

Abstract
We describe libmolgrid, a general-purpose library for representing three-dimensional molecules using multidimensional arrays of voxelized molecular data. libmolgrid provides functionality for sampling batches of data suited to machine learning workflows, and it also supports temporal and spatial recurrences over that data to facilitate work with convolutional and recurrent neural networks. It was designed for seamless integration with popular deep learning frameworks and features optimized performance by leveraging graphics processing units (GPUs). libmolgrid is a free and open source project (GPLv2) that aims to democratize grid-based modeling in computational chemistry.
Graphical Abstract
INTRODUCTION
Deep learning has emerged as an important area of research in computational chemistry. It holds great promise for unprecedented improvements in predictive capabilities for such problems as virtual screening,1 binding affinity prediction,2,3 pose prediction,4,5 and lead optimization.6–9 The representation of input data can fundamentally limit or enhance the performance and applicability of machine learning algorithms.10–12 Deep learning can derive class-defining features directly from training examples. Common input representations include molecular formats like SMILES and/ or InChi strings,12,13 molecular graphs,10,11,14–17 and voxelized spatial grids18,19 representing the locations of atoms.
Compared with other representation schemes, spatial grids possess certain virtues including minimal overt featurization by the user (theoretically permitting greater model expressiveness) and full representation of three-dimensional spatial interactions in the input. For regular cubic grids, this comes at the cost of coordinate frame dependence, which can be ameliorated by data augmentation19 and can also be theoretically addressed with various types of inherently equivariant network architectures20–24 or by using other types of multidimensional grids.25,26 Spatial grids have been applied successfully to tasks relevant to computational chemistry like virtual screening,18,19,27 pharmacophore generation,28 molecular property prediction,29,30 molecular classi-fication,29,31 protein binding site prediction,32–34 molecular autoencoding,35 and generative modeling.36–38
Chemical data sets have many physical and statistical properties that prove problematic for machine learning, and special care must be taken to manage them. Classes are typically highly imbalanced with many more known inactive than active compounds for a given protein target; regression tasks may span many orders of magnitude with nonuniform representation of the underlying chemical space at particular ranges of the regressor, and examples with matching class labels or regression target values may also be unequally sampled from other underlying classes (e.g., there may be significantly more binding affinity data available for specific proteins that have been the subject of greater investigation, such as the estrogen receptors, or for protein classes like kinases). By offloading data processing tasks required to manage these problems to an open source library specialized for chemical data, computational chemists can systematically obtain better results in a transparent manner.
Using multidimensional grids (e.g., voxels) to represent atomic locations (and potentially distributions) is computationally efficient - their generation is embarrassingly parallel and therefore readily amenable to modern GPU architectures - and preserves three-dimensional spatial relationships present in the original input. Coordinate frame dependence can be removed or circumvented. However, commonly available molecular parsing and conversion libraries do not yet provide gridding functionality, nor do they implement the other tasks required to obtain good performance on typical chemical data sets such as strategic resampling and data augmentation. Thus, we abstracted the gridding and batch preparation functionality from our past work, gnina,19 into a library that can be used for general molecular modeling tasks but also interfaces naturally with popular Python deep learning libraries. Implemented in C++/CUDA with Python bindings, libmolgrid is a free and open source project (distributed under the GNU General Public License, version 2) intended to accelerate advances in molecular modeling via multidimensional spatial arrays.
IMPLEMENTATION
Key libmolgrid functionality is implemented in a modular fashion to ensure maximum versatility. Essential library features are abstracted into separate classes to facilitate use independently or in concert as required by a particular application.
Atom Typing.
Several atom typing schemes are supported, featuring flexibility in the ways types are assigned and represented. Atoms may be typed according to XS atom typing, atomic element, or a user-provided callback function (an example of this use-case is shown in Table S1). Types may be represented by a single integer or a vector encoding. For a typical user, typing (with either index or vector types) can be performed automatically via an ExampleProvider.
Examples.
Examples consist of typed coordinates that will be analyzed together, along with their labels. An Example may consist of multiple CoordinateSets (which may each utilize a different scheme for atom typing) and may be one of a sequence of Examples within a group. For example, a single Example may have a CoordinateSet for a receptor and another CoordinateSet for a ligand to be scored with that receptor, or perhaps multiple CoordinateSets corresponding to multiple poses of a particular ligand. Examples may be part of a group that will be processed in sequence, for example as input to a recurrent network; in that case distinct groups are identified with a shared integer value, and a sequence continuation flag indicates whether a given Example is a continuation of a previously observed sequence or is initiating a new one.
ExampleProvider.
To obtain strategically sampled batches of data for training, a user can employ an ExampleProvider. The desired sampling options are specified to the ExampleProvider constructor, which can then be populated with one or more files specifying examples. Properly sampled Examples are obtained via ExampleProvider::next or ExampleProvider::next_batch. Figure 1 shows graphically how an ExampleProvider might obtain a batch of 10 shuffled, class-balanced, receptor-stratified Examples from a larger data set, with accompanying code.
Figure 1.
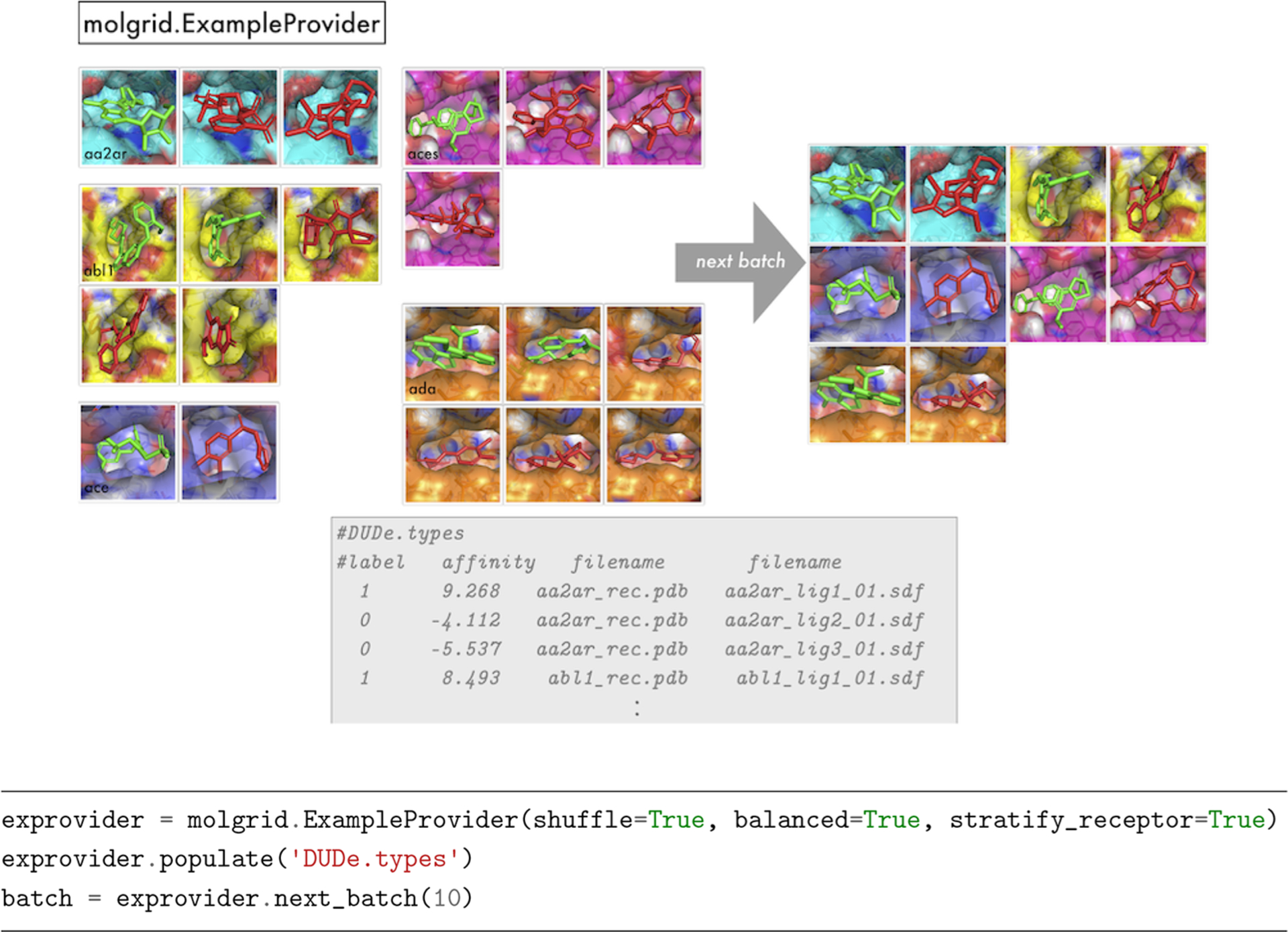
An illustration of molgrid::ExampleProvider usage, sampling a batch of 10 randomized, balanced, and receptor-stratified examples from a data set.
Currently, the simplest way to initialize a provider is to populate it with one or more files that specify metadata for Examples, with one Example per line. At a high level, that line will specify class and regression target values for the Example, any group identification associated with the Example (i.e., a shared integer label identifying Examples to be processed sequentially, as with temporal data provided as input to a recurrent network), and one or more strings identifying filenames of molecules corresponding to that Example. Each molecule file can have distinct typing rules applied. A typical use case is to have a receptor and a ligand file, but a single receptor file could be provided, e.g., when learning properties of binding sites, or multiple files could be provided, e.g., when learning properties of ensembles. The default line layout is [(int)group][(float)label]*-[molfile]+. An example is shown in Figure 1. In the examples we provide with our project, these files have a .types suffix.
Table S2 shows all the available options at the time of construction. These options are described in more detail in the Supporting Information and online documentation.
Grids.
The fundamental object used to represent data in libmolgrid is a multidimensional array which the API generically refers to as a grid. Grids are typically used during training to represent voxelized input molecules or matrices of atom coordinates and types. They can be constructed in two flavors, Grids and ManagedGrids. ManagedGrids manage their own underlying memory, while Grids function as views over a preexisting memory buffer. Grids and ManagedGrids are convertible to NumPy arrays as well as Torch tensors. Additional exposition is available in Figure SS1.
Because of automatic conversions designed for PyTorch interoperability, a user intending to leverage basic batch sampling, grid generating, and transformation capabilities provided by libmolgrid in tandem with PyTorch for neural network training can simply use Torch tensors directly, with little to no need for explicit invocation of or interaction with libmolgrid grids. Memory allocated on a GPU via a Torch tensor will remain there, with grids generated in-place. An example of this type of usage is shown in the first example in Table 1.
Table 1.
Examples of Gridand ManagedGrid Usage
|
# Usage 1: molgrid functions taking Grid objects can be passed Torch tensors directly, # with conversions managed internally tensor = torch.zeros(tensor_shape, dtype=torch.float32, device='cuda') molgrid.gmaker.forward(batch, tensor) |
|
# Usage 2: construct Grid as a view over a Torch tensor with provided helper function tensor = torch.zeros((2,2), dtype=torch.float32, device='cuda') grid=molgrid.tensor_as_grid(tensor) # dimensions and data location are inferred # alternatively, construct Grid view over Torch tensor directly grid = molgrid.Grid2fCUDA(tensor) |
|
# Usage 3: copy ManagedGrid data to NumPy array # first, construct a ManagedGrid mgrid = molgrid.MGridlf(batch_size) # copy to GPU and do work on it there mgrid.gpu() # (do work) # copy ManagedGrid data to a NumPy array with helper function; # this copies data back to the CPU if necessary array1 = mgrid. tonumpyO # alternatively, construct NumPy array with a copy of ManagedGrid CPU data; # must sync to CPU first mgrid.cpu() array2 = np.array(mgrid) |
|
# Usage 4: construct Grid from NumPy array array3 = np.zeros((2,2), dtype=np.float32) # must match source and destination dtypes tensor = molgrid.Grid2f(array3) |
A Grid may also be constructed explicitly from a Torch tensor, a NumPy array, or, if necessary, from a pointer to a memory buffer. Examples of constructing a Grid from a Torch tensor are shown in the second usage section in Table 1. The third usage section shows provided functionality for copying NumPy array data to ManagedGrids, while the fourth usage section shows functionality for constructing Grid views over NumPy array data buffers. In the fourth example, note that in recent NumPy versions the default floating-point data type is float64, so the user should take care to match the data type between arrays and Grids.
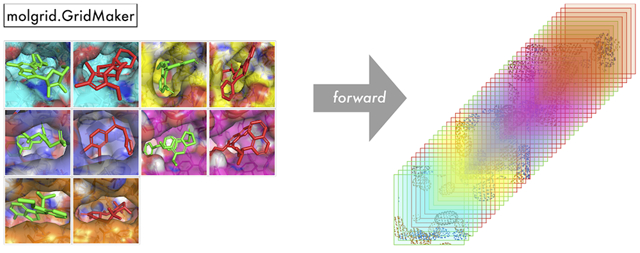
GridMaker.
A GridMaker is used to generate a voxel grid from an Example, an ExampleVec, a CoordinateSet, or paired Grids of coordinates and types. GridMaker can operate directly on a user-provided Torch tensor or Grid, or it can return into a new NumPy array via GridMaker::make_ndarray or Torch tensor via GridMaker::make_tensor. GridMaker features GPU-optimized gridding that will be used if a compatible device is available. GridMaker options pertaining to the properties of the resulting grid are specified when the GridMaker is constructed, while the examples from which a grid will be generated and their instantiation properties (including any transformations) are specified by a particular invocation of GridMaker::forward. Specifically, Table S3 shows the possible constructor arguments, which are described in more detail in the Supporting Information. Figure 2 shows an example of basic GridMaker usage, default-constructing a GridMaker and using it to populate a grid with densities for a batch of molecules. If desired, the values of random_translation and random_rotation can be set in the call to gmaker::forward, thereby applying random data augmentation to each example in the batch. If it is desirable to retain the applied transformation, then transformations can be created explicitly, as shown in Figure S2. The GridMaker class also defines a backward function that computes atomic gradients, which can be used for tasks ranging from visualizing what a network has learned to using a trained network to optimize the coordinates and types of input molecules.
Figure 2.
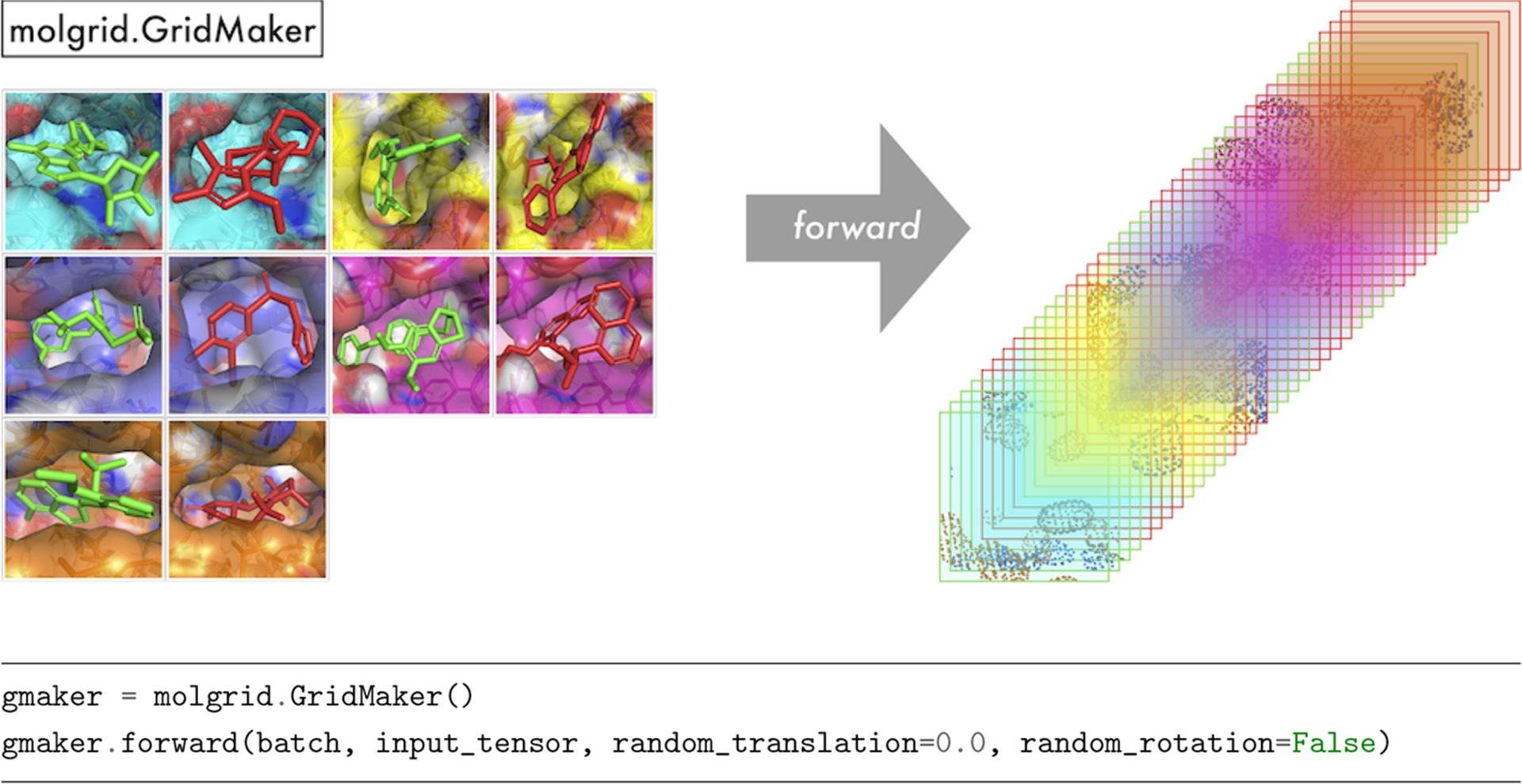
An illustration of molgrid::GridMaker usage, generating a 4-dimensional grid from a batch of molecules, with data layout examples×channels×length×width×height.
Transformations.
Data augmentation in the form of random rotations and translations of input examples can be performed by passing the desired options to GridMaker::forward, as described in the previous section. Specific translations and rotations can also be applied to arbitrary Grids, CoordinateSets, or Examples by using the Transform class directly. Transforms can store specific rotations, described by a libmolgrid::Quaternion, an origin around which to rotate, described by a libmolgrid::float3, which is also interconvertible with a Python tuple, and a specific translation, expressed in terms of Cartesian coordinates and also described by a float3. These prove useful for sophisticated networks such as the spatial transformer.39 Additional examples of Transform constructor invocation are shown in Table S4, and information about Transform::forward is shown in Figure S2.
RESULTS
We demonstrate model training with input tensors populated by libmolgrid and neural networks implemented using Caffe, PyTorch, and Keras with a Tensorflow backend (code available at https://gnina.github.io/libmolgrid/tutorials.html). Training loss performance is similar across all three frameworks, as shown in Figure S3. libmolgrid is fully functional with any of these popular libraries. Its overall speed and memory footprint vary significantly with the user’s chosen library, however. As shown in Figure 3(a) and (b), the performance when using a GPU for gridding and neural network training is much faster when using Caffe and PyTorch than it is when using Tensorflow via Keras, with modest improvements in performance for Caffe and PyTorch when using the newer Titan V GPU rather than the older GTX Titan X. This is due to libmolgrid’s ability to directly access underlying data buffers when interoperating with Caffe and PyTorch, thus avoiding unnecessary data migration between the CPU and GPU; this is not currently possible with Tensorflow, and so passes through the network involve grids being generated on the GPU by libmolgrid, copied into a NumPy array on the CPU, and then copied back onto the GPU by Tensorflow when training begins. This results in a significant performance penalty, with memory transfers fundamentally limiting performance; future versions of libmolgrid will seek to mitigate this issue with Tensorflow 2.0. The discrepancy in memory utilization shown in Figure 3(c) is somewhat less dramatic, but similarly, memory utilization when doing neural network training with Tensorflow is less efficient than using the other two libraries.
Figure 3.
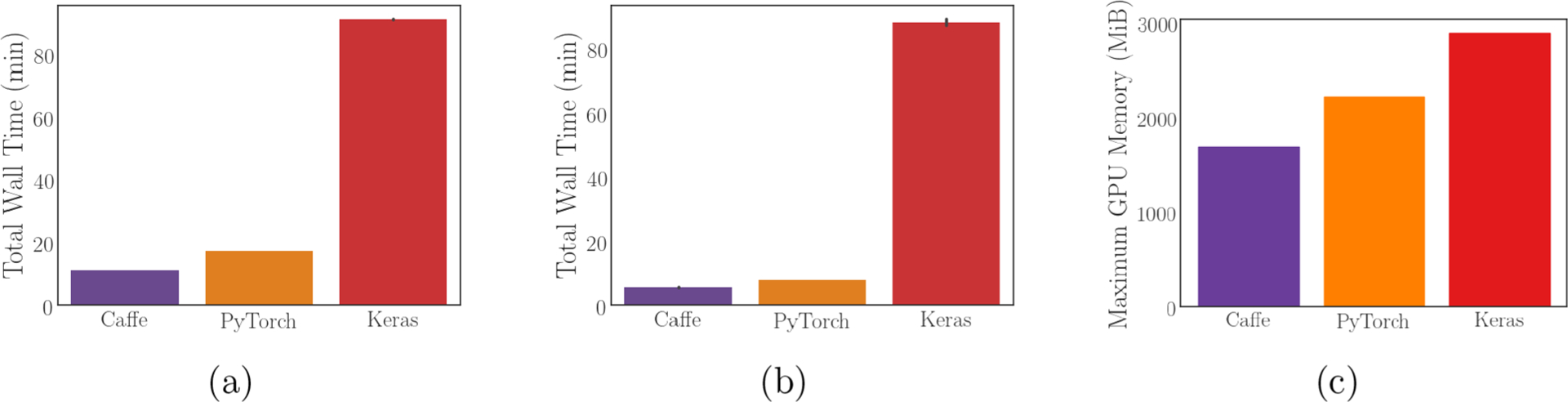
Performance information for using libmolgrid with each major supported neural network library. All error bars are 98% confidence intervals computed via bootstrap sampling of five independent runs. (a) Walltime for training the simple model shown training above using a GTX Titan X. (b) Walltime for training the same simple model using a Titan V. (c) Maximum GPU memory utilization while training.
As an example of a more specialized task that uses the backward gradients computed by GridMaker, we demonstrate training a CNN to convert voxelized atomic densities to Cartesian coordinates. Each training example consists of a single atom provided to the network as a voxelized grid for which the network will output Cartesian coordinates. The loss function is a simple mean squared error grid loss for coordinates that fall within the grid, and a hingelike loss for coordinates outside. As shown in Figure 4(a), the model initially has difficulty learning because the atomic gradients only receive information from the parts of the grid that overlap an atom, but eventually converges to an accuracy significantly better than the grid resolution of 0.5 Å. Example predictions are shown in Figure 4(b). This task could be applicable to a generative modeling workflow and also demonstrates libmolgrid’s versatility as a molecular modeling tool.
Figure 4.
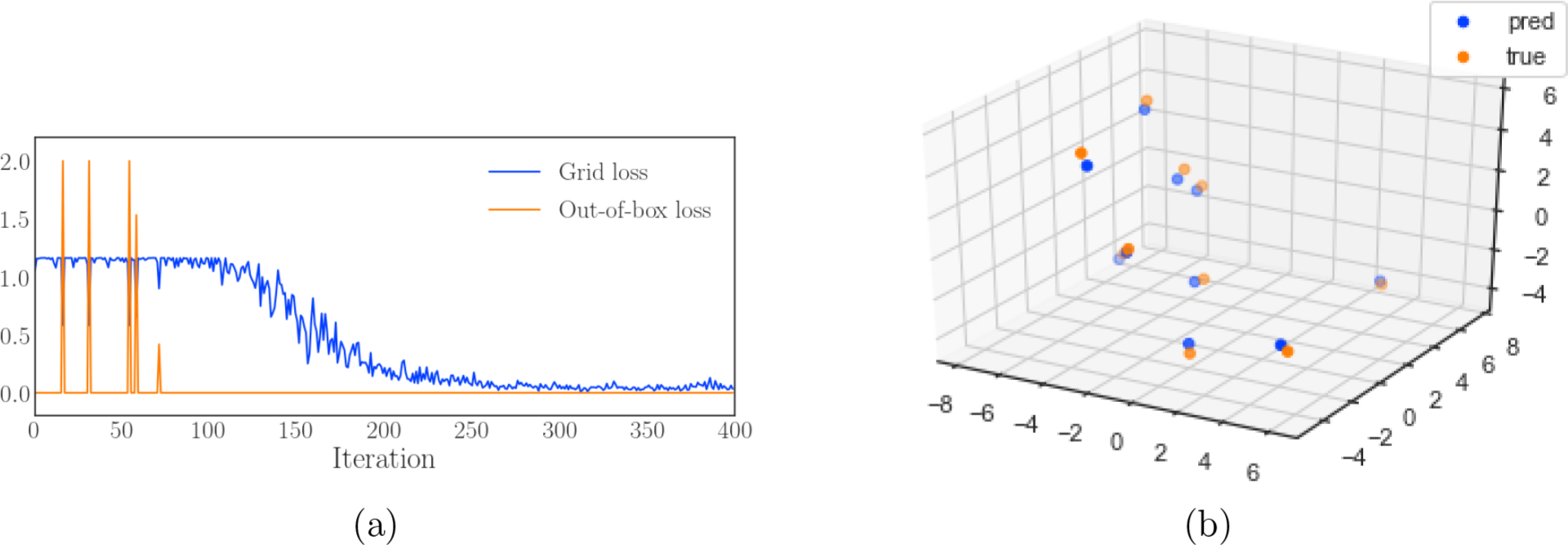
Cartesian coordinates from grid densities. (a) Loss per iteration for both the grid loss and out-of-box loss for training with naively initialized coordinates, showing libmolgrid’s utility for converting between voxelized grids and Cartesian coordinates. (b) Sampled coordinate predictions compared with the true coordinates, demonstrating a root mean squared accuracy of 0.09 Å.
CONCLUSION
Machine learning is a major research area within computational chemistry and drug discovery, and grid-based representation methods have been applied to many fundamental problems with great success. No standard library exists for automatically generating voxel grids or spatial array representations more generally from molecular data or for performing the basic tasks such as data augmentation that typically must be done to achieve high predictive capability on chemical data sets using these methods. This means that researchers hoping to pursue methodological advances using grid-based methods must reproduce the work of other groups and waste time with redundant programming. libmolgrid empowers researchers to pursue advances in grid-based machine learning for molecular data by providing an efficient, concise, and natural C++/CUDA and Python API for data resampling, grid generation, and data augmentation. It also supports spatial and temporal recurrences over input, allowing for size extensiveness even while using cubic grids (by performing a subgrid decomposition), and processing of simulation data such as molecular dynamics trajectories while preserving temporal ordering of frames, if desired. With adoption, it will also help standardize performance, enhance reproducibility, and facilitate experimentation among computational chemists interested in machine learning methods. libmolgrid support for Caffe and PyTorch is complete, while we plan to enhance Tensorflow support by taking advantage of the Tensorflow 2.0 programming model and avoiding the unnecessary data transfers that currently limit combined libmolgrid-Tensorflow performance. Other future enhancements will include the ability to generate other types of grids, for example, spherical ones. Documentation, tutorials, and installation instructions are available at http://gnina.github.io/libmolgrid, while the source code along with active support can be found at https://github.com/gnina/libmolgrid.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Grant R01GM108340 from the National Institute of General Medical Sciences.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.9b01145.
Additional API documentation and information required to reproduce results and figures (PDF)
Contributor Information
Jocelyn Sunseri, Department of Computational and Systems Biology, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, United States;.
David R. Koes, Department of Computational and Systems Biology, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, United States;.
■ REFERENCES
- (1).Jorissen RN; Gilson MK Virtual Screening of Molecular Databases Using a Support Vector Machine. J. Chem. Inf. Model 2005, 45, 549–561. [DOI] [PubMed] [Google Scholar]
- (2).Ballester PJ; Mitchell JBO A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics 2010, 26, 1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zilian D; Sotriffer CA SFCscore RF: a random forest-based scoring function for improved affinity prediction of protein–ligand complexes. J. Chem. Inf. Model 2013, 53, 1923–1933. [DOI] [PubMed] [Google Scholar]
- (4).Ashtawy HM; Mahapatra NR Machine-learning scoring functions for identifying native poses of ligands docked to known and novel proteins. BMC Bioinf. 2015, 16, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chupakhin V; Marcou G; Baskin I; Varnek A; Rognan D Predicting ligand binding modes from neural networks trained on protein-ligand interaction fingerprints. J. Chem. Inf. Model 2013, 53, 763–772. [DOI] [PubMed] [Google Scholar]
- (6).Ekins S; Freundlich JS; Hobrath JV; White EL; Reynolds RC Combining computational methods for hit to lead optimization in Mycobacterium tuberculosis drug discovery. Pharm. Res 2014, 31, 414–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yasuo N; Watanabe K; Hara H; Rikimaru K; Sekijima M Predicting Strategies for Lead Optimization via Learning to Rank. IPSJ. Transactions on Bioinformatics 2018, 11, 41–47. [Google Scholar]
- (8).Zhou Z; Kearnes S; Li L; Zare RN; Riley P Optimization of molecules via deep reinforcement learning. Sci. Rep 2019, 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jiménez-Luna J; Pérez-Benito L; Martínez-Rosell G; Sciabola S; Torella R; Tresadern G; De Fabritiis G DeltaDelta neural networks for lead optimization of small molecule potency. Chemical Science 2019, 10, 10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lusci A; Pollastri G; Baldi P Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules. J. Chem. Inf. Model 2013, 53, 1563–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Duvenaud DK; Maclaurin D; Iparraguirre J; Bombarell R; Hirzel T; Aspuru-Guzik A; Adams RP Convolutional networks on graphs for learning molecular fingerprints. Advances in neural information processing systems 2015, 2224–2232. [Google Scholar]
- (12).Gómez-Bombarelli R; Wei JN; Duvenaud D; Hernández-Lobato JM; Sánchez-Lengeling B; Sheberla D; Aguilera-Iparraguirre J; Hirzel TD; Adams RP; Aspuru-Guzik A Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci 2018, 4, 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Winter R; Montanari F; Noé F; Clevert D-A Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chemical Science 2019, 10, 1692–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Urban G; Subrahmanya N; Baldi P Inner and outer recursive neural networks for chemoinformatics applications. J. Chem. Inf. Model 2018, 58, 207–211. [DOI] [PubMed] [Google Scholar]
- (15).Kearnes S; McCloskey K; Berndl M; Pande V; Riley P Molecular graph convolutions: moving beyond fingerprints. J. Comput.-Aided Mol. Des 2016, 30, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Pham T; Tran T; Venkatesh S Graph Memory Networks for Molecular Activity Prediction; 2018 24th International Conference on Pattern Recognition (ICPR) 2018; pp 639–644. [Google Scholar]
- (17).Feinberg EN; Sur D; Wu Z; Husic BE; Mai H; Li Y; Sun S; Yang J; Ramsundar B; Pande VS Potentialnet for molecular property prediction. ACS Cent. Sci 2018, 4, 1520–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wallach I; Dzamba M; Heifets A AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. arXiv preprint arXiv:1510.02855 2015. [Google Scholar]
- (19).Ragoza M; Hochuli J; Idrobo E; Sunseri J; Koes DR Protein–Ligand scoring with Convolutional neural networks. J. Chem. Inf. Model 2017, 57, 942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cohen T; Welling M Group Equivariant Convolutional Networks; International Conference on Machine Learning 2016; pp 2990–2999. [Google Scholar]
- (21).Zaheer M; Kottur S; Ravanbakhsh S; Poczos B; Salakhutdinov RR; Smola AJ In Advances in Neural Information Processing Systems 30; Guyon I, Luxburg UV, Bengio S, Wallach H, Fergus R, Vishwanathan S, Garnett R, Eds.; Curran Associates, Inc.: 2017; pp 3391–3401. [Google Scholar]
- (22).Weiler M; Hamprecht FA; Storath M Learning Steerable Filters for Rotation Equivariant CNNs; Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition 2018; pp 849–858. [Google Scholar]
- (23).Cohen TS; Weiler M; Kicanaoglu B; Welling M Gauge equivariant convolutional networks and the icosahedral CNN. arXiv preprint arXiv:1902.04615 2019. [Google Scholar]
- (24).Brown RC; Lunter G An equivariant Bayesian convolutional network predicts recombination hotspots and accurately resolves binding motifs. Bioinformatics 2019, 35, 2177–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Boomsma W; Frellsen J Spherical convolutions and their application in molecular modelling. Advances in Neural Information Processing Systems 2017, 3433–3443. [Google Scholar]
- (26).Cohen TS; Geiger M; Köhler J; Welling M Spherical CNNs. arXiv preprint arXiv:1801.10130 2018. [Google Scholar]
- (27).Skalic M; Martínez-Rosell G; Jiménez J; De Fabritiis G; Valencia A PlayMolecule BindScope: Large scale CNN-based virtual screening on the web. Bioinformatics 2019, 35, 1237. [DOI] [PubMed] [Google Scholar]
- (28).Skalic M; Varela-Rial A; Jiménez J; Martínez-Rosell G; De Fabritiis G LigVoxel: Inpainting binding pockets using 3D-convolutional neural networks. Bioinformatics 2019, 35, 243. [DOI] [PubMed] [Google Scholar]
- (29).Kajita S; Ohba N; Jinnouchi R; Asahi R A universal 3D voxel descriptor for solid-state material informatics with deep convolutional neural networks. Sci. Rep 2017, 7, 16991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Jiménez Luna J; Skalic M; Martinez-Rosell G; De Fabritiis G K DEEP: Protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks. J. Chem. Inf. Model 2018, 58, 287. [DOI] [PubMed] [Google Scholar]
- (31).Amidi A; Amidi S; Vlachakis D; Megalooikonomou V; Paragios N; Zacharaki EI EnzyNet: enzyme classification using 3D convolutional neural networks on spatial representation. PeerJ 2018, 6, e4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hendlich M; Rippmann F; Barnickel G LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graphics Modell 1997, 15, 359–363. [DOI] [PubMed] [Google Scholar]
- (33).Jiménez J; Doerr S; Martínez-Rosell G; Rose A; De Fabritiis G DeepSite: protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 2017, 33, 3036–3042. [DOI] [PubMed] [Google Scholar]
- (34).Jiang M; Li Z; Bian Y; Wei Z A novel protein descriptor for the prediction of drug binding sites. BMC Bioinf. 2019, 20, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Kuzminykh D; Polykovskiy D; Kadurin A; Zhebrak A; Baskov I; Nikolenko S; Shayakhmetov R; Zhavoronkov A 3D molecular representations based on the wave transform for convolutional neural networks. Mol. Pharmaceutics 2018, 15, 4378–4385. [DOI] [PubMed] [Google Scholar]
- (36).Brock A; Lim T; Ritchie JM; Weston N Generative and Discriminative Voxel Modeling with Convolutional Neural Networks. arXiv preprint arXiv:1608.04236 2016. [Google Scholar]
- (37).Brock A; Donahue J; Simonyan K Large Scale GAN Training for High Fidelity Natural Image Synthesis. arXiv preprint arXiv:1809.11096 2018. [Google Scholar]
- (38).Thomas N; Smidt T; Kearnes S; Yang L; Li L; Kohlhoff K; Riley P Tensor Field Networks: Rotation- and Translation-Equivariant Neural Networks for 3D Point Clouds. arXiv preprint arXiv:1802.08219 2018. [Google Scholar]
- (39).Jaderberg M; Simonyan K; Zisserman A; et al. Spatial transformer networks. Advances in Neural Information Processing Systems 2015, 2017–2025. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.