

Keywords: diabetes, metabolomics, microbiota, xenometabolites
Abstract
The gut microbiome has the potential to create or modify xenometabolites (i.e., nonhost-derived metabolites) through de novo synthesis or modification of exogenous and endogenous compounds. While there are isolated examples of xenometabolites influencing host health and disease, wide-scale characterization of these metabolites remains limited. We developed a metabolomics platform (“XenoScan”) using liquid chromatography-mass spectrometry to characterize a range of known and suspected xenometabolites and their derivatives. This assay currently applies authentic standards for 190 molecules, enriched for metabolites of microbial origin. As a proof-of-principle, we characterized the cecal content xenometabolomics profile in adult male lean Sprague-Dawley (LSD) and University of California, Davis type 2 diabetes mellitus (UCD-T2DM) rats at different stages of diabetes. These results were correlated to specific bacterial species generated via shotgun metagenomic sequencing. UCD-T2DM rats had a unique xenometabolite profile compared with LSD rats, regardless of diabetes status, suggesting that at least some of the variation is associated with host genetics. Furthermore, modeling approaches revealed that several xenometabolites discriminated UCD-T2DM rats at early stages of diabetes versus those at 3 mo postdiabetes onset. Several xenometabolite hubs correlated with specific bacterial species in both LSD and UCD-T2DM rats. For example, indole-3-propionic acid negatively correlated with species within the Oscillibacter genus in UCD-T2DM rats considered to be prediabetic or recently diagnosed diabetic, in contrast to gluconic acid and trimethylamine, which were positively correlated with Oscillibacter species. The application of a xenometabolite-enriched metabolomics assay in relevant milieus will enable rapid identification of a wide variety of gut-derived metabolites, their derivatives, and their potential biochemical origins of xenometabolites in relationship to host gastrointestinal microbial ecology.
NEW & NOTEWORTHY We debut a liquid chromatography-mass spectrometry (LC/MS) platform called the XenoScan, which is a metabolomics platform for xenometabolites (nonself-originating metabolites). This assay has 190 in-house standards with the majority enriched for microbe-derived metabolites. As a proof-of-principle, we used the XenoScan to discriminate genetic differences from cecal samples associated with different rat lineages, in addition to characterizing diabetes progression in rat model of type 2 diabetes. Complementing microbial sequencing data with xenometabolites uncovered novel microbial metabolism in targeted organisms.
INTRODUCTION
A great deal of research has focused on the role of the gut microbiome in modulating host physiology and health, including associations between specific intestinal microbe populations and metabolites with the function of gastrointestinal tract and systemic immune systems (37, 55), liver function and steatosis (10), amino acid metabolism (27), gut hormone release (11), brain development and behavior (13a), kidney function (28), and other physiological systems. Some of the strongest evidence that specific bacteria or groups of bacteria impact physiological function stems from fecal/cecal content transfer studies, in which donor obesity phenotypes were transferred to recipients (47, 52). Recently, clinical application of this strategy has shown promise, with fecal transfer from healthy donors to patients suffering from inflammatory bowel disease (IBD) helping mitigate IBD-associated symptoms (54).
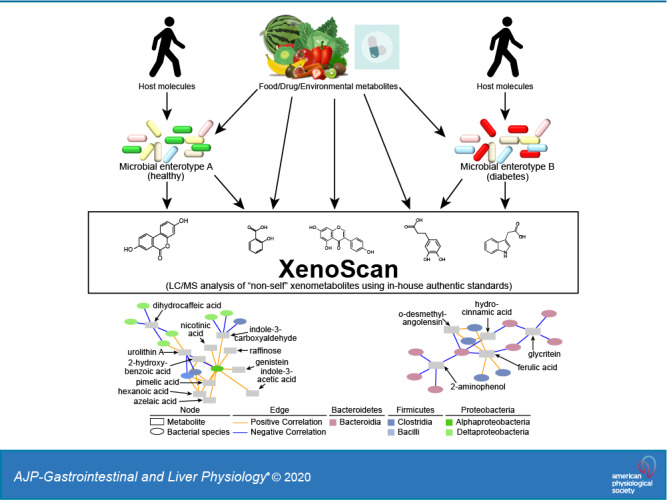
Most studies of gut microbiome-host interactions have focused on correlations between abundances of specific microbes and specific (patho)physiological variables, which allows for hypothesis-generation in terms of the potential bacterial taxa that are involved. However, the specific molecular signals underlying microbe-to-host communication and gut-derived metabolites that reflect shifts in microbial populations, ecology, and biochemistry remain to be fully elaborated. There is a massive genetic potential (number of genes and gene variants) for the gut microbiome; when coupled to the large biomass of microbes in the gastrointestinal tract, and the complex molecule mixtures in foods, there is tremendous enzymatic and nonenzymatic capacity for generating xenometabolites. This can occur via de novo synthesis, through microbial conversion of exogenous components (e.g., phytonutrients, fibers, oligosaccharides, pharmacological agents, etc.), and via microbial modification of host-derived molecules (primary bile acids, urea, etc.). The latter may also be referred to as “cometabolites” since they involve metabolism by both host and microbe(s). Much work remains to fully categorize xenometabolites and their nomenclature, to determine which of these metabolites serve as modulators of mammalian physiology, and to understand their specific microbial and food-derived origins.
Metabolomics has proven quite valuable to identify molecular biomarkers of microbial metabolism; however, few laboratories have dedicated authentic standard libraries that encompass the wide breadth of metabolites associated with microbial activities. Furthermore, most large-scale metabolomics platforms have standard and spectral libraries that generally encompass human and mammalian metabolic pathways, which complicates identification of microbial specific metabolites. To address this issue, we have developed a metabolomics platform that enables a greater focus on “nonhost” xenometabolites, xenometabolite derivatives, cometabolites, and related molecules, which can be applied to better understand alterations in microbial environments. This platform, which we term “XenoScan,” currently has >190 authentic in-house xenometabolite standards curated from published literature and is continuously growing. As a proof-of-concept, we utilized the XenoScan to extend our previous work in the UC Davis type 2 diabetes mellitus (UCD-T2DM) Rat model (45). Our results, herein, demonstrate that the XenoScan can detect genetic/strain-specific differences in microbial metabolism, distinguish early and late states of diabetes, and identify species-specific relationships between bacterial taxa and xenometabolites.
MATERIALS AND METHODS
Animals
Male UCD-T2DM rats were utilized for the current study. The UCD-T2DM rat model spontaneously develops diabetes while consuming a standard low-fat, low-sugar rodent chow diet, has a polygenic origin of obesity, has an inherited β-cell defect, and retains functional leptin signaling. Full description of the UCD-T2DM rat lineage has been previously described (12, 30). This study was approved by the University of California, Davis Institutional Animal Care and Use Committee (Protocol No. 18267) and all methods involving animals were carried out in accordance with the institutional guidelines and regulations.
Study Design
In-depth study details have been previously described (45). Briefly, all rats were singly housed within the same room at the animal facility in the Department of Nutrition, had Carefresh bedding and ad libitum access to standard chow (2018 Telkad Global; Harlan Laboratories), and were on a 14:10-h light-dark schedule. Male UCD-T2DM rats were selected for inclusion if ~180 days old and had nonfasting blood glucose <200 mg/dL (i.e., prediabetic, referred to as PD), nonfasting blood glucose >200 mg/dL for the previous 7–14 days (i.e., recent onset of diabetes, referred to as RD), and nonfasting blood glucose >200 mg/dL for the previous 90 days (i.e., 3-mo post-onset of diabetes, referred to as D3M). Male LSD rats aged 180 days were also studied as a metabolic healthy nonobese control group. Microbial analysis (16S rRNA and shotgun metagenomic sequencing) of animals used in this study has previously been conducted and a major effect of collection year was observed even though UCD-T2DM rats had been maintained at the same animal facility for >12 yr (45). Thus the current study only reports results from rats collected during 2016. Final sample numbers (n) were as follows: LSD = 7, PD = 9, RD = 10, and D3M = 5. Since only two animals were available from 6-mo diabetic rats (D6M) from 2016, these animals were not included for analyses herein.
Sample Collection
As previously described (45), rats were given a 200 mg/kg ip dose of pentobarbital sodium after a 13-h fast. Fully anesthetized rats were exsanguinated via cardiac puncture between ca. 0800 and 1100. Cecal tissue was immediately removed after exsanguination and blotted dry. A hole was punched into the cecal tissue, and a cecal content sample was squeezed into a sterile 1.5-mL microcentrifuge tube. Samples were immediately placed in a −80°C freezer until analysis.
Metagenomic Sequencing
Total DNA was extracted from cecal content samples (0.1–0.3 g) using the DNeasy PowerSoil HTP 96 Kit (Qiagen, Germantown, MD) following the manufacturer’s instructions. Sequencing libraries were generated from extracted DNA samples using Nextera XT reagents including dual indexes. Libraries were quantitated using Qubit dsDNA reagents and pooled and sequenced using NextSeq 500 high-output reagents (150-bp paired reads). Alignment of reads and taxonomic binning was conducted with MEGAN Community Edition software. Reads were aligned against the National Center for Biotechnology Information nonredundant database (NCBI-nr) with the DIAMOND program, and taxonomic analysis was then performed with Meganizer. Median sample depth for samples used in this study herein was 2.38 Gbp. Species level sequencing count data were exported from MEGAN as a csv file and further analyzed using the R Statistical Language (version 3.6.0). Sequencing data are publicly available at the NCBI Sequence Read Archive (Accession No. SRP140861; https://www.ncbi.nlm.nih.gov/sra).
Metabolomics Analyses
In-depth description of analytical workflow has been previously published (46). For each animal, duplicate cecal samples were prepared and subjected to analysis. Cecal contents (~25 mg) were resuspended in 500 µL 50% aqueous methanol plus 25 μL recovery standard [13C]valine (42 μM) and homogenized using a Precellys 24 homogenizer (Bertin Corp) at 5,300 rpm for two 30-s cycles. Homogenates were then extracted in 1 mL of ice-cold acetonitrile. Experimental pools, used for quality control (QC) samples, were prepared by pooling equal volumes (100 μL) of each sample extract. Samples and QC extracts were evaporated to dryness under a nitrogen stream and reconstituted in 300 µL 5% aqueous methanol containing an internal standard (lorazepam, 5.1 μM final concentration; Sigma Aldrich, St. Louis, MO). Chromatography was performed on a Dionex Ultimate 3000 UHPLC as using an XSelect CSH C18 reversed phase column (2.1 × 100 mm, 2.5 μm) kept at 49°C as previously described with minor modification (46). All samples, including duplicates, from each diet group assayed in mixed and random order. Detection was carried out on a Q-Exactive Hybrid Quadrupole-Orbitrap mass spectrometer with data acquisition executed using Xcalibur 4.0 software as previously described (46). All samples were analyzed by positive and negative electrospray ionization (ESI+/−) full-mass spectrometry (MS) scan mode.
The acquired data set, composed of full MS and data-dependent MS2 raw files, was processed using Compound Discoverer 3.0 using an untargeted metabolomics workflow including retention time alignment, unknown compound detection, compound-grouping across all samples, gap filling and metabolite identification using online, and “in-house” data-dependent MS2-fragmentation spectral databases (ddMS2). Software parameters for alignment were as follows: adaptive curve model, 5-ppm mass tolerance, and 0.2-min maximum shift for alignment. Software parameters for detecting unknown compounds were as follows: mass tolerance for detection of 10 ppm, intensity tolerance of 30%, S/N threshold of 3, and minimum peak height of 1e6, and parameters for compound groups were a mass tolerance of 10 ppm and retention time tolerance 0.15 min. Gap filling was performed across all samples using the Real Peak detection method, a mass tolerance of 10 ppm and S/N threshold of 1.5. Metabolites were identified by using MassList [accurate mass ± 5 ppm, retention time (RT) ± 15 s to a known standard], mzCloud (online ddMS2 database), and mzVault (in house ddMS2 database) and given the following confidence levels: Level 1 identification if accurate mass, retention time, and MS2 spectra matching to mzVault or mzCloud; and Level 2 identification if accurate mass, retention time matching to known standard, and no ddMS2 information. For the current paper, only specific, Level 1 metabolites were utilized for final analysis.
The term “xenometabolites” refers to “nonself” metabolites/compounds and their derivatives (i.e., derived from microbes, food/plants, pharmaceutical, industry, etc.). The current xenometabolomics platform (XenoScan) was constructed using 190 authentic standards (Supplemental Table S1: https://doi.org/10.6084/m9.figshare.11419383.v1). Identification of library components was based largely on an exhaustive survey of the extant literature related to known gut microbial and other xenometabolites, plus in some cases deduction of microbial origins of select metabolites based on conditions in which literature reports characterized the molecules (Supplemental Table S1). In addition to strict xenometabolites, we expanded our library to include metabolites that would not be identified in a host organism without comodification by a foreign enzyme/organism (e.g., bacterial enzymatic deconjugation of bile acids in cecum and large intestines).
Peak area values were generated on metabolites identified in the cecal content samples within our in-house library (i.e., not limited to only xenometabolites). All detected metabolites were normalized using vector normalization on log transformed data. Average relative standard deviation (RSD) of duplicate samples was 24.1% and 23.0% in negative and positive modes, respectively. The average of duplicate samples was used. RSDs of QC samples were calculated for all detected metabolites. A total of 7 and 13 metabolites from negative and positive modes, respectively, had mean RSD >40% and were removed from statistical analyses. Metabolites were further filtered to include only xenometabolites and their derivatives including cometabolites (Supplemental Table S2), resulting in a total of 82 metabolites that were ultimately used for statistical analysis (47 and 35 in negative and positive modes, respectively).
Data Preprocessing and Statistical Analysis
All statistical analyses were conducted in R version 3.6.0. Metabolites were assessed individually for outliers using an iterative application of Grubbs’ test (33) on log-transformed data. Identified outliers were removed, which affected <0.7% of the total data. Imputation of all missing data, including missing data due to technical and outlier assessment, was conducted using the K-nearest neighbor algorithm from the impute package (22). Sample outliers were visually assessed using sample boxplots and principal component analysis (PCA). No samples were removed from statistical analysis due to sample outlier assessments. Group differences in metabolites were assessed with Kruskal-Wallis tests followed by correction for multiple comparisons using Benjamini and Hochberg’s false discovery rate (FDR) methodology (4). Mann-Whitney U tests were used when two groups were compared, followed by FDR correction. PCA was used to reduce the dimensionality of metabolomics data and then summarized in two- and/or three-dimensional plots. Data were log transformed and scaled to unit variance before PCA assessment. Partial least squares-discriminant analysis (PLS-DA) was used to determine whether the global metabolome could accurately predict group classification. PLS-DA was determined using the caret package (35) with training parameters set to fourfold cross-validation with five repeats. Average classification accuracy across each cross-validation repeat was assessed to determine performance of PLS-DA models. The optimal number of PLS-DA latent variables (LVs) was chosen based on which LV had the highest average classification accuracy. Variable importance in projection (VIP) was calculated by fitting a PLS-DA model in the pls package (40) using the parameters determined in the caret workflow, followed by VIP assessment using the plsVarSel package (39). Confidence intervals (95%) were then assessed for VIP calculations using the boot package (8, 13). Metabolites with bootstrapped VIP confidence intervals >0.8 were selected as metabolites contributing to class discrimination in PLS-DA models (39). PLS-DA scores were visualized on the first three LVs using the rgl package (2). Full details of preprocessing steps related to shotgun metagenomic taxonomy data, including removal of poorly sequenced samples and low abundant species, have previously been reported (45). Functions from the phyloseq (38) and vegan (41) packages were routinely used in this workflow. The centered log-ratio transformation was applied to the taxonomy data before analyses. Taxonomy and metabolomics data were combined, and Spearman’s correlations were assessed using the rcorr() function in Hmisc (21). Correlations were conducted on LSD rats and combined PD and RD rat groups. We analyzed PD and RD groups together to increase our overall power and due to the similarity in microbiota and metabolomics phenotypes (herein and Refs. 44, 45). Correlations were not conducted on D3M (i.e., later stage diabetic rats) due to the small sample size. Data that had a significant group difference were used in the correlation analysis. Significant correlations were then imported into Cytoscape (version 3.7.1) for network visualization. Supplemental material is available at https://doi.org/10.6084/m9.figshare.11419383.v1 including the data sets, R code, and supplemental files generated during the current study.
RESULTS
The Cecal Xenometabolome Discriminates UCD-T2DM Rats from Lean Sprague-Dawley Controls
The UCD-T2DM rat spontaneously develops a diabetic phenotype, characterized by a polygenic origin of obesity and peripheral insulin resistance, while maintaining functional leptin signaling (12). We previously assessed the cecal contents from age-matched UCD-T2DM rats at differing stages of diabetes progression for shotgun metagenomic sequencing and global, untargeted metabolomics enriched for metabolites associated with pathways related to energy metabolism (45). Although the previous metabolomics assay demonstrated that the cecal metabolome is altered with diabetes progression, that analysis platform was not optimized to fully characterize metabolites directly associated with microbial metabolism. Therefore, we leveraged samples from the prior cohort to determine whether the xenometabolome and related metabolites are altered by the progression of diabetes. Of the 190 metabolites available as authentic standards in our XenoScan platform, 82 were detected in either UCD-T2DM rat groups (regardless of diabetes status) or lean Sprague-Dawley (LSD) control rats (Supplemental Table S2). Of these, 39 metabolites were significantly different (FDR <0.05) in pairwise comparisons of cecal concentrations (quantifier ion peak areas) between the UCD-T2DM rat groups and LSD controls (Fig. 1A). Furthermore, only seven of these did not intersect with metabolites identified in pairwise assessments between LSD and UCD-T2DM rats considered to be at an early stage of diabetes development (PD: prediabetic; RD: recent diabetic, diabetes onset <2 wk; Fig. 1B), suggesting the majority of the variation in the assay was due to differences between LSD rats and UCD-T2DM rats (indicating a genetic strain effect contributing at least some of the metabolite variance). This was highlighted by PCA, which showed discrimination between LSD and UCD-T2DM rats along PC1 and PC2, accounting for 39% of the overall variance in the data (Fig. 1C).
Fig. 1.

A: differential abundant metabolites between LSD rats (n = 7), UCD-T2DM rats before the onset of diabetes (PD; n = 9), recent onset (RD; n = 10), and 3 mo post-onset (D3M; n = 5). Metabolites were selected if found to have a statistically altered group after FDR correction (Kruskal-Wallis Test, FDR < 0.05). Adjacent bargraph to heatmap shows the log-transformed median peak area for each metabolite. Cyan bar indicates xenometabolite, and beige bar indicates microbial modified metabolite. B: Venn diagram indicating the intersection of metabolites with FDR < 0.05 under the following tests: 1) all experimental groups (LSD, PD, RD, and D3M) using Kruskal-Wallis test, 2) LSD vs PD groups using Mann-Whitney U test, and 3) LSD vs. RD groups using Mann-Whitney U tests. C: PCA of xenometabolomics data. All metabolites were included in the PCA. Data were scaled to unit variance before PCA assessment. CA, cholic acid; CDCA, chenodeoxycholic acid; FDR, false discovery rate; LSD, lean Sprague-Dawley; PCA, principal component analysis; TDCA, taurodeoxycholic acid; UCD-T2DM, UC Davis Type 2 Diabetes Mellitus; UDCA, ursodeoxycholic acid.
We next investigated whether the xenometabolome could discriminate all groups using a multivariate approach. Modeling with PLS-DA showed good discrimination with four-group classification (Fig. 2A; 82.6% prediction accuracy with 4-fold cross-validation). With the use of a VIP cutoff of 0.8, acetic acid, crotonobetaine, ectoine, tetradecanedioic acid, 4-hydroxybenzoic acid, α-hydroxyisobutyric acid, azelaic acid, glycodeoxycholic acid, and quinic acid were identified as discriminant metabolites (Fig. 2B and Supplemental Table S3). Of these, only crotonobetaine, ectoine, and glycodeoxycholic acid had an FDR >0.2 in univariate analyses. In terms of directionality, acetic acid, 4-hydroxbenzoic acid, α-hydroxyisobutyric acid, and quinic acid were found to be lower in LSD rats compared with UCD-T2DM groups, while tetradecanedioic acid was higher in LSD rats compared with UCD-T2DM rats (Fig. 2C).
Fig. 2.

A: PLS-DA of untargeted xenometabolomics in LSD (n = 7), UCD-T2DM rats before the onset of diabetes (PD; n = 9), recent onset (RD; n = 10), and 3 mo post-onset (D3M; n = 5). PLS-DA scores dimension shown for first 3 latent variables. B: VIP for metabolites that had a bootstrapped VIP 95% confidence interval > 0.08. Lines indicate bootstrap percentile interval using 500 bootstrap replicates. C: Boxplots display group differences of metabolites identified in B. Pairwise differences in groups were assessed by Tukey’s test. Groups differing from LSD rats: #P value < 0.05 and ##P value < 0.01. &Groups differing from PD rats: P value < 0.05. GDCA, glycodeoxycholic acid; LSD, lean Sprague-Dawley; PLS-DA, partial least squares-discriminant analysis; UCD-T2DM, UC Davis type 2 diabetes mellitus; VIP, variable importance in projection; LV, latent variable.
The data described above strongly suggest that the variance is mostly attributed to differences between LSD and UCD-T2DM rats. As we are unable to conclusively distinguish which differences between LSD and UCD-T2DM rats are due to genetic, metabolic, or microbiota effects, we next considered whether the XenoScan could differentiate different stages of diabetes within UCD-T2DM rats (prediabetic, recent onset of diabetes, and 3-mo post-onset of diabetes). When comparing differences among the 3 UCD-T2DM rat groups, 24 metabolites were altered before FDR correction; however, none maintained statistical significance at FDR ≤0.05 (Supplemental Table S2). In contrast to the univariate results, PLS-DA modeling with jasmonic acid, ω-hydroxyhippuric acid, cholic acid, indole-3-carboxaldehyde, ω-desmethylangolensin, ursodeoxycholic acid, ergothioneine, and ectoine resulted in robust cross-validation prediction accuracy (Fig. 3, A and B and Supplemental Table S4; 80.4% accuracy; 4-fold cross-validation; 6 latent variables), suggesting that a multivariate model is necessary to uncover variation associated with diabetes status in this model. While the metabolites selected by PLS-DA modeling were not determined to be significant at FDR <0.05 in univariate analysis, they still had FDR <0.2 except for ectoine and ω-hydroxyhippuric acid (Supplemental Table S2). All selected metabolites showed within-group differences with post-hoc analyses (Dunn’s test), mainly driven by differences at 3 mo post-onset of diabetes. Abundances of cholic acid, ursodeoxycholic acid, indole-3-carboxaldehyde, and jasmonic acid were all lower in D3M rats relative to PD and RD rats (Fig. 3C). Ectoine trended higher in D3M rats relative to PD and RD rats (P = 0.061 and 0.056 to PD and RD, respectively). Only ergothioneine showed a different pattern than the others, with PD rats having higher abundances relative to RD and D3M groups.
Fig. 3.

A: PLS-DA of untargeted xenometabolomics in UCD-T2DM rats before the onset of diabetes (PD; n = 9), recent onset (RD; n = 10), and 3 mo post-onset (D3M; n = 5). PLS-DA scores dimension shown for first 3 latent variables. B: VIP for metabolites that had a bootstrapped VIP 95% confidence interval >0.08. Lines indicate bootstrap percentile interval using 500 bootstrap replicates. C: boxplots display raw metabolomics data for metabolites identified in B group differences of metabolites. Pairwise differences in groups were assessed by Tukey’s test. Groups differing from D3M rats: *P value < 0.05 and **P value < 0.01. &Groups differing from RD rats P value < 0.05. CA, cholic acid; PLS-DA, partial least squares-discriminant analysis; UCD-T2DM, UC Davis type 2 diabetes mellitus; UDCA, ursodeoxycholic acid; VIP, variable importance in projection; LV, latent variable.
Xenometabolites Correlate Strongly to Subsets of Bacterial Species within an Experimental Group
Correlations within LSD rats.
We next wanted to identify bacterial species and xenometabolite correlations that could signal which bacterial species regulate the cecal metabolite pool (e.g., through production, degradation/metabolism or metabolite conversions). Due to the disparate metabolic states of LSD and UCD-T2DM rats, and the confounding “strain effect” on metabolome and microbiome (discussed above), analyses focused only within-group (i.e., correlations between xenometabolites and bacterial species within LSD or within UCD-T2DM groups). We used strict criteria to filter low abundant taxa to combat the high prevalence of zeros in microbial sequencing data, understanding that this potentially increases type II error rate. In LSD rats, the majority of correlations of xenometabolites were among species within the Bacteroidia and Clostridia classes (Fig. 4 and Supplemental Fig. S1). Based on the correlation network (Fig. 4 and Supplemental Fig. S1), several metabolites served as major intersection points for select bacteria. For example, taurodeoxycholic acid, 2-aminophenol, 3,5-dihydroxybenzoic acid, protocatechuic acid, N-ω-acetylhistamine, cholic acid, and trimethylamine-N-oxide (TMAO) had at least 6 negative correlations with bacterial species primarily in the class of Clostridia. For example, Faecalibacterium prausnitzii, Intestinimonas butyriciproducen, Flavonifractor plautii, and Oscillibacter valericigenes were bacterial species negatively correlated to taurodeoxycholic acid, 2-aminophenol, and 3,5-dihydroxybenzoic acid. Blautia schinki, B. obeum, and Anaerotruncus colihominis were negatively correlated with 3,5-dihydroxybenzoic acid and N-ω-acetylhistamine, whereas p-aminobenzoic acid, hexanoic acid, and gluconic acid were metabolites that positively correlated with Clostridia-grouped bacteria (Fig. 4, panels 1 and 2). Examples include correlations among hexanoic acid and gluconic acid, with Ruminococcus albus, R. callidus, and Butyrivibrio fibrisolven.
Fig. 4.

Spearman’s correlation network of xenometabolites and bacterial species in LSD (n = 7). Edges (i.e., correlations) connect xenometabolites (rectangles) and bacterial species (ovals) at P < 0.05. Orange edges indicate Spearman’s rho > 0 and blue edges indicate Spearman’s rho < 0. Network in top left of figure is the full network. Labeled boxes in the full network are enlarged and adjacent to the full network. Xenometabolites are only annotated in the enlarged panels for clarity. The full network is provided in a larger format in Supplemental Fig. S1. Xenometabolite data were peak areas, and bacterial species data were centered-log ratios of sequencing count data. CA, cholic acid; CDCA, chenodeoxycholic acid; GDCA, glycolithocholic acid; TDCA, taurodeoxycholic acid.
There were fewer total correlations among xenometabolites and bacteria within the Bacteroidia class compared with the amount observed with the Clostridia class. Major xenometabolite hubs of negative correlations with species within the Bacteroidia class included hydroxyphenyl lactic acid, p-cresol sulfate, equol, and indole-3-propionic acid. Indole-3-propionic acid and equol were negatively correlated to Bacteroides cecimuris, B. thetaiotaomicron, and Parabacteroides goldsteinii. Sterocobilin, pyrocatechol, and 2-hydroxybenzoic acid served as intersection points for positive correlations within the Bacteroidia class (Fig. 4, panels 3 and 4). Within the Bacteroidia class, several Bacteroides spp. were positively correlated to both pyrocatechol and 2-hydroxybenzoic acid, including B. fragilis, B. finegoldii, B. coprophilus, and B. dorei.
Only 12 correlations were maintained in LSD rats after adjusting all correlations for multiple comparisons using Benjamini and Hochberg’s false discovery rate correction (Fig. 5). All of these correlations were perfectly monotonic (i.e., Spearmans rho = 1) and were among species from the orders Bacteroidales, Clostridiales, and Spirochaetales. In LSD rats, tryptophan-related metabolites, indole-3-propionic acid and p-cresol sulfate were negatively correlated to Parabacteroides goldsteinii and a Prevotella sp., respectively. Benzoic acid-related xenometabolites, pyrocatechol and 2-hydroxybenzoic acid, were positively correlated to Bacteroides dorei. Other correlations within the Bacteroidales order included a positive correlation between crotonobetaine and a Prevotella sp. and sterocobilin and Alloprevotella tannerae. Clostridium clostridioforme and the Lachnospiraceae bacterium A4 were both negatively correlated to monosaccharides, hexanoic acid and gluconic acid, respectively. Marvinbryantia formatexigens and Acetivibrio ethanolgignens were negatively correlated to N-acetylhistamine derivatives, while Oscillibacter valericigenes, Intestinimonas butyriciproducens, and Flavonifractor sp. were negatively correlated to 3,5-dihydroxybenzoic acid.
Fig. 5.

Linear relationship among xenometabolites and bacterial species found to have Spearman’s correlation with false discovery rate (FDR) < 0.05 in lean Sprague-Dawley rats (n = 7). Graphs are colored based on bacterial taxonomy order. Xenometabolite data were log peak areas, and bacterial species data were centered-log ratios of sequencing count data.
Correlations within UCD-T2DM rats.
We next assessed relationships between xenometabolites and bacterial species within UCD-T2DM rats. Similar to LSD rats, correlations in UCD-T2DM rats classified at an early stage of diabetes consisted of primarily the Bacteroidia and Clostridia classes; however, a noticeable cluster of correlations between xenometabolites and species within the Proteobacteria phyla was also observed (Fig. 6 and Supplemental Fig. S2). For example, dihydrocaffeic acid and indole-3-carboxyaldehyde intersected with several species within the Deltaproteobacteria class (including Desulfovibrio desulfuricans, D. fairfieldensis, D. piger, Bilophila wadsworthia, and Mailhella masiliensis) (Fig. 6, panel 1). A bacterium within the Alphaproteobacteria class, Acidiphilium sp., correlated with multiple xenometabolites, including indole-3-carboxyaldehyde, nicotinic acid, raffinose, genistein, indole-3-acetic acid, azelaic acid, hexanoic acid, pimelic acid, urolithin A, and 2-hydroxybenzoic acid (Fig. 6, panel 1 and Supplemental Fig. S2). Gluconic acid, trimethylamine, homoprotocatechuic acid, and dimethyalanine served as hubs for several positive correlations among species within the Clostridia class, whereas indole-3-propionic acid, 2-phenylacetamide, and protocatechuic acid were intersection points for negative correlations among species within the Clostridia class (Fig. 6, panels 2 and 4). Indole-3-propionic acid, in particular, was negatively correlated with Oscillibacter valericigenes, O. ruminantium, Butyricicoccus pullicecorum, Eubacterium plexicaudatum, and Flavonifractor plautii. Additionally, glycitein and 2-aminophenol served as hubs for negative correlations among several Bacteroidia species (Fig. 6, panel 3), including negative correlations between glycitein and Alloprevotella rava and A. tannerae and between 2-aminophenol and Parabacteroides chinchilla, P. goldsteinii, and Prevotella copri. In contrast to the LSD rats, no correlations remained after adjusting for multiple comparisons.
Fig. 6.

Spearman’s correlation network of xenometabolites and bacterial species in UCD-T2DM rats before the onset of diabetes (PD; n = 9) and recent onset (RD; n = 10). PD and RD rats were combined to increase statistical power before correlation analysis. Edges (i.e., correlations) connect xenometabolites (rectangles) and bacterial species (ovals) at P < 0.05. Orange edges indicate Spearman’s rho > 0 and blue edges indicate Spearman’s rho < 0. Network at top left is the full network. Labeled boxes in the full network are enlarged and adjacent to the full network. Xenometabolites are only annotated in the enlarged panels for clarity. The full network is provided in a larger format in Supplemental Fig. S2.
DISCUSSION
Microbes produce or modify a myriad of small molecules that have the potential to influence host health and disease. For example, microbiota-derived xenometabolites (e.g., short chain fatty acids, indoles) and microbe-modified endogenous metabolites (e.g., secondary bile acids) have a wide range of effects on host immunity (3, 29, 58), energy metabolism (5, 50), and hormone secretion (11, 32), and other functions. Furthermore, a wide range of microbe-regulated xenometabolites have been hypothesized to contribute to disease sequelae of obesity [e.g., lipopolysaccharide (13a)], diabetes [e.g., hydrocinnamic acid and indole-3-lactic acid (43)], cardiovascular disease [e.g., trimethylamine N-oxide (51)], kidney disease [e.g., cresol- and indoxyl-sulfate (60)], and various other diseases (24, 49, 61). In addition, our work in the UCD-T2DM Rat model and human controlled feeding studies has suggested that changes in the host’s metabolic health status can drive changes in the xenometabolome (6, 45). While mechanisms behind many of these relationships have yet to be fully identified, much work is being done to identify xenometabolites and characterize their relationship with host physiology. In this study, we describe a metabolomics platform, the “XenoScan,” which is uniquely enriched in “nonself” xenometabolites, their derivatives, and associated molecules. By strict definition, exogenously derived metabolites can emanate from microbial metabolism, foods and food components, environmental and industrial sources, or microbe modification of host metabolites. The latter may also be termed “cometabolites” since they stem from metabolism of host and microbe(s).
Importantly, results from the XenoScan assay of cecal contents correctly classified disparate rat strains and diabetes stages. Many of these changes were concurrent with genetic- and diabetes stage-dependent shifts in the microbiome. These findings concur with previous reports that have documented differences in microbial composition due to disparate mouse genetics (9, 16, 17, 34, 42, 53). The latter studies used amplicon and genomic sequencing-based methods, which do not address functional alterations of the gut microbiota; thus, there has been a push to combine metabolomics and microbiomics technologies. With much interest focused on human health, many core metabolomics laboratories have libraries enriched for mammalian metabolism. As such, metabolomics results from microbiota-related studies have a large proportion of host-derived or metabolically shared metabolites that may not be directly related to microbial alterations or exogenous metabolite sources. To our knowledge, the XenoScan is the first liquid chromatography-mass spectrometry (LC/MS)-based metabolomics platform with a library specifically enriched in metabolites known or suspected microbial or “nonself” origins, which can be a strong complement to microbial taxonomic survey methods. As knowledge of microbe-associated metabolites grows, the XenoScan serves as a dynamic and evolving platform that can accommodate increasing numbers of xenometabolites verified through authentic chemical standards.
As previously noted, the gut microbiota has been implicated in the pathogenesis of insulin resistance and diabetes (7, 14, 47, 52). Thus archived cecal content samples from our previous work in the UCD-T2DM rat to leverage the XenoScan platform and further examine how diabetes alters the composition and metabolic function of the gut microbiota. When applying “global” metabolomics approaches to cecal contents (45) or blood plasma (44) in this model, we have observed a potential genetic effect when comparing LSD to UCD-T2DM rats, since nondiabetic LSD and prediabetic UCD-T2DM rats had distinct metabolite signatures. Using the XenoScan platform herein, we again were unable to distinguish whether cecal xenometabolite differences between LSD and UCD-T2DM rats are due to genetics/strain or due to differences in metabolic status. Focusing just within UCD-T2DM rat groups, we found a handful of xenometabolites that discriminated rats considered as prediabetic or recently diagnosed as diabetic versus rats with fully developed and untreated diabetes. Most of these are derived from plant sources (e.g., jasmonic acid, ergothioneine, and genestein). However, diets were not different in these animals, so changes in these diet-derived xenometabolites are likely the result of altered host or microbial metabolism. We acknowledge that potential differences in food intake across groups, and potential differences in total cecal contents (which would alter the absolute pool size) should be taken into account and controlled experimentally in future studies of these rat models. Regardless, the xenometabolites that discriminated the spectrum of diabetes status in the current experiment are interesting to consider. Jasmonic acid, for instance, acts as a hormone in plants, akin to mammalian eicosanoids, and is typically synthesized in response to pathogens and insects (15, 20). While the current study cannot determine a specific role for jasmonic acid, there is also evidence suggesting that jasmonic acid is produced by fungi to subvert plant hormonal response (18), which underlies the complex behaviors that microbes and colonies utilize to compete with other microbes. Two nonxenometabolite bile acids, cholic acid and urosodeoxycholic acid, were also found to be lower in D3M rats compared PD and RD rats. These bile acids are primary and secondary bile acids, respectively, and their abundance in cecal contents are likely the result of bacterial enzymatic deconjugation. Thus their reduction in advanced diabetes may suggest either decreased synthesis due to diabetes-associated hepatic injury (1) or alterations in microbial metabolism (45). Further assessment will be required to fully characterize these findings in this model.
Very little is known about the microecological environment within the gastrointestinal tract and the interaction between microbes and small molecules in vivo. A major goal of the XenoScan metabolomics assay is to assist in the identification of microbe-metabolite relationships and then generate new hypotheses that can be further investigated. In the current study, several xenometabolites and associated molecules appear to serve as metabolic hubs where clades of bacteria cluster around certain metabolites. The correlations may signal that these microbes contribute to the metabolism of the hub metabolite and/or that the metabolite milieu regulates microbe ecology. In LSD rats, for instance, taurodeoxycholic acid, 2-aminophenol, 3,5-dihydroxybenzoic acid, N-omega-acetylhistamine, protocatechuic acid, cholic acid, and trimethylamine-N-oxide had at least five species within the Clostridia class that were negatively correlated to each metabolite, whereas p-aminobenzoic acid, hexanoic acid, and gluconic acid had several positive correlations among Clostridia-specific species. The combination of these xenometabolites together do not indicate a specific metabolic pathway, but their close association may suggest how clusters of metabolites promote specific niches for certain clades. An example of this can be gleaned from short-chain fatty acid fermentation, which lowers pH levels and in turn, increases butyrate-producing bacteria, and suppresses Bacteroides spp. (56). Thus combinations of xenometabolites could alter the intestinal chemical environment (e.g., pH, osmolality) and produce favorable conditions for certain microbes. Furthermore, clusters of correlations among chemically similar xenometabolites provide evidence of microbial metabolism. Many of the strongest correlations within LSD rats were among hydroxybenzoic acids (2-hydroxybenzoic acid, pyrocatechol, and 3,5-dihydroxybenzoic acid), which had positive correlations with species within the Bacteroidales order and negative correlations with species in the Clostridiales order, suggesting that these metabolites promote Bacteroidales and suppress Clostridiales. Intriguingly, these hydroxybenzoic metabolites are thought to be derived from plants but are also synthesized by intestinal bacteria from polyphenols and flavonoids (26). Additional investigations will be required to determine whether the hydroxybenzoic acids and other xenometabolites possess biological activity and contribute to the physiological function of these animals. The current results provide an important framework and foundation for these efforts.
We focused our correlation analysis in the UCD-T2DM rats on the combined PD and RD groups to increase statistical power and because the study design has continually demonstrated that these groups have similar microbial and metabolic profiles (44, 45). While the PD group is not considered diabetic based on their nonfasting glucose levels, prediabetic UCD-T2DM rats are hyperinsulinemic, have enlarged pancreatic islets, and have increased liver triglycerides when compared with LSD controls (12); thus, our categorization of PD rats likely differs from RD classified rats based mainly on nonfasting glucose concentrations. When we derived a correlation network using these combined groups, we found a wide variety of xenometabolites that associated with species primarily in the Bacteroidetes, Firmicutes, and Proteobacteria phyla. With the focus on indole-3-propionic acid as an example (Fig. 5, panel 2), this metabolite was negatively correlated to several species within the Oscillibacter genus, including O. valericigenes and O. ruminantium. The genus Oscillibacter was identified in 2007 (25), and bacteria within this genus are gram-negative obligate anaerobes that are considered to be beneficial to mammals due to their putative butyrate production (19, 36). Oscillibacter spp. are not known to carry the reductase needed for indole-3-propionic acid and its negative relationship with this metabolite would further suggest that these bacteria do not produce it. Oscillibacter spp. were also positively correlated to gluconic acid and trimethylamine, the latter of which is a bacteria-derived metabolite proposed to be detrimental to host health due to association of its derivative TMAO with cardiovascular disease (31). While there are likely many other taxonomy-metabolite relationships that can be highlighted within the correlation network, this particular example demonstrates how a single metabolite (e.g., butyrate) may not fully encompass the metabolic process of a microbial ecosystem and its interaction with the host organism. The ability to generate a more comprehensive metabolite landscape reflective of microbe metabolism and ecology is a major strength of analytical platforms such as the XenoScan.
Bacterial production of indole-3-propionic acid has generally been indicated with Clostridium sporogenes (59), but has been noted for other Clostridium and Peptostreptococcus species (48). Paradoxically, the XenoScan identified abundances of indole-3-propionic acid even though C. sporogenes was not identified in the metagenomic analysis (data not shown) nor were any other bacterial species previously known to produce indole-3-propionic acid. To date, indole-3-propionic acid producers have been identified using culture (59) or bioinformatic prediction methods (14). In our correlation analysis, the majority of correlations among bacterial species and indole-3-propionic acid were negative, suggesting that none of the correlations were based on metabolite production. We speculate that indole-3-propionic acid may be a marker of resource competition, where its abundance may signal bacterial species that have a competition disadvantage. Alternatively, negative correlations for metabolites may signal that certain bacteria catabolize or further metabolize parent molecules, thus lowering the cecal pool size of the latter. More work will need to be done in this model to confirm this hypothesis and to determine how this metabolite and other metabolites can be modified when known producers are not apparent.
We report herein the first LC-MS-based untargeted metabolomics platform with an authentic metabolic library enriched in xenometabolites and related molecules, but several limitations should be discussed. We acknowledge that the term “xenometabolite” can technically apply to a wider class of molecules that are not necessarily microbe-derived (e.g., vitamins, pharmaceuticals, plant-derived molecules, etc.). The terminology itself is derived from the Latin “xeno,” thus defining metabolites/compounds that emanate wholly or in part from “nonself” or “foreign” sources. As the field evolves, some xenometabolites may be categorized based on compartmentalization and context. For example, short-chain fatty acids (SCFAs) can also be produced by mammals during combustion of macronutrients in the mitochondria and in that specific context are not xenometabolites. Yet, this class of molecules is produced robustly by gut bacteria and so the gut luminal SCFAs are predominantly xenometabolites. It should also be acknowledged that large spectral repositories, like the Global Natural Products Social Molecular Networking (GNPS) (57), have the ability to predict the identification of a greater number of xenometabolites than our platform, but annotating and identifying many metabolites (especially microbial metabolites) without access to authentic standards remains a challenge. Another limitation to the current study of gut content metabolomics is that total cecal content weight was not determined, so the total cecal pool sizes of metabolites (e.g., concentration multiplied by total content) cannot be accurately known. Lastly, in the current analyses we largely excluded host-derived metabolites to highlight the xenometabolomics profile of our samples, but of course the gastrointestinal environment contains significant metabolic inputs from the host. Thus other factors than the xenometabolome will associate with or impact the microbial ecology in addition to the metabolites profiled here.
In addition to the classification and technological limitations, our study highlighted the current statistical challenges associated with microbiota-metabolome exploratory analyses. We were careful to utilize appropriate methods to minimize type I errors; however, our penalization for multiple hypothesis-based tests either contradicted the multivariate results or rendered our correlations analysis insignificant. While we understand the need to minimize type I errors, we also point that 1) correlations noted herein are not causative and should be considered hypothesis generating until confirmed with targeted studies and 2) our type II error rate was likely significantly increased due to stringent abundance criteria in the shotgun metagenome taxonomy data combined with the multiple comparison correction. Also, it is important to note that FDR assumptions typically consider each comparison as independent from one another; yet, many metabolites are related in pathways in other aspects of metabolism and thus traditional FDR approaches have limitations in “omics” studies.
In conclusion, the application of a new metabolomics platform enriched in xenometabolites discriminated LSD rats from UCD-T2DM rats, concurrent with microbial composition differences associated with host genetics/strain. In addition, we were able to discriminate UCD-T2DM rats with 3 mo post-onset of diabetes from those without diabetes or <2 wk post-onset of diabetes, providing yet another example of host metabolic health influencing the gut microbial ecosystem. Examples of how this method can be used in combination with taxonomy data to generate new hypotheses were provided: future studies will be needed to confirm which specific microbes regulate levels of specific metabolites and vice versa. We feel this is the next evolutionary step in combining metabolomics and microbiomics to delineate fully the complex metabolic ecology in the mammalian intestine.
GRANTS
This work was funded by United States Department of Agriculture-Agricultural Research Services Projects 6026-51000-010-05S and 6026-51000-010-06S. In addition, L. Yeruva is also supported by National Institutes of Health (NIH) Grants P20-GM-121293 and R21-AI-146521. Additional funding support from the University of Arkansas for Medical Sciences College of Medicine Research Council and the Sturgis Foundation for Diabetes Research. P. J. Havel’s laboratory also received funding during the project period from NIH Grants R01-HL-091333, R01-HL-107256, R01-HL-121324, U24-DK-092993, RC1-DK-087307, and R01-DK-095060 and a Multicampus Award from the University of California, Office of the President (Award No.142691).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.E.M., L.Y., S.V.C., P.J.H., S.H.A., and B.D.P. conceived and designed research; K.E.M., L.P., J.L.G., K.L.S., U.D.W., K.S., and B.D.P. performed experiments; K.E.M., S.V.C., and B.D.P. analyzed data; K.E.M., L.Y., S.H.A., and B.D.P. interpreted results of experiments; B.D.P. prepared figures; B.D.P. drafted manuscript; K.E.M., L.Y., S.H.A., and B.D.P. edited and revised manuscript; K.E.M., L.Y., L.P., J.L.G., K.L.S., S.V.C., U.D.W., K.S., P.J.H., S.H.A., and B.D.P. approved final version of manuscript.
REFERENCES
- 1.Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol 42: 132–138, 2005. doi: 10.1016/j.jhep.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 2.Adler D, Murdoch D. rgl: 3D Visualization Using OpenGL. R package version 0.100.3. https://CRAN.R-project.org/package=rgl, 2019.
- 3.Alexeev EE, Lanis JM, Kao DJ, Campbell EL, Kelly CJ, Battista KD, Gerich ME, Jenkins BR, Walk ST, Kominsky DJ, Colgan SP. Microbiota-derived indole metabolites promote human and murine intestinal homeostasis through regulation of interleukin-10 receptor. Am J Pathol 188: 1183–1194, 2018. doi: 10.1016/j.ajpath.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57: 289–300, 1995. doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 5.Byndloss MX, Olsan EE, Rivera-Chávez F, Tiffany CR, Cevallos SA, Lokken KL, Torres TP, Byndloss AJ, Faber F, Gao Y, Litvak Y, Lopez CA, Xu G, Napoli E, Giulivi C, Tsolis RM, Revzin A, Lebrilla CB, Bäumler AJ. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357: 570–575, 2017. doi: 10.1126/science.aam9949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campbell C, Grapov D, Fiehn O, Chandler CJ, Burnett DJ, Souza EC, Casazza GA, Gustafson MB, Keim NL, Newman JW, Hunter GR, Fernandez JR, Garvey WT, Harper ME, Hoppel CL, Meissen JK, Take K, Adams SH. Improved metabolic health alters host metabolism in parallel with changes in systemic xeno-metabolites of gut origin. PLoS One 9: e84260, 2014. doi: 10.1371/journal.pone.0084260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57: 1470–1481, 2008. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 8.Canty A, Ripley B. boot: Bootstrap R (S-Plus) Functions. R package version 1.3-22. http://CRAN.R-project.org/package=boot, 2019.
- 9.Carmody RN, Gerber GK, Luevano JM Jr, Gatti DM, Somes L, Svenson KL, Turnbaugh PJ. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17: 72–84, 2015. doi: 10.1016/j.chom.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caussy C, Hsu C, Lo MT, Liu A, Bettencourt R, Ajmera VH, Bassirian S, Hooker J, Sy E, Richards L, Schork N, Schnabl B, Brenner DA, Sirlin CB, Chen CH, Loomba R; Genetics of NAFLD in Twins Consortium . Link between gut-microbiome derived metabolite and shared gene-effects with hepatic steatosis and fibrosis in NAFLD. Hepatology 68: 918–932, 2018. doi: 10.1002/hep.29892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chimerel C, Emery E, Summers DK, Keyser U, Gribble FM, Reimann F. Bacterial metabolite indole modulates incretin secretion from intestinal enteroendocrine L cells. Cell Reports 9: 1202–1208, 2014. doi: 10.1016/j.celrep.2014.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cummings BP, Digitale EK, Stanhope KL, Graham JL, Baskin DG, Reed BJ, Sweet IR, Griffen SC, Havel PJ. Development and characterization of a novel rat model of type 2 diabetes mellitus: the UC Davis type 2 diabetes mellitus UCD-T2DM rat. Am J Physiol Regul Integr Comp Physiol 295: R1782–R1793, 2008. doi: 10.1152/ajpregu.90635.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davison A, Hinkley D. Bootstrap Methods and Their Application. Cambridge, UK: Cambridge University Press, 1997. [Google Scholar]
- 13a.Diaz Heijtz R, Wang S, Anuar F, Qian Y, Björkholm B, Samuelsson A, Hibberd ML, Forssberg H, Pettersson S. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci USA 108: 3047–3052, 2011. doi: 10.1073/pnas.1010529108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM, Cani PD. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA 110: 9066–9071, 2013. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farmer EE, Johnson RR, Ryan CA. Regulation of expression of proteinase inhibitor genes by methyl jasmonate and jasmonic acid. Plant Physiol 98: 995–1002, 1992. doi: 10.1104/pp.98.3.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friswell MK, Gika H, Stratford IJ, Theodoridis G, Telfer B, Wilson ID, McBain AJ. Site and strain-specific variation in gut microbiota profiles and metabolism in experimental mice. PLoS One 5: e8584, 2010. doi: 10.1371/journal.pone.0008584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujisaka S, Avila-Pacheco J, Soto M, Kostic A, Dreyfuss JM, Pan H, Ussar S, Altindis E, Li N, Bry L, Clish CB, Kahn CR. Diet, genetics, and the gut microbiome drive dynamic changes in plasma metabolites. Cell Reports 22: 3072–3086, 2018. doi: 10.1016/j.celrep.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gimenez-Ibanez S, Chini A, Solano R. How microbes twist jasmonate signaling around their little fingers. Plants (Basel) 5: 9, 2016. doi: 10.3390/plants5010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gophna U, Konikoff T, Nielsen HB. Oscillospira and related bacteria - From metagenomic species to metabolic features. Environ Microbiol 19: 835–841, 2017. doi: 10.1111/1462-2920.13658. [DOI] [PubMed] [Google Scholar]
- 20.Hamberg M, Gardner HW. Oxylipin pathway to jasmonates: biochemistry and biological significance. Biochim Biophys Acta 1165: 1–18, 1992. doi: 10.1016/0005-2760(92)90069-8. [DOI] [PubMed] [Google Scholar]
- 21.Harrell Jr. FE. Hmisc: Harrell Miscellaneous. R package version 4.2-0. http://CRAN.R-project.org/package=Hmisc, 2019.
- 22.Hastie T, Tibshirani R, Narasimhan B, Chu G. impute: Imputation for Microarray Data. R Package Version 1.58.0, 2019.
- 24.Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, Patterson PH, Mazmanian SK. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155: 1451–1463, 2013. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iino T, Mori K, Tanaka K, Suzuki KI, Harayama S. Oscillibacter valericigenes gen. nov., sp. nov., a valerate-producing anaerobic bacterium isolated from the alimentary canal of a Japanese corbicula clam. Int J Syst Evol Microbiol 57: 1840–1845, 2007. doi: 10.1099/ijs.0.64717-0. [DOI] [PubMed] [Google Scholar]
- 26.Juurlink BH, Azouz HJ, Aldalati AM, AlTinawi BM, Ganguly P. Hydroxybenzoic acid isomers and the cardiovascular system. Nutr J 13: 63, 2014. doi: 10.1186/1475-2891-13-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kieffer DA, Piccolo BD, Marco ML, Kim EB, Goodson ML, Keenan MJ, Dunn TN, Knudsen KE, Martin RJ, Adams SH. Mice fed a high-fat diet supplemented with resistant starch display marked shifts in the liver metabolome concurrent with altered gut bacteria. J Nutr 146: 2476–2490, 2016. doi: 10.3945/jn.116.238931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kieffer DA, Piccolo BD, Vaziri ND, Liu S, Lau WL, Khazaeli M, Nazertehrani S, Moore ME, Marco ML, Martin RJ, Adams SH. Resistant starch alters gut microbiome and metabolomic profiles concurrent with amelioration of chronic kidney disease in rats. Am J Physiol Renal Physiol 310: F857–F871, 2016. doi: 10.1152/ajprenal.00513.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim M, Qie Y, Park J, Kim CH. Gut microbial metabolites fuel host antibody responses. Cell Host Microbe 20: 202–214, 2016. doi: 10.1016/j.chom.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleinert M, Clemmensen C, Hofmann SM, Moore MC, Renner S, Woods SC, Huypens P, Beckers J, de Angelis MH, Schürmann A, Bakhti M, Klingenspor M, Heiman M, Cherrington AD, Ristow M, Lickert H, Wolf E, Havel PJ, Müller TD, Tschöp MH. Animal models of obesity and diabetes mellitus. Nat Rev Endocrinol 14: 140–162, 2018. doi: 10.1038/nrendo.2017.161. [DOI] [PubMed] [Google Scholar]
- 31.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19: 576–585, 2013. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koh A, Molinaro A, Ståhlman M, Khan MT, Schmidt C, Mannerås-Holm L, Wu H, Carreras A, Jeong H, Olofsson LE, Bergh PO, Gerdes V, Hartstra A, de Brauw M, Perkins R, Nieuwdorp M, Bergström G, Bäckhed F. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell 175: 947–961.e17, 2018. doi: 10.1016/j.cell.2018.09.055. [DOI] [PubMed] [Google Scholar]
- 33.Komsta L. Outliers: Tests for outliers. R Package Version 0.14. http://CRAN.R-project.org/package=outliers, 2011.
- 34.Kovacs A, Ben-Jacob N, Tayem H, Halperin E, Iraqi FA, Gophna U. Genotype is a stronger determinant than sex of the mouse gut microbiota. Microb Ecol 61: 423–428, 2011. doi: 10.1007/s00248-010-9787-2. [DOI] [PubMed] [Google Scholar]
- 35.Kuhn M. caret: Classification and Regression Training. R package version 6.0-84. https://CRAN.R-project.org/package=caret, 2019.
- 36.Lee GH, Rhee MS, Chang DH, Lee J, Kim S, Yoon MH, Kim BC. Oscillibacter ruminantium sp. nov., isolated from the rumen of Korean native cattle. Int J Syst Evol Microbiol 63: 1942–1946, 2013. doi: 10.1099/ijs.0.041749-0. [DOI] [PubMed] [Google Scholar]
- 37.Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, David E, Savidor A, Korem T, Herzig Y, Pevsner-Fischer M, Shapiro H, Christ A, Harmelin A, Halpern Z, Latz E, Flavell RA, Amit I, Segal E, Elinav E. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163: 1428–1443, 2015. doi: 10.1016/j.cell.2015.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8: e61217, 2013. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mehmood T, Liland KH, Snipen L, Saebo S. A review of variable selection methods in Partial Least Squares Regression. Chemom Intel Lab 118: 62–69, 2012. doi: 10.1016/j.chemolab.2012.07.010. [DOI] [Google Scholar]
- 40.Mevik BH, Wehrens R. The pls package: principal component and partial least squares regression in R. J Stat Softw 18: 1–23, 2007. doi: 10.18637/jss.v018.i02. [DOI] [Google Scholar]
- 41.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MH, Szoecs E, Wagner H. . vegan: Community Ecology Package R package version 2.5-6. https://cran.r-project.org/package=vegan, 2019.
- 42.Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, Pan C, Civelek M, Rau CD, Bennett BJ, Mehrabian M, Ursell LK, He A, Castellani LW, Zinker B, Kirby M, Drake TA, Drevon CA, Knight R, Gargalovic P, Kirchgessner T, Eskin E, Lusis AJ. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab 17: 141–152, 2013. doi: 10.1016/j.cmet.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BAH, Forslund K, Hildebrand F, Prifti E, Falony G, Le Chatelier E, Levenez F, Doré J, Mattila I, Plichta DR, Pöhö P, Hellgren LI, Arumugam M, Sunagawa S, Vieira-Silva S, Jørgensen T, Holm JB, Trošt K, Kristiansen K, Brix S, Raes J, Wang J, Hansen T, Bork P, Brunak S, Oresic M, Ehrlich SD, Pedersen O; MetaHIT Consortium . Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 535: 376–381, 2016. doi: 10.1038/nature18646. [DOI] [PubMed] [Google Scholar]
- 44.Piccolo BD, Graham JL, Stanhope KL, Fiehn O, Havel PJ, Adams SH. Plasma amino acid and metabolite signatures tracking diabetes progression in the UCD-T2DM rat model. Am J Physiol Endocrinol Metab 310: E958–E969, 2016. doi: 10.1152/ajpendo.00052.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Piccolo BD, Graham JL, Stanhope KL, Nookaew I, Mercer KE, Chintapalli SV, Wankhade UD, Shankar K, Havel PJ, Adams SH. Diabetes-associated alterations in the cecal microbiome and metabolome are independent of diet or environment in the UC Davis Type 2 Diabetes Mellitus Rat model. Am J Physiol Endocrinol Metab 315: E961–E972, 2018. doi: 10.1152/ajpendo.00203.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piccolo BD, Mercer KE, Bhattacharyya S, Bowlin AK, Saraf MK, Pack L, Chintapalli SV, Shankar K, Adams SH, Badger TM, Yeruva L. Early postnatal diets affect the bioregional small intestine microbiome and ileal metabolome in neonatal pigs. J Nutr 147: 1499–1509, 2017. doi: 10.3945/jn.117.252767. [DOI] [PubMed] [Google Scholar]
- 47.Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341: 1241214, 2013. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roager HM, Licht TR. Microbial tryptophan catabolites in health and disease. Nat Commun 9: 3294, 2018. doi: 10.1038/s41467-018-05470-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, Chao CC, Patel B, Yan R, Blain M, Alvarez JI, Kébir H, Anandasabapathy N, Izquierdo G, Jung S, Obholzer N, Pochet N, Clish CB, Prinz M, Prat A, Antel J, Quintana FJ. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med 22: 586–597, 2016. doi: 10.1038/nm.4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17: 225–235, 2013. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 51.Senthong V, Li XS, Hudec T, Coughlin J, Wu Y, Levison B, Wang Z, Hazen SL, Tang WH. Plasma trimethylamine N-oxide, a gut microbe-generated phosphatidylcholine metabolite, is associated with atherosclerotic burden. J Am Coll Cardiol 67: 2620–2628, 2016. doi: 10.1016/j.jacc.2016.03.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027–1031, 2006. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 53.Ussar S, Griffin NW, Bezy O, Fujisaka S, Vienberg S, Softic S, Deng L, Bry L, Gordon JI, Kahn CR. Interactions between gut microbiota, host genetics and diet modulate the predisposition to obesity and metabolic syndrome. Cell Metab 22: 516–530, 2015. doi: 10.1016/j.cmet.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, Speelman P, Dijkgraaf MG, Keller JJ. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med 368: 407–415, 2013. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- 55.Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, Kolde R, Vlamakis H, Arthur TD, Hämäläinen A-M, Peet A, Tillmann V, Uibo R, Mokurov S, Dorshakova N, Ilonen J, Virtanen SM, Szabo SJ, Porter JA, Lähdesmäki H, Huttenhower C, Gevers D, Cullen TW, Knip M, Xavier RJ; DIABIMMUNE Study Group . Variation in microbiome lps immunogenicity contributes to autoimmunity in humans. Cell 165: 842–853, 2016. doi: 10.1016/j.cell.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walker AW, Duncan SH, McWilliam Leitch EC, Child MW, Flint HJ. pH and peptide supply can radically alter bacterial populations and short-chain fatty acid ratios within microbial communities from the human colon. Appl Environ Microbiol 71: 3692–3700, 2005. doi: 10.1128/AEM.71.7.3692-3700.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, Porto C, Bouslimani A, Melnik AV, Meehan MJ, Liu W-T, Crüsemann M, Boudreau PD, Esquenazi E, Sandoval-Calderón M, Kersten RD, Pace LA, Quinn RA, Duncan KR, Hsu CC, Floros DJ, Gavilan RG, Kleigrewe K, Northen T, Dutton RJ, Parrot D, Carlson EE, Aigle B, Michelsen CF, Jelsbak L, Sohlenkamp C, Pevzner P, Edlund A, McLean J, Piel J, Murphy BT, Gerwick L, Liaw CC, Yang YL, Humpf HU, Maansson M, Keyzers RA, Sims AC, Johnson AR, Sidebottom AM, Sedio BE, Klitgaard A, Larson CB, Boya P CA, Torres-Mendoza D, Gonzalez DJ, Silva DB, Marques LM, Demarque DP, Pociute E, O’Neill EC, Briand E, Helfrich EJ, Granatosky EA, Glukhov E, Ryffel F, Houson H, Mohimani H, Kharbush JJ, Zeng Y, Vorholt JA, Kurita KL, Charusanti P, McPhail KL, Nielsen KF, Vuong L, Elfeki M, Traxler MF, Engene N, Koyama N, Vining OB, Baric R, Silva RR, Mascuch SJ, Tomasi S, Jenkins S, Macherla V, Hoffman T, Agarwal V, Williams PG, Dai J, Neupane R, Gurr J, Rodríguez AM, Lamsa A, Zhang C, Dorrestein K, Duggan BM, Almaliti J, Allard PM, Phapale P, Nothias LF, Alexandrov T, Litaudon M, Wolfender JL, Kyle JE, Metz TO, Peryea T, Nguyen DT, VanLeer D, Shinn P, Jadhav A, Müller R, Waters KM, Shi W, Liu X, Zhang L, Knight R, Jensen PR, Palsson BØ, Pogliano K, Linington RG, Gutiérrez M, Lopes NP, Gerwick WH, Moore BS, Dorrestein PC, Bandeira N. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat Biotechnol 34: 828–837, 2016. doi: 10.1038/nbt.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ward JB, Lajczak NK, Kelly OB, O’Dwyer AM, Giddam AK, Ní Gabhann J, Franco P, Tambuwala MM, Jefferies CA, Keely S, Roda A, Keely SJ. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am J Physiol Gastrointest Liver Physiol 312: G550–G558, 2017. doi: 10.1152/ajpgi.00256.2016. [DOI] [PubMed] [Google Scholar]
- 59.Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci USA 106: 3698–3703, 2009. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu IW, Hsu KH, Lee CC, Sun CY, Hsu HJ, Tsai CJ, Tzen CY, Wang YC, Lin CY, Wu MS. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol Dial Transplant 26: 938–947, 2011. doi: 10.1093/ndt/gfq580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Honda K, Ishikawa Y, Hara E, Ohtani N. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499: 97–101, 2013. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]