

Abstract
Synthetic assembly within living cells represents an innovative way to explore purely chemical tools that can direct and control cellular behavior. We use a simple and modular platform that is broadly accessible and yet incorporates highly intricate molecular recognition, immolative, and rearrangement chemistry. Short bimodular peptide sequences undergo a programmed sequence of events that can be tailored within the living intracellular environment. Each sequential stage of the pathways beginning with the cellular uptake, intracellular transport, and localization imposes distinct structural changes that result in the assembly of fibrillar architectures inside cells. The observation of apoptosis, which is characterized by the binding of Annexin V, demonstrates that programmed cell death can be promoted by the peptide assembly. Higher complexity of the assemblies was also achieved by coassembly of two different sequences, resulting in intrinsically fluorescent architectures. As such, we demonstrate that the in situ construction of architectures within cells will broaden the community’s perspective toward how structure formation can impact a living system.
Introduction
Supramolecular interactions govern core aspects of cellular life where they are omnipresent in every biological pathway. On the molecular level, noncovalent forces guide structure formation and biomolecular interactions, which can be seen within the DNA double helix, the secondary to quaternary structures of proteins, and the dipoles of lipids. Systematically, the individual assemblies propagate into interconnecting systems to perform DNA replication/transcription, protein folding/receptor interactions, and shuttling molecules in and out of cells.1 The resulting dynamics between these biological processes would thus define the fundamental elements of life (i.e., proliferation, homeostasis, metabolism).
As a whole, it is critical to realize that many of these assemblies elicit their function at the nanometer level, while their separate constituents are seemingly nonfunctional (i.e., nucleotides/DNA, fatty acids/vesicles, rRNA/ribosome).1 Therefore, instead of using intrinsically bioactive components like proteins or DNA, the impact of nanoscience toward biology can also be realized through structure formation. Application wise, there has been growing interest in methods to enrich and accumulate drug molecules within cells to circumvent efflux-based drug resistance.2,3 As the rate of efflux of molecules is directly dependent on size,4,5 significant efforts have been made to direct drug/imaging molecules to form aggregates within cells6,7 and with promising in vivo results.8,9
Nonetheless, the bioactivity of these systems often originates from known small molecule interactions such as from a chemotherapeutic agent or singlet oxygen production by metal complexes.10,11 In contrast, specific biological responses, like programmed cell death, driven purely by the formed self-assembled nanostructures are less known. We envisioned that the assembly of nonfunctional constituents into functional architectures directly in a living cell would not only be a significant milestone in nanobiotechnology but also provide a platform to integrate synthetic chemistry with living processes.
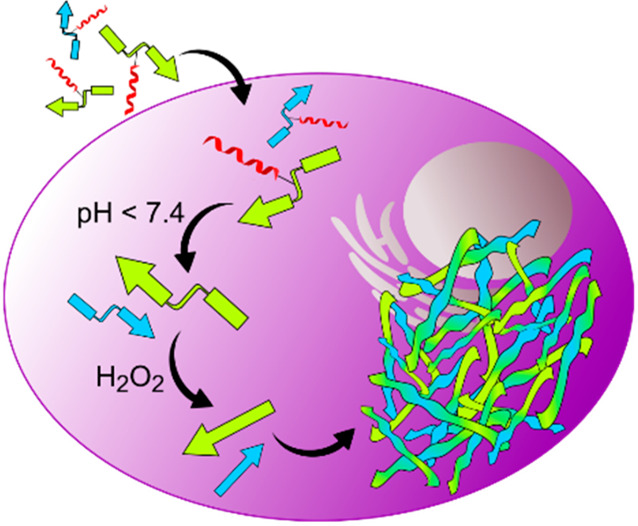
Herein, we report the construction of two peptide sequences designed to undergo a multistage transformation that results in the assembly of fibrillar architectures inside cancer cells (Figure 1). The first stage, comprising the cellular entry process, is gated by a pH-dependent boronic acid–salicylhydroxamate complexation.12 This chemistry links a transporter “TAT” sequence (trans-activator of transcription), derived from the human immunodeficiency virus (HIV),13 together with a pro-assembling sequence (henceforth referred as depsipeptide). As such, upon successful endocytosis, acidification within the intracellular vesicles releases the pro-assembling sequence. Here, the second and third stage are incorporated into the pro-assembling sequence with the second stage guarded by an immolative boronic acid cage sensitive toward elevated or endogenous H2O2 within cancer cells.14−16 Upon the immolation of the cage, the third stage is triggered by the O,N-acyl rearrangement of the depsipeptide that generates the linear isoleucine-serine-alanine (ISA) self-assembling motif.17,18 Production of ISA promotes the final stage of self-assembly into fibrillar architectures and in the process triggers apoptosis.
Figure 1.

Intracellular coassembly of peptides. (a) Depsipeptides (kinked arrows) are uptaken by cells due to dynamic covalently bound salicylhydroxamate-TAT (SHA-TAT, red coil, step A). After hydrolysis of the complex in an acidic environment (step B), the boronic acid headgroup of the depsipeptides is cleaved by intracellular hydrogen peroxide (step C). Subsequent O,N-acyl shift forms the linear coassembling peptides (step D). Linear peptides (straight arrows) form fibrillar networks inside A549 cells (step E), which are visible by transmission electron microscopy (TEM, scale bar 500 nm). (b) Chemical reactions that lead to cellular uptake, peptide linearization, and peptide coassembly of Fmoc (green) and coumarin 343 (blue) functionalized ISA.
In essence, the design comprises three modular components: (1) the pro-assembling depsi unit and its pH-reversible functionalization with TAT, (2) the peroxide-triggered cleavage of the boronic acid masking group, and the (3) pH-controlled O,N-acyl rearrangement to generate the self-assembling peptide sequence. In this way, intracellular transport, release, and supramolecular assembly into peptide fibrils is individually and sequentially programmed inside different cellular compartments by consecutive chemical reactions (Figure 1).
In addition, we demonstrate coassembly as a strategy to increase the level of functionality by imparting fluorescence into the fibrillar structures to allow imaging.19,20 Coassembly, i.e., assembly of more than a single component, is prevalent in Nature, and important examples include the assembly of α-/β-tubulin in microtubules,21 cholesterol/phospholipids in membranes,22 or the Arp2/3 complex in actins.23 While Nature uses highly specific proteins to transport and program these assemblies, synthetic methods are advantageous as they can be bioorthogonal and also be specifically tailored. By incorporating sophisticated chemical designs into a simple bimodular peptide sequence, we demonstrate that synthetic architectures can be formed directly within living systems using natural triggers.
Results and Discussion
Solid-phase peptide synthesis using fluorenylmethoxycarbonyl (Fmoc) chemistry was conducted with alanine-preloaded Wang resin (Scheme 1). Fmoc-serine was added as the second amino acid using (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP) and N,N-diisopropylethylamine (DIPEA).24 Importantly, Fmoc-serine was used without a protecting group on the hydroxyl group in order to create an ester bond with isoleucine later in the synthesis. After Fmoc deprotection of serine, modification of the N-terminus using 4-(nitrophenyl)phenylboronic acid pinacol ester was performed. Fmoc-isoleucine was coupled on the amino acid side chain of serine using N,N′-diisopropylcarbodiimide (DIC) and 4-(dimethylamino)pyridine (DMAP), forming the ester and therefore the so-called depsipeptide.17 The peptide was removed from the solid support, and the boronic acid was deprotected simultaneously using a cleavage cocktail based on trifluoroacetic acid (TFA). After purification by high-performance liquid chromatography (HPLC), the pure peptide 1a was characterized by electrospray ionization mass spectrometry (ESI-MS, Figure S6). In order to synthesize the fluorescent Depsi(C343-I)pba-SA (Depsi(coumarin 343-isoleucine)phenylboronic acid-serine-alanine), the Fmoc group was removed using piperidine and the peptide was modified with coumarin 343 using PyBOP/DIPEA for activation before cleavage of the peptide from the solid support. The identity of the product 1b was confirmed after HPLC purification by ESI-MS (Figure S9), which also showed that the peptide was isolated in high purity.
Scheme 1. Synthesis of Depsipeptides Depsi(Fmoc-I)pba-SA 1a and Depsi(C343-I)pba-SA 1b by Solid-Phase Peptide Synthesis.
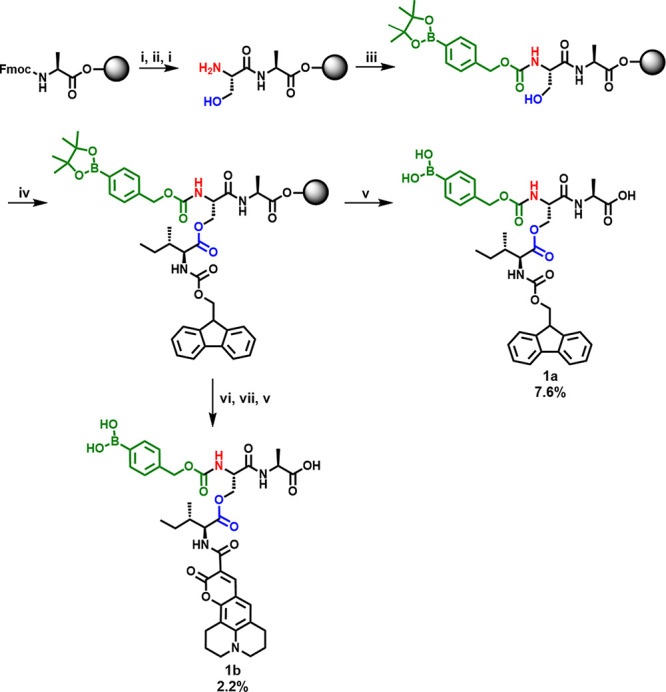
(i) Piperidine, (ii) Fmoc-Serine, PyBOP, DIPEA, (iii) 4-(nitrophenyl)phenylboronic acid pinacol ester, DIPEA, (iv) Fmoc-isoleucine, DIC, DMAP, (v) TFA, triisopropylsilane, H2O, (vi) piperidine, (vii) coumarin 343, PyBOP, DIPEA.
In order to prove the hydrogen peroxide-induced removal of the PBA protecting group and the subsequent O,N-acyl shift in solution outside cells, Depsi(Fmoc-I)pba-SA 1a was incubated in NH4HCO3 buffer pH 7.4 with and without hydrogen peroxide (Figure 2a–c, Figure S15). The peptide (RT = 15.6 min) was stable in the absence of H2O2 and led to only 3% conversion into Fmoc-ISA 3a (RT = 14.6 min) within 45 h due to slow hydrolysis of the carbamate bond. In contrast, addition of hydrogen peroxide led to a yield of 94% Fmoc-ISA 3a in the same time. The intermediate Depsi(Fmoc-I)-SA 2a could also be observed by the appearance of a peak at 14.0 min, which started to decrease after 8 h, while the product peak of 3a increased. The same study was performed using Depsi(C343-I)pba-SA 1b, and the results showed that both the removal of the phenylboronic acid by H2O2 as well as the O,N-acyl shift were successful for the coumarin derivative of the peptide (Figures 2e and S16). After 4 h, Depsi(C343-I)pba-SA 1b (RT = 6.9 min) was no longer present, while the peaks for Depsi(C343-I)-SA 2b (RT = 5.0 min) and the linear peptide C343-ISA 3b (RT = 6.2 min) increased. After 24 h only 3b was found in the sample. Incubation of Depsi(C343-I)pba-SA 1b in NH4HCO3 buffer without H2O2 led to formation of only 3% of the linear peptide 3b, which proves the stability of the peptide under these conditions (Figure 2 d).
Figure 2.

Hydrogen peroxide induced peptide fiber formation. (a) Phenylboronic acid removal of Depsi(R*-I)pba-SA peptides 1a and 1b and subsequent O,N-acyl shift to give R*-ISA 3a and 3b (R* = Fmoc or coumarin 343). (b) HPLC spectra at different time points showing the stability of Depsi(Fmoc-I)pba-SA 1a (15.6 min) in NH4HCO3 buffer and reference spectrum of Fmoc-ISA 3a (14.6 min). (c) H2O2 (2 mM) induced removal of the boronic acid to give Depsi(Fmoc-I)SA 2a (14.0 min) and linearization of the peptide to Fmoc-ISA 3a (14.6 min). (d) Stability of Depsi(C343-I)pba-SA 1b (6.9 min) in buffer, and HPLC spectrum of C343-ISA 3b (6.2 min). (e) H2O2 (1 mM) induced PBA removal and O,N-acyl shift of Depsi(C343-I)pba-SA 1b (6.9 min) to first give Depsi(C343-I)SA 2b (5.0 min) and finally C343-ISA 3b (6.2 min). TEM images of (f) Depsi(Fmoc-I)pba-SA 1a, (g) Fmoc-ISA 3a, (h) Depsi(C343-I)pba-SA 1b, (i) C343-ISA 3b, (j) coincubation of Fmoc-ISA 3a and C343-ISA 3b 5:1. Scale bars 500 nm. (k) Fluorescence microscope images of Fmoc-ISA:C343-ISA 3a:3b 5:1. (Left) Coumarin 343 channel (cyan). (Middle) Proteostat stained peptide fibers (yellow). (Right) Merged Proteostat and coumarin 343 channel showing the overlay of the fluorescent dyes (green). Scale bars 20 μm. (l) Circular dichroism spectra of Fmoc-ISA (orange), C434-ISA (red), and their differently prepared assemblies in H2O (coassembled, green; mixed, blue). Fmoc/C343-ISA (calculated, dashed purple line) is the sum of the separately recorded spectra of Fmoc-ISA and C343-ISA at a ratio of 5:1 to match the experimental conditions. Region of interest is highlighted in gray.
TEM measurements of Depsi(Fmoc-I)pba-SA 1a in phosphate buffer pH 7.4 after 24 h incubation with and without hydrogen peroxide showed the oxidation-triggered self-assembly of the peptide due to formation of the linear fibrillating sequence Fmoc-ISA 3a, while the corresponding boronic acid-modified depsipeptide 1a did not form peptide fibers (Figure 2f and 2g). In order to determine the critical fibrillation concentration of Fmoc-ISA 3a, which is important for intracellular fiber formation, Fmoc-ISA was incubated at different concentrations ranging from 2 mM to 15 μM for 24 h in phosphate-buffered saline (PBS). In TEM measurements, the lowest detectable concentration of peptide fibers was 62.5 μM (Figure S26).
The fluorescent peptide Depsi(C343-I)pba-SA 1b was synthesized in order to enable live cell imaging of the peptide fibers by coassembly of both intracellularly rearranged peptides. While incubation of Depsi(C343-I)pba-SA 1b in phosphate buffer did not lead to fiber formation, addition of H2O2 led to the appearance of amorphous aggregates in TEM (Figure 2h and 2i). Coincubation of both PBA depsipeptides upon hydrogen peroxide treatment at a ratio of 5:1 of 1a:1b led to a mixture of fibers and some aggregates (Figure 2j). Preliminary confirmation of coassembly was accomplished by fluorescence microscopy, demonstrating the colocalization of the coumarin 343 signal with Proteostat, which detects cross-β-sheet-containing peptide fibers (Figure 2k).19,20,24,25
Next, we elucidated the secondary structure of Fmoc-ISA as it is the primary driving force for fiber formation. To address this question, we performed Fourier transform infrared spectroscopy measurements (Figure S29). The results indicated formation of β-sheets due to the appearance of a maximum at 1634 cm–1. Another maximum at 1688 cm–1 is usually assigned to antiparallel β-sheets in proteins but recently has been reported to derive from the carbamate bond of the Fmoc group. A shoulder which appeared at 1653 cm–1 in the FT-IR spectrum might be assigned to either α-helices or disordered structures.26−2913C (1H) CP-MAS NMR spectra (cross-polarization-magic angle spinning nuclear magnetic resonance) confirmed formation of three different competing structures by showing two sharp peaks and one broad peak for the Fmoc carbonyl as well as several overlapping peaks for the C=O groups of the amino acids (Figure S30).
For additional proof of coassembly of both peptides, circular dichroism spectroscopy was used to visualize changes in the secondary structure (Figure 2l). The CD spectrum of Fmoc-ISA 3a in water showed a maximum at 218 nm, which can be attributed to a n → π* transition, and a shoulder peak at ∼209 nm was observed in the CD spectrum as well as a minimum at 190 nm.30 Notably, a strong positive Cotton effect was observed with a maximum at 267 nm, which corresponds to the π → π* transition of the Fmoc groups in the self-assembled peptide fibers.18 In contrast, the CD spectrum of C343-ISA 3b revealed a maximum at 195 nm and a minimum at 215 nm with opposite ellipticity compared to 3a. In proteins these signals are attributed to β-sheets and correspond to π → π* and n → π* transitions, respectively.31 For coassembly, the study was accomplished using two different sample preparation methods: (1) the individual peptides (3a and 3b) were first mixed before triggering the coassembly in H2O, and (2) the separate peptide assemblies are preformed in H2O, and the resultant nanostructures are combined. Mixing of the preformed nanostructures leads to the appearance of an additional shoulder peak at ∼235 nm and an overall decrease in signal intensities. This effect is larger than expected from the spectral sum of a 5:1 mixture of 3a:3b, which represents the hypothetical spectrum in the absence of interactions between 3a and 3b. Hence, the results indicate interactions at the nanostructure level, causing a decrease in chirality of the assemblies. Upon coincubation of both peptides to induce coassembly, the intensities of the maxima and minima are further decreased significantly, which also includes signals attributed to electronic transitions of the Fmoc groups. This is especially visible in the change of peak proportions at 218 and at 206 nm where the latter was previously observed as a shoulder at ∼209 nm. We conclude that upon coassembly of 3a and 3b, the overall structural chirality of the peptide assemblies is decreased, leading to a comparable CD spectrum of 3a due to the 5-fold excess but with distinct differences in ellipticity.32−34
In order to provide cellular uptake of depsipeptides 1a/b, a salicylhydroxamate-functionalized TAT peptide 12 was needed. TAT, which has the amino acid sequence YGRKKRRQRRR, was synthesized by standard SPPS methods and was modified with 4-pentynoic acid on its N-terminus to give molecule 11 (Figure S11). After a copper-catalyzed azide–alkyne cycloaddition (CuAAC) with trityl (Trt)- and (2-methoxyethoxy)methoxy (MEM)-protected 4-azido salicylhydroxamate 10, which was synthesized in seven synthetic steps (Figure S1), the peptide was cleaved from the solid support and purified by HPLC. After isolation of the pure peptide SHA-TAT 12, it was characterized by MALDI-TOF mass spectrometry (matrix-assisted laser desorption/ionization-time-of-flight, Figure S13).
Attachment of the transporter peptide TAT was accomplished by boronic acid/salicyl hydroxamate chemistry. Formation of the dynamic covalent bond between the boronic acid-functionalized depsipeptides 1a and 1b and SHA-TAT 12 in phosphate buffer pH 7.4 was proven by MALDI-TOF measurements for both Fmoc- and C343-modified peptides 1a and 1b. Both products were formed by condensation of PBA and SHA, and multiple cation adducts were observed in the spectra (Figure 3a and 3b). Fluorescence quenching experiments with Depsi(C343-I)pba-SA 1b and SHA-TAT 12 gave an KD of 2.67 μM, which was expected due to previously reported results for SHA-PBA complexes (Figure 3c).35 Reversibility of the complexation was accomplished at pH 5, which results in the recovery of fluorescence (Figure S31). The binding and release were confirmed by previous reports by our group to transport proteins into cells.36,37
Figure 3.

Complexes of SHA-TAT 12 and the boronic acid depsipeptides 1a and 1b. (a) Chemical structure of dynamic covalently bound SHA-TAT and boronic acid-modified depsipeptide conjugates 13a and 13b. (b) MALDI spectra showing the successful formation of complexes of SHA-TAT 12 with Depsi(Fmoc-I)pba-SA 1a (top) and Depsi(C343-I)pba-SA 1b (bottom). [M‘] corresponds to the molecular weight after loss of OH–. [M‘] = [M–OH–]. (c) Determination of the KD of the SHA-TAT complex with Depsi(C343-I)pba-SA (13b) by fluorescence quenching.
Alongside the acidification-induced release of the pro-assembling sequence 1a/b from the TAT complexes 13a/b inside endosomes, the H2O2 stimulus is required for assembly. The average concentration of H2O2 within A549 cells was assayed using the Intracellular Hydrogen Peroxide Assay Kit from Sigma-Aldrich and found to be 1.64 ± 0.16 μM (Figure S 57). Stimulation of H2O2 production can be performed with 100 nM of phorbol-12-myristat-13-acetate (PMA),38 affording a 49% increase to 2.45 ± 0.37 μM. Using these conditions, the TAT-complexed depsipeptides 13a/b were incubated with A549 cells, and successful cellular uptake was shown by confocal microscopy (Figure 4a). The concentration of the depsipeptides was adjusted to 150 μM in total to meet the requirements of the critical fibrillation concentration. The depsipeptides were used at a ratio of 5:1 (13a:13b) in order to receive a sufficient fluorescence signal by coumarin 343 inside cells while maintaining an excess of the Fmoc peptides to direct fiber formation. Incubation of cells with only the depsipeptides 1a and 1b did not show internalization of the peptides, which proves the necessity of SHA-TAT 12 for cellular uptake (Figure 4a, top row). The increase in fluorescence intensity of coumarin 343 in the presence of PMA suggests local accumulation of fluorescent 3b upon fiber formation, indicating that the coassembly was more pronounced at higher concentrations of H2O2, although as shown in Figures S47 and S48, peptide fiber formation also occurs without PMA. Significant cell deformation and nuclear condensation imply that cell viability is significantly affected (Figures S38, S39, and S41). Incubation of the peptide samples with the cells for 2 h showed less cellular uptake, while extending it to 6 h led to more internalization of the peptides (Figure S34). Peptide-treated cells were also examined under higher magnification, where fibrillar structures inside the cell were visible in the coumarin 343 channel (Figure S35). A striking observation was the inefficient staining of the nucleus in cells where the postulated assembly has occurred. This phenomenon has already been described in the literature, where formation of peptide fibers inside cells prevented the staining of nuclei as nuclear stains were trapped inside fibrillar networks.39
Figure 4.

Cellular uptake and intracellular coassembly of peptides. (a) Confocal laser scanning micrographs of A549 cells treated for 4 h with only the depsipeptide mixture of 1a and 1b (top row) and treated with addition of SHA-TAT 12 to induce cellular uptake without (middle row) and with PMA (bottom row). Scale bars 20 μm. (b) Förster resonance energy transfer studies on A549 cells with 15a/b using coumarin343 (donor, cyan) and 5-FAM (acceptor, red) as FRET pairs. Scale bars 20 μm, λex = 405 nm.
We subsequently explored the mechanisms involved in each step of intracellular transport using time-lapsed Förster resonance energy transfer (FRET) and colocalization studies. TAT was labeled with 5-FAM (compound 14) serving as the FRET acceptor for coumarin 343 (Figure S14). Using this FRET-labeled variant of 13a/b, named 15a/b, we performed confocal microscopy at 1 and 4 h time points (Figure 4 c). At 1 h, uptake of 15a/b can be visualized as punctuated spots characteristic of vesicle-based intracellular transport. A significant proportion of these vesicles exhibits FRET signals (red), suggesting that most of the uptaken 15a/b has not undergone dissociation. Nonetheless, a first indication of release into the cytosol was observed for a small proportion of these vesicles. At 4 h, among cells that are still intact, assembly has been accomplished in large areas of the cells. In these regions, FRET signals are absent, suggesting that 15a/b has been mostly dissociated into 1a/b and TAT 14 followed by the peroxide-driven cascade. However, remaining amounts of undissociated 15a/b could be observed to proceed further along the TAT transportation pathway into the nuclear region. Here, the complex remains bound as the pH within the nucleus is no longer acidic.40 The transport pathway through TAT-promoted endocytosis was confirmed using early/late endosome studies with CellLight, which saw major colocalization (Figure S37).
In addition, endosomal escape could also be visualized as diffused coumarin 343 signals in the cytosol. Furthermore, nuclear transport of undissociated peptides 13a/b, demonstrated by SYTO RNASelect Green, was found to be localized inside the nucleolus, most likely due to the electrostatic attraction between TAT and DNA/RNA or perhaps hydrophobic interactions with the depsipeptides (Figure S40).41 A previous report showed that self-assembling peptides might interact with RNA located inside the nucleoli.42 In order to ascertain that the H2O2-driven assembly is specific toward the cytosol, we monitored the fiber formation using TEM at pH 5.0, 6.0, and 7.4 (Figures S23 and S25). At both acidic pH values, corresponding to the early and late endosomes as well as lysosomes, H2O2 does not possess enough oxidative strength to initiate the reaction cascade for assembly. This pH-dependent activity of H2O2 was reported in the literature.43 As such, the acidic conditions within the endosomes specifically trigger the dissociation of 13a/b, while the cytosolic environment provides the condition for the oxidative cascade to initiate fiber formation. To demonstrate the molecular rearrangement of the depsipeptides inside cells, we analyzed cell lysates by HPLC and showed the H2O2-induced conversion of 1b to 3b (Figure S49).
The biological response of the cells to the intracellular assembly was subsequently investigated by Annexin-V/propidium iodide assay (Figure 5a). Annexin-V is a protein that binds to phosphatidylserine, which is located on the external leaflet of the membrane structures exclusively during apoptosis.44 The assay was conducted after 2 and 4 h on A549 cells treated with 13a/b.
Figure 5.

(a) Apoptosis assay using Annexin V-FITC/propidium iodide on 13a/b and PMA-treated A549 cells over 2 and 4 h. Binding of Annexin V (green) toward the cell membrane was observed prominently at 2 h, demonstrating cells undergoing apoptosis due to inversion of the phosphatidylserine motifs. Membrane collapse at 4 h was detected with the entry of propidium iodide (red) into the nucleus. Scale bars 20 μm. (b) Cell viability of depsipeptides 1a and 1b and/or TAT 12-treated A549 cells determined by CellTiter-Glo Luminescent Cell Viability Assay. Cells were treated for different incubation times from 2 to 6 h with only the depsipeptides 1a and 1b, only SHA-TAT 12, or both to create 13a and 13b at a concentration of 150 μM. Significant cytotoxic effect was observed for A549 cells treated with the depsipeptide–TAT complexes 13a and 13b. All samples were coincubated with 100 nM PMA.
At 2 h, Annexin-V was found to bind prominently to the cell membranes, indicating that affected cells are undergoing apoptosis. At this time frame, the integrity of the cell membrane still remains intact as propidium iodide failed to enter the cells. In contrast, at 4 h, propidium iodide signals were observed in the nuclear region due to its affinity toward DNA.44 This observation represented the membrane permeabilization process associated with late-stage apoptosis. Analysis of cytotoxicity was accomplished using CellTiter-Glo luminescent cell viability assay, which is based on quantification of adenosine triphosphate (ATP) and therefore actively metabolizing cells.45 No significant toxic effects were observed after incubation of 1a/b at a concentration of 150 μM for 6 h with A549 cells (Figure 5b). However, upon complexing with TAT to form 13a/b, a significant impact toward cell viability was observed. Only 14% of cells was viable after 6 h, while incubation for 4 or 2 h led to a viability of 34% and 66%, respectively. As the A549 cancer cells do produce intrinsic H2O2, transformation of 1a/b into 3a/b still occurs in cells which are not treated with PMA, and therefore, the cell viability without PMA was 40% after 4 h (Figure S56), which is similar to the cell viability with addition of PMA. As the increase of hydrogen peroxide concentration by PMA stimulation (49%) does not cause a significant difference in cell viability, we conclude that nonstimulated cells already offer enough H2O2 to generate sufficient peptide fibers leading to effective apoptosis. In addition, as known from β-amyloid structures, we believe that shorter fibers/protofibrils can already contribute to the apoptotic effects.46
Lastly, to visualize the intracellular fiber formation directly, 13a/b-treated cells were fixed by high-pressure freezing, and subsequently, freeze substitution was performed using acetone. After infiltration with epoxy resin and its polymerization, sample blocks were sectioned into slices. The slices of the cells were examined in TEM (Figure 6a and 6b). Formation of many dense peptide fiber networks was clearly visible inside the cells. Peptide fibrils were distributed inside the cell and were also observed to form next to the nucleus and mitochondria. To further show the formation of fibers, cells were lysed after treatment with the sample and the cell lysate was analyzed by TEM and fluorescence microscopy (Figure 6c and 6d).
Figure 6.

Intracellular peptide fiber formation. (a and b) TEM micrographs of peptide fibers inside A549 cells (marked with dashed lines) after coassembly of intracellularly generated Fmoc-ISA 3a and C343-ISA 3b and images of a cell, which were received after stitching TEM images together. Scale bars 500 nm. (c) Fluorescence microscopy images of Proteostat-stained peptide fibers after formation inside cells and extraction from the cells by lysis. Scale bars 5 μm. (d) TEM micrographs of peptide fibers, which were received after lysis of depsipeptide TAT complex 13a- and 13b-treated A549 cells. Scale bars 500 nm.
Fibers were observed in TEM, which further proved the successful assembly of 3a/b inside cells. Furthermore, Proteostat staining of the cell lysate clearly showed formation of amyloid, cross-ß-sheet structures to which Proteostat is known to bind, and coumarin 343 fluorescent fibers could also be observed in the lysate (Figures S45 and S46). Due to the resolution in the fluorescence microscope, only very thick fibers could be found in the cell lysate. Cell lysates of cells which were not treated with PMA but only with the complexes 13a/b also contained fibers, which shows that enhancing the intracellular H2O2 concentration with PMA is not necessary to induce peptide assembly, as cells naturally already produce hydrogen peroxide (Figures S47 and S48).33 TEM images of cells as well as cell lysate of A549 cells, which were not treated with the depsipeptide-TAT complexes 13a and 13b, did not show fibrillar structures, which demonstrates that fibers derived from the coassembly of the intracellularly rearranged peptides Fmoc-ISA 3a and C343-ISA 3b (Figures S44, S54, and S55).
Conclusions
In summary, we designed a peptide sequence that provides synthetic components for controlling cellular entry, intracellular dissociation, and supramolecular assembly. The reactivity of each synthetic component within the peptide sequence is dictated by the intracellular localization where its immediate environment defines its chemistry and subsequent transport pathway. Consecutive reactions are initiated in a controlled fashion to afford coassembling sequences that form fibrillar structures within the cytosol. Formation of these superstructures was imaged by fluorescence and electron microscopy and led to programmed cell death accompanied by nuclear fragmentation, actin disruption, and membrane collapse. Furthermore, coassembly features the potential of the nanosystem to dynamically customize functions and/or components to tune additional biological behavior. Collectively, the platform provides a broad accessibility and expands the domain of nanotechnology to directly impact living systems through structure formation.
Acknowledgments
This project received funding from the Max Planck-Bristol Centre for Minimal Biology and the Volkswagen Foundation under project number 89943. We thank Dr. Anke Kaltbeitzel for help with planning of the experiment, Christoph Sieber for sample preparation for TEM of cells, Dr. Gönül Kizilsavas for assigning NMR peaks, Jutta Schnee for measuring MALDI spectra, and Jessica Wagner for measuring an APCI spectrum. We thank the reviewers for their efforts in stimulating additional experiments that improved the quality of the manuscript. Open Access funding provided by the Max Planck Society.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c05261.
Materials and methods as well as extensive supplementary figures and data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Cragg P. J.Supramolecular Chemistry; Springer Science+Business Media B.V.: Dordrecht, Heidelberg, London, New York, 2010. [Google Scholar]
- Eckford P. D. W.; Sharom F. J. ABC Efflux Pump-Based Resistance to Chemotherapy Drugs. Chem. Rev. 2009, 109 (7), 2989–3011. 10.1021/cr9000226. [DOI] [PubMed] [Google Scholar]
- Nikaido H. Prevention of drug access to bacterial targets: permeability barriers and active efflux. Science 1994, 264 (5157), 382. 10.1126/science.8153625. [DOI] [PubMed] [Google Scholar]
- Brownlee W. J.; Seib F. P. Impact of the hypoxic phenotype on the uptake and efflux of nanoparticles by human breast cancer cells. Sci. Rep. 2018, 8 (1), 12318. 10.1038/s41598-018-30517-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z.; Wu J.; Yu Y.; Liu S.; Jiang W.; Nurmamat H.; Wu B. Comparison of cytotoxicity and membrane efflux pump inhibition in HepG2 cells induced by single-walled carbon nanotubes with different length and functional groups. Sci. Rep. 2019, 9 (1), 7557. 10.1038/s41598-019-43900-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng D. Y. W.; Vill R.; Wu Y.; Koynov K.; Tokura Y.; Liu W.; Sihler S.; Kreyes A.; Ritz S.; Barth H.; Ziener U.; Weil T. Directing intracellular supramolecular assembly with N-heteroaromatic quaterthiophene analogues. Nat. Commun. 2017, 8 (1), 1850. 10.1038/s41467-017-02020-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao S.-L.; Ma Y.; Wang Y.; Lin Y.-X.; An H.-W.; Li L.-L.; Wang H. General Approach of Stimuli-Induced Aggregation for Monitoring Tumor Therapy. ACS Nano 2017, 11 (7), 7301–7311. 10.1021/acsnano.7b03375. [DOI] [PubMed] [Google Scholar]
- Nakamura H.; Lee A. A.; Afshar A. S.; Watanabe S.; Rho E.; Razavi S.; Suarez A.; Lin Y.-C.; Tanigawa M.; Huang B.; DeRose R.; Bobb D.; Hong W.; Gabelli S. B.; Goutsias J.; Inoue T. Intracellular production of hydrogels and synthetic RNA granules by multivalent molecular interactions. Nat. Mater. 2018, 17 (1), 79–89. 10.1038/nmat5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye D.; Shuhendler A. J.; Cui L.; Tong L.; Tee S. S.; Tikhomirov G.; Felsher D. W.; Rao J. Bioorthogonal cyclization-mediated in situ self-assembly of small-molecule probes for imaging caspase activity in vivo. Nat. Chem. 2014, 6 (6), 519–526. 10.1038/nchem.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blunden B. M.; Stenzel M. H. Incorporating ruthenium into advanced drug delivery carriers – an innovative generation of chemotherapeutics. J. Chem. Technol. Biotechnol. 2015, 90 (7), 1177–1195. 10.1002/jctb.4507. [DOI] [Google Scholar]
- Cheng Z.; Cheng Y.; Chen Q.; Li M.; Wang J.; Liu H.; Li M.; Ning Y.; Yu Z.; Wang Y.; Wang H. Self-assembly of pentapeptides into morphology-adaptable nanomedicines for enhanced combinatorial chemo-photodynamic therapy. Nano Today 2020, 33, 100878. 10.1016/j.nantod.2020.100878. [DOI] [Google Scholar]
- Shin S. B. Y.; Almeida R. D.; Gerona-Navarro G.; Bracken C.; Jaffrey S. R. Assembling ligands in situ using bioorthogonal boronate ester synthesis. Chem. Biol. 2010, 17 (11), 1171–1176. 10.1016/j.chembiol.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herce H. D.; Garcia A. E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (52), 20805–20810. 10.1073/pnas.0706574105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen H.; Marzenell P.; Jentzsch E.; Wenz F.; Veldwijk M. R.; Mokhir A. Aminoferrocene-based prodrugs activated by reactive oxygen species. J. Med. Chem. 2012, 55 (2), 924–934. 10.1021/jm2014937. [DOI] [PubMed] [Google Scholar]
- Jourden J. L. M.; Daniel K. B.; Cohen S. M. Investigation of self-immolative linkers in the design of hydrogen peroxide activated metalloprotein inhibitors. Chem. Commun. 2011, 47 (28), 7968–7970. 10.1039/c1cc12526e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstain R.; Savariar E. N.; Felsen C. N.; Tsien R. Y. In Vivo Targeting of Hydrogen Peroxide by Activatable Cell-Penetrating Peptides. J. Am. Chem. Soc. 2014, 136 (3), 874–877. 10.1021/ja411547j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coin I.; Dölling R.; Krause E.; Bienert M.; Beyermann M.; Sferdean C. D.; Carpino L. A. Depsipeptide Methodology for Solid-Phase Peptide Synthesis: Circumventing Side Reactions and Development of an Automated Technique via Depsidipeptide Units. J. Org. Chem. 2006, 71 (16), 6171–6177. 10.1021/jo060914p. [DOI] [PubMed] [Google Scholar]
- Tao K.; Levin A.; Adler-Abramovich L.; Gazit E. Fmoc-modified amino acids and short peptides: simple bio-inspired building blocks for the fabrication of functional materials. Chem. Soc. Rev. 2016, 45 (14), 3935–3953. 10.1039/C5CS00889A. [DOI] [PubMed] [Google Scholar]
- Liu X.; Cole J. M.; Low K. S. Molecular origins of dye aggregation and complex formation effects in coumarin 343. J. Phys. Chem. C 2013, 117 (28), 14723–14730. 10.1021/jp4024266. [DOI] [Google Scholar]
- Rode S.; Hayn M.; Röcker A.; Sieste S.; Lamla M.; Markx D.; Meier C.; Kirchhoff F.; Walther P.; Fändrich M.; Weil T.; Münch J. Generation and characterization of virus-enhancing peptide nanofibrils functionalized with fluorescent labels. Bioconjugate Chem. 2017, 28 (4), 1260–1270. 10.1021/acs.bioconjchem.7b00079. [DOI] [PubMed] [Google Scholar]
- Lewis S. A.; Tian G.; Cowan N. J. The α- and β-tubulin folding pathways. Trends Cell Biol. 1997, 7 (12), 479–484. 10.1016/S0962-8924(97)01168-9. [DOI] [PubMed] [Google Scholar]
- Ohvo-Rekilä H.; Ramstedt B.; Leppimäki P.; Slotte J. P. Cholesterol interactions with phospholipids in membranes. Prog. Lipid Res. 2002, 41 (1), 66–97. 10.1016/S0163-7827(01)00020-0. [DOI] [PubMed] [Google Scholar]
- Goley E. D.; Welch M. D. The ARP2/3 complex: an actin nucleator comes of age. Nat. Rev. Mol. Cell Biol. 2006, 7 (10), 713–726. 10.1038/nrm2026. [DOI] [PubMed] [Google Scholar]
- Coste J.; Le-Nguyen D.; Castro B. PyBOP®: A new peptide coupling reagent devoid of toxic by-product. Tetrahedron Lett. 1990, 31 (2), 205–208. 10.1016/S0040-4039(00)94371-5. [DOI] [Google Scholar]
- Navarro S.; Ventura S. Fluorescent dye proteostat to detect and discriminate intracellular amyloid-like aggregates in escherichia coli. Biotechnol. J. 2014, 9 (10), 1259–1266. 10.1002/biot.201400291. [DOI] [PubMed] [Google Scholar]
- Fleming S.; Frederix P. W. J. M.; Ramos Sasselli I.; Hunt N. T.; Ulijn R. V.; Tuttle T. Assessing the Utility of Infrared Spectroscopy as a Structural Diagnostic Tool for β-Sheets in Self-Assembling Aromatic Peptide Amphiphiles. Langmuir 2013, 29 (30), 9510–9515. 10.1021/la400994v. [DOI] [PubMed] [Google Scholar]
- Jackson M.; Mantsch H. H. The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 1995, 30 (2), 95–120. 10.3109/10409239509085140. [DOI] [PubMed] [Google Scholar]
- Micsonai A.; Wien F.; Kernya L.; Lee Y.-H.; Goto Y.; Réfrégiers M.; Kardos J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (24), E3095–E3103. 10.1073/pnas.1500851112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei G.; Su Z.; Reynolds N. P.; Arosio P.; Hamley I. W.; Gazit E.; Mezzenga R. Self-assembling peptide and protein amyloids: from structure to tailored function in nanotechnology. Chem. Soc. Rev. 2017, 46 (15), 4661–4708. 10.1039/C6CS00542J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argudo P. G.; Contreras-Montoya R.; Álvarez de Cienfuegos L.; Cuerva J. M.; Cano M.; Alba-Molina D.; Martín-Romero M. T.; Camacho L.; Giner-Casares J. J. Unravelling the 2D self-assembly of Fmoc-dipeptides at fluid interfaces. Soft Matter 2018, 14 (46), 9343–9350. 10.1039/C8SM01508B. [DOI] [PubMed] [Google Scholar]
- Jiang J.; Abramavicius D.; Bulheller B. M.; Hirst J. D.; Mukamel S. Ultraviolet spectroscopy of protein backbone transitions in aqueous solution: combined QM and MM simulations. J. Phys. Chem. B 2010, 114 (24), 8270–8277. 10.1021/jp101980a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horgan C. C.; Rodriguez A. L.; Li R.; Bruggeman K. F.; Stupka N.; Raynes J. K.; Day L.; White J. W.; Williams R. J.; Nisbet D. R. Characterisation of minimalist co-assembled fluorenylmethyloxycarbonyl self-assembling peptide systems for presentation of multiple bioactive peptides. Acta Biomater. 2016, 38, 11–22. 10.1016/j.actbio.2016.04.038. [DOI] [PubMed] [Google Scholar]
- Mu X.; Eckes K. M.; Nguyen M. M.; Suggs L. J.; Ren P. Experimental and Computational Studies Reveal an Alternative Supramolecular Structure for Fmoc-Dipeptide Self-Assembly. Biomacromolecules 2012, 13 (11), 3562–3571. 10.1021/bm301007r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. K. Lost in translation? Chirality effects in the self-assembly of nanostructured gel-phase materials. Chem. Soc. Rev. 2009, 38 (3), 684–694. 10.1039/b800409a. [DOI] [PubMed] [Google Scholar]
- Zegota M. M.; Wang T.; Seidler C.; Wah Ng D. Y.; Kuan S. L.; Weil T. Tag and Modify” protein conjugation with dynamic covalent chemistry. Bioconjugate Chem. 2018, 29 (8), 2665–2670. 10.1021/acs.bioconjchem.8b00358. [DOI] [PubMed] [Google Scholar]
- Seidler C.; Ng D. Y. W.; Wu Y.; Weil T. pH responsive supramolecular core-shell protein hybrids. Supramol. Chem. 2016, 28 (9–10), 742–746. 10.1080/10610278.2016.1140764. [DOI] [Google Scholar]
- Seidler C.; Zegota M. M.; Raabe M.; Kuan S. L.; Ng D. Y. W.; Weil T. Dynamic core–shell bioconjugates for targeted protein delivery and release. Chem. - Asian J. 2018, 13 (22), 3474–3479. 10.1002/asia.201800843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwabara W. M. T.; Zhang L.; Schuiki I.; Curi R.; Volchuk A.; Alba-Loureiro T. C. NADPH oxidase-dependent production of reactive oxygen species induces endoplasmatic reticulum stress in neutrophil-like HL60 cells. PLoS One 2015, 10 (2), e0116410. 10.1371/journal.pone.0116410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Du X.; Yamagata N.; Xu B. Enzyme-instructed self-assembly of small D-peptides as a multiple-step process for selectively killing cancer cells. J. Am. Chem. Soc. 2016, 138 (11), 3813–3823. 10.1021/jacs.5b13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llopis J.; McCaffery J. M.; Miyawaki A.; Farquhar M. G.; Tsien R. Y. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. U. S. A. 1998, 95 (12), 6803–6808. 10.1073/pnas.95.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audas T. E.; Jacob M. D.; Lee S. Immobilization of Proteins in the Nucleolus by Ribosomal Intergenic Spacer Noncoding RNA. Mol. Cell 2012, 45 (2), 147–157. 10.1016/j.molcel.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Wang H.; Feng Z.; Tan W.; Xu B. Assemblies of D-peptides for targeting cell nucleolus. Bioconjugate Chem. 2019, 30 (10), 2528–2532. 10.1021/acs.bioconjchem.9b00524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruice T. C.; Zipplies M. F.; Lee W. A. The pH dependence of the mechanism of reaction of hydrogen peroxide with a nonaggregating, non-mu-oxo dimer-forming iron (III) porphyrin in water. Proc. Natl. Acad. Sci. U. S. A. 1986, 83 (13), 4646. 10.1073/pnas.83.13.4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demchenko A. P. Beyond annexin V: fluorescence response of cellular membranes to apoptosis. Cytotechnology 2013, 65 (2), 157–172. 10.1007/s10616-012-9481-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannah R.; Beck M.; Moravec R.; Riss T. CellTiter-Glo luminescent cell viability assay: A sensitive and rapid method for determining cell viability. Promega Cell Notes 2001, 2, 11–13. [Google Scholar]
- Jan A.; Adolfsson O.; Allaman I.; Buccarello A.-L.; Magistretti P. J.; Pfeifer A.; Muhs A.; Lashuel H. A. Aβ42 Neurotoxicity Is Mediated by Ongoing Nucleated Polymerization Process Rather than by Discrete Aβ42 Species. J. Biol. Chem. 2011, 286 (10), 8585–8596. 10.1074/jbc.M110.172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.