

Abstract
PURPOSE
Axicabtagene ciloleucel (axi-cel) was approved by the Food and Drug Administration for relapsed aggressive B-cell non-Hodgkin lymphoma in part on the basis of durable remission rates of approximately 40% in a clinical trial population. Whether this efficacy, and the rates of toxicity, would be consistent in a postcommercial setting, with relaxed eligibility criteria and bridging therapy, is unknown. This study describes the efficacy and safety correlates and outcomes in this setting.
PATIENTS AND METHODS
One hundred twenty-two patients from 7 medical centers in the United States were treated with axi-cel and were included in a modified intent-to-treat (mITT) analysis. Seventy-six patients (62%) were ineligible for the ZUMA-1 trial. Response and toxicity rates, duration of response (DOR), survival, and covariates are described on the basis of the mITT population. Correlative studies on blood and tumor samples were performed to investigate potential biomarkers of response and resistance.
RESULTS
Median follow-up was 10.4 months. In the mITT population, the best overall and complete response (CR) rates were 70% and 50%, respectively. Median DOR and progression-free survival (PFS) were 11.0 and 4.5 months in all patients and were not reached (NR) in CR patients. Median overall survival (OS) was NR; 1-year OS was 67% (95% CI, 59% to 77%). Although response rates were similar in the ZUMA-1–eligible and ZUMA-1–ineligible groups (70% v 68%), there was a statistically significant improvement in CR rate (63% v 42%, P = .016), DOR (median, NR v 5.0 months; P = .014), PFS (median, NR v 3.3 months; P = .020), and OS (1-year OS, 89% v 54%; P < .001) in patients who were ZUMA-1 eligible. Rates of grade ≥ 3 cytokine release syndrome and neurotoxicty were 16% and 35%, respectively.
CONCLUSION
Axi-cel yields similar rates of overall response and toxicity in commercial and trial settings, although CR rates and DOR were more favorable in patients eligible for ZUMA-1.
INTRODUCTION
Before the Food and Drug Administration (FDA) approval of axicabtagene ciloleucel (axi-cel), an anti–cluster of differentiation (CD)19 chimeric antigen receptor (CAR) T-cell therapy, outcomes in refractory diffuse large B-cell lymphoma (DLBCL) were dismal.1-3 Large registries demonstrated a 4- to 6-month median overall survival (OS) with fewer than one third of patients responding to therapies.4,5 In ZUMA-1, 82% of patients with refractory DLBCL, high-grade B-cell lymphoma (HGBL), primary mediastinal B-cell lymphoma (PMBL), and transformed follicular lymphoma (tFL) responded to axi-cel, and 54% had a complete response (CR).6 Median OS had not been reached at 27.1 months.7 The FDA has since approved a second product, tisagenlecleucel, for DLBCL, HGBL, and tFL on the basis of the results of the JULIET trial.8 In subsequent follow-up of both trials, the majority of responses are durable,7,8 with approximately 40% continued response beyond the time of expected relapse.
CONTEXT
Key Objectives
Assess the safety and efficacy of axicabtagene ciloleucel (axi-cel) in a nonclinical trial population and identify correlates of response and toxicity.
Knowledge Generated
Axi-cel maintains a comparable safety and efficacy profile in a broader patient population with additional comorbidities; however, patients who were ineligible for clinical trials—including those ineligible only because they received bridging therapy—do less well and represent an ongoing unmet need for whom we need better products or combinations.
Relevance
Anti–cluster of differentiation cluster of differentiation chimeric antigen receptor T-cell therapy has had considerable impact on the treatment of chemotherapy refractory aggressive B-NHL and this data supports its ongoing use in a broader and potentially less fit patient population than included in the pivotal clinical trials.
ZUMA-1 and JULIET have been criticized for including highly selected patients. These therapies involve the ex vivo engineering of autologously collected T cells to express an antitumor CAR. These cells are then reinfused into the patient, where they are further activated and expand. The toxicities resulting from T-cell activation, namely cytokine release syndrome (CRS) and neurologic toxicity (NT), coupled with the time it takes to manufacture these personalized “drugs,” raises the question: is this therapy restricted to a subset of patients with minimal comorbidities and tumors indolent enough to withstand the engineering process? The assumption is that included patients and diseases do not reflect the real world. The FDA approval of these products for relapsed/refractory aggressive B-cell non-Hodgkin lymphoma (B-NHL) creates an opportunity to study their safety and efficacy in a nontrial setting, where eligibility criteria may be broadened and where decisions regarding bridging therapy are per the treating physician. Here, we report the experience of commercially available axi-cel in the standard-of-care setting from 7 academic centers in the United States.
PATIENTS AND METHODS
Patients
We performed a retrospective, multicenter study of adult patients with relapsed/refractory aggressive B-NHL who were treated with axi-cel at 7 centers (Appendix, online only). All patients were treated between December 2017 and October 2018. Patient selection, supportive care, toxicity assessment/management, and response assessment followed institutional practice. The use of bridging therapy and the timing of pretreatment imaging were per the treating physician. Information on tumor bulk was taken from the most recent scan before axi-cel. Assessment of performance status (PS), International Prognostic Index (IPI), and ZUMA-1 eligibility was at lymphodepletion. All patients received axi-cel in the hospital, and this was followed by observation. CRS was graded according to the modified Lee criteria,9 and NT grading was per Common Terminology Criteria for Adverse Events (version 4). First response assessment and subsequent imaging was per institutional practice. First response was assessed by Lugano criteria at individual centers without centralized review.10 Response assessment was performed on 116 of 122 patients at 1 month (n = 86), 2 months (n = 8), and 3 months (n = 22). Six patients died as a result of toxicity before response assessment.
Statistical Analysis
Response and toxicity were reported in a modified intent-to-treat (mITT) analysis on all patients who received axi-cel with 95% exact binomial CI. This was the primary analysis in ZUMA-1 and JULIET.6,8 An intent-to-treat (ITT) analysis was performed on all patients with T cells collected. OS for the mITT and ITT populations was defined as from infusion date and leukaphersis, respectively, to death from any cause, censoring for patients alive at last contact. Progression-free survival (PFS) was analyzed by mITT and was defined as from infusion to the earlier of progression or death, censoring for patients alive and progression free at last contact. Survival distributions were estimated using the Kaplan-Meier method, and differences between groups were evaluated with log-rank tests. Categoric data were summarized as proportions with 95% exact binomial CI. Associations between continuous and binary variables were assessed with Wilcoxon rank-sum tests. In an exploratory analysis, C-reactive protein (CRP) day 0, peak CRP, ferritin day 0, and peak ferritin were evaluated for association with survival outcomes. For each, we used recursive partitioning to fit a survival tree using the R package “rpart” v4.1-15, with PFS as the outcome; the first split was taken as the “best” cut point to separate patients into superior versus inferior PFS. To reduce the bias of our sample and to provide a more extrapolable cut point, we resampled patients with replacement and repeated the model 5,000 times to obtain a distribution of cut points for each variable. We used the median of this distribution as a cut point to separate patients into inferior versus superior groups. The cut points were used to evaluate our patients for duration of response (DOR), PFS, and OS. CyTOF analyses were performed on resistant and responding patients using 38 different markers. The Wilcoxon matched-pairs signed-rank test was used to compare paired samples at stated time points. This analysis was exploratory, and P values were not corrected for multiple testing.
RESULTS
Patients
Table 1 outlines patient characteristics. mITT analysis included 122 patients treated with axi-cel. Median age was 62 years (range, 21-79 years). Most patients (91%) had an Eastern Cooperative Oncology Group (ECOG) PS of 0-1 at lymphodepletion. DLBCL was the most common lymphoma (43%), followed by tFL (27%), HGBL (14%), and PMBL (7%). Seven patients had transformation from marginal zone lymphoma (MZL; n = 5) or chronic lymphocytic leukemia (CLL; n = 2). One quarter had double/triple-hit lymphomas. More than one half had an IPI of 0-2, and 20% had an IPI of 4-5 at lymphodepletion. Of 59 patients for whom tumor measurements were available, 53% had tumors > 5 cm in maximal diameter before treatment. Twenty-eight percent had a prior stem-cell transplantation; the majority (25%) were autologous, but 4 patients had had an allogeneic transplantation. Median absolute lymphocyte count (ALC) at leukapheresis was 710 cells/μL (range, 100-4,200 cells/μL). Median CRP at axi-cel infusion was 26.0 mg/dL (range, 0-300 mg/dL). Sixty-two percent (76 of 112) were ineligible for ZUMA-1 because of patient and/or disease characteristics and/or the use of bridging therapy. Forty-two patients were ineligible because of bridging therapy alone; reasons for ineligibility for the remaining 34 patients are listed in Table 1. In addition, 13 patients had T cells collected but were not treated with commercial axi-cel; reasons are in Appendix Fig A1A (online only).
TABLE 1.
Patient Characteristics

ZUMA-1–ineligible patients were significantly more likely to have a higher ECOG PS (P = .008) and to have received bridging therapy (P < .001), factors that defined ineligibility in ZUMA-1. They also had a significantly higher IPI at lymphodepletion (P = .008) and a higher median CRP at infusion (33.4 v 17.0 mg/dL; P = .033).
Efficacy Outcomes
Median follow-up from infusion is 10.4 months. Table 2 reviews efficacy outcomes by mITT. The best overall response rate (ORR) was 70%; 61 patients (50%) achieved a CR, and 24 patients (20%) achieved a PR as best response. When including the 13 patients who had cells collected but were not treated under commercial specifications, the best ORR by ITT was 65% and the best CR rate was 47%; 2 patients had durable CRs after out-of-specification axi-cel in a clinical trial, and 1 patient had a CR to bridging therapy. Of patients in PR at first restaging with subsequent imaging (n = 31), 32% had a subsequent CR (Appendix Fig A1). Of the 84% of patients (43 of 51) in CR at first restaging with ≥ 6 months of follow-up, 79% (34 of 43) maintained their CR. By mITT, the 6-month CR rate was 41% (38% by ITT).
TABLE 2.
Efficacy and Toxicity

Figure 1 shows the DOR and survival estimates. Median DOR among all responding patients was 11.0 months (95% CI, 7.9 months to not reached [NR]), but among patients who achieved a CR at first restaging, it was NR (Fig 1A). Median PFS for treated patients was 4.5 months (95% CI, 3.2 to 12.1 months), and among patients who achieved a CR at first restaging, it was NR (Fig 1B). Median OS has not been reached; 12-month OS was 67% (95% CI, 59% to 77%). One-year OS by ITT analysis was 65% (95% CI, 57% to 74%; Appendix Fig A1B).
FIG 1.
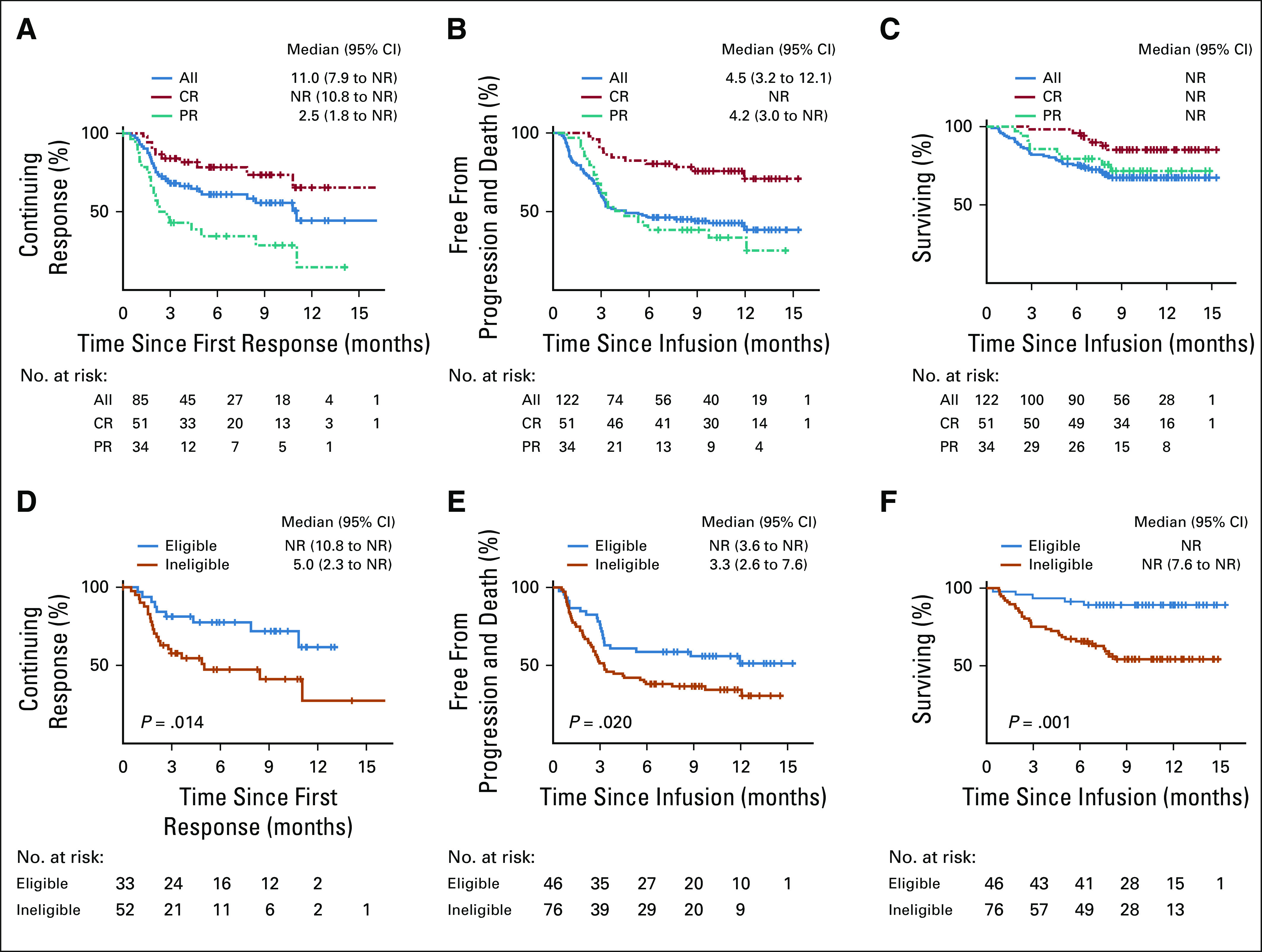
Efficacy outcomes of axicabtagene ciloleucel (axi-cel) overall and by ZUMA-1 eligibility. (A) Duration of response (DOR) curves for patients with overall response at first restage after chimeric antigen receptor (CAR) T-cell therapy. (B) Progression-free survival (PFS) curves for all patients. (C) Overall survival (OS) curves for all patients who underwent infusion of axi-cel. (D) DOR curves for patients who would have been eligible for ZUMA-1 (blue) and those who were ineligible for ZUMA-1 (orange). (E) PFS curves for patients who would have been eligible for ZUMA-1 (blue) and those who were ineligible for ZUMA-1 (orange). (F) OS curves for patients who would have been eligible for ZUMA-1 (blue) and those who were ineligible for ZUMA-1 (orange). All, all patients who underwent infusion of axi-cel; CR, complete response at first restage; NR, not reached; PR, partial response at first restage.
Toxicity
CRS occurred in 93% of patients, with 16% being grade ≥ 3 (Table 2). One patient (1%) died as a result of CRS. Median time to CRS onset was 3 days (range, 0-20 days); median duration was 6 days (range, 1-27 days). NT occurred in 70% of patients, and for 35% this was grade ≥ 3. One death occurred as a result of NT (1%); this was not cerebral edema, but fatal complications related to depressed consciousness. The median time to NT onset was 5 days (range, 0-34 days); median duration was 7 days (range, 1-52 days). The rate of nonrelapse mortality was 6%; 6 of 7 patients died before response assessment (Appendix Fig A1A). The causes of death are listed in Table 2. Intensive care unit (ICU) transfer occurred for 28% of patients, and 18% required hospital readmission, primarily for CAR T-cell–related complications. Tocilizumab was administered to 66% of patients, with 39% receiving ≥ 2 doses. Steroids were administered to 53% of patients, with 14% receiving a high dose (> 40 mg dexamethasone or equivalent per day).
Univariate Analysis for Response and Toxicity
Univariate analyses for response are shown in Fig 2C and Appendix Fig A2A (online only). ORR did not differ significantly by lymphoma histology, IPI, cell of origin, double/triple-hit status, grade ≥ 3 NT, number of prior therapies, tumor bulk, bridging therapy, tocilizumab or steroid use, or ZUMA-1 eligibility. Patients with an ECOG PS of ≥ 2 or high-grade CRS seemed to have a nonsignificantly inferior response (Appendix Fig A2A). Biomarkers of T-cell activation/expansion (peak CRP/ferritin and ALC), T-cell health (ALC at leukapheresis), and pretreatment inflammation (CRP/ferritin pretreatment) were analyzed. Patients with a lower day 0 CRP and higher ALC at leukapheresis were more likely to respond; peak CRP, ALC, and ferritin had no association with response (Appendix Fig A2C). After this, we used a recursive partitioning algorithm to identify variables predictive of superior outcomes; day 0 CRP and peak ferritin identified groups the most disparate for survival (Fig 3). Day 0 CRP of < 30 mg/L correlated with improved DOR (median, NR v 3.6 months; P = .003), PFS (median, NR v 2.5 months; P < .001), and OS (median, NR v 6.5 months; P < .001; Figs 3A-3C), whereas peak ferritin of < 5,000 µg/L correlated with improved PFS (median, 6.8 v 2.2 months; P = .020) and OS (median, NR v 2.7 months; P < .001; Figs 3D-3E).
FIG 2.
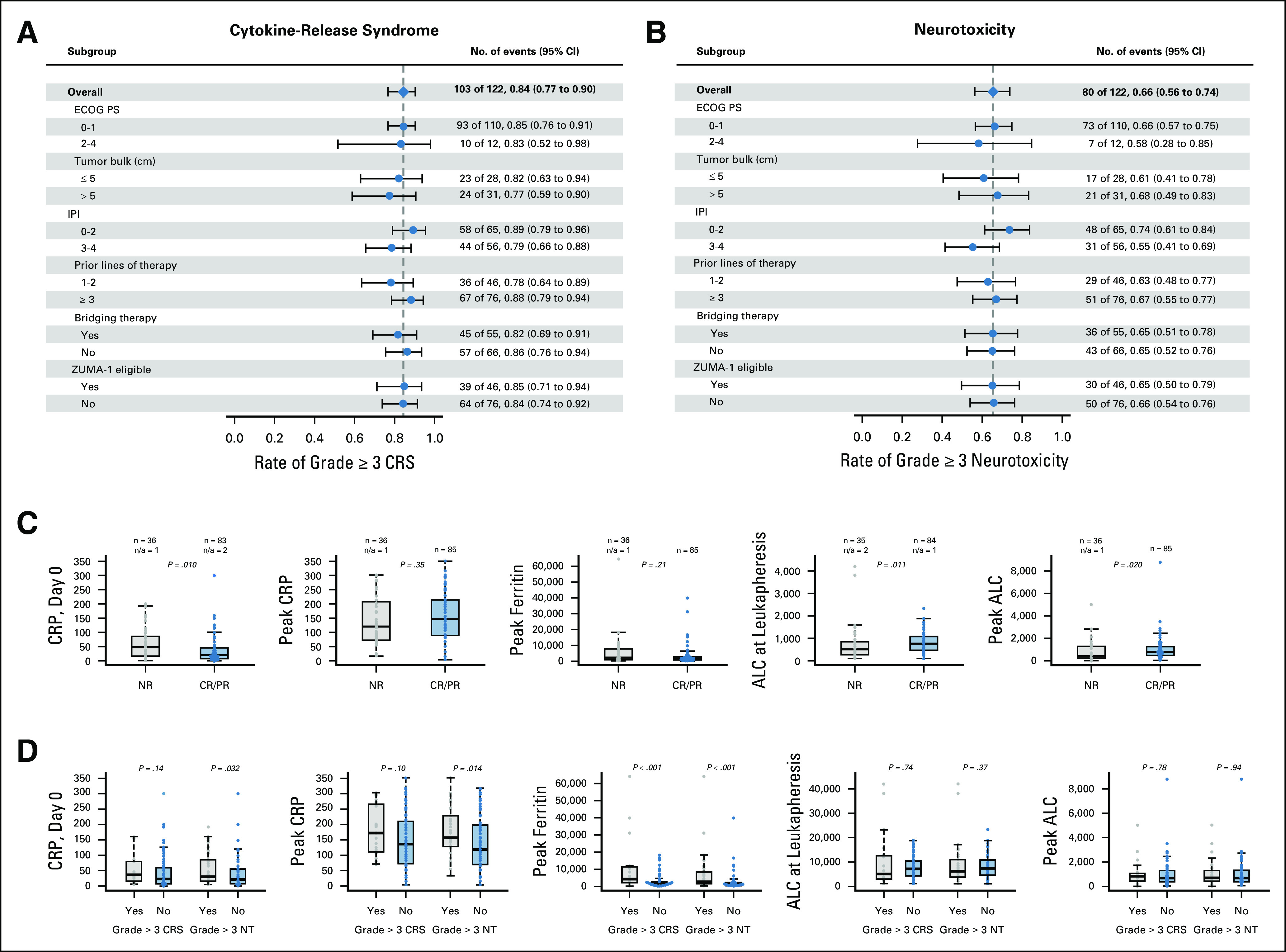
Univariate analysis of response and toxicity. (A) Presence of grade ≥ 3 cytokine release syndrome (CRS) stratified by multiple covariates. (B) Presence of grade ≥ 3 neurotoxicity (NT) stratified by multiple covariates. (C) Overall response rate stratified by levels of key cytokines and absolute lymphocyte count (ALC). (D) Presence of grade ≥ 3 CRS or NT stratified by multiple covariates. CR, complete response; CRP, C-reactive protein; ECOG PS, Eastern Cooperative Oncology Group performance status; IPI, International Prognostic Index; n/a, not applicable; NR, no response; PR, partial response.
FIG 3.
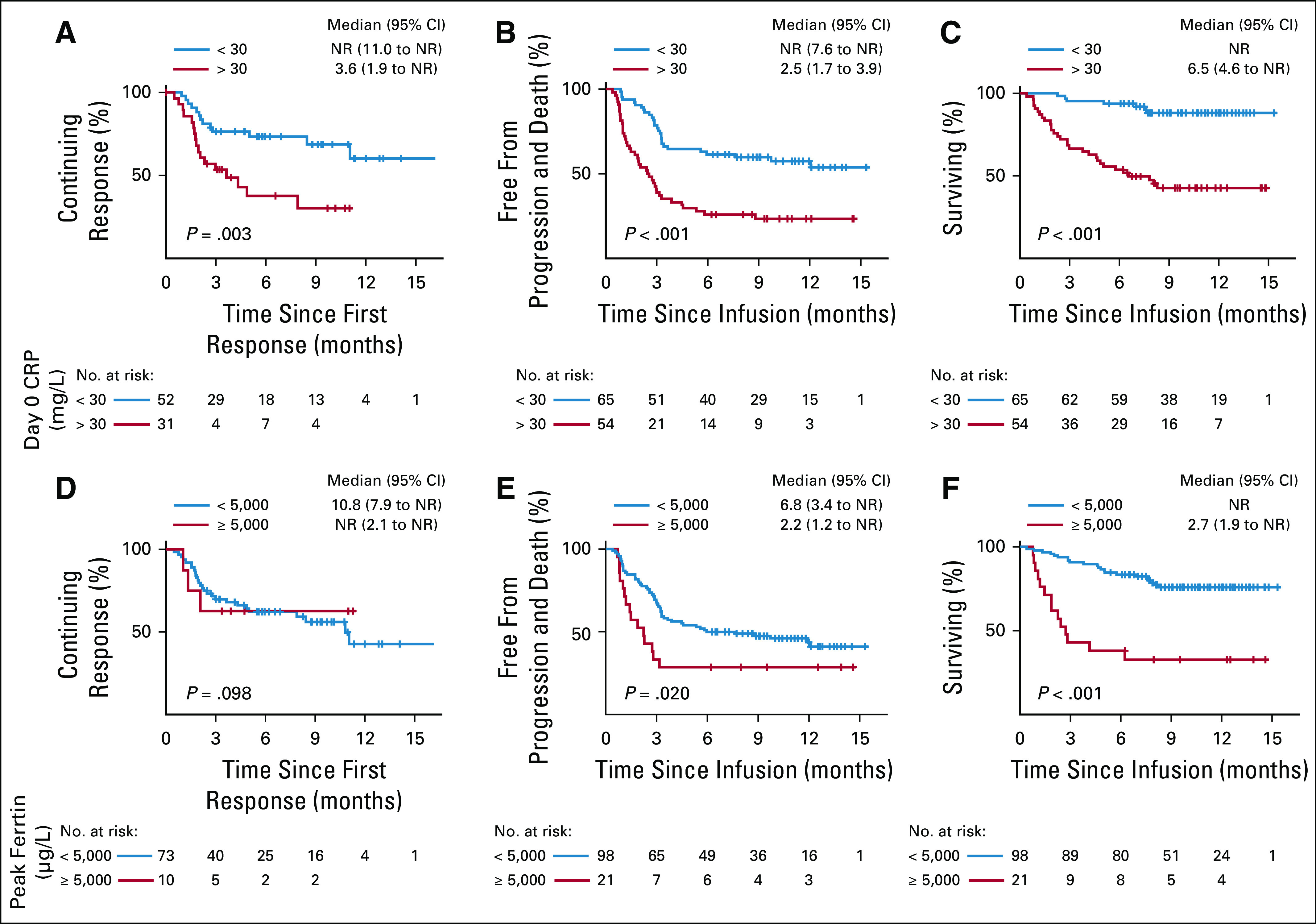
Relationship between day 0 C-reactive protein (CRP) and peak ferritin and outcome with axi-cel. (A) Duration of response (DOR) stratified by CRP (units, milligrams per liter) at day 0. (B) Progression-free survival (PFS) stratified by CRP at day 0. (C) Overall survival (OS) stratified by CRP at day 0. (D) DOR stratified by peak ferritin (units, micrograms per liter). (E) PFS stratified by peak ferritin. (F) OS stratified by peak ferritin. NR, not reached.
Figure 2 shows univariate analyses for toxicity. There was no correlation between ECOG PS, tumor bulk, IPI, number of prior therapies, bridging therapy, or ZUMA-1 eligibility and high-grade CRS or NT (Figs 2A and 2B). An increased day 0 and peak CRP were associated with grade ≥ 3 NT but not CRS, whereas an increased peak ferritin was associated with high-grade NT and CRS (Fig 2D). There was no association between ALC, peak or at leukapheresis, and high-grade CRS or NT (P < .001). Prophylactic use of tocilizumab in a nonpivotal ZUMA-1 cohort was associated with a trend toward increased high-grade NT.11 We assessed for differences in rates of NT after ≥ 2 doses of tocilizumab (38%), compared with 0-1 dose, because most patients received at least 1 dose (66%). Rates of all-grade and high-grade NT were significantly increased after ≥ 2 doses (91% v 55%, P < .001, and 60% v 19%, P < .001, respectively). These patients, however, were also significantly more likely to have had any-grade and high-grade CRS (100% v 89%, P = .023, and 32% v 7%, P < .001, respectively).
ZUMA-1 Eligibility and Outcomes
To analyze the effect of bridging therapy and ZUMA-1 eligibility on response duration and survival, we considered 3 groups: ZUMA-1 eligible (n = 46); ZUMA-1 ineligible because of bridging therapy alone (n = 42); and ZUMA-1 ineligible for other reasons (n = 34), some of whom also received bridging therapy (n = 13). Survival and DOR curves by ZUMA-1 eligibility are shown in Figs 1D-1F. Although ORR was similar in the ZUMA-1–eligible and ZUMA-1–ineligible groups, there was a statistically significant improvement in CR rates overall and at 6 months in eligible patients (63% v 42%, P = .016, and 53% v 30%, P = .047, respectively; Table 2). ZUMA-1–eligible patients had significantly improved DOR (median, NR v 5.0 months; P = .014), PFS (median, NR v 3.3 months; P = .020), and OS (12-month OS, 89% v 54%; P < .001; Figs 1D-1F). DOR, PFS, and OS were similar for patients who were ZUMA-1 ineligible because of bridging therapy alone and those who were ZUMA-1 ineligible for other reasons, and these were inferior to those who were ZUMA-1 eligible (Appendix Figs A2B-A2D).
ZUMA-1–ineligible patients had significantly higher rates of death (43% v 11%, P < .001). The majority of these deaths (26 of 33) were a result of PD, but all treatment-related deaths (n = 7) occurred in ineligible patients. Rates of CRS and NT did not differ by ZUMA-1 eligibility, but ineligible patients were significantly more likely to have received high-dose steroids (21% v 2%, P = .003). Ineligible patients were more likely to be transferred to the ICU, but this difference was not significant (34% v 17%, P = .058).
Immunohistochemical Markers and Resistance
Of 14 biopsies performed at relapse, 5 (36%) were programmed death ligand 1 (PD-L1)+. Five relapsed tumors (36%) were CD19−. Post-treatment biopsy specimens from 2 primary-refractory patients were analyzed by multiplex immunofluorescence (IF) and immunohistochemistry (Figs 4A and 4B). Known markers of resistance differed between these patients. The first had CAR+ T cells within the tumor at day37, but the tumor was CD19− and strongly PD-L1+ (Fig 4A), whereas the second had no intratumoral CAR+ T cells at day 58 but had retained CD19 and was PD-L1− (Fig 4B).
FIG 4.
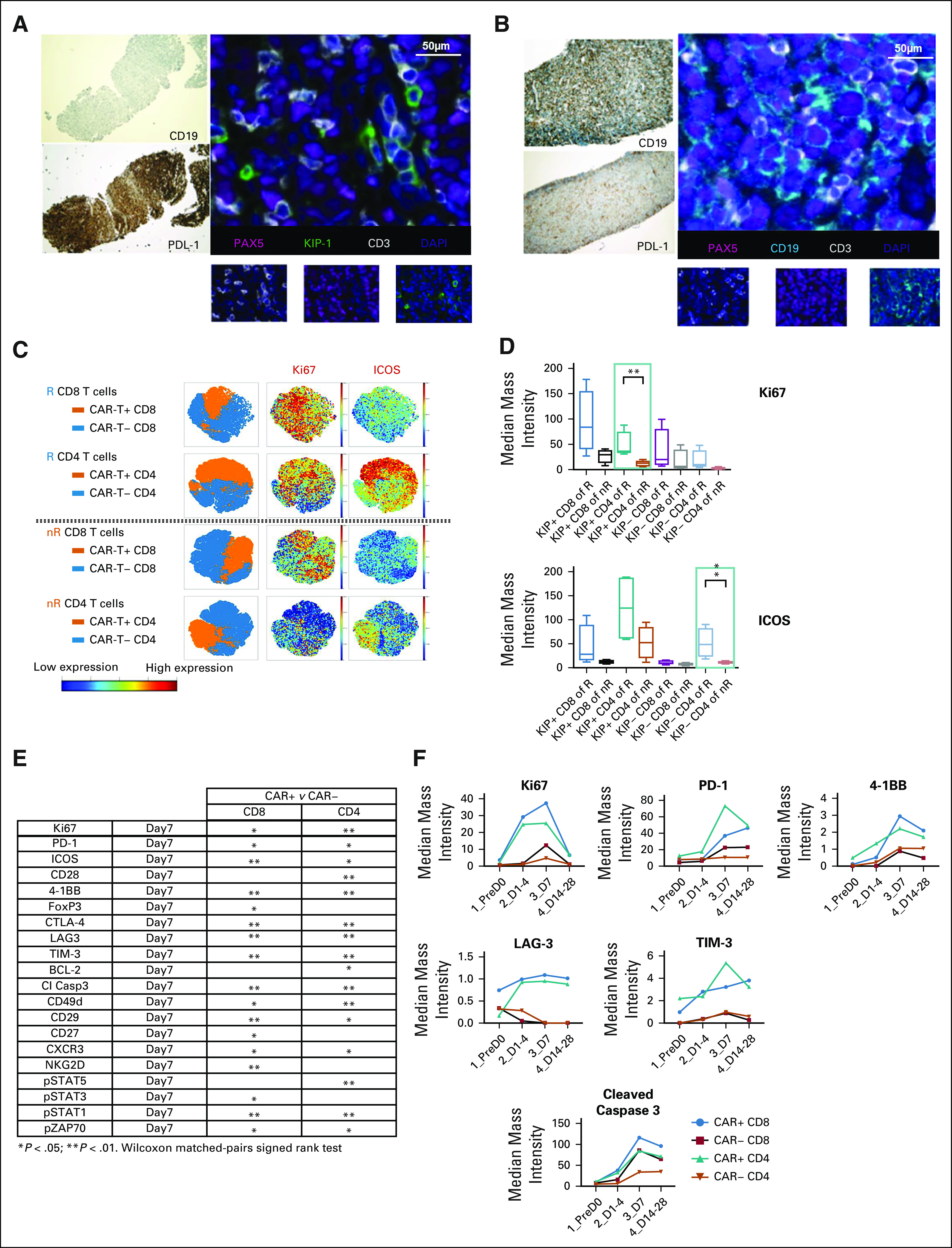
Correlative studies of tumor tissue and of blood from patients treated with axi-cel demonstrate potential mechanisms of resistance. (A and B) Two post-treatment tumor biopsy specimens stained for cluster of differentiation (CD) 19 and programmed death ligand 1 (PD-L1) by immunohistochemistry, as well as for paired box 5 (PAX5), kinesin-like protein 1 (KIP-1), CD3, and 4′,6-diamidino-2-phenylindole (DAPI) by multiplex immunofluorescence. Biopsy specimens were obtained on day 37 (A) and on day 58 (B). (C) CyTOF analysis of expression of CD4, CD8, Ki67, and iducible T cell costimulator (ICOS) of chimeric antigen receptor (CAR) T+ and CAR T− cells from 4 responding (R) and 4 nonresponding (nR) patients. (D) CyTOF comparison of intensity of expression of Ki67 and ICOS among CAR+ (KIP+) and CAR− (KIP−) CD4 as well as CD8 cells from 4 R and 4 nR patients. (E) Differences in expression of multiple cell markers between CAR+ and CAR− T cells from blood samples taken on day 7 of axicabtagene ciloleucel therapy. (F) Differences in expression of multiple cell markers stratified by CAR positivity and CD4/8 expression from serial blood samples.
We analyzed serial blood samples before and after axi-cel for 38 immunomodulatory markers by CyTOF in 4 responders and 4 nonresponders (Fig 4C). Both CD4 and CD8+ CAR+ T cells peaked on day 7, so day 7 samples were used for comparison. Markers of T-cell activation, including Ki67 and inducible T cell costimulator (ICOS), were significantly higher in CAR+ and CAR− T cells among responders (Figs 4C and 4D). Univariate analysis of CAR+ versus CAR− T cells on day 7 demonstrated upregulation of markers of T-cell activation, trafficking, and immunomodulation in CAR+ T cells (Figs 4E and 4F).
DISCUSSION
In our multicenter, off-trial experience of axi-cel in aggressive B-NHL, a best ORR of 70% and a best CR rate of 50% are similar to those found in ZUMA-1 (ORR, 82%; CR, 54%).6 The results of ZUMA-1 would predict that many of the 44 patients in CR at 6 months will have durable remissions beyond 2 years.7 Median DOR in our series was 11.0 months, comparable to that of ZUMA-1 (11.1 months). Our median PFS of 4.5 months was also comparable to that of ZUMA-1 (5.8 months). In both series, median DOR and PFS were NR in CR patients. High response rates were seen despite a majority being ZUMA-1 ineligible. The true denominator of this population, including the patients never considered for axi-cel, is unknown, but our analysis demonstrates that relaxation of eligibility had no effect on ORR. Similar rates of high-grade CRS (16% v 13%) and NT (35% v 28%), despite the inclusion of potentially higher-risk patients, are notable. Furthermore, equally significant is the presence of preserved ORR, DOR, and survival despite a much higher use of tocilizumab and steroids.
Although ORR did not differ by ZUMA-1 eligibility, CR rates, DOR, and survival were inferior in ineligible patients. These differences are important, because this therapy’s power is its response durability. This warrants additional investigation into a possible set of patient/disease characteristics that could predict treatment failure; novel combinations or consolidation strategies for these patients are needed. In addition, treatment-related and all-cause mortality were increased in ineligible patients after axi-cel. These patients also had a significantly higher IPI and pretreatment CRP and a trend toward increased tumor bulk compared with eligible patients, although only an elevated day 0 CRP was associated with worse outcomes; in composite, each may contribute to reduced response durability and survival and increased toxicity. DOR and PFS for ZUMA-1–eligible patients in our study seem to be superior to those in ZUMA-1; these outcome differences may reflect a statistical aberration related to the better-than-expected outcomes in the ZUMA-1–eligible cohort.
In ZUMA-1, there was a direct correlation between tumor bulk and high-grade NT.12 A similar relationship between tumor volume and CRS and NT risk was noted in JULIET, which allowed bridging therapy and which reported lower rates of high-grade NT than did ZUMA-1.8,13 Bridging therapy could debulk tumors, and we were therefore interested in its impact on toxicity and efficacy. Surprisingly, we saw no difference in toxicity after bridging therapy or in patients with increased pretreatment tumor bulk. We did observe an unexpected negative impact of bridging therapy on CR rate, DOR, and survival. Because reasons for bridging therapy in this study are not readily available, possible explanations remain elusive. An early response may reflect a response to bridging, rather than to axi-cel, as reflected in a higher proportion of PRs at first restaging; these are expected to be less durable. This question warrants additional study.
Less heavily pretreated patients in ZUMA-1 had improved ORR, with a trend toward better CAR T-cell expansion after < 5 lines of therapy.12 In addition, axi-cel products with a shorter doubling time in culture were associated with improved CAR T-cell expansion and ORR.14 Given this, we examined the relationship between ALC at pheresis and peak ALC on outcomes and found a positive association between ALC at pheresis and response. Ongoing studies will further investigate this relationship through analyses of immune-cell subsets at leukapheresis to assess whether ALC at pheresis is a surrogate for T-cell fitness/phenotype and a predictor of response. The inverse relationship between both day 0 CRP and peak ferritin and response duration and survival is also intriguing. It is likely that these serum biomarkers are surrogates for aspects of the patient’s disease or immune response, although which aspects it reflects remains to be elucidated.
We saw responses in histologies such as transformed MZL, and in patients with CNS involvement. To our knowledge, these are the first responses to axi-cel among these subtypes, although responses were seen after other CAR T-cell products.15-17 In addition, responses were seen after allogeneic stem-cell transplantation, after other CD19-directed and anti-CD20 CAR T-cell therapies, and in 1 patient with HIV. We saw no durable responses in Richter’s transformation from CLL (n = 2) and only 1 durable response (of 4) in T-cell/histiocyte-rich DLBCL, the latter with increased expression of programmed cell death protein 1 (PD-1) ligands. Given the small numbers, additional study is needed to confirm these observations.
We demonstrate that multiplex IF on post-treatment biopsies and serial blood T-cell profiling with CyTOF may provide useful insights into potential mechanisms of resistance and immunologic targets for combination therapy. In contrasting postprogression biopsies from 2 patients, we corroborate known and postulated resistance mechanisms, namely CD19 loss18,19 and PD-L1 upregulation20,21; however, mechanisms that impede CAR T-cell trafficking and/or persistence may also be potentially important. In comparing a panel of immunomodulatory markers on CAR+/− T cells at time points before and after therapy, we identified differences in T-cell activation markers (Ki67, ICOS) that were associated with response. In this small subset, CAR+/− T cells were more highly activated and proliferative in responders. In addition, we found differential upregulation of immunomodulatory markers (PD-1, 4-1BB) in CAR+ versus CAR− T cells at maximal expansion. These studies support the use of immunomodulatory drugs targeting ICOS, LAG3, 4-1BB, and PD-1/PD-L1 in combination with CAR T cells to improve activation and/or persistence.
Axi-cel in the nontrial setting retains its efficacy, with a similar safety profile. Patients eligible for ZUMA-1 do better than do ineligible patients, but CAR T cells do yield durable responses in this latter group as well, beyond that predicted by the SCHOLAR-1 study.4 This then is not a group to exclude from CAR T-cell therapy, but rather, defines a group for whom there is an unmet need with our currently available treatments. Our analyses identify biomarkers that are associated with efficacy, including day 0 CRP, ALC at pheresis, and peak ferritin. Additional investigation, incorporating additional functional dissection of lymphocyte and immune cell subsets before and after CAR T cells, as well as investigation of the tumor and microenvironment at these time points, will be performed to understand how these biomarkers relate to host and disease factors. It is through analyses such as these that we aim to understand the mechanisms of resistance in all, but also in the ZUMA-1 ineligible population, to potentially inform new combinations and cellular therapy constructs to improve outcomes.
APPENDIX
Supplementary Methods
Seven treating centers: Dana-Farber Cancer Institute/Brigham and Women’s Hospital (DFCI/BWH; Boston, MA); Massachusetts General Hospital (MGH; Boston, MA); University of Chicago (Chicago, IL); University of Washington/Fred Hutchinson Cancer Research Center/Seattle Cancer Care Alliance (UW/FH/SCCA; Seattle, WA); Ohio State University (OSU; Columbus, OH); Emory University (Atlanta, GA); and City of Hope National Medical Center (COH; Duarte, CA). Protocols that allow for these retrospective analyses were approved by institutional review boards at each site.
Single-Cell Mass Cytometry
Single-cell mass cytometry (CyTOF) was performed on frozen peripheral blood mononuclear cells from a convenience subset of 8 adult patients treated at DFCI/BWH (4 responders and 4 nonresponders). A panel of 38 metal-tagged monoclonal antibodies was used to simultaneously examine the phenotypic and functional effects of chimeric antigen receptor T-cell therapy on lymphocyte subsets in vivo. Preconjugated antibodies were purchased from Fluidigm or conjugated with metal isotopes using the MaxPAR antibody conjugation kit (Fluidigm) according to the manufacturer’s recommended protocol. The method for staining cells has been described previously (Hirakawa M, et al: JCI Insight 1: e89278, 2017; Sievers SA, et al: AAACR Annual Meeting 2019; a1204). Cells were analyzed on a CyTOF 2 mass cytometer (Fluidigm) at an event rate of approximately 500 cells/s. To normalize CyTOF data over different days, EQ Four Element Calibration Beads (Fluidigm) were added to all samples. The resulting data were analyzed with software available through Cytobank (www.cytobank.org). To remove debris and doublets, single cells were gated on the basis of cell length and DNA content as described by Bendall et al (Bendall SC, et al: Science 332:687-696, 2011). To interpret high-dimensional single-cell data that were produced by mass cytometry, we used a visualization tool that was based on the viSNE algorithm, which allows visualization of high-dimensional cytometry data on a 2-dimensional map at a single-cell resolution.
Multiplex Immunofluorescence and Standard Immunohisto‐chemistry
Multiplex immunofluorescence was performed on formalin-fixed, paraffin-embedded whole tissue sections with standard, primary antibodies sequentially and paired with a unique fluorochrome per published protocols (Sievers SA, et al: AAACR Annual Meeting 2019; a1204; Carey CD, et al: Blood 130:2420-2430, 2017). Standard immunohistochemistry was performed on formalin-fixed, paraffin-embedded whole tissues using standard laboratory protocols.
FIG A1.
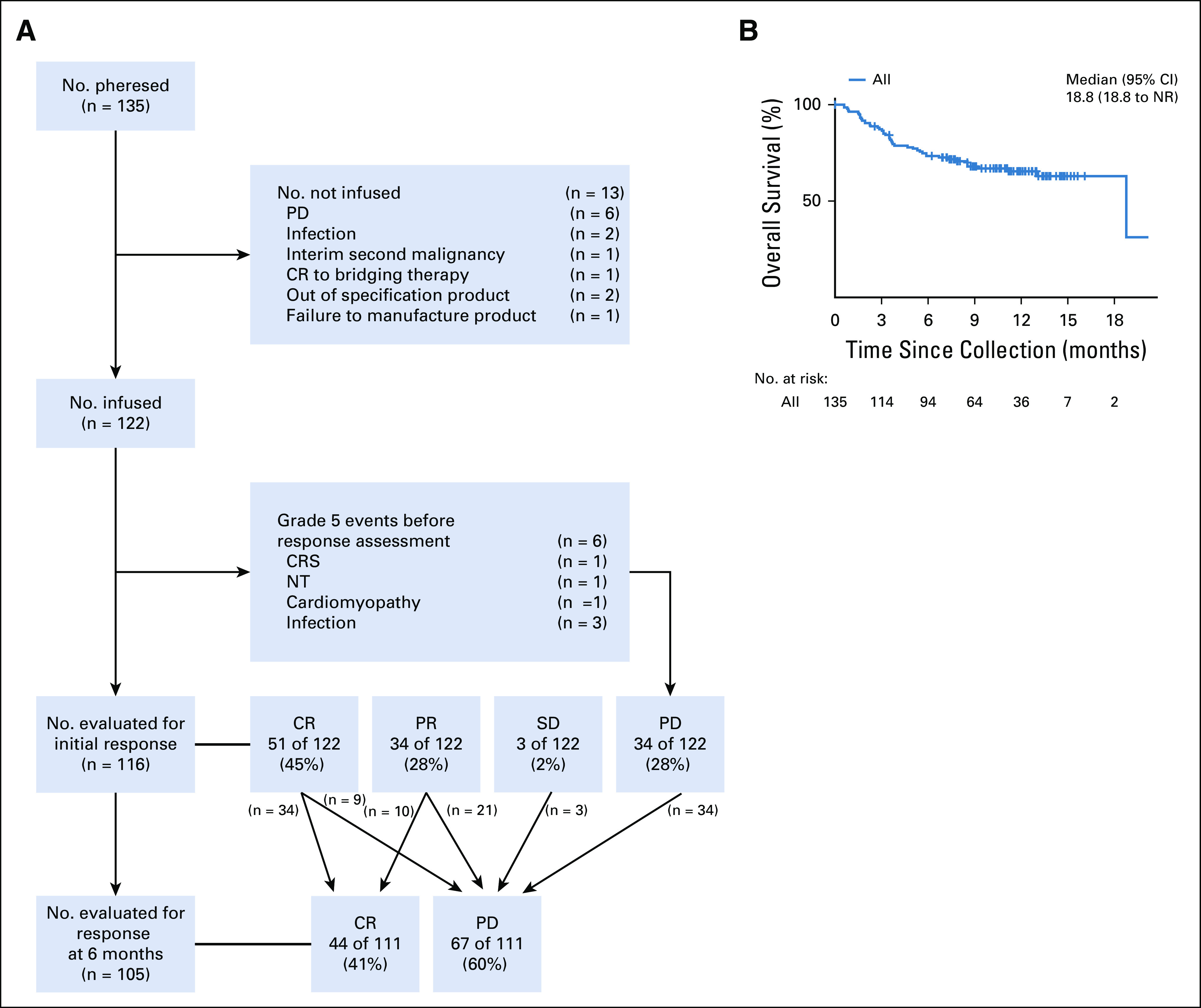
Disposition of all patients, including those leuakapheresed and not treated (intent-to-treat population). (A) Consort diagram of patient disposition throughout the course of the study. (B) Overall survival of the intent-to-treat population from the day of leuakapheresis. CR, complete response; CRS, cytokine release syndrome; NR, not reached; NT, neurotoxicity; PD, progressive disease; PR, partial response; SD, stable disease.
FIG A2.
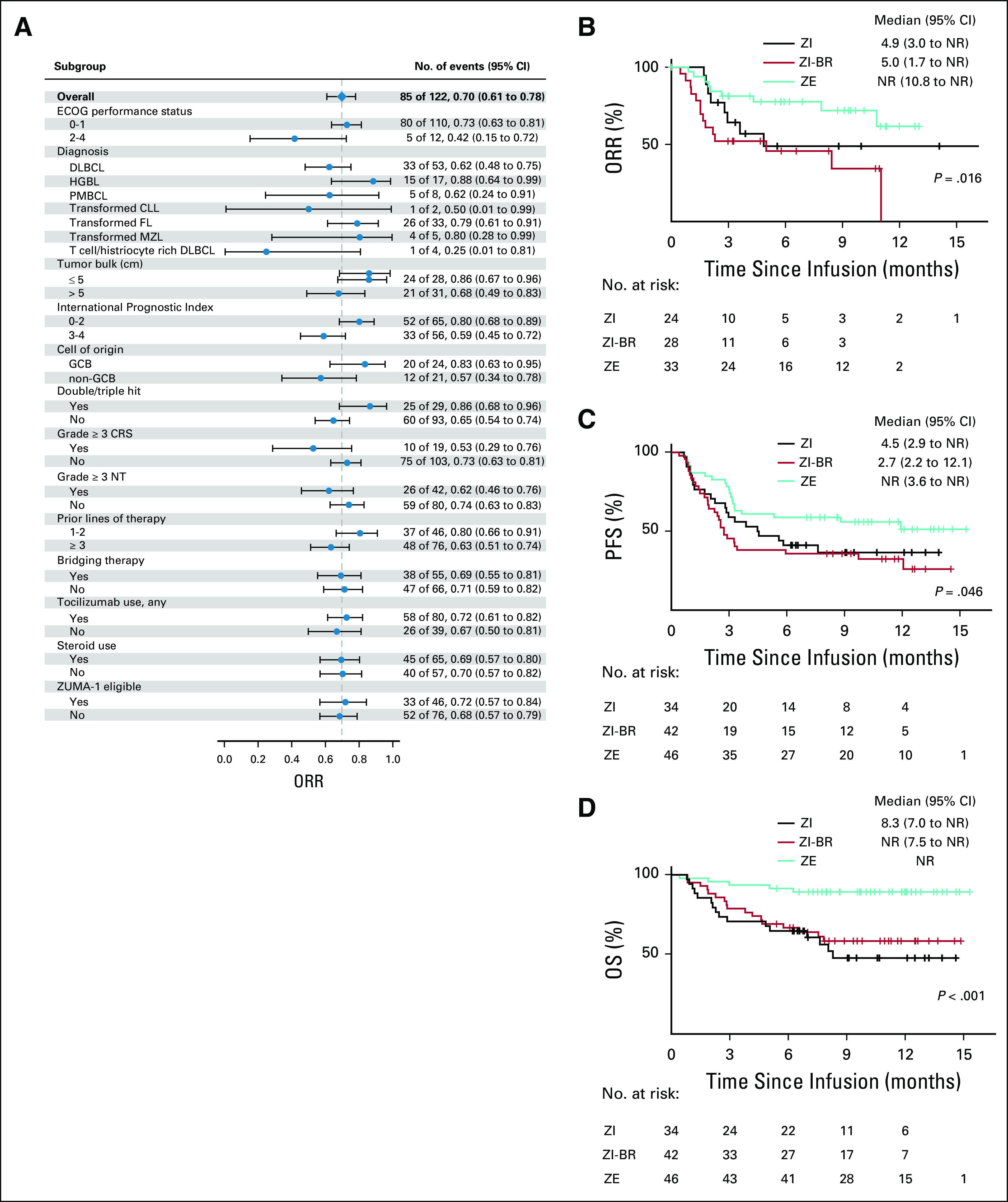
Key variates and efficacy outcomes of axicabtagene ciloleucel. (A) Overall response rate (ORR) stratified by multiple covariates. (B) Duration of response curves stratified by ZUMA-1 eligibility and use of bridging therapy. (C) Progression-free survival (PFS) stratified by ZUMA-1 eligibility and use of bridging therapy. (D) Overall survival (OS) stratified by ZUMA-1 eligibility and use of bridging therapy. CLL, chronic lymphocytic leukemia; CRS, cytokine release syndrome; DLBCL, diffuse large B cell lymphoma; ECOG, Eastern Cooperative Oncology Group; FL, follicular lymphoma; GCB, germinal center B cell; HGBL, high-grade B-cell lymphoma; MZL, marginal zone lymphoma; NR, not reached; NT, neurotoxicity; PMBCL, primary mediastinal large B-cell lymphoma; ZE, ZUMA-1 eligible; ZI, ZUMA-1 ineligible for reasons other than bridging therapy; ZI-BR, ZUMA-1 ineligible solely because of the use of bridging therapy.
PRIOR PRESENTATION
Presented in part at the American Society of Hematology Annual Meeting, San Diego, CA, December 1, 2018.
SUPPORT
Supported in part by funding from the Cancer Clinical Investigator Team Leadership Award from the National Cancer Institute though a supplement to P30CA006516.
See accompanying editorial on page 3085
AUTHOR CONTRIBUTIONS
Conception and design: Caron A. Jacobson, Bradley D. Hunter, Jerome Ritz, Philippe Armand, Alex Herrera, Ajay K. Gopal, Matthew J. Frigault, Utkarsh H. Acharya
Financial support: Caron A. Jacobson, Jerome Ritz, Marcela V. Maus
Administrative support: Caron A. Jacobson, Matthew J. Frigault
Provision of study material or patients: Caron A. Jacobson, Jerome Ritz, Samantha Jaglowski, Marcela V. Maus, Justin Kline, Jonathon B. Cohen, Stephen D. Smith, David G. Maloney, Ajay K. Gopal, Matthew J. Frigault
Collection and assembly of data: Caron A. Jacobson, Bradley D. Hunter, Scott J. Rodig, Pei-Hsuan Chen, Kyle Wright, Mikel Lipschitz, Jerome Ritz, Yusuke Kamihara, Philippe Armand, Sarah Nikiforow, Michael Rogalski, Joseph Maakaron, Samantha Jaglowski, Marcela V. Maus, Yi-Bin Chen, Jeremy S. Abramson, Justin Kline, Elizabeth Budde, Alex Herrera, Matthew Mei, Jonathon B. Cohen, Stephen D. Smith, David G. Maloney, Ajay K. Gopal, Matthew J. Frigault, Utkarsh H. Acharya
Data analysis and interpretation: Caron A. Jacobson, Bradley D. Hunter, Robert Redd, Scott J. Rodig, Jerome Ritz, Yusuke Kamihara, Philippe Armand, Joseph Maakaron, Samantha Jaglowski, Yi-Bin Chen, Jeremy S. Abramson, Justin Kline, Elizabeth Budde, Alex Herrera, Jonathon B. Cohen, Stephen D. Smith, David G. Maloney, Ajay K. Gopal, Matthew J. Frigault, Utkarsh H. Acharya
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Axicabtagene Ciloleucel in the Non-Trial Setting: Outcomes and Correlates of Response, Resistance, and Toxicity
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Caron A. Jacobson
Honoraria: Kite/Gilead, Novartis, Celgene, Bristol Myers Squibb, Nkarta, Precision Biosciences, Humanigen
Consulting or Advisory Role: Kite/Gilead, Precision Biosciences, Novartis, Celgene, Humanigen, Bristol Myers Squibb, Nkarta, Lonza
Speakers' Bureau: Clinical Care Options, Axis Bioservices
Research Funding: Pfizer
Travel, Accommodations, Expenses: Kite/Gilead, Novartis, Precision Biosciences
Bradley D. Hunter
Stock and Other Ownership Interests: Aprea Therapeutics, Forty Seven
Honoraria: Curio Sciences
Speakers' Bureau: Kite Pharma
Scott J. Rodig
Honoraria: Perkin Elmer, Bristol Myers Squibb
Consulting or Advisory Role: Bristol Myers Squibb
Research Funding: Bristol Myers Squibb, Merck, Affimed Therapeutics, Kite Pharma
Patents, Royalties, Other Intellectual Property: Patent pending for use of anti-galectin1 antibodies for diagnostic use.
Travel, Accommodations, Expenses: Roche, Bristol Myers Squibb
Jerome Ritz
Stock and Other Ownership Interests: LifeVault Bio, TScan Therapeutics
Consulting or Advisory Role: Celgene, Draper Labs, Avrobio, Aleta Biotherapeutics, TScan Therapeutics, Talaris
Research Funding: Kite/Gilead, Equillium, Amgen
Philippe Armand
Honoraria: Merck, Bristol Myers Squibb
Consulting or Advisory Role: Bristol Myers Squibb, Merck Sharp & Dohme, Adaptive Biotechnologies, Affimed Therapeutics, ADC Therapeutics, Celgene, Daiichi Sankyo, C4 Therapeutics, GenMab, Miltenyi Biotec, Enterome, Tessa Therapeutics, MorphoSys
Research Funding: Merck Sharp & Dohme (Inst), Bristol Myers Squibb (Inst), Tensha Therapeutics (Inst), Roche (Inst), Adaptive (Inst), Affimed (Inst), Genentech (Inst), IGM (Inst)
Travel, Accommodations, Expenses: Bristol Myers Squibb, Merck Sharp & Dohme
Sarah Nikiforow
Consulting or Advisory Role: Kite/Gilead, Novartis, Nkarta
Travel, Accommodations, Expenses: Kite/Gilead, Novartis, Nkarta
Mark Rogalski
Employment: Celldex Therapeutics, Alexion Pharmaceuticals (I)
Stock and Other Ownership Interests: Celldex Therapeutics, Alexion Pharmaceuticals (I)
Joseph Maakaron
Research Funding: Forty Seven (Inst)
Samantha Jaglowski
Consulting or Advisory Role: Novartis, Kite/Gilead, Juno Therapeutics, Crispr Therapeutics
Research Funding: Novartis, Kite/Gilead, Unum Therapeutics
Marcela V. Maus
Stock and Other Ownership Interests: Agenus, Century Therapeutics, TCR2 Therapeutics
Honoraria: BD Biosciences
Consulting or Advisory Role: Agenus, TCR2 Therapeutics, Windmill, Adaptimmune, Arcellx, Bluebird Bio, Crispr Therapeutics, EMD Serono, Kite Pharma, Novartis, Takeda, Torque, Cellectis, GlaxoSmithKline, Incysus, Allogene, MicroMedicine
Patents, Royalties, Other Intellectual Property: Inventor on patents held by University of Pennsylvania with and without Novartis; inventor on patents held by Massachusetts General Hospital/Partners Health Care; all related to CAR T cells and/or gene therapy
Travel, Accommodations, Expenses: GlaxoSmithKline, BD Biosciences
(OPTIONAL) Open Payments Link: https://openpaymentsdata.cms.gov/physician/xxxxxxx/summary
Yi-Bin Chen
Consulting or Advisory Role: Magenta Therapeutics, Takeda, Incyte, Kiadis, AbbVie, Equillium, Pharmacyclics
Jeremy S. Abramson
Consulting or Advisory Role: Gilead Sciences, Celgene, Novartis, Juno Therapeutics, Verastem, Bayer, AbbVie, Janssen, Merck, Kite Pharma, Genentech, EMD Serono, MorphoSys, Allogene, Karyopharm Therapeutics, Bristol Myers Squibb
Research Funding: Seattle Genetics (Inst), Celgene (Inst), AI Therapeutics (Inst)
Justin Kline
Honoraria: Cardinal Health
Consulting or Advisory Role: Merck, Seattle Genetics, Verastem
Speakers' Bureau: Kite/Gilead
Research Funding: Merck, ITeos Therapeutics, Verastem
Travel, Accommodations, Expenses: Merck, Bristol Myers Squibb, ITeos Therapeutics
Elizabeth Budde
Honoraria: Gilead Sciences, Kite/Gilead, AstraZeneca
Consulting or Advisory Role: Roche/Genentech, Kite/Gilead
Speakers' Bureau: Kite Pharma, AstraZeneca
Research Funding: Merck, Amgen, Mustang Bio
Patents, Royalties, Other Intellectual Property: Patents pending
Travel, Accommodations, Expenses: Roche/Genentech, Kite/Gilead, AstraZeneca
Alex Herrera
Consulting or Advisory Role: Bristol Myers Squibb, Merck, Kite Pharma, Seattle Genetics
Research Funding: Pharmacyclics (Inst), Bristol Myers Squibb (Inst), Merck (Inst), Genentech/Roche (Inst), Kite Pharma (Inst), Immune Design (Inst), AstraZeneca (Inst), Seattle Genetics (Inst), Gilead Sciences (Inst)
Travel, Accommodations, Expenses: Bristol Myers Squibb
Matthew Mei
Consulting or Advisory Role: Sanofi
Jonathon B. Cohen
Consulting or Advisory Role: Celgene, Seattle Genetics, AbbVie, Janssen, Loxo Oncology, Kite/Gilead, AstraZeneca, Cellectar, Aptitude
Research Funding: Bristol Myers Squibb (Inst), Janssen (Inst), Novartis (Inst), Takeda (Inst), AI Therapeutics (Inst), Genentech (Inst), ASH (Inst), Lymphoma Research Foundation (Inst), Loxo Oncology (Inst), Bioinvent (Inst), AstraZeneca (Inst)
Stephen D. Smith
Consulting or Advisory Role: AstraZeneca, BeiGene, Takeda
Research Funding: Acerta Pharma/AstraZeneca (Inst), Ayala (I), Bristol Myers Squibb (I), Genentech/Roche (Inst), Ignyta (I), Incyte (Inst), Merck Sharp & Dohme (Inst), Pharmacyclics (Inst), Portola Pharmaceuticals (Inst), Seattle Genetics (Inst), De Novo Pharmaceuticals (Inst), BeiGene (Inst), Bayer (Inst)
David G. Maloney
Honoraria: Juno Therapeutics, Celgene, Kite Pharma, Gilead Sciences, Novartis, Pharmacyclics, Genentech/Roche
Consulting or Advisory Role: A2 Biotherapeutics
Research Funding: Juno Therapeutics (Inst), Kite Pharma (Inst), Celgene (Inst)
Patents, Royalties, Other Intellectual Property: Inventor on 4 provisional patents
Ajay K. Gopal
Honoraria: Millennium, Seattle Genetics, Pfizer, Gilead Sciences, Janssen Oncology, ADC Therapeutics, Amgen, I-mab, Actinium Pharmaceuticals, Cellectar, Nurix
Consulting or Advisory Role: Pfizer, Seattle Genetics, Janssen Oncology, Millennium, Gilead Sciences, Nurix, Cellectar
Speakers' Bureau: Seattle Genetics
Research Funding: Merck (Inst), Bristol Myers Squibb (Inst), Gilead Sciences (Inst), Seattle Genetics (Inst), Teva (Inst), Pfizer (Inst), Janssen Oncology (Inst), Millennium (Inst), IgM (Inst)
Matthew J. Frigault
Consulting or Advisory Role: Novartis, Gilead Sciences, Juno Therapeutics, Arcellx
Patents, Royalties, Other Intellectual Property: Own IP related to chimeric antigen receptors and T cell manufacturing
Utkarsh H. Acharya
Honoraria: Prometheus Laboratories
Consulting or Advisory Role: Kite Pharma, Teva, Karyopharm Therapeutics
Research Funding: Juno Therapeutics, Bristol Myers Squibb
No other potential conflicts of interest were reported.
REFERENCES
- 1.Seshadri T, Stakiw J, Pintilie M, et al. Utility of subsequent conventional dose chemotherapy in relapsed/refractory transplant-eligible patients with diffuse large B-cell lymphoma failing platinum-based salvage chemotherapy. Hematology. 2008;13:261–266. doi: 10.1179/102453308X343527. [DOI] [PubMed] [Google Scholar]
- 2.Hitz F, Connors JM, Gascoyne RD, et al. Outcome of patients with primary refractory diffuse large B cell lymphoma after R-CHOP treatment. Ann Hematol. 2015;94:1839–1843. doi: 10.1007/s00277-015-2467-z. [DOI] [PubMed] [Google Scholar]
- 3.Nagle SJ, Woo K, Schuster SJ, et al. Outcomes of patients with relapsed/refractory diffuse large B-cell lymphoma with progression of lymphoma after autologous stem cell transplantation in the rituximab era. Am J Hematol. 2013;88:890–894. doi: 10.1002/ajh.23524. [DOI] [PubMed] [Google Scholar]
- 4.Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study Blood 1301800–1808.2017[Erratum: Blood 131:587-588, 2018] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Den Neste E, Schmitz N, Mounier N, et al. Outcome of patients with relapsed diffuse large B-cell lymphoma who fail second-line salvage regimens in the International CORAL study. Bone Marrow Transplant. 2016;51:51–57. doi: 10.1038/bmt.2015.213. [DOI] [PubMed] [Google Scholar]
- 6.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019;20:31–42. doi: 10.1016/S1470-2045(18)30864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 9.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome Blood 124188–195.2014[Erratum: Blood 128:1533, 2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: The Lugano classification. J Clin Oncol. 2014;32:3059–3068. doi: 10.1200/JCO.2013.54.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Locke FL NS, Bartlett NL, et al. Preliminary results of prophylactic tocilizumab after axicabtageneciloleucel (axi-cel; KTE-C19) treatment for patients with refractory aggressive non-Hodgkin lymphoma (NHL) Blood. 2017;031 (suppl; abstr 1547) [Google Scholar]
- 12.Locke FL, Ghobadi A, Jacobson CA, et al. Durability of response in ZUMA-1, the pivotal phase 2 study of axicabtagene ciloleucel (Axi-Cel) in patients (Pts) with refractory large B-cell lymphoma. J Clin Oncol. 2018;36:3003–3003. [Google Scholar]
- 13. Borchmann P, Tam C, Jager U, et al: An updated analysis of JULIET, a global pivotal phase 2 trial of tisagenlecleucel in adult patients with relapsed or refractory (r/r) diffuse large B-cell lymphoma (DLBCL). 2018 EHA Congress, Stockholm, Sweden, June 14-17, 2018 (abstr S799) [Google Scholar]
- 14. Locke FL, Rossi JM, Jacobson CA, et al: Preinfusion product doubling time is associated with CAR T cell expansion and outcomes in ZUMA-1, the pivotal study of axicabtagene ciloleucel (axi-cel) in refractory large B cell lymphoma. SITC 2018 Meeting, Washington, DC, November 7-11, 2018 (abstr P212) [Google Scholar]
- 15.Abramson JS, McGree B, Noyes S, et al. Anti-CD19 CAR T cells in CNS diffuse large-B-cell lymphoma. N Engl J Med. 2017;377:783–784. doi: 10.1056/NEJMc1704610. [DOI] [PubMed] [Google Scholar]
- 16.Turtle CJ, Hay KA, Hanafi LA, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified T cells after failure of ibrutinib. J Clin Oncol. 2017;35:3010–3020. doi: 10.1200/JCO.2017.72.8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frigault MJ, Dietrich J, Martinez-Lage M, et al. Tisagenlecleucel CAR T-cell therapy in secondary CNS lymphoma. Blood. 2019;134:860–866. doi: 10.1182/blood.2019001694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacoby E, Nguyen SM, Fountaine TJ, et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun. 2016;7:12320. doi: 10.1038/ncomms12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. doi: 10.1097/CJI.0000000000000169. Fischer J, Paret C, El Malki K, et al: CD19 isoforms enabling resistance to CART-19 immunotherapy are expressed in B-ALL patients at initial diagnosis. J Immunother 40:187-195, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherkassky L, Morello A, Villena-Vargas J, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130–3144. doi: 10.1172/JCI83092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zolov SN, Rietberg SP, Bonifant CL. Programmed cell death protein 1 activation preferentially inhibits CD28.CAR-T cells. Cytotherapy. 2018;20:1259–1266. doi: 10.1016/j.jcyt.2018.07.005. [DOI] [PubMed] [Google Scholar]