

Abstract
This modeling and simulation analysis was aimed at selecting doses of cinpanemab (BIIB054), a monoclonal antibody targeting aggregated α‐synuclein, for a phase II study in Parkinson’s disease (PD). Doses and regimens were proposed based on anticipated target concentration in brain interstitial fluid (ISF); in vitro/in vivo data on the affinity of monoclonal antibodies to the target protein; and safety, tolerability, and pharmacokinetic data (1–135 mg/kg intravenous administration) from a phase I single ascending dose (SAD) study. A population pharmacokinetic modeling approach was used to select intravenous doses of 250, 1,250, and 3,500 mg every 4 weeks, to maintain 50%, 90%, and > 90% of target binding in ISF of PD participants. A favorable safety profile from the SAD study—which showed that cinpanemab was generally well‐tolerated at doses up to 90 mg/kg, supported by modeling and simulations of the anticipated safety margins—allowed implementation of a fixed‐dose approach.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ The safety, tolerability, and pharmacokinetics (PK) of cinpanemab (BIIB054) in a single ascending dose (SAD) study have been reported.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ In the absence of human efficacy and target engagement information, what should be the dosing rationale for a phase II study of cinpanemab in participants with Parkinson’s disease? Is switching from a weight‐based to fixed‐dose option feasible?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ We identify fixed doses that target the anticipated efficacious cinpanemab concentration in brain interstitial fluid using a population PK model based on the SAD study.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This study showcases the integration of human study data, in vitro information, and in vivo information for dose selection in the target population, in the absence of an established efficacy marker in humans. Thus, time and cost could be reduced during clinical development. This concept could be applied to other neurological disease areas, for which human efficacy information is often lacking.
Parkinson’s disease (PD) is one of the most common neurodegenerative diseases in the world and it is clinically diagnosed by the constellation of rest tremor, bradykinesia, and cogwheel rigidity. 1 In the United States alone, its estimated prevalence ranges from 430,000 to ~ 1 million, 2 and its economic burden was estimated to be approximately US $15 billion in 2010. 3 The incidence of PD increases with age, with the disease being rare before age 50 years but affecting up to 4% of the population in the oldest age groups. 4 Men may be 1.5 times more likely to be affected than women, especially among those older than 70 years in Western populations. 5 Severe disability or death may be expected in 35% of patients within 5 years of onset, in 65% of patients within 10 years, and in 80% of patients within 15 years. 6 , 7 , 8 , 9
The pathophysiology of PD has been linked to aggregation of α‐synuclein (α‐syn), with stereotypic cell‐to‐cell spreading of synuclein pathology believed to contribute to disease progression. Immunotherapy with antibodies directed against α‐syn is considered a promising therapeutic approach for slowing disease progression. 10 Cinpanemab (BIIB054) is a human‐derived α‐syn antibody and is highly selective for aggregated forms of α‐syn, with an ≥ 800‐fold higher apparent affinity for fibrillar vs. monomeric recombinant α‐syn. 11 The half maximal effective concentration (EC50) of cinpanemab for aggregated α‐syn was estimated at ~ 0.25 nM and the 90% maximal effective concentration (EC90) was ~ 2.1 nM (0.0375 and 0.315 µg/mL, respectively).
Recently, a phase I study (NCT02459886) was conducted with the primary objective to assess the safety and tolerability of single doses of cinpanemab in healthy volunteers (HVs) and participants with early PD. Secondary objectives included assessment of the pharmacokinetics (PKs) and immunogenicity of cinpanemab. This was a two‐part, phase I, randomized, double‐blind, placebo‐controlled, single ascending dose (SAD) study. Part 1 included HVs (n = 48, age 40–65 years), whereas part 2 included participants with early PD (n = 18, age 47–75 years), Hoehn and Yahr stage ≤ 2.5, and disease duration of ≤ 5 years. In this study, cinpanemab exhibited linear PK when administered at i.v. dose levels of 1–135 mg/kg. The mean clearance values were 0.00401–0.00542 L/h. The volume of distribution at steady‐state ranged from 4.34–5.25 L. The serum half‐life of cinpanemab was 28–35 days; the cerebrospinal fluid (CSF)‐to‐serum ratio ranged from 0.13–0.56%. Details related to study design and results have been previously reported. 12
Drug level in the CSF is widely used as a surrogate for assessment of central nervous system (CNS) exposure. CSF is the only biofluid that can be sampled to provide insights into the CNS and the biochemical processes taking place in the brain. 13 The increase in CSF cinpanemab concentrations with an increase in dose indicates that cinpanemab can reach the target site of action in brain interstitial fluid (ISF). At 4 weeks postinfusion, the mean CSF‐to‐serum ratios were reported to be in the range of 0.273–0.559%, with slightly higher ratios for patients with PD than HVs. Incidence of neutralizing anti‐cinpanemab antibodies was reported as < 1%. 12
The objectives of this modeling and simulation analysis were to (i) develop the first population PK (PopPK) model of cinpanemab for characterizing the PK and distribution of cinpanemab from the central compartment (serum) to CSF, using serum and CSF cinpanemab concentrations in HVs from the SAD study; (ii) utilize the developed model to propose three dose levels for a phase II proof‐of‐concept (POC) study (NCT03318523) as well as a Japanese first‐in‐human (FIH) phase I SAD and multiple ascending dose study (NCT03716570) in PD patients; and (iii) evaluate if fixed dosing instead of weight‐based dosing can be implemented in the above two studies of participants with PD.
METHODS
Clinical data
Clinical data for modeling purposes were obtained from a phase I, randomized, double‐blind, placebo‐controlled, SAD study (NCT02459886), in which a single i.v. dose of cinpanemab or placebo was administered. In part 1, HVs (n = 34) were assigned to one of six cohorts and randomized to receive either a single i.v. dose of cinpanemab (1, 5, 15, 45, 90, or 135 mg/kg) or placebo. Serial serum PK samples were collected postinfusion at the following time points: < 10 minutes, 0.5 hours, 1 hour, 2 hours, 4 hours, 8 hours, 12 hours, 24 hours, 48 hours, 72 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 6 weeks, 9 weeks, 12 weeks, and 16 weeks. CSF samples for the measurement of cinpanemab were collected at baseline, 8 or 24 hours postinfusion, and 3 weeks postdose. In part 2 of the study, participants with PD were randomly assigned to a single i.v. dose of 15 or 45 mg/kg or placebo; however, the study was still ongoing for participants with PD at the time of this analysis, hence part 2 data were unavailable and not included. Additional details of the study design and results have been previously reported. 12
The study was sponsored by Biogen and conducted in accordance with the ethical standards of the institutional and/or national research committee and with the principles for human experimentation as defined in the 1964 Declaration of Helsinki and its later amendments, or comparable ethical standards, and the International Conference on Harmonisation Good Clinical Practice Guideline. The trial was approved by the respective institutional review boards and informed consent was obtained from all individual participants included in the studies.
Bioanalysis
The concentration of cinpanemab in human serum and CSF samples was determined using a validated enzyme‐linked immunosorbent assay. Microtiter plates coated with truncated α‐syn (1–20 amino acid) peptide were used to capture cinpanemab from serum or CSF. After sample incubation and washing steps, bound cinpanemab was detected by horseradish peroxidase (HRP)‐conjugated goat anti‐human F(ab’)2 Fc fragment antibody. At the end of the incubation with the HRP‐antibody conjugate, the plates were washed, and the bound HRP‐conjugate was detected by reaction with a chromogenic substrate tetramethylbenzidine. The color development was stopped with acid stop solution (H2SO4). The intensity of the color, which was directly proportional to the amount of cinpanemab present in serum or CSF, was measured in a microplate reader at 450 nm with a reference wavelength of 650 nm. The validated assay range was 375–5,000 ng/mL for serum and 15–750 ng/mL for CSF.
Software for PopPK analysis
Serum and CSF concentration‐time profiles were used for nonlinear mixed‐effect modeling (NONMEM) by extended least‐squares regression using NONMEM version 7.4 (ICON Development Solutions, Ellicott City, MD). 14 The first‐order conditional estimation with interaction method was used. Graphical and all other statistical analyses, including evaluation of NONMEM outputs, were performed using R version 3.0.1 or higher.
PopPK modeling
The analysis included three steps: (i) construction of serum and CSF base model; (ii) covariate analysis; and (iii) model evaluation.
The schematic for the serum CSF PK model is shown in Figure 1 . A linear two‐compartment model, including first‐order elimination from the central compartment with proportional residual error, was selected as the final base serum model. The model was characterized using systemic clearance (CL), intercompartmental clearance, volume of distribution of the central compartment (V1), and volume of distribution of the peripheral compartment (V2). CSF‐related parameters included volume of the CSF compartment (V3), the rate constant from the central compartment to the CSF (K13), and the rate constant from the CSF compartment to the central compartment (K31). Likelihood ratio test was used to discriminate hierarchical models with a significance level of P < 0.05 (change in minimum value of objective function = 3.84 for 1 degree of freedom). V3 was attempted to be estimated during model building steps, however, due to limited CSF data, there were difficulties in estimating this parameter reliably, hence V3 was fixed to 0.175 L (which is within the reported ranges of CSF in human). 15 , 16
Figure 1.
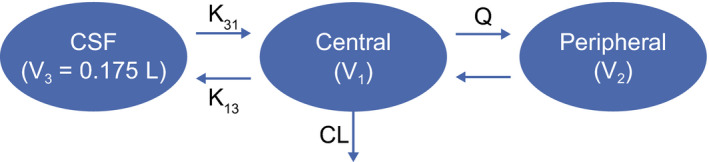
Structure of the serum cerebrospinal fluid (CSF) population pharmacokinetic model for cinpanemab. CL, systemic clearance; K13, rate constant from the central compartment to the cerebrospinal fluid; K31, rate constant from the cerebrospinal fluid compartment to the central compartment; Q, intercompartmental clearance; V1, volume of distribution of the central compartment; V2, volume of distribution of the peripheral compartment; V3, volume of the cerebrospinal fluid compartment.
The correlations between random effects were explored and incorporated into the model if deemed necessary.
Interindividual variability (IIV) was modeled assuming a log‐normal distribution as shown below:
where P is a PK parameter, tvP is a typical value of PK parameter, and eta distribution is assumed to follow normal distribution N(0, ). Due to insufficient data, the IIV term for K31 was not determined. In the final model, K31 was fixed to the typical value K31. Due to the limited sample size of participants in the analysis, and all being HVs, only body weight was used in covariate analysis. Volume of distribution and/or CL of monoclonal antibodies generally increase with body size, which results in a decrease of their concentrations. Because distribution and elimination of monoclonal antibodies are often proportional to body weight, 3 , 17 CL and V1 were scaled by body weight (normalized to 70 kg) using a power function. Once the phase II study (NCT03318523) data from early PD participants are available, the covariate model will be updated to reflect the covariate effect from a larger population dataset, which will include disease‐related and demographic‐related covariates.
The goodness‐of‐fit of NONMEM analyses was assessed by examination of scatterplots of observed concentrations vs. population‐predicted concentrations and vs. individual‐predicted concentrations, and scatterplots of conditional weighted residuals and normalized prediction distribution error vs. population‐predicted concentrations and vs. time since last dose.
In addition, the estimated shrinkage of random effects was also assessed, as previously described. 17 The covariance step was examined and the asymptotic standard errors of fixed and random effects produced by NONMEM were used to calculate the percent of relative standard errors. In addition, correlations between population parameters and the condition number were evaluated to verify that the model was not ill‐conditioned.
The final PopPK model was internally evaluated using visual predictive checks.
Model‐based simulations for selection of phase II doses
The final model was used for simulation of a range of dosing scenarios. Estimated PK parameters, as well as between‐participant variability and residual variability estimates from the PopPK model, were used to simulate 1,000 serum and CSF steady‐state profiles. Simulations of CSF profiles were conducted for several doses between 1 and 50 mg/kg to enable dose selection for phase II. To account for weight differences between HVs and the target PD population in phase II, the Parkinson’s Progression Markers Initiative database 18 was used as a source of weight distribution (Figure S1 ) in participants with PD. To account for weight differences between HVs and the target Japanese PD population in the Japanese FIH study, Biogen internal study data were used as a source of weight distribution for Japanese participants with PD (Figure S2 ). CSF and ISF concentrations of cinpanemab were assumed to be equal. The in vitro binding potency of cinpanemab to aggregated α‐syn (0.0375 and 0.315 µg/mL for EC50 and EC90, respectively) was used as the target value for dose selection purposes. For the low dose in the study, simulated cinpanemab trough concentrations in CSF at steady‐state were expected to be above EC50 for the majority of participants. The highest dose was selected to maintain trough steady‐state CSF levels above EC90 for 95% of participants. The intermediate dose was selected to provide reasonable separation between low and high dose and to maintain trough CSF concentrations at approximately EC90 for 50% of participants (Figure 2 ). For a final dose selection, simulations were also conducted to evaluate the feasibility of switching from body weight‐based dosing to fixed dosing in the phase II and Japanese FIH studies. For the highest dose level, steady‐state serum area under the concentration‐time curve from time zero to the time of next dosing (AUCtau), maximum observed concentration (Cmax), and corresponding safety margins were calculated for both populations using a weight‐based vs. fixed‐dose approach.
Figure 2.
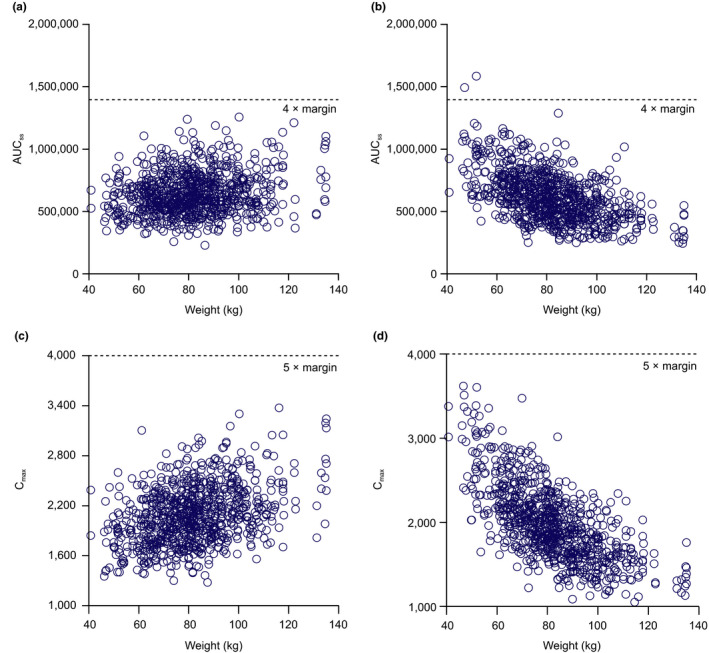
Simulated steady‐state pharmacokinetics for assessing weight‐based vs. fixed‐dose approach. Area under the concentration‐time curve at steady‐state (AUCss) using a weight‐based 45 mg/kg (a) vs. a fixed‐dose 3,375 mg approach (b). Maximum concentration at steady‐state (Cmaxss, µg/mL) using a weight‐based 45 mg/kg (c) vs. fixed‐dose 3,375 mg approach (d).
RESULTS
Summary of data for PopPK analysis
The PopPK analysis dataset had cinpanemab serum and CSF data for 34 HVs, of which 62% were men. Among the HVs, mean (SD) age was 50 (6.3) years and mean (SD) weight was 76 (10.5) kg. Available PK data included 583 serum and 66 CSF concentrations. Of these samples, 8 CSF measurements were below the limit of quantification in the lower dose groups of 1 and 5 mg/kg. Overall, ~ 11% of CSF PK samples were below the limit of quantification, with all 8 samples being from the lower dose groups of 1 and 5 mg/kg, for which concentrations are expected to be low, due to < 1% of the dose being able to penetrate the CSF compartment from serum. Hence, the final analysis dataset consisted of 583 serum and 58 CSF PK samples. Graphical analysis showed that the CSF cinpanemab concentrations generally increased with increased serum cinpanemab concentrations across doses (Figure S3 ). Some overlap was seen at higher doses (90 and 135 mg/kg, for which doses were less than twofolds apart) due to high variability observed.
Model
The PopPK model adequately described the serum and CSF PK data in HVs (Figure S4 ). Population parameter estimates from the final serum CSF PK model using HV data are shown in Table 1 . The linear clearance was estimated to be 0.00465 L/h (IIV, 25%) and the central volume of distribution was 2.43 L (IIV, 14%). The precision of fixed‐effect and random‐effect parameter estimates was acceptable, with a relative standard error < 37% for the fixed‐effect and < 45% for the random‐effect parameters. The largest IIV estimate was in rate constant (45%). In general, the shrinkage for IIV was low, with the largest eta shrinkage estimated as 33% for the intercompartmental clearance. Residual (proportional) variability in cinpanemab serum and CSF concentrations were: ~ 31% and ~ 54%, respectively, of the predicted concentrations. The sparse nature of the CSF sampling may have contributed to the moderately high residual variability associated with the CSF concentrations. The objective function value dropped by ~ 24 points for the model, with CL and V1 adjusted by body weight (P < 0.001). The exponent of the body‐weight effect on CL was estimated as 0.851, whereas the exponent of body‐weight effect on V1 was estimated as 0.588. Using the estimated exponents of body weight, it can be calculated that a 10% increase in body weight was associated with an 8% and 6% increase in CL and V1, respectively. Model fitting was not significantly improved (P > 0.05) when exponents were fixed to allometric exponents of 0.75 for CL and 1 for V1, vs. estimated.
Table 1.
Cinpanemab population pharmacokinetic parameter estimates
| Parameter | Estimate | SE (%RSE) | Shrinkage |
|---|---|---|---|
| Serum | |||
| tvCL. L/h | 0.00465 | 2.37E−04 (5.1) | |
| tvV1, L | 2.43 | 6.96E−02 (2.9) | |
| tvQ, L/h | 0.0183 | 1.15E−03 (6.3) | |
| tvV2, L | 2.21 | 6.67E−02 (3.0) | |
| Proportional error | 0.0975 | 3.25E−03 (3.3) | |
| CSF | |||
| tvV3, L | 0.175 | ||
| tvK13, 1/h | 7.38E−07 | 7.56E−08 (10.2) | |
| tvK31, 1/H | 0.00732 | 6.16E−04 (8.4) | |
| Body‐weight exponent on V1 | 0.588 | 3.13E−01 (36.8) | |
| Body‐weight exponent on CL | 0.851 | 1.76E−01 (29.9) | |
| Proportional error | 0.296 | 4.04E−02 (1.37) | 9.6 |
| IIV | |||
| tvCL | 0.0645 (25.4) | 0.0161 (25) | 1.1 |
| tvV1 | 0.0192 (13.9) | 0.00508 (26.4) | 3.3 |
| tvQ | 0.0544 (23.3) | 0.0294 (54) | 32.6 |
| tvV2 | 0.0169 (13.0) | 0.00742 (43.9) | 24.0 |
| tvK13 | 0.2 (44.7) | 0.0639 (32) | 12.0 |
Shrinkage is the residual error shrinkage standard deviation percent reported for residual error using EPSSHRINKSD from nonlinear mixed‐effect modeling (NONMEM) output and interparticipant shrinkage for eta reported in percent using ETASHRINKSD from NONMEM output.
%RSE, percent of relative standard errors; CL, systemic clearance; CSF, cerebrospinal fluid; IIV, interindividual variability; K13, rate constant from the central compartment to the cerebrospinal fluid; K31, rate constant from the cerebrospinal fluid compartment to the cerebrospinal fluid compartment; Q, intercompartmental clearance; tv, typical value; V1, volume of distribution of the central compartment; V2, volume of distribution of the peripheral compartment; V3, volume of cerebrospinal fluid compartment.
The visual predictive check (Figure S5 ) showed that the PopPK model developed was appropriate in describing the time course of serum and CSF cinpanemab concentrations and the variability in HVs in the SAD study.
Simulations for dose selection
After a first round of simulations, conducted to identify appropriate weight‐based doses, the doses of 3, 15, and 45 mg/kg were selected for the phase II study based on predefined CSF target concentrations. Further evaluation showed that for the expected weight distribution, the safety margin for AUC steady‐state (AUCss) at a fixed dose of 3,375 mg is expected to be similar to the body weight‐based dose of 45 mg/kg (Figure 2 ). The fixed dose was calculated assuming a mean body weight of 75 kg. With either of these approaches, AUCtau and Cmax at steady‐state are expected to be above a fivefold safety margin, based on exposure observed in the 6‐month toxicology study in rats (Table 2 ). Finally, fixed doses of 250, 1,250, and 3,500 mg were selected for the phase II study. The simulated CSF profiles and summary statistics of the simulated steady‐state trough CSF concentrations for proposed phase II doses are shown in Figure 3 and Table 3 .
Table 2.
Projected steady‐state cinpanemab serum AUCtau, steady‐state Cmax, and safety margins for phase II doses
| Dose, mg | Projected parameters | Safety margins a | ||
|---|---|---|---|---|
|
Median (q5–q95) AUCtau, h*µg/mL |
Median (q5–q95) Cmax, µg/mL |
Median (q5‐q95) AUCtau, h*µg/mL |
Median (q5–q95) Cmax, µg/mL |
|
| Phase II study | ||||
| 250 | 42,700 (27,400–68,200) | 175 (111–276) | 131 (204–82) | 114 (180–72) |
| 1,250 | 214,000 (135,000–337,000) | 882 (573–1,450) | 26 (41–17) | 23 (35–14) |
| 3,500 | 612,000 (386,000–960,000) | 2,480 (1,630–3,930) | 9 (15–6) | 8 (12–5) |
| Japanese FIH study | ||||
| 1,250 | 371,580 (276,340–504,370) | 1,020 (790–1,360) | 15 (20–11) | 20 (15–25) |
| 3,500 | 1,015,600 (761,000–1,347,000) | 2,840 (2,160–3,700) | 5 (7–4) | 7 (9–5) |
AUCtau, area under the concentration‐time curve from time zero to the time of next dosing; Cmax, maximum observed concentration; FIH, first‐in‐human; q, percentile.
Calculated based on mean AUC0–168h and Cmax after the last dose of cinpanemab in the 26‐week rat toxicology study. AUCtau at no‐observed‐adverse‐effect level = AUC0–168h*4 = 5,580,000 h*µg/mL.; Cmax = 20,000 µg/mL.
Figure 3.
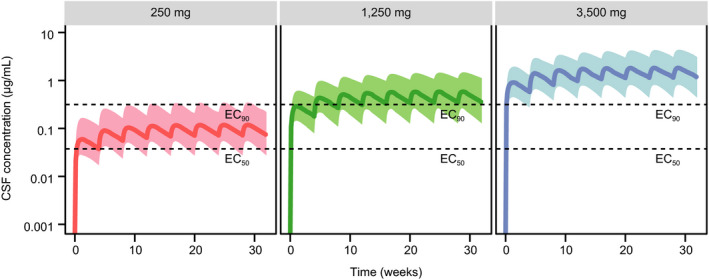
Simulated cinpanemab cerebrospinal fluid (CSF) concentration‐time profiles for proposed doses in phase II. Solid line = population median; shaded area = 90% prediction interval. EC50, half maximal effective concentration; EC90, 90% maximal effective concentration.
Table 3.
Summary statistics of simulated steady‐state trough CSF concentrations (µg/mL) for proposed phase II doses
| Dose, mg | Simulated steady‐state trough CSF concentrations, μg/mL | ||
|---|---|---|---|
| Median, q50 | q5 | q95 | |
| 250 | 0.076 | 0.029 | 0.191 |
| 1,250 | 0.369 | 0.128 | 0.936 |
| 3,500 | 1.07 | 0.403 | 2.79 |
CSF, cerebrospinal fluid; q, percentile.
DISCUSSION
A modeling and simulation‐based approach was used to select three doses of cinpanemab for a phase II POC study in early PD. The dose levels of cinpanemab were selected based on the safety, tolerability, and PK of cinpanemab in HVs; exposure of cinpanemab in nonclinical toxicology studies; cinpanemab affinity to aggregated α‐syn in vitro; and simulated cinpanemab CSF concentration‐time profiles in steady‐state.
Although ISF data exist for some biomarkers, such as tau and α‐syn proteins, the current technology does not allow measurements of monoclonal antibodies in human ISF. Furthermore, it is assumed that monoclonal antibodies largely penetrate through the blood‐brain barrier and then move toward CSF with the bulk flow of ISF.
The cynomolgus monkey is generally considered the most relevant species for predicting human PK of monoclonal antibodies. Recent Biogen internal studies of monoclonal antibodies in cynomolgus monkeys suggest that CSF and ISF concentrations equilibrate over several days following dosing. During the distribution phase, the CSF and ISF concentrations may demonstrate different transient profiles; however, similar drug concentrations and half‐lives are achieved in both fluids throughout the clearance phase. In the absence of human ISF data, it is thus assumed that similar mechanisms underlie the brain distribution in both species. Penetration of antibodies into the brain is low across species and is generally in the range of 0.1–0.35%. 19 Because only a fraction of CSF comes from ISF, antibody concentration in CSF can be used as a conservative estimate of brain ISF levels. Hence, in both preclinical and clinical settings, CSF has been widely accepted as a surrogate for assessing CNS drug levels. 20 , 21 , 22
Cinpanemab is designed to bind aggregated species of α‐syn in extracellular fluid; however, no reliable quantitative assay exists to date that measures aggregated α‐syn in CSF. Thus, no direct target engagement measurements are available to inform clinical doses. Monomeric α‐syn in CSF has been measured extensively in both HVs and participants with PD. Based on the Parkinson’s Progression Markers Initiative dataset, mean α‐syn CSF levels are ~ 2,200 pg/mL for HV and 2,000 pg/mL for participants with PD. Assessment of α‐syn secretion in the brain showed α‐syn presence in healthy human brain ISF, and α‐syn in ISF collected via microdialysis was found to be in the range of 0.5–8 ng/mL. 18 No information is available on levels of α‐syn in the ISF of participants with PD. Limited experiments measuring aggregated α‐syn in human CSF suggest that its concentration is very low and represents a small fraction of monomeric α‐syn levels. Overall, concentration of aggregated α‐syn in CSF and ISF is expected to be below EC50 of cinpanemab (0.25 nM); therefore, only the potency of cinpanemab and its concentration determine the extent of target binding in the brain.
Efficacious doses for the phase II study were estimated using four major assumptions. First, it was assumed that a decrease in levels of unbound toxic aggregated α‐syn in brain ISF would slow progression of PD pathology and that the relative decrease in aggregated α‐syn is related to disease progression. Second, it was assumed that in vitro binding of cinpanemab to aggregated α‐syn is a reasonable estimate of its binding in vivo. Third, it was assumed that PK is similar between HVs and participants with PD, which was unknown at the time of the phase II dose selection but confirmed later from the PK of participants with PD (n = 12) in the FIH study. 12
Fourth, it was assumed that the CSF drug concentrations approximate ISF drug concentrations. The estimated efficacious exposure in mice was ~1,317 day*μg/mL. Clearance of cinpanemab in HVs is, on average, 0.0052 or 0.1248 L/d. 12 Thus, the projected mean minimum pharmacologically efficacious dose is ~164 mg. As in other neurodegenerative diseases, there is no reliable preclinical model for PD; therefore, the available exposure data were used as supportive information to set the lower exposure limit in the study.
Based on preclinical mouse data, the minimal efficacy is expected at a dose that corresponds to a decrease of ~50% in aggregated α‐syn brain ISF level; therefore, the low dose in the phase II study was selected to maintain the cinpanemab concentration in ISF at or above EC50 at steady‐state. Similarly, the highest dose targeted levels above EC90, and the middle dose was selected to maintain binding at around EC90.
PK data from HVs in the SAD study 12 were used for the dose selection. It was later demonstrated that PK parameters in serum and CSF are similar between HVs and participants with PD, and, therefore, dose adjustment for a phase II study is not required. In addition, a small cohort of participants in a phase II study had intense PK sampling after doses one and three, which allowed the elimination of a separate multiple ascending dose study.
Switching to fixed doses
At the time of protocol development for the phase II study, information about the safety of cinpanemab in participants with PD was limited to the SAD study only. Hence, one of the major concerns for implementing fixed dose in the phase II study was ensuring good safety margins using the fixed‐dose approach for participants of lower weight because lighter participants on a fixed‐dose regimen are expected to have higher exposure compared with those on a weight‐based regimen. Here, we utilized a modeling and simulation approach to compare a weight‐based vs. fixed‐dose approach. Given the favorable safety profile of cinpanemab, as demonstrated in preclinical toxicology studies and in the SAD study for HVs and participants with PD, a fixed‐dose approach was implemented in this phase II study as well as in the Japanese FIH study. Phase II study participants are receiving an i.v. infusion of cinpanemab (250, 1,250, or 3,500 mg) once every 4 weeks.
Based on safety margins and preclinical data, all three doses are expected to be safe and well‐tolerated in participants with PD, both Japanese and non‐Japanese. The highest planned dose (3,500 mg) is expected to yield mean steady‐state AUCtau and Cmax values approximately eightfold to ninefold lower than those observed at the no‐observed‐adverse‐effect level in the 26‐week toxicology study in rats (Table 2 ).
Limitations of this work include lack of efficacy data of PD in humans for this novel α‐syn pathway and lack of PK data in participants with PD at the time of this analysis. Hence, the dose selection here relies heavily on the in vitro drug receptor binding information. PK parameters of cinpanemab in individuals with PD, although unavailable at the time of the modeling work, were later reported to be similar to those of HVs. 12 When data from the phase II study become available, the PK model will be further refined and used to characterize the exposure‐response relationship in the phase II population, with the aim of providing further insight on future phase III study designs. In addition, it has to be emphasized that this study can only simulate the anticipated CSF concentration of the antibody in patients with PD and is unable to make any prediction regarding removal of aggregated a‐syn in the ISF.
In conclusion, a three‐compartment PopPK model with linear elimination from the central compartment was used to describe the time course of serum and CSF cinpanemab concentrations following i.v. administration of doses ranging from 1 to 135 mg/kg in HVs in the SAD study. For the POC phase II study, dose levels of 250, 1,250, and 3,500 mg every 4 weeks were selected based on this modeling and simulation work. These doses are expected to allow exploration of dose and exposure response as well as evaluation of maximum response. The proposed doses were implemented for the POC phase II study and for the FIH study in Japanese participants with PD. The data from these two ongoing studies will validate the exposure and efficacy predictions from this modeling and simulation work.
Conflicts of Interest
M.K., M.M., K.K.M., C.W., and N.P. are employees of and hold stock/stock options in Biogen.
Funding
This study was funded by Biogen. Biogen designed the study and collected, analyzed, and interpreted the data.
Author Contributions
M.K. and N.P. wrote the manuscript. All authors designed the research, conducted the research, and analyzed the data.
Supporting information
Fig S1‐S5
Supplementary Material
Acknowledgments
The authors thank Excel Scientific Solutions for editorial help, which was supported by Biogen. The authors also thank the entire cinpanemab clinical team for their timely support toward this work.
References
- 1. Bertram, L. & Tanzi, R.E. The genetic epidemiology of neurodegenerative disease. J. Clin. Invest. 115, 1449–1457 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beck, J.C. et al Abstract P0. 01.04: Prevalence of Parkinson’s disease in North America: a nationwide epidemiological study sharing available databases. J. Parkinsons Dis. 6 (suppl. 1), 53 (2016).26891177 [Google Scholar]
- 3. Kowal, S.L. , Dall, T.M. , Chakrabarti, R. , Storm, M.V. & Jain, A. The current and projected economic burden of Parkinson's disease in the United States. Mov. Disord. 28, 311–318 (2013). [DOI] [PubMed] [Google Scholar]
- 4. de Lau, L.M. & Breteler, M.M. Epidemiology of Parkinson's disease. Lancet Neurol. 5, 525–535 (2006). [DOI] [PubMed] [Google Scholar]
- 5. Twelves, D. , Perkins, K.S. & Counsell, C. Systematic review of incidence studies of Parkinson's disease. Mov. Disord. 18, 19–31 (2003). [DOI] [PubMed] [Google Scholar]
- 6. Poewe, W. The natural history of Parkinson’s disease. J. Neurol. 253(suppl. 7), vii2–vii6 (2006). [DOI] [PubMed] [Google Scholar]
- 7. Schrag, A. & Banks, P. Time of loss of employment in Parkinson's disease. Mov. Disord. 21, 1839–1843 (2006). [DOI] [PubMed] [Google Scholar]
- 8. Schrag, A. , Jahanshahi, M. & Quinn, N. How does Parkinson's disease affect quality of life? A comparison with quality of life in the general population. Mov. Disord. 15, 1112–1118 (2000). [DOI] [PubMed] [Google Scholar]
- 9. Shulman, L.M. et al Subjective report versus objective measurement of activities of daily living in Parkinson's disease. Mov. Disord. 21, 794–799 (2006). [DOI] [PubMed] [Google Scholar]
- 10. Liu‐Seifert, H. et al Delayed‐start analysis: mild Alzheimer's disease patients in solanezumab trials, 3.5 years. Alzheimers Dement. (N Y) 1, 111–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weihofen, A. et al Development of an aggregate‐selective, human‐derived alpha‐synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson's disease models. Neurobiol. Dis. 124, 276–288 (2019). [DOI] [PubMed] [Google Scholar]
- 12. Brys, M. et al Randomized phase I clinical trial of anti‐α‐synuclein antibody BIIB054. Mov. Disord. 34, 1154–1163 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eusebi, P. et al Diagnostic utility of cerebrospinal fluid alpha‐synuclein in Parkinson's disease: a systematic review and meta‐analysis. Mov. Disord. 32, 1389–1400 (2017). [DOI] [PubMed] [Google Scholar]
- 14. Beal, S.L. , Sheiner, L.B. , Boeckmann, A. & Bauer, R.J. NONMEM User's Guides. (Icon Development Solutions, Ellicott City, MD, 2009). [Google Scholar]
- 15. Chazen, J.L. et al Automated segmentation of MR imaging to determine normative central nervous system cerebrospinal fluid volumes in healthy volunteers. Clin. Imaging 43, 132–135 (2017). [DOI] [PubMed] [Google Scholar]
- 16. Sakka, L. , Coll, G. & Chazal, J. Anatomy and physiology of cerebrospinal fluid. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 128, 309–316 (2011). [DOI] [PubMed] [Google Scholar]
- 17. Savic, R.M. & Karlsson, M.O. Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J. 11, 558–569 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marek, K. et al Parkinson progression marker initiative. The Parkinson progression marker initiative (PPMI). Prog. Neurobiol. 95, 629–635 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shah, D.K. & Betts, A.M. Antibody biodistribution coefficients: inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. MAbs 5, 297–305 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Lange, E.C. Utility of CSF in translational neuroscience. J. Pharmacokinet. Pharmacodyn. 40, 315–326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johanson, C.E. , Duncan, J.A. 3rd , Klinge, P.M. , Brinker, T. , Stopa, E.G. & Silverberg, G.D. Multiplicity of cerebrospinal fluid functions: new challenges in health and disease. Cerebrospinal Fluid Res. 5, 10 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin, J.H. CSF as a surrogate for assessing CNS exposure: an industrial perspective. Curr. Drug Metab. 9, 46–59 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S5
Supplementary Material