). Another probe set (SARS-CoV-2-BR3) predicted two additional positives out of three that were incorrectly predicted as negatives by the thermal lysis SENSR workflow (c, right). Among the samples, a total of 20 (ten positives and ten negatives) were tested using the chemical lysis SENSR workflow (d). Positive samples were numbered according to their Ct values, sorted from lowest to highest (Supplementary Table 6). Negative samples were numbered according to SENSR fluorescence intensity, sorted from highest to lowest. The dashed lines indicate fluorescence intensities from SENSR reactions with reference samples containing one copy of synthetic target RNA (top) or no target RNA (bottom). SENSR reactions were run for 50 min. e,f, Detection of SARS-CoV-2 by 30-min SENSR reaction. Four positive samples with the highest Ct values (the lowest target RNA concentrations) and four negative samples with the highest fluorescence values from 50-min SENSR reactions were tested to validate the feasibility of SENSR to detect SARS-CoV-2 from clinical samples in 30 min. The dashed lines are as in c and d. The clinical and reference sample tests were performed with four biological replicates (two-tailed Student’s t-test for comparison between the results from clinical samples and the results from reference samples with one copy of synthetic target RNA; *P < 0.05, **P < 0.01; ***P < 0.001; ****P < 0.0001; horizontal lines represent means ± s.d).
The updated SENSR workflows were validated using clinical samples obtained from patients with suspected COVID-19 in the Republic of Korea. Before the SENSR assays, the samples were analysed by standard rRT–PCR assay to acquire reference diagnostic test results (Supplementary Table 6). First, 40 samples (20 positives and 20 negatives) were tested through the thermal lysis workflow (Fig. 8c). The non-purified nasopharyngeal swab samples in the universal transport medium (UTM) were heated to 95 °C for 5 min, cooled and mixed directly with the SENSR master mix containing a SARS-CoV-2-specific probe pair (SARS-CoV-2-BR1; Supplementary Fig. 8 and Supplementary Table 1) for a 50-min SENSR reaction. To set a standard for diagnostic decision, we included a reference test with a sample containing a single copy of synthetic target RNA in UTM. A sample was classified as positive if it showed a higher fluorescence intensity than the reference test result, with a statistically significant difference. Otherwise, a sample was classified as negative. The SENSR assay showed 85% (17 out of 20) positive predictive agreement and 100% (20 out of 20) negative predictive agreement relative to the rRT–PCR results (Fig. 8c, left). SENSR with another probe pair (SARS-CoV-2-BR3; Supplementary Fig. 8 and Supplementary Table 1) was able to predict two additional positives out of three that were incorrectly predicted as negatives (positive sample numbers 8, 13 and 18 in Fig. 8c, right), increasing the collective positive predictive agreement to 95% (19 out of 20). We have shown that the SENSR probe design is straightforward and that two orthogonal probe pairs can be used simultaneously. Therefore, parallel individual assays with distinct probe pairs or a dual-SENSR assay with orthogonal probe pairs would allow for a more confirmative test in the future.
Next, the chemical lysis workflow was validated using 20 samples (ten positives and ten negatives). For a rigorous validation, we used positive samples with the highest Ct values (the lowest target RNA amount) from the rRT–PCR test. The non-purified clinical samples in UTM were mixed with Nonidet P-40. After a 5-min incubation at room temperature, the SENSR master mix was added to the sample tube for a 50-min SENSR reaction. The assay showed 80% (eight out of ten) positive predictive agreement and 100% (ten out of ten) negative predictive agreement relative to the rRT–PCR result (Fig. 8d). Intriguingly, two positive samples (numbers 13 and 18) were falsely classified as negative in both the thermal and chemical lysis workflow, suggesting some factors associated with the samples as possible sources for the incorrect predictions.
Finally, we examined the feasibility of SENSR as a rapid diagnostic test. Following the results obtained using a synthetic MRSA RNA (Fig. 3b), we performed 30-min SENSR reactions with the clinical samples. Correct predictive agreements were derived from the 30-min SENSR results through either thermal (Fig. 8e) or chemical lysis (Fig. 8f) workflows, indicating that the updated workflows are sufficient to distinguish between positives and negatives of the clinical samples. Collectively, we have demonstrated rapid SENSR workflows for clinical samples with a short sample-to-result time (35–55 min), including the sample preparation step.
Discussion
Rapid, simple, economical and sensitive diagnostic tests are needed to detect and manage infectious diseases at the earliest possible time. However, conventional approaches lack one or more of these features. Culture-based methods are time consuming (>24 h)33 while PCR-based methods, including real-time PCR, require a complex procedure, expensive instruments and skilled expertise. Various isothermal amplification methods for RNA have been introduced to replace traditional methods7,8. Isothermal nucleic acid amplification testing provides unique advantages for point-of-care detection of pathogens over PCR-based methods, particularly in resource-limited settings. First, isothermal methods can be performed with simple, portable and cheap equipment34. PCR requires repeated steps of heating and cooling under a precise temperature control mechanism. In contrast, isothermal methods require only single, constant temperature throughout the reaction. A simple heat block or even water bath could be used to maintain the reaction temperature needed35. Furthermore, an isothermal reaction can reduce the overall reaction time by eliminating the time necessary for temperature adjustment. Therefore, many isothermal techniques are faster than PCR-based methods36. Based on these advantages, isothermal methods have been applied to the point-of-care detection of pathogens. For example, isothermal amplification techniques were performed with a commercially available portable fluorescence reader36 or custom-designed optical reader9 to demonstrate their feasibility for field testing. Still, they generally require numerous reaction components, often making them expensive and incompatible with the complex clinical sample. Thus, separate RNA purification is often a prerequisite for diagnostic assays based on isothermal RNA amplification methods.
In contrast, SENSR satisfies many desirable requirements for onsite diagnostic tests for pathogens, such as having a short turnaround time (30 min), a low limit of detection (0.1 aM), inexpensive instrumentation and reagents and a simple, one-pot diagnostic procedure. SENSR integrates all component reaction steps using the specially designed probes that contain all of the required functional parts: a promoter, a hybridization sequence to target and an aptamer template. Even with the multifaceted features of the SENSR probes, the design process is systematic and straightforward. Therefore, the SENSR assay can be promptly developed for emerging pathogens, as exemplified by the successful design of the SENSR assay for SARS-CoV-2.
The probe design is unique in that two DNA probes have been designed to expose single-stranded target recognition parts, enabling hybridization of the target RNA and the probes at 37 °C. The hybridization sequences were systematically selected using the nucleic acid design software Primer-BLAST and NUPACK to minimize any structure formation while maximizing hybridization to the target RNA. The efficient hybridization between the probes and target RNA is one of the reasons for enabling high sensitivity during the isothermal reaction.
The promoter probe is programmed to form a stem–loop structure and the stem part forms a double-stranded T7 promoter sequence that initiates transcription by recruiting T7 RNA polymerase. Since the two strands of the T7 promoter part are physically connected by the loop, the probability of formation of a functional double-stranded promoter is higher in the stem–loop structured design than when each strand of the promoter is not connected by the loop. Thus, the hairpin-structured, self-assembling promoter sequence in the promoter probe can facilitate hybridization and subsequent transcription more efficiently.
The initiated transcription elongates through the single-stranded DNA as a template to amplify target RNAs containing aptamer RNAs. The use of a fluorogenic RNA aptamer facilitated SENSR development by enabling fast and straightforward signal generation. Compared with conventional fluorescent protein outputs, the use of RNA aptamers as reporters can reduce the time it takes to observe the signal37.
The simple enzyme composition is another reason to enable one-step and one-pot detection. The fewer enzymes there are, the easier it is to optimize in terms of temperature and buffer composition. In designing the detection scheme, we deliberately tried to reduce the number of enzymes, thus creating one of the simplest isothermal detection schemes based on two enzymes: SplintR ligase for target detection; and T7 RNA polymerase for amplification.
In addition to the results shown in this study, we expect that SENSR has a broad range of applications for pathogen detection. First, SENSR can be easily implemented in the initial screening of infectious diseases at places where a large number of people gather and transfer38,39. With a short turnaround time and a simple reaction composition, SENSR is an ideal diagnostic test for rapid and economical screening. SENSR can be implemented in any diagnostic laboratory with commonly available instruments such as a water bath and a spectrophotometer. In addition, the full potential of SENSR would be realized through the development of field-deployable devices for isothermal incubation and fluorescence measurement37,40–43. Due to the simplicity of the operating mechanism, SENSR can be further developed into a portable, easy-to-use test of paper-based or lateral-flow type, as exemplified by recent developments in nucleic acid diagnostics9,44–47. Second, SENSR will be a valuable method for the immediate development of diagnostic tests for emerging pathogens1,48 because of the simple probe design process and broad adaptability of SENSR. In this work, we demonstrated the successful application of SENSR to six pathogens, using minimal redesign based on the highly modular structure of the probes. In theory, SENSR detection probes can be designed for any RNA as long as the target nucleic acid sequence is available. This feature provides SENSR with a substantial advantage over antibody-based diagnostics to rapidly respond to the outbreak of infectious disease. The nucleic acid probe synthesis is more scalable than animal antibody production. Therefore, SENSR is more suitable for rapid mass production of diagnostic kits than antibody-based diagnostics. Future efforts on automated probe design will be needed to accelerate the development of SENSR assays for newly emerging pathogens.
In conclusion, SENSR is a powerful diagnostic technology for RNA detection that offers a short turnaround time, high sensitivity and specificity and a simple assay procedure, and eliminates the need for expensive instrumentations and diagnostic specialists. Given the simple probe design process and its rapid development, SENSR will be a suitable diagnostic method for emerging infectious diseases.
Methods
Materials
SplintR ligase, T7 RNA polymerase, extreme thermostable single-stranded DNA binding protein (ET-SSB), DNase I (RNase-free) and ribonucleotide solution mix were obtained from New England Biolabs. Recombinant RNase inhibitor was obtained from Takara. Malachite green oxalate was purchased from Sigma–Aldrich. DFHBI-1T was purchased from Tocris Bioscience. Dithiothreitol was acquired from Thermo Fisher Scientific. Tris-HCl, potassium chloride, sodium chloride, magnesium chloride and 5′ phosphate modified oligonucleotides were obtained from Bioneer. Full-length probe oligonucleotides were synthesized by Integrated DNA Technologies. Oligonucleotides other than these were synthesized by Cosmogenetech.
Preparation of synthetic target RNA
Target RNA was synthesized using an in vitro transcription process. To accomplish this, the template DNA containing the target RNA region was amplified by PCR with primers (Supplementary Table 7). The PCR amplicon containing the T7 promoter sequence was used as a template for in vitro transcription. A transcription reaction mixture containing 1 μg template DNA, 2 μl 10× T7 RNA polymerase reaction buffer, 1 μl dithiothreitol (100 mM), 0.8 μl NTPs (25 mM for each), 0.5 μl Recombinant RNase Inhibitor (20 U μl−1), 2 μl T7 RNA polymerase (50 U μl−1) and RNase-free water (up to 20 μl) was incubated at 37 °C for 16 h. The resulting reaction products were treated with 1 μl DNase I (RNase-free) for 1 h at 37 °C. The transcript was purified using the Riboclear (plus!) RNA kit (GeneAll) and quantified using an absorbance at 260 nm. The purified RNA was used immediately for the downstream reaction or stored at −80 °C.
Preparation of MSSA and MRSA cell lysates
MRSA (NCCP 15919) and MSSA (NCCP 11488) were obtained from the Korea Centers for Disease Control and Prevention. Single colonies were isolated by cultivating on 5% sheep blood agar (Hanil Komed) at 37 °C for 24 h. Single colonies were cultured in Luria–Bertani liquid medium at 37 °C for 24 h with shaking (300 r.p.m.). Cells were resuspended in RNase-free water and heat lysed at 95 °C for 2 min.
Preparation of the proxy clinical sample
Human serum was purchased from EMD Millipore Corporation. MRSA (NCCP 15919) and MSSA (NCCP 11488) cells were spiked into the human serum. Human serum was diluted at a 1/7 ratio in RNase-free water9 and the diluted human serum was heat lysed at 95 °C for 2 min.
Preparation of the real clinical sample
Nasopharyngeal swabs were acquired from patients suspected of COVID-19 and collected in UTM (Noble Biosciences) by Seoul Clinical Laboratories. We obtained samples that had been analysed using a standard rRT–PCR to confirm the presence of SARS-CoV-2 (Supplementary Table 6). The remaining non-purified samples in UTM were used for the SENSR tests. Informed consent exemption and the protocol for this study were approved by the Seoul Clinical Laboratories Institutional Review Board (under IRB-20-010) for the use of nasopharyngeal swab remainder samples of COVID-19 rRT–PCR tests.
RNA-splinted single-stranded DNA ligation assay
The ligation reaction was performed according to a previously reported method17. In brief, 0.2 μl promoter probe (10 μM), 0.22 μl reporter probe (10 μM), 0.22 μl target RNA (10 μM) and 1 μl annealing buffer (100 mM Tris-HCl (pH 7.4) and 500 mM KCl) were added to 6.86 μl RNase-free water. The mixture was heated to 95 °C for 3 min, then slowly cooled to room temperature. This step was followed by the addition of 1 μl 10× SplintR buffer and 0.5 μl SplintR ligase (25 U μl−1), then incubation of the mixture at 37 °C for 30 min. The reaction was terminated by heating at 95 °C for 10 min. Next, 2 μl of the ligated product was amplified through 20 μl PCR reaction with LigChk_F and LigChk_R primers (Supplementary Table 7). The PCR products were assessed using an Agilent 2100 Bioanalyzer with a DNA 1000 Kit (Agilent Technologies), according to the manufacturer’s protocol.
In vitro transcription assay
The ligated products were used directly as templates for the in vitro transcription assay. A transcription reaction mixture containing 5 μl of the ligated product, 5 μl 10× T7 RNA polymerase reaction buffer, 2.5 μl dithiothreitol (100 mM), 2 μl NTPs (25 mM each NTP), 1.25 μl Recombinant RNase Inhibitor (20 U μl), 5 μl T7 RNA polymerase (50 U μl−1) and 29.25 μl RNase-free water was incubated at 37 °C for 16 h. The reaction mixture was purified following the protocol described in a previous section for the preparation of synthetic target RNA. The purified RNAs were analysed using an Agilent 2100 Bioanalyzer (Agilent Technologies) with an RNA 6000 Nano Kit (Agilent Technologies), following the manufacturer’s instructions.
Malachite green and aptamer binding assay
First, 500 ng of the purified RNA solution was mixed with 20 μl 5× malachite green buffer (250 mM Tris-HCl (pH 7.5), 5 mM ATP, 50 mM NaCl and 700 mM KCl) and RNase-free water up to 90 μl. The mixture was heated to 95 °C for 10 min and left at room temperature for 20 min. Then, 5 μl MgCl2 (200 mM) was added to the mixture and allowed to stabilize at room temperature for 15 min, followed by the addition of 5 μl malachite green solution (320 μM) to produce a total volume of 100 μl. The mixture was incubated at room temperature for 30 min. After incubation, the fluorescence intensity was measured using a Hidex Sense 425-301 microplate reader (Hidex) in 384-well black/clear flat-bottom polystyrene microplates (Corning). For the malachite green aptamer fluorescence, the excitation wavelength was 616 nm with a slit width of 8.5 nm, and the emission wavelength was 665 nm with a slit width of 7.5 nm. The background intensity from a 100 μl 1× malachite green buffer containing 16 μM malachite green was subtracted from all fluorescence intensities.
SENSR protocol
Detailed step-by-step protocols for the SENSR assay are described in Supplementary Note 4. The one-pot isothermal reaction master mix consisted of the following components: 2 μl promoter probe (10 μM), 2.2 μl reporter probe (10 μM), 5 μl malachite green solution (320 μM) (or 20 μl DFHBI-1T solution (50 μM)), 10 μl NTPs (each 25 mM), 0.8 μl ET-SSB (500 ng μl−1), 0.5 μl Recombinant RNase Inhibitor (20 U μl−1), 10 μl SplintR ligase (25 U μl−1), 5 μl T7 RNA polymerase (50 U μl−1) and 10 μl 10× SENSR buffer (500 mM Tris-HCl (pH 7.4) and 100 mM MgCl2). The reaction master mix was adjusted to 99.22 μl in RNase-free water, and 0.78 μl target RNA was added to produce a total volume of 100 μl. The reaction mixture was incubated at 37 °C for 0.5–2 h. After incubation, the fluorescence intensity was measured using a Hidex Sense 425-301 microplate reader (Hidex), as described above. For the malachite green aptamer, a background intensity from 100 μl 1× SENSR buffer containing 16 μM malachite green was subtracted from all malachite green SENSR fluorescence intensities. For the Broccoli aptamer fluorescence, a background intensity from 100 μl 1× SENSR buffer containing 10 μM DFHBI-1T was subtracted from all Broccoli SENSR fluorescence intensities. For the Broccoli aptamer fluorescence, the excitation wavelength was 460 nm with a slit width of 20 nm, and the emission wavelength was 520 nm with a slit width of 14 nm.
For dual detection, we used the following reaction mixture: 2 μl promoter probe 1 (10 μM), 2.2 μl reporter probe 1 (10 μM), 2 μl promoter probe 2 (10 μM), 2.2 μl reporter probe 2 (10 μM), 5 μl malachite green solution (320 mM), 20 μl DFHBI-1T solution (50 μM), 10 μl NTPs (each 25 mM), 0.8 μl ET-SSB (500 ng μl−1), 0.5 μl Recombinant RNase Inhibitor (20 U μl−1), 10 μl SplintR ligase (25 U μl−1), 5 μl T7 RNA polymerase (50 U μl−1) and 10 μl 10× SENSR buffer (500 mM Tris-HCl (pH 7.4) and 100 mM MgCl2). The reaction master mix was adjusted to 99.22 μl in RNase-free water, and 0.78 μl target RNA was added, producing a total volume of 100 μl. The remaining steps were identical to the single-target detection. A background intensity from 100 μl 1× SENSR buffer containing 16 μM malachite green and 10 μM DFHBI-1T was subtracted from all dual-SENSR fluorescence intensities.
For the clinical sample tests, samples were pre-treated either by thermal or chemical lysis. For the thermal lysis, a non-purified clinical sample was heated at 95 °C for 5 min in a PCR tube and placed on ice to cool. For the chemical lysis, a 2.5-μl non-purified clinical sample was mixed with 2.5 μl Nonidet P-40 (2%; Sigma–Aldrich) in a PCR tube and left at room temperature for 5 min. We used the following reaction mixture for SENSR: 1 μl promoter probe (10 μM), 1.1 μl reporter probe (10 μM), 10 μl DFHBI-1T solution (50 μM), 5 μl NTPs (each 25 mM), 0.4 μl ET-SSB (500 ng μl−1), 0.25 μl Recombinant RNase Inhibitor (20 U μl−1), 5 μl SplintR ligase (25 U μl−1), 2.5 μl T7 RNA polymerase (50 U μl−1) and 5 μl 10× SENSR buffer (500 mM Tris-HCl (pH 7.4) and 100 mM MgCl2). The reaction master mix was adjusted to 47.5 μl (for the thermal lysis workflow) or 45 μl (for the chemical lysis workflow) with RNase-free water, and 2.5 μl of thermally lysed or 5 μl of chemically lysed clinical sample in UTM was mixed to produce a total volume 50 μl. A background fluorescence intensity from 50 μl 1× SENSR buffer containing 10 μM DFHBI-1T and 2.5 μl UTM was subtracted from the SENSR fluorescence intensities obtained from the thermal lysis workflow. A background fluorescence intensity from 50 μl 1× SENSR buffer containing 10 μM DFHBI-1T, 2.5 μl UTM and 0.1% Nonidet P-40 was subtracted from the SENSR fluorescence intensities obtained from the chemical lysis workflow. The remaining steps were identical to the single-target detection.
Statistics
All data in this work were obtained by measurements from distinct samples. Two-tailed Student’s t-tests were performed using Microsoft Excel 2016. The linear regression in Fig. 3a and the reported R2 values associated with the data for Figs. 3 and 4 and Supplementary Fig. 5a were computed using SigmaPlot 12.0.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Supplementary notes 1–4, Figs. 1–10, Tables 1–7 and references.
Acknowledgements
This research was supported by the C1 Gas Refinery Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT (NRF-2015M3D3A1A01064926 and NRF-2018M3D3A1A01055754). This work was also supported by an NRF grant funded by the Korean government (Ministry of Science and ICT; numbers 2018R1C1B3007409 and 2019R1A2C2008680). This research was also supported by the Human Resources Program in Energy Technology of the Korea Institute of Energy Technology Evaluation and Planning (KETEP), which granted financial resources from the Ministry of Trade, Industry and Energy, Republic of Korea (number 20194030202330). We thank C.-K. Kim, Y. Sun, C. Lee and S.-W. Kim at Seoul Clinical Laboratories for assistance with the clinical study. We also thank S. J. Kim and C. Ihm at Eulji University School of Medicine and M. K. Kim at Gimcheon Medical Center for assistance with obtaining access to the clinical samples.
Author contributions
J.W.L. conceived of the project. C.H.W., S.J., G.S., G.Y.J. and J.W.L. designed the experiments. C.H.W., S.J. and G.S. performed the experiments. C.H.W., S.J., G.S., G.Y.J. and J.W.L. analysed the results. C.H.W., S.J., G.S., G.Y.J. and J.W.L. wrote the manuscript.
Data availability
The authors declare that all of the data supporting the findings of this study are available within the paper and its Supplementary Information file. Raw pre-processed data for Figs. 2–8 and Supplementary Figs. 1–9 are available from Figshare with the identifier 10.6084/m9.figshare.12547391. RefSeq messenger RNA of Homo sapiens (ftp://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/) was used in the primer pair specificity-checking parameters section of the primer design process through Primer-BLAST.
Competing interests
Patent applications have been submitted by J.W.L., G.Y.J., C.H.W., S.J. and G.S. based on the results of this study (PCT/KR2020/005331 (Patent Cooperation Treaty) and 10-2020-0048912 (Republic of Korea)).
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Chang Ha Woo, Sungho Jang.
Contributor Information
Gyoo Yeol Jung, Email: gyjung@postech.ac.kr.
Jeong Wook Lee, Email: jeongwook@postech.ac.kr.
Supplementary information
is available for this paper at 10.1038/s41551-020-00617-5.
References
- 1.Caliendo AM, et al. Better tests, better care: improved diagnostics for infectious diseases. Clin. Infect. Dis. 2013;57:S139–S170. doi: 10.1093/cid/cit578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rajapaksha P, et al. A review of methods for the detection of pathogenic microorganisms. Analyst. 2019;144:396–411. doi: 10.1039/c8an01488d. [DOI] [PubMed] [Google Scholar]
- 3.Sintchenko V, Gallego B. Laboratory-guided detection of disease outbreaks: three generations of surveillance systems. Arch. Pathol. Lab. Med. 2009;133:916–925. doi: 10.5858/133.6.916. [DOI] [PubMed] [Google Scholar]
- 4.O’Connor L, Glynn B. Recent advances in the development of nucleic acid diagnostics. Expert Rev. Med. Devices. 2010;7:529–539. doi: 10.1586/erd.10.22. [DOI] [PubMed] [Google Scholar]
- 5.Pavšič J, et al. Standardization of nucleic acid tests for clinical measurements of bacteria and viruses. J. Clin. Microbiol. 2015;53:2008–2014. doi: 10.1128/JCM.02136-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong J, Olano JP, McBride JW, Walker DH. Emerging pathogens: challenges and successes of molecular diagnostics. J. Mol. Diagn. 2008;10:185–197. doi: 10.2353/jmoldx.2008.070063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao Y, Chen F, Li Q, Wang L, Fan C. Isothermal amplification of nucleic acids. Chem. Rev. 2015;115:12491–12545. doi: 10.1021/acs.chemrev.5b00428. [DOI] [PubMed] [Google Scholar]
- 8.Yan L, et al. Isothermal amplified detection of DNA and RNA. Mol. Biosyst. 2014;10:970–1003. doi: 10.1039/c3mb70304e. [DOI] [PubMed] [Google Scholar]
- 9.Pardee K, et al. Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell. 2016;165:1255–1266. doi: 10.1016/j.cell.2016.04.059. [DOI] [PubMed] [Google Scholar]
- 10.Gootenberg JS, et al. Nucleic acid detection with CRISPR–Cas13a/C2c2. Science. 2017;356:438–442. doi: 10.1126/science.aam9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gootenberg JS, et al. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018;360:439–444. doi: 10.1126/science.aaq0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao W. Recent developments in ligase-mediated amplification and detection. Trends Biotechnol. 2004;22:38–44. doi: 10.1016/j.tibtech.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Stuppia L, Antonucci I, Palka G, Gatta V. Use of the MLPA assay in the molecular diagnosis of gene copy number alterations in human genetic diseases. Int. J. Mol. Sci. 2012;13:3245–3276. doi: 10.3390/ijms13033245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shin GW, et al. Stuffer-free multiplex ligation-dependent probe amplification based on conformation-sensitive capillary electrophoresis: a novel technology for robust multiplex determination of copy number variation. Electrophoresis. 2012;33:3052–3061. doi: 10.1002/elps.201200334. [DOI] [PubMed] [Google Scholar]
- 15.Chung B, et al. Rapid and sensitive detection of lower respiratory tract infections by stuffer-free multiplex ligation-dependent probe amplification. Electrophoresis. 2014;35:511–514. doi: 10.1002/elps.201300374. [DOI] [PubMed] [Google Scholar]
- 16.Chung B, et al. Precise H1N1 swine influenza detection using stuffer-free multiplex ligation-dependent probe amplification in conformation-sensitive capillary electrophoresis. Anal. Biochem. 2012;424:54–56. doi: 10.1016/j.ab.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 17.Jin J, Vaud S, Zhelkovsky AM, Posfai J, McReynolds LA. Sensitive and specific miRNA detection method using SplintR Ligase. Nucleic Acids Res. 2016;44:e116. doi: 10.1093/nar/gkw399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ying Z-M, et al. Spinach-based fluorescent light-up biosensors for multiplexed and label-free detection of microRNAs. Chem. Commun. 2018;54:3010–3013. doi: 10.1039/c8cc00123e. [DOI] [PubMed] [Google Scholar]
- 19.Kukarin A, Rong M, McAllister WT. Exposure of T7 RNA polymerase to the isolated binding region of the promoter allows transcription from a single-stranded template. J. Biol. Chem. 2003;278:2419–2424. doi: 10.1074/jbc.M210058200. [DOI] [PubMed] [Google Scholar]
- 20.Zadeh JN, et al. NUPACK: analysis and design of nucleic acid systems. J. Comput. Chem. 2011;32:170–173. doi: 10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
- 21.Ouellet J. RNA fluorescence with light-up aptamers. Front. Chem. 2016;4:29. doi: 10.3389/fchem.2016.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marlowe EM, Bankowski MJ. Conventional and molecular methods for the detection of methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 2011;49:S53–S56. [Google Scholar]
- 23.Woolhouse MEJ, Brierley L. Epidemiological characteristics of human-infective RNA viruses. Sci. Data. 2018;5:180017. doi: 10.1038/sdata.2018.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alsolamy S, Arabi YM. Infection with Middle East respiratory syndrome coronavirus. Can. J. Respir. Ther. 2015;51:102. [PMC free article] [PubMed] [Google Scholar]
- 25.Mackay IM, Arden KE. MERS coronavirus: diagnostics, epidemiology and transmission. Virol. J. 2015;12:222. doi: 10.1186/s12985-015-0439-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corman VM, et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. 2020;25:2000045. doi: 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouhedda F, Autour A, Ryckelynck M. Light-up RNA aptamers and their cognate fluorogens: from their development to their applications. Int. J. Mol. Sci. 2017;19:44. doi: 10.3390/ijms19010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mulcahy ME, McLoughlin RM. Staphylococcus aureus and influenza A virus: partners in coinfection. mBio. 2016;7:e02068-16. doi: 10.1128/mBio.02068-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Filonov GS, Moon JD, Svensen N, Jaffrey SR. Broccoli: rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J. Am. Chem. Soc. 2014;136:16299–16308. doi: 10.1021/ja508478x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filonov GS, Jaffrey SR. RNA imaging with dimeric Broccoli in live bacterial and mammalian cells. Curr. Protoc. Chem. Biol. 2016;8:1–28. doi: 10.1002/9780470559277.ch150174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okuda M, Fourmy D, Yoshizawa S. Use of baby spinach and Broccoli for imaging of structured cellular RNAs. Nucleic Acids Res. 2017;45:1404–1415. doi: 10.1093/nar/gkw794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Torelli E, et al. Isothermal folding of a light-up bio-orthogonal RNA origami nanoribbon. Sci. Rep. 2018;8:6989. doi: 10.1038/s41598-018-25270-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sinha M, et al. Emerging technologies for molecular diagnosis of sepsis. Clin. Microbiol. Rev. 2018;31:e00089-17. doi: 10.1128/CMR.00089-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niemz A, Ferguson TM, Boyle DS. Point-of-care nucleic acid testing for infectious diseases. Trends Biotechnol. 2011;29:240–250. doi: 10.1016/j.tibtech.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martzy R, et al. Challenges and perspectives in the application of isothermal DNA amplification methods for food and water analysis. Anal. Bioanal. Chem. 2019;411:1695–1702. doi: 10.1007/s00216-018-1553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel JC, et al. Field evaluation of a real-time fluorescence loop-mediated isothermal amplification assay, RealAmp, for the diagnosis of malaria in Thailand and India. J. Infect. Dis. 2014;210:1180–1187. doi: 10.1093/infdis/jiu252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung, J. K. et al. Cell-free biosensors for rapid detection of water contaminants. Nat. Biotechnol. 10.1038/s41587-020-0571-7 (2020). [DOI] [PMC free article] [PubMed]
- 38.Gaber W, Goetsch U, Diel R, Doerr HW, Gottschalk R. Screening for infectious diseases at international airports: the Frankfurt model. Aviat. Space Environ. Med. 2009;80:595–600. doi: 10.3357/asem.2360.2009. [DOI] [PubMed] [Google Scholar]
- 39.Khan K, et al. Entry and exit screening of airline travellers during the A(H1N1) 2009 pandemic: a retrospective evaluation. Bull. World Health Organ. 2013;91:368–376. doi: 10.2471/BLT.12.114777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bridgeman J, Baker A, Brown D, Boxall JB. Portable LED fluorescence instrumentation for the rapid assessment of potable water quality. Sci. Total Environ. 2015;524:338–346. doi: 10.1016/j.scitotenv.2015.04.050. [DOI] [PubMed] [Google Scholar]
- 41.Katzmeier F, et al. A low-cost fluorescence reader for in vitro transcription and nucleic acid detection with Cas13a. PLoS ONE. 2019;14:e0220091. doi: 10.1371/journal.pone.0220091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zanoli LM, Spoto G. Isothermal amplification methods for the detection of nucleic acids in microfluidic devices. Biosensors. 2013;3:18–43. doi: 10.3390/bios3010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang H, et al. LAMP-on-a-chip: revising microfluidic platforms for loop-mediated DNA amplification. Trends Anal. Chem. 2019;113:44–53. doi: 10.1016/j.trac.2019.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Myhrvold C, et al. Field-deployable viral diagnostics using CRISPR–Cas13. Science. 2018;360:444–448. doi: 10.1126/science.aas8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, et al. Clustered regularly interspaced short palindromic repeats/Cas9-mediated lateral flow nucleic acid assay. ACS Nano. 2020;14:2497–2508. doi: 10.1021/acsnano.0c00022. [DOI] [PubMed] [Google Scholar]
- 46.Broughton JP, et al. CRISPR–Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020;38:870–874. doi: 10.1038/s41587-020-0513-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xia S, Chen X. Single-copy sensitive, field-deployable, and simultaneous dual-gene detection of SARS-CoV-2 RNA via modified RT-RPA. Cell Discov. 2020;6:37. doi: 10.1038/s41421-020-0175-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y, Yu X. Identification and diagnosis of newly emerging pathogens. Infect. Dis. Transl. Med. 2017;3:12–16. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary notes 1–4, Figs. 1–10, Tables 1–7 and references.
Data Availability Statement
The authors declare that all of the data supporting the findings of this study are available within the paper and its Supplementary Information file. Raw pre-processed data for Figs. 2–8 and Supplementary Figs. 1–9 are available from Figshare with the identifier 10.6084/m9.figshare.12547391. RefSeq messenger RNA of Homo sapiens (ftp://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/) was used in the primer pair specificity-checking parameters section of the primer design process through Primer-BLAST.