

Abstract
Abemaciclib is an oral anticancer drug that inhibits cyclin dependent kinases 4 and 6 and is metabolized by cytochrome P450 3A in the intestines and liver to active metabolites. The objectives were (1) to develop a mechanistic model to characterize the pharmacokinetics (PK) of the active moieties and investigate the effect of patient factors and (2) apply the model to dat from two phase III breast cancer trials of abemaciclib in combination with endocrine therapy. To develop the model, data from seven phase I studies and two phase II studies including 421 patients with cancer and 65 healthy individuals were pooled for nonlinear mixed effects modeling. The PK was similar between patients and healthy subjects, and the effects of diarrhea, formulation, race, and patient covariates on exposure were negligible. Application of the model confirmed its predictive performance and that abemaciclib PK did not change when coadministered with endocrine therapy.
Abemaciclib is an orally administered small molecule inhibitor of cyclin‐dependent kinases (CDKs) 4 and 6. 1 Inhibition of CDK4 and CDK6 prevents cell‐cycle progression through the G1 restriction point that controls entry into S phase, thus arresting tumor growth. 2 A phase I human study (ClinicalTrials.gov identifier NCT01394016) demonstrated antitumor activity with abemaciclib as a single agent on a continuous dosing schedule for the first time. 2 Abemaciclib has been approved in several geographies (including the United States, Japan, Australia, Canada, and Europe) for the treatment of metastatic breast cancer. 3
The global clinical development program for abemaciclib in cancer patients included oral twice daily dosing of 200 mg as a single agent on a continuous dosing schedule (MONARCH 1 study) 4 and 150 mg in combination with endocrine therapy, with dose reduction permitted in 50 mg units to as low as 50 mg twice daily, as needed for individual tolerability (MONARCH 2 and 3 studies, respectively). 5 , 6 One of the most common adverse events in patients taking abemaciclib is diarrhea.
The population pharmacokinetics (PK) of abemaciclib as parent drug in a phase I cancer patient population has been previously described empirically. 7 Abemaciclib exhibits a slow absorption phase with a median time to maximum plasma concentration of approximately 8 hours and mean terminal elimination half‐life in patients of approximately 24 hours. Abemaciclib clearance is dose proportional across the therapeutic dose range (50–200 mg). Abemaciclib is primarily metabolized by cytochrome P450 (CYP) 3A4 to active and equipotent metabolites, and 3% of the parent drug is renally excreted. Coadministration with rifampicin (CYP3A inducer) reduced abemaciclib area under the curve (AUC) and maximum plasma concentration (Cmax) by 95% and 92%, respectively, whereas coadministration with clarithromycin (CYP3A inhibitor) increased abemaciclib AUC and Cmax by 237% and 30%, respectively. 8
Strong CYP3A inhibitors increase the exposure of abemaciclib and its active M2 and M20 metabolites, leading to increased toxicity. These inhibitors are predicted to increase the relative potency‐adjusted unbound AUC of abemaciclib and metabolites by 4‐fold to 16‐fold. 3 , 9 Because of the need to appropriately account for the total active species (abemaciclib plus its active metabolites from intestinal and hepatic metabolism), we sought to develop a mechanistic population model to describe the slow and highly variable absorption kinetics as well as the formation and disposition of abemaciclib, M2, and M20. The model was then used to determine the impact of intrinsic (demographics, diarrhea) and extrinsic factors (drug formulation) on the PK of abemaciclib. The model was subsequently applied to a phase III data set, which contained more data around drug formulation and diarrhea to further evaluate the impact of covariates on the PK of abemaciclib. Finally, the model’s predictive performance was validated in a separate phase III data set, as part of an external evaluation.
METHODS
The clinical trials conducted in healthy subjects and patients with cancer were registered at clinicaltrials.gov (NCT01394016, NCT01739309, NCT02014129, NCT01913314, NCT02117648, NCT02256267, NCT02059148, NCT02102490, and NCT02327143). The trial designs and data characteristics are summarized in Table 1 (model development) and Table S1 (model application). All studies were conducted in accordance with the Helsinki declaration and with approval from the corresponding institutional review boards.
Table 1.
Summary of clinical studies in healthy subjects and cancer patients used to develop and validate the semimechanistic population PK model
| Clinical Trials.gov | NCT01394016 | NCT01739309 | NCT02014129 | NCT01913314 | NCT02117648 | NCT02256267 | NCT02059148 | NCT02102490 | NCT02327143 |
|---|---|---|---|---|---|---|---|---|---|
| Study alias | JPBA | JPBB | JPBC | JPBD | JPBE | JPBF | JPBG | JPBN | JPBS |
| Phase | I | II | I | I | I | I | I | II | I |
| Population | Cancer patients | Cancer patients | Cancer patients | Healthy subjects | Cancer patients | Healthy subjects | Healthy subjects | Cancer patients | Healthy subjects |
| Description | Multicenter, dose‐escalation study of a CDK4/6 dual inhibitor in patients with advanced cancer | Multicenter, nonrandomized, open‐label study of a CDK4/6 inhibitor for patients with relapsed or refractory mantle cell lymphoma | Nonrandomized, open‐label, dose‐escalation study of LY2835219 in Japanese patients with advanced cancer | Nonrandomized, open‐label, single‐dose study—Disposition of [14C]‐LY2835219 following oral administration in healthy subjects | Open‐label, 2‐period, fixed‐sequence study—Effects of CYP3A inhibition by clarithromycin on the PK of LY2835219 and its metabolites in cancer patients | Open‐label, 2‐period, fixed‐sequence study—Effects of CYP3A induction by rifampin on the PK of LY2835219 and its metabolites in healthy subjects | Open‐label, randomized, single‐dose, three‐period crossover study—Effect of food on the PK of LY2835219 in healthy subjects | Multicenter, nonrandomized, open‐label study of LY2835219 for patients with previously treated hormone receptor–positive, HER2‐negative metastatic breast cancer | Single‐center, open‐label, single‐period study—An absolute bioavailability study of LY2835219 in healthy subjects using the intravenous tracer method |
| Total n (n with PK) | 225 (224) | 28 (27) | 12 (12) | 6 (6) | 26 (26) | 24 (24) | 24 (24) | 132 (128) | 11 (11) |
| N PK observations | 3993 | 282 | 232 | 96 | 296 | 308 | 977 | 579 | 177 |
| Age, y | 61.0 (24–85) | 70.0 (53.0–83.0) | 61.5 (37–73) | 46.5 (28–59) | 59.5 (37–78) | 58 (44–70) | 57.5 (30–64) | 58 (36–89) | 56.0 (45–65) |
| Weight, (kg) | 70.5 (43.6–175.1) | 73.0 (46.8–107.0) | 55.9 (37.3–74.7) | 71.55 (61.7–94.6) | 72 (46.8–138.2) | 71.95 (50.3–93.7) | 73.95 (56.2–95.3) | 64.4 (44–127.4) | 76.6 (62.2‐103.2) |
| % Female | 66.7 | 39.29 | 58.3 | 50 | 73.1 | 87.5 | 87.5 | 100 | 27.3 |
| Abemaciclib formulation | 25 mg and 150 mg capsules (C1) | 25 mg and 150 mg capsules (C1) | 25 mg and 150 mg capsules (C1) | Oral solution | 25 mg and 150 mg capsules (C1) | 50 mg 25% w/w capsules (C3) | 50 mg 50% w/w capsules (C2) | 50 mg 25% w/w capsules (C3) | 50 mg 25% w/w capsules (C3) and IV solution |
| Sampling | Intensive | Intensive | Intensive | Intensive | Intensive | Intensive | Intensive | Sparse | Intensive |
| N PK samples per patient per protocol | 23 samples in cycle 1 (days 1–29) | 14 samples in cycle 1 (days 1–15) | 23 samples in cycle 1 (days 1–29) | 25 samples after a single dose (days 1–15) |
Period 1–14 samples after a single dose (days 1–8) Period 2—17 samples after a single dose on day 5 (days 5–15) |
Period 1–15 samples after a single dose (days 1–9) Period 2—15 samples after a single dose on day 7 (days 7–15) |
Period 1–18 samples after a single dose (days 1–9) Period 2—18 samples after a single dose (days 1–9) Period 2—18 samples after a single dose (days 1–9) |
3 samples cycle 1—days 1 and 15 2 samples cycle 2 day 1 1 sample cycle 3 day 1 |
20 samples after a single dose (days 1–9) |
Data in age = median (range), weight = median (range), and % female are calculated from total n.
CDK, cyclin dependent kinase; CYP, cytochrome P450; HER2, human epidermal growth factor receptor 2; JPBN also known as MONARCH 1; PK, pharmacokinetic.
Patients
The analysis data set to develop the model contained data from seven phase I studies and two phase II studies (studies JPBB and JPBN [MONARCH 1]) and included both healthy subjects and patients with cancer. The cross‐study application of the model was evaluated with data from two phase I studies in healthy subjects (studies JPBU and JPBV) and two phase III studies in breast cancer patients also receiving endocrine therapy (studies JPBL [MONARCH 2] and JPBM [MONARCH 3]).
Abemaciclib doses ranging from 50 to 275 mg were evaluated in these 13 studies. Dosing frequency in studies was either single dose or multiple dose (generally every 12 hours). Three formulations of oral capsules were used depending on the study and stage of development. Study JPBS was a single‐dose study that used the 25% w/w capsule (C3) formulation (bioequivalent to the commercial formulation administered orally) and a 13C8 tracer that was administered intravenously 6 hours after the oral dose in the same individuals to determine absolute bioavailability.
Information related to the time to onset of the first diarrhea event was included from 6 studies in which abemaciclib was administered as multiple doses in cancer patients: Studies JPBA, JPBB, JPBC, and JPBN for model development and studies JPBL and JPBM for model application. Whether an individual had diarrhea as a preexisting condition or as an adverse event on study was accounted for in the data set.
PK sampling and drug measurement
Various PK sampling times were used, depending on the study, and as a result the pooled data set had both rich and sparse sampling. Plasma samples obtained during the studies were analyzed for abemaciclib and metabolites using liquid chromatography with tandem mass spectrometry detection at Charles River Laboratories and Q2 Solutions, formerly Quintiles (Ithaca, NY). The lower limit of quantitation (LOQ) was 1 ng/mL, and the upper LOQ was 500 ng/mL. Interassay accuracy (% relative error) during validation ranged from −4.3% to 7.8%. Interassay precision (percent relative SD) during validation ranged from 2.9% to 6.1%. For the 13C8 tracer assay, the lower LOQ was 0.002 ng/mL.
Data analysis and model development
A nonlinear mixed effect PK model approach was used to analyze the PK data. Various model structures were explored to determine which best described the PK of abemaciclib and its two major metabolites, M2 and M20. Because of the expectation of both gastrointestinal metabolism and hepatic metabolism contributing to a substantial first‐pass effect, a mechanistic model was necessary to adequately describe the appearance of the metabolites following oral dosing. Secondary peaks occurred following oral dosing, which were not present for intravenous dosing, suggesting biphasic drug absorption. Estimates of the PK parameters were obtained by fitting models to the concentration‐time data using Nonlinear Mixed Effect Model (NONMEM) version 7.3.0 (ICON Development Solutions, Hanover, MD). PsN 4.2.0 10 was used for the NONMEM analysis ( Supplementary Text ). The stochastic approximation expectation maximization estimation algorithm was used for the analyses. After stochastic approximation expectation maximization estimation, an evaluation step using importance sampling was implemented to obtain reliable SEs and objective function value. Because abemaciclib, M2, and M20 were measured in the same PK samples, correlations between the residual errors for these dependent variables were tested using the L2 data item. In addition, interindividual variability in the residual error, which eases the assumption that magnitude of the residual error is the same in all patients, 11 was tested.
Various absorption models, including first‐order, zero‐order, saturable, Weibull‐like, transit compartment, and inclusion of lag time, were investigated.
Various combinations of these models were also investigated in attempts to describe the multiple peaks and the slow and long absorption duration. As the active metabolites M2 and M20 are formed by CYP3A, which occurs in the gut in addition to the liver, the formation of M2 and M20 in the gut was also included. The well‐stirred model for hepatic elimination was applied to describe abemaciclib, M2, and M20 elimination in relation to hepatic blood flow, protein‐binding (in blood and plasma), and enzyme activity. 12 Body weight was included a priori using allometric relationships 13 for some PK parameters (intrinsic clearance (CLint) and volumes) and on liver volume (through using body‐surface area to predict liver volume).
Standard goodness‐of‐fit plots, visual predictive checks (VPCs), and a bootstrap were used for model evaluation.
Covariates
As a result of computational intensity, an empiric abemaciclib population PK model 14 built without the metabolite data but that accounted for the effects of diarrhea using a time‐to‐event approach was used to screen for significant covariates. Covariates were screened for significance based on parameter precision, plausibility, magnitude, and >10% relative decrease in the interindividual variance estimates. 15 Subsequently, the significant covariates identified in the empiric model were then tested on the semimechanistic model. Patient factors assessed in the empiric model included age, sex, race, population (patients/healthy), baseline creatinine clearance, albumin, liver function test (aspartate aminotransferase, alanine transaminase, alkaline phosphatase, total bilirubin), concomitant medications (loperamide, opioids, CYP3A inducers, CYP3A inhibitors), fed status, and formulation. Plots of random effects (ETAs) vs. covariates were created (data not shown) using the final model to determine that all necessary covariates had been included in the model.
Model applications
The final model was applied to an augmented data set that included more data for abemaciclib when administered as the C3 formulation in healthy subjects (richly sampled data from studies JPBU and JPBV) and sparse sampling in breast cancer patients (JPBL) and diarrhea events at lower doses in breast cancer patients (study JPBL: Combination with fulvestrant). The model parameters were reestimated to update the model without any additional model development. Finally, the model was applied (importance sampling with expectation‐only step) to sparsely sampled data in breast cancer patients (study JPBM: Combination with letrozole or anastrozole) to confirm the predictive properties of the model in cancer patients.
To compare abemaciclib exposures between East Asian (Japanese, Korean, or Taiwanese patients) and non–East Asian patients, simulations were performed with each patient’s post hoc PK parameters and dosage averaged over their respective study duration in MONARCH 2 and MONARCH 3, respectively. Individual post hoc PK parameters were used to generate steady‐state exposure metrics after 56 days of dosing every 12 hours to match the collection of the last per‐protocol PK samples collected at cycle 3 day 1. Single‐dose exposures were generated for cycle 1 day 1 using the assigned starting dosage (i.e., either 150 mg or 200 mg). The geometric mean (percent coefficient of variation) of the abemaciclib, M2, M20, and total active species exposure metrics (Cmax, minimum observed drug concentration, and AUC during 1 dosing interval after a single dose and at steady state) were calculated from the simulated concentration‐time profiles.
RESULTS
A total of 8633 abemaciclib, 8637 M2, and 8570 M20 concentration data‐time data from 486 individuals were used for model development. About 4% of the postdose sample results were below the lower LOQ and were treated as missing and not included in the analysis. Ages ranged from 24 to 89, and weight ranged between 37.2 and 175.1 kg. The majority were female (75.3%), 86.6% were patients with cancer, and White was the most common race (84%).
The mechanistic model included an estimated fraction of abemaciclib that is absorbed via a route without metabolism while another fraction is absorbed via a route that results in formation of 2 major metabolites (M2 and M20). The amounts of these 3 moieties that escape intestinal metabolism are then presented to the liver, where further metabolism can take place, i.e., formation of M2 and M20 from abemaciclib as well as elimination of M2 and M20 that had been already formed from the intestines. From the liver follows entry into the general circulation where abemaciclib could distribute into a peripheral compartment (two‐compartment model) while distribution of the metabolites was described using one‐compartment models. Figure 1 shows the model that best described the PK of abemaciclib, M2, and M20. The figure shows the separate compartments for parent abemaciclib (LY) and the metabolites M2 and M20. The numbers on the boxes correspond to the compartments in the control stream, which can be found in the supplementary material. Model equations can also be found in the control stream. The parameter estimates and their precision (from the NONMEM SEs) are reported in Table 2 . Because of their mechanistic nature, the parameter estimates can be difficult to interpret. Therefore, they have been translated into more common PK metrics using equations.
Figure 1.
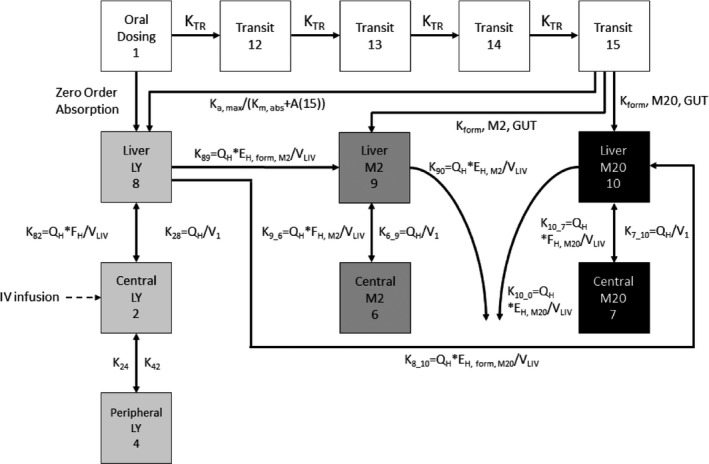
Final model schematic. The compartment numbers correspond to those in the control stream. The light grey boxes represent parent abemaciclib (LY), the dark grey boxes are for M2, and the black boxes are for M20. A(15), amount in last transit compartment; EH, extraction ratio of formation of metabolite; FH,Mx, fraction of parent or metabolite presented to the liver that escapes hepatic metabolism; IV = intravenous; Ka,max, maximum absorption rate constant; Km,abs, saturable absorption rate constant; KNY, the first order rate constant from compartment N to compartment Y; KTR, transit rate constant; LY, abemaciclib; M2, metabolite LSN2839567; M20, metabolite LSN3106726; QH, hepatic blood flow; V1, volume of the central compartment; VLIV, liver volume.
Table 2.
Pharmacokinetic and covariate parameters in final population model
| Parameter | Estimate (%SEE) | % Variability a , a (%SEE) | Bootstrap median (95% CI) | Bootstrap median % variability a , a (95% CI) |
|---|---|---|---|---|
| Fraction of dose entering gut compartment, F a | 0.868 (0.0583) | 7.08 (fixed) b , b | 0.882 (0.874–0.890) | 7.08 (fixed) b , b |
| Fraction of F a absorbed via transit compartment | 0.458 (0.336) | 7.08 (fixed) | 0.479 (0.453–0.507) | 7.08 (fixed) |
| Maximum rate of absorption, ka,max (µmol/h) | 91.1 (3.96) | 77.3 (20.6) | 97.6 (85.0–111) | 71.5 (60.6–83.7) |
| Km for saturable absorption (µmol) | 52.7 (0.410) | 7.08 (fixed) | 51.8 (47.8–56.2) | 7.08 (fixed) |
| MTT (h) | 1.44 (3.68) | 71.9 (8.73) | 1.52 (1.43–1.62) | 70.1 (64.2–77.9) |
| Central volume (L) | 588 (2.81) | 68.6 (6.68) | 604 (563–635) | 65.7 (59.9–72.8) |
| Intercompartmental clearance (L/h) | 4.57 (0.449) | 7.08 (fixed) | 3.66 (3.10–4.29) | 7.08 (fixed) |
| Peripheral volume (L) | 159 (0.421) | 7.08 (fixed) | 152 (132–184) | 7.08 (fixed) |
| Zero‐order absorption duration (h) | 3.18 (10.3) | 638 (5.79) | 3.04 (2.52–3.84) | 810 (533–1278) |
| Absorption lag time (h) | 1.74 (0.538) | 7.08 (fixed) | 1.76 (1.69–1.80) | 7.08 (fixed) |
| Rate constant for formation of M2 from gut (/h) | 0.361 (0.479) | 7.08 (fixed) | 0.401 (0.366–0.432) | 7.08 (fixed) |
| Rate constant for formation of M20 from gut (/h) | 0.284 (0.468) | 7.08 (fixed) | 0.306 (0.276–0.339) | 7.08 (fixed) |
| CLint to form M2 in liver (L/h) c , c | 280 (0.436) | BSV of 44.9 (27.1) for healthy and 57.9 (39.5) for patients. WSV of 68.1 (5.12) for patients | 235 (199–284) | BSV of 44.3 (36.1–54.5) for healthy and 56.7 (44.8–71.2) for patients. WSV of 68.7 (59.6–80.7) for patients |
| CLint to form M20 in liver (L/h) c , c | 705 (0.421) | BSV of 44.9 (27.1) for healthy and 57.9 (39.5) for patients. WSV of 68.1 (5.12) for patients | 700 (627–775) | BSV of 44.3 (36.1–54.5) for healthy and 56.7 (44.8–71.2) for patients. WSV of 68.7 (59.6–80.7) for patients |
| CLint of M2 in liver (L/h) | 254 (2.06) | 41.9 (20.3) | 241 (218–271) | 42.2 (32.9–50.4) |
| CLint of M20 in liver (L/h) | 431 (2.00) | 42.5 (18.9) | 454 (417–489) | 42.1 (32.6–51.4) |
| Box‐cox shape for zero‐order duration ETAs | 0.19 (3.16) | – | 0.145 (0.116–0.184) | – |
| Diarrhea effect on Fa d | −0.417 (7.99) | – | −0.0079 (−1.29, 2.65 × 10−7) | – |
| C2 formulation on absorption lag e | 0.846 (2.99) | – | 0.872 (0.618–1.06) | – |
| C3 formulation on absorption lag e | 0.968 (1.82) | – | 0.982 (0.770–1.12) | – |
| Formulation on ka,max f | −0.513 (6.84) | – | −0.487 (−0.587 to −0.376) | – |
| Correlation between variability for MTT and zero‐order duration | 0.474 (16.9) | 0.474 (16.9) | ||
| Correlation between variability in CLint of M2 and M20 | 0.909 (15.6) | 0.908 (0.869–0.927) |
| Residual error | Estimate (%SEE) | Bootstrap median (95% CI) |
|---|---|---|
| Interindividual variability for residual error | 45.7 (17.5) | 44.7 (39.5–51.0) |
| Proportional error parent drug (oral) (%) | 21.7 (3.65) | 20.6 (19.1–22.1) |
| Proportional error M2 (%) | 21.9 (2.74) | 20.7 (19.1–22.1) |
| Proportional error M20 (%) | 13.8 (3.00) | 13.1 (12.3–14.1) |
| Proportional error 13C8‐labeled parent i.v. (%) | 61.6 (12.2) | 62.2 (51.7–71.4) |
| Additive error parent (oral) (µmol/L) | 0.000758 (28.9) | 0.000833 (0.000398–0.00261) |
| Additive error M2 (µmol/L) | 0.00122 (8.11) | 0.00121 (0.000985–0.00154) |
| Additive error M20 (µmol/L) | 0.00154 (10.0) | 0.00158 (0.00113–0.00204) |
ALAG8, absorption lag time; BSV, between subject variability; CI, confidence interval; CLint, intrinsic clearance; CV, coefficient of variation; i.v., intravenous; Ka,max, maximum absorption rate; Km, saturable rate constant; MTT, mean transit time; SEE, standard error of the estimate; TVCL, typical value of the clearance; WSV, within subject variability (no WSV for healthy individuals).
Reported as %CV, calculated by the following equation: , where OMEGA(N) is the NONMEM output for the intersubject variability of the Nth parameter.
Fixed OMEGA to 0.005 to facilitate efficiency of the stochastic approximation expectation maximization algorithm.
Variability incorporated in model on total CLint, which was sum of the typical values of CLint to form M2 and CLint to form M20. CLint = (TVCLint,form,m2 + TVCLint,form,m20)*e(BSV+WSV).
Estimate is on the logit parameter for F a. This translates to a 6% decrease in F a when there is diarrhea.
Absorption lag time (ALAG8) = Typical value (TV)ALAG8*(1+0.846*FORM1)*(1+0.968*FORM2), where FORM1 has a value of zero except for individuals with C2 formulation, and FORM2 has a value of zero except for individuals with the C3 formulation where the values are then set to 1. ALAG8= absorption lag time.
Maximum absorption rate (ka,max) = TVka,max *(1‐0.513*FORM2), where FORM2 has a value of zero except for individuals with the C3 formulation where it is set to 1.
Calculation of bioavailability (F):
| (1) |
where F a is the fraction absorbed, F h is the fraction escaping hepatic metabolism, and F g is defined according to Eq. (2).
Fraction escaping gut metabolism:
| (2) |
A abs,tot was the total amount of abemaciclib that was absorbed through the gut compartment following a single dose. This amount was determined as the sum of the fraction absorbed through the zero‐order route () and the transit route (F trans is the fraction absorbed via transit compartments). For the transit compartment route, as a result of the loss of some abemaciclib through gut metabolism to M2 and M20, the time integral of abemaciclib absorbed via this route following a single dose was used to obtain the amount absorbed through this mechanism:
Amount of abemaciclib absorbed through transit compartments:
| (3) |
where k a,max is the maximum rate of absorption, A trans is the amount of drug in the fourth (last) transit compartment, and K m,abs is the amount of drug that results in half the maximal absorption rate. The value 720 hours (representing 30 days after the dose is administered) was used to reflect infinity for the integration and capture the totality of the process. The previous calculation process to determine the total amount of abemaciclib absorbed through the gut wall can be understood in the context of Figure 1 .
F a was a parameter estimated in the model, and F h was calculated using the model parameter estimates applied to the well‐stirred model.
Because F could now be determined as shown previously, and the plasma clearance calculated from the well‐stirred model, the AUC0−∞ for abemaciclib could then be obtained according to Eq. (4).
AUC0‐∞:
| (4) |
The AUC0−∞ for M2 and M20 were obtained from the time integral of their predicted concentrations up to 720 hours after a single dose. Peak plasma concentrations were also obtained from the predicted concentration‐time profile. Table 3 shows the geometric mean and geometric percent coefficient of variation of various PK metrics in healthy and patient populations. These were obtained for a 200‐mg single dose using the individual post hoc PK parameters from all individuals in the final model. The metabolites were not administered intravenously and it was not possible to identify the fractions of their formation, their volumes of distribution and their clearances simultaneously. Therefore, the volumes were fixed to that of the parent drug, meaning that the fractions of metabolite formation and clearances are apparent values. The derived PK metrics were also compared with results from eight healthy subjects (who were a subset of the healthy population in the population PK data set) that were previously analyzed using a noncompartmental approach for a 200‐mg single dose as well. The VPC (Figure 2 ) demonstrated the adequacy of the mechanistic population PK model to describe abemaciclib, M2, and M20 concentrations. There appeared to be a slight overprediction of variability at the later times for M2 and M20, but this was likely a result of the limited amount of observed data in the later regions evidenced by the widening confidence intervals.
Table 3.
Geometric mean (%CV) for derived PK parameters after a single oral dose in MONARCH 1 (JPBN), MONARCH 2 (JPBL), and MONARCH 3 (JPBM) and comparison with previous analyses
| Parameter | Semimechanistic model development | External validation with phase III data | NCA(200 mg) | ||||
|---|---|---|---|---|---|---|---|
|
200 mg N = 421 |
Healthy subjects 200 mg N = 65 |
JPBN (patients) 200 mg N = 132 |
JPBL (patients) 200 mg N = 142 |
JPBL (patients) 150 mg N = 326 |
JPBM (patients) 150 mg N = 322 |
N = 8 |
|
| Parent | |||||||
| Bioavailability (F) | 0.482 (20.1) | 0.462 (17.9) | 0.426 (15.3) | 0.466 (16.8) | 0.494 (14.5) | 0.516 (12.9) | 0.448 (19) |
| Hepatic clearance (L/h) | 21.8 (39.8) | 23.8 (30.3) | 24.3 (24.9) | 26 (25.7) | 25.1 (21) | 23 (26.7) | 24.0 (27) |
| Half‐life (h) c , c | 24.8 (52.1) | 25.5 (25.8) | 25.8 (55.4) | 17.7 (54) | 17.5 (46) | 20.3 (43) | 29.3 (18) |
| AUC0−∞ (ng·h/mL) | 4420 (58.6) | 3890 (47.1) | 3510 (38.0) | 3580 (39.5) | 2960 (32.2) | 3360 (36.6) | 3730 (31) |
| Cmax (ng/mL) | 123 (77.1) | 113 (48.4) | 86.9 (59.4) | 141 (67.5) | 123 (59.9) | 125 (54.6) | 114 (32) |
| M2 | |||||||
| Apparent hepatic clearance (L/h) | 21.5 (54.3) | 22.9 (28.4) | 23.9 (35.3) | 20.9 (48.1) | 18.3 (49.9) | 21.6 (50.4) | NA |
| Half‐life (h) | 19.3 (65.4) | 21.2 (25.8) | 21.3 (73.5) | 16 (73.6) | 17.2 (65.1) | 16 (68.9) | 72.6 (23) |
| AUC0−∞ (ng.h/mL) | 1790 (71.6) | 1690 (39.9) | 1620 (55.0) | 1820 (72) | 1640 (70.2) | 1290 (75.8) | 1450 (19) |
| Cmax (ng/mL) | 30.3 (75.7) | 28.8 (36.1) | 26.9 (54.7) | 40.2 (73.9) | 33.6 (70.4) | 26.8 (68) | 31.7 (22) |
| M20 | |||||||
| Apparent hepatic clearance (L/h) | 23.7 (52.1) | 23.9 (23.7) | 26.2 (33.5) | 25.8 (43.3) | 22.5 (44.3) | 26 (46.4) | NA |
| Half‐life (h) | 17.5 (62.5) | 20.3 (21.3) | 19.4 (72.7) | 13 (70.1) | 14 (62.5) | 13.3 (65.6) | 35.3 (16) |
| AUC0−∞ (ng·h/mL) | 3240 (74.7) | 3450 (34.4) | 2750 (55.0) | 3020 (72.8) | 2870 (69.6) | 2300 (74.4) | 3260 (26) |
| Cmax (ng/mL) | 48.8 (83.0) | 49.1 (32.5) | 39.4 (56.0) | 66.1 (82.8) | 59.3 (78.7) | 45.7 (72) | 55.7 (26) |
%CV, coefficient of variation; AUC0−∞, area under the curve from 0 to infinity; Cmax, maximum plasma concentration; i.v., intravenous; JPBL, MONARCH 2; JPBM, MONARCH 3; JPBN, MONARCH 1; NA, not applicable; NCA, noncompartmental analysis.
Absolute bioavailability study that had 13C8‐abemaciclib administered i.v.
Absolute bioavailability study that had 13C8‐abemaciclib administered i.v.
Approximated as 0.693*(Vc+Vp)/CL, where Vc is the central volume, Vp is peripheral volume, and CL is the total hepatic clearance.
Figure 2.
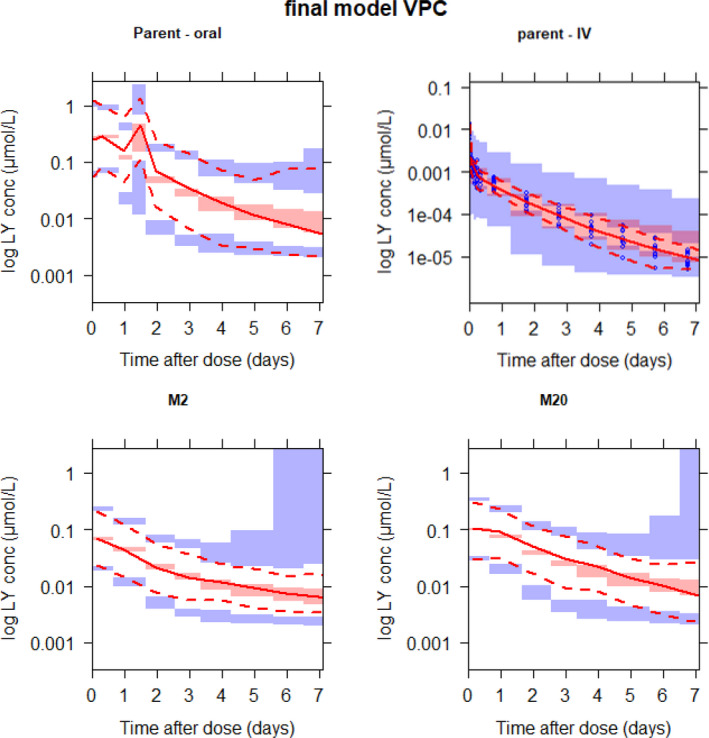
Prediction‐corrected visual predictive checks (VPC) up to 1 week after the dose for the final model. The images below demonstrate a simulated mechanistic population pharmacokinetic model predicting concentrations of abemaciclib (top) and its active metabolites M2 (bottom left) and M20 (bottom right). Intravenous (IV) data are only included for abemaciclib (top right). The blue dots (top right) are observations (shown only for intravenous data because they were too dense for the other panels and overshadowed the plot). The solid red line in each panel depicts median observed data, and the pink shaded areas define 95% confidence intervals around the median of simulated data. The dashed lines represent the observed 5th and 95th percentiles, and the blue shaded areas represent simulated 95% confidence intervals of the same. conc, concentration.
Absorption
The absorption of abemaciclib was best described through a parallel dual‐absorption process. One absorption pathway was a zero‐order process from the depot compartment into the liver, which had a typical duration of 3.18 hours (with high variability) preceded by a lag time of 1.74 hours. Formation of M2 and M20 through the zero‐order absorption pathway could not be identified. The other absorption pathway was described by a series of four transit compartments with a typical mean transit time of 1.44 hours. Abemaciclib absorbed through these transit pathways was susceptible to metabolism to form M2 and M20 in the gut. In addition, the absorption process from the final transit compartment to the liver was saturable, with a typical k a,max of 91.1 µmol/h (46.2 mg/h) and an amount that results in half maximal absorption rate of 52.7 µmol (27 mg). This translates to an intrinsic k a (absorption rate constant) of 1.73 h−1 in the absence of drug for the metabolism‐susceptible pathway. An estimated 86.8% of the total administered dose is absorbed either as abemaciclib, M2, or M20. Of this absorbed amount, 54.2% is absorbed via the zero‐order and 45.8% via first‐order transit compartments through the gut wall. The remaining 13.2% of the dose was either not absorbed or lost as other metabolites. Overall, the absorption process of abemaciclib was slow, saturable, and biphasic and was adequately captured using a parallel dual‐absorption model. The saturable absorption of parent drug through the transit compartments confers a dose ‐dependency, with higher doses leading to reduced overall absorption but more metabolism to M2 and M20. However, the overall impact on bioavailability and total exposure is minimal, as seen from model simulations in Figure 3 .
Figure 3.
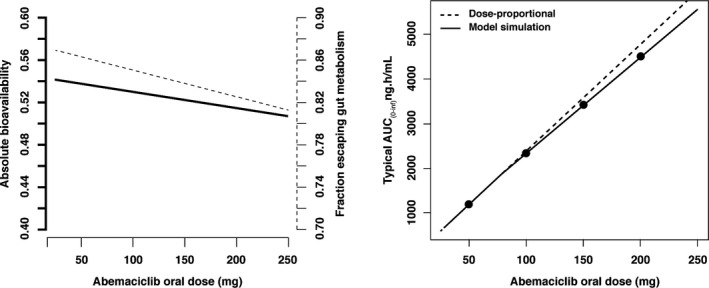
Impact of administered abemaciclib dose on bioavailability and exposure. The left panel shows the relationship between absolute bioavailability (solid line, left‐hand axis) and dose and also fraction escaping gut metabolism (dashed line, right hand axis) and dose. The right panel shows the relationship between AUC0−∞ and dose under the model simulation (solid line) and the assumption of dose proportionality (dashed line). AUC0−∞, area under the curve from 0 to infinity.
Significant covariates affecting absorption parameters were formulation, where the 50% w/w capsule and 25% w/w capsule (C3) have a longer lag time (almost 2‐fold) than the oral solution and the drug in capsule (C1). In addition, the 25% w/w capsule had a k a,max that was 51% lower than the other formulations. However, these differences did not translate to clinically meaningful differences in exposure as shown in Figure S1 . The effect of diarrhea on F a (fraction of dose entering the portal vein from the intestines) was minimal, with a reduction of 6%.
Hepatic elimination and first‐pass metabolism
The well‐stirred model provided an adequate fit to the data and successfully characterized the elimination of abemaciclib and the formation of metabolites. The typical total CLint of unbound abemaciclib that resulted in formation of M2 and M20 was 985 L/h. Taking these values, protein binding, and the blood‐to‐plasma ratio into account gives a plasma clearance of 22.8 L/h for a 70 kg individual. The hepatic extraction ratio was calculated to be 0.281, meaning that the fraction of drug entering the liver that subsequently escaped hepatic metabolism, F h, was 0.719. A third elimination pathway of abemaciclib, outside the formation of M2 and M20, could not be identified, and the parameter estimate for this pathway reduced to the lower boundary of zero. Between‐subject variability (BSV) in the CLint was estimated to be lower in healthy subjects than in patients. In addition, within‐subject variability (WSV) in CLint was estimated to be 68.1% in patients. Because the studies in healthy subjects were mainly single dose, it was not possible to separately identify WSV in CLint. Therefore, BSV for the healthy individuals more accurately represents the sum of BSV and WSV, which results in an even greater difference in the total population variability between healthy and patient populations.
Covariates
The effects of formulation on abemaciclib absorption (formulation on the lag time and absorption rate constant) appeared significant when screened using the empiric model. Inspection of covariate‐η plots showed no outstanding potential covariates upon inclusion of this covariate, except for slight trends for alkaline phosphatase and population (healthy vs. patient) on clearance parameters. However, upon testing these in the mechanistic model, the difference between healthy and patient populations was negligible, as was the effect of alkaline phosphate. There was no significant difference in absorption parameters between the fasted state, a standard meal, and a high‐fat meal in the crossover food effect study. Notably, covariates such as age, race, sex, and renal function were not significant or included in the final mechanistic model.
Model application
The addition of the sparsely sampled data from study JPBL to the meta‐analysis data set substantially increased the amount of data available on diarrhea and the effect of the C3 formulation on abemaciclib absorption. The parameters informed by these data (e.g., WSV in CLint, effects of C3 formulation on absorption, effect of diarrhea on F a) did not change significantly, and the covariate relationships remained statistically significant based on the bootstrap evaluation (Table S2 ). Therefore, no clinically meaningful differences in bioavailability, hepatic clearance, half‐life, AUC0−∞, or Cmax (Table 3 ) were observed when the model was extended to include JPBL. The model was also used to determine individual exposure metrics from the sparsely sampled JPBL and JPBM populations via simulation (Table S3 and Table S4 ).
This updated model (based on 13 studies including one phase III study) was successfully used to predict a separate phase III study (MONARCH 2/3) without requiring reestimation of the model parameters, as confirmed by VPCs (Figure 4 ).
Figure 4.
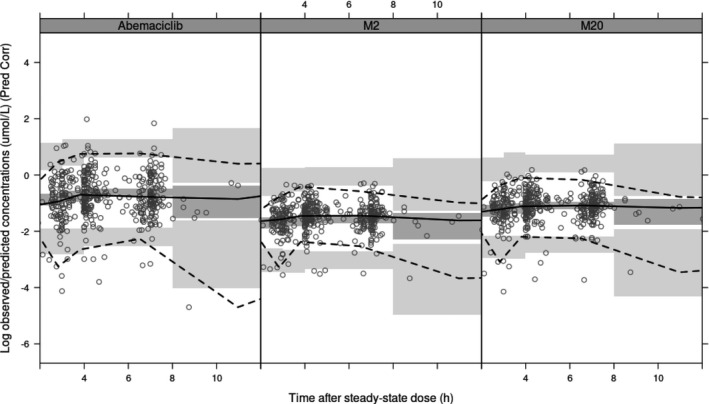
Model application: Prediction‐corrected (Pred Corr) visual predictive checks for Study JPBM (MONARCH 3). The model predicts concentrations of abemaciclib (left panel) and its active metabolites M2 (middle panel) and M20 (right panel) over time after steady‐state dose. The circles show the observed data. The solid lines in each panel depict median observed data, and the dark gray shaded areas define 95% confidence intervals around the median of the simulated data. The dashed lines represent the observed 5th and 95th percentiles, and the light gray shaded areas represent 95% confidence intervals of the same percentiles. h, hour.
To compare PK in East Asian and non–East Asian patients, single‐dose and steady‐state exposure metrics (Cmax, minimum observed drug concentration, AUC during 1 dosing interval) of abemaciclib, M2, M20, and total active species were simulated (Table S5 (MONARCH 2) and Table S6 (MONARCH 3)). Exposures for East Asian and non–East Asian patients with metastatic breast cancer in MONARCH 2 and MONARCH 3 were highly comparable across all exposure metrics of abemaciclib, M2, M20, and total active species, thus demonstrating no effect of East Asian ethnicity on abemaciclib PK.
DISCUSSION
We have employed a population PK modeling approach that includes mechanistic components to describe the PK of abemaciclib and its major metabolites. The approach included a combination of top‐down (data driven) and bottom‐up (based on physicochemical properties to estimate blood‐plasma ratios and protein binding). Standard model diagnostics demonstrated the adequacy and robustness of the model. The VPC appears to overpredict the variability for the intravenous data. This is likely a result of the small number of study participants with intravenous data (N = 8).
Furthermore, a comparison of model‐derived PK metrics with those from a noncompartmental analysis of rich data showed good agreement, further supporting model performance. Inclusion of data for intravenous dosing was key for model identifiability and issues with identifiability were resolved by fixing metabolite volumes to the values for parent. In addition, this mechanistic population PK model was developed in tandem with a physiologically‐based PK model, and the consistency between these two models further speaks to identifiability. 9 The population PK model was subsequently used to investigate the impact of intrinsic and extrinsic factors on various PK parameters. The model building process involved a combination of integration of data from multiple studies (some with rich sampling) with some more sparsely sampled data from a single pivotal trial (JPBN). However, sequentially adding data from a second pivotal trial (JPBL) did not result in any significant changes in model parameters. This gave us confidence in the ability to predict a future study (JPBM) using the same model parameters. Other pooled analyses have also found that a sequential approach of adding new studies in a stepwise manner and assessing model performance and compatibility of the new data proved more efficient than pooling all data at once at the outset. 16
Body weight was included using allometric relationships 13 for some PK parameters (CLint and volumes) and on liver volume (through using body‐surface area as a predictor of liver volume). However, there was no clinically significant effect of body weight on abemaciclib exposure in adults (Figure [Link] , [Link] ), supporting a flat dosing paradigm. This conclusion is strengthened by the fairly wide distribution of weights in the data set (Table 1 ). The low impact of body weight is likely attributed to the relatively low extraction ratio of abemaciclib, such that body weight–related changes in hepatic metabolic capacity translate to small changes in elimination. In addition, age, race, sex, or renal function were not significant covariates, indicating that no dose adjustments are necessary based on these patient factors in adults. The small change in bioavailability attributed to diarrhea is not expected to be of clinical significance. However, a wide confidence interval (from bootstrap) of the diarrhea effect means that the result should be interpreted cautiously. The uncertainty may be a consequence of the sampling scheme where most patients would not have both prediarrhea and postdiarrhea PK samples as PK sampling was limited to the first three cycles in patients and diarrhea tended to occur quite early. Drug formulation was a statistically significant covariate, but the effect concerned rate rather than the extent of absorption. Therefore, differences would be noted for the time to peak concentration, but not overall drug exposure. The application of the model to new data, and especially in the target population, i.e., patients with advanced breast cancer, confirmed the robustness assessment, covariate relationships, and the predictive performance of the model.
In summary, we present a mechanistic population PK model that incorporated intestinal metabolism and hepatic metabolism (including first‐pass effect) using the well‐stirred liver model. The parallel biphasic absorption model captured the slow, long, and complex absorption process of abemaciclib. The model was used to investigate the effect of potentially clinically significant covariates, although none were found. As part of future work, the model can be used as part of an exposure–response model to determine the exposure–response relationship of abemaciclib in patients with cancer. The model was able to predict a separate study as part of an external evaluation process without having to refit the model parameters, adequately predicting both the central tendency and population variability. This means that the model can be used to predict the PK of abemaciclib and its two metabolites without the need of intensive PK sampling in future studies.
Funding
The study was funded by Eli Lilly and Company.
Conflict of Interest
All authors are current or former employees and shareholders of Eli Lilly and Company
Author Contributions
E.C., A.K.S., M.M.P., J.S.W., and P.K.T. wrote the manuscript. E.C and P.K.T. designed the research. E.C., S.R.P.K., M.M.P., and J.S.W. analyzed the data.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Abemaciclib is primarily metabolized by cytochrome P450 3A4 to several active metabolites. The best measure of the active moiety is total exposure of abemaciclib plus its metabolites. The published pharmacokinetics of abemaciclib is currently limited to parent abemaciclib from a single phase I study.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ A semimechanistic pharmacokinetic model was developed to characterize the absorption kinetics and intestinal and hepatic biotransformation of abemaciclib and its active metabolites to evaluate the importance of patient characteristics on changes in the active moiety.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Abemaciclib undergoes parallel, dual, saturable absorption processes as well as intestinal and hepatic metabolism. Dose adjustment is not necessary for race, gender, bodyweight, or age. Diarrhea has a minimal impact on the bioavailability of abemaciclib.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ A platform that extends the population approach to more mechanistic PK modeling enables the characterization of drug pharmacokinetics through quantification of intestinal and hepatic first‐pass effects to evaluate the effects of patient characteristics on the active moiety.
Supporting information
Fig S1
Fig S2
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Supplementary Material
Acknowledgments
The authors would like to thank Stephen D. Hall, Michael Heathman, and Sonya C. Chapman for expert opinion and insightful advice. Editorial support was provided by Rangan Gupta, Eli Lilly Services India Private Limited.
References
- 1. Gelbert, L.M. et al Preclinical characterization of the CDK4/6 inhibitor LY2835219: in‐vivo cell cycle‐dependent/independent anti‐tumor activities alone/in combination with gemcitabine. Invest. New Drugs 32, 825–837 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Patnaik, A. et al Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non‐small cell lung cancer, and other solid tumors. Cancer Discov. 6, 740–753 (2016). [DOI] [PubMed] [Google Scholar]
- 3. Eli Lilly and Company . Verzenio prescribing information <https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208716s000lbl.pdf> (2017). Accessed April 16, 2020.
- 4. Dickler, M.N. et al MONARCH 1, A phase II study of abemaciclib, a CDK4 and CDK6 inhibitor, as a single agent, in patients with refractory HR+/HER2‐ metastatic breast cancer. Clin. Cancer Res. 23, 5218–5224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Goetz, M.P. et al MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 35, 3638–3646 (2017). [DOI] [PubMed] [Google Scholar]
- 6. Sledge, G.W. Jr. et al MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2‐ advanced breast cancer who had progressed while receiving endocrine therapy. J. Clin. Oncol. 35, 2875–2884 (2017). [DOI] [PubMed] [Google Scholar]
- 7. Tate, S.C. , Sykes, A.K. , Kulanthaivel, P. , Chan, E.M. , Turner, P.K. & Cronier, D.M. A population pharmacokinetic and pharmacodynamic analysis of abemaciclib in a phase I clinical trial in cancer patients. Clin. Pharmacokinet. 57, 335–344 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kulanthaivel, P. et al Pharmacokinetic drug interactions between abemaciclib and CYP3A inducers and inhibitors [abstract CT153]. Cancer Res. 76 (14 suppl.), CT153–CT153 (2016). [Google Scholar]
- 9. Posada, M.M. , Morse, B.L. , Turner, P.K. , Kulanthaivel, P. , Hall, S.D. & Dickinson, G.L. Predicting clinical effects of CYP3A4 modulators on abemaciclib and active metabolites exposure using physiologically based pharmacokinetic modeling. J. Clin. Pharmacol. 60, 915–930 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lindbom, L. , Pihlgren, P. & Jonsson, E.N. PsN‐Toolkit–a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput. Meth. Programs Biomed. 79, 241–257 (2005). [DOI] [PubMed] [Google Scholar]
- 11. Karlsson, M.O. , Beal, S.L. & Sheiner, L.B. Three new residual error models for population PK/PD analyses. J. Pharmacokinet Biopharm. 23, 651–672 (1995). [DOI] [PubMed] [Google Scholar]
- 12. Pond, S.M. & Tozer, T.N. First‐pass elimination. Basic concepts and clinical consequences. Clin. Pharm. 9, 1–25 (1984). [DOI] [PubMed] [Google Scholar]
- 13. Holford, N.H. A size standard for pharmacokinetics. Clin. Pharma. 30, 329–332 (1996). [DOI] [PubMed] [Google Scholar]
- 14. Kambhampati, S.R.P. , Chigusta, E. , Sykes, A.K. & Turner, P.K. A simultaneous PK‐diarrhea model to assess the impact of diarrhea on bioavailability of abemaciclib [M‐002]. The Eighth American Conference on Pharmacometrics, Fort Lauderdale, FL, October 14–19, 2017 <https://www.go‐acop.org/assets/Legacy_ACOPs/ACOP8/Abstracts/M‐02.pdf>. Accessed April 15, 2020.
- 15. Duffull, S.B. , Wright, D.F. & Winter, H.R. Interpreting population pharmacokinetic‐pharmacodynamic analyses—a clinical viewpoint. Br. J. Clin. Pharmacol. 71, 807–814 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Svensson, E. et al Integration of data from multiple sources for simultaneous modelling analysis: experience from nevirapine population pharmacokinetics. Br. J. Clin. Pharmacol. 74, 465–476 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Supplementary Material