

Abstract
Because of the fast increase in deaths due to Corona Viral Infection in majority region in the world, the detection of drugs potent of this infection is a major need. With this idea, docking study was executed on eighteen imidazole derivatives based on 7-chloro-4-aminoquinoline against novel Coronavirus (SARS-CoV-2).
In this study, we carried out a docking study of these molecules in the active site of SARS-CoV-2 main protease. The result indicate that Molecules N° 3, 7 and 14 have more binding energy with SARS-CoV-2 main protease recently crystallized (pdb code 6LU7) in comparison with the other imidazole derivatives and the two drug; Chloroquine and hydroxychloroquine. Because of the best energy of interaction, these three molecules could have the most potential antiviral treatment of COVID-19 than the other studied compounds. The structures with best affinity in the binding site of the protease have more than 3 cycles and electronegative atoms in the structure. This may increase the binding affinity of these molecules because of formation of π-bonds, halogen interactions and/or Hydrogen bond interactions between compounds and the enzyme. So, compounds with more cycles and electronegative atoms could have a potent inhibition of SARS-CoV-2 main protease.
Keywords: Coronavirus, COVID-19, Imidazole, Chloroquine, Hydroxychloroquine, Molecular docking
1. Introduction
Infection with coronavirus (SARS-CoV-2) first reported in Wuhan (China) in December 2019. This contagion has spread throughout the world [[1], [2], [3], [4], [5]]. SARS-CoV-2 is a member of Betacoronaviruses like the Severe Acute Respiratory Syndrome Human coronavirus (SARS-CoV-1) and the Middle-East Respiratory Syndrome Human coronavirus (MERS HCoV) [6].
The sequence of coronavirus (SARS-CoV-2) was entered into Genbank as entry QHR63300.1 and in the protein data bank (PDB) as entry 6LU7 (DOI: https://doi.org/10.2210/pdb6LU7/pdb) [7], this crystallographic structure present the three dimensional form of the crystal structure of the SARS-CoV-2 main protease that is encoded within the viral genome, in complex with an inhibitor (N3) [8].
The antimalarial drugs chloroquine and hydroxychloroquine are currently used to treat autoimmune diseases for their immunomodulatory and anti-thrombotic properties [9]. Although chloroquine and hydroxychloroquine have also been proposed for the treatment of several viral infections, due to their antiviral effects in cell cultures and animal models [9], and, currently, for the treatment of COVID-19 [3,[10], [11], [12]]. The mechanism of action of chloroquine and its derivatives, both an antiviral and an anti-inflammatory, have not been fully elucidated. Certainly, there are many ways in which these drugs could exert their anti-SARS-CoV-2 effects. Chloroquine and hydroxychloroquine act as weak bases which can have several intracellular effects, including affecting intracellular traffic and disrupting enzymes [13]. Studies of molecular docking and mechanic molecular simulation of Baildya et al. suggest that hydroxychloroquine could have a considerable effect on SARS-COV-2 main protease, which could have a significant inhibitory effect on it [14]. Unfortunately these two drugs have toxic effects and their use in high doses can create major risks for patients [14,15]; that is why the use of these drugs to treat covid-19 is not recommended by World Health Organization (WHO) [16].
Imidazole is a heterocyclic aromatic molecule; its structure has been of great importance to scientists in recent years due to its divergent synthetic strategies and different pharmacological activities, e.g. antivirals, anticancer, anti-inflammatory, antimicrobials, anti-ulcer, etc. Due to the distinct mechanisms of actions, this area of research is always expanding [17].
Molecular docking is an effective method for predicting the structure of ligand/macromolecule complexes. Its use has grown over the years to become one of the most widely used techniques for the discovery and development of biologically active molecules. The general principle is to minimize a simplified potential energy function which measures the interaction between a ligand and a target macromolecule.
Repositioning medications may be the only response to the epidemic of unexpected infectious diseases, due to the long time of producing new medicines. Among the drugs proposed to test the antiviral effect of COVID-19, we selected imidazole derivatives [3,18]. To date, imidazole derivatives has been the first choice as an effective treatment for infections with Malaria, the principle is to replace diethylamine function of chloroquine by substituted imidazole derivatives containing tertiary terminal nitrogen [19]. This kind of compounds has been tested against Malaria infection to explore more active compounds than chloroquine [19]. Many papers reported the effect of chloroquine and hydroxychloroquine in the treatment of COVID-19 [3,10,11,20], today these two drugs are used in different hospitals around the world as antiviral treatment of COVID-19 disease caused by SARS-CoV-2 virus [12,21].
Based on all these effects, the study of interactions between chloroquine, hydroxychloroquine and the eighteen imidazole derivatives against the SARS-CoV-2 main protease are recommended.
In this paper, the modeling interaction of eighteen imidazole derivatives against novel Coronavirus are performed using the molecular docking method followed by comparison with chloroquine, hydroxychloroquine interactions formed in the same binding site of SARS-CoV-2 main protease.
2. Material and methods
2.1. Data set
The studied compounds were evaluated against novel Coronavirus (SARS-CoV-2). The chemical compounds reported as potent Antiplasmodial inhibitors of imidazole derivatives based on 7-chloro-4-aminoquinoline were taken from literature [19].
2.2. Ligand preparation
Each structure of 18 studied compounds is sketched and optimized with SYBYL program [22], using the standard tripos molecular mechanics force field and energy gradient convergence criterion (0.01 kcal/mol), the partial atomic charges required for calculation of the electrostatic potential are assigned using the Gasteiger_Huckel formation [23,24].
2.3. Molecular docking
We performed a docking study of 18 imidazole derivatives (Table 1 ), our research is based on crystal structures of studied enzyme with bound ligand molecules. This structure has been obtained from X-ray crystal data of RCSB Protein Data Bank (PDB) [25], and we have determined the binding site location. The docking study was carried out with Autodock vina program [26]. The crystallographic structure of SARS-CoV-2 main protease (Code PDB: 6LU7; Resolution of 2.16 Å) is imported into “work space” of Discovery Studio 2016 program [27] for detect binding site location. The center of the active site has been determined and it corresponds to the coordinates: x = −10.782, y = 15.787 and z = 71.277 on the basis of the co-crystallized bound peptidomimetic ligand [28]. The grid size was set at 20 × 34 × 20 xyz points with a grid spacing of 1 Å to cover the folic acid binding site in the enzyme and was generated by using the co-crystallized ligand (N3) as the center for docking [28]. For ligand and enzyme preparations; an extended PDB format, termed PDBQT, is used for coordinate files, which includes atomic partial charges and atom types using Autodock tools 1.5.6. Torsion angles were calculated to assign the flexible and non-bonded rotation of molecules. The results were subsequently analyzed using Discovery studio 2016 [29].
Table 1.
Chemical structures of 18 imidazole derivatives based on 7-chloro-4-aminoquinoline.

3. Results and discussions
3.1. Molecular docking results
Molecular docking was performed to find a type of interactions and binding affinity between 18 different imidazole derivatives and studied enzyme. Fig. 1 shows the position of the best conformation of all studied compounds in the binding pocket of SARS-CoV-2 main protease (the co-crystallized ligand N3 is presented by yellow color) [30]. The result of molecular docking is presented in Table 2 .
Fig. 1.

Position of the best conformation of all studied compounds in the binding pocket of SARS-CoV-2 main protease (the co-crystallized ligand N3 is presented by yellow color). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Table 2.
The results of docking study: Affinity of the best conformation.
| N° | Affinity (kcal/mol) | N° | Affinity (kcal/mol) | N° | Affinity (kcal/mol) |
|---|---|---|---|---|---|
| 1 | −7.4 | 8 | −7.8 | 15 | −7.8 |
| 2 | −7.4 | 9 | −7.1 | 16 | −7.9 |
| 3 | −8.5 | 10 | −7.4 | 17 | −7.5 |
| 4 | −7.7 | 11 | −8.1 | 18 | −7.9 |
| 5 | −7.6 | 12 | −7.3 | Chloroquine | −5.9 |
| 6 | −7.4 | 13 | −7.6 | Hydroxychloroquine | −6.6 |
| 7 | −8.3 | 14 | −8.3 | N3 | −8.7 |
All studied compounds have a significant affinity in the binding pocket of SARS-CoV-2 main protease in comparison with chloroquine and hydroxychloroquine. The best energies of interaction with the SARS-CoV-2 main protease are observed for molecules N° 3, 7 and 14 (Table 2), because of their affinity in the active site of studied enzyme; these molecules could have more potential antiviral treatment of COVID-19 than chloroquine, hydroxychloroquine. In addition, their affinity is close to that of the N3 inhibitor.
The interaction results of chloroquine and SARS-CoV-2 main protease (Fig. 2 ) shows Carbon Hydrogen Bond with Glu166 and Arg188, π-alkyl bond with Cys145 and Met49 residues, and alkyl interaction with Met49, Met165, Pro168 and Leu167 residues.
Fig. 2.

Interactions between Chloroquine and the SARS-CoV-2 main protease.
The docking study of hydroxychloroquine in the SARS-CoV-2 main protease (Fig. 3 ) shows more type and number of interactions in comparison with chloroquine (π-alkyl, alkyl, Halogen interaction, π-Sulfure, π-π Stached and Hydrogen Bond interaction).
Fig. 3.

Interactions between Hydroxychloroquine and the SARS-CoV-2 main protease.
The interaction results of molecules N° 3, 7 and 14 in the SARS-CoV-2 main protease (Fig. 4, Fig. 5, Fig. 6 ) shows more type and number of interactions in comparison with chloroquine and hydroxychloroquine (π-alkyl, π-Donor Hydrogen bond, π-Sigma, π-π T-shaped, π-Sulfure, Carbon-Hydrogen bond and Hydrogen bond interaction).
Fig. 4.

Interactions between compound N° 3 and the SARS-CoV-2 main protease.
Fig. 5.

Interactions between compound N° 7 and the SARS-CoV-2 main protease.
Fig. 6.

Interactions between compound N° 14 and the SARS-CoV-2 main protease.
The results indicate that the structures with best affinity in the binding site of the protease have four or more than 4 cycles and electronegative atoms in the structure. This may increase the binding affinity of these molecules because of formation of π-bonds, halogen interactions and/or Hydrogen bond interactions between compounds and the enzyme; compounds with more cycles and electronegative atoms could have more potent inhibition of SARS-CoV-2 main protease than the other compounds.
So these compounds (Molecules N° 3, 7 and 14) could have potential antiviral treatment of COVID-19 than chloroquine and hydroxychloroquine, because of their interactions and the best affinity in the binding pocket of SARS-CoV-2 main protease.
3.2. Docking validation protocol
Re-docking of the Co-Crystallized ligand was applied to validate the accuracy of the docking procedure. Fig. 7 clarifies the superimposed view between the docked ligand conformation and the co-crystallized ligand (inhibitor N3).
Fig. 7.
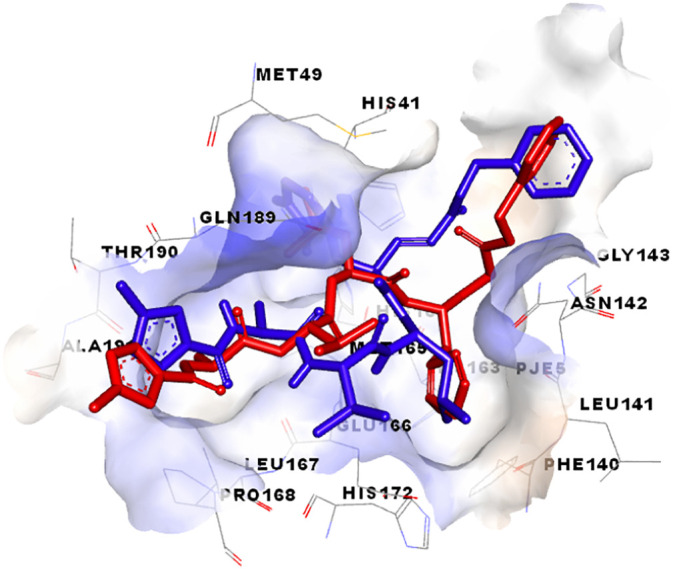
Overlay of default conformation (red colored) on docked conformation (blue colored) of the co-crystallized ligand validating docking protocol. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
As observed in Fig. 7, the co-crystallized ligand (inhibitor N3) within the active cavity of SARS-CoV-2 in a similar position and interacted with similar residues, compared to that observed for the docked ligand conformation. Thus, the docking process in this study was successfully validated.
It could be seen from Fig. 8 that they are present of Conventional Hydrogen Bonds interactions with Glu 166, His 164, Gly 143, Thr 190, Gln 189, His 163 and Phe 140 residues, amide-π Staked interactions with Leu 141 residue, Alkyl and/or π-Alkyl interactions with Leu 167, Ala 191, Met 167 and Pro 168, Met 49 and His 41 residues, Carbon Hydrogen Bond with Met 165 and His 172 residues and Van der Waals interaction with Asn142. The selected molecules with the best affinity present the interactions with the same residues. Thus, these molecules could have a potent inhibition of SARS-CoV-2 main protease.
Fig. 8.

Interactions between inhibitor N3 (co-crystallized ligand) and SARS-CoV-2 main protease.
4. Conclusion
In this study, we have tried to carry out a docking study of chemical compounds reported as potent Antiplasmodial inhibitors of imidazole derivatives based on 7-chloro-4-aminoquinoline and analogues in the active site of SARS-Cov-2 main protease, flowed by comparison with two drugs; chloroquine and hydroxychloroquine.
The result indicate that molecules N° 3, 7 and 14 have the best affinity in the binding pocket of studied enzyme than all studied molecules especially than chloroquine and hydroxychloroquine; that can indicate that these molecules could have the most potential antiviral treatment of COVID-19. The structures with best affinity in the binding site of the protease have more than 3 cycles and electronegative atoms in the structure. This may increase the binding affinity of these molecules because of formation of π-bonds, halogen interactions and/or Hydrogen bond interactions between compounds and the enzyme. So, compounds with more cycles and electronegative atoms could have more potent inhibition of SARS-CoV-2 main protease than the reference compounds. So in this study, we describe the optimal binding features of imidazole derivatives based on 7-chloro-4-aminoquinoline and analogues were with the SARS-CoV-2 main protease (Code PDB: 6LU7) for further consideration. The synthesis of these molecules and the evaluation of their in vitro activity against COVID-19 could be interesting.
Declaration of competing interest
The authors declare that they have no competing interests.
References
- 1.Tosepu R., Gunawan J., Effendy D.S., Ahmad L.O.A.I., Lestari H., Bahar H., Asfian P. Correlation between weather and Covid-19 pandemic in Jakarta, Indonesia. Sci. Total Environ. 2020:138436. doi: 10.1016/j.scitotenv.2020.138436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B. 2020;10(5):766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh A.K., Singh A., Shaikh A., Singh R., Misra A. Chloroquine and hydroxychloroquine in the treatment of COVID-19 with or without diabetes: a systematic search and a narrative review with a special reference to India and other developing countries. Diabetes Metab. Syndr. Clin. Res. Rev. 2020;14:241–246. doi: 10.1016/j.dsx.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang D., Choi H., Kim J.-H., Choi J. Spatial epidemic dynamics of the COVID-19 outbreak in China. Int. J. Infect. Dis. 2020;94:96–102. doi: 10.1016/j.ijid.2020.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhai P., Ding Y., Wu X., Long J., Zhong Y., Li Y. The epidemiology, diagnosis and treatment of COVID-19. Int. J. Antimicrob. Agents. 2020:105955. doi: 10.1016/j.ijantimicag.2020.105955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elfiky A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020;248 doi: 10.1016/j.lfs.2020.117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robson B. Computers and viral diseases. Preliminary bioinformatics studies on the design of a synthetic vaccine and a preventative peptidomimetic antagonist against the SARS-CoV-2 (2019-nCoV, COVID-19) coronavirus. Comput. Biol. Med. 2020;119 doi: 10.1016/j.compbiomed.2020.103670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatada R., Okuwaki K., Mochizuki Y., Fukuzawa K., Komeiji Y., Okiyama Y., Tanaka S. 2020. Fragment Molecular Orbital Based Interaction Analyses on COVID-19 Main Protease - Inhibitor N3 Complex (PDB ID:6LU7) (ChemRxiv) [DOI] [PubMed] [Google Scholar]
- 9.Quiros Roldan E., Biasiotto G., Magro P., Zanella I. The possible mechanisms of action of 4-aminoquinolines (chloroquine/hydroxychloroquine) against Sars-Cov-2 infection (COVID-19): a role for iron homeostasis? Pharmacol. Res. 2020;158 doi: 10.1016/j.phrs.2020.104904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahraei Z., Shabani M., Shokouhi S., Saffaei A. Aminoquinolines against coronavirus disease 2019 (COVID-19): chloroquine or hydroxychloroquine. Int. J. Antimicrob. Agents. 2020;55(4) doi: 10.1016/j.ijantimicag.2020.105945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsang A.C., Ahmadi S., Hamilton J., Gao J., Virgili G., Coupland S.G., Gottlieb C.C. The diagnostic utility of multifocal electroretinography in detecting chloroquine and hydroxychloroquine retinal toxicity. Am J. Ophthalmol. 2019;206:132–139. doi: 10.1016/j.ajo.2019.04.025. [DOI] [PubMed] [Google Scholar]
- 12.Radke J.B., Kingery J.M., Maakestad J., Krasowski M.D. Diagnostic pitfalls and laboratory test interference after hydroxychloroquine intoxication: a case report. Toxicol. Rep. 2019;6:1040–1046. doi: 10.1016/j.toxrep.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu T.Y., Frieman M., Wolfram J. Insights from nanomedicine into chloroquine efficacy against COVID-19. Nat. Nanotechnol. 2020;15:247–249. doi: 10.1038/s41565-020-0674-9. Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baildya N., Ghosh N.N., Chattopadhyay A.P. Inhibitory activity of hydroxychloroquine on COVID-19 main protease: an insight from MD-simulation studies. J. Mol. Struct. 2020;1219 doi: 10.1016/j.molstruc.2020.128595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marmor M.F., Kellner U., Lai T.Y., Melles R.B., Mieler W.F. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision) Ophthalmology. 2016;123:1386–1394. doi: 10.1016/j.ophtha.2016.01.058. (Elsevier) [DOI] [PubMed] [Google Scholar]
- 16.https://www.who.int
- 17.Soni J., Sethiya A., Sahiba N., Agarwal D.K., Agarwal S. Contemporary progress in the synthetic strategies of imidazole and its biological activities. Curr. Org. Synth. 2019;16:1078–1104. doi: 10.2174/1570179416666191007092548. [DOI] [PubMed] [Google Scholar]
- 18.Tang X., Du R., Wang R., Cao T., Guan L., Yang C., Zhu Q., Hu M., Li X., Li Y., et al. Comparison of hospitalized patients with acute respiratory distress syndrome caused by COVID-19 and H1N1. Chest. 2020;158(1):195–205. doi: 10.1016/j.chest.2020.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kondaparla S., Manhas A., Dola V.R., Srivastava K., Puri S.K., Katti S.B. Design, synthesis and antiplasmodial activity of novel imidazole derivatives based on 7-chloro-4-aminoquinoline. Bioorg. Chem. 2018;80:204–211. doi: 10.1016/j.bioorg.2018.06.012. [DOI] [PubMed] [Google Scholar]
- 20.Fantini J., Scala C.D., Chahinian H., Yahi N. Structural and molecular modeling studies reveal a new mechanism of action of chloroquine and hydroxychloroquine against SARS-CoV-2 infection. Int. J. Antimicrob. Agents. 2020;55(5) doi: 10.1016/j.ijantimicag.2020.105960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gautret P., Lagier J.-C., Parola P., Hoang V.T., Meddeb L., Mailhe M., Doudier B., Courjon J., Giordanengo V., Vieira V.E., et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents. 2020;56(1) doi: 10.1016/j.ijantimicag.2020.105949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tripos Inc., St. Louis, MO, USA. SYBYL-X 2.0.
- 23.Clark M., Cramer R.D., Van Opdenbosch N. Validation of the general purpose Tripos 5.2 force field. J. Comput. Chem. 1989;10:982–1012. [Google Scholar]
- 24.Purcell W.P., Singer J.A. A brief review and table of semiempirical parameters used in the Hueckel molecular orbital method. J. Chem. Eng. Data. 1967;12:235–246. [Google Scholar]
- 25.Protein Data Bank PDB. http://www.rcsb.org
- 26.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dassault Systèmes BIOVIA Discovery Studio Modeling Environment . 2016. Release 2017 Dassault Systèmes, San Diego. [Google Scholar]
- 28.Hakmi M., Bouricha E.M., Kandoussi I., El Harti J., Ibrahimi A. Repurposing of known anti-virals as potential inhibitors for SARS-CoV-2 main protease using molecular docking analysis. Bioinformation. 2020;16:301. doi: 10.6026/97320630016301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aanouz I., Belhassan A., El Khatabi K., Lakhlifi T., El Idrissi M., Bouachrine M. Moroccan medicinal plants as inhibitors of COVID-19: computational investigations. J. Biomol. Struct. Dyn. 2020:1–12. doi: 10.1080/07391102.2020.1758790. (Taylor & Francis) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020:1–5. doi: 10.1038/s41586-020-2223-y. (Nature Publishing Group) [DOI] [PubMed] [Google Scholar]