

Abstract
The complex and heterogeneous pathophysiology of schizophrenia can be deconstructed by integration of large-scale datasets encompassing genes through behavioral phenotypes. Genome-wide datasets are now available for genetic, epigenetic and transcriptomic variations in schizophrenia, which are then analyzed by newly devised systems biology algorithms. A missing piece, however, is the inclusion of information on the proteome and its dynamics in schizophrenia. Proteomics has lagged behind omics of the genome and transcriptome since analytic platforms of proteins were previously not as robust as those for nucleic acids. In recent years, however, there has been an unprecedented progress in the instrumentation (liquid chromatography (LC) and mass spectrometry (MS)), experimental paradigms, and bioinformatics of the proteome. Large-scale analyses of the schizophrenia proteome are now possible and ought to be pursued vigorously and integrated with other omics results. With that in our view, we review proteomics studies that have been conducted in schizophrenia to date, present a summary of methodological innovations of recent years in MS based proteomics and the power of new generation proteomics, and propose how such data can be analyzed and integrated with other omics results. Unlike DNAs or RNAs, the function of protein is determined by multiple molecular properties, i.e., subcellular localization, posttranslational modification (PTMs) and protein-protein interactions (PPIs). Thus, how to assess and incorporate these properties poses additional challenges in proteomics and their integration with other omics; yet will be a critical next step to close the loop of multi-omics integration.
Keywords: schizophrenia, proteomics, omics
1. Introduction:Proteomics and multi-omics integration in schizophrenia
One of the most exciting developments in recent neuropsychiatric research is the introduction of omics technologies and perspectives (Manzoni et al., 2018; Wang et al., 2018a; Willsey et al., 2018). These methodologies enable us to generate genome-wide datasets (Fromer et al., 2014; Schizophrenia Psychiatric Genome-Wide Association Study, 2011; Schizophrenia Working Group of the Psychiatric Genomics, 2014), link genes to transcriptome to phenotypes and consider pathophysiologic mechanisms (Fromer et al., 2016; Jaffe et al., 2016; Jaffe et al., 2018). In schizophrenia, genome-wide studies of common and rare genetic variants have been conducted in tens of thousands of patients and controls (Schizophrenia Working Group of the Psychiatric Genomics, 2014) and epigenomic changes and their impact on chromatin conformation are also being extensively studied on large scale samples (Girdhar et al., 2018; Hwang et al., 2016; Jaffe et al., 2016). Aided by newly devised systems biology algorithms, these massive datasets are now integrated to identify specific pathways bearing susceptibility to the illness (Gandal et al., 2018a; Gandal et al., 2018b; Li et al., 2018). Perhaps the most challenging element in schizophrenia studies lies with the complex makeup of the pathophysiology and its heterogeneity among patients. To that end, multi-omics investigations on large patient populations and their integration are essential.
Notably, presently available omics data are predominantly those of nucleic acids, i.e., genomics, epigenomics and transcriptomics, while inclusion of proteomics is relatively limited. Proteins are the biological entity that perform the function of genes and as such reflect functional consequences of most variations of genome, epigenome and transcripts. Transcripts are most often studied as proxies of protein levels or indicators of gene function, while it is understood that protein and mRNA levels correlate between 40 to 60% at best (Nagaraj et al., 2011; Schwanhausser et al., 2011). Moreover, the function of proteins is determined not just by abundance but by their physical and biochemical properties, including subcellular localization, posttranslational modifications and protein – protein interactions (PPIs) (Larance and Lamond, 2015; Yugi et al., 2016). Thus, functional genomics investigations should be inclusive of in-depth analyses of the proteome and analyses of the physical and biochemical properties of proteins.
Proteomic investigations have been hampered by a slower pace in the development of assay platforms compared to those for DNAs and RNAs (Arora and Somasundaram, 2019; Arrington et al., 2017; Li et al., 2017a). Over the last decade, however, there has been a dramatic improvement in mass spectrometry (MS)- based proteomics assay platforms (Hada et al., 2018; Mardamshina and Geiger, 2017). Newly devised mass spectrometers are equipped with the resolution, accuracy and speed that were unimaginable even a few years ago (Baker et al., 2017; Cai et al., 2016; Hada et al., 2018; Mardamshina and Geiger, 2017). In parallel, there has been a remarkable development in software and search engines, which permit rapid and detailed data processing as well as bioinformatic analyses of data. These together mark a new era in MS based proteomics, in which assessment of the proteome on a genome-wide basis is within reach. Large-scale investigations of the proteome should therefore be fully incorporated into multi-omics investigations of schizophrenia.
The goal of this article is to review the current status of proteomics investigations of schizophrenia, consider future investigations in the field and the potential impact of such studies to our pathophysiologic understanding of schizophrenia. To that end, we will first review proteomics studies in schizophrenia (primarily postmortem investigations) to date and provide a summary of new technologies, which permit future proteomic investigations of a deeper and broader scope. Finally, we will discuss how these proteomics results have been and will be analyzed and integrated in a multi-omics context.
2. Proteomics, phosphoproteomics and PPI analyses in schizophrenia
Proteins are the pillars for the structural fabric of cells and underpin most metabolic and regulatory processes (Larance and Lamond, 2015). Such functions of proteins are determined not only by the abundance of the proteins but also several properties of the molecules. To perform their cellular functions, proteins should travel to specific subcellular microdomains, interact with other proteins (protein-protein interactions, PPIs, and potentially also undergo chemical modifications (posttranslational modifications. PTMs) (Larance and Lamond, 2015). These properties therefore should be incorporated into comprehensive analyses of protein function. Multiple groups have conducted proteomic studies in schizophrenia by examining postmortem brains of patients and mouse models. The majority of previous studies have so far examined the abundance of proteins, either in the whole cell or subcellular locale, while PTMs and PPIs are yet to be further investigated.
2.1. Protein abundance in whole cells and subcellular microdomains
Among the early postmortem proteomic studies in schizophrenia (detailed in Table 1) were analyses of whole cell extracts. The anterior cingulate cortex (ACC), prefrontal cortex (PFC) or hippocampus, was extracted as a whole in detergents or chaotropic agents. The extracts were then analyzed by 2D gel electrophoresis (2D GE) followed by MS analyses employing MALDI-TOF or LTQ (Beasley et al., 2006; Clark et al., 2007; Focking et al., 2011; Martins-De-Souza et al., 2010; Nesvaderani et al., 2009; Pennington et al., 2008). In most of these studies, the sample size ranged between 8 to 15 in each group and the assessment was semi-quantitative based on spectral counting. Results were reported typically in two ways: differentially expressed proteins and/or their enrichment in specific pathways. Notably, the lists of proteins that were found to be altered in the patient group were not highly congruent between studies. However, the pathways enriched among differentially regulated proteins were more similar between studies. For instance, three different groups (Beasley et al., 2006; Clark et al., 2006; Martins-De-Souza et al., 2010) examined the ACC using 2DE/ MALDI-TOF. These groups found vastly different lists of proteins altered in the patient group, while pathways for synaptic signaling or metabolism were found to be shared between these studies.
Table 1:
Proteomic Studies in Schizophrenia.
| cohort | region | extracts | Proteomic method | # altered proteins | altered proteins | Protein groups/pathways | |
|---|---|---|---|---|---|---|---|
| Whole cells | |||||||
| Clark et al., 2006 | 10 SCZ1 10 CTRL1 |
ACC | whole cell | 2D-E MALDI-TOF MS |
SCZ −36 | Table 4 in Clark et al., 2006 | synaptic, signaling, metabolic and oxidative stress, trafficking, cytoskeletal and glial-specific proteins |
| Clark et al., 2007 | 10 SCZ1 10 CTRL1 |
ACC | whole cell | 2D-E MALDI-TOF MS |
SCZ - 27 | Table 3 in Clark et al., 2007 | functionally classified as metabolism, cytoskeleton, synapse |
| Sivagnanasu et al., 2007 | 10 SCZ1 10 CTRL1 |
CC genu | whole cell | 2D-E Maldi-TOF |
SCZ −34 21 altered L/R hemisphere |
Table 2 in Sivagnasundaram et al., 2007 |
Human Proteome Database cytoskeletal structure and function, neuroprotective function and energy metabolism |
| Pennington et al., 2008a | 35 SCZ2 35 BPD2 35 CTRL2 |
DLPFC | whole cell | 2D DIGE 2D linear Ion trap |
SCZ −9 BPD - 45 Both - 6 |
PASCIN 1 DYN NF-L |
SCZ: synaptic proteins (7 of the 15) in septin family |
| Martins-de-Souza et al., 2010 | 11 SCZ3 (6F/5M) 8 CTRL4 (2F/6M) |
ACC | whole cell | 2D-E MALDI-TOF MS |
SCZ - 28 Males 11 Females 7 |
WB validation – SCZ-CTRL DPYSL2, CRYABPRDX6 WB validation –M/F GLUL |
Human Protein Reference Database
Communication/signal transduction Metabolism/energy |
| Saia-Cereda et al., 2105 | 9 SCZ3 5 CTRL4 |
CC | Cytoplasmic | LTQ orbitrap XL spectral counting |
SCZ - 65 | Table 2 in Saia-Cereda et al., 2015 |
Ingenuity:
energy metabolism, cell communication and signaling and cell growth and maintenance |
| MacDonald et al., 2015 | 22 SCZ5 23 CTRL5 |
AC Gray matter |
whole cell | LC-SRM/MS | SCZ- 155 | Table 2 in MacDonald et al., 2015 |
DAVID: Glutamate signaling pathway Co-expression network analysis Clathrin coated vesicle membrane, NADH binding |
| Saia-Cereda et al., 2106 | 5 SCZ6 5 CTRL4 |
CC | whole cell | 2D-RP/RP multiplexed (DIA) |
SCZ - 56 differentially phosphorylated - 68 | Tables 2 and 3 in Saia-Cereda et al., 2016 | Ingenuity: Phosphorylation CTNF pathway incl. PI3K, mTOR ephrinB |
| Whole cell laser dissected | |||||||
| Pennington et al., 2008b | 12 SCZ2 13 CTRL2 |
Insular cortex layer 2 | whole cell Laser dissected |
2D-DIGE 2D linear ion trap |
in SCZ or BPD or both - 19 | Table 2 in Pennington et al., 2008b WB validation: DRP-2 a-synuclein |
Most affected categories: cell communication/signal transduction protein metabolism |
| Focking et al., 2011 | 20 SCZ2 20 BPD2 20 CTRL2 |
Mid-HC cornu ammonis regions 2 and 3 |
whole cell Laser dissected |
2D-DIGE mass spectrometry |
common to more than one HC region SCZ - 32 BPD - 38 |
WB Validation:
PCMT1, SPTAN1, ARMCX1, ANXA6 SEPT11, FSCN1 |
Ingenuity (SCZ) Cellular assembly and organization Cellular compromise Cell morphology Cell signaling Cell to cell signaling and interaction |
| Nuclear | |||||||
| Saia-Cereda et al., 2107 | 12 SCZ3 8 CTRL4 |
CC (white matter) ATL (gray matter) |
Nuclear enrichment | 2D-E LTQ orbitrap |
SCZ Nuclear proteins CC – 552 ATL – 224 |
Table 2 in Saia-Cereda et al., 2017 |
Reactome for proteins commonly regulated between the two regions: Cellular stress response Heat shock proteins STRING:Nuclear protein specific: CC-calcium-calmodulin ATL spliceosome |
| Membrane | |||||||
| Behan et al., 2009 | Proteomic studies : pooled samples 10 SCZ2 10 BPD2 10 CTRL2 Validation Studies: 20 subjects/grp2 10 subjects/ group7 |
DLPFC | Proteomics: membrane microdomains Validations: whole cell |
2D-DIGE RP-LC-MS/MS |
in one or both disorders −16 |
WB Validation
Incr. BPD and SCZ in: Stanley Brains STXBP1, BASP1 LAMP Harvard Brains STXBP1, BASP1 |
NA |
| Synaptic | |||||||
| Smalla et al., 2008 | 8 SCZ8 8 CTRL8 |
DLPFC | synaptic structures | 2D-E MALDI-TOF |
SCZ – 41 |
WB validation: Prohibitin common alteration between SCZ AND ketamine treated rats |
NA |
| Velasquez et al., 2017 | 8 SCZ6 8 CTRLS4 pooled |
PFC | synaptosomes | iTRAQ LTQ orbitrap Q-Exactive |
SCZ iTRAQ −12 label free −55 |
Limbic system associated protein Alpha-calcium/calmodulin-dependent protein kinase II confirmed by PRM |
DAVID and Reactome:
Dysregulated by both methods: synaptic activity signaling pathways associated with calcium |
| Post synaptic density | |||||||
| Focking, Lopez, et al., 2015 | 20 SCZ2
20 CTRL2 2 sample pooled |
ACC | PSD | label free LC-MS Q Exactive | SCZ-143 (25 after correction for FDR) |
WB validation: AP2B1 DNM1 MAPK3 SYNPO |
DAVID NIH: KEGG Endocytosis Calcium signaling Long-term potentiation (FDR signif) Neurotrophin signaling (FDR signif) |
NSW Tissue Resource Centre (University of Sydney, NSW, Australia),
Stanley Brain Collection,
Norbaden, Germany,
Heidelberg Germany,
University of Pittsburgh,
State Mental Hospital Weisloch, Germany,
Harvard,
New Magdeberg Brain Collection. Abbreviations: SCZ, Schizophrenia; BPD, Bipolar Disorder; CTRL, Control; DLPFC, Dorsolateral Prefrontal Cortex; ACC, anterior cingulate cortex; AC, auditory cortex; CC, corpus collosum; HC, hippocampus; ATL, anterior temporal lobe.
Study of subcellular fractions permits a detailed analysis of the proteome enriched in the microdomain and can offer a basis for understanding the intracellular trafficking of those molecules. Among various subcellular microdomains, synaptic membranes (or synaptosomes), postsynaptic density (PSD) and nuclear enrichments have been studied.
Multiple lines of evidence have implicated synaptic signaling in schizophrenia (Kirov et al., 2012; Schizophrenia Psychiatric Genome-Wide Association Study, 2011; Schizophrenia Working Group of the Psychiatric Genomics, 2014) and synaptic membranes harbor these signaling pathways. Several groups have examined postmortem brains by extracting synaptosome (or synaptic membranes) as detergent insoluble fractions (Smalla et al., 2008) or by ultracentrifugation of sucrose density gradients (Velasquez et al., 2017). Smalla et al. reported enrichment of altered proteins in metabolic pathways, synaptic signaling and also prohibitin (Smalla et al., 2008). Velasquez et al employed iTRAQ and label free quantification using a hybrid orbitrap platform and reported signaling transduction, cell adhesion and calcium signaling as dysregulated in the patient group (Table 1). Given the small sample sizes therein, the results of these studies are to be verified in future investigations.
The postsynaptic density (PSD) is a specialized microdomain and a hub for postsynaptic signaling including glutamatergic and trophic pathways. Many genome wide association studies of common or rare variants, have demonstrated that the PSD and signaling therein are associated with schizophrenia (Fromer et al., 2014; Kirov et al., 2012; Purcell et al., 2014; Szatkiewicz et al., 2014). Notably, the molecular architecture of the PSD offers a landscape in which these pathways interact with each other and are integrated in a concerted fashion (Li et al., 2017b). Thus, the PSD is a microdomain where molecular alterations of pathophysiologic significance can converge (Banerjee et al., 2010; Hahn, 2011).
PSD enriched fractions can be isolated reproducibly from postmortem brains, when procedures are carefully conducted and verified (Hahn et al., 2009; MacDonald et al., 2012). Focking et al isolated PSD enrichments and conducted label free quantification employing a hybrid orbitrap platform (Focking et al., 2015). About 25% of the identified proteins were altered in the patient group before multiple comparison corrections. The pathway analysis indicated long term potentiation, calcium signaling and trophin signaling, which are well aligned with the pathways implicated in genetics studies (Kirov et al., 2012; Schizophrenia Psychiatric Genome-Wide Association Study, 2011; Schizophrenia Working Group of the Psychiatric Genomics, 2014). In parallel, the same group examined the ACC of BD patients and found altered proteins enriched in metabolic pathways including oxidative phosphorylation, mitochondrial function and protein translation (Focking et al., 2016).
Several developmental rodent models relevant to schizophrenia have been investigated in proteomics studies. These include methylazoxymethanol acetate (MAM) (Chalkiadaki et al., 2019; Hradetzky et al., 2012; Kisby et al., 2006), maternal immune activation (MIA) with Poly I:C (Deng et al., 2011; Oh-Nishi et al., 2016), prenatal stress (Lee et al., 2015) and social isolation rearing (Roncada et al., 2009). These studies employed various platforms; most commonly 2D gel electrophoresis (2D GE) followed by MS analyses employing MALDI-TOF (Deng et al., 2011; Kisby et al., 2006; Roncada et al., 2009), Q-TOF (Hradetzky et al., 2012), or nano LC-orbitrap (Chalkiadaki et al., 2019; Oh-Nishi et al., 2016). Studies have involved differing brain regions; the frontal cortex, hippocampus, cerebellum and striatum. Some of these findings parallel those already seen in schizophrenia brains, such as alterations in glutamatergic pathways in a recent study in mouse prefrontal cortex (Chalkiadaki et al., 2019). Notably, these studies are have little overlap with respect to the rodent species, age, sex or brain region, leaving opportunities for replication before such findings provide a solid basis for studies in schizophrenia.
Postmortem transcriptomics data are already available from large cohorts of schizophrenia investigations (Fromer et al., 2016; Hwang et al., 2016; Jaffe et al., 2018). Previous postmortem proteomic studies of whole cell extracts studies suffered from small sample size, suboptimal quantitative accuracy and also limited coverage of the brain proteome. The next step should be extending these studies to large postmortem cohorts using cutting edge MS platforms (as described below). Assessment of protein abundance will have functional valence only when it is conducted in a specific locale in the brain. This includes specific cortical layers, subcellular microdomains and specific cell types beyond brain areas and sub regions. This will be discussed further below.
2.2. Phosphoproteomics
Protein phosphorylation is a posttranslational modification (PTM) that affects serine, threonine and tyrosine residues (Arrington et al., 2017). Phosphorylation incorporates a negatively charged phosphate group, which in turn affects the conformation of the proteins as well as its interactions with other proteins (Junger and Aebersold, 2014). As such, protein phosphorylation is involved with virtually all cellular functions and its alterations are involved with many illnesses.
Historically, analysis of phosphorylation of a large-scale proteome (phosphoproteomics) had been a methodological challenge. However, this is no longer the case since the development of methodologies for enrichment of phosphopeptides and the recent advances in MS instrumentation; liquid chromatography (LC), software and search engines described below.
As in proteomics, phosphoproteomics can be conducted in the discovery (global) or targeted mode. In global phosphoproteomics, cell/tissue homogenates harboring the phosphoproteome are first digested, then enriched for phosphopeptides using the immobilized metal affinity chromatography (IMAC) (Wolf-Yadlin et al., 2007a), metal oxide affinity chromatography (MOAC), or immunoprecipitation (IP) with antibodies for phosphorylated residue (e.g., pTyr) (Leitner, 2016). Phosphopeptides can now be fractionated and analyzed by nano LC-MS/MS. Targeted phosphoproteomics are employed for more accurate quantification of phosphorylation dynamics for specific sets of proteins targets. Here, the workflow aims for quantification of site-specific phosphorylation using SRM/PRM or data-independent acquisition (DIA) for specific signaling proteins (Adachi et al., 2016; Lawrence et al., 2016; Wolf-Yadlin et al., 2007b).
Presently, the workflow combining a phosphopeptide enrichment method with LC-MS/MS can identify 10,000 phosphorylation sites (de Graaf et al., 2014; Huttlin et al., 2010). Added with multi-dimensional separations, such as strong cation exchange (SCX) fractionation and IMAC or TiO2 enrichment, the coverage rises to 30,000 phosphosites (Huttlin et al., 2010; Sharma et al., 2014). In targeted phosphoproteomics analyses, IMAC-SRM based quantification of up to hundreds of phosphorylated sites in various signaling pathways is readily feasible (de Graaf et al., 2014; Huttlin et al., 2010).
Large scale phosphoproteomic analyses have been introduced to pathophysiologic and biomarker studies of various illnesses (Ke et al., 2016; Venerando et al., 2017). In cancer biology, for instance, there have been many studies and some of their findings are utilized for clinical application (Blume-Jensen and Hunter, 2001; Huttlin et al., 2010; Zanivan et al., 2013). More recently, these methods have been extensively applied to brain disorders including neurodegenerative illnesses (Johnston-Wilson et al., 2000; McGuire et al., 2017; Swatton et al., 2004). Schizophrenia and other psychiatric illnesses, however, are in a very early stage of phosphoproteomic investigation.
With the recent advancement of MS technologies, we envision the following steps in developing phosphoproteomics of postmortem studies in schizophrenia. These studies may entail a) identification and quantification of phosphorylation sites in specific brain regions, b) extending them to their temporal and spatial dynamics and c) integrating them with proteomics and then with other omics results. Presently, there are several groups in the field who have launched this line of investigation in postmortem cohorts of various sources (Saia-Cereda et al., 2016) and the field will soon be populated with the results. The challenge, however, is how to address postmortem confounds and their impact on protein phosphorylation and its stability. For instance, Li et al has shown that 90% of serine phosphorylation of GSK-3 can be lost in two minutes (Li et al., 2005). An important next step will be to establish how the rapid dephosphorylation in the postmortem condition can be controlled experimentally and how the results should be interpreted accordingly.
2.3. Protein – protein interactions (PPIs)
Protein function is modulated by its association with binding partners most often within the context of a specific protein complex. As such, the protein complexes and PPIs therein constitute a molecular context that determines a specific cellular function of the molecule. Therefore, PPIs and the protein composition of protein complexes should be incorporated into our understanding of the functional properties of proteins.
Historically, PPIs were studied with methodologies by which singular association between the two proteins was analyzed one at a time, e.g., by Western analysis. The results were thus binary and inclusive of only a small number of proteins in protein complexes. This limitation has been overcome with the advent of multiple new methodologies, including immunoprecipitation- mass spectrometry (IP-MS), cross linking – mass spectrometry (XL-MS), as well as cryo electron microscopy (EM) (Smits and Vermeulen, 2016). These methods now enable us to identify and quantify many proteins within protein complexes, assess stoichiometry and topology of PPIs and their temporal and spatial dynamics under various physiological or disease conditions.
The current IP-MS methodology, allows us to delineate many PPIs in a protein complex. In this method, protein complexes are captured by antibodies for the core molecule and immune-precipitates (IPs) are typically analyzed by LC-MS/MS. A few challenges previously hampered accurate identification and quantification of PPIs using this method. First, antibodies for proteins of interest are not always of the optimal sensitivity and specificity. As a result, IP – MS results often show hundreds of proteins that bind to the support matrix constituting a high non-specific background. Specificity or sensitivity of antibodies can be increased using a revolutionized design of antibodies, so called nanobodies. Nanobodies contain single antigen-binding domains (VHH) and have much enhanced affinity and specificity for target proteins (Beghein and Gettemans, 2017). Epitope tagging has also been used to capture protein complexes with enhanced sensitivity and specificity. Epitopes could be FLAG, TAP, as examples, or biotin, which can be captured by antibodies of very strong affinity or streptavidin (for biotin) (Natividad et al., 2018; Smits and Vermeulen, 2016; Varnaite and MacNeill, 2016; Yang et al., 2015).
PPIs and their dysregulations are important in the current conceptualization of the pathophysiology of schizophrenia (Fernandez et al., 2017; Ganapathiraju et al., 2016; Jia et al., 2018; Liu et al., 2018). Genetic studies have shown enrichment of risk genes of the illness in various PPI networks (Fromer et al., 2014; Kirov et al., 2012; Purcell et al., 2014; Szatkiewicz et al., 2014). Conversely, PPI networks are now routinely used to prioritize candidate genes toward the goal of building hypotheses about pathophysiology (Schwarz et al., 2016). Consistent with this, postmortem studies have demonstrated a number of PPI alterations in specific brain regions of patients with schizophrenia. In GluN complexes, PLCg, nNos, rPTPa were decreased while PSD-95 and erbB4 were increased in the dorsolateral prefrontal cortex (DLPFC) of patients (Banerjee et al., 2015; Hahn et al., 2006). In mGluR5 complexes, Preso 1, Norbin and tamalin were decreased while RGS4 was increased in the DLPFC of patients (Wang et al., 2018b). Furthermore, PPI networks of GluN or mGluR pathways were indeed found to be highly enriched for risk variants of schizophrenia compared to non-psychiatric illnesses (Banerjee et al., 2015; Wang et al., 2018b). These together indicate that PPI networks enriched for risk variants could be a point of convergence for genetic and epigenetic variations. If so, the interactions among these proteins and functional valence of their dynamic changes may serve as mechanisms that integrate disease risks of seemingly unrelated molecular alterations.
3. New generation proteomics methodologies.
The last decade has seen an unprecedented advancement in mass spectrometry (MS) based methodologies, which has opened a new era in proteomics, phosphoproteomics and analyses of PPIs. The number of proteins that can be identified in one sample has grown from hundreds to about ten thousands (Monaci et al., 2018; Uzozie and Aebersold, 2018); and the dynamic range of quantitative accuracy has been extended by orders of magnitude (Al Shweiki et al., 2017). This has affected all major domains of MS based proteomics, namely bottom -up and top-down proteomics, or the discovery and targeted analyses. Below, we briefly summarize various domains of MS – proteomics methodology and how their capabilities progressed recently aided by cutting edge equipment, software and experimental paradigms.
3.1. Bottom up proteomics
In this category of proteomics methodologies, the proteins are first digested by proteases to peptides, which are then analyzed by LC and MS to assess the mass-to charge ratios of each peptide (Aebersold and Mann, 2003; Zhang et al., 2013). There are two forms of bottom-up proteomics: the discovery (“shotgun”) or targeted mode. In the former, many ions are scanned in an unbiased fashion; in the latter, specific ions are searched for and quantified, typically with isotope labeled standards. In the early 2000’s, the Quadrupole-Time of Flight (Q-TOF) MS was a popular platform in discovery proteomics. This platform was highly capable of fast full-scan data acquisition; yet had limitations in resolution and identification of analytes. Subsequently, a hybrid of LTQ mass filter and Orbitrap analyzer was introduced, which provided high mass resolution and accuracy over a wider mass range (Zubarev and Makarov, 2013). More recently, this hybrid Orbitrap set up has been extensively upgraded (Scheffler et al., 2018). With the cutting-edge equipment set up, discovery proteomics now can identify up to 10,000 proteins with much improved accuracy of quantification. In these platforms, the dynamic range can be achieved up to 5000 with resolving power of >50000 at m/z 200 and the data accuracy of ~0.03 Da (Al Shweiki et al., 2017; Iwamoto and Shimada, 2018).
Targeted proteomics has also seen a remarkable advancement in MS and LC instrumentation. Targeted proteomics include the SRM/MRM, Isobaric Tags for Relative and Absolute Quantitation (iTRAQ) and the Tandem Mass Tag (TMT) methods. In the SRM/MRM method, the sample is combined with stable isotope labeled standards and both the absolute and relative quantification are conducted for both the sample and heavy labeled standard peptides (Lange et al., 2008; Picotti and Aebersold, 2012). As such, the SRM/MRM method offers the most accurate quantification of analytes. The isobaric Tags for Relative and Absolute Quantitation (iTRAQ) method (Wiese et al., 2007) is used in targeted and discovery quantitative proteomics to determine the quantity of proteins from different sources in a single experiment. The Tandem Mass Tag (TMT) method quantifies data obtained from fragmented tags in MS/MS spectra. Each reporter contains isotopes substituted at various positions but they are identical in chemical structure (Thompson et al., 2003). The recently advanced Mascot, Maxquant and Paragon search algorithms have the capability to quantify those TMT tagged as well as other isobaric tagged proteins.
The parallel reaction monitoring (PRM) method is another innovation in targeted proteomics. The PRM method offers quantitative accuracy close to that of SRM but method development is much less labor intensive (Güzel et al., 2018; Peterson et al., 2012; Picotti and Aebersold, 2012). In the PRM method, target precursor ions are isolated in the 1st quadrupole, fragmented in the 2nd quadrupole and then identified and quantified by the Orbitrap mass analyzer (Güzel et al., 2018; Peterson et al., 2012; Picotti and Aebersold, 2012). Compared to the SRM method, PRM has an advantage in high-throughput quantification with confident targeted peptide confirmation. It is most suitable for quantifying hundreds of targeted proteins in a complex mixture with attomole level of limits of detection (Güzel et al., 2018; Peterson et al., 2012; Picotti and Aebersold, 2012).
It is important to note recent progress in a form of data independent acquisition (DIA), specifically SWATH-MS (Sequential Windowed Acquisition of all Theoretical Fragment ion Mass Spectra) method. Discovery proteomics can be performed in a data dependent acquisition (DDA) mode, where the precursor ions are selected in the order of their intensity to generate the product ions (Kalli et al., 2013; Schwudke et al., 2007) or DIA mode, where all precursor ions are fragmented. The DIA mode combines the advantages of DDA and SRM/PRM (Doerr, 2014) and repeatedly surveys every peptide. This can have the scope of identification similar to DDA while achieving the reproducible quantification characteristic of the MRM/SRM or PRM (Barkovits et al., 2018). In SWATH-MS, targeted extraction of a fragment ion is used to identify and quantify the proteins in the fragment ion spectra. A recent study shows that the SWATH-MS label-free quantification can quantify ~5,000 proteins reproducibly in high precision and accuracy for a highly complex mixture of proteins (Navarro et al., 2016).
3.2. Top down proteomics strategies.
Methods in this category allow in-depth characterization of proteins by fragmenting entire proteins inside a MS (Catherman et al., 2014; Skinner et al., 2017) and thus the ion masses of intact and fragmented proteins can be measured and quantified. This approach can provide up to 98 % sequence coverage and full characterization of proteins, while maintaining PTMs (Catherman et al., 2014; Skinner et al., 2017). Previously, top down proteomics had suboptimal detection sensitivity and accuracy for molecules larger than 30 kD. Aided by incorporating versatile fragmentation techniques, detection sensitivity, of larger proteins (up to 70 kD), in particular, significantly improved as shown in a recent study (Scheffler et al., 2018). Using the high-field quadrupole orbitrap tribrid mass spectrometer (Christoforou et al., 2016a; Zhang et al., 2016), they achieved resolution settings of >500,000 FWHM at m/z 200 and can have mass accuracy of ~10 ppm.
3.3. Software, search engines and data consortia.
Critical to the recent progress in MS technologies is the development of algorithm/search engines to identify and quantify peptides based on mass spectra and software for proteome data processing. Several algorithms/search engines have changed the field and rendered MS spectra readily usable towards identification of peptides and their modifications. Among widely utilized proteome searching tools are Sequest (Thermo Scientific, US) (Eng et al., 1994), Mascot (Matrix Science, UK) (Koenig et al., 2008), MaxQuant (Tyanova et al., 2016) and Paragon (Shilov et al., 2007). The Sequest algorithm, developed by Yates and colleagues, along with the algorithm X!TANDEM, can identify ~2,000,000 peptides which can be matched with over 14000 proteins (Dufresne et al., 2018). Likewise the MaxQuant developed by Mann and colleague (Cox and Mann, 2008) identifies and quantifies of >4,000 proteins in mammalian proteome. For instance, the MaxQuant algorithm was employed to generate as much as 9700 unique proteins from a single experiment (Mathieson et al., 2018; Tyanova et al., 2016). In addition, Proteome Discoverer (Thermo Fisher, US) (Xiao et al., 2016), ProteinPilot (Sciex, US) (Ernoult et al., 2008), ProteinProspector (UCSF, US) (Chalkley et al., 2005) and, ProteinScape (Bruker, DE) (Hinneburg et al., 2016) are widely used proteome data processing software. For targeted proteomics data analysis and protein quantification, Skyline (http://skyline.ms/project/home/software/Skyline) is the most widely used open source software. The software supports multiple workflows like SRM/MRM, PRM, and DDA etc. (Frewen et al., 2010).
In addition, web-based consortia have been established, where proteomics data and raw files can be deposited and accessed by the scientific community. A prime example of this is the ProteomeXchange consortium (http://www.proteomexchange.org/)(He et al., 2019), which supports submission of experimental data from all proteomics data workflows. The data can be submitted mainly in three data depository partners ;“PRIDE - PRoteomics IDEntifications Database” that supports DIA and DDA discovery data, “MassIVE” that supports specifically DDA shotgun proteomics data and “PeptideAtlas – PASSEL” that supports targeted proteomics data. In addition,another consortium, ProteomicsDB (https://www.ProteomicsDB.org), released in 2014, is a protein-centric in-memory database for the exploration of large collections of quantitative mass spectrometry-based proteomics data (Frejno et al., 2017).
4. Integration of proteomics and its dynamics into the multi-omics in schizophrenia
The ultimate goals of omics investigations are to discover pathophysiologic mechanisms and to form therapeutic strategies (Yugi et al., 2016; Yurkovich and Palsson, 2018). Considering its complex and heterogeneous nature, the pathophysiology of schizophrenia (Fromer et al., 2016), (Boyle et al., 2017) will be delineated not by a single omics alone, e.g., genomics or transcriptomics (Geschwind and State, 2015; Prinz et al., 2004; Willsey and State, 2015) but by integration of multi-omics outcomes. Figure 1 depicts multiple layers of omics ranging from genes to disease phenotypes. These include alterations at the level of nucleic acids, i.e., DNAs and RNAs, proteins, metabolites and microbiomes. In complex trait disorders, each layer can contribute to pathophysiologic mechanisms via complex interactions amongst them (Figure 1). Many algorithms have been developed to analyze and integrate the data from genomics, epigenomics, and transcriptomics and other omics investigations. Such studies have been conducted in various diseases including cancers (Bian et al., 2018; Chaudhary et al., 2018; Dimitrakopoulos et al., 2018; Doostparast Torshizi and Petzold, 2018; Le Van et al., 2016; Ritchie et al., 2015) and neuropsychiatric illnesses (Cattaneo and Pariante, 2018; Chakraborty et al., 2017; Crowther et al., 2018; Higdon et al., 2015; Wu et al., 2018). It is important to note, however, that each domain of omics has different configuration to their assessment and thus should be integrated into the whole construct with considerations for specific characteristics. Here, we will briefly summarize current methods on multi-omics integration and consider how proteome data can be better integrated into a multi-omics framework.
Figure 1:
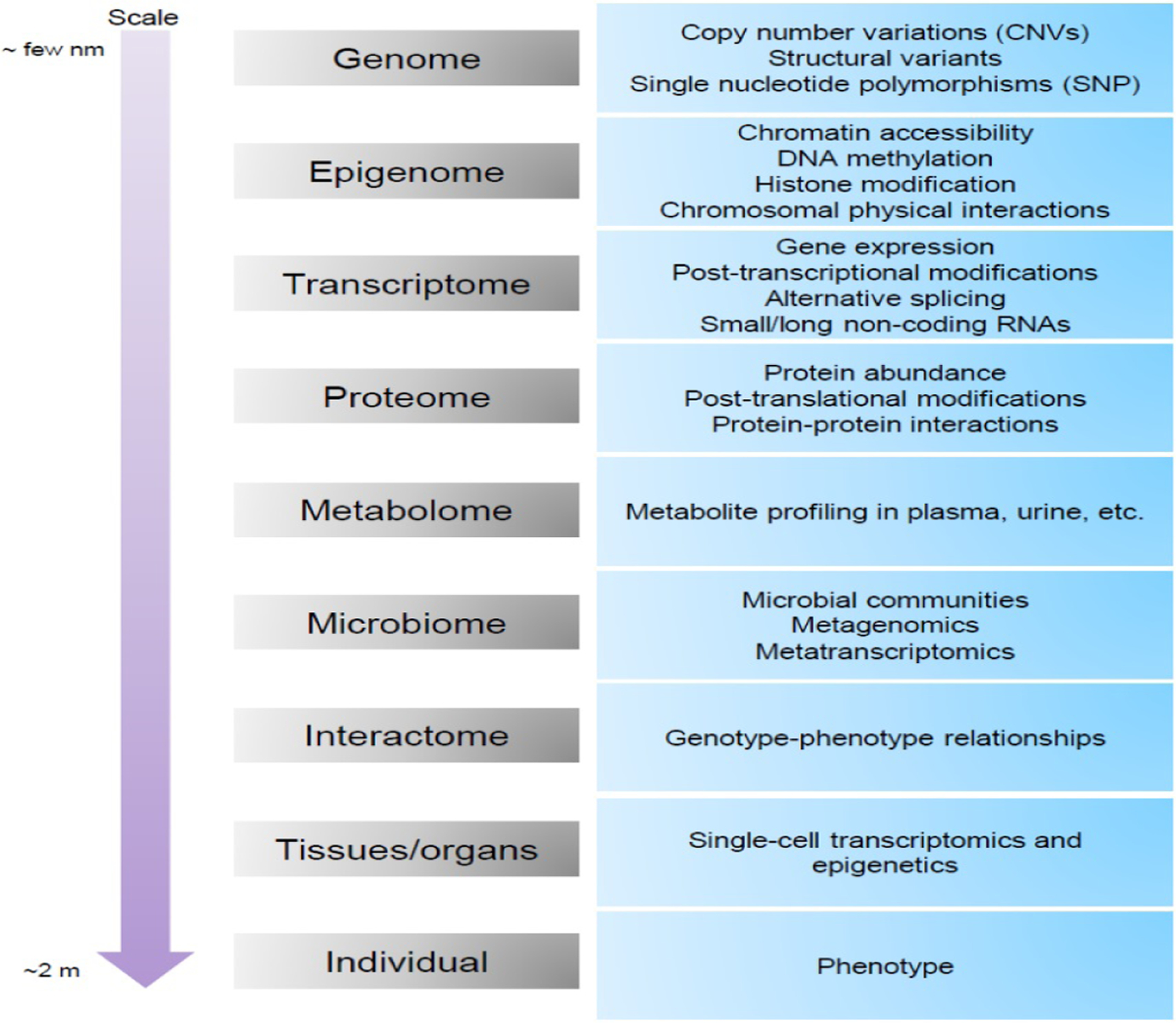
Omics data types from Genome to an Individual (Phenome).
4.1. Adapting preexisting approaches developed for transcriptomics to proteomics analysis.
Some of well-established approaches for analysis of DNA - and RNA - omics can be adapted to proteomics data. Figure 2 summarizes widely used systems biology approaches that have been employed for omics of DNAs and RNAs. These algorithms can be grossly categorized into three distinct groups: supervised methods, unsupervised methods, and semi-supervised methods (Figure 2). Of these multi-omics data analysis methods, many can be directly applied to proteomic analyses. For example, pathway-based methods are utilized for all levels of omics to assign biological functions to risk variants as they have guided interpretations of genomic and transcriptomic studies. The same paradigm could be applied to proteins as in many previous proteomic studies (Clark et al., 2006; Focking et al., 2016; Martins-de-Souza et al., 2009). Definition of each pathway and their memberships reflect biases of the field. Given that, a number of algorithms have been developed to find functional clusters of the genes that are relevant to pathophysiologic mechanisms by leveraging the concept of transcriptional networks.
Figure 2:
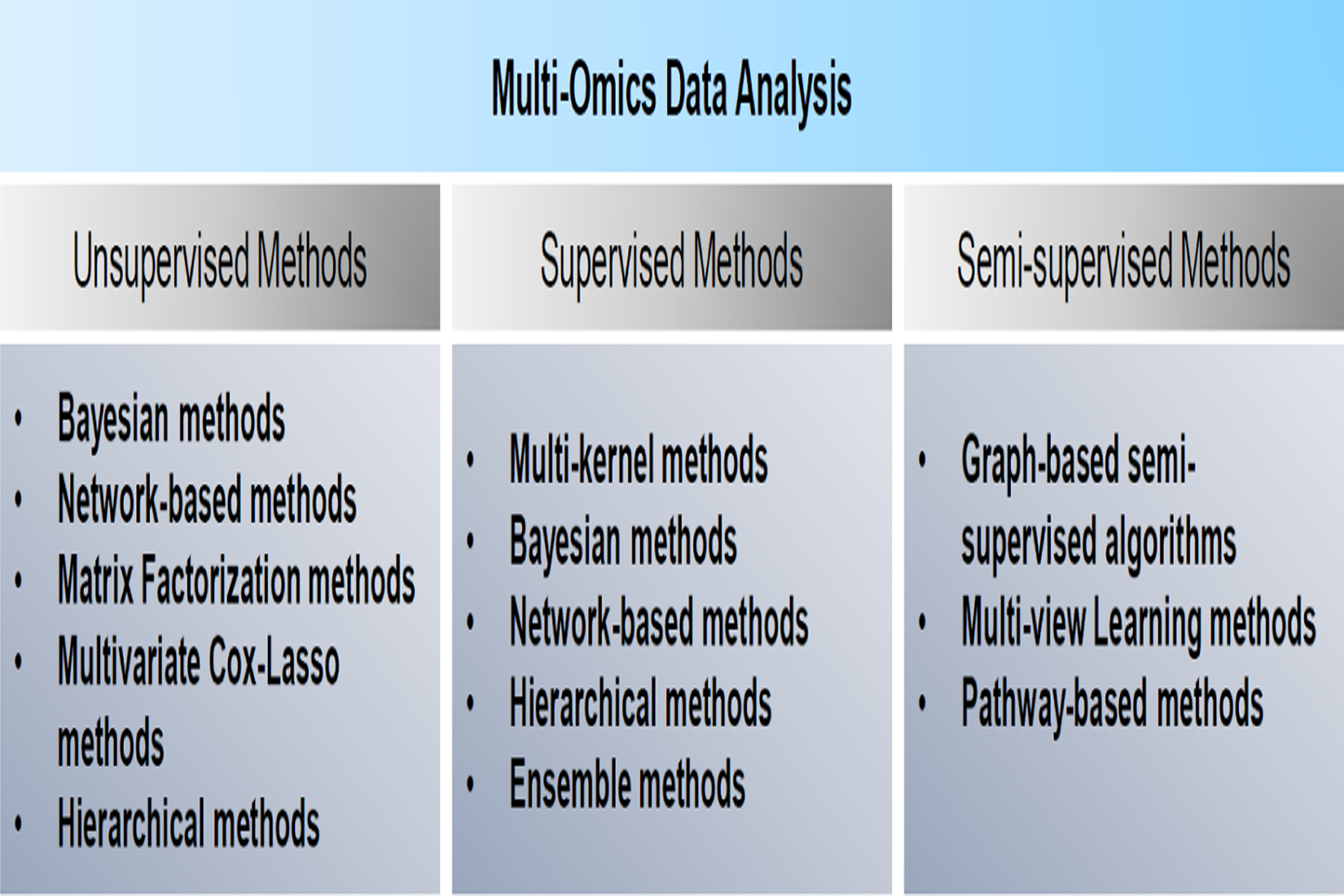
The overall categorization of the available multi-omics data analysis methods.
Transcriptional networks are harmonized orchestrations of genomic and regulatory interactions with a definitive role in mediating cellular processes through regulating gene expression (Doostparast Torshizi et al., 2018a). Scale-free co-expression networks (Jordan et al., 2004; Lukashin et al., 2003) are among the most commonly used network methods for modeling cellular functions. Despite vast adoption of co-expression networks, they may not fully recapitulate the underlying molecular interactions driving the disease phenotype (Basso et al., 2005). This is due to multiple limitations (Margolin et al., 2006) including the lack of incorporation of causal regulatory relationships and the presence of high false positive rates due to indirect connections (Doostparast Torshizi et al., 2018b; Doostparast Torshizi and Wang, 2017). In contrast, information-theoretic deconvolution techniques (Basso et al., 2005) are capable of compensating for such deficiencies with successful applications in complex diseases such as cancers (Alvarez et al., 2016), prostate differentiation (Dutta et al., 2016), and neurodegenerative diseases (Brichta et al., 2015).
Among these, co-expression analyses with weighted correlation network analysis (WGCNA) is an important example (Langfelder and Horvath, 2008; Zhang and Horvath, 2005). It provides a network identification scheme based on the similarity of genetic transcription-level profiles across individuals by defining clusters of co-expressed genes (Langfelder and Horvath, 2008; Zhang and Horvath, 2005). This has been routinely utilized for transcriptomics and proteomics studies for various psychiatric illnesses including autism, schizophrenia and others. One remaining challenge, however, is how to incorporate function-defining molecular properties of proteins (described above). For instance, the WGCNA is based on the notion that co-regulated genes may have common functionality at some level of biological function. Should protein extracts from whole cell homogenate or specific subcellular fractions be studied? How about posttranslational modifications and PPIs? How should these to be integrated with WGCNA analyses of transcriptomics of the same cells/tissues?
4.2. Novel approaches needed to incorporate proteomics data into multi-omics analyses.
To generate pathophysiologic hypotheses, it is necessary to integrate data from multiple omics investigations. In this regard, the concept of “convergence” figures greatly into how integration algorithms have been conceived (Geschwind and State, 2015; Prinz et al., 2004; State and Sestan, 2012; Willsey and State, 2015). In complex trait disorders, risk carrying alterations in different dimensions may interact and conspire to impact specific pathways. If so, identifying points of convergence may pinpoint the disease mechanisms. There are indeed multiple examples where genomic variants, transcriptomic alterations, co-expression modules and epigenomic changes are stratified and found to be overlapping on specific pathways. How to integrate proteomics data with their multiple dimensions incorporated will be the next challenge as the MS based proteomics field grows rapidly.
Unlike gene expression data, proteomics data sets provide additional dimensions, such as protein complex information, protein-protein interactions (PPI), sub-cellular physical location in the cell, phosphorylation and other PTMs. An important question then is how these should be incorporated in multi-omics approaches to facilitate our understanding of schizophrenia. In the following we will consider how such multi-dimensional features can be incorporated into a proteome map and then be integrated into multi-omics analyses.
Subcellular localization places proteins in intracellular locale where their function is needed (Itzhak et al., 2016). For instance, transcription factors can only function via proper regulation of nucleo-cytoplasmic shuttling (Itzhak et al., 2016; Plotnikov et al., 2011). Therefore, considering sub-compartmental location (Lee et al., 2016) and spatial distribution is essential in understanding cellular mechanisms and their contribution to the disease. With the advent of high-throughput proteomic readouts, rich sources of known proteome localization data are available to be used in integrated studies (Christoforou et al., 2016b; Thul et al., 2017; Uhlen et al., 2017).
PPI networks represent direct physical interactions between proteins are another dimension that need to be integrated for functional assessment of proteins. PPIs show more direct relationships between gene products than those from co-expression approaches such as the WGCNA or Algorithm for the reconstruction of Accurate Cellular Networks (ARACNE) that are based on the “statistical correlation” between genes. Indeed, PPIs are commonly used for prioritizing and identifying disease genes and pathways (Karczewski and Snyder, 2018) . For instance, in patients with autism spectrum disorder (ASD), genes harboring de novo missense or nonsense mutations are enriched for genes with high degrees of connectivity in PPI networks to all other genes and particularly previously ASD-implicated genes (Li et al., 2014; Neale et al., 2012). It is of note, however, that the prevalent assumption that dense regions of PPI networks constitute protein complexes was found not to be true (Dong et al., 2018). Therefore, it will be important to further refine available databases on PPI networks by obtaining experimental results of PPIs in relevant tissues, i.e., postmortem human brains of the regions of interest.
Toward the goal of integration with diverse other omics data-types, there is a need to construct a multi-dimensional proteome map. This will require an algorithm to incorporate the information on subcellular localization, PTMs, PPIs and other functionally relevant properties. Once the proteome map is created, it can be integrated to other data types using graph-integration methods. An example could be the method, which uses different types of edges to denote different types of interactions between genes in a network structure (Doostparast Torshizi and Petzold, 2018). Another approach is to combine and “amplify” signals from individual genes from various dimensions of omics. Here, prior information of genes associated with the illness in multiple domains, e.g., transcriptomics or epigenomics, are superimposed on the nodes of the network. The information is then propagated through the edges to nearby nodes in an iterative manner until convergence is observed. There are now a number of algorithms designed to amplify signals from multi-dimensional omics results (Cowen et al., 2017). Among these are based on the strategies of “Random walks’ (Voevodski et al., 2009), “electrical resistance” (Suthram et al., 2008) and “information diffusion” (Cao et al., 2014).
5. Conclusion
Multi-omics approaches and their integration are essential for deconstructing the complex nature of the pathophysiologic makeup of schizophrenia. Such investigations are now possible as genome wide analyses of risk variants, transcripts and epigenome have been or are being conducted on large scale. (Fromer et al., 2016; Wang et al., 2018a). Rapidly expanding bioinformatics algorithms now enable us to analyze these massive data and connect dots between genes to transcripts. One missing piece is incorporation of information on proteome changes and their dynamic properties, such as subcellular localization, PTMs and PPIs in the illness.
The new generation proteomics methodologies offer analyses of a scale close to the whole proteome and of highly quantitative accuracy. Importantly, they enable us to analyze not only the abundance of proteins, but also their functional properties, their subcellular locale, PTMs and PPIs. The transcriptome and epigenome have been extensively investigated in postmortem brain regions of subjects with schizophrenia and controls of a large cohort. A compelling next step will be to apply the new generation proteomics methodologies to postmortem brains, which will permit us to close the loop of functional genomics at the molecular level.
There are a few steps that need to be addressed in doing so. First, how to address the effects of postmortem confounds and medication effects on all proteomics parameters. How do we monitor the stability of proteins in various postmortem conditions? For transcripts, there are universally accepted measures of their stability, such as RIN, while there are no such measures for proteins. How should the stability of posttranslational modifications be monitored? To what extent is protein phosphorylation is maintained in postmortem brains and what measures should be employed? Do they reflect in vivo phosphorylation or ex vivo postmortem phosphatase activity? Similar lines of questions should be addressed for subcellular localization and PPIs.
Another important challenge is how to integrate various aspects of proteome and its dynamics, the abundance, subcellular localization, PTMs and PPIs. Transcriptomic or epigenomic changes are distilled to singular quantitative measures before they integrate with genomic variations. The proteome and its dynamics represent multi-dimensional measures reflecting protein functionality. How to capture their interactions and how to incorporate this into measures for the integration with other omics is the next challenge.
Integrative multi-omics approaches enable us to consider the pathophysiologic understanding of complex trait disorders, such as schizophrenia, as an attainable goal. The new generation instrumentation and software/search engines can now be utilized to conduct a large-scale investigation of proteome and its multi-dimensional properties in schizophrenia relevant tissue or cell samples. While protein and its dynamics carry out the gene function, the majority of multi-omics in schizophrenia have been biased for nucleic acids. Future proteomic investigations will add a new dimension to our conceptualization of the complex pathophysiology of schizophrenia.
References
- Adachi J, Narumi R, Tomonaga T, 2016. Targeted Phosphoproteome Analysis Using Selected/Multiple Reaction Monitoring (SRM/MRM), in: Reinders J (Ed.), Proteomics in Systems Biology: Methods and Protocols. Springer New York, New York, NY, pp. 87–100. [Google Scholar]
- Aebersold R, Mann M, 2003. Mass spectrometry-based proteomics. Nature 422(6928), 198–207. [DOI] [PubMed] [Google Scholar]
- Al Shweiki MHDR, Mönchgesang S, Majovsky P, Thieme D, Trutschel D, Hoehenwarter W, 2017. Assessment of Label-Free Quantification in Discovery Proteomics and Impact of Technological Factors and Natural Variability of Protein Abundance. Journal of Proteome Research 16(4), 1410–1424. [DOI] [PubMed] [Google Scholar]
- Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, Califano A, 2016. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nature Genetics 48(8), 838-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora A, Somasundaram K, 2019. Targeted Proteomics Comes to the Benchside and the Bedside: Is it Ready for Us? Bioessays 41(2), e1800042. [DOI] [PubMed] [Google Scholar]
- Arrington JV, Hsu CC, Elder SG, Andy Tao W, 2017. Recent advances in phosphoproteomics and application to neurological diseases. The Analyst 142(23), 4373–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TC, Han J, Borchers CH, 2017. Recent advancements in matrix-assisted laser desorption/ionization mass spectrometry imaging. Current opinion in biotechnology 43, 62–69. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Macdonald ML, Borgmann-Winter KE, Hahn CG, 2010. Neuregulin 1-erbB4 pathway in schizophrenia: From genes to an interactome. Brain Res Bull 83(3–4), 132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Wang HY, Borgmann-Winter KE, MacDonald ML, Kaprielian H, Stucky A, Kvasic J, Egbujo C, Ray R, Talbot K, Hemby SE, Siegel SJ, Arnold SE, Sleiman P, Chang X, Hakonarson H, Gur RE, Hahn CG, 2015. Src kinase as a mediator of convergent molecular abnormalities leading to NMDAR hypoactivity in schizophrenia. Mol Psychiatry 20(9), 1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkovits K, Linden A, Galozzi S, Schilde L, Pacharra S, Mollenhauer B, Stoepel N, Steinbach S, May C, Uszkoreit J, Eisenacher M, Marcus K, 2018. Characterization of Cerebrospinal Fluid via Data-Independent Acquisition Mass Spectrometry. Journal of Proteome Research 17(10), 3418–3430. [DOI] [PubMed] [Google Scholar]
- Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A, 2005. Reverse engineering of regulatory networks in human B cells. Nat Genet 37(4), 382–390. [DOI] [PubMed] [Google Scholar]
- Beasley CL, Pennington K, Behan A, Wait R, Dunn MJ, Cotter D, 2006. Proteomic analysis of the anterior cingulate cortex in the major psychiatric disorders: Evidence for disease-associated changes. Proteomics 6(11), 3414–3425. [DOI] [PubMed] [Google Scholar]
- Beghein E, Gettemans J, 2017. Nanobody Technology: A Versatile Toolkit for Microscopic Imaging, Protein-Protein Interaction Analysis, and Protein Function Exploration. Frontiers in immunology 8, 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian S, Hou Y, Zhou X, Li X, Yong J, Wang Y, Wang W, Yan J, Hu B, Guo H, Wang J, Gao S, Mao Y, Dong J, Zhu P, Xiu D, Yan L, Wen L, Qiao J, Tang F, Fu W, 2018. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 362(6418), 1060–1063. [DOI] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T, 2001. Oncogenic kinase signalling. Nature 411(6835), 355–365. [DOI] [PubMed] [Google Scholar]
- Boyle EA, Li YI, Pritchard JK, 2017. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169(7), 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brichta L, Shin W, Jackson-Lewis V, Blesa J, Yap EL, Walker Z, Zhang J, Roussarie JP, Alvarez MJ, Califano A, Przedborski S, Greengard P, 2015. Identification of neurodegenerative factors using translatome-regulatory network analysis. Nature Neuroscience 18(9), 1325-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Tucholski TM, Gregorich ZR, Ge Y, 2016. Top-down Proteomics: Technology Advancements and Applications to Heart Diseases. Expert review of proteomics 13(8), 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, Pietras CM, Feng X, Doroschak KJ, Schaffner T, Park J, Zhang H, Cowen LJ, Hescott BJ, 2014. New directions for diffusion-based network prediction of protein function: incorporating pathways with confidence. Bioinformatics 30(12), i219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catherman AD, Skinner OS, Kelleher NL, 2014. Top Down proteomics: Facts and perspectives. Biochemical and Biophysical Research Communications 445(4), 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo A, Pariante CM, 2018. Integrating ‘Omics’ Approaches to Prioritize New Pathogenetic Mechanisms for Mental Disorders. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 43(1), 227–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty N, Meyerhoff J, Jett M, Hammamieh R, 2017. Genome to Phenome: A Systems Biology Approach to PTSD Using an Animal Model. Methods in molecular biology (Clifton, N.J.) 1598, 117–154. [DOI] [PubMed] [Google Scholar]
- Chalkiadaki K, Velli A, Kyriazidis E, Stavroulaki V, Vouvoutsis V, Chatzaki E, Aivaliotis M, Sidiropoulou K, 2019. Development of the MAM model of schizophrenia in mice: Sex similarities and differences of hippocampal and prefrontal cortical function. Neuropharmacology 144, 193–207. [DOI] [PubMed] [Google Scholar]
- Chalkley RJ, Baker PR, Huang L, Hansen KC, Allen NP, Rexach M, Burlingame AL, 2005. Comprehensive Analysis of a Multidimensional Liquid Chromatography Mass Spectrometry Dataset Acquired on a Quadrupole Selecting, Quadrupole Collision Cell, Time-of-flight Mass Spectrometer. Molecular & Cellular Proteomics 4(8), 1194. [DOI] [PubMed] [Google Scholar]
- Chaudhary K, Poirion OB, Lu L, Garmire LX, 2018. Deep Learning-Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clin Cancer Res 24(6), 1248–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforou A, Mulvey CM, Breckels LM, Geladaki A, Hurrell T, Hayward PC, Naake T, Gatto L, Viner R, Arias AM, Lilley KS, 2016a. A draft map of the mouse pluripotent stem cell spatial proteome. Nature Communications 7, 9992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoforou A, Mulvey CM, Breckels LM, Geladaki A, Hurrell T, Hayward PC, Naake T, Gatto L, Viner R, Martinez Arias A, Lilley KS, 2016b. A draft map of the mouse pluripotent stem cell spatial proteome. Nat Commun 7, 8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark D, Dedova I, Cordwell S, Matsumoto I, 2006. A proteome analysis of the anterior cingulate cortex gray matter in schizophrenia. Mol Psychiatry 11(5), 459–470, 423. [DOI] [PubMed] [Google Scholar]
- Clark D, Dedova I, Cordwell S, Matsumoto I, 2007. Altered proteins of the anterior cingulate cortex white matter proteome in schizophrenia. Proteomics Clin Appl 1(2), 157–166. [DOI] [PubMed] [Google Scholar]
- Cowen L, Ideker T, Raphael BJ, Sharan R, 2017. Network propagation: a universal amplifier of genetic associations. Nat Rev Genet 18(9), 551–562. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M, 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology 26, 1367. [DOI] [PubMed] [Google Scholar]
- Crowther LM, Poms M, Plecko B, 2018. Multiomics tools for the diagnosis and treatment of rare neurological disease. J Inherit Metab Dis 41(3), 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf EL, Giansanti P, Altelaar AF, Heck AJ, 2014. Single-step enrichment by Ti4+-IMAC and label-free quantitation enables in-depth monitoring of phosphorylation dynamics with high reproducibility and temporal resolution. Mol Cell Proteomics 13(9), 2426–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng MY, Lam S, Meyer U, Feldon J, Li Q, Wei R, Luk L, Chua SE, Sham P, Wang Y, McAlonan GM, 2011. Frontal-subcortical protein expression following prenatal exposure to maternal inflammation. PLoS One 6(2), e16638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrakopoulos C, Hindupur SK, Hafliger L, Behr J, Montazeri H, Hall MN, Beerenwinkel N, 2018. Network-based integration of multi-omics data for prioritizing cancer genes. Bioinformatics 34(14), 2441–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerr A, 2014. DIA mass spectrometry. Nature Methods 12, 35. [Google Scholar]
- Dong Y, Sun Y, Qin C, 2018. Predicting protein complexes using a supervised learning method combined with local structural information. PLoS One 13(3), e0194124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doostparast Torshizi A, Armoskus C, Zhang S, Zhang H, Souaiaia T, Forrest MP, Evgrafov OV, Knowles JA, Duan J, Wang K, 2018a. Deconvolution of Transcriptional Networks Identifies TCF4 as a Master Regulator in Schizophrenia. bioRxiv, 133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doostparast Torshizi A, Duan J, Wang K, 2018b. Transcriptional network analysis on brains reveals a potential regulatory role of PPP1R3F in autism spectrum disorders. BMC Res Notes 11(1), 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doostparast Torshizi A, Petzold LR, 2018. Graph-based semi-supervised learning with genomic data integration using condition-responsive genes applied to phenotype classification. J Am Med Inform Assoc 25(1), 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doostparast Torshizi A, Wang K, 2017. Deconvolution of Transcriptional Networks in Post-Traumatic Stress Disorder Uncovers Master Regulators Driving Innate Immune System Function. Sci Rep 7(1), 14486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufresne J, Bowden P, Thavarajah T, Florentinus-Mefailoski A, Chen ZZ, Tucholska M, Norzin T, Ho MT, Phan M, Mohamed N, Ravandi A, Stanton E, Slutsky AS, dos Santos CC, Romaschin A, Marshall JC, Addison C, Malone S, Heyland D, Scheltens P, Killestein J, Teunissen CE, Diamandis EP, Michael Siu KW, Marshall JG, 2018. The plasma peptides of ovarian cancer. Clinical Proteomics 15(1), 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta A, Le Magnen C, Mitrofanova A, Ouyang XS, Califano A, Abate-Shen C, 2016. Identification of an NKX3.1-G9a-UTY transcriptional regulatory network that controls prostate differentiation. Science 352(6293), 1576–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR, 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry 5(11), 976–989. [DOI] [PubMed] [Google Scholar]
- Ernoult E, Gamelin E, Guette C, 2008. Improved proteome coverage by using iTRAQ labelling and peptide OFFGEL fractionation. Proteome Science 6(1), 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez E, Collins MO, Frank RAW, Zhu F, Kopanitsa MV, Nithianantharajah J, Lempriere SA, Fricker D, Elsegood KA, McLaughlin CL, Croning MDR, McLean C, Armstrong JD, Hill WD, Deary IJ, Cencelli G, Bagni C, Fromer M, Purcell SM, Pocklington AJ, Choudhary JS, Komiyama NH, Grant SGN, 2017. Arc Requires PSD95 for Assembly into Postsynaptic Complexes Involved with Neural Dysfunction and Intelligence. Cell Rep 21(3), 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focking M, Dicker P, English JA, Schubert KO, Dunn MJ, Cotter DR, 2011. Common proteomic changes in the hippocampus in schizophrenia and bipolar disorder and particular evidence for involvement of cornu ammonis regions 2 and 3. Arch Gen Psychiatry 68(5), 477–488. [DOI] [PubMed] [Google Scholar]
- Focking M, Dicker P, Lopez LM, Hryniewiecka M, Wynne K, English JA, Cagney G, Cotter DR, 2016. Proteomic analysis of the postsynaptic density implicates synaptic function and energy pathways in bipolar disorder. Transl Psychiatry 6(11), e959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focking M, Lopez LM, English JA, Dicker P, Wolff A, Brindley E, Wynne K, Cagney G, Cotter DR, 2015. Proteomic and genomic evidence implicates the postsynaptic density in schizophrenia. Mol Psychiatry 20(4), 424–432. [DOI] [PubMed] [Google Scholar]
- Frejno M, Samaras P, Schmidt T, Wilhelm M, Gessulat S, Kuster B, Ehrlich H-C, Aiche S, Kienegger H, Krcmar H, Barnert M, Schlegl J, 2017. ProteomicsDB. Nucleic Acids Research 46(D1), D1271–D1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frewen B, MacLean B, Liebler DC, Tomazela DM, Tabb DL, Finney GL, Chambers M, MacCoss MJ, Shulman N, Kern R, 2010. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26(7), 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, Carrera N, Humphreys I, Johnson JS, Roussos P, Barker DD, Banks E, Milanova V, Grant SG, Hannon E, Rose SA, Chambert K, Mahajan M, Scolnick EM, Moran JL, Kirov G, Palotie A, McCarroll SA, Holmans P, Sklar P, Owen MJ, Purcell SM, O’Donovan MC, 2014. De novo mutations in schizophrenia implicate synaptic networks. Nature 506(7487), 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, Ruderfer DM, Oh EC, Topol A, Shah HR, Klei LL, Kramer R, Pinto D, Gumus ZH, Cicek AE, Dang KK, Browne A, Lu C, Xie L, Readhead B, Stahl EA, Xiao J, Parvizi M, Hamamsy T, Fullard JF, Wang YC, Mahajan MC, Derry JM, Dudley JT, Hemby SE, Logsdon BA, Talbot K, Raj T, Bennett DA, De Jager PL, Zhu J, Zhang B, Sullivan PF, Chess A, Purcell SM, Shinobu LA, Mangravite LM, Toyoshiba H, Gur RE, Hahn CG, Lewis DA, Haroutunian V, Peters MA, Lipska BK, Buxbaum JD, Schadt EE, Hirai K, Roeder K, Brennand KJ, Katsanis N, Domenici E, Devlin B, Sklar P, 2016. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci 19(11), 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathiraju MK, Thahir M, Handen A, Sarkar SN, Sweet RA, Nimgaonkar VL, Loscher CE, Bauer EM, Chaparala S, 2016. Schizophrenia interactome with 504 novel protein-protein interactions. NPJ Schizophr 2, 16012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, Schork AJ, Appadurai V, Buil A, Werge TM, Liu C, White KP, CommonMind C, Psych EC, i, P.-B.W.G., Horvath S, Geschwind DH, 2018a. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359(6376), 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, Shieh AW, Haney J, Parhami S, Belmont J, Kim M, Moran Losada P, Khan Z, Mleczko J, Xia Y, Dai R, Wang D, Yang YT, Xu M, Fish K, Hof PR, Warrell J, Fitzgerald D, White K, Jaffe AE, Psych EC, Peters MA, Gerstein M, Liu C, Iakoucheva LM, Pinto D, Geschwind DH, 2018b. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362(6420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, State MW, 2015. Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol 14(11), 1109–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girdhar K, Hoffman GE, Jiang Y, Brown L, Kundakovic M, Hauberg ME, Francoeur NJ, Wang YC, Shah H, Kavanagh DH, Zharovsky E, Jacobov R, Wiseman JR, Park R, Johnson JS, Kassim BS, Sloofman L, Mattei E, Weng Z, Sieberts SK, Peters MA, Harris BT, Lipska BK, Sklar P, Roussos P, Akbarian S, 2018. Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat Neurosci 21(8), 1126–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güzel C, Govorukhina NI, Stingl C, Dekker LJM, Boichenko A, van der Zee AGJ, Bischoff RPH, Luider TM, 2018. Comparison of Targeted Mass Spectrometry Techniques with an Immunoassay: A Case Study for HSP90α. PROTEOMICS – Clinical Applications 12(1), 1700107. [DOI] [PubMed] [Google Scholar]
- Hada V, Bagdi A, Bihari Z, Timari SB, Fizil A, Szantay C Jr., 2018. Recent advancements, challenges, and practical considerations in the mass spectrometry-based analytics of protein biotherapeutics: A viewpoint from the biosimilar industry. Journal of pharmaceutical and biomedical analysis 161, 214–238. [DOI] [PubMed] [Google Scholar]
- Hahn CG, 2011. A Src link in schizophrenia. Nat Med 17(4), 425–427. [DOI] [PubMed] [Google Scholar]
- Hahn CG, Banerjee A, Macdonald ML, Cho DS, Kamins J, Nie Z, Borgmann-Winter KE, Grosser T, Pizarro A, Ciccimaro E, Arnold SE, Wang HY, Blair IA, 2009. The post-synaptic density of human postmortem brain tissues: an experimental study paradigm for neuropsychiatric illnesses. PLoS One 4(4), e5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn CG, Wang HY, Cho DS, Talbot K, Gur RE, Berrettini WH, Bakshi K, Kamins J, Borgmann-Winter KE, Siegel SJ, Gallop RJ, Arnold SE, 2006. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med 12(7), 824–828. [DOI] [PubMed] [Google Scholar]
- He B, Shi J, Wang X, Jiang H, Zhu H-J, 2019. Label-free absolute protein quantification with data-independent acquisition. Journal of Proteomics. [DOI] [PMC free article] [PubMed]
- Higdon R, Earl RK, Stanberry L, Hudac CM, Montague E, Stewart E, Janko I, Choiniere J, Broomall W, Kolker N, Bernier RA, Kolker E, 2015. The promise of multi-omics and clinical data integration to identify and target personalized healthcare approaches in autism spectrum disorders. OMICS 19(4), 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinneburg H, Stavenhagen K, Schweiger-Hufnagel U, Pengelley S, Jabs W, Seeberger PH, Silva DV, Wuhrer M, Kolarich D, 2016. The Art of Destruction: Optimizing Collision Energies in Quadrupole-Time of Flight (Q-TOF) Instruments for Glycopeptide-Based Glycoproteomics. Journal of The American Society for Mass Spectrometry 27(3), 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hradetzky E, Sanderson TM, Tsang TM, Sherwood JL, Fitzjohn SM, Lakics V, Malik N, Schoeffmann S, O’Neill MJ, Cheng TM, Harris LW, Rahmoune H, Guest PC, Sher E, Collingridge GL, Holmes E, Tricklebank MD, Bahn S, 2012. The methylazoxymethanol acetate (MAM-E17) rat model: molecular and functional effects in the hippocampus. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 37(2), 364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP, 2010. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143(7), 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang T, Park CK, Leung AK, Gao Y, Hyde TM, Kleinman JE, Rajpurohit A, Tao R, Shin JH, Weinberger DR, 2016. Dynamic regulation of RNA editing in human brain development and disease. Nat Neurosci 19(8), 1093–1099. [DOI] [PubMed] [Google Scholar]
- Itzhak DN, Tyanova S, Cox J, Borner GH, 2016. Global, quantitative and dynamic mapping of protein subcellular localization. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto N, Shimada T, 2018. Recent advances in mass spectrometry-based approaches for proteomics and biologics: Great contribution for developing therapeutic antibodies. Pharmacology & Therapeutics 185, 147–154. [DOI] [PubMed] [Google Scholar]
- Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, Kleinman JE, 2016. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci 19(1), 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AE, Straub RE, Shin JH, Tao R, Gao Y, Collado-Torres L, Kam-Thong T, Xi HS, Quan J, Chen Q, Colantuoni C, Ulrich WS, Maher BJ, Deep-Soboslay A, BrainSeq C, Cross AJ, Brandon NJ, Leek JT, Hyde TM, Kleinman JE, Weinberger DR, 2018. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat Neurosci 21(8), 1117–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia P, Chen X, Fanous AH, Zhao Z, 2018. Convergent roles of de novo mutations and common variants in schizophrenia in tissue-specific and spatiotemporal co-expression network. Transl Psychiatry 8(1), 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston-Wilson NL, Sims CD, Hofmann JP, Anderson L, Shore AD, Torrey EF, Yolken RH, 2000. Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. Molecular Psychiatry 5, 142. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Marino-Ramirez L, Wolf YI, Koonin EV, 2004. Conservation and coevolution in the scale-free human gene coexpression network. Mol Biol Evol 21(11), 2058–2070. [DOI] [PubMed] [Google Scholar]
- Junger MA, Aebersold R, 2014. Mass spectrometry-driven phosphoproteomics: patterning the systems biology mosaic. Wiley Interdiscip Rev Dev Biol 3(1), 83–112. [DOI] [PubMed] [Google Scholar]
- Kalli A, Smith GT, Sweredoski MJ, Hess S, 2013. Evaluation and Optimization of Mass Spectrometric Settings during Data-dependent Acquisition Mode: Focus on LTQ-Orbitrap Mass Analyzers. Journal of Proteome Research 12(7), 3071–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Snyder MP, 2018. Integrative omics for health and disease. Nat Rev Genet 19(5), 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke M, Shen H, Wang L, Luo S, Lin L, Yang J, Tian R, 2016. Identification, Quantification, and Site Localization of Protein Posttranslational Modifications via Mass Spectrometry-Based Proteomics. Advances in experimental medicine and biology 919, 345–382. [DOI] [PubMed] [Google Scholar]
- Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, Moran J, Chambert K, Toncheva D, Georgieva L, Grozeva D, Fjodorova M, Wollerton R, Rees E, Nikolov I, van de Lagemaat LN, Bayes A, Fernandez E, Olason PI, Bottcher Y, Komiyama NH, Collins MO, Choudhary J, Stefansson K, Stefansson H, Grant SG, Purcell S, Sklar P, O’Donovan MC, Owen MJ, 2012. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry 17(2), 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisby GE, Standley M, Park T, Olivas A, Fei S, Jacob T, Reddy A, Lu X, Pattee P, Nagalla SR, 2006. Proteomic analysis of the genotoxicant methylazoxymethanol (MAM)-induced changes in the developing cerebellum. J Proteome Res 5(10), 2656–2665. [DOI] [PubMed] [Google Scholar]
- Koenig T, Menze BH, Kirchner M, Monigatti F, Parker KC, Patterson T, Steen JJ, Hamprecht FA, Steen H, 2008. Robust Prediction of the MASCOT Score for an Improved Quality Assessment in Mass Spectrometric Proteomics. Journal of Proteome Research 7(9), 3708–3717. [DOI] [PubMed] [Google Scholar]
- Lange V, Picotti P, Domon B, Aebersold R, 2008. Selected reaction monitoring for quantitative proteomics: a tutorial. Molecular Systems Biology 4(1), 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S, 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larance M, Lamond AI, 2015. Multidimensional proteomics for cell biology. Nat Rev Mol Cell Biol 16(5), 269–280. [DOI] [PubMed] [Google Scholar]
- Lawrence RT, Searle BC, Llovet A, Villén J, 2016. Plug-and-play analysis of the human phosphoproteome by targeted high-resolution mass spectrometry. Nature Methods 13, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Van T, van Leeuwen M, Carolina Fierro A, De Maeyer D, Van den Eynden J, Verbeke L, De Raedt L, Marchal K, Nijssen S, 2016. Simultaneous discovery of cancer subtypes and subtype features by molecular data integration. Bioinformatics 32(17), i445–i454. [DOI] [PubMed] [Google Scholar]
- Lee H, Joo J, Nah SS, Kim JW, Kim HK, Kwon JT, Lee HY, Kim YO, Kim HJ, 2015. Changes in Dpysl2 expression are associated with prenatally stressed rat offspring and susceptibility to schizophrenia in humans. International journal of molecular medicine 35(6), 1574–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Kang MG, Park JS, Lee G, Ting AY, Rhee HW, 2016. APEX Fingerprinting Reveals the Subcellular Localization of Proteins of Interest. Cell Rep 15(8), 1837–1847. [DOI] [PubMed] [Google Scholar]
- Leitner A, 2016. Enrichment Strategies in Phosphoproteomics. Methods in molecular biology (Clifton, N.J.) 1355, 105–121. [DOI] [PubMed] [Google Scholar]
- Li H, Han J, Pan J, Liu T, Parker CE, Borchers CH, 2017a. Current trends in quantitative proteomics - an update. Journal of mass spectrometry : JMS 52(5), 319–341. [DOI] [PubMed] [Google Scholar]
- Li J, Shi M, Ma Z, Zhao S, Euskirchen G, Ziskin J, Urban A, Hallmayer J, Snyder M, 2014. Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Mol Syst Biol 10, 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang W, Yang H, Howrigan DP, Wilkinson B, Souaiaia T, Evgrafov OV, Genovese G, Clementel VA, Tudor JC, Abel T, Knowles JA, Neale BM, Wang K, Sun F, Coba MP, 2017b. Spatiotemporal profile of postsynaptic interactomes integrates components of complex brain disorders. Nat Neurosci 20(8), 1150–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, Sunkin SM, Li Z, Shin Y, Zhu Y, Sousa AMM, Werling DM, Kitchen RR, Kang HJ, Pletikos M, Choi J, Muchnik S, Xu X, Wang D, Lorente-Galdos B, Liu S, Giusti-Rodriguez P, Won H, de Leeuw CA, Pardinas AF, BrainSpan C, Psych EC, Psych EDS, Hu M, Jin F, Li Y, Owen MJ, O’Donovan MC, Walters JTR, Posthuma D, Reimers MA, Levitt P, Weinberger DR, Hyde TM, Kleinman JE, Geschwind DH, Hawrylycz MJ, State MW, Sanders SJ, Sullivan PF, Gerstein MB, Lein ES, Knowles JA, Sestan N, 2018. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362(6420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Friedman AB, Roh MS, Jope RS, 2005. Anesthesia and post-mortem interval profoundly influence the regulatory serine phosphorylation of glycogen synthase kinase-3 in mouse brain. Journal of neurochemistry 92(3), 701–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Li M, Luo XJ, Su B, 2018. Systems-level analysis of risk genes reveals the modular nature of schizophrenia. Schizophr Res 201, 261–269. [DOI] [PubMed] [Google Scholar]
- Lukashin AV, Lukashev ME, Fuchs R, 2003. Topology of gene expression networks as revealed by data mining and modeling. Bioinformatics 19(15), 1909–1916. [DOI] [PubMed] [Google Scholar]
- MacDonald ML, Ciccimaro E, Prakash A, Banerjee A, Seeholzer SH, Blair IA, Hahn CG, 2012. Biochemical fractionation and stable isotope dilution liquid chromatography-mass spectrometry for targeted and microdomain-specific protein quantification in human postmortem brain tissue. Mol Cell Proteomics 11(12), 1670–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni C, Kia DA, Vandrovcova J, Hardy J, Wood NW, Lewis PA, Ferrari R, 2018. Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences. Briefings in bioinformatics 19(2), 286–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardamshina M, Geiger T, 2017. Next-Generation Proteomics and Its Application to Clinical Breast Cancer Research. The American journal of pathology 187(10), 2175–2184. [DOI] [PubMed] [Google Scholar]
- Margolin AA, Wang K, Lim WK, Kustagi M, Nemenman I, Califano A, 2006. Reverse engineering cellular networks. Nat Protoc 1(2), 662–671. [DOI] [PubMed] [Google Scholar]
- Martins-De-Souza D, Dias-Neto E, Schmitt A, Falkai P, Gormanns P, Maccarrone G, Turck CW, Gattaz WF, 2010. Proteome analysis of schizophrenia brain tissue. World J Biol Psychiatry 11(2), 110–120. [DOI] [PubMed] [Google Scholar]
- Martins-de-Souza D, Gattaz WF, Schmitt A, Novello JC, Marangoni S, Turck CW, Dias-Neto E, 2009. Proteome analysis of schizophrenia patients Wernicke’s area reveals an energy metabolism dysregulation. BMC Psychiatry 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson T, Franken H, Kosinski J, Kurzawa N, Zinn N, Sweetman G, Poeckel D, Ratnu VS, Schramm M, Becher I, Steidel M, Noh K-M, Bergamini G, Beck M, Bantscheff M, Savitski MM, 2018. Systematic analysis of protein turnover in primary cells. Nature Communications 9(1), 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JL, Depasquale EA, Funk AJ, O’Donnovan SM, Hasselfeld K, Marwaha S, Hammond JH, Hartounian V, Meador-Woodruff JH, Meller J, McCullumsmith RE, 2017. Abnormalities of signal transduction networks in chronic schizophrenia. npj Schizophrenia 3(1), 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaci L, De Angelis E, Montemurro N, Pilolli R, 2018. Comprehensive overview and recent advances in proteomics MS based methods for food allergens analysis. TrAC Trends in Analytical Chemistry 106, 21–36. [Google Scholar]
- Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, Mann M, 2011. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natividad LA, Buczynski MW, McClatchy DB, Yates JR 3rd, 2018. From Synapse to Function: A Perspective on the Role of Neuroproteomics in Elucidating Mechanisms of Drug Addiction. Proteomes 6(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro P, Kuharev J, Gillet LC, Bernhardt OM, MacLean B, Röst HL, Tate SA, Tsou C-C, Reiter L, Distler U, Rosenberger G, Perez-Riverol Y, Nesvizhskii AI, Aebersold R, Tenzer S, 2016. A multicenter study benchmarks software tools for label-free proteome quantification. Nature Biotechnology 34, 1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, Cai G, Lihm J, Dannenfelser R, Jabado O, Peralta Z, Nagaswamy U, Muzny D, Reid JG, Newsham I, Wu Y, Lewis L, Han Y, Voight BF, Lim E, Rossin E, Kirby A, Flannick J, Fromer M, Shakir K, Fennell T, Garimella K, Banks E, Poplin R, Gabriel S, DePristo M, Wimbish JR, Boone BE, Levy SE, Betancur C, Sunyaev S, Boerwinkle E, Buxbaum JD, Cook EH Jr., Devlin B, Gibbs RA, Roeder K, Schellenberg GD, Sutcliffe JS, Daly MJ, 2012. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485(7397), 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvaderani M, Matsumoto I, Sivagnanasundaram S, 2009. Anterior hippocampus in schizophrenia pathogenesis: molecular evidence from a proteome study. Aust N Z J Psychiatry 43(4), 310–322. [DOI] [PubMed] [Google Scholar]
- Oh-Nishi A, Koga K, Maeda T, Suhara T, 2016. A possible serologic biomarker for maternal immune activation-associated neurodevelopmental disorders found in the rat models. Neuroscience research 113, 63–70. [DOI] [PubMed] [Google Scholar]
- Pennington K, Beasley CL, Dicker P, Fagan A, English J, Pariante CM, Wait R, Dunn MJ, Cotter DR, 2008. Prominent synaptic and metabolic abnormalities revealed by proteomic analysis of the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder. Mol Psychiatry 13(12), 1102–1117. [DOI] [PubMed] [Google Scholar]
- Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ, 2012. Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Molecular & Cellular Proteomics 11(11), 1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Aebersold R, 2012. Selected reaction monitoring–based proteomics: workflows, potential, pitfalls and future directions. Nature Methods 9, 555. [DOI] [PubMed] [Google Scholar]
- Plotnikov A, Zehorai E, Procaccia S, Seger R, 2011. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim Biophys Acta 1813(9), 1619–1633. [DOI] [PubMed] [Google Scholar]
- Prinz AA, Bucher D, Marder E, 2004. Similar network activity from disparate circuit parameters. Nat Neurosci 7(12), 1345–1352. [DOI] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kahler A, Duncan L, Stahl E, Genovese G, Fernandez E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PK, Banks E, Shakir K, Garimella K, Fennell T, DePristo M, Grant SG, Haggarty SJ, Gabriel S, Scolnick EM, Lander ES, Hultman CM, Sullivan PF, McCarroll SA, Sklar P, 2014. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506(7487), 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie MD, Holzinger ER, Li R, Pendergrass SA, Kim D, 2015. Methods of integrating data to uncover genotype-phenotype interactions. Nat Rev Genet 16(2), 85–97. [DOI] [PubMed] [Google Scholar]
- Roncada P, Bortolato M, Frau R, Saba P, Flore G, Soggiu A, Pisanu S, Amoresano A, Carpentieri A, Devoto P, 2009. Gating deficits in isolation-reared rats are correlated with alterations in protein expression in nucleus accumbens. Journal of neurochemistry 108(3), 611–620. [DOI] [PubMed] [Google Scholar]
- Saia-Cereda VM, Cassoli JS, Schmitt A, Falkai P, Martins-de-Souza D, 2016. Differential proteome and phosphoproteome may impact cell signaling in the corpus callosum of schizophrenia patients. Schizophr Res 177(1–3), 70–77. [DOI] [PubMed] [Google Scholar]
- Saia-Cereda VM, Santana AG, Schmitt A, Falkai P, Martins-de-Souza D, 2017. The Nuclear Proteome of White and Gray Matter from Schizophrenia Postmortem Brains. Mol Neuropsychiatry 3(1), 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffler K, Viner R, Damoc E, 2018. High resolution top-down experimental strategies on the Orbitrap platform. Journal of Proteomics 175, 42–55. [DOI] [PubMed] [Google Scholar]
- Schizophrenia Psychiatric Genome-Wide Association Study, C., 2011. Genome-wide association study identifies five new schizophrenia loci. Nat Genet 43(10), 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics, C., 2014. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511(7510), 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M, 2011. Global quantification of mammalian gene expression control. Nature 473(7347), 337–342. [DOI] [PubMed] [Google Scholar]
- Schwarz E, Izmailov R, Lio P, Meyer-Lindenberg A, 2016. Protein Interaction Networks Link Schizophrenia Risk Loci to Synaptic Function. Schizophrenia bulletin 42(6), 1334–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwudke D, Liebisch G, Herzog R, Schmitz G, Shevchenko A, 2007. Shotgun Lipidomics by Tandem Mass Spectrometry under Data‐Dependent Acquisition Control, Methods in Enzymology. Academic Press, pp. 175–191. [DOI] [PubMed] [Google Scholar]
- Sharma K, D’Souza RC, Tyanova S, Schaab C, Wisniewski JR, Cox J, Mann M, 2014. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep 8(5), 1583–1594. [DOI] [PubMed] [Google Scholar]
- Shilov IV, Seymour SL, Patel AA, Loboda A, Tang WH, Keating SP, Hunter CL, Nuwaysir LM, Schaeffer DA, 2007. The Paragon Algorithm, a Next Generation Search Engine That Uses Sequence Temperature Values and Feature Probabilities to Identify Peptides from Tandem Mass Spectra. Molecular & Cellular Proteomics 6(9), 1638. [DOI] [PubMed] [Google Scholar]
- Sivagnanasundaram S, Crossett B, Dedova I, Cordwell S, Matsumoto I, 2007. Abnormal pathways in the genu of the corpus callosum in schizophrenia pathogenesis: a proteome study. Proteomics Clin Appl 1(10), 1291–1305. [DOI] [PubMed] [Google Scholar]
- Skinner OS, Haverland NA, Fornelli L, Melani RD, Do Vale LHF, Seckler HS, Doubleday PF, Schachner LF, Srzentić K, Kelleher NL, Compton PD, 2017. Top-down characterization of endogenous protein complexes with native proteomics. Nature Chemical Biology 14, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalla KH, Mikhaylova M, Sahin J, Bernstein HG, Bogerts B, Schmitt A, van der Schors R, Smit AB, Li KW, Gundelfinger ED, Kreutz MR, 2008. A comparison of the synaptic proteome in human chronic schizophrenia and rat ketamine psychosis suggest that prohibitin is involved in the synaptic pathology of schizophrenia. Mol Psychiatry 13(9), 878–896. [DOI] [PubMed] [Google Scholar]
- Smits AH, Vermeulen M, 2016. Characterizing Protein-Protein Interactions Using Mass Spectrometry: Challenges and Opportunities. Trends Biotechnol 34(10), 825–834. [DOI] [PubMed] [Google Scholar]
- State MW, Sestan N, 2012. Neuroscience. The emerging biology of autism spectrum disorders. Science 337(6100), 1301–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthram S, Beyer A, Karp RM, Eldar Y, Ideker T, 2008. eQED: an efficient method for interpreting eQTL associations using protein networks. Mol Syst Biol 4, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swatton JE, Sellers LA, Faull RLM, Holland A, Iritani S, Bahn S, 2004. Increased MAP kinase activity in Alzheimer’s and Down syndrome but not in schizophrenia human brain. European Journal of Neuroscience 19(10), 2711–2719. [DOI] [PubMed] [Google Scholar]
- Szatkiewicz JP, O’Dushlaine C, Chen G, Chambert K, Moran JL, Neale BM, Fromer M, Ruderfer D, Akterin S, Bergen SE, Kahler A, Magnusson PK, Kim Y, Crowley JJ, Rees E, Kirov G, O’Donovan MC, Owen MJ, Walters J, Scolnick E, Sklar P, Purcell S, Hultman CM, McCarroll SA, Sullivan PF, 2014. Copy number variation in schizophrenia in Sweden. Mol Psychiatry 19(7), 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Hamon C, 2003. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Analytical Chemistry 75(8), 1895–1904. [DOI] [PubMed] [Google Scholar]
- Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Bjork L, Breckels LM, Backstrom A, Danielsson F, Fagerberg L, Fall J, Gatto L, Gnann C, Hober S, Hjelmare M, Johansson F, Lee S, Lindskog C, Mulder J, Mulvey CM, Nilsson P, Oksvold P, Rockberg J, Schutten R, Schwenk JM, Sivertsson A, Sjostedt E, Skogs M, Stadler C, Sullivan DP, Tegel H, Winsnes C, Zhang C, Zwahlen M, Mardinoglu A, Ponten F, von Feilitzen K, Lilley KS, Uhlen M, Lundberg E, 2017. A subcellular map of the human proteome. Science 356(6340). [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Cox J, 2016. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols 11, 2301. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnstrom H, Glimelius B, Sjoblom T, Edqvist PH, Djureinovic D, Micke P, Lindskog C, Mardinoglu A, Ponten F, 2017. A pathology atlas of the human cancer transcriptome. Science 357(6352). [DOI] [PubMed] [Google Scholar]
- Uzozie AC, Aebersold R, 2018. Advancing translational research and precision medicine with targeted proteomics. Journal of Proteomics 189, 1–10. [DOI] [PubMed] [Google Scholar]
- Varnaite R, MacNeill SA, 2016. Meet the neighbors: Mapping local protein interactomes by proximity-dependent labeling with BioID. Proteomics 16(19), 2503–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasquez E, Nogueira FCS, Velasquez I, Schmitt A, Falkai P, Domont GB, Martins-de-Souza D, 2017. Synaptosomal Proteome of the Orbitofrontal Cortex from Schizophrenia Patients Using Quantitative Label-Free and iTRAQ-Based Shotgun Proteomics. J Proteome Res 16(12), 4481–4494. [DOI] [PubMed] [Google Scholar]
- Venerando A, Cesaro L, Pinna LA, 2017. From phosphoproteins to phosphoproteomes: a historical account. The FEBS journal 284(13), 1936–1951. [DOI] [PubMed] [Google Scholar]
- Voevodski K, Teng SH, Xia Y, 2009. Spectral affinity in protein networks. BMC Syst Biol 3, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, Clarke D, Gu M, Emani P, Yang YT, Xu M, Gandal MJ, Lou S, Zhang J, Park JJ, Yan C, Rhie SK, Manakongtreecheep K, Zhou H, Nathan A, Peters M, Mattei E, Fitzgerald D, Brunetti T, Moore J, Jiang Y, Girdhar K, Hoffman GE, Kalayci S, Gumus ZH, Crawford GE, Psych EC, Roussos P, Akbarian S, Jaffe AE, White KP, Weng Z, Sestan N, Geschwind DH, Knowles JA, Gerstein MB, 2018a. Comprehensive functional genomic resource and integrative model for the human brain. Science 362(6420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, MacDonald ML, Borgmann-Winter KE, Banerjee A, Sleiman P, Tom A, Khan A, Lee KC, Roussos P, Siegel SJ, Hemby SE, Bilker WB, Gur RE, Hahn CG, 2018b. mGluR5 hypofunction is integral to glutamatergic dysregulation in schizophrenia. Mol Psychiatry. [DOI] [PMC free article] [PubMed]
- Wiese S, Reidegeld KA, Meyer HE, Warscheid B, 2007. Protein labeling by iTRAQ: A new tool for quantitative mass spectrometry in proteome research. PROTEOMICS 7(3), 340–350. [DOI] [PubMed] [Google Scholar]
- Willsey AJ, Morris MT, Wang S, Willsey HR, Sun N, Teerikorpi N, Baum TB, Cagney G, Bender KJ, Desai TA, Srivastava D, Davis GW, Doudna J, Chang E, Sohal V, Lowenstein DH, Li H, Agard D, Keiser MJ, Shoichet B, von Zastrow M, Mucke L, Finkbeiner S, Gan L, Sestan N, Ward ME, Huttenhain R, Nowakowski TJ, Bellen HJ, Frank LM, Khokha MK, Lifton RP, Kampmann M, Ideker T, State MW, Krogan NJ, 2018. The Psychiatric Cell Map Initiative: A Convergent Systems Biological Approach to Illuminating Key Molecular Pathways in Neuropsychiatric Disorders. Cell 174(3), 505–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, State MW, 2015. Autism spectrum disorders: from genes to neurobiology. Curr Opin Neurobiol 30, 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM, 2007a. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proceedings of the National Academy of Sciences of the United States of America 104(14), 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM, 2007b. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proceedings of the National Academy of Sciences 104(14), 5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zeng J, Zhang F, Zhu Z, Qi T, Zheng Z, Lloyd-Jones LR, Marioni RE, Martin NG, Montgomery GW, Deary IJ, Wray NR, Visscher PM, McRae AF, Yang J, 2018. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun 9(1), 918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Zhang Y, Kim Y, Kim S, Kim JJ, Kim KM, Yoshizawa J, Fan L-Y, Cao C-X, Wong DTW, 2016. Differential Proteomic Analysis of Human Saliva using Tandem Mass Tags Quantification for Gastric Cancer Detection. Scientific Reports 6, 22165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wagner SA, Beli P, 2015. Illuminating Spatial and Temporal Organization of Protein Interaction Networks by Mass Spectrometry-Based Proteomics. Frontiers in genetics 6, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yugi K, Kubota H, Hatano A, Kuroda S, 2016. Trans-Omics: How To Reconstruct Biochemical Networks Across Multiple ‘Omic’ Layers. Trends Biotechnol 34(4), 276–290. [DOI] [PubMed] [Google Scholar]
- Yurkovich JT, Palsson BO, 2018. Quantitative -omic data empowers bottom-up systems biology. Current opinion in biotechnology 51, 130–136. [DOI] [PubMed] [Google Scholar]
- Zanivan S, Meves A, Behrendt K, Schoof EM, Neilson LJ, Cox J, Tang HR, Kalna G, van Ree JH, van Deursen JM, Trempus CS, Machesky LM, Linding R, Wickstrom SA, Fassler R, Mann M, 2013. In vivo SILAC-based proteomics reveals phosphoproteome changes during mouse skin carcinogenesis. Cell Rep 3(2), 552–566. [DOI] [PubMed] [Google Scholar]
- Zhang B, Horvath S, 2005. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4, Article17. [DOI] [PubMed] [Google Scholar]
- Zhang T, Shen S, Qu J, Ghaemmaghami S, 2016. Global Analysis of Cellular Protein Flux Quantifies the Selectivity of Basal Autophagy. Cell Reports 14(10), 2426–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Fonslow BR, Shan B, Baek M-C, Yates JR, 2013. Protein Analysis by Shotgun/Bottom-up Proteomics. Chemical Reviews 113(4), 2343–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev RA, Makarov A, 2013. Orbitrap Mass Spectrometry. Analytical Chemistry 85(11), 5288–5296. [DOI] [PubMed] [Google Scholar]