

In this study, we examined the inactivation mechanisms of peracetic acid (PAA), a sanitizer commonly used for postharvest vegetable washing, for two enteric viruses: Tulane virus (TV) as a human norovirus surrogate and rotavirus (RV). PAA disinfection mechanisms for RV were mainly due to genome damage. In contrast, PAA disinfection in TV was due to damage of the proteins important for binding to its host receptor. We also observed that PAA triggered aggregation of TV to a much greater extent than RV. These studies demonstrate that different viruses are inactivated via different PAA mechanisms. This information is important for designing an optimal sanitation practice for postharvest vegetable washing to minimize foodborne viral diseases.
KEYWORDS: RNA virus, disinfectants, noroviruses, rotavirus, sanitation
ABSTRACT
We determined the disinfection efficacy and inactivation mechanisms of peracetic acid (PAA)-based sanitizer using pH values relevant for vegetable sanitation against rotavirus (RV) and Tulane virus (TV; a human norovirus surrogate). TV was significantly more resistant to PAA disinfection than RV: for a 2-log10 reduction of virus titer, RV required 1 mg/liter PAA for 3.5 min of exposure, while TV required 10 mg/liter PAA for 30 min. The higher resistance of TV can be explained, in part, by significantly more aggregation of TV in PAA solutions. The PAA mechanisms of virus inactivation were explored by quantifying (i) viral genome integrity and replication using reverse transcription-quantitative PCR (RT-qPCR) and (ii) virus-host receptor interactions using a cell-free binding assay with porcine gastric mucin conjugated with magnetic beads (PGM-MBs). We observed that PAA induced damage to both RV and TV genomes and also decreased virus-receptor interactions, with the latter suggesting that PAA damages viral proteins important for binding its host cell receptors. Importantly, the levels of genome-versus-protein damage induced by PAA were different for each virus. PAA inactivation correlated with higher levels of RV genome damage than of RV-receptor interactions. For PAA-treated TV, the opposite trends were observed. Thus, PAA inactivates each of these viruses via different molecular mechanisms. The findings presented here potentially contribute to the design of a robust sanitation strategy for RV and TV using PAA to prevent foodborne disease.
IMPORTANCE In this study, we examined the inactivation mechanisms of peracetic acid (PAA), a sanitizer commonly used for postharvest vegetable washing, for two enteric viruses: Tulane virus (TV) as a human norovirus surrogate and rotavirus (RV). PAA disinfection mechanisms for RV were mainly due to genome damage. In contrast, PAA disinfection in TV was due to damage of the proteins important for binding to its host receptor. We also observed that PAA triggered aggregation of TV to a much greater extent than RV. These studies demonstrate that different viruses are inactivated via different PAA mechanisms. This information is important for designing an optimal sanitation practice for postharvest vegetable washing to minimize foodborne viral diseases.
INTRODUCTION
Fresh produce consumption has caused a large number of foodborne illnesses in the United States (1). Sanitation of postharvest fresh produce is essential to prevent foodborne illnesses associated with fresh produce. Vegetable sanitizers are commonly used to prevent cross-contamination of fresh produce and to inactivate pathogens on produce surfaces (2). Free chlorine, a highly reactive and low-cost sanitizer, is one of the most commonly used sanitizers for postharvest produce (3). However, the efficacy of free chlorine depends on pH. Specifically, the pH of free chlorine needs to be adjusted to 6 to 7 to ensure sufficient reactivity and avoid its corrosiveness (4, 5). Moreover, chlorine-based vegetable sanitation produces potentially harmful disinfection by-products (DBPs) (6). For this reason, some European countries (e.g., Germany, Switzerland, and the Netherlands) already prohibit the use of chlorine for washing fresh vegetables (7, 8). Thus, there is an interest and need to use or develop nonchlorine sanitizers (9).
Peracetic acid (PAA) is a highly effective antimicrobial agent and is increasingly being used to sanitize fresh vegetables (2). PAA is synthesized through the reaction between acetic acid and hydrogen peroxide and can be purchased commercially as a stable mixture of PAA, acetic acid, and hydrogen peroxide (10). As a vegetable sanitizer, PAA is preferred to chlorine because pH adjustment is not necessary (11), and fewer chlorinated and aldehyde DBPs are formed by PAA than by chlorination of vegetables (12).
Although PAA possesses strong antimicrobial activity, PAA is not effective at inactivating some viruses (2, 13, 14). For example, PAA at 80 ppm inactivates only about 2.8 log10 of MS2 bacteriophage and 2.9 log10 of feline calicivirus (FCV) attached to salad vegetables (13). Additionally, PAA does not inactivate all viruses to the same degree. For example, 100 ppm of PAA inactivates 3.2 log10 FCV and 2.3 log10 murine norovirus-1 (MNV-1) attached to lettuce, as determined by 50% tissue culture infective dose (TCID50) assays, but only 0.7 log10 hepatitis A virus as determined by plaque assay (2). PAA at 100 ppm for 2 min inactivates only 1.2 log10 polioviruses attached to strawberries in plaque assays but has a higher efficacy at this same concentration and time for bacteriophages MS2 (1.8 log10), ΦX174 (2.2 log10), and PRD1 (1.7 log10) in plaque assays (14). These studies suggest that the PAA efficacy may be virus specific and based on the different physical properties of viruses. Therefore, to design an optimal PAA sanitation practice for virus-contaminated postharvest fresh produce, it is important to understand how PAA mechanistically inactivates different viruses under conditions that are relevant for fresh produce sanitation.
In general, disinfectants or sanitizers inactivate viruses by destabilizing or degrading the viral capsid proteins, including capsid proteins responsible for host cell attachment (15, 16), or by damaging or degrading the encapsidated viral genome (17, 18). PAA is a strong oxidant; thus, it is possible that PAA could oxidize and damage virus capsid proteins and/or genomes (19). In support of this possibility, publications from our research group and from others have shown that PAA decreases rotavirus (RV) binding to MA104 cells and human norovirus (HuNoV) binding to porcine gastric mucin conjugated with magnetic beads (PGM-MBs) (19, 20), suggesting that PAA damages the proteinaceous capsids of these viruses. There are other publications showing that PAA treatment damages genomes of RNA viruses (e.g., feline calicivirus, murine norovirus, and hepatitis A virus) as revealed by reverse transcription-quantitative PCR (RT-qPCR) (2). However, it is not known if PAA causes both genome and protein damage simultaneously in these viruses and whether this may affect rates of viral inactivation. This gap in the knowledge is important to fill because without this information, scientists cannot design conditions most effective for virus disinfection. For this reason, comprehensive studies on the PAA inactivation mechanisms for viruses are critical.
This study aims to identify PAA mechanisms of inactivation for two enteric viruses, Tulane virus (TV), as a HuNoV surrogate, and RV. HuNoV, a member of the Caliciviridae family, is a nonenveloped virus with a single-stranded RNA genome (21). HuNoV is the major cause of foodborne diseases in the United States and is responsible for 5.5 million cases of foodborne illness every year (22). Although there are publications showing in vitro propagation of HuNoV (23, 24), cultivation of HuNoV remains difficult. For this reason, we examined TV, a virus often used as a surrogate of HuNoV (21). Indeed, 22 to 26% of the TV genome is identical to that of HuNoV (25), and both viruses recognize histo-group blood antigens (HBGAs) as attachment receptors (26). RV, a member of Reoviridae, is a nonenveloped virus with a double-stranded, segmented RNA genome (27). RV is the major cause of acute gastroenteritis worldwide for children under 5 years of age (28). RV has been detected in irrigation water (29, 30) and from fresh produce like lettuce and spinach (31, 32). Therefore, there could be potential RV transmission via fresh produce consumption. In this study, TV and RV were exposed to PAA and then subjected to several assays to identify how PAA inactivated each virus. A cell-free binding assay was used to quantify the loss of virus-host receptor interactions. A two-step reverse transcription-quantitative PCR (RT-qPCR) was used to determine genome integrity and genome replication. Traditional plaque assays were used to determine virus titers as a measure of virus replication.
RESULTS
Inactivation kinetics of TV and RV by PAA.
We fitted the inactivation kinetics of PAA-treated TV and RV to pseudo-first-order kinetics according to Chick’s law of disinfection to obtain the inactivation rate constants, as shown in Fig. 1. The RV inactivation rate constant (1.03 ± 0.08), calculated based on natural log reduction, was significantly larger than the inactivation rate constant for TV (0.02 ± 0.0005) (P < 0.01), suggesting that RV was more susceptible to PAA disinfection than TV. Because of the substantial differences in the PAA susceptibilities of these two viruses, we used different concentrations of PAA and different exposure times to inactivate RV and TV. For TV, we exposed viruses to PAA at 10 ppm for 5, 15, 30, 45, and 60 min. For RV OSU, we exposed viruses to PAA at 1 to 3.5 ppm for 3 min. CT is the product of the PAA concentration (C) and time of incubation (T). At a CT of 10 (ppm×min), a 4-log10 inactivation of RV in PFU was observed as determined by plaque assays. However, a CT of over 430 (ppm×min) was required for the same level of inactivation for TV.
FIG 1.
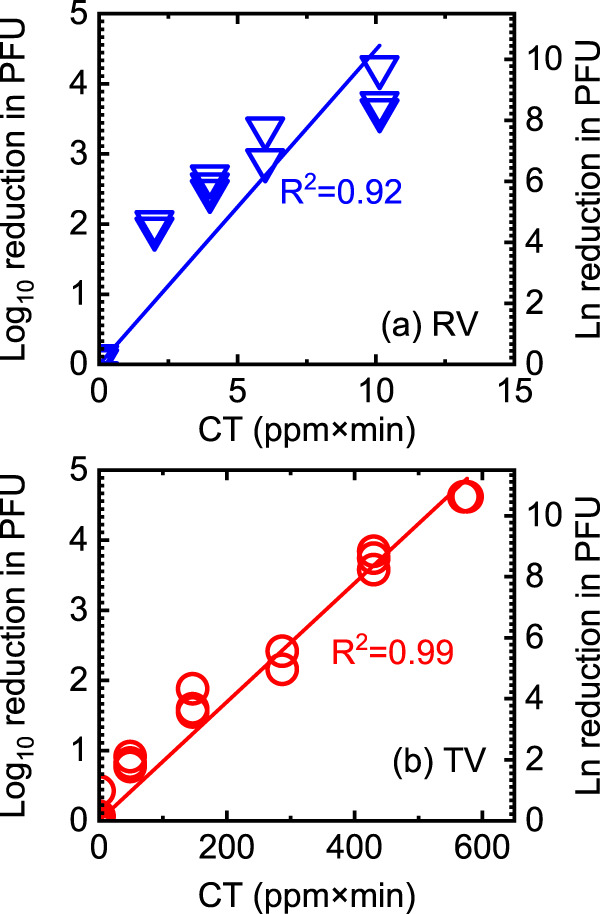
Inactivation of RV (a) and TV (b) exposed to PAA. The log10 reduction in PFU was calculated as log10 PFU0/PFU (or −log10 PFU/PFU0) in which PFU0 is the titer of untreated TV and RV, and PFU is the titer of each virus post-PAA exposure. The natural log reduction was calculated similarly. All data from three separate experiments are plotted. The slopes of the regression lines using the natural log were 1.03 ± 0.08 and 0.02 ± 0.0005 for RV and TV, respectively.
Aggregation kinetics of TV and RV.
It was shown previously that virus aggregation protects virions from the neutralizing effects of disinfectants (33–35). In these cases, aggregation may prevent optimal interactions between PAA and the surface area of a virion, or aggregation may decrease PAA diffusion inside aggregated virions. Thus, it was important to determine if PAA triggered RV or TV aggregation to determine how PAA might affect virus inactivation.
First, we measured the aggregate diameter of TV and RV suspended in 10 ppm of PAA at pH 3. The diameter of a TV particle, as determined by a transmission electron microscope, is approximately 36 nm (25). As seen in Fig. 2a, TV aggregation increased gradually from 250 nm to 1,100 nm in diameter over 30 min. In addition to PAA, low-pH also triggered TV aggregation because particle diameters were increased when TV was incubated in a low-pH solution devoid of PAA, as shown in Fig. 2b. This aggregation appeared to be pH dependent because TV aggregates became smaller (250 nm in aggregate diameter) when the pH of the PAA-containing solution was raised to 7.0 by adding a quencher solution (600 mg/liter sodium thiosulfate containing 16 mg/liter catalase in 0.1 M phosphate buffer [PB]).
FIG 2.
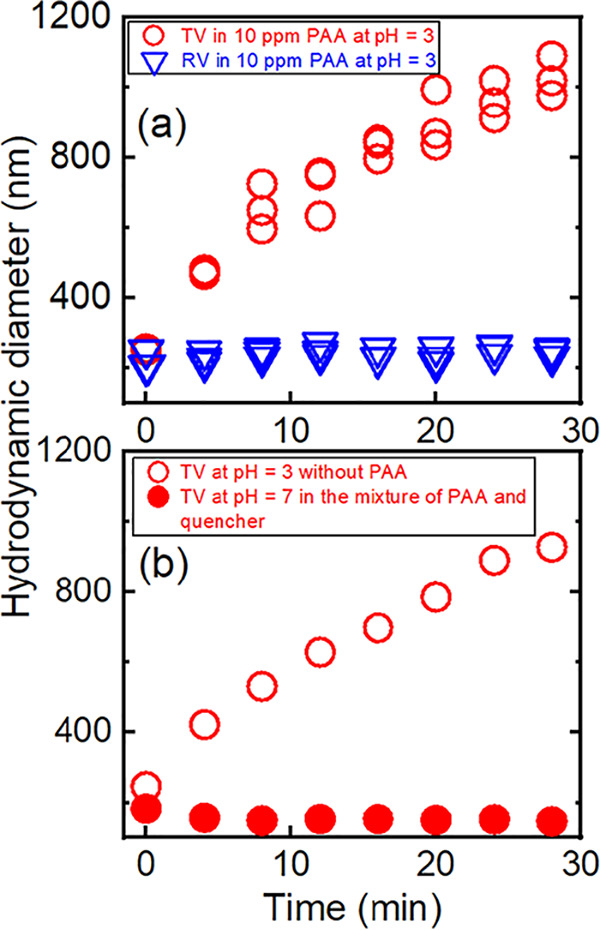
(a) Aggregate diameters of TV and RV, as indicated, suspended in 10 ppm PAA at pH 3. (b) Aggregate diameters of TV suspended in the mixture of PAA and quencher (600 mg/liter sodium thiosulfate containing 16 mg/liter catalase in 0.1 M PB) at a final pH of 7 and in 0.01 M PB at pH 3 without PAA, as indicated.
In contrast to TV, the measured aggregate diameter of RV remained constant (200 nm) in PAA solution (Fig. 2a). This RV stability is explained by steric interactions measured by atomic force microscopy among the virions (36). We suggest that, as a result of this lack of aggregation, PAA more easily accesses nonaggregated RV than aggregated TV and that this may account, in part, for RV’s increased susceptibility to PAA (Fig. 1). As previously reported (37, 38), the pH near the viral isoelectric point (pI) theoretically would favor virus aggregation. The pI values of RV and TV are 4.5 and 4.8, respectively (39, 40). Thus, at a pH 3 in the experiments, both viruses were in conditions below the pI, and theoretically they should have similar aggregation behaviors. The observed differences in levels of aggregation of TV and RV is likely related to differences in protein capsid properties and is beyond the scope of this study.
Impacts of exposure to PAA on TV and RV binding to host receptors.
Despite the aggregation effects observed above, which is expected to protect virions from the disinfecting effects of PAA, PAA still inactivated both TV and RV. To understand how PAA inactivated these RNA viruses on a molecular level, we first examined if PAA affected the ability of the virus to bind to its cognate host receptor. To this end, we used a cell-free binding assay in which untreated or PAA-treated virions were incubated with porcine gastric mucin conjugated with magnetic beads (PGM-MBs). PGM-MBs contain cellular attachment receptors (histo-blood group antigens [HBGAs] and sialic acid) for TV and RV strain OSU (41, 42). RNA from viruses that bound to PGM-MBs were then isolated. RT-qPCR was used to determine the quantities of the TV nonstructural polyprotein (NSP) or RV NSP3 genes as a measure of the numbers of virions bound to the PGM-MBs. By comparing the results from untreated and PAA-treated viruses, we could extrapolate whether PAA altered a virus such that it could no longer interact with its cognate receptor proteins. Note that this virus binding assay with PGM-MBs is an indirect assay to measure the virus binding to PGM-MBs by quantifying the viral RNA gene of bound viruses instead of quantifying the bound viral capsids. Using PGM-MBs affords the opportunity to examine if virus-host receptor interactions are negatively affected by PAA. However, this method cannot distinguish between binding loss due to binding protein degradation and binding loss caused by capsid degradation.
First, we examined TV binding under several conditions. Results are shown in Fig. 3. For this first set of experiments, there were four sets of data that were collected: viruses that did not receive PAA or RNase treatment, viruses that received PAA but not RNase treatment, viruses that did not receive PAA but received RNase treatment, and viruses that received PAA and RNase treatment. These treatments all occurred before incubation with PGM-MBs. If PAA treatment altered the integrity of the TV capsid, then we expected that RNase would permeate the damaged capsid and degrade viral RNA. Thus, the viral RNA would not be qPCR amplified from PGM-MB-bound virions that lacked integrity. As a corollary, only genomes from intact capsids (capsids that remain impermeable to RNase) would be qPCR amplified in the binding assays. The efficacy of the RNase solution to degrade TV and RV genomes was established in our previous study (15), where we observed a 6.4-log10 reduction in the copy number of the TV NSP gene and a 7.8-log10 reduction in the copy number of the RV NSP3 gene in RNase-treated RNAs compared to levels in untreated RNAs.
FIG 3.
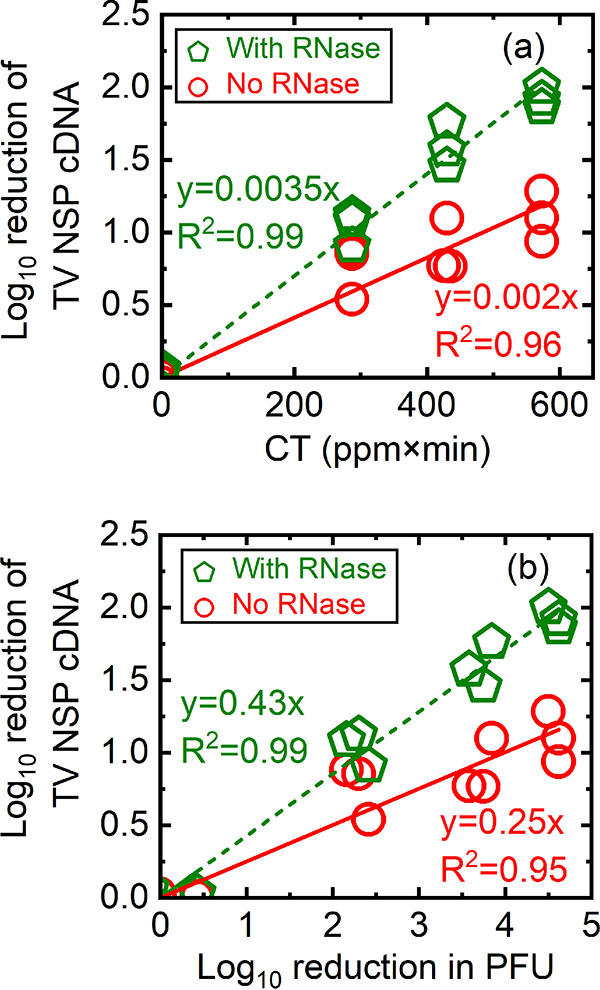
Log10 reduction of TV NSP cDNA quantified from RNA extracted from PGM-MB-bound viruses that were either untreated or treated with PAA only, or untreated/treated with PAA followed by RNase. The log10 reduction of TV NSP cDNA was calculated as log10 N0/N (or −log10 N/N0) in which N0 is the copy number of untreated TV, and N is the copy number of TV post-PAA exposure and was plotted as a function of CT (a) or as a function of log10 reduction in PFU (b). Green pentagons, untreated TV and PAA-treated TV samples treated with RNase prior to PGM-MB binding assays; red circles, untreated TV and PAA-treated TV samples not treated with RNase and directly exposed to PGM-MBs. All data from three separate experiments are plotted.
In Fig. 3a, we plotted the log10 reduction of TV NSP cDNA quantified from PGM-MB-bound virions (y axis) as a function of CT (ppm×min) (x axis). Remember that the TV NSP cDNAs are due to RNA that was isolated from virions that were captured by the PGM-MBs. As shown in Fig. 3a, there was a reduction of NSP copies as the CT values (ppm×min) increased. The log10 reduction of the NSP copy numbers linearly correlated with PAA exposure, suggesting that PAA altered TV in a manner that prevented TV from binding to its host receptors. A similar experiment was performed in which untreated and PAA-treated viruses were next incubated with RNase. As discussed above, the purpose of this experiment was to identify whether PAA treatment also altered the integrity of the PGM-MB-bound TV capsid to the extent that the capsid would be permeable to RNase molecules. Thus, only genomes from intact capsids (capsids that remained impermeable to RNase) would be qPCR amplified in the binding assays. A similar trend was observed when TV was treated with RNase post-PAA treatment (Fig. 3a, green line) in contrast to results with RNase treatment (Fig. 3a, red line): as the CT value increased, the reduction of NSP copy numbers increased. However, the slope of the regression line for RNase-treated samples was significantly larger than the slope of the regression line for samples not treated with RNase (Fig. 3a) (0.0035 ± 0.000085 versus 0.0021 ± 0.00012; P < 0.01, respectively). Specifically, after TV was treated with PAA at a CT of 570 (ppm×min), the reduction in TV bound to PGM-MBs was a 1-log10 (90%) reduction for conditions without RNase treatment versus a 2-log10 (99%) reduction from assays containing RNase treatment. A conclusion to draw from the data shown in Fig. 3a is that PAA treatment reduced TV binding integrity in a manner that prevents TV from interacting with its cognate receptor. In addition, since the reduction of NSP cDNA is greater in the presence of RNase, it is likely that PAA exposure also compromised the integrity of the PGM-MB-bound TV capsid.
In Fig. 3b, the log10 reduction of TV NSP cDNA quantified from PGM-MB-bound virions (y axis) and log10 reduction in PFU (x axis) were plotted. We also found that the log10 reduction of the TV NSP cDNA linearly correlated with the log10 reduction in PFU. Similar to the data shown in Fig. 3a, the slope of the regression line for the binding loss of TV with RNase was significantly larger than the slope of binding loss of TV without RNase (0.43 ± 0.01 versus 0.25 ± 0.02, P < 0.01). Because of a greater reduction in cDNA quantified from RNA isolated from PGM-MB-captured virions in the presence versus that in the absence of RNase, the conclusion is that PAA exposure disrupted not only interactions between TV and its host receptor but also compromised the integrity of the PGM-MB-bound TV capsid to the extent that the capsid became permeable to molecules at least as large as RNase. We noted that the slopes of these lines (0.43 and 0.25) were less than 1. This implied that PAA-induced reduction of TV binding integrity was not completely responsible for TV inactivation. Thus, PAA may have additional mechanisms to inactivate TV.
The same approach was then used to examine how PAA disinfected RV particles. For RV binding assays, we used conditions with CTs higher than those shown in Fig. 1a. This high-CT range was necessary because RV exposed to PAA at CTs of 0 to 10 did not have sufficient loss of binding that could be detected in the PGM-MBs binding assay used here. For this reason, RV samples were exposed to PAA at higher CTs under conditions that completely inactivated RV. Thus, data from RV binding assays were plotted as a function of CT (ppm×min), not as a function of a log10 reduction in PFU.
Similar to TV, untreated or PAA-exposed RV was either untreated or RNase treated before incubation with PGM-MBs. The log10 reduction of RV NSP3 cDNA copies (y axis) was plotted versus PAA exposure at various CTs (ppm×min) (x axis), as shown in Fig. 4a. Similar to results with TV, there was a reduction in RV NSP3 cDNA copy numbers as PAA exposure increased, suggesting that PAA altered RV in a manner that reduced its capacity to bind to sialic acid (Fig. 4a). Also, RNase treatment increased this loss of cDNA copies. For example, after PAA exposure, at a CT of 38 (ppm×min), the loss in binding of RV without RNase treatment was significantly lower than that with RNase treatment (0.5 ± 0.07 versus 0.73 ± 0.06, P < 0.05). A linear correlation was observed for viruses that were incubated with or without RNase (Fig. 4a). The fitted slope for RV binding in the absence of RNase (0.011 ± 0.00089) was significantly lower than the slope for RV binding in the presence of RNase (0.016 ± 0.00124) (P < 0.01). This pattern suggests that PAA not only damaged the RV VP8* (RV binding protein) to compromise virus-host receptor interactions but also disrupted the three-layered RV capsids sufficiently to allow RNase penetration.
FIG 4.
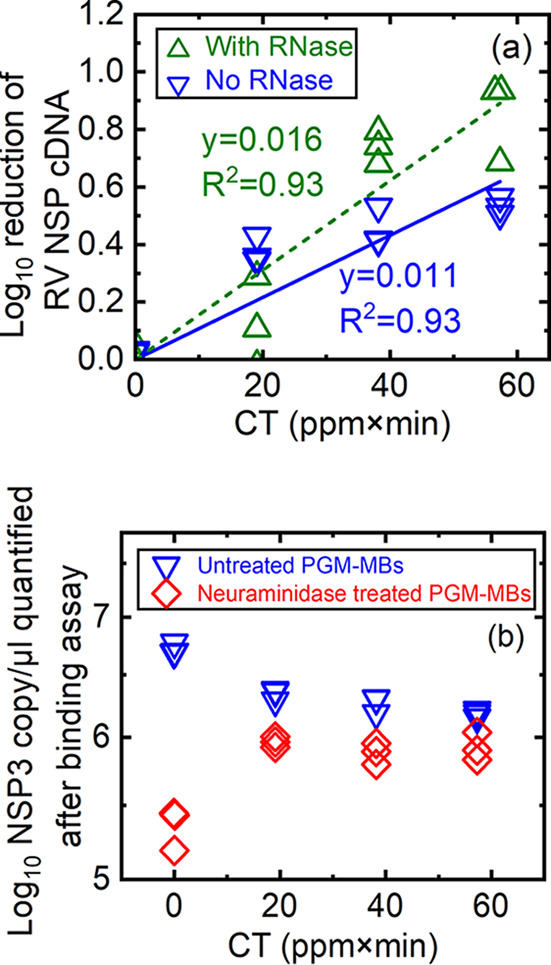
(a) Log10 reduction of RV NSP3 cDNA quantified from RNA extracted from PGM-MB-bound viruses that were either untreated, treated with PAA only, or untreated/treated with PAA followed by RNase treatment, as indicated. Untreated or PAA-exposed RV was treated with trypsin before incubation of viruses with PGM-MBs. The log10 reduction of RV NSP cDNA was calculated as log10 N0/N (or −log10 N/N0) in which N0 is the copy number of untreated RV, and N is the copy number of RV post-PAA exposure and was plotted as a function of CT. (b) Log10 NSP3 gene in copy number/μl quantified from either untreated or PAA-treated RV bound to PGM-MBs pretreated with 0 and 63.1 mU/ml Vibrio cholerae neuraminidase. All data from three separate experiments were plotted.
The VP8* protein of RV strain OSU binds to sialic acid to initiate host cell infection (43, 44). To quantify the specificity of the binding of PAA-exposed RV to sialic acid, we conducted an additional assay in which PGM-MBs were pretreated with Vibrio cholerae neuraminidase to digest sialic acid moieties (20) before PGM-MBs were incubated with untreated or PAA-exposed viruses. The data were plotted as values of log10 RV NSP3 cDNA copies/μl (y axis) and CT (ppm×min) of PAA exposure (x axis) (Fig. 4b). For RV that did not receive PAA (CT of 0), neuraminidase treatment of PGM-MBs resulted in a 1.4-log10 decrease of the NSP3 copy number compared to that of untreated PGM-MBs (5.35 ± 0.14 versus 6.72 ± 0.05 log10 NSP3 copies; P < 0.01) (see Table S1 in the supplemental material). This decrease in RV binding confirmed that RV VP8*-PGM-MB interactions were mainly due to sialic acid (44). For PAA-treated RV, the differences between the number of virions bound to untreated versus neuraminidase-treated PGM-MBs became smaller (e.g., a 0.4-log10 decrease in the NSP3 gene at a CT of 19) (Table S1). This smaller difference (0.4 log10 at a CT of 19 versus 1.4 log10 at a CT of 0) suggested that PAA exposure altered the RV VP8* in a manner that compromises known VP8*-sialic acid interactions. PAA altered the conformation of RV in a manner that increases nonspecific binding in the PGM substrate.
In this study, we used RV strain OSU, which binds to sialic acid to initiate host cell infection (43). Of course, PGM also contains HBGAs, rotavirus-binding receptors that are resistant to neuraminidase treatment. However, HBGAs are receptors for other RV strains such as K8 and HAL1166 but not for strain OSU (45, 46). Thus, it is unlikely that RV strain OSU used here would interact with HBGAs to a large degree.
Based on data from the binding assays, it appears that PAA alters TV and RV in a manner that reduced its capacity to bind to host receptor moieties (e.g., sialic acid for RV). Without this interaction, the subsequent steps in the virus replication cycle (e.g., virus entry into the host cell, viral gene expression, viral genome replication, and virus maturation) would not be initiated. Note that for both viruses, the observed loss in binding was less than the observed loss in PFU caused by PAA, suggesting that there are PAA mechanisms of virus inactivation in addition to the ones that alter virus-host cell receptor interactions.
Impacts of exposure to PAA on TV and RV genomes.
We hypothesized that PAA altered the virus capsids to the extent that small molecules (including PAA) could now access and inactivate the viral RNA genomes. Once damaged, the RNA can no longer be amplified or translated during the virus infection life cycle. We conducted two sets of experiments to test this hypothesis that PAA could directly damage viral RNA genomes. First, we measured the PAA-induced decay rate of purified viral RNA. After purified viral genomes were exposed to PAA, a 1,324-bp region of the nonstructural polyprotein (NSP) gene for TV or a 1,052-bp region in the VP7 gene for RV was reverse transcribed, and the resultant cDNA was amplified and quantified in the qPCR step. If PAA damaged the viral RNA to the point where it could not be reverse transcribed by reverse transcriptase, then there should be a decrease in RT-qPCR amplicons. In Fig. 5a, we plotted the log10 reduction in copy numbers of the TV NSP gene or RV VP7 gene. In the x axis, C was the PAA concentration at the exposure time, and T was the exposure time. As shown in Fig. 5a, increasing PAA exposure to viral RNA decreased copy numbers for both the TV NSP and RV VP7 genes. At CT values of 0.5 and 9.4 (ppm×min), the log10 reduction in copy numbers of the TV NSP gene was significantly higher than that of the RV VP7 gene for viral RNAs that were not exposed to PAA. At a CT of 0.5, there was a 0.49 ± 0.07-log10 reduction of TV NSP and a 0.25 ± 0.03-log10 reduction of RV VP7, as shown in Table S2 (P < 0.01). At a CT of 9.4, there was a 1.02 ± 0.12-log10 reduction of TV NSP and a 0.78 ± 0.06-log10 reduction of RV VP7 (P < 0.05). At CT values of 2.3 and 4.7 (ppm×min), the log10 reductions in copy numbers were similar between the TV NSP gene and the RV VP7 gene, implying that PAA directly damaged the naked viral genomes. Thus, at lower CT values, PAA may have a higher reactivity with single-stranded TV RNA genomes than with double-stranded RV RNA genomes.
FIG 5.
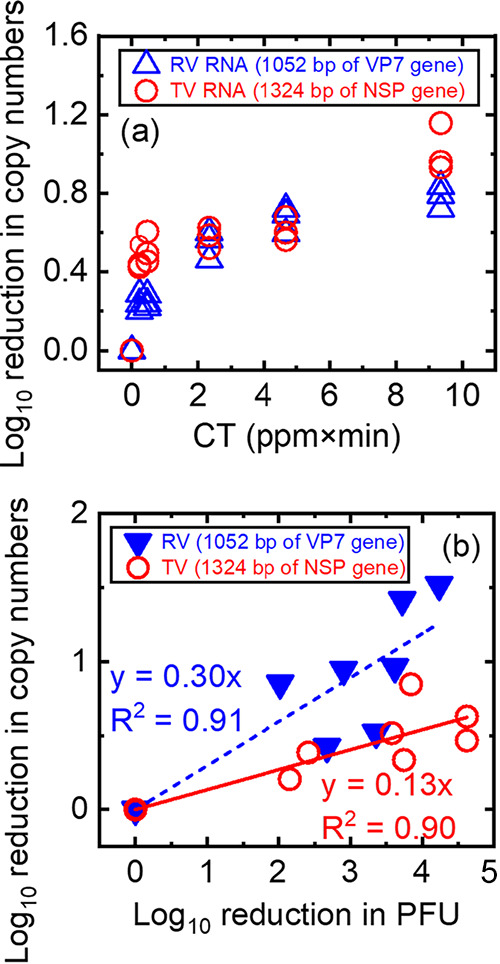
(a) Log10 reduction in copy numbers of the TV NSP gene or the RV VP7 gene after the naked viral genome was exposed to PAA. Log10 reduction in copy numbers was calculated as log10 N0/N (or −log10 N/N0) in which N0 is the copy number of the cDNA amplified from RNA isolated from untreated TV and RV, and N is the copy number of the cDNA amplified from RNA isolated from each virus post-PAA exposure. Data from three independent experiments were plotted. (b) Log10 reduction in copy numbers of the TV NSP gene and the RV VP7 gene as a function of the log10 reduction in PFU of TV and RV exposed to PAA.
Data in Fig. 5a showed that PAA could indeed damage naked viral genomes. The next question was to ask if PAA could damage capsid-associated viral RNA. To answer this question, we exposed whole virions to PAA, extracted RNA from these virions, and then used these virions as templates for RT-qPCR as a proxy for genome integrity. Again, the TV NSP and RV VP7 genes were used as the templates for RT-qPCR. The log10 reduction in PFU and log10 reduction in copy numbers were plotted for both TV and RV (Fig. 5b). Linear correlations were found between the log10 reduction in PFU (x axis) and log10 reduction in copy numbers (y axis) for both studied viruses. The slopes of these regression lines give quantitative information about the genome damage of these viruses after exposure to PAA. The significantly higher slope observed for RV data than for TV data (RV, 0.3 ± 0.03; TV, 0.13 ± 0.0; P < 0.01) suggested that PAA induced more RV genome damage than TV genome damage at the same levels of inactivation. For example, a 4-log10 reduction in RV PFU was associated with approximately a 1-log10 reduction in RV VP7 cDNA, but a 4-log10 reduction in TV PFU was associated with only a 0.5-log10 reduction for TV NSP cDNA. This finding suggested that PAA was more permeable to the capsids of RV than to those of TV such that PAA now has greater access to the genome of RV than of TV through the capsids.
In Fig. 5b, the slopes of the regression lines for both RV and TV are less than 1, implying that genome damage was not the sole cause of virus inactivation. Based on this, we propose that PAA may have multiple mechanisms to inhibit RV and TV replication by both targeting virus-host receptor interactions and by damaging viral genomes. Specifically, the slope of TV genome damage (0.13 ± 0.00) shown in Fig. 5b was smaller than the slope of TV binding loss (0.25 ± 0.02) shown in Fig. 3b, suggesting that PAA affected binding integrity of TV more than genome integrity, leading to TV inactivation. In contrast, for RV, we detected genome damage of RV exposed to PAA at CT values of 0 to 10 (ppm×min), as shown in Fig. 5b, while we employed a higher CT range (up to 60 ppm×min) for RV binding assays to detect binding loss of RV. This indicates that RV inactivation by PAA was caused more by genome damage than by binding damage.
DISCUSSION
Viruses are a major cause of foodborne diseases (22) and responsible for many reported outbreaks associated with fresh vegetables in many countries (47). For this reason, it is important to elucidate how viruses like TV and RV become inactivated by sanitizers. We demonstrated that TV has a higher resistance to PAA at pH 3 than RV (Fig. 1), and this resistance correlates with TV aggregation in PAA solution (Fig. 2).
The PAA used in this study was Tsunami 100. Undiluted Tsunami 100 as sold from Ecolab is composed of 15.2% peracetic acid, 11.2% hydrogen peroxide, and 30 to 60% acetic acid (48). PAA is created by the reaction between acetic acid and hydrogen peroxide. The equilibrium solution containing PAA, acetic acid, and hydrogen peroxide is the sanitizer commercially sold as peracetic acid. It is important to note that pure PAA is not available commercially because it is an explosive (49). For this reason, pure PAA is not used in the food industry, and it is not possible to examine the effect of pure PAA on viruses. PAA studies, including ours, examined virus inactivation mechanisms using commercially available PAA solutions, which are composed of PAA, hydrogen peroxide, and acetic acid; however, the percentage composition for each of these chemical components might differ depending on the manufacturer of PAA (2, 19). One question is whether the other components of Tsunami 100 may have antiviral effects either independently or synergistically with PAA. Such possibilities were not examined here. However, in three other publications, the addition of a surfactant such as sodium dodecyl sulfate (SDS) into a sanitizer (chlorine and levulinic acid) yielded a higher inactivation efficacy of viruses than surfactant alone (50–52).
When PAA is used for vegetable washing in the food industry, a high concentration of PAA from 30 to 80 ppm for a short period of time (up to 90 s) is typically used (53). The reason why such high PAA concentrations are used is that produce surface-attached viruses are more difficult to inactivate than suspended viruses (4, 15, 20). This difficulty to inactivate produce-attached viruses could be explained by many possible reasons, including specific interactions between viruses and produce surfaces (20). The reason for a short exposure time of PAA to vegetables is to avoid oxidation of vegetable surfaces. In this study, to examine the virus inactivation mechanisms caused by PAA, we intentionally set PAA concentrations lower than the established guidelines for vegetable washing. Specifically, we used PAA concentrations of 1 to 3.5 ppm for RV and 10 ppm for TV.
As a strong oxidant, PAA oxidizes virus capsid proteins and genetic material, resulting in virus inactivation (2, 19). The oxidizing potential of PAA is stronger than chlorine but less that of than ozone (10). The protonated form of PAA, PAAH, is a stronger oxidant than the deprotonated form PAA− (10). At pH 3, which is lower than a pKa of 8.2, PAAH is the dominant species (5, 54). In this study, we used Tsunami 100, and the pH of Tsunami 100 in undiluted form is 1.8; once diluted, the pH is designed to be around 2.8 to 4.6 (48), depending on the target concentration for vegetable washing. In this study, we found that PAA solution at pH 3 induced TV aggregation, resulting in high resistance of TV to PAA. For this reason, balancing the pH of PAA and aggregation of viruses is important in designing effective PAA sanitation.
In the food industry, acidified sanitizers other than PAA, such as acidified sodium chlorite (ASC), lactic acid, acetic acid, and malic acid, are also used for their antimicrobial effect (5). The pH of these acid-based sanitizers ranges from 2.1 to 3.1, depending on the concentrations (3, 5). Moreover, previous studies also reported the inactivation of RV and TV at low pH. At pH 2, TV had less than a 1-log10 reduction in infectivity after 1 h (55) although RV (strains C486, WC3, and NCDV) had approximately a 3-log10 inactivation after 10 min (56). At pH 3, TV did not have a significant reduction in infectivity after 1 h (55), while RV (same strains as above) had a 2-log10 inactivation after 10 min (56). Both TV and HuNoV are members of the Caliciviridae family (21), and TV proteins are 22 to 26% similar to HuNoV proteins (25). HuNoV virus-like particles (VLPs) also are reported to aggregate at pH 3 (57). We also observed TV aggregation at pH 3 without PAA (Fig. 2b). Based on our findings and previous studies (57), the use of acidified disinfectants may generate favorable conditions for HuNoV to aggregate, which may prevent HuNoV from being inactivated. Thus, the potential aggregation of HuNoV in solutions of other organic acid sanitizers should be considered.
We determined that PAA treatment of TV or RV compromised the ability of each virus to bind to PGM-MBs and also destabilized portions of each genome. For example, treatment with PAA at a CT of 287 (ppm×min) reduced TV PGM-MB interactions by 1 log10 (Fig. 3a). These data agree with the previously reported results that a 1-log10 reduction of HuNoV binding to PGM-MBs was observed after PAA treatment at 195 ppm for 1 min (19). When HuNoV was treated for up to 1 h with 4% hydrogen peroxide (H2O2), a component of the PAA mixture, only a 0.1-log10 binding loss to PGM-MBs was observed (19). Thus, the loss of TV-PGM-MBs interactions would be minimally affected by H2O2, and PAA was more responsible than H2O2 for reduced binding integrity of TV.
For RV, a 0.4-log10 binding loss was observed when RV was treated with PAA at a CT of 19 (ppm×min) compared to the level for untreated RV (Fig. 4a). Under these conditions, only 58% of PAA-exposed RV bound to sialic acid (Fig. 4b and Table S1 in the supplemental material). The current hypothesis is that PAA may oxidize RV VP8* in such a manner that VP8* no longer interacts with sialic acid, and this is an area of future investigations. Please note that VP8* is the protein exposed after trypsin cleavage of VP4 (43). When we used the same strain of RV in our previous study, where intact VP4 instead of VP8* was used, we observed lower inactivation efficacy of RV with PAA (20). A 4-log10 inactivation of RV with VP4 intact required exposure to PAA at 50 ppm for 30 s to 8 min. For the same inactivation level of RV with VP8*, only 3.5 ppm of PAA over 3 min was required. This observation suggests that the cleavage of RV VP4 allowed higher inactivation, indicating that damaging VP8* facilitates the inactivation of RV.
To examine the capsid integrity of viruses bound to PGM-MBs, we incubated untreated and PAA-treated viruses with RNase prior to the binding assay. An alternative method to identify capsid integrity uses propidium monoazide (PMA) instead of RNase. In this case, PMA would bind only to viral RNA that is either not associated with capsids or when capsids are permeable. PMA-RNA complexes cannot be amplified by RT-qPCR (58). However, difficulties with the PMA method have been reported (58–61). These difficulties include optimization (58), conflicting results between bacteriophage MS2 and MNV (59), and inconsistent outcomes depending on the secondary structure of the viral nucleic acid (60). In addition, RNase treatments yield more consistent results than PMA in examinations of the capsid integrity of RV and norovirus (62). For these reasons, we chose to use RNase treatment to determine the capsid integrity of viruses bound to PGM-MBs.
We determined that PAA treatment of TV and RV caused genome damage of each virus in this study. In addition, previously it was shown that PAA at higher pHs (6.5, 7.5, and 8.5) also caused genome damage to both MNV and MS2 (63), suggesting that PAA at higher pHs also damages viral genomes. Moreover, the same amount of exposure to PAA inactivated more MNV than MS2 and also caused more damage on MNV (63), indicating that resistance to PAA depends on the virus type, which is in agreement with our findings in this study.
In summary, we found that Tulane virus (TV, a surrogate of human norovirus) formed aggregates in PAA at pH 3 (conditions relevant to commercial vegetable washing), and this likely contributes to TV being more resistant to PAA disinfection than rotavirus (RV). In contrast, RV formed much smaller aggregates and remained susceptible to PAA disinfection. We have examined how RV and TV become inactivated by PAA. Interestingly, in the proposed PAA inactivation mechanisms shown in Fig. 6, RV inactivation by PAA is caused more by genome than by binding damage. In contrast, PAA caused TV inactivation as a result of primarily binding loss rather than genome damage. These findings illustrate that no one condition or concentration of PAA has the same effect on viruses, and therefore PAA may not be effective against all viral contaminants. We propose that TV may be a favorable model virus for HuNoV because TV has shown increased resistance to PAA in this study and to other various treatments, including chlorine and alcohols, in previous studies (21, 64), in addition to the capsid similarity of TV and HuNoV (25, 61).
FIG 6.

Proposed PAA inactivation mechanisms of TV and RV.
MATERIALS AND METHODS
RV and TV propagation.
Rotavirus (RV) strain OSU (VR-892) was purchased from the ATCC (Manassas, VA). Trypsin-activated RV was propagated in MA104 cells with minimal essential medium (MEM) containing trypsin at a final concentration of 0.5 μg/ml at 37°C in a 5% CO2 incubator, as described previously (20). Tulane virus (TV) was kindly provided by Xi Jiang (Cincinnati Children’s Hospital). TV was propagated in the monkey kidney epithelial MA104 cell line, which is similar to the monkey epithelial LLC-MK2 cell line widely used to propagate TV. MA104 cells are likely permissive to TV replication because MA104 cells express the sialic acid moiety that TV interacts with (65). Two previous publications from our research group described the use of MA104 cells to propagate TV and the confirmation of the viruses by Sanger sequencing (15, 66). For either RV or TV, infected MA104 cellular monolayers were incubated at 37°C in a 5% CO2 incubator in T175 cell culture flasks (15, 66). Infected cells and their supernatants were collected at 3 to 5 days postinfection, a time when cytopathic effect (CPE) has occurred and infected cells are detached from flask surfaces. This solution was subjected to three cycles of freezing and thawing to release viruses from infected cells. Suspensions were transferred to centrifuge tubes and centrifuged at 1,500 × g for 12 min. Virus-containing supernatants were collected. Viruses were purified from supernatants by the sucrose cushion method using ultracentrifugation as described previously (15, 16). Purified virus stocks were aliquoted and stored at –80°C. The RV VP4 and TV nonstructural polyprotein (NSP) genes were sequenced by the high-throughput sequencing and genotyping unit at the University of Illinois at Urbana-Champaign using the Sanger method (primers are shown in Table 1), as previously described (16, 66). Sequences for each virus were aligned using Sequencer software (Gene Codes Corporation, Ann Arbor, MI), and the aligned sequence of each virus was compared with the sequence in the GenBank database (accession number KJ450845.1 for the RV strain OSU VP4 gene and EU391643 for the TV NSP gene) to ensure no cross-contamination of virus stocks.
TABLE 1.
Primer sequences for used for Sanger sequencing
Quantification of virus infectivity using plaque assays.
The titers of infectious RV and TV particles were determined by using plaque assays, and results were recorded as the number of PFU. For RV, virus was activated with trypsin at a final concentration of 10 μg/ml at 37°C for 30 min, and plaque assays were conducted as described previously (20). For TV, serially diluted TV solutions were added to confluent monolayers of MA104 cells in a six-well plate and incubated at 37°C for 1 h in a CO2 incubator. Next, the viral solutions were aspirated and then replaced with an overlay of MEM containing 1% agarose, 7.5% sodium bicarbonate, 15 mM HEPES, and antibiotic-antimycotic (10,000 U/ml of penicillin, 10,000 μg/ml of streptomycin, and 25 μg/ml of Gibco amphotericin B) (Thermo Fisher Scientific, Waltham, MA). The six-well plates were incubated at 37°C for 48 h in a 5% CO2 incubator. Next, cellular monolayers were fixed with 10% formaldehyde in phosphate-buffered saline (PBS) at room temperature for 1 h, and then stained with 0.05% crystal violet in 10% ethanol for 20 min at room temperature. Visualized plaques were counted.
PAA disinfection experiments.
A commercially available PAA-containing solution, Tsunami 100 (Ecolab, Saint Paul, MN), was used, and this product is approved by the U.S. Food and Drug Administration (FDA) for postharvest fruit and vegetable washing (67). PAA is also permitted for use in organic vegetable sanitation (4). A high concentration of PAA (30 to 80 ppm) is used for vegetable washing in the food industry (53). PAA is an organic peroxide-based, colorless liquid with a low pH. PAA is often used without pH adjustment for produce washing (68) as the effectiveness of PAA is less dependent on pH than chlorine (5). Thus, to use PAA in a way similar to its use in the food industry, the pH of PAA was not adjusted in this study. Tsunami 100, the sanitizer used in this study, contains 15.2% peracetic acid, 30 to 60% acetic acid, and 11.2% hydrogen peroxide (48). Peracetic acid (CH3COOOH) is generated by the reaction between acetic acid (CH3COOH) and hydrogen peroxide (H2O2) (10). The protonated form of peracetic acid (CH3COOH; also expressed as PAAH) is the reactive form of PAA (10). Below the pH of PAA used in this study (3.0 to 3.33), the speciation of PAA, which has a pKa of 8.2, is 100% PAAH (5, 54). PAA was prepared by diluting Tsunami 100 with water purified with a Milli-Q water purification system (i.e., nanopure water). The concentration of PAA-based solutions was measured by a peracetic acid test kit (Vacu-vials instrumental kit; CHEMetrics, Midland, VA).
Purified RV and TV were used for these studies, and starting titers were 1.2 × 108 PFU/ml for RV and 9.3 × 107 PFU/ml for TV. Ten-microliter aliquots of each virus were incubated with 990 μl of PAA-containing solutions for a total volume of 1 ml. For RV, a solution containing 1 to 3.5 ppm of PAA at pH 3.2 to 3.3 was used. For TV, a solution containing 10 ppm of PAA at pH 3.0 was used. Incubations occurred between 0 and 3 min for RV and 0 to 60 min for TV, as indicated for different experiments. To quench the PAA reaction, 495 μl of 600 mg/liter sodium thiosulfate containing 16 mg/liter catalase in 0.1 M phosphate buffer (PB; a mixture of 0.1 M NaH2PO4 and 0.1 M Na2HPO4) at pH 7.1 was immediately added to each tube. As a control, separate aliquots of RV and TV were treated with nanopure water lacking PAA and the quencher. Three replicates were conducted for each condition. Following disinfection, RV was activated with trypsin at a final concentration of 10 μg/ml for 30 min at 37°C. Plaque assays for both TV and trypsin-activated RV were then conducted as described above.
To test if the reaction by-product of PAA and the quencher in 0.1 M PB can inactivate RV, which was more sensitive to PAA than TV, we conducted a preliminary control experiment wherein we mixed 990 μl of PAA (2.2 ppm, pH 3.25) and 495 μl of the quencher in 0.1 M PB (pH 7.1) and exposed this mixture to 10 μl of RV for 30 min at room temperature. For the control, instead of the mixture of PAA and quencher, a corresponding volume of nanopure water at pH 7.2 was added to 10 μl of RV. When the quencher in 0.1 M PB was used, the final pH of the mixture of PAA and the quencher was pH 7. Based on results from the plaque assays, a reduction of infectivity compared to the level of the control was not observed (P = 0.235, n = 3) (control, 106.3±0.2 PFU/ml; samples, 106.5±0.2). Thus, we used the quencher dissolved in 0.1 M PB for this study.
Measurement of RV and TV aggregates in PAA solution by using DLS.
The extent of RV and TV aggregation in PAA-containing solutions was measured by using a dynamic light scattering (DLS) ZS90 Zetasizer (Malvern, UK). This measurement gives the aggregate diameter of the virions. Ten microliters RV or TV virus stocks was added to 990 μl of a solution containing 10 ppm PAA at pH 3. The final concentration of RV or TV was approximately 106 PFU/ml. This concentration of RV or TV was sufficient to give a single peak centered at the aggregate diameter of the virions measured in three separate experiments (see Fig. S1a to c in the supplemental material). This mixture was immediately subjected to the measurement for aggregate diameters every 4 min for 28 min. Three biological replicates were conducted for each virus.
Binding assays using PGM-MBs.
We previously published assays that detect the ability of RV and TV to bind to their host receptors using a cell-free binding assay (15, 16). Specifically, we used a cell-free binding matrix (porcine gastric mucin conjugated with magnetic beads [PGM-MBs]), which contains host receptors (sialic acid and HBGAs) for RV and TV (41, 42). PGM-MBs were prepared as described previously (16). Three types of binding assays were conducted as described below.
(i) PGM-MBs binding assay with PAA-exposed RV and TV.
To determine the ability of PAA-exposed RV and TV to bind to PGM-MBs, we added 50 μl of PAA-exposed RV (treated with trypsin) or TV to a 1.5-ml low-adhesion centrifuge tube containing 50 μl of PGM-MBs and 900 μl of PBS. This ratio of virions to PGM-MBs was determined previously using calibration curves (15, 16). For the control, 50 μl of untreated RV (treated with trypsin) or TV was used. Virus and PGM-MBs were incubated on a VWR rocking platform shaker at room temperature for 30 min at 8 rpm. PGM-MBs were then collected by using a magnetic separation rack (New England Biolabs, Ipswich, MA). PGM-MBs were washed three times with 1 ml of PBS to remove unbound virions. Washed PGM-MBs were resuspended in 140 μl of nuclease-free water; then viral RNA was extracted from the resuspended PGM-MBs using a QIAmp viral RNA minikit (Qiagen, Hilden, Germany) according to the manufacturer's protocol.
(ii) PGM-MB binding assay with PAA-exposed RV and TV treated with RNase.
We conducted a second set of binding assays in which PAA-exposed viruses subsequently were treated with RNase. After RNase treatment, these viruses were incubated with PGM-MBs. The logic is as follows: if PAA treatment altered capsids to the extent that the capsids became permeable to RNase, then RNase should degrade viral RNA (15, 16). Using this approach, RNase treatment versus no treatment can differentiate between viruses with intact capsids versus virus capsids that have lost their integrity.
RNase treatment of viruses was conducted as previously described (16). Briefly, 50 μl of untreated or PAA-exposed virus was incubated with 50 μl of RNase A/T1 mix (Thermo Fisher Scientific) at a concentration of 40 μg/ml RNase-A and 100 U/ml RNase-T1 for 30 min at 37°C. RNase activity was halted by adding 50 μl of SUPERase·In RNase inhibitor (Thermo Fisher Scientific) at a concentration of 2 U/μl to each reaction mixture for 20 min at room temperature. After the RNase treatment, the virions (without or with previous exposure to PAA) were incubated with PGM-MBs as described above. Viruses bound to PGM-MBs were resuspended in 140 μl of nuclease-free water and subjected to RNA extraction using a QIAmp viral RNA minikit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. Only the RNA from viruses that (i) have intact capsids and (ii) attach to PGM-MBs would be quantified by RT-qPCR (see below for the methodology). These values were then compared to PAA-treated viruses that were not incubated with RNase to identify the quantity of PGM-MB-bound viruses with intact capsids.
(iii) Neuraminidase-treated PGM-MB binding assay with PAA-exposed RV.
To examine the interactions between PAA-exposed RV and sialic acid, we conducted a binding assay wherein PGM-MBs were pretreated with neuraminidase, an enzyme that cleaves sialic acids (20), before incubation with RV. This treatment digests sialic acids in the PGM-MBs, providing a means to show the specificity of virus-sialic acid receptor interactions in the cell-free binding assays. Fifty microliters of PGM-MBs was incubated with 54 μl of a PBS-based solution containing 63 mU/ml of V. cholerae neuraminidase for 1 h at 37°C. Next, PGM-MBs were washed with 1 ml of PBS to remove residual neuraminidase and then collected by using a magnetic separation rack. PGM-MBs were suspended in 950 μl of PBS, and 50 μl of untreated or PAA-treated RV activated with trypsin was added to this suspension, followed by incubation at 37°C for 30 min at 8 rpm. PGM-MBs were washed and resuspended in the same manner as described above, and then RNA extraction was performed for each sample, and samples were subjected to one-step RT-qPCR. The differences in untreated versus PAA-treated virions bound to PGM-MBs gave information as to whether PAA compromised virus binding to sialic acid receptors.
(iv) One-step RT-qPCR to quantify viruses bound to PGM-MBs.
RNA from the above-described assays was used for one-step RT-qPCR assays to amplify virus genomes. For RV RNA samples, 5 μl of the extracted double-stranded RNA (dsRNA) genomes was denatured at 95°C for 5 min and then immediately incubated on ice. This denaturation step was not required for TV genomes because TV has a single-stranded RNA genome. Three microliters of RNA (denatured RNA for RV and nondenatured RNA for TV) was incubated with 0.125 μl of iScript reverse transcriptase, 5 μl of 2× iTaq universal SYBR green reaction mix (Bio-Rad Laboratories, Hercules, CA), 0.3 μl of 10 μM forward primer, 0.3 μl of 10 μM reverse primer, and 1.275 μl of nuclease-free water, for a total volume of 10 μl for each RT-qPCR. To generate a standard curve for each viral gene of interest, we serially diluted a known concentration of cDNA containing the NSP3 gene for RV or the NSP gene for TV (Integrated DNA Technologies, Coralville, IA) and quantified these serially diluted samples in parallel with the experimental viral RNA samples. Thermal cycling conditions for both the NSP3 gene of RV and the NSP gene of TV were as follows: 1 cycle at 50°C for 10 min and 95°C for 1 min and then 40 cycles of 95°C for 10 s and 60°C for 30 s, followed by melting curve analysis. The qPCR efficiency ranged from 90 to 95%. In addition, we previously have observed qPCR amplification inhibition from binding samples (15). Thus, to avoid qPCR inhibition, we routinely dilute RNA extracts 100-fold with nuclease-free water prior to subjecting RNA to one-step RT-qPCR (69). The sequences of primer and standards used for the binding assays are shown in Table 2. Note that the target amplicon sizes for both TV and RV were approximately 110 and 120 bp, respectively, and we have found that this product is too short of a region to accurately assess if the viral genome is damaged (66). Thus, data quantified by one-step RT-qPCR indicate the number of viruses bound to PGM-MBs, but not necessarily genome damage.
TABLE 2.
Primers and standard sequences used for one-step RT-qPCR to quantify viruses bound to PGM-MBs
| Target gene | Primer name | Sequence (5′–3′) | Position in the gene (nt) | Amplicon size (bp) | Reference | RT-qPCR conditions |
|---|---|---|---|---|---|---|
| RV strain OSU NSP3a | JVKF | CAGTGGTTGATGCTGAAGAT | 17–37 | 122 | 72 | 1 cycle at 50°C for 10 min and 95°C for 1 min; then 40 cycles at 95°C for 10 s and 60°C for 30 s |
| JVKR | TCATTGTAATCATATTGAATACCCA | 122–138 | ||||
| TV NSPb | TV-NSP-qPCR-F2 | GGCAGCTGGGAAGAAATCTG | 2774–2793 | 111 | 15 | 1 cycle at 50°C for 10 min and 95°C for 1 min; then 40 cycles at 95°C for 10 s and 60°C for 30 s |
| TV-NSP-qPCR-R2 | CCTGCTGTGTGAATGCCTAC | 2865–2884 |
qPCR standard sequence (5′–3′) acquired from GenBank accession number DQ146697.1: GGCTTTTAATGCTTTTCAGTGGTTGATGCTCAAGATGGAGTCTACTCAACAGATGGCATCTTCTATTATTAACTCTTCTTTTGAAGCTGCAGTTGTCGCTGCAACTTCTACATTAGAATTAATGGGTATTCAATATGATTATAATGAAGTATACACTAGAGTTAAAAGTAAGTTTGATTTTGTAATGGATGATTCTGGTG. The forward (F) primer attaches to the sequence indicated in boldface, and the reverse primer (R) attaches to the complementary strand indicated by underlining.
qPCR standard sequence (5′–3′) acquired from GenBank accession number EU391643.1: CCGTGGTTGTGCGCAGTATTGGAAACACAAACATTGCTGGGAAATTCCTCAACGTCTTCACAGGTACAGTTGTGGCAGCTGGGAAGAAATCTGACGGCCTCGGGTCTGAACCAGGAGACTGTGGCTCACCATATCTTAAATTTGTTAATGGAAAACCAACTCTTGTAGGCATTCACACAGCAGGCAGCTACACTACCAACCAGGTTGCAGGCTTAGTGATACCTTCTAGATTCAACCTTG. The forward (F) primer attaches to the sequence indicated in boldface, and the reverse primer (R) attaches to the complementary strand indicated by underlining.
Effect of PAA on naked viral TV and RV RNA genomes.
To determine if PAA can damage viral RNA genomes that are not encapsidated, we extracted viral RNA from 140 μl of intact RV or TV virions using a QIAmp viral RNA minikit (Qiagen). Next, 1.7 μl of RV RNA (106.7 VP7 copy/μl) or TV RNA (106.9 NSP copy/μl) was exposed to 3.5 μl of 1.3 ppm PAA at pH 3.2 for 0.5, 1, 5, 10, or 20 min. Next, 1.7 μl of 600-mg/liter sodium thiosulfate containing 16 mg/liter catalase in 0.1 M PB was immediately added to each solution to quench the PAA reaction. In a separate set of tubes used for controls, instead of PAA and the quencher, a corresponding volume of nuclease-free water was added to viral RNAs. Three replicates were conducted for each condition. The integrity of the PAA-exposed viral RNA was evaluated by using two-step RT-qPCR, as described previously (70). Briefly, during the first RT step, only undamaged/intact viral RNA can be reverse transcribed by reverse transcriptase to cDNA (15). Then, cDNA from this first step can be quantified by the second qPCR step.
Before the RT step, rotavirus dsRNA was denatured at 95°C for 5 min and immediately incubated on ice. For the RT step, we used a ProtoScript First Strand cDNA Synthesis kit (New England BioLabs, Ipswich, MA) to amplify a 1,052-bp segment of the RV VP7 gene and a 1,324-bp segment for the nonstructural polyprotein (NSP) gene of TV. Each RT reaction mix included the following: 3 μl of RNA, 2 μl of 10 μM either OSU-VP7-R or TV-NSP-R primer, 10 μl of Moloney murine leukemia virus (M-MuLV) reaction mix (2×), 2 μl of M-MuLV enzyme mix (10×), and 3 μl of nuclease-free water, for a 20-μl volume total. Reaction mixtures were incubated at 42°C for 60 min and then at 80°C for 5 min. After the RT step, we added 30 μl of nuclease-free water to each reaction mixture, according to the manufacturer's protocol. To quantify cDNA that was generated during the RT reaction, we used qPCR. Each qPCR consisted of 2 μl of cDNA, 7.5 μl of SYBR green PCR Master Mix (Applied Biosystems, Foster City, CA), 0.3 μl of 10 μM forward primer (either OSU-VP7-qPCR-F1 or TV-NSP-qPCR-F), 0.3 μl of 10 μM reverse primer (either OSU-VP7-qPCR-R1 or TV-NSP-qPCR-R), and 4.9 μl of nuclease-free water. The thermal cycle conditions for both the TV NSP gene and the RV VP7 gene were as follows: 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 1 min, followed by melting curve analysis. The qPCR efficiency ranged from 90% to 93%. Primer and standard sequences for the genome damage assays are in Table 3.
TABLE 3.
Primers and standard sequences used for two-step RT-qPCR to detect PAA-induced genome damage of RV and TV
| Target gene | Step no.a | Primer name | Sequence (5′–3′) | Position in the gene (nt) | Amplicon size (bp) | PCR conditions |
|---|---|---|---|---|---|---|
| RV strain OSU VP7b | 1 | OSU-VP7-R | GTTAGAACTGTATGATGTGACC | 1041–1062 | 1,052 | 42°C for 60 min and 80°C for 5 min |
| 2 | OSU-VP7-qPCR-F1 | GAGAGAATTTCCGACTGG | 11–28 | 131 | 95°C for 10 min; then 40 cycles of 95°C for 15 s and 60°C for 1 min | |
| OSU-VP7-qPCR-R1 | GTCCATTGTTCTAGTAACTGA | 121–141 | ||||
| TV NSPc | 1 | TV-NSP-R | TGAGGTCTTCTTCAACGCCC | 4078–4098 | 1,324 | 42°C for 60 min and 80°C for 5 min |
| 2 | TV-NSP-qPCR-F | GGCAGCTGGGAAGAAATCTG | 2774–2794 | 111 | 95°C for 10 min; then 40 cycles of 95°C for 15 s and 60°C for 1 min | |
| TV-NSP-qPCR-R | CCTGCTGTGTGAATGCCTAC | 2865-2885 |
Step 1 consists of reverse transcription of the target gene, and step 2 consists of qPCR of the target gene.
qPCR standard sequence (5′–3′) acquired from GenBank accession number KJ450849.1: GGCTTTAAAAGAGAGAATTTCCGACTGGCTATCGGATAGCCTTTTTAATGTATGGTATTGAATATACCACAGTTCTAACTTTTTTGATATCGCTTGTATTTGTCAATTATATACTGAAATCAGTTACTAGAACAATGGACTTTATCATTTATAGATTCTTATTGGTTATAGTCGTACTTGCACCGCTCA. The forward (F) primer attaches to the sequence indicated in boldface, and the reverse primers (R) attach to the complementary strand indicated by underlining.
qPCR standard sequence (5′–3′) acquired from GenBank accession number EU391643.1: CCGTGGTTGTGCGCAGTATTGGAAACACAAACATTGCTGGGAAATTCCTCAACGTCTTCACAGGTACAGTTGTGGCAGCTGGGAAGAAATCTGACGGCCTCGGGTCTGAACCAGGAGACTGTGGCTCACCATATCTTAAATTTGTTAATGGAAAACCAACTCTTGTAGGCATTCACACAGCAGGCAGCTACACTACCAACCAGGTTGCAGGCTTAGTGATACCTTCTAGATTCAACCTTG. The forward (F) primer attaches to the sequence indicated in boldface, and the reverse primers (R) attach to the complementary strand indicated by underlining.
Effect of PAA on encapsidated RNA within TV and RV virions.
To detect PAA-induced viral genomic damage, RNA was extracted from 10 μl of PAA-exposed RV or TV by QIAmp viral RNA minikit (Qiagen). RNA extracts were subjected to two-step RT-qPCR as described above. As we exposed PAA to TV and RV before this experiment, the concentration of virus (either in PFU or genomic copies) varied depending on the sample; for RV, the concentration was 4.7 × 101 to 8 × 105 PFU/ml, and that for TV was 1.5 × 101 to 6.2 × 105 PFU/ml.
Statistical analyses.
Data from PAA virus inactivation, binding assays, and genome damage on PAA-exposed viruses were fitted by a linear regression model and then compared by an F test in OriginPro, version 2018b. For genome damage of PAA-exposed naked viral RNA, we conducted two-way nonparametric tests using the R software package with the WRS2 package (71).
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by USDA 2017-68007-26307, EPA/USDA 2017-39591-27313 to T.H.N. and J.L.S.
We thank Xi Jiang from Cincinnati Children’s Hospital for providing Tulane virus, and Hao Feng and Sindy Paola Palma-Salgado from the Department of Food Science and Human Nutrition at the University of Illinois at Urbana-Champaign for providing the sanitizer Tsunami 100.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Carstens CK, Salazar JK, Darkoh C. 2019. Multistate outbreaks of foodborne illness in the United States associated with fresh produce from 2010 to 2017. Front Microbiol 10:2667. doi: 10.3389/fmicb.2019.02667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fraisse A, Temmam S, Deboosere N, Guillier L, Delobel A, Maris P, Vialette M, Morin T, Perelle S. 2011. Comparison of chlorine and peroxyacetic-based disinfectant to inactivate Feline calicivirus, Murine norovirus and Hepatitis A virus on lettuce. Int J Food Microbiol 151:98–104. doi: 10.1016/j.ijfoodmicro.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 3.Martínez-Sánchez A, Allende A, Bennett RN, Ferreres F, Gil MI. 2006. Microbial, nutritional and sensory quality of rocket leaves as affected by different sanitizers. Postharvest Biol Technol 42:86–97. doi: 10.1016/j.postharvbio.2006.05.010. [DOI] [Google Scholar]
- 4.Sapers GM. 2014. Dsinfection of contaminated produce with conventional washing and sanitizing technology, p 389–431. In Matthews KR, Sapers GM, Gerba CP (ed), The produce contamination problem: causes and solutions. Elsevier, San Diego, CA. [Google Scholar]
- 5.Herdt J, Feng H. 2009. Aqueous antimicrobial treatments to improve fresh and fresh-cut produce safety, p 167–190. In Fan X, Niemira BA, Doona DJ, Feeherry FE, Gravani RB (ed), Microbial safety of fresh produce. Blackwell Publishing, Ames, IA. [Google Scholar]
- 6.Komaki Y, Simpson AM, Choe JK, Plewa MJ, Mitch WA. 2018. Chlorotyrosines versus volatile byproducts from chlorine disinfection during washing of spinach and lettuce. Environ Sci Technol 52:9361–9369. doi: 10.1021/acs.est.8b03005. [DOI] [PubMed] [Google Scholar]
- 7.Van Haute S, Sampers I, Holvoet K, Uyttendaele M. 2013. Physicochemical quality and chemical safety of chlorine as a reconditioning agent and wash water disinfectant for fresh-cut lettuce washing. Appl Environ Microbiol 79:2850–2861. doi: 10.1128/AEM.03283-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ölmez H, Kretzschmar U. 2009. Potential alternative disinfection methods for organic fresh-cut industry for minimizing water consumption and environmental impact. LWT Food Sci Technol 42:686–693. doi: 10.1016/j.lwt.2008.08.001. [DOI] [Google Scholar]
- 9.Ölmez H. 2017. Environmental impacts of minimally processed refrigerated fruits and vegetables’ industry, p 747–756. In Yildiz F, Wiley RC (ed), Minimally processed refrigerated fruits and vegetables. Springer, Boston, MA. [Google Scholar]
- 10.Zhang K, Zhou X, Du P, Zhang T, Cai M, Sun P, Huang C-H. 2017. Oxidation of beta-lactam antibiotics by peracetic acid: Reaction kinetics, product and pathway evaluation. Water Res 123:153–161. doi: 10.1016/j.watres.2017.06.057. [DOI] [PubMed] [Google Scholar]
- 11.Block SS. 2000. Disinfection, sterilization, and preservation. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 12.Lee W-N, Huang C-H. 2019. Formation of disinfection byproducts in wash water and lettuce by washing with sodium hypochlorite and peracetic acid sanitizers. Food Chem X 1:100003. doi: 10.1016/j.fochx.2018.100003. [DOI] [Google Scholar]
- 13.Allwood PB, Malik YS, Hedberg CW, Goyal SM. 2004. Effect of temperature and sanitizers on the survival of feline calicivirus, Escherichia coli, and F-specific coliphage MS2 on leafy salad vegetables. J Food Prot 67:1451–1456. doi: 10.4315/0362-028x-67.7.1451. [DOI] [PubMed] [Google Scholar]
- 14.Lukasik J, Bradley ML, Scott TM, Dea M, Koo A, Hsu WY, Bartz JA, Farrah SR. 2003. Reduction of poliovirus 1, bacteriophages, Salmonella montevideo, and Escherichia coli O157:H7 on strawberries by physical and disinfectant washes. J Food Prot 66:188–193. doi: 10.4315/0362-028x-66.2.188. [DOI] [PubMed] [Google Scholar]
- 15.Fuzawa M, Araud E, Li J, Shisler JL, Nguyen TH. 2019. Free chlorine disinfection mechanisms of rotaviruses and human norovirus surrogate Tulane virus attached to fresh produce surfaces. Environ Sci Technol 53:11999–12006. doi: 10.1021/acs.est.9b03461. [DOI] [PubMed] [Google Scholar]
- 16.Araud E, Shisler JL, Nguyen TH. 2018. Inactivation mechanisms of human and animal rotaviruses by solar UVA and visible light. Environ Sci Technol 52:5682–5690. doi: 10.1021/acs.est.7b06562. [DOI] [PubMed] [Google Scholar]
- 17.Brie A, Bertrand I, Meo M, Boudaud N, Gantzer C. 2016. The effect of heat on the physicochemical properties of bacteriophage MS2. Food Environ Virol 8:251–261. doi: 10.1007/s12560-016-9248-2. [DOI] [PubMed] [Google Scholar]
- 18.Wigginton KR, Pecson BM, Sigstam T, Bosshard F, Kohn T. 2012. Virus inactivation mechanisms: impact of disinfectants on virus function and structural integrity. Environ Sci Technol 46:12069–12078. doi: 10.1021/es3029473. [DOI] [PubMed] [Google Scholar]
- 19.Kingsley DH, Vincent EM, Meade GK, Watson CL, Fan X. 2014. Inactivation of human norovirus using chemical sanitizers. Int J Food Microbiol 171:94–99. doi: 10.1016/j.ijfoodmicro.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 20.Fuzawa M, Ku KM, Palma-Salgado SP, Nagasaka K, Feng H, Juvik JA, Sano D, Shisler JL, Nguyen TH. 2016. Effect of leaf surface chemical properties on efficacy of sanitizer for rotavirus inactivation. Appl Environ Microbiol 82:6214–6222. doi: 10.1128/AEM.01778-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cromeans T, Park GW, Costantini V, Lee D, Wang Q, Farkas T, Lee A, Vinje J. 2014. Comprehensive comparison of cultivable norovirus surrogates in response to different inactivation and disinfection treatments. Appl Environ Microbiol 80:5743–5751. doi: 10.1128/AEM.01532-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Jones JL, Griffin PM. 2011. Foodborne illness acquired in the United States—major pathogens. Emerg Infect Dis 17:7–15. doi: 10.3201/eid1701.P11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, Gonzalez-Hernandez MB, Iovine NM, Wobus CE, Vinje J, Tibbetts SA, Wallet SM, Karst SM. 2014. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346:755–759. doi: 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, Neill FH, Blutt SE, Zeng XL, Qu L, Kou B, Opekun AR, Burrin D, Graham DY, Ramani S, Atmar RL, Estes MK. 2016. Replication of human noroviruses in stem cell-derived human enteroids. Science 353:1387–1393. doi: 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farkas T, Sestak K, Wei C, Jiang X. 2008. Characterization of a rhesus monkey calicivirus representing a new genus of Caliciviridae. J Virol 82:5408–5416. doi: 10.1128/JVI.00070-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang D, Huang P, Zou L, Lowary TL, Tan M, Jiang X. 2015. Tulane virus recognizes the A type 3 and B histo-blood group antigens. J Virol 89:1419–1427. doi: 10.1128/JVI.02595-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Angel J, Franco MA, Greenberg HB. 2007. Rotavirus vaccines: recent developments and future considerations. Nat Rev Microbiol 5:529–539. doi: 10.1038/nrmicro1692. [DOI] [PubMed] [Google Scholar]
- 28.Tate JE, Burton AH, Boschi-Pinto C, Parashar UD, World Health Organization-Coordinated Global Rotavirus Surveillance Network. 2016. Global, regional, and national estimates of rotavirus mortality in children <5 years of age, 2000–2013. Clin Infect Dis 62(Suppl 2):S96–S105. doi: 10.1093/cid/civ1013. [DOI] [PubMed] [Google Scholar]
- 29.Prez VE, Martinez LC, Victoria M, Giordano MO, Masachessi G, Re VE, Pavan JV, Colina R, Barril PA, Nates SV. 2018. Tracking enteric viruses in green vegetables from central Argentina: potential association with viral contamination of irrigation waters. Sci Total Environ 637–638:665–671. doi: 10.1016/j.scitotenv.2018.05.044. [DOI] [PubMed] [Google Scholar]
- 30.van Zyl WB, Page NA, Grabow WO, Steele AD, Taylor MB. 2006. Molecular epidemiology of group A rotaviruses in water sources and selected raw vegetables in southern Africa. Appl Environ Microbiol 72:4554–4560. doi: 10.1128/AEM.02119-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aw TG, Wengert S, Rose JB. 2016. Metagenomic analysis of viruses associated with field-grown and retail lettuce identifies human and animal viruses. Int J Food Microbiol 223:50–56. doi: 10.1016/j.ijfoodmicro.2016.02.008. [DOI] [PubMed] [Google Scholar]
- 32.Quiroz-Santiago C, Vázquez-Salinas C, Natividad-Bonifacio I, Barrón-Romero BL, Quiñones-Ramírez EI. 2014. Rotavirus G2P[4] detection in fresh vegetables and oysters in Mexico City. J Food Prot 77:1953–1959. doi: 10.4315/0362-028X.JFP-13-426. [DOI] [PubMed] [Google Scholar]
- 33.Mattle MJ, Crouzy B, Brennecke M, Wigginton KR, Perona P, Kohn T. 2011. Impact of virus aggregation on inactivation by peracetic acid and implications for other disinfectants. Environ Sci Technol 45:7710–7717. doi: 10.1021/es201633s. [DOI] [PubMed] [Google Scholar]
- 34.Gerba CP, Betancourt WQ. 2017. Viral aggregation: impact on virus behavior in the environment. Environ Sci Technol 51:7318–7325. doi: 10.1021/acs.est.6b05835. [DOI] [PubMed] [Google Scholar]
- 35.Feng Z, Lu R, Yuan B, Zhou Z, Wu Q, Nguyen TH. 2016. Influence of solution chemistry on the inactivation of particle-associated viruses by UV irradiation. Colloids Surf B Biointerfaces 148:622–628. doi: 10.1016/j.colsurfb.2016.09.025. [DOI] [PubMed] [Google Scholar]
- 36.Gutierrez L, Nguyen TH. 2012. Interactions between rotavirus and Suwannee River organic matter: aggregation, deposition, and adhesion force measurement. Environ Sci Technol 46:8705–8713. doi: 10.1021/es301336u. [DOI] [PubMed] [Google Scholar]
- 37.Wang Q, Zhang Z, Saif LJ. 2012. Stability of and attachment to lettuce by a culturable porcine sapovirus surrogate for human caliciviruses. Appl Environ Microbiol 78:3932–3940. doi: 10.1128/AEM.06600-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.da Silva AK, Kavanagh OV, Estes MK, Elimelech M. 2011. Adsorption and aggregation properties of norovirus GI and GII virus-like particles demonstrate differing responses to solution chemistry. Environ Sci Technol 45:520–526. doi: 10.1021/es102368d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gutierrez L, Li X, Wang J, Nangmenyi G, Economy J, Kuhlenschmidt TB, Kuhlenschmidt MS, Nguyen TH. 2009. Adsorption of rotavirus and bacteriophage MS2 using glass fiber coated with hematite nanoparticles. Water Res 43:5198–5208. doi: 10.1016/j.watres.2009.08.031. [DOI] [PubMed] [Google Scholar]
- 40.Esseili MA, Saif LJ, Farkas T, Wang Q. 2015. Feline calicivirus, murine norovirus, porcine sapovirus, and Tulane virus survival on post-harvest lettuce. Appl Environ Microbiol 81:5085–5092. doi: 10.1128/AEM.00558-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shah HN, Gharbia SE. 2017. MALDI-TOF and tandem MS for clinical microbiology. John Wiley & Sons, Hoboken, NJ. [Google Scholar]
- 42.Li X, Chen H. 2015. Evaluation of the porcine gastric mucin binding assay for high-pressure-inactivation studies using murine norovirus and Tulane virus. Appl Environ Microbiol 81:515–521. doi: 10.1128/AEM.02971-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu X, Dang VT, Fleming FE, von Itzstein M, Coulson BS, Blanchard H. 2012. Structural basis of rotavirus strain preference toward N-acetyl- or N-glycolylneuraminic acid-containing receptors. J Virol 86:13456–13466. doi: 10.1128/JVI.06975-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baker M, Prasad BVV. 2010. Rotavirus cell entry, p 121–148. In Johnson EJ. (ed), Cell entry by non-enveloped viruses. Springer, Berlin, Germany. [Google Scholar]
- 45.Haselhorst T, Fleming FE, Dyason JC, Hartnell RD, Yu X, Holloway G, Santegoets K, Kiefel MJ, Blanchard H, Coulson BS, von Itzstein M. 2009. Sialic acid dependence in rotavirus host cell invasion. Nat Chem Biol 5:91–93. doi: 10.1038/nchembio.134. [DOI] [PubMed] [Google Scholar]
- 46.Coulson BS. 2015. Expanding diversity of glycan receptor usage by rotaviruses. Curr Opin Virol 15:90–96. doi: 10.1016/j.coviro.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 47.Li D, De Keuckelaere A, Uyttendaele M. 2015. Fate of foodborne viruses in the “farm to fork” chain of fresh produce. Compr Rev Food Sci Food Saf 14:755–770. doi: 10.1111/1541-4337.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.ECOLAB. 2017. Safety data sheet: Tsunami 100. www.ecolab.com.
- 49.U.S. Department of Agriculture. 2016. Peracetic acid handling/processing. https://www.ams.usda.gov.
- 50.Aydin A, Cannon JL, Zhao T, Doyle MP. 2013. Efficacy of a levulinic acid plus sodium dodecyl sulfate (SDS)-based sanitizer on inactivation of influenza a virus on eggshells. Food Environ Virol 5:215–219. doi: 10.1007/s12560-013-9129-x. [DOI] [PubMed] [Google Scholar]
- 51.Kamarasu P, Hsu HY, Moore MD. 2018. Research progress in viral inactivation utilizing human norovirus surrogates. Front Sustain Food Syst 2:00089. doi: 10.3389/fsufs.2018.00089. [DOI] [Google Scholar]
- 52.Predmore A, Li J. 2011. Enhanced removal of a human norovirus surrogate from fresh vegetables and fruits by a combination of surfactants and sanitizers. Appl Environ Microbiol 77:4829–4838. doi: 10.1128/AEM.00174-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.U.S. Environmental Protection Agency. 2015. Tsunami 100 label notification. https://www3.epa.gov.
- 54.Yuan Z, Ni Y, Van Heiningen ARP. 1997. Kinetics of peracetic acid decomposition: Part I: Spontaneous decomposition at typical pulp bleaching conditions. Can J Chem Eng 75:37–41. doi: 10.1002/cjce.5450750108. [DOI] [Google Scholar]
- 55.Arthur SE, Gibson KE. 2015. Physicochemical stability profile of Tulane virus: a human norovirus surrogate. J Appl Microbiol 119:868–875. doi: 10.1111/jam.12878. [DOI] [PubMed] [Google Scholar]
- 56.Weiss C, Clark HF. 1985. Rapid inactivation of rotaviruses by exposure to acid buffer or acidic gastric juice. J Gen Virol 66:2725–27230. doi: 10.1099/0022-1317-66-12-2725. [DOI] [PubMed] [Google Scholar]
- 57.Samandoulgou I, Fliss I, Jean J. 2015. Zeta potential and aggregation of virus-like particle of human norovirus and feline calicivirus under different physicochemical conditions. Food Environ Virol 7:249–260. doi: 10.1007/s12560-015-9198-0. [DOI] [PubMed] [Google Scholar]
- 58.Escudero-Abarca BI, Rawsthorne H, Goulter RM, Suh SH, Jaykus LA. 2014. Molecular methods used to estimate thermal inactivation of a prototype human norovirus: more heat resistant than previously believed? Food Microbiol 41:91–95. doi: 10.1016/j.fm.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 59.Kim SY, Ko G. 2012. Using propidium monoazide to distinguish between viable and nonviable bacteria, MS2 and murine norovirus. Lett Appl Microbiol 55:182–188. doi: 10.1111/j.1472-765X.2012.03276.x. [DOI] [PubMed] [Google Scholar]
- 60.Parshionikar S, Laseke I, Fout GS. 2010. Use of propidium monoazide in reverse transcriptase PCR to distinguish between infectious and noninfectious enteric viruses in water samples. Appl Environ Microbiol 76:4318–4326. doi: 10.1128/AEM.02800-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manuel CS, Moore MD, Jaykus LA. 2018. Predicting human norovirus infectivity - Recent advances and continued challenges. Food Microbiol 76:337–345. doi: 10.1016/j.fm.2018.06.015. [DOI] [PubMed] [Google Scholar]
- 62.Oristo S, Lee HJ, Maunula L. 2018. Performance of pre-RT-qPCR treatments to discriminate infectious human rotaviruses and noroviruses from heat-inactivated viruses: applications of PMA/PMAxx, benzonase and RNase. J Appl Microbiol 124:1008–1016. doi: 10.1111/jam.13737. [DOI] [PubMed] [Google Scholar]
- 63.Dunkin N, Coulter C, Weng S, Jacangelo JG, Schwab KJ. 2019. Effects of pH variability on peracetic acid reduction of human norovirus GI, GII RNA, and infectivity plus RNA reduction of selected surrogates. Food Environ Virol 11:76–89. doi: 10.1007/s12560-018-9359-z. [DOI] [PubMed] [Google Scholar]
- 64.Hirneisen KA, Kniel KE. 2013. Comparing human norovirus surrogates: murine norovirus and Tulane virus. J Food Prot 76:139–143. doi: 10.4315/0362-028X.JFP-12-216. [DOI] [PubMed] [Google Scholar]
- 65.Tan M, Wei C, Huang P, Fan Q, Quigley C, Xia M, Fang H, Zhang X, Zhong W, Klassen JS, Jiang X. 2015. Tulane virus recognizes sialic acids as cellular receptors. Sci Rep 5:11784. doi: 10.1038/srep11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Araud E, Fuzawa M, Shisler JL, Li J, Nguyen TH. 2019. UV inactivation of rotavirus and Tulane virus targets different components of the virions. Appl Environ Microbiol 86:e02436-19. doi: 10.1128/AEM.02436-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blakistone B, Chuyate R, Kautter D Jr, Charbonneau J, Suit K. 1999. Efficacy of oxonia active against selected spore formers. J Food Prot 62:262–267. doi: 10.4315/0362-028x-62.3.262. [DOI] [PubMed] [Google Scholar]
- 68.Davidson GR. 2013. Impact of organic load on sanitizer efficacy against Escherichia coli O157:H7 during pilot-plant production of fresh-cut lettuce. PhD dissertation. Michigan State University, East Lansing, MI. [Google Scholar]
- 69.European Network of GMO Laboratories. 2011. Verification of analytical methods for GMO testing when implementing interlaboratory validated methods. European Commission Joint Research Centre, Ispra, Italy. [Google Scholar]
- 70.Beck SE, Rodriguez RA, Hawkins MA, Hargy TM, Larason TC, Linden KG. 2015. Comparison of UV-induced inactivation and RNA damage in MS2 phage across the germicidal UV spectrum. Appl Environ Microbiol 82:1468–1474. doi: 10.1128/AEM.02773-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mair P, Wilcox R. 2017. WRS2: a collection of robust statistical methods. https://cran.r-project.org/web/packages/WRS2/index.html. [DOI] [PubMed]
- 72.Jothikumar N, Kang G, Hill VR. 2009. Broadly reactive TaqMan assay for real-time RT-PCR detection of rotavirus in clinical and environmental samples. J Virol Methods 155:126–131. doi: 10.1016/j.jviromet.2008.09.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.