

Keywords: CFTR, glucocorticoids, PDK1, PI3K, SGK1
Abstract
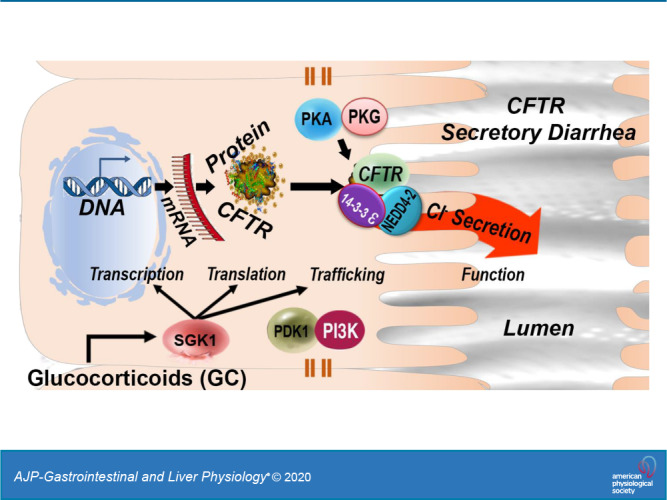
Nongenomic glucocorticoid (GC) and serum- and glucocorticoid-inducible kinase 1 (SGK1) signaling regulate ion transport, but CFTR has not been investigated in the intestine. We examined GC, SGK1, and phosphatidylinositol 3-kinase (PI3K) kinase signaling of CFTR ion transport in native intestine and the role of GCs on mRNA, protein, surface expression, and cyclic guanosine monophosphate (cGMP)-elicited diarrhea. Rats were treated with dexamethasone (DEXA; 2 mg/kg ip) or DMSO for 1, 4, and 24 h. Cyclic adenosine monophosphate (cAMP)-activated ion transport was examined in the presence or absence of SGK1 and PI3K inhibitors. Phosphorylation of SGK1, phosphoinositide-dependent kinase 1, and Akt kinases was confirmed by immunoblots using phosphor-specific antibodies. Tissue lysates were analyzed by mass spectrometry. CFTR and SGK1 mRNA were measured by quantitative PCR. Changes in total and surface CFTR protein were determined. The role of GC in cGMP-activated CFTR ion transport was examined. GC synergistically increased CFTR ion transport by SGK1 and PI3K signaling and increased CFTR protein without altering SGK1 or CFTR mRNA. GC induced highest levels of CFTR protein at 4 h that were associated with marked increase in surface CFTR, phosphorylation of the ubiquitin ligase neural precursor cell expressed developmentally downregulated 4-like (Nedd4-2), and 14-3-3ε, supporting their roles in surface retention and stability. Coimmunoprecipitation of CFTR, Nedd4-2, and 14-3-3ε indicated that assembly of this complex is a likely effector of the SGK and Akt pathways. Mass spectrometry identified phosphorylated peptides in relevant proteins. GC-SGK1 potently regulates CFTR in the intestine and is implicated in diarrheal disease.
NEW & NOTEWORTHY This is the first study to examine the mechanisms of glucocorticoid, serum- and glucocorticoid-inducible kinase 1, and nongenomic kinase signaling of CFTR in the native intestine. We identified unique and druggable intestine-specific factors of the pathway that are targets for treating stress-induced diarrhea.
INTRODUCTION
Cystic fibrosis transmembrane conductance regulator (CFTR) channels are present on the apical membrane of epithelia, where they play a critical role in fluid transport. CFTR regulation is cell- and tissue-specific (3, 23, 24, 32, 44). cAMP-dependent phosphorylation regulates CFTR function in most epithelial cells. In the intestine, however, CFTR is regulated by cAMP/protein kinase A (PKA)- and cGMP/PKG-dependent phosphorylation and traffics into and out of the apical membrane of enterocytes, mechanisms that are critical in acute secretory diarrhea (4, 20).
Absence of functional CFTR in the genetic disease cystic fibrosis (CF) leads to obstructive lung disease and intestinal obstruction (meconium ileus) in newborns (7). The pathogenesis of CF lung disease in newborns is linked to high levels of maternal circulating glucocorticoids (GCs) at the time of birth that stimulate rapid nongenomic kinase signaling of the epithelial Na+ channel (ENaC) and CFTR in the newborn (32, 47). GCs, stress-induced steroids produced by the adrenal cortex, exert their actions through genomic and nongenomic mechanisms (35). The more widely understood genomic effects are observed after 24 h to days (34). Genomic regulation is mediated by cytosolic glucocorticoid receptors (cGCRs) that undergo nuclear translocation for activation or transrepression of genes through binding of the cGCR complex to transcription factors, resulting in protein expression (29, 35). In contrast, nongenomic regulation involves rapid cellular changes resulting from specific interactions with membrane-associated proteins and receptors and the release of proteins from the cGCR complex (16, 46) that does not involve modulation of gene expression.
Nongenomic actions of GCs are mediated by serum- and glucocorticoid-inducible kinase 1 (SGK1). SGK1, a serine and threonine kinase, cloned as an immediate early gene, regulates many functions, including apoptosis, cell volume, electrolyte transport, and ion channel activation (50, 51). SGK1 is activated by serum, glucocorticoids, cytokines, and phosphorylation through a phosphatidylinositol 3-kinase (PI3K)- and phosphoinositide-dependent kinase 1 (PDK1)-dependent mechanism (29, 50, 51). SGK1, in turn, mediates the stimulating effects on several transport systems by multiple mechanisms, including kinase signaling, transcription, translation, membrane traffic, and membrane protein retention by ubiquitin ligases (6, 22, 26, 30, 31, 48, 49).
GC-activated nongenomic pathways regulate ion channels and contribute to diseases (5, 17, 47, 53). CFTR is regulated by GC-activated kinase signaling in airway and pancreatic cells, but data on GC-activated kinase regulation of CFTR in the intestine have not been reported (6, 8, 10, 32). However, this pathway is an important regulator of sodium/hydrogen exchanger 3 (NHE3), the major ion transport counterpart to CFTR in the intestine that regulates Na+ absorption (48, 49).
We hypothesized that GC and SGK1 pathways regulate CFTR in the intestine with features distinct from its regulation in other epithelia. Elucidation of this pathway could provide a unique opportunity to identify druggable kinases for treating CFTR-mediated diseases in the intestine. This study utilized native rat intestine (jejunum) to examine physiological mechanisms employed by GC-SGK1 and PI3K to regulate CFTR and identify tissue-specific factors key to CFTR regulation. CFTR expression, localization, membrane traffic, and function were examined in tissues. The relevance of this pathway to cGMP-PKG-elicited diarrhea was examined.
MATERIALS AND METHODS
Animals and treatments.
Sprague-Dawley male rats (6–8 wk) were purchased from Charles River and housed and fed in the Yale Animal Research Facility. Rats were fasted overnight (O/N) before intraperitoneal (IP) administration of vehicle control dimethyl sulfoxide (DMSO; equivalent to 2 mg/kg) or dexamethasone (DEXA; 2 mg/kg) for 1, 4, or 24 h. Animals were anesthetized using Inactin (thiobutabarbital) hydrate (120 mg/kg ip), and intestinal tissues (jejunum) were harvested following laparotomy. Mucosal scrapings were collected for immunoblot analysis, intestinal segments prepared for immunofluorescence and short-circuit current measurements, and intestinal loops secured and prepared for surface biotinylation. Yale University’s Institutional Animal Care and Use Committee approved all animal protocols.
Antibodies and reagents.
The reagents dimethyl sulfoxide (DMSO; D4540), Inactin hydrate (T-133), forskolin (F6886), CFTR-inh172 (C2992), and agarose (A9539) were purchased from Sigma-Aldrich. DEXA (cat. no. 1126), 8-bromoguanosine 3′,5′-cyclic monophosphate (8-Br-cGMP), sodium salt (cat. no. 1089), and phenylmethylsulfonylfluoride (PMSF) were purchased from Tocris Bioscience. Protease inhibitors used for lysate preparation included PhosSTOP EASYpack (cat. no. 04906837001) and cOmplete EDTA-free (cat. no. 11873580001), purchased from Roche. Antibodies against phosphorylated (phospho-) NEDD4LSer342 (phospho-Nedd4-2; cat. no. 12146), NEDD4L (Nedd4-2; cat. no. 4103), phospho-n-myc downregulated gene 1 (NDRG1)Thr346 (cat. no. 5482), NDRG1 (cat. no. 5196), phospho-AktSer473 (cat. no. 4060), Akt (cat. no. 9272), phospho-PDK1Thr346 (cat. no. 3061), PDK1 (cat. no. 3062), and GAPDH (cat. no. 8884) and Protein A Agarose for immunocoprecipitation were purchased from Cell Signaling Technology. Antibodies against 14-3-3 (pan; cat. no. 1657), 14-3-3ε (cat. no. 393177), and SGK1 inhibitor (inh) GSK 650394 (cat. no. 361201) were purchased from Santa Cruz Biotechnology. Additional antibodies were CFTR (AME-4991, affinity-purified polyclonal antibody raised against rat CFTR; produced by N. A. Ameen), anti-SGK1 (cat. no. 07-315; Millipore), phospho-SGK1Thr256 (cat. no. 44-1260G; Invitrogen), NHE3-3H3 (cat. no. MABN1813; Millipore), and phospho-PIK3R1/PIK3R3 (Tyr458, Tyr199; cat. no. PA5-17387; Invitrogen). Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from BD Pharmingen (cat. no. 554021, HRP goat anti-rabbit Ig; and cat. no. 554002; HRP-labeled polyclonal anti-mouse Ig). Solutions used for short-circuit current (Isc) studies included LY 294002-PI3K inh (cat. no. 440202; Millipore), Ringer solution (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM glucose, and 10 mM HEPES, pH 7.4), and 3 M KCl (cat. no. 3040-19; J. T. Baker).
Tissue preparation and immunofluorescence labeling.
Tissues from DEXA- or DMSO-treated animals were removed, cut into 2-mm-thick segments, and fixed in 2% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 45 min at room temperature (RT shaking), changed to 0.2% PFA for 1 h at RT, and rinsed three times with PBS. Intestinal segments were then cryoprotected in 20% sucrose O/N at 4°C, shaking as previously described (15). Tissue segments were embedded in molds containing optimal cutting temperature (cat. no. 4583; Tissue-Tek) compound and then frozen by immersing molds in liquid nitrogen-cooled isopentane. Tissue blocks were stored at −20°C and sectioned (5 μm) onto polylysine-coated slides before immunolabeling as previously described (15, 24).
All immunolabeling steps were carried out in a humidified chamber at RT as previously described (24), except for O/N incubation with primary antibodies, which was carried out at 4°C. Mounted tissue cryosections (5 µm) were rehydrated with warm PBS and then treated with 1% Na borohydride for 10 min. Sections were washed with each solution: PBS, PBS-0.15% glycine (solution A), and PBS-1% BSA-0.15% glycine (solution B). Subsequent steps were carried out in solution B. To reduce nonspecific binding, sections were blocked for 1 h at RT with normal goat serum (1:20) diluted in solution B and then washed. Cryosections were incubated with primary antibodies diluted in solution B plus 0.2% Triton X-100 O/N at 4°C. Negative control sections were labeled in the absence of primary antibodies. Sections were washed and then incubated for 1 h with Alexa Fluor 488 (green)-conjugated secondary antibodies (mouse or rabbit) diluted 1:200 in solution B. Tissue sections were washed, treated with DAPI for 20 s, and then washed with PBS. Tissue sections were coverslipped with ProLong Diamond Antifade Mountant media (cat. no. P36961; Invitrogen) and allowed to dry O/N at 4°C.
For heat-stable enterotoxin (STa) studies, O/N-fasted rats (250–300 g) were anesthetized, intestinal loops (jejunum, 5 cm) were isolated, ligatures were placed, and loops were injected with either PBS alone or Escherichia coli STa toxin (0.5 µM) prepared in PBS (500 µL) for 30 min as previously described (20). Tissues were prepared for immunolocalization as described above.
Fluorescence microscopy.
Immunolabeled sections were examined on a Zeiss Axio Observer epifluorescence inverted microscope equipped with a Hamamatsu ORCA-R2 C10600 digital camera. The acquisition parameters were standardized in relation to the highest-intensity regions to avoid oversaturation of pixel intensity. Digital images (8 bits/channel; 1,344 × 1,024 pixels) were taken at ×10, ×40, and ×63 magnifications and at the same exposure time. Images were captured using Zeiss Efficient Navigation (ZEN) 2.0 software. Scale bars were added to each image based on the objective at which the image was taken.
Western blot and densitometry analysis.
Mucosal scrapings from rat jejunum were homogenized in TGH lysis buffer [25 mM HEPES, 10% (vol/vol) glycerol, 1% (vol/vol) Triton X-100, pH 7.4] containing a complete EDTA-free protease inhibitor cocktail (1×), 1 mM PMSF, and PhosSTOP EASYpack cocktail (1×) for 30 min on ice. Homogenates were solubilized, subject to centrifugation at 13,200 g for 15 min at 4°C, and cleared supernatants were recovered. Protein concentrations were determined with Coomassie Plus (Bradford) Assay reagent (cat. no. 23236; Thermo Fisher Scientific), and 2 sets of samples (1 set was warmed at 37°C and used to detect CFTR, and another set was boiled and used to detect kinases) were prepared and analyzed as previously described (1). Densitometry was performed on scanned immunoblot images using the ImageJ gel analysis tool. Experimental band intensity for each sample was standardized as a ratio of the net band value over GAPDH, the net loading control, for each lane. At least 3 independent Western blots per antibody were used for densitometry analysis.
Surface biotinylation.
Surface biotinylation was performed in the rat intestine (jejunum) as previously described (1). In brief, immediately after completion of the time interval of treatment with or without DEXA (2 mg/kg body wt), rats were injected with the anesthetic reagent Inactin. On confirmation of deep anesthesia, the abdomen was opened, and tissues were collected in a petri dish containing ice-cold PBS. The intestinal lumen was flushed cleaned several times with ice-cold PBS. A small portion of intestine was used to prepare nonbiotinylated control samples, and the remaining tissue was used to prepare loops for surface biotinylation. Intestinal loops were incubated with freshly prepared Sulfo-NHS-LC-Biotin (1 mg/mL; cat. no. PG82075; Thermo Fisher Scientific) in ice-cold PBS conditioned media (pH 8.0) for 30 min (rotating) in the cold room and processed as previously described (1, 20). Biotinylated proteins were dissociated from streptavidin agarose by 2× SDS sample buffer. Total lysates (30 µg) and biotinylated samples were separated by SDS-PAGE to detect CFTR and NHE3 by Western blot analysis using the rat-specific CFTR AME-4991 rabbit polyclonal antibody and mouse monoclonal NHE3-3H3 antibody, respectively.
Coimmunoprecipitation.
Following treatment with or without DEXA, rat intestines (jejunum) were collected in a petri dish containing ice-cold PBS, and the lumen was washed extensively with PBS. Mucosal scrapings were lysed in TGH lysis buffer containing a complete EDTA-free protease inhibitor cocktail (1×), 1 mM PMSF, and PhosSTOP EASYpack cocktail (1×) for 30 min on ice. Homogenates were solubilized, subject to centrifugation at 13,200 g for 15 min at 4°C, and cleared supernatants were recovered. Immunoprecipitation of CFTR-NEDD4-2-14-3-3ε complexes was then performed in 1.5 mg of total protein with phospho-NEDD4-2Ser342 rabbit monoclonal antibody or with an isotype-matched rabbit IgG control O/N at 4°C. Protein was then prewashed for 1 h with Protein A-Sepharose beads. Immunoprecipitates, including 30 µg of input protein, were electrophoresed on SDS-PAGE under reducing conditions and blotted onto membranes. Immunoblots were analyzed using CFTR 217 mouse monoclonal antibody (MAb) and 14-3-3ε (F-3) mouse MAb to detect CFTR and 14-3-3ε, respectively.
Semiquantitative RT-PCR and quantitative RT-PCR.
To analyze mRNA expression in jejunal tissues, total RNA was isolated from mucosal scrapings of rat intestines following treatment with or without DEXA (2 mg/kg body wt) using TRIzol reagent (cat. no. T9424; Invitrogen). Three micrograms of mRNA was used to synthesize cDNA using the SuperScript First-Strand Synthesis System (cat. no. 11904018; Invitrogen) with oligo(dT)12–18 according to manufacturer’s protocol. The cDNA was amplified by normal PCR using a Taq DNA polymerase (cat. no. 201203; QIAGEN) for the semiquantitative reverse transcription-polymerase chain reaction (RT-PCR) analysis with a standard protocol where PCR band intensities were not saturated. PCR was performed under the following conditions: 31 cycles for NHE3 (denaturing at 94°C for 15 s, annealing at 55°C for 15 s, and extension at 72°C for 20 s) and 20 cycles for GAPDH (denaturing at 94°C for 20 s, annealing at 58°C for 30 s, and extension at 72°C for 30 s). The PCR products were visualized by electrophoresis on 2% agarose gel (Sigma-Aldrich) containing 1 µg/mL ethidium bromide (cat. no. 1610433EDU; Bio-Rad). The mRNA expression levels of CFTR and SGK1 were also analyzed by quantitative (q) RT-PCR using Power SYBR Green PCR Master Mix (cat. no. 4344463; Applied Biosystems) with GAPDH as a loading control in an Applied Bioscience qPCR machine. The test was repeated three times using independent samples. Data were displayed as means ± standard deviation (SD), and data analysis was employed by 2-ΔΔCt comparative cycle threshold method. The primers used for the amplification are listed in Table 1.
Table 1.
Primers used for QRT-PCR and semi-QRT-PCR to amplify CFTR, SGK1, NHE3, and GAPDH
| Gene | Forward | Reverse | Product, bp |
|---|---|---|---|
| CFTR | 5′-CTGGACCACACCAATTTTGAGA-3′ | 5′-GCGTGGATAAGCTGGGGCT-3′ | 162 |
| SGK1 | 5′-CAAGGAGAACATCGAGCACA-3′ | 5′-TTTCAGCTGGAGAGGCTTGT-3′ | 220 |
| NHE3 | 5′-GTCACCCAGGATGTAGCCTCTG-3′ | 5′-GGTGGCACCCTGGATAGGAT-3′ | 101 |
| GAPDH | 5′-GATTTGGCCGTATCGGAC-3′ | 5′-GAAGACGCCAGTAGACTC-3′ | 278 |
bp, Base pairs; NHE3, sodium/hydrogen exchanger 3; QRT-PCR, quantitative reverse transcription-polymerase chain reaction; semi-QRT-PCR, semiquantitative reverse transcription-polymerase chain reaction; SGK1, serum- and glucocorticoid-inducible kinase 1.
Short-circuit current measurements.
Following treatment with or without DEXA, rat intestines (jejunum) were removed, and Cl− transport was determined by measuring short-circuit currents in Ussing chambers (Physiologic Instruments, San Diego, CA) as before (28). Ringer solution along with 100% O2 was used to bathe and oxygenate both sides of mounted tissues, respectively. To eliminate electrical bias, a series of steps were taken to set electrical measurements to baseline in the absence of intestinal tissue. These included 1) electrode voltage offset potential correction, 2) fluid resistance compensation, and 3) liquid resistance correction. Intestinal tissues were mounted on slides with an exposed area of 0.5 cm2 and placed in the chambers. Ringer solution, which was circulated with 100% O2, was maintained at 37°C with external water bath. Transepithelial Isc was measured via voltage-clamp. After 10-min recording of baseline current, tissues were pretreated on the basolateral side with kinase inhibitors GSK 650394-SGK1 (10 µM) or LY 294002-PI3K (10 µM) for 30 min. Forskolin (10 µM) was added to the basolateral side of each chamber. To determine the role of SGK1/PI3K in cGMP-activated CFTR ion transport, tissues were preincubated with the SGK1 or PI3K inhibitors (10 μM) for 30 min, and then 8-Br-cGMP sodium salt (1 mM) was added to the basolateral side of each chamber. Transepithelial current tracings were plotted, and the change in short-circuit current (ΔIsc) was calculated [ΔIsc = −(Iscmax − Iscmin)] and analyzed for significance using GraphPad Prism 7.
Phosphosite functional annotation.
Phosphopeptides passing the 95% confidence cutoff (42) and found in at least three independent observations were scanned for the presence of biologically relevant phosphosites by using PhosphoSitePlus (https://www.phosphosite.org/), an open, comprehensive, manually curated posttranslational modifications database (21). Ingenuity Pathway Analysis (IPA) software (QIAGEN; https://www.ingenuity.com/) was used to find association between the experimental data and significant biological functions. The IPA knowledge base, derived from literature, computed a score based on one-tailed Fisher exact test. Final significance threshold was set to P < 0.01. Identified phosphorylated sites in three independent pairs of samples are listed in Table 2.
Table 2.
Identified phosphorylated sites from ≥3 independent observations
| Swiss-Prot Acc. | Gene Name | Identified Phosphorylated Sites | Mascot Expectation Value‡ | Mass Error, ppm |
|---|---|---|---|---|
| 1433E_RAT | Ywhae | AAFDDAIAELDTLSEESYK | 1.90e−07 | 0.086 |
| 1433Z_RAT | Ywhaz | TAFDEAIAELDTLSEESYK | 3.30e−07 | 0.75 |
| KAP2_RAT | Prkar2a | RVSVCAETFNPDEEEDNDPR | 1.20e−06 | 0.42 |
| NDRG1_RAT* | Ndrg1 | SRTASGSSVTSLEGTR | 4.30e−06 | 0.82 |
| PDPK1_RAT | Pdpk1 | ANSFVGTAQYVSPELLTEK | 8.70e−10 | 1.77 |
| SL9A3_RAT* | Slc9a3 | RGSLAFIR | 5.50e−03 | 0.18 |
| PI3R4_RAT | Pik3r4 | SESSAGVCVPLSTSPQVSEAAHIPSK† | 2.90e−02 | 0.0081 |
Phosphorylated amino acids S and T are underlined and bold-highlighted.
Note there are other phosphorylated sites identified (see the supplemental file).
Uncertain of which Ser or Thr is phosphorylated, but 1 (and only 1) of the bold is phosphorylated.
Statistical analysis.
To compare the expression and phosphorylation status of NDRG1/phosphorylated (p) NDRG1, SGK1/pSGK1, NEDD4L/pNEDD4L (Nedd4-2), and CFTR in tissues from DMSO- versus DEXA-treated animals at 1, 4, and 24 h, data were analyzed using Student’s t test with a significance level of P < 0.05. To determine the change in Isc in tissues from DMSO/DEXA-treated animals treated with/without kinase inhibitors within each group (1, 4, and 24 h), data were analyzed using pairwise Student’s t test with a significance level of P < 0.025. To determine the change in Isc between DMSO and DEXA within each group (1, 4, and 24 h), data were analyzed using unpaired Student’s t test with a significance level of P < 0.05. Transformed data were used for statistical analysis and raw data for the graphical presentation. Each specified hypothesis of interest was stated a priori to running analysis.
RESULTS
GC, SGK1, and PI3K regulate CFTR ion transport in rat jejunum.
PI3K has been shown to participate at an early step in the pathway, whereas SGK1 exerts its action downstream to activate ion transporters. To determine the roles of SGK1, PI3K, and GC in CFTR ion transport, specific inhibitors of SGK1 and PI3K were used to examine their effects on forskolin (fsk)-stimulated CFTR currents (Isc) using Ussing chambers. Following treatment of rats for 1 or 4 h with DEXA (2 mg/kg ip) or DMSO, jejunum was removed, baseline Isc was recorded, tissues were pretreated with the SGK1 inhibitor (GSK 650394; 10 µM), PI3K inhibitor (LY-294002; 10 µM), or diluent for 30 min in the chamber, and CFTR Isc was activated with the cAMP agonist fsk. Activation of kinases by phosphorylation was confirmed by immunoblot analysis of tissue lysates and phosphor-specific antibodies where possible. Increased phosphorylation of NDRG1, a specific substrate of SGK1, is confirmatory for increased SGK1 activity and was used to confirm changes in SGK1 phosphorylation in jejunal tissue lysates (8, 22, 40).
DEXA treatment alone significantly increased baseline Isc at 1, 4, and 24 h by 30–60 μA that was highest at 4 h as shown in Fig. 1A. Fsk-activated CFTR Isc trended higher in tissues from DEXA-treated rats after 1 or 4 h, consistent with a synergistic effect, but the difference was not statistically significant, likely because of elevated DEXA-stimulated baseline Isc. Importantly, both SGK1 and PI3K inhibitors significantly decreased the fsk-stimulated Isc at 1 and 4 h in tissues from DEXA-treated rats (Fig. 1, B and C), indicating that both kinases play a role in CFTR function regulation during DEXA treatment. The PI3K inhibitor also reduced the fsk-stimulated Isc in DMSO-treated rats, consistent with the presence of endogenous circulating cortisol. Immunoblot analysis of jejunum lysates from rats treated with DEXA for 1 h (Fig. 1D) revealed a small but not statistically significant increase in NDRG1 phosphorylation at Thr346, the specific site that is phosphorylated by SGK1 but not by other closely related kinases (22, 40). Analysis of immunoblots for phosphorylated SGK1 at Ser256, using a phospho-SGK1 antibody, did not reveal increased phosphorylation after 1 h with DEXA treatment (Fig. 1, D and F). However, immunoblot analysis of jejunum lysates with antibodies against SGK1Ser256 and NDRG1Thr346 revealed statistically significant increase in phosphorylation of DEXA-treated tissues at 4 h, whereas total SGK1 and NDRG1 levels were unchanged (Fig. 1, E, G, and I).
Fig. 1.

Dexamethasone (DEXA) synergistically increases baseline and forskolin (FSK)-activated CFTR current in rat jejunum and is reduced by inhibition of serum- and glucocorticoid-inducible kinase 1 (SGK1) and phosphatidylinositol 3-kinase (PI3K). Rats were treated with DEXA (2 mg/kg ip) or DMSO, and intestines were harvested after 1, 4, and 24 h. Tissues were mounted in Ussing chambers, and baseline and FSK-activated short-circuit currents (Isc) were recorded in the presence and absence of SGK1 inhibitor GSK 650394 or PI3K inhibitor (inh) LY 294002 as described in materials and methods. A: basal current (Isc) in DEXA/DMSO-treated jejunum at 1, 4, and 24 h. B: changes in FSK-stimulated CFTR ion transport (Isc) in DEXA/DMSO-treated jejunum at 1 h. C: changes in FSK-stimulated CFTR ion transport (Isc) in DEXA/DMSO-treated jejunum at 4 h. N = 6 pairs of rats for each time point. D and E: tissue lysates were prepared from jejunum of rats treated with DEXA/DMSO for 1 or 4 h, and immunoblots were analyzed using phosphor-specific antibodies to detect changes in total and phosphorylated (p) SGK1 (Ser256) and total and phosphorylated n-myc downregulated gene 1 (NDRG1; Thr346) relative to GAPDH. N = 3 pairs of rats for each time point examined. F and G: Western blot band intensities of total and phosphorylated SGK1 (Ser256) normalized by GAPDH in the 1- and 4-h samples, respectively. H and I: Western blot band intensities of total and phosphorylated NDRG1 (Thr346) normalized against GAPDH in the 1- and 4-h samples, respectively. Values are means ± SE. *P < 0.05, **P < 0.01, and ***P < 0.001.
Changes in phosphorylation of PDK1 and Akt in rat jejunum by immunoblot.
GC-stimulated PI3K activates Akt independently of PDK1 via mechanistic target of rapamycin complex 2 through phosphorylation in Akt Ser473 (36). Rat mucosa stimulated with DEXA showed a 1.8-fold statistically significant increase in PDK1 pSer241 (Fig. 2, A and D), at 1 h, an autophosphorylation reporter of PDK1 activity (41). pSer473 Akt, on the other hand, increased substantially 24 h after DEXA treatment (Fig. 2C), supporting a late engagement of the PI3K/Akt pathway.
Fig. 2.

Immunoblot analysis of jejunum lysates from rats treated with DMSO/dexamethasone (DEXA) for 1, 4, or 24 h detects total and phosphorylated (p) phosphoinositide-dependent kinase 1 (PDK1; Ser241) and Akt (Ser473). Kinase activation was determined by changes in phosphorylation. Shown are immunoblots of total and phosphorylated PDK1 and Akt relative to GAPDH at 1 h (A), 4 h (B), and 24 h (C). D: Western blot band intensities of phosphorylated PDK1 (Ser241) normalized by GAPDH in the 1-, 4-, and 24-h samples. N = 3 pairs of rats for each time point examined. Values are means ± SE. *P < 0.05.
GC treatment does not alter transcription of CFTR and SGK1 at 1 or 4 h.
GC is reported to regulate SGK1 by genomic mechanisms (29), but GC regulates CFTR in airway cells in an SGK1-independent manner (8). We examined acute (30 min, 1 h, and 4 h) transcriptional changes in SGK1 and CFTR to understand the role of transcriptional regulation of GC synergy of CFTR function. qPCR was used to examine changes in mRNA in mucosa taken from neighboring segments of jejunum used for Isc measurements. There was no appreciable change in SGK1 or CFTR mRNA in the jejunum between DMSO versus DEXA-treated animals at 30 min, 1 h, or 4 h (Fig. 3A). CFTR transcripts also remained unchanged at 24 h (data not shown).
Fig. 3.

Change in mRNA expression of serum- and glucocorticoid-inducible kinase 1 (SGK1), CFTR, and sodium/hydrogen exchanger 3 (NHE3) in intestine (jejunum) of dexamethasone (DEXA)-treated rats. A: quantitative PCR (qPCR) analysis of relative fold changes in mRNA for CFTR and SGK1 30 min, 1 h, or 4 h after DEXA treatment. B: semi-qPCR analysis of NHE3 mRNA in jejunum from DEXA/DMSO-treated rats at 1 and 4 h. N = 3 pairs of rats for each time point examined.
GC treatment modulates NHE3 transcripts differently in native tissues and cultured cells.
NHE3 (SLC9A3) is the main counterpart to CFTR in the small intestine. This ion exchanger regulates Na+ absorption on villus enterocytes and, like CFTR, is regulated by PKA, PKG, and membrane traffic (12, 19). GC and SGK1 regulate NHE3 in cultured intestinal cells (48, 49), but whether the observations in cultured cells mimic those in native intestine is unknown. To assess whether the effect of GC on NHE3 is similar in native intestine and cultured cells, we examined NHE3 transcripts in DEXA/DMSO-treated rat jejunum at 1 and 4 h. NHE3 mRNA was unchanged at 1 h but decreased at 4 h (Fig. 3B). This is in stark contrast with published evidence showing that in DEXA-treated (1 µM) intestinal cells in culture, NHE3 mRNA increased by 220% at 4 h (49). Thus there appear to be major differences in the effects of GC on NHE3 transcript between tissues and cultured cells.
GC potently stimulates CFTR protein expression in the native intestine.
Two independent studies demonstrated that dexamethasone treatment of cultured airway cells increased CFTR expression almost twofold after 16 and 24 h (6, 43). In contrast to airway cells, we found that jejunum from DEXA-treated rats displayed robust increases in CFTR protein more rapidly than cultured cells. DEXA treatment increased CFTR protein in the jejunum almost twofold after 1 h and approximately fourfold at 4 h (Fig. 4, A and B) but was also upregulated after 24 h (Fig. 4C). The greatest increase in CFTR protein induced by DEXA was observed during the 1st 4 h of treatment (Fig. 4, B and D). We compared DEXA-induced changes in CFTR protein with NHE3 in native jejunum. Like CFTR, DEXA treatment increased NHE3 protein at 1 h, but unlike CFTR, NHE3 decreased at 4 h but increased again at 24 h (Fig. 4). This contrasts with data in cultured intestinal cells that showed GC did not alter NHE3 protein at 1 or 4 h (49). These findings suggest that, similar to our observed transcriptional effects, GC affects NHE3 protein differently in cultured cells and tissues. The robust increase in CFTR protein following DEXA treatment that was detected in tissue lysates prompted us to examine whether GC alters CFTR subcellular distribution in tissues. Immunolocalization of CFTR in jejunum cryosections from DEXA-treated rats at 1, 4, and 24 h revealed increased fluorescence in the apical domain of crypts at all time points (Fig. 5, C, F, and I), supporting the increased protein observed by immunoblots.
Fig. 4.

Dexamethasone (DEXA) treatment to rats significantly increases CFTR protein expression at 1, 4, and 24 h. In contrast, sodium/hydrogen exchanger 3 (NHE3) protein is increased at 1 and 24 h but is reduced at 4 h. Shown are immunoblot analysis of intestinal (jejunum) lysates from DMSO/DEXA-treated rats probed to detect CFTR and NHE3 relative to GAPDH. Appearance of CFTR and NHE3 bands are at 1 h (A), 4 h (B), and 24 h (C). N = 3 pairs of rats for each time point examined. D: Western blot band intensities of CFTR normalized by GAPDH. Values are means ± SE. *P < 0.05, and **P < 0.001.
Fig. 5.

Dexamethasone (DEXA) treatment to rats increases CFTR immunolocalization at the apical domain at 1, 4, and 24 h. A–I: jejunal tissue sections from DMSO- and DEXA-treated rats were immunolabeled to detect CFTR (green, arrowhead) and counterstained with DAPI nuclear stain (blue). A, D, and G: negative controls were stained with only secondary antibody. B, E, and H: CFTR staining in apical domain of crypts of DMSO-treated rats at 1, 4, and 24 h. C, F, and I: CFTR staining in the apical domain of crypt cells in DEXA-treated rats at 1, 4, and 24 h.
GC stimulates robust CFTR traffic to the surface of the intestine.
Traffic of subapical pools of CFTR to the surface increases the number of channels available for anion transport. This mechanism underlies cAMP- and cGMP-dependent diarrhea and CFTR activation by drugs for treatment of constipation (1, 20). GC-stimulated traffic of CFTR to the intestinal surface could underlie acute stress-induced diarrhea in the absence of cyclic nucleotides, but this mechanism has not been studied. To determine whether GCs activate CFTR membrane traffic, we examined changes in surface biotinylated CFTR in rat jejunum following treatment with DEXA (2 mg/kg ip) or DMSO for 1 or 4 h (Fig. 6). DEXA treatment for 4 h stimulated a robust increase in surface CFTR by 3.6-fold, consistent with increased membrane traffic or apical membrane retention/stabilization (Fig. 6B). Surface NHE3 increased after 1 h of DEXA (Fig. 6A) but, unlike CFTR, did not change at 4 h (Fig. 6B).
Fig. 6.

Dexamethasone (DEXA) stimulates CFTR traffic to the surface of the intestine (jejunum) and reduces surface sodium/hydrogen exchanger 3 (NHE3) at 4 h. Shown are representative immunoblots of surface biotinylated samples and total lysates analyzed to detect CFTR and NHE3 and GAPDH loading controls from rats treated with DMSO/DEXA for 1 h (A) and 4 h (B). N = 2 pairs of rats for each time point examined.
CFTR interacts with GC-stimulated phosphorylated Nedd4-2 and 14-3-3.
To understand the mechanisms responsible for GC-stimulated increase in CFTR protein in the jejunum, we examined the roles of E3 ubiquitin (Ub) ligase Nedd4-2 and 14-3-3 adaptor proteins on CFTR protein and traffic to the intestinal surface. Nedd4-2 is phosphorylated by SGK1 and on activation, functions with 14-3-3 proteins to prevent degradation and stabilization of ion transporters on the plasma membrane (13, 26, 37, 38). Ion transporters such as ENaC that participate in the GC-SGK1 pathway undergo ubiquitination, a posttranslational modification that conjugates Ub to Lys residues of target proteins to control protein degradation and their intracellular fate, by sequential actions of Ub-activating enzymes (E1, E2, and E3; Refs. 5, 13). E3 ligases play a pivotal role in dictating the specificity of ubiquitination reactions. Nedd4-2, a member of the Nedd4 family of E3 Ub ligases, can potently inhibit ion channel activity by mediating increased endocytosis from the plasma membrane and enhanced proteasomal and/or lysosomal degradation (13). Phosphorylation in Ser342 is an important inhibitory mechanism regulating Nedd4-2. SGK1 and cAMP-activated protein kinase upregulate ion transporters such as ENaC by phosphorylation of Nedd4-2. This is a potential mechanism to stabilize CFTR and increase the overall cellular CFTR independently of transcription.
Coimmunoprecipitation experiments were conducted (Fig. 7) using jejunum lysates from DEXA/DMSO-treated (4 h) rats since highest levels of GC-induced protein and surface CFTR were observed at this time point. These experiments identified CFTR interaction with phosphorylated (but not nonphosphorylated) Nedd4-2 at Ser342 and 14-3-3ε in DEXA-treated tissues (Fig. 7A). Immunoblots of jejunum lysates confirmed increased phosphorylation of Nedd4-2 in DEXA-treated rats at 4 h (Fig. 7B). These data support GC-induced traffic linked to SGK1 activation of Nedd4-2 and 14-3-3ε phosphorylation to retain plasma membrane CFTR and increase its function.
Fig. 7.

Dexamethasone (DEXA)-treated rat intestinal (jejunum) lysates using phosphorylated (phospho-) neural precursor cell expressed developmentally downregulated 4-like (NEDD4-2)Ser342 antibody confirms interaction of NEDD4-2-CFTR-14-3-3ε when NEDD4-2 is highly phosphorylated. A: immunoprecipitation (IP) with phospho-NEDD4-2Ser342 antibody followed by immunoblots (IB) of CFTR and 14-3-3ε relative to GAPDH in the 4-h DMSO/DEXA-treated rat intestinal lysates. N = 2 pairs of rats. B: immunoblots of phosphorylated (p) and total NEDD4-2 relative to GAPDH in the 4-h DMSO/DEXA-treated rat intestinal lysates. N = 3 pairs of rats. Ctrl, control.
Expression of 14-3-3 proteins in native intestine.
14-3-3 Adaptor proteins are highly conserved from yeast to mammals. They play a role in processing, biogenesis, and stability of channels and receptors, stabilize a protein’s phosphorylated state, and promote plasma membrane expression of ion channels (52). There are seven isoforms of 14-3-3, five of which were previously identified in intestinal epithelial cells (39). Studies in cultured nonintestinal cells revealed that 14-3-3β and -ε isoforms bind to CFTR within its regulatory domain on phosphorylation (33) to regulate its trafficking (9). Since CFTR interaction with 14-3-3ε was increased in DEXA-treated tissues (Fig. 7A), we examined endogenous expression of 14-3-3ε and used pan 14-3-3 antibodies to detect 14-3-3 isoforms in jejunum lysates from DEXA/DMSO-treated rats at 1, 4, and 24 h. Antibodies against 14-3-3ε and pan 14-3-3 detected proteins in tissue lysates at all time points, with mild increase in 14-3-3ε expression in DEXA-treated tissues at 4 h (Supplemental Fig. S2; all supplemental material is available at https://doi.org/10.6084/m9.figshare.12420695.v1). Independent mass spectrometry analysis of jejunum lysates from DEXA- and DMSO-treated animals at 4 h confirmed phosphorylation changes in rat isoforms of proteins relevant to the pathway, including 14-3-3ε and -ζ, NDRG1, NHE3 (SL93A), PDK1, and PI3K (Table 2 and supplemental Excel file).
GC exacerbates changes in NHE3 and CFTR membrane localization following STa.
Net fluid secretion and diarrhea following STa result from increased CFTR activation and fluid secretion and simultaneous reduced Na+ absorption. These combined activities result from NHE3 internalization by endocytosis from the apical plasma membrane in villus enterocytes that occurs in association with increased CFTR traffic into the enterocyte apical plasma membrane from apical recycling endosomes (14, 20). To determine whether GCs alter transporter localization in acute enterotoxin diarrhea, we examined NHE3 and CFTR localization following DMSO/DEXA (24 h) and STa treatment in a closed-loop model of diarrhea (Fig. 8, A–E). As expected, STa treatment to DMSO-treated rats resulted in increased CFTR fluorescence in the apical domain of crypt and villus enterocytes compared with PBS (Fig. 8, B and C). CFTR fluorescence was more intense and compact in the apical domain of DEXA-treated tissues following STa (Fig. 8E). Conversely, NHE3 internalization from the apical plasma membrane was more pronounced in DEXA-treated tissues following STa (Fig. 8E). These localization changes support the findings that GCs exert an additive effect on traffic of transporters to exacerbate enterotoxin-induced diarrhea.
Fig. 8.

Dexamethasone (DEXA) exacerbates changes in apical CFTR and sodium/hydrogen exchanger 3 (NHE3) immunolocalization in the jejunum in heat-stable enterotoxin (STa)-induced diarrhea (A–E) and exacerbates cGMP-stimulated CFTR short-circuit (Isc) currents (F). Rats were treated with DEXA/DMSO for 24 h, and intestinal loops were treated with STa or PBS for 30 min. Tissues were removed, and cryostat sections were immunostained to detect CFTR (green), NHE3 (green), and F-actin (red). A: negative controls (Ctrl) stained with secondary antibody. B and C: tissues from phosphate-buffered saline (PBS) or STa-treated loops from DMSO-treated rats. D and E: tissues from PBS or STa-treated loops from DEXA-treated rats. Shown are CFTR (green) localizations in the apical domain of crypts (top) and villi (middle), indicated by white arrows, in DMSO/STa (C)-, DEXA/PBS (D)-, and DEXA/STa (E)-treated loops compared with DMSO/PBS (B). Shown as well are NHE3 (green) staining in villi (bottom) in DMSO/STa (C)- and DEXA/STa (E)-treated loops compared with DMSO/PBS loops (B). D: DEXA-treated PBS loops. Tissues were counterstained with DAPI nuclear stain (blue). F: baseline Isc was recorded in jejunum from DMSO- or DEXA-treated rats (24 h) as shown in Fig. 1A. Jejuna from DMSO- or DEXA-treated rats (24 h) were pretreated with serum- and glucocorticoid-inducible kinase 1 (SGK1; GSK 650394; 10 μM) or phosphatidylinositol 3-kinase (PI3K; LY 294002; 10 μM) inhibitors for 30 min in Ussing chambers. After 30 min, cGMP was added to all chambers, and short-circuit currents (Isc) were recorded. The cGMP-stimulated Isc was significantly increased (*P = 0.0184; n = 4 pairs of rats) in DEXA- vs. DMSO-treated rats. Statistical significance was determined using an unpaired Student’s t test. Additionally, the cGMP-stimulated Isc was significantly decreased in the presence of the SGK1 inhibitor (*P = 0.025; n = 4 pairs of rats) and the PI3K inhibitor (**P = 0.0042; n = 3 pairs of rats). Statistical significance was determined using a paired Student’s t test.
GC exacerbates cGMP-activated CFTR-mediated secretion.
cAMP/PKA and cGMP/PKG activate CFTR to result in fluid secretion in the intestine. Increased international travel is associated with higher rates of travelers’ diarrhea due to enterotoxigenic E. coli (14). We hypothesized that GC exacerbates enterotoxin-mediated diarrhea with involvement of SGK1 or PI3K kinase signaling. We examined the roles of GC, SGK1, and PI3K on cGMP-activated CFTR ion transport in the jejunum. As shown in Fig. 8F, we confirmed that cGMP activates CFTR Isc. CFTR Isc was significantly (~2-fold) higher in tissues from DEXA-treated rats and was reduced in the presence of the PI3K inhibitor and SGK1 inhibitor (Fig. 8F). These data indicate that GC exacerbates enterotoxin-mediated electrogenic anion secretion by SGK1 and PI3K kinase signaling at 24 h.
DISCUSSION
Nongenomic GC-activated SGK1 regulates ion transporters, including ENaC, NHE3, and the Na+-K+-2Cl− cotransporter NKCC1 (30, 31, 48). GC-SGK1 regulation of CFTR was examined in independent studies of cultured airway (6, 8, 32) and pancreatic cells (10, 11) but not in native tissues, intestinal cells, or native intestine. The aim of this study was to understand the physiological role of GC-SGK1 kinase signaling of CFTR in the native intestine, identify features in the pathway that are unique to the intestine, and understand its potential relevance to diarrheal disease. We demonstrated that nongenomic kinase signaling by GC-SGK1-PI3K and downstream mediators exert potent effects on CFTR function, protein expression, and membrane traffic into the surface of the native intestine that differ from its regulation in other CFTR-expressing epithelia. The observed differences in how CFTR is regulated by GC and downstream kinase signaling in the intestine compared with other epithelia is consistent with cell-type specificity of CFTR regulation and the physiological role that CFTR plays in different tissues. Moreover, activation of this pathway exacerbates CFTR-mediated anion secretion in diarrhea. This pathway is druggable and thus provides new opportunities and therapeutic targets for treating conditions linked to stress-induced diarrhea.
To understand the physiological changes in CFTR elicited by GC in native intestine, we determined the time course of changes in mRNA, protein, and ion transport function over 24 h. We also examined GC-stimulated CFTR traffic to the cell surface similar to airway cells (6) and examined whether GC-SGK1 and PI3K kinase signaling was relevant to CFTR function in cGMP-activated diarrhea. Where possible, we compared GC-induced changes in CFTR with NHE3 (SL9A3) in the jejunum, a major site of action for both transporters in the intestine. The idea was to identify differences in regulation between tissues and cultured intestinal cells given the existing body of literature on NHE3 regulation by this pathway in cultured intestinal cells (48, 49, 54).
We found that GC significantly increased baseline Isc over the short (1 and 4 h) and long term (24 h) in rat intestine. These data are consistent with previous observations in animal models that confirmed the effect of stress in elevating Isc, Cl− secretion, and eliciting diarrhea (25, 45). Furthermore, the GC-induced synergistic increase in CFTR ion transport at all time points (1, 4, and 24 h) was mediated by SGK1 and PI3K, confirming the role of kinase signaling in regulating GC-stimulated CFTR function. The findings here differ from the results of studies in rat primary airway cells and cultured human bronchial submucosal gland-derived Calu-3 cells that found no requirement for SGK1 but a role for PI3K in DEXA-stimulated increase in CFTR function (8). Our findings also contrast with observations of GC regulation of CFTR function in cultured fetal distal lung airway cells, where DEXA decreased CFTR activity (32), findings that are consistent with the role of GC in regulating fluid transport in the lung during development and the transition to extrauterine life.
DEXA treatment did not alter CFTR mRNA at 1, 4, or 24 h in the intestine, and SGK1 mRNA was not altered at 30 min to 4 h, but SGK1 activity was evident at all time points examined (1, 4, and 24 h). Our findings contrast with data from Calu-3 airway cells where treatment with DEXA for 24–48 h reduced CFTR mRNA (0.33-fold), whereas ENaC mRNA increased 3 h after treatment (43). In CFBE41o human CF airway epithelial cells, DEXA increased SGK1 mRNA within 15 min (6), indicating rapid transcriptional regulation by DEXA in this cell model. Our data on DEXA’s effect on NHE3 mRNA in native jejunum also differ from cultured intestinal cells. In the intestine, NHE3 mRNA was unchanged 1 h after DEXA but was markedly reduced at 4 h. In cultured Caco-2 cells, NHE3 mRNA was unchanged at 1 h but increased 220-fold at 4 h (49).
CFTR mRNA did not correlate with changes in protein in the intestine of DEXA-treated rats. DEXA potently increased CFTR at all time points (1, 4, and 24 h) with the greatest increase (~4-fold) observed by immunoblot at 4 h. DEXA-induced changes in CFTR protein were confirmed by increase in apical CFTR immunolocalization in tissue sections. DEXA also stimulated CFTR protein in cultured airway cells, but the increase was incremental (~1.8-fold) after 24 h compared with the findings in the native intestine of a ~3.6-fold increase at the same time interval (6, 43). Changes in NHE3 protein in the intestine also differed from cultured intestinal cells and did not correlate with mRNA. NHE3 protein did not increase in DEXA-treated cultured cells until 24 h, whereas it increased quickly after 1 h in the intestine (49). The observed differences in DEXA-induced changes in intestine versus cultured cells can be accounted for by differences in the model systems, dose, and mode of administration but also point to cell- and tissue-specific regulation of CFTR and ion transporters. More importantly, the accumulation of CFTR protein without changes in mRNA strongly suggested the possibility of stabilization of CFTR as a mechanism for its increase on the cell surface.
DEXA-induced CFTR protein was associated with robust increase (4-fold) in surface CFTR at 4 h, similar to our observations of cGMP- and cAMP-stimulated traffic of CFTR to the surface of the intestine in enterotoxin diarrhea (20). This is the first observation of robust CFTR traffic in the intestine in the absence of cyclic nucleotides, although this has been observed in other epithelia. Silencing of SGK1 in cultured human CFBE airway cells confirmed SGK1-mediated DEXA-induced increase in surface CFTR (6). The increase in Nedd4-2 phosphorylation at Ser-342 by DEXA at 4 h in the intestine and its association with CFTR and 14-3-3ε are consistent with known mechanisms involving DEXA-induced SGK1, phosphorylation of Nedd4-2, and interaction with 14-3-3 proteins to increase CFTR (10, 11) and ENaC surface abundance (13). This mechanism involves activation of PDK1/SGK1 resulting in phosphorylation and inactivation of Nedd4-2 and decreased ubiquitination and degradation of ion transporters as shown (13, 52). Alternate mechanisms for DEXA-induced CFTR protein and surface abundance were proposed that involve SGK1-dependent phosphorylation of PDZ domain proteins (27) but were not examined in this study.
Mass spectrometry (MS) analysis of intestinal lysates independently validated expression of rat isoforms of proteins relevant to this pathway and identified relevant phosphorylation sites in peptides including NHE3 (SL9A3), PI3K, PDK1, NDRG1, and 14-3-3. Studies in cultured cells found that 14-3-3β and -ε isoforms bind to CFTR within the regulatory domain upon its phosphorylation (33) to regulate its trafficking (9). We also found that 14-3-3ε is complexed with phosphorylated Nedd4-2 and CFTR in DEXA-treated intestine. Our manually validated MS/MS fragmentation (see supplemental Excel file) confirmed expression of intestinal isoforms in tissue lysates; however, 14-3-3ε and -ζ but not 14-3-3β peptides were phosphorylated, suggesting some intestine specificity in adaptors. Ingenuity Pathway Analysis revealed concordance and relationships between identified proteins in this pathway and relevance to ion transport as shown in Fig. 9.
Fig. 9.

Ingenuity pathway-generated network showing the relationship between identified phosphorylated proteins (red) and associated functions. Fisher exact test P values of the overlap between proteins and functions were calculated by Ingenuity knowledge base.
Our observations that GCs exacerbate CFTR traffic and ion secretion in cGMP-activated diarrhea and are reduced by inhibiting SGK1 and PI3K are new and support the notion that stress, GCs, and downstream kinases play a role in diarrheal diseases. Our findings align with published data that stress increases cGMP-activated fluid secretory responses in animals (2). This pathway is also involved in genetic forms of diarrhea as we (18) recently showed involvement of GCs and downstream activation of PKA and CFTR in microvillus inclusion disease, a lethal genetic disease affecting newborns. The findings here provide previously unrecognized opportunities for targeting SGK1, PI3K, and relevant kinases in diarrheal diseases.
GRANTS
This study was supported by National Institute of General Medical Sciences Grant 5R01GM127953 to P. J. Salas and Department of Internal Medicine, Yale University National Institute of Diabetes and Digestive and Kidney Diseases Grant T32 NIH DK007017-41 and Yale School of Medicine funds to N. A. Ameen. Proteomics data were collected on a mass spectrometer supported by NIH SIG S10ODOD018034 and Yale School of Medicine.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.J.S. and N.A.A. conceived and designed research; M.K.A., L.F.-H., V.B., T.T.L., and K.H. performed experiments; M.K.A., L.F.-H., R.G.-M., T.T.L., and K.H. analyzed data; M.K.A., L.F.-H., and T.T.L. interpreted results of experiments; M.K.A., L.F.-H., R.G.-M., and T.T.L. prepared figures; M.K.A., L.F.-H., and N.A.A. drafted manuscript; M.K.A., L.F.-H., V.B., R.G.-M., T.T.L., K.H., P.J.S., and N.A.A. edited and revised manuscript; M.K.A., L.F.-H., V.B., R.G.-M., T.T.L., K.H., P.J.S., and N.A.A. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Florine Collin and Jean Kanyo from the Mass Spectrometry and Proteomics Resource at Yale University for their assistance with the proteomics sample preparation and data collection.
REFERENCES
- 1.Ahsan MK, Tchernychev B, Kessler MM, Solinga RM, Arthur D, Linde CI, Silos-Santiago I, Hannig G, Ameen NA. Linaclotide activates guanylate cyclase-C/cGMP/protein kinase-II-dependent trafficking of CFTR in the intestine. Physiol Rep 5: e13299, 2017. doi: 10.14814/phy2.13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Balool FY. Fluid secretory responses to enterotoxin STa and 8-bromo-cyclic GMP in fed and nutrionally-deprived gerbils: jejunum, ileum and colon in vivo. Physiol Res 53: 669–674, 2004. [PubMed] [Google Scholar]
- 3.Ameen N, Silvis M, Bradbury NA. Endocytic trafficking of CFTR in health and disease. J Cyst Fibros 6: 1–14, 2007. doi: 10.1016/j.jcf.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ameen NA, Martensson B, Bourguinon L, Marino C, Isenberg J, McLaughlin GE. CFTR channel insertion to the apical surface in rat duodenal villus epithelial cells is upregulated by VIP in vivo. J Cell Sci 112: 887–894, 1999. [DOI] [PubMed] [Google Scholar]
- 5.Bhalla V, Daidié D, Li H, Pao AC, LaGrange LP, Wang J, Vandewalle A, Stockand JD, Staub O, Pearce D. Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Mol Endocrinol 19: 3073–3084, 2005. doi: 10.1210/me.2005-0193. [DOI] [PubMed] [Google Scholar]
- 6.Bomberger JM, Coutermarsh BA, Barnaby RL, Sato JD, Chapline MC, Stanton BA. Serum and glucocorticoid-inducible kinase1 increases plasma membrane wt-CFTR in human airway epithelial cells by inhibiting its endocytic retrieval. PLoS One 9: e89599, 2014. doi: 10.1371/journal.pone.0089599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borowitz D, Gelfond D. Intestinal complications of cystic fibrosis. Curr Opin Pulm Med 19: 676–680, 2013. doi: 10.1097/MCP.0b013e3283659ef2. [DOI] [PubMed] [Google Scholar]
- 8.Bossmann M, Ackermann BW, Thome UH, Laube M. Signaling cascade involved in rapid stimulation of cystic fibrosis transmembrane conductance regulator (CFTR) by dexamethasone. Int J Mol Sci 18: 1807, 2017. doi: 10.3390/ijms18081807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bozoky Z, Krzeminski M, Muhandiram R, Birtley JR, Al-Zahrani A, Thomas PJ, Frizzell RA, Ford RC, Forman-Kay JD. Regulatory R region of the CFTR chloride channel is a dynamic integrator of phospho-dependent intra- and intermolecular interactions. Proc Natl Acad Sci USA 110: E4427–E4436, 2013. doi: 10.1073/pnas.1315104110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caohuy H, Jozwik C, Pollard HB. Rescue of ΔF508-CFTR by the SGK1/Nedd4-2 signaling pathway. J Biol Chem 284: 25241–25253, 2009. doi: 10.1074/jbc.M109.035345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caohuy H, Yang Q, Eudy Y, Ha TA, Xu AE, Glover M, Frizzell RA, Jozwik C, Pollard HB. Activation of 3-phosphoinositide-dependent kinase 1 (PDK1) and serum- and glucocorticoid-induced protein kinase 1 (SGK1) by short-chain sphingolipid C4-ceramide rescues the trafficking defect of ΔF508-cystic fibrosis transmembrane conductance regulator (ΔF508-CFTR). J Biol Chem 289: 35953–35968, 2014. doi: 10.1074/jbc.M114.598649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cha B, Kim JH, Hut H, Hogema BM, Nadarja J, Zizak M, Cavet M, Lee-Kwon W, Lohmann SM, Smolenski A, Tse CM, Yun C, de Jonge HR, Donowitz M. cGMP inhibition of Na+/H+ antiporter 3 (NHE3) requires PDZ domain adapter NHERF2, a broad specificity protein kinase G-anchoring protein. J Biol Chem 280: 16642–16650, 2005. doi: 10.1074/jbc.M500505200. [DOI] [PubMed] [Google Scholar]
- 13.Chandran S, Li H, Dong W, Krasinska K, Adams C, Alexandrova L, Chien A, Hallows KR, Bhalla V. Neural precursor cell-expressed developmentally down-regulated protein 4-2 (Nedd4-2) regulation by 14-3-3 protein binding at canonical serum and glucocorticoid kinase 1 (SGK1) phosphorylation sites. J Biol Chem 286: 37830–37840, 2011. doi: 10.1074/jbc.M111.293233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen T, Lin R, Avula L, Sarker R, Yang J, Cha B, Tse CM, McNamara G, Seidler U, Waldman S, Snook A, Bijvelds MJC, de Jonge HR, Li X, Donowitz M. NHERF3 is necessary for Escherichia coli heat-stable enterotoxin-induced inhibition of NHE3: differences in signaling in mouse small intestine and Caco-2 cells. Am J Physiol Cell Physiol 317: C737–C748, 2019. doi: 10.1152/ajpcell.00351.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collaco A, Marathe J, Kohnke H, Kravstov D, Ameen N. Syntaxin 3 is necessary for cAMP- and cGMP-regulated exocytosis of CFTR: implications for enterotoxigenic diarrhea. Am J Physiol Cell Physiol 299: C1450–C1460, 2010. doi: 10.1152/ajpcell.00029.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Croxtall JD, Choudhury Q, Flower RJ. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br J Pharmacol 130: 289–298, 2000. doi: 10.1038/sj.bjp.0703272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fejes-Tóth G, Frindt G, Náray-Fejes-Tóth A, Palmer LG. Epithelial Na+ channel activation and processing in mice lacking SGK1. Am J Physiol Renal Physiol 294: F1298–F1305, 2008. doi: 10.1152/ajprenal.00579.2007. [DOI] [PubMed] [Google Scholar]
- 18.Forteza R, Ahsan MK, Cartón-García F, Arango D, Ameen NA, Salas PJ. Glucocorticoids and myosin5b loss of function induce heightened PKA signaling in addition to membrane traffic defects. Mol Biol Cell 30: 3076–3089, 2019. doi: 10.1091/mbc.E18-07-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foulke-Abel J, In J, Yin J, Zachos NC, Kovbasnjuk O, Estes MK, de Jonge H, Donowitz M. Human enteroids as a model of upper small intestinal ion transport physiology and pathophysiology. Gastroenterology 150: 638–649.e8, 2016. doi: 10.1053/j.gastro.2015.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Golin-Bisello F, Bradbury N, Ameen N. STa and cGMP stimulate CFTR translocation to the surface of villus enterocytes in rat jejunum and is regulated by protein kinase G. Am J Physiol Cell Physiol 289: C708–C716, 2005. doi: 10.1152/ajpcell.00544.2004. [DOI] [PubMed] [Google Scholar]
- 21.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43: D512–D520, 2015. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ismail NA, Baines DL, Wilson SM. The phosphorylation of endogenous Nedd4-2 In Na+-absorbing human airway epithelial cells. Eur J Pharmacol 732: 32–42, 2014. doi: 10.1016/j.ejphar.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jakab RL, Collaco AM, Ameen NA. Cell-specific effects of luminal acid, bicarbonate, cAMP, and carbachol on transporter trafficking in the intestine. Am J Physiol Gastrointest Liver Physiol 303: G937–G950, 2012. doi: 10.1152/ajpgi.00452.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jakab RL, Collaco AM, Ameen NA. Physiological relevance of cell-specific distribution patterns of CFTR, NKCC1, NBCe1, and NHE3 along the crypt-villus axis in the intestine. Am J Physiol Gastrointest Liver Physiol 300: G82–G98, 2011. doi: 10.1152/ajpgi.00245.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang SJ, Kang SS, Son SJ, Lee JY, Kim G, Choi SH. Cortisol levels and gastrointestinal disorders after stressful surgery in rabbits. In Vivo 31: 637–640, 2017. doi: 10.21873/invivo.11105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang C, Kawabe H, Rotin D. The ubiquitin ligase Nedd4L regulates the Na/K/2Cl co-transporter NKCC1/SLC12A2 in the colon. J Biol Chem 292: 3137–3145, 2017. doi: 10.1074/jbc.M116.770065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koeppen K, Coutermarsh BA, Madden DR, Stanton BA. Serum- and glucocorticoid-induced protein kinase 1 (SGK1) increases the cystic fibrosis transmembrane conductance regulator (CFTR) in airway epithelial cells by phosphorylating Shank2E protein. J Biol Chem 289: 17142–17150, 2014. doi: 10.1074/jbc.M114.555599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kravtsov DV, Ahsan MK, Kumari V, van Ijzendoorn SC, Reyes-Mugica M, Kumar A, Gujral T, Dudeja PK, Ameen NA. Identification of intestinal ion transport defects in microvillus inclusion disease. Am J Physiol Gastrointest Liver Physiol 311: G142–G155, 2016. doi: 10.1152/ajpgi.00041.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lang F, Böhmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev 86: 1151–1178, 2006. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- 30.Lang F, Shumilina E. Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1. FASEB J 27: 3–12, 2013. doi: 10.1096/fj.12-218230. [DOI] [PubMed] [Google Scholar]
- 31.Lang F, Stournaras C, Alesutan I. Regulation of transport across cell membranes by the serum- and glucocorticoid-inducible kinase SGK1. Mol Membr Biol 31: 29–36, 2014. doi: 10.3109/09687688.2013.874598. [DOI] [PubMed] [Google Scholar]
- 32.Laube M, Bossmann M, Thome UH. Glucocorticoids distinctively modulate the CFTR channel with possible implications in lung development and transition into extrauterine life. PLoS One 10: e0124833, 2015. doi: 10.1371/journal.pone.0124833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang X, Da Paula AC, Bozóky Z, Zhang H, Bertrand CA, Peters KW, Forman-Kay JD, Frizzell RA. Phosphorylation-dependent 14-3-3 protein interactions regulate CFTR biogenesis. Mol Biol Cell 23: 996–1009, 2012. doi: 10.1091/mbc.e11-08-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lösel RM, Falkenstein E, Feuring M, Schultz A, Tillmann HC, Rossol-Haseroth K, Wehling M. Nongenomic steroid action: controversies, questions, and answers. Physiol Rev 83: 965–1016, 2003. doi: 10.1152/physrev.00003.2003. [DOI] [PubMed] [Google Scholar]
- 35.Löwenberg M, Stahn C, Hommes DW, Buttgereit F. Novel insights into mechanisms of glucocorticoid action and the development of new glucocorticoid receptor ligands. Steroids 73: 1025–1029, 2008. doi: 10.1016/j.steroids.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 36.Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell 169: 381–405, 2017. doi: 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manning JA, Henshall TL, Kumar S. NEDD4-2-dependent control of Na+ homeostasis and renal disease. Cell Cycle 17: 1–2, 2018. doi: 10.1080/15384101.2017.1386514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manning JA, Kumar S. Physiological functions of Nedd4-2: lessons from knockout mouse models. Trends Biochem Sci 43: 635–647, 2018. doi: 10.1016/j.tibs.2018.06.004. [DOI] [PubMed] [Google Scholar]
- 39.Monroy FP. Toxoplasma gondii: effect of infection on expression of 14-3-3 proteins in human epithelial cells. Exp Parasitol 118: 134–138, 2008. doi: 10.1016/j.exppara.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murray JT, Campbell DG, Morrice N, Auld GC, Shpiro N, Marquez R, Peggie M, Bain J, Bloomberg GB, Grahammer F, Lang F, Wulff P, Kuhl D, Cohen P. Exploitation of KESTREL to identify NDRG family members as physiological substrates for SGK1 and GSK3. Biochem J 384: 477–488, 2004. doi: 10.1042/BJ20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park J, Li Y, Kim SH, Kong G, Shrestha R, Tran Q, Hong J, Hur GM, Hemmings BA, Koo BS, Park J. Characterization of fragmented 3-phosphoinsitide-dependent protein kinase-1 (PDK1) by phosphosite-specific antibodies. Life Sci 93: 700–706, 2013. doi: 10.1016/j.lfs.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 42.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20: 3551–3567, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 43.Prota LF, Cebotaru L, Cheng J, Wright J, Vij N, Morales MM, Guggino WB. Dexamethasone regulates CFTR expression in Calu-3 cells with the involvement of chaperones HSP70 and HSP90. PLoS One 7: e47405, 2012. doi: 10.1371/journal.pone.0047405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Silvis MR, Bertrand CA, Ameen N, Golin-Bisello F, Butterworth MB, Frizzell RA, Bradbury NA. Rab11b regulates the apical recycling of the cystic fibrosis transmembrane conductance regulator in polarized intestinal epithelial cells. Mol Biol Cell 20: 2337–2350, 2009. doi: 10.1091/mbc.e08-01-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith F, Clark JE, Overman BL, Tozel CC, Huang JH, Rivier JE, Blisklager AT, Moeser AJ. Early weaning stress impairs development of mucosal barrier function in the porcine intestine. Am J Physiol Gastrointest Liver Physiol 298: G352–G363, 2010. doi: 10.1152/ajpgi.00081.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stahn C, Buttgereit F. Genomic and nongenomic effects of glucocorticoids. Nat Clin Pract Rheumatol 4: 525–533, 2008. doi: 10.1038/ncprheum0898. [DOI] [PubMed] [Google Scholar]
- 47.Wagner CA, Ott M, Klingel K, Beck S, Melzig J, Friedrich B, Wild KN, Bröer S, Moschen I, Albers A, Waldegger S, Tümmler B, Egan ME, Geibel JP, Kandolf R, Lang F. Effects of the serine/threonine kinase SGK1 on the epithelial Na+ channel (ENaC) and CFTR: implications for cystic fibrosis. Cell Physiol Biochem 11: 209–218, 2001. doi: 10.1159/000051935. [DOI] [PubMed] [Google Scholar]
- 48.Wang D, Sun H, Lang F, Yun CC. Activation of NHE3 by dexamethasone requires phosphorylation of NHE3 at Ser663 by SGK1. Am J Physiol Cell Physiol 289: C802–C810, 2005. doi: 10.1152/ajpcell.00597.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang D, Zhang H, Lang F, Yun CC. Acute activation of NHE3 by dexamethasone correlates with activation of SGK1 and requires a functional glucocorticoid receptor. Am J Physiol Cell Physiol 292: C396–C404, 2007. doi: 10.1152/ajpcell.00345.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Webster MK, Goya L, Firestone GL. Immediate-early transcriptional regulation and rapid mRNA turnover of a putative serine/threonine protein kinase. J Biol Chem 268: 11482–11485, 1993. [PubMed] [Google Scholar]
- 51.Webster MK, Goya L, Ge Y, Maiyar AC, Firestone GL. Characterization of sgk, a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Mol Cell Biol 13: 2031–2040, 1993. doi: 10.1128/MCB.13.4.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wiemuth D, Lott JS, Ly K, Ke Y, Teesdale-Spittle P, Snyder PM, McDonald FJ. Interaction of serum- and glucocorticoid regulated kinase 1 (SGK1) with the WW-domains of Nedd4-2 is required for epithelial sodium channel regulation. PLoS One 5: e12163, 2010. doi: 10.1371/journal.pone.0012163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang L, Frindt G, Lang F, Kuhl D, Vallon V, Palmer LG. SGK1-dependent ENaC processing and trafficking in mice with high dietary K intake and elevated aldosterone. Am J Physiol Renal Physiol 312: F65–F76, 2017. doi: 10.1152/ajprenal.00257.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yun CC, Chen Y, Lang F. Glucocorticoid activation of Na+/H+ exchanger isoform 3 revisited. The roles of SGK1 and NHERF2. J Biol Chem 277: 7676–7683, 2002. doi: 10.1074/jbc.M107768200. [DOI] [PubMed] [Google Scholar]