

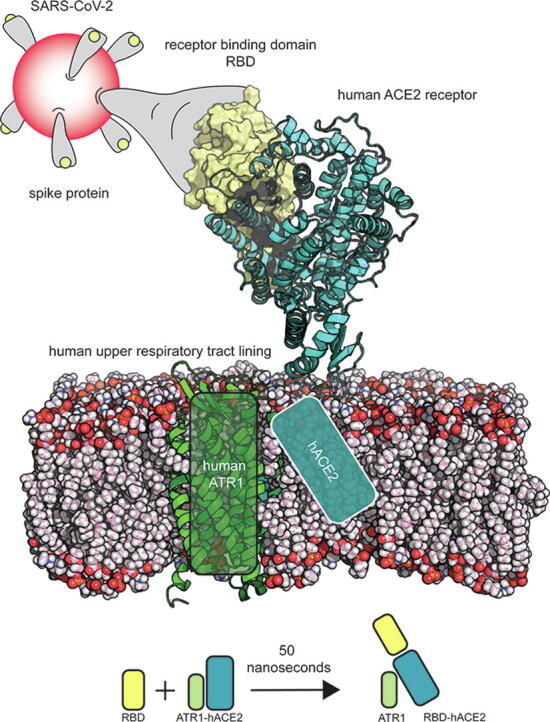
Graphical abstract
Keywords: Biophysics, SARS CoV-2, COVID 19, ATR1, Human ACE2
Abstract
SARS-CoV-2 is a novel highly virulent pathogen which gains entry to human cells by binding with the cell surface receptor – angiotensin converting enzyme (ACE2). We computationally contrasted the binding interactions between human ACE2 and coronavirus spike protein receptor binding domain (RBD) of the 2002 epidemic-causing SARS-CoV-1, SARS-CoV-2, and bat coronavirus RaTG13 using the Rosetta energy function. We find that the RBD of the spike protein of SARS-CoV-2 is highly optimized to achieve very strong binding with human ACE2 (hACE2) which is consistent with its enhanced infectivity. SARS-CoV-2 forms the most stable complex with hACE2 compared to SARS-CoV-1 (23% less stable) or RaTG13 (11% less stable). Notably, we calculate that the SARS-CoV-2 RBD lowers the binding strength of angiotensin 2 receptor type I (ATR1) which is the native binding partner of ACE2 by 44.2%. Strong binding is mediated through strong electrostatic attachments with every fourth residue on the N-terminus alpha-helix (starting from Ser19 to Asn53) as the turn of the helix makes these residues solvent accessible. By contrasting the spike protein SARS-CoV-2 Rosetta binding energy with ACE2 of different livestock and pet species we find strongest binding with bat ACE2 followed by human, feline, equine, canine and finally chicken. This is consistent with the hypothesis that bats are the viral origin and reservoir species. These results offer a computational explanation for the increased infection susceptibility by SARS-CoV-2 and allude to therapeutic modalities by identifying and rank-ordering the ACE2 residues involved in binding with the virus.
1. Introduction
The causative agent of coronavirus disease 2019 (COVID-19) was identified in January 2020 to be a novel β-coronavirus of the same subgenus as SARS-CoV-1. SARS-CoV-2 strain has caused a dramatically greater number of infections and fatalities and an effective antiviral treatment and vaccine remains elusive to this day. It has been reported that the first step to viral entry is association between the viral spike RBD and human ACE2 protein [1]. There have been several structural analyses [2], [3] of both SARS-CoV-1 and SARS-CoV-2 binding interactions with human ACE2 (hACE2) but no quantitative assessment of the contribution of different residues in the spike RBD towards tight binding or comparisons with its native receptor ATR1. It has been suggested [2], [4] that viral spike binding to hACE2 prevents ATR1 binding with hACE2 but no computational binding energy comparisons have been drawn. Experimental and computational investigations have focused on the RBD-hACE2 interaction for SARS-CoV-1 [5] and CoV-2 [7], the role of glycosylated spike residues [8], and the potential impact of the CoV-2’s furin cleavage site [6].
Wang et al. [9] using all atom MD simulations showed that the SARS-CoV-2 RBD forms a more stable complex with hACE2 compared to SARS-CoV-1 by recruiting an enhanced hydrogen bonding network and greater electrostatic complementarity. Several other studies also corroborate the increased affinity of SARS-CoV-2 [10], [11], [12]. In another recent study [13], deep mutational scanning of SARS-CoV-2 RBD revealed that out of 21 interface residues, 18 residues upon substitution to alanine lead to a loss in binding affinity (10 of those leading to more than 50% loss in binding affinity) indicating that most interface amino acid changes are detrimental to ACE2 binding. This study also showed that the binding affinity of SARS-CoV-2 RBD with hACE2 is about 2 times greater than that of SARS-CoV-1 which in turn is about 4 times greater than that of RaTG13. These results are in agreement with our in silico alanine scan except for one case (Q493A) which exhibits ~60% loss of binding between SARS-CoV-2 RBD and hACE2 though in vitro results show no loss.
In this paper, we first assess the molecular interactions between the three spike RBDs with the hACE2 complex. We also provide a comparative analysis of the most important RBD residues from all three viral spike proteins that drive binding with hACE2. Using the Rosetta binding energy function to score interactions, we find that SARS-CoV-2 reduces the human ATR1 surface receptor protein binding affinity for hACE2 by 44.2%. A recent study [14] explained interactions between hACE2 and SARS-CoV-1 vs. SARS-CoV-2 RBDs using a homology modeled structure of SARS-CoV-2 RBD and only considering five residues from the spike RBDs. Building on these results, we used an experimentally confirmed atomic scale map (cryo-EM structures) for the SARS-CoV-1 and CoV-2 RBD in complex with hACE2. Because no experimentally resolved RaTG13-hACE2 complex structure is available, we computationally reconstructed a putative one using flexible protein–protein docking (see Methods). We find that the RBD of SARS-CoV-2 binds hACE2 23% stronger than SARS-CoV-1 and 11% compared to RaTG13 quantified using the Rosetta energy function. 50 ns MD simulation trajectories demonstrated that hACE2 shows preferential binding to RBD of SARS-CoV-2 than human ATR1 by 3.4 kcal/mol in terms of binding energy forming ~7 more interface hydrogen bonds. This suggests that in the presence of spike RBD, the ATR1-bound hACE2 is energetically disfavored as a 36.2% stronger competing electrostatic attachment with the spike RBD is predicted. Extending this analysis to include non-human ACE2 orthologues, we calculated a descending order of binding strength starting with bats and followed by human, feline, canine, equine, bovine, and finally poultry. This rank order is consistent with a recent experimental report that finds that mammals especially felines are susceptible to SARS-CoV-2, whereas birds, fish, and reptiles are not [15].
2. Results
Using Rosetta binding energy, we calculated that SARS-CoV-2 RBD exhibits higher binding affinity (by more than 5 kcal/mol) to hACE2 in comparison to the SARS-CoV-1 or bat-specific RaTG13 strains. This is accomplished by establishing a greater number of interfacial electrostatic contacts which is detailed in a contact map analysis (See Fig. 1). Subsequently, though the use of a computational alanine scan analysis we pinpoint key residues at these interfaces that drive the binding of these three RBDs with hACE2 (see Fig. 1). Results indicate that SARS-CoV-2 recruits nearly 90% of the interface residues to bind to hACE2. Interface residue composition reveals the key biophysical role played by tyrosine and glycine amino acids at the hACE2 binding interface of SARS-CoV-2 RBD (see Fig. 1). Finally, an MD simulation on membrane bound hACE2 computationally corroborates that native ATR1-hACE2 transmembrane complex stability can be weakened by 44.2% in the presence of SARS-CoV-2 RBD in the extracellular space, which establishes nearly seven more interface hydrogen-bonded contacts with hACE2.
Fig. 1.

SARS-CoV-2 RBD causes the greatest disruption to the original intra-residue contacts of hACE2 achieving the strongest-binding complex. Shown in the figure are the residue contact maps of the hACE2 receptor in the unbound state and when bound with the viral spike protein RBDs from SARS-CoV-1, SARS-CoV-2, and RaTG13, respectively. Filled dots (in green) represent electrostatic (i.e., circles) or hydrophobic (i.e., squares) intra-residue contacts within hACE2. Open circles and squares in the bound state of hACE2 with RBD signify the lost intra-residue contacts within hACE2 upon binding with the three spikes. Shown in yellow, pink and purple filled circles and squares are the inter-residues contacts formed upon binding with the three spike RBDs. Filled circles or squares in the light blue region show indirect/contact-map mediated interactions between hACE2 residues (region 1) and the spike RBD (region 3). SARS-CoV-2 disrupts and co-opts the most intra-hACE2 residue contacts forming the most residue contacts between hACE2 and RBD. RBD self-stabilizing contact information and weak (long-range) electrostatic interactions (between 4.5 Å and 6.0 Å) between the spike and hACE2 are not shown in the figure. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.1. Analysis of human ACE2 in complex with spike RBDs from the three different coronavirus strains
Rosetta-based energy minimization of the hACE2-RBD complexes with RBDs from SARS-CoV-1, SARS-CoV-2, and RaTG13 reveals that SARS-CoV-2 exhibits the strongest Rosetta binding score (−48.312 ± 3.4 kcal/mol). SARS-CoV-1 and RaTG13 Rosetta binding energy scores with hACE2 are −37.308 ± 2.3 and −43.168 ± 2.1 kcal/mol, respectively. In an uninfected human cell, the ATR1 receptor binds to ACE2 to form a receptor complex. Upon infection, the coronavirus presents the RBD of its spike protein to the human ACE2 forming an electrostatically-driven association between the two. Rosetta binding results indicate that hACE2 can bind with either human ATR1 or the viral spike (but not both simultaneously) as the binding domains overlap. hACE2 forms hydrophobic and strong electrostatic (including pi-pi, and cation-pi) interactions with the binding domain of ATR1 with a Rosetta binding energy of −31.4 kcal/mol which is 35% less strong than the one with the SARS-CoV-2 RBD. The CoV-2 RBD maximally co-opts these interactions to gain entry via strong non-covalent attachment (see Fig. 1).
To understand the role of the inter-residue interaction network formed during viral entry, we first constructed a contact map depicting all such interactions for the spike-binding interface of unbound hACE2 (see Fig. 1). We then computed the changes in this contact map upon binding with the RBD of SARS-CoV-1, SARS-CoV-2, and RaTG13. We observe that SARS-CoV-2 more radically co-opts the original contact map of unbound hACE2 to form a highly stabilized hACE2-RBD interface (see Fig. 1).
We observe that SARS-CoV-2 forms the greatest number of effective hACE2 contacts (11 hydrogen-bonded, eight electrostatic and two hydrophobic) with sixteen RBD residues at the hACE2 binding interface (see Fig. 1). For example, SARS-CoV-2 RBD residue Phe456 simultaneously forms a hydrophobic contact with hACE2 residue Thr27 (using the side-chain) and an electrostatic stabilization with hACE2 residue Asp30 (using the backbone) (see Fig. 2). The RaTG13 RBD only forms the hydrophobic interaction whereas the SARS-CoV-1 RBD forms neither (see Fig. 2). Consequently, a computational alanine scan (see Fig. 3) reveals that an amino acid substitution to alanine at this position leads to significant loss of hACE2 binding in both SARS-CoV-2 (~61% reduction) and RaTG13 (~59% reduction) but not in SARS-CoV-1 (only ~ 12% reduction). The spike protein RBD for SARS-CoV-1 (and RaTG13) are only able to form eight (and eleven) strong electrostatic contacts using seven (and ten) RBD residues, respectively. This does not imply that SARS-CoV-1 and RaTG13 only use these residues to bind to hACE2. More than fifteen additional interface residues either form weak electrostatic contacts or are simply non-interacting. Table 1 lists the hydrogen-bonded interactions between the RBDs and hACE2 along with the corresponding distances. SARS-CoV-2 reforms the original contact map with hACE2 by leveraging 34.1% (15 out of 44) of self-stabilizing contacts around the spike-binding domain to form 21 new complex-stabilizing contacts. SARS-CoV-1 and RaTG13 show weaker attachments as they are able to co-opt only 13.6% and 20.4% contacts, respectively.
Fig. 2.

Leu443 present in the SARS-CoV-1 spike RBD is aligned with Phe456 present in SARS-CoV-2 and RaTG13. In SARS-CoV-2, Phe456 simultaneously interacts with hACE2 residues Thr27 and Asp30 whereas only the hydrophobic contact is observed in RatG13. In SARS-CoV-1, Leu443 is unable to establish neither the backbone electrostatic contact nor the hydrophobic stabilization of the methyl group of Thr27 present in hACE2. The thickness of the dashed lines denotes the strength of interaction.
Fig. 3.

Alanine scan on hACE2 binding residues of spike RBDs of SARS-CoV-2, SARS-CoV-1, and RaTG13 coronavirus. Bars represent the hACE2 Rosetta binding energies upon alanine substitution at the indicated site as a percentage of binding score prior to substitution. SARS-CoV-2 spike RBD appears to be highly optimized for binding hACE2 as the single substitution to more than 90% of the residues forming the RBD to alanine causes more than 50% reduction in binding energy. The symbol × refers to dashes (or gaps) in local sequence alignment of the corresponding RBD chains of the SARS, SARS-CoV-2, and RaTG13, respectively.
Table 1.
List of hydrogen-bonded contacts between the spike RBDs from (SARS-CoV-1, SARS-CoV-2, and RaTG13) and hACE2.
| Sequence ID | Spike residue | hACE2 residue | Distance (Å) |
|---|---|---|---|
| NC_004718_SARS-CoV-1 | Y450 | Q42 | 2.5 |
| Y456 | H34/D30 | 2.8/2.7 | |
| N487 | Q24 | 2.0 | |
| G496 | K353 | 1.8 | |
| T500 | Y41/D355 | 2.6/1.8 | |
| G502 | K353 | 1.9 | |
| NC_045512_SARS-CoV-2 | Y449 | Q42 | 2.0 |
| Q474 | Q24 | 2.9 | |
| Q493 | H34 | 2.8 | |
| S494 | D38 | 1.9 | |
| T500 | Y41 | 1.8 | |
| G502 | K353/Q325/G354 | 2.0/2.4/3.0 | |
| Y505 | R393 | 2.1 | |
| Q506 | Q325 | 2.0 | |
| A475 | S19 | 1.9 | |
| N487 | Q24 | 2.3 | |
| K417 | D30 | 1.9 | |
| MN996532_RaTG13 | K417 | D30 | 1.8 |
| Y473 | T27 | 2.4 | |
| N487 | Q24 | 2.1 | |
| Y493 | H34 | 2.6 | |
| Y498 | Q42 | 1.9 | |
| T500 | Y41 | 1.8 | |
| G502 | K353 | 1.9 |
2.2. In silico alanine scanning to identify spike residues most important for hACE2 binding
Each one of the hACE2 binding residues from the three viral spike RBDs was computationally mutated to alanine (one at a time) and the resultant hACE2-RBD complexes were energy minimized and scored using the Rosetta energy function. This procedure assesses how important is the identity of the native residues by defaulting them to alanine and observing whether this significantly affects binding. The percent loss of hACE2 binding upon substitution to alanine was used as a proxy score for assessing the importance of each RBD residue in binding and subsequent pathogenesis. The results from the alanine scan study (see Fig. 3) reveal that ~90% (19 out of 21) of the hACE2-binding residues of SARS-CoV-2 are important for complex formation. Even a single amino acid substitution to alanine of any of these residues lowers the binding score by more than 60%. These results imply that the SARS-CoV-2 RBDs of the spike protein are highly optimized for binding with hACE2. We note that positions Lys417 and Gly502 have one of the strongest impacts on binding (78% and 63.2% reduction upon changing to Ala, respectively). This is because they help establish one strong electrostatic contact with Asp30, and three with Gln325, Lys353, and Gly354 (as listed in Table 1). The computational alanine scanning results identify the same three residues Phe486, Gln493, and Asn501 to be important for hACE2 binding as proposed by Wan et al. [14]. We find that Phe486, Gln493, and Asn501 each establish three new contacts, consequently their substitution to Ala (even for only one of them) leads to loss of ACE2 binding by more than ~62.5%.
Alanine scanning results of the spike protein RBD of SARS-CoV-1 show less significant penalty to the binding score upon substitution to alanine. Only twelve residues are involved in strong electrostatic coupling with hACE2 residues, out of which six are hydrogen bonded (indicated in Table 1). In summary, alanine scans indicate that SARS-CoV-2 has the highest number of “effectively” interacting residues at the ACE2 binding interface whereas the SARS-CoV-1 spike forms only a few strong hACE2 connectors with a large number of “idle” interface residues (43% – 9 out of 21) which do not affect hACE2 binding upon substitution to alanine. RatG13 appears to be between the two with 13 strong electrostatic interactors (61% – 13 out of 21), out of which seven are hydrogen bonded, and only four idle residues at the interface (i.e., residues Thr484, Leu486, Gly496, and Tyr505).
2.3. Presence of tyrosine and glycine residues in the hACE2 binding domains of these spike proteins
All three viral RBDs are enriched in tyrosine residues. As many as 26.3% (5 out of 19 residues) of the SARS-CoV-1 RBD residues, 25% (4 out of 16 residues) for SARS-CoV-2, and 29% (5 out of 17 residues) for RaTG13 are tyrosine residues. We have not explored the phylogenetic basis for the presence of tyrosine residues but they do seem to be important for conferring high binding affinity spike and hACE2 for both SARS-CoV-2 and RaTG13, as alluded to by the alanine scan results (see Fig. 3). In contrast, the tyrosine residues in SARS-CoV-1 (Tyr442, Tyr475, and Tyr491) only constitute self-stabilizing electrostatic contacts. We use Fig. 4a to explain one representative case of interface tyrosine residues from all three RBDs: SARS-CoV-1 (Tyr442 and Asn473), SARS-CoV-2 (Tyr473 and Tyr489), and RaTG13 (Tyr473 and Tyr489).
Fig. 4.

(a). The role of tyrosine residues in SARS-CoV-2 and RaTG13 RBD is to form strong contacts with hACE2 residues while in SARS-CoV-1 they are primarily responsible for forming stabilizing contacts within the spike and are hence unavailable for hACE2 binding. (b) The role of glycine residues in both all three RBDs is to provide a xGzGx motif for binding hACE2 Lys353 using a strong electrostatic (or cation-π interaction). Here, ‘x’ is a polar residue, and ‘z’ a short chain hydrophobic residue (Ile or Val). The glycine residues along with residue ‘z’ offer a hinge to present polar residue ‘x’ for strong electrostatic interactions with hACE2 residue Lys353.
The SARS-CoV-2 and RaTG13 Tyr473 and Tyr489 backbones, even though present in a loop, are mutually stabilized by hydrogen bonding and the side chains are locked in place by a pi-pi aromatic interaction between the phenyl rings. This enables both of these tyrosine side-chains to form a strong electrostatic contact with the Thr27 side-chain of hACE2. It is thus unsurprising that substitution of either Tyr473 or Tyr489 (in both SARS-CoV-2 and RaTG13) to alanine results in a similar (>58%, respectively as shown in Fig. 3) reduction in binding with hACE2. In contrast, in the energy minimized complex of SARS-CoV-1 RBD with hACE2 both Tyr442 and Tyr475 (see Fig. 4a) only contribute to internal stability of the spike by forming strong electrostatic contacts with RBD residues Trp476 and Asn473. They are therefore unavailable (or too far >6.0 Å) for binding with the neighboring hACE2 residues.
Next, we focus on the role of glycine residues (see Fig. 4b) in all three spike RBDs which form important electrostatic contacts with hACE2 as they lead to more than 55% loss of binding (on average) upon substitution to alanine. We chose to study in detail one such representative glycine from all three spike protein RBDs –Gly488 and Gly490 from SARS-CoV-1 and Gly502 and Gly504 from SARS-CoV-2 and RaTG13.
Interestingly, for all three variants the interaction with the hACE2 residue Lys353 with glycine residues in the spike protein is the same. Atomic coordinates of both these complexes were independently, and experimentally confirmed by Song et al. [16] in 2018 and Wang et al. in 2020 (manuscript unpublished but structure deposited at – www.rcsb.org/structure/6lzg). Both SARS spike RBDs use a combination of a cation-π and a strong electrostatic interaction to bind with Lys353 whereas RaTG13 uses two electrostatic contacts. One electrostatic interaction is mediated by Thr487 in SARS-CoV-1 and Asn501 (and Asp501) in SARS-CoV-2 (and RaTG13). Two glycine residues and a short hydrophobic residue (‘z’ – Val or Ile) brings Thr487, Asn501, and Asp501 for SARS-CoV-1, SARS-CoV-2, and RaTG13, respectively, within strong electrostatic reach of Lys353 while ensuring another cation-π or an electrostatic interaction between Tyr491, Tyr505, and His505 residues, respectively (see Fig. 4b). Amino acid substitution Y491A for SARS-CoV-1 has no effect on hACE2 binding but Y505A (and H505A) in SARS-CoV-2 (and RaTG13) reduces binding by more than 40%. However, alanine substitution to any of the hinge glycine residues leads to >70% loss of hACE2 binding in all three RBD-hACE2 complexes. Thus, we recover the strong functional motif xGzGx in the spike RBD which is conserved between all three SARS-CoV strains.
Analysis of the three hACE2 binding interfaces (see Fig. 5a–c) demonstrate that even though all three spike proteins have a similar number of total interface residues (see Fig. 5f), SARS-CoV-2 establishes more hydrogen bonded contacts (see Fig. 5g) followed by RaTG13 and SARS-CoV-1. Consequently, SARS-CoV-2 exhibits the strongest Rosetta binding energy with hACE2 (see Fig. 5d) calculated using ten unique Rosetta energy minimization trajectories. Interestingly, RaTG13 spike residues occupy the largest number of hACE2 residues resulting in the highest reduction (~14% more than SARS-CoV-2) of solvent accessible surface area (SASA) (see Fig. 5e). Nevertheless, the associated Rosetta binding energy is 11.2% less than the one for SARS-CoV-2 which forms overall stronger hydrogen-bonded contacts.
Fig. 5.

(a–c). hACE2 binding interfaces of the three spike proteins with six hydrogen-bonded contacts from each of them indicated. (d) Rosetta binding energies between spike RBD and hACE2 averaged from ten independent binding energy minimization trajectories. (e) RaTG13 shows the highest reduction of hACE2 solvent accessible surface area (SASA). (f and g) Even though RaTG13 recruits the highest number of interface residues, SARS-CoV-2 forms the most hydrogen-bonded contacts with hACE2. (h) The sequence alignment of the three RBDs is shown and the residues establishing hydrogen bonds with hACE2 are highlighted in cyan. The mean and median binding energies for each batch of ten RBD-hACE2 simulations are denoted with a horizontal line and a cross (×), respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.4. Competitive hACE2 binding of the spike RBDs and angiotensin receptor (ATR1)
Up to this point we examined the biophysical characterization of hACE2 binding with the spike protein. However, in an uninfected cell, through the action of the renin angiotensin system (RAS), hACE2 forms a complex with the angiotensin 2 receptor type I (ATR1) [17]. Deshotels et al. [18] and Lindrooth [19] report that hACE2 forms a complex with ATR1 based on co-immunoprecipitation experiments. Due to the lack of an experimentally resolved structure for the hACE2-ATR1 complex, we used the membrane-bound conformation of hACE2 (in complex with B0AT1 amino acid transporter – PDB id: 6M17; reported by Yan et al. [20]) where the RBD binding region of hACE2 is solvent exposed. We ran 50 ns all-atom MD simulations with the membrane-bound conformation of hACE2 in complex with ATR1 first in the absence of viral spike RBD in the extracellular space, and next with the spike RBD bound to hACE2 as shown in Fang et al. [21] MD results suggest a lowering of hACE2-ATR1 complex stability by 44.2% when RBD was bound to hACE2. Even though there are no shared hACE2 interacting residues between ATR1 and RBD, binding with RBD is predicted to introduce a reversible conformational change (RMSD 5.8 Å) in the seven-residue long Q728-P734 flexible loop that is connected to the C-terminal helix of hACE2 bound to ATR1. This leads to the loss of ~3 hydrogen bonded interactions (and 7.89 kcal/mol of Rosetta binding energy) between hACE2 and ATR1 (see Fig. 6). Residue-level details about the interactions at the hACE2-ATR1 interface (both when RBD is bound or unbound to hACE2) and also at the hACE2-RBD interface, were informed using the Prodigy server [22] and have been listed in Supplementary Data 1.
Fig. 6.

(a–d) Membrane bound conformation of hACE2-ATR1 complex with and without RBD bound to the extracellular domain of hACE2. The orange highlighted region shows the ordered, tightly bound, C-terminal helix of hACE2 that binds to ATR1 which becomes disordered and poorly bound in the presence of RBD. (e and f) The number of hydrogen bonded contacts at the hACE2-ATR1 interface reduces from 5.17 to 2.71 (averaged across a 50 ns MD trajectory) when the RBD binds to hACE2. (g) The RBD-hACE2 interface shows an average of 9 H-bonded interactions. (h–j) The ATR1-hACE2 complex energy is reduced by 120 kcal/mol when RBD is bound to the extracellular domain of hACE2. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Rosetta binding calculations therefore support that SARS-CoV-2 can more effectively bind to hACE2 than ATR1 and also significantly lower the stability of the hACE2-ATR1 complex. We computationally explored the potentially available margin of improvement for the binding affinity of SARS-CoV-2 with hACE2 using the IPRO [23], [24] protein design software. We allowed all 21 contacting residues of the RBD of the spike protein to simultaneously mutate. We run two separate design trajectories and, in both cases the best design achieved an approximately 23% improvement in binding affinity using the Rosetta scoring function. We find (see also Supplementary Fig. 1) seven (G446R, Y449R, Y453K, Y484R, N487D, Q489H, and Q498H) amino acid changes on the RBD that contribute the most in further enhancing hACE2. These predictions are largely in agreement with the exhaustive mutagenesis study by Wang et al. [9] in terms of the strengthening contribution to binding energy of each amino acid change separately. In contrast, for the three remaining residues (Y440, L455, and Y505) of the RBD we predict amino acid changes Y440D, L455K, and Y505E to improve hACE2 binding, whereas Wang et al. [9] report Y440K, L455M, and Y505F to be the best point mutations at these loci. We do recover all single amino acid changes Y440K, L455M, and Y505F (with hACE2 binding improvements of – 2.1%, 2.5%, and 3.3%, respectively) from our simulations but they do not show the highest improvements in hACE2 binding, and hence are not featured in our top predictions. Nevertheless, the overall predicted binding improvement is less than the difference between the calculated binding scores of SARS-CoV-1 and SARS-CoV-2 implying that SARS-CoV-2 has already achieved most of the theoretically possible binding affinity gain with hACE2 compared to SARS-CoV-1. Interestingly, the network of glycine residues in SARS-CoV-2 is conserved in all redesigned RBDs.
A recent report [25] analyzes that humans can transfer SARS-CoV-2 to domesticated animals such as dogs, cats, ducks, and chickens in varying degrees. However, animal-to-human transmission has not been observed [26]. Similar to SARS-CoV-1 [27], felines are more susceptible to SARS-CoV-2 followed by canines [28] whereas chickens and ferrets are less susceptible [28]. The calculated Rosetta binding energies do not follow the trends (R2 = 0.383) expected from simply their respective sequence identities with the human ACE2. Interestingly, even though the ACE2 (Uniprot Entry: G1PKW9_MYOLU) of the little brown bat (Myotis lucifugus) is quite different from human (similarity 84.5%, identity 66.7%), we predict a stronger Rosetta binding energy (by about ~5.6%). This is due to the formation of nine electrostatic contacts and one pi-pi stacking. Strong binding with bat ACE2 may be a consequence of the SARS-CoV-2 origins. In all other cases, the Rosetta binding energies of ACE2 with the spike protein were at most 78.3% of the one calculated with hACE2. We found that feline ACE2 had the closest (78.3% of hACE2-CoV-2) Rosetta binding energy with the spike compared to other pet or livestock animals. Our findings qualitatively agree with Qiu et al. [29] as both imply highest SARS-CoV-2 susceptibility to felines followed by canines. Note that Qiu et al. [29] used an ad hoc metric to rank animal ACE2 susceptibility as a proxy of interaction strengths with SARS-CoV-2 RBD. Thus it is difficult to resolve discrepancies as their result shows birds can serve as hosts for SARS-CoV-2 while our binding calculations say otherwise which corroborates experimental data by Mallapaty et al. [28].
Chan et al. [30] have reported all point mutations in the hACE2 that have been experimentally shown to enhance binding with SARS-CoV-2 RBD. We carried out a computational study using IPRO [23], [24] to identify amino acid substitutions in the 30 hACE2 residues present at the hACE2-RBD binding interface that lead to enhanced RBD binding affinities (see Table 2). We recovered 40 out of 77 binding improving single amino acid substitutions reported by Chan et al. [30] By exploring simultaneous mutations we identify two interface residues changes Phe32 and Ala36 which upon change (i.e., F32A, F32E, A36E, and A36S) can establish electrostatic contacts with the RBD residue Gln493 (see Supplementary Fig. 2). In addition, we computationally predicted eleven double and seven triple amino acid substitutions that lead to improvements in binding interactions by as much as 5.4% (or 2.7 kcal/mol) quantified by Rosetta energy.
Table 2.
Comparison of hACE2 mutations that show enhanced RBD binding from experiments and computational predictions.
| hACE2 residues | Point mutations to amino acids that show enhanced binding with RBD experimentally **(log2 enrichment ratio) |
Computationally predicted hACE2 variants that improve binding to RBD |
||
|---|---|---|---|---|
| Single # (ΔGwt kcal/mol) ↑ |
Double # (ΔGwt kcal/mol) ↑ |
Triple # (ΔGwt kcal/mol) ↑ |
||
| S19 | P > F > Y > W > V (2.1 > 1.2 > 1.1 > 1.1 > 1.1) |
P* > F* > L (2.3 > 2.0 > 1.9) |
S19K + T20H > S19K + T20E (2.4 > 2.2) |
S19K + T20H + I21T > S19K + T20H + E22Q (3.2 > 2.9) |
| T20 | S > E (0.1 > 1.0) |
S* > D > E* (0.5 > 0.3) |
S19K + T20H > S19K + T20E (2.4 > 2.2) |
S19K + T20H + I21T > S19K + T20H + E22Q (3.2 > 2.9) |
| I21 | V > G > T (0.7 > 0.2 > 0.2) |
L > V* (1.2 > 0.8) |
none | S19K + T20H + I21T (3.2) |
| E22 | T > Q > D (1.1 > 0.6 > 0.5) |
Q* > S (1.6 > 1.2) |
none | S19K + T20H + E22Q (2.9) |
| E23 | F > C > M > Q (2.1 > 0.9 > 0.8 > 0.8) |
Y* > F > T (2.2 > 2.0 > 1.9) |
none | None |
| Q24 | T > P (1.8 > 0.1) |
S > T* (2.0 > 1.2) |
none | None |
| A25 | V > T > F > I (2.8 > 1.4 > 1.2 > 1.1) |
S > T* > L (3.2 > 3.1 > 2.2) |
none | None |
| K26 | D > I > V > A > R (2.2 > 1.7 > 1.4 > 1.4 > 1.3) |
D* > E > R* (2.0 > 1.8 > 1.8) |
none | None |
| T27 | Y > L > M > H (2.8 > 2.8 > 2.4 > 2.2) |
S > M* > W > A (3.4 > 3.1 > 1.9 > 1.3) |
T27I + F28D (2.1) |
None |
| F28 | Y (0.8) |
Y* > V (1.2 > 0.4) |
F28Y + D30E > F28D + L29E > T27I + F28D (2.8 > 2.4 > 2.1) |
None |
| L29 | F > E (1.9 > 0.15) |
T > F* (2.3 > 2.1) |
F28D + L29E > L29F + D30I > L29E + A36E (2.4 > 2.2 > 2.1) |
None |
| D30 | E > I > V > T (2.3 > 1.7 > 1.3 > 0.8) |
E* > V* > T* (2.5 > 2.1 > 1.2) |
F28Y + D30E > L29F + D30I (2.4 > 2.2) |
None |
| K31 | W > Y > F (2.6 > 2.0 > 0.8) |
R > W* (3.1 > 1.4) |
none | None |
| F32 | not reported | A > E (2.5 > 1.9) |
F32E + A36E (2.1) |
F32E + A36E + D37P (2.7) |
| N33 | D > E > H > S (1.9 > 0.8 > 0.8 > 0.8) |
E* > D* > H* (1.3 > 1.2 > 1.1 > 1.1) |
none | None |
| H34 | V > A > P > S > W (2.8 > 2.7 > 2.6 > 1.7 > 0.3) |
S* > T > G (3.2 > 3.1 > 2.2) |
H34V + E35D (2.2) |
None |
| E35 | C > D > M > A (1.8 > 1.4 > 1.2 > 0.7) |
D* > M* > V (1.5 > 1.2 > 1.1) |
H34V + E35D (2.2) |
None |
| A36 | not reported | E > S (2.4 > 2.1) |
L29E + A36E > F32E + A36E (2.1 > 2.1) |
F32E + A36E + D37P > A36E + D37P + D38E (2.7 > 2.7) |
| E37 | P (0.9) |
V > P* > A (1.9 > 1.8 > 1.4) |
none | F32E + A36E + D37P > A36E + D37P + D38E (2.7 > 2.7) |
| D38 | E (0.3) |
E* > S (1.2 > 1.0) |
none | A36E + D37P + D38E (2.7) |
| L39 | K > R > I > V > A (2.3 > 2.2 > 1.4 > 1.1 > 1.0) |
R* > K* (2.7 > 2.1) |
L39A + F40H (2.3) |
None |
| F40 | D > R > K > C > H (1.7 > 1.5 > 1.3 > 0.9 > 0.13) |
E > R* (2.0 > 1.8) |
L39A + F40H (2.3) |
F40E + Y41A + Q42K > D40E + Y41A + Q42K (2.8 > 2.3) |
| Y41 | R (1.7) |
F > R* (2.2 > 2.1) |
Y41A + Q42K (2.2) |
F40E + Y41A + Q42K > D40E + Y41A + Q42K (2.8 > 2.3) |
| Q42 | C > L > M > V > I (2.8 > 2.6 > 2.4 > 1.9 > 1.7) |
M* > V* > I* > L* (3.1 > 2.8 > 2.2 > 1.7) |
Y41A + Q42K (2.2) |
F40E + Y41A + Q42K > F40D + Y41A + Q42K (2.8 > 2.3) |
| Q325 | P > K (2.7 > 2.0) |
K* > R (2.8 > 2.2) |
none | Q325K + G326E + A387H > Q325K + G354A + D355E (2.7 > 2.6) |
| G326 | E (1.3) |
D > E* (2.6 > 2.2) |
none | Q325K + G326E + A387H (2.7) |
| K353 | none | none | none | none |
| G354 | none | A (2.4) |
none | Q325K + G354A + D355E (2.6) |
| D355 | none | E (1.7) |
none | Q325K + G354A + D355E (2.6) |
| A387 | H (1.4) |
Q > H* (1.1 > 0.4) |
none | Q325K + G326E + A387H (2.7) |
**log2 enrichment ratios reflect increase in experimentally binding affinity with RBD upon point mutation with respect to wild-type. ^ Increase in RBD binding energy (in kcal/mol) of an hACE2 variant with respect to WT hACE2. * RBD-binding enhancing hACE2 mutations identified from experiments which were recovered from in silico predictions as well.
3. Discussion
In this effort we applied Rosetta binding analysis to gain insight onto possible biophysical factors that may explain the difference in infectivity of SARS-CoV-2 in comparison to SARS-CoV-1 and RaTG13. Multiple lines of computational evidence indicate that the spike RBD binds hACE2 through electrostatic attachment with every fourth residue on the N-terminal alpha-helix (starting from Ser19 to Asn53) as the turn of the helix makes these residues solvent accessible. Results from computational models of canine, feline, bovine, equine, and chicken ACE2 in complex with SARS-CoV-2 spike RBD recapitulates infectivity potential observed so far and pinpoint bat ACE2 as the most highly optimized for binding the SARS-CoV-2 spike protein.
Our computational results are mostly consistent with the exhaustive mutagenesis study on SARS-CoV-2 RBD residues (to assess their effects on hACE2 binding) by Wang et al. [9] where eight out of eleven of our predicted RBD single amino acid substitutions were shown to improve binding. Higher affinity with ACE2 may enable infection of epithelial cell in the upper respiratory track (where ACE2 expression is lower) in contrast to SARS-CoV that primarily infects epithelial cells in the deeper respiratory track (lungs) where ACE2 expression is higher [31], [32], [33], [34]. This is consistent with enhanced person to person infection rates of SARS-CoV-2 through exhaled droplets or aerosols. The prevalence of amino acid change D614G has been widely reported [35], [36] to have increased from less 10% originally to over 95% in late spring of 2020 [37]. Even though a founder effect cannot be excluded as the reason, increased conformational flexibility [37] of the spike may enhance SARS-CoV-2 ability to access cells. It is clear that binding with ACE2 remains unchanged as neither our computational analysis nor the exhaustive mutagenesis effort by Wang et al. [9] showed any difference. This is expected as residue 614 is more than 16 Å away from the residues that directly interact with the spike. Zhang et al. [36] recently reported that the amino acid substitution D614G leads to less S1 shedding and easier incorporation of S-protein into a pseudo-virion (retrovirus pseudo-typed with spikeD614G) leading to more efficient infection of hACE2-expressing cells. Their binding experiments agree with our computational finding that the D614G mutation does not cause stronger binding with hACE2.
4. Methods
4.1. Binding energy calculations
We have used experimentally determined coordinates of SARS-CoV-1 and SARS-CoV-2 in complex with ACE2 (PDB accessions: 6ACG [16] and 6LZG – www.rcsb.org/structure/6lzg, respectively). RaTG13 RBD model was built using the iTasser program [38]. Similarly, unbound ATR1 structure (PDB: 4YAY [39]) was also separately downloaded and docked against hACE2 using protein–protein docking scripts from Z-DOCK 3.0 [40]. ZDOCK uses pairwise shape-complementarity, electrostatics, and implicit solvation terms in scoring the docked poses. Implicit solvation treats the water as a dielectric continuum. The rotational sampling interval was set to 10°. Clustering of the docked poses were done at an 8 Å cutoff. Subsequently, PyRosetta [41] scripts were written to rank and identify the most stable complexes from each cluster which were then energy-minimized and re-ranked. Finally, the complex which ranked high in stability and binding scores was chosen as the model. An alanine scan was again performed using PyRosetta scripts, where the computational models of the alanine variants were first generated, energy minimized, and hACE2 binding scores computed. The hACE2 interface definitions for each binding partner (RBDs and ATR1) were obtained by feeding the energy minimized protein–protein complexes through the find_contacts module of OptMAVEn-2.0 [42]. We used the three-dimensional atomic coordinates of the experimentally determined human ACE2 (hACE2) in complex with SARS-CoV-2 spike RBD (PDB id: 6ZLG https://www.rcsb.org/structure/6lzg) as a backbone template to repackage the updated residue side-chains of bat, feline, canine, bovine, equine, and chicken ACE2. A python script was prepared to execute multiple times the iTasser program [38]. First, a fragment structure assembly was performed using replica-exchange Monte Carlo [43] followed by clustering of decoy ACE2 structures generated using the SPICKER protocol [44]. Finally atomic-level backbone and side chain refinement was performed using fragment-guided molecular dynamics simulations (FG-MD) [45] for 50 ns for each structure. All five ACE2s were subsequently docked with the SARS-CoV-2 spike RBD protein whose 3D coordinates were downloaded from the hACE2-spike complex (PDB id: 6LZG). Rosetta binding energy calculations were performed by first running the relax application with the ref2015 [46] energy function and extracting binding energy (dG_binding) using the InterfaceAnalyzer [47] application.
4.2. Molecular dynamics simulations
The ATR1-hACE2 (with and without RBD) complex models were prepared for molecular dynamics (MD) simulation using protein preparation wizard [48] of Maestro wherein the hydrogen bonding network was optimized and a heavy atom-restrained minimization with the OPLS_2005 [49] force field was carried out. MD simulations were performed using the Desmond [50] application (v2.3) within the Schrodinger software suite (v2019.4). The initial positioning of the structure in the membrane was obtained from .opm file for ATR1 downloaded from the OPM database [51]. Then, the membrane-bound structure was prepared using Desmond system builder. 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) membrane per-equilibrated at 310 K was chosen and SPC solvent molecules were added to the top and bottom of membrane. The residual charges from the system were neutralized by adding Na+ and Cl− ions to maintain 0.15 M salt concentration. The default relaxation protocol of Desmond was performed followed by a 50 ns of production simulation at 1 atm pressure and 310 K temperature, using the NPT ensemble under a periodic boundary condition using particle mesh Ewald. Time step of 2.0 fs and nonbonded cutoff threshold of 9 Å were imposed. Finally, the SHAKE [50] algorithm was used to keep all bonds involving hydrogen atoms rigid.
Author contributions
RC, and CDM conceived, and designed the study. VSB helped in performing the MD simulations. All three authors wrote the study.
CRediT authorship contribution statement
Ratul Chowdhury: Conceptualization, Methodology, Software, Investigation, Writing - original draft, Writing - review & editing, Visualization. Veda Sheersh Boorla: Software, Investigation, Writing - review & editing. Costas D. Maranas: Conceptualization, Writing - review & editing, Supervision, Project administration, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
This activity was primarily supported by the United States Department of Agriculture (USDA) NIFA Award 2020-67015-32175 and also partially enabled by funding provided by The Center for Bioenergy Innovation a U.S. Department of Energy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science (DE-SC0018420DE-AC05-000R22725) and NSF award CBET1703274. All simulations were performed on the Institute for Computational and Data Sciences Advanced CyberInfrastructure (ICDS-ACI) high-performance computing (HPC) facility at the Pennsylvania State University. RC thanks Debolina Sarkar for advice on the renin angiotensin system and also editing the paper.
Competing financial interests
The authors declare no competing financial interests.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.09.019.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Hoffmann M. SARS-CoV-2 Cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020 doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shang J. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;1–8 doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Q. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020 doi: 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asai A. COVID-19 drug discovery using intensive approaches. Int J Mol Sci. 2020 doi: 10.3390/ijms21082839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuba K. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walls A.C. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020 doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan R. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science (80-) 2020 doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe Y., Allen J.D., Wrapp D., McLellan J.S., Crispin M. Site-specific analysis of the SARS-CoV-2 glycan shield. bioRxiv. 2020 doi: 10.1101/2020.03.26.010322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y., Liu M., Gao J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc Natl Acad Sci. 2020;117:202008209. doi: 10.1073/pnas.2008209117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brielle E.S., Schneidman-Duhovny D., Linial M. The SARS-CoV-2 exerts a distinctive strategy for interacting with the ACE2 human receptor. Viruses. 2020;12:497. doi: 10.3390/v12050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spinello A., Saltalamacchia A., Magistrato A. Is the rigidity of SARS-CoV-2 spike receptor-binding motif the hallmark for its enhanced infectivity? insights from All-atom simulations. J Phys Chem Lett. 2020;11:4785–4790. doi: 10.1021/acs.jpclett.0c01148. [DOI] [PubMed] [Google Scholar]
- 12.Amin M., Sorour M.K., Kasry A. Comparing the binding interactions in the receptor binding domains of SARS-CoV-2 and SARS-CoV. J Phys Chem Lett. 2020;11:4897–4900. doi: 10.1021/acs.jpclett.0c01064. [DOI] [PubMed] [Google Scholar]
- 13.Starr TN. et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. bioRxiv 2020.06.17.157982 (2020) doi:10.1101/2020.06.17.157982. [DOI] [PMC free article] [PubMed]
- 14.Wan Y., Shang J., Graham R., Baric R.S., Li F. Receptor recognition by novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS. J Virol. 2020 doi: 10.1128/jvi.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lam S.D. SARS-CoV-2 spike protein predicted to form stable complexes with host receptor protein orthologues from mammals, but not fish, birds or reptiles. biorxiv. 2020 doi: 10.1038/s41598-020-71936-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song W., Gui M., Wang X., Xiang Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLOS Pathog. 2018;14 doi: 10.1371/journal.ppat.1007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tikellis Chris, Thomas M.C. Angiotensin-converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int J Peptides. 2012 doi: 10.1155/2012/256294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deshotels M.R., Xia H., Sriramula S., Lazartigues E., Filipeanu C.M. Angiotensin II mediates angiotensin converting enzyme type 2 internalization and degradation through an Angiotensin II type I receptor-dependent mechanism. Hypertension. 2014 doi: 10.1161/HYPERTENSIONAHA.114.03743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charis Lindrooth. The role of ACE2 and treatment implications for COVID-(2020).
- 20.Yan R. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science (80-) 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang L., Karakiulakis G., Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Resp Med. 2020 doi: 10.1016/S2213-2600(20)30116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xue L.C., Rodrigues J.P., Kastritis P.L., Bonvin A.M., Vangone A. PRODIGY: A web server for predicting the binding affinity of protein-protein complexes. Bioinformatics. 2016 doi: 10.1093/bioinformatics/btw514. [DOI] [PubMed] [Google Scholar]
- 23.Pantazes R.J., Grisewood M.J., Li T., Gifford N.P., Maranas C.D. The Iterative Protein Redesign and Optimization (IPRO) suite of programs. J Comput Chem. 2015;36:251–263. doi: 10.1002/jcc.23796. [DOI] [PubMed] [Google Scholar]
- 24.Chowdhury R. IPRO+/−: Computational protein design tool allowing for insertions and deletions. Structure. 2020;3 doi: 10.1016/j.str.2020.08.003. [DOI] [PubMed] [Google Scholar]
- 25.Chen, H. Susceptibility of ferrets, cats, dogs, and different domestic animals to SARS-coronavirus-2. bioRxiv (2020) doi:10.1101/2020.03.30.015347. [DOI] [PMC free article] [PubMed]
- 26.Xu J. Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses. 2020 doi: 10.3390/v12020244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peiris JSM., Poon LLM. Severe acute respiratory syndrome (SARS). in Encyclopedia of virology (2008). doi:10.1016/B978-012374410-4.00780-9.
- 28.Mallapaty S. Coronavirus can infect cats - dogs, not so much. Nature. 2020 doi: 10.1038/d41586-020-00984-8. [DOI] [PubMed] [Google Scholar]
- 29.Qiu Y. Predicting the angiotensin converting enzyme 2 (ACE2) utilizing capability as the receptor of SARS-CoV-2. Microbes Infect. 2020 doi: 10.1016/j.micinf.2020.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan K.K. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science (80-) 2020;1265:eabc0870. doi: 10.1126/science.abc0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li G. Assessing ACE2 expression patterns in lung tissues in the pathogenesis of COVID-19. J Autoimmun. 2020 doi: 10.1016/j.jaut.2020.102463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jia H.P. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol. 2005 doi: 10.1128/jvi.79.23.14614-14621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu H. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int J Oral Sci. 2020 doi: 10.1038/s41368-020-0074-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pinto B.G.G. ACE2 expression is increased in the lungs of patients with comorbidities associated with severe COVID-19. J Infect Dis. 2020 doi: 10.1093/infdis/jiaa332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grubaugh N.D., Hanage W.P., Rasmussen A.L. Journal pre-proof making sense of mutation: what D614G means for the COVID-19 pandemic remains unclear. Cell. 2020 doi: 10.1016/j.cell.2020.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang L. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv. 2020 doi: 10.1101/2020.06.12.148726. [DOI] [Google Scholar]
- 37.Korber B. Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020 doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J., Zhang Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015 doi: 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Towler P. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem. 2004 doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mintseris J. Integrating statistical pair potentials into protein complex prediction. Proteins Struct Funct Genet. 2007 doi: 10.1002/prot.21502. [DOI] [PubMed] [Google Scholar]
- 41.Chaudhury S., Lyskov S., Gray J.J. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics. 2010;26(5):689–691. doi: 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chowdhury R., Allan M.F., Maranas C.D. OptMAVEn-2.0: De novo design of variable antibody regions against targeted antigen epitopes. Antibodies. 2018;7:23. doi: 10.3390/antib7030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Swendsen R.H., Wang J.S. Replica Monte Carlo simulation of spin-glasses. Phys Rev Lett. 1986 doi: 10.1103/PhysRevLett.57.2607. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y., Skolnick J. SPICKER: A clustering approach to identify near-native protein folds. J Comput Chem. 2004 doi: 10.1002/jcc.20011. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J., Liang Y., Zhang Y. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure. 2011 doi: 10.1016/j.str.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alford R.F. The Rosetta all-atom energy function for macromolecular modeling and design. J Chem Theory Comput. 2017;13:3031–3048. doi: 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benjamin Stranges P., Kuhlman B. A comparison of successful and failed protein interface designs highlights the challenges of designing buried hydrogen bonds. Protein Sci. 2013;22:74–82. doi: 10.1002/pro.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Madhavi Sastry G., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 49.Kaminski G.A., Friesner R.A., Tirado-Rives J., Jorgensen W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 50.Bowers, K. J. et al. Scalable Algorithms for molecular dynamics simulations on commodity clusters. in 43–43 (Institute of Electrical and Electronics Engineers (IEEE), 2007). doi:10.1109/sc.2006.54.
- 51.Lomize M.A., Pogozheva I.D., Joo H., Mosberg H.I., Lomize A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370–D376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.