

Abstract
Additive manufacturing is a promising method for producing customized three-dimensional (3D) bioactive constructs for regenerative medicine. Here, we report 3D printed highly osteogenic scaffolds using nanoengineered ionic-covalent entanglement ink (NICE) for bone tissue engineering. This NICE ink consists of ionic-covalent entanglement reinforced with Laponite, a two-dimensional (2D) nanosilicate (nSi) clay, allowing for the printing of anatomic-sized constructs with high accuracy. In addition, the 3D printed structure is able to maintain high structural stability in physiological conditions without any significant swelling or deswelling. While the presence of nSi imparts osteoinductive characteristics to the NICE scaffolds, this was further augmented by depositing pluripotent stem cell-derived extracellular matrix (ECM) on the surface of the scaffolds. This was achieved by stimulating human induced pluripotent stem cell-derived mesenchymal stem cells (iP-hMSCs) with 2-chloro-5-nitrobenzanilide, a PPARγ inhibitor that enhances Wnt pathway, resulting in the deposition of an ECM characterized by high levels of collagens VI and XII found in anabolic bone. The osteoinductive characteristics of these bioconditioned NICE (bNICE) scaffolds is demonstrated through osteogenic differentiation of bone marrow derived human mesenchymal stem cells (hMSCs). A significant increase in the expression of osteogenic gene markers including bone morphogenic protein-2, osteocalcin and osteopontin was observed on ECM-coated scaffolds compared to bare scaffolds, as well as improved mineralization. This approach of augmenting the bioactivity of 3D printed scaffolds by depositing an anabolic bone ECM will provide a unique strategy to design personalized bone graft geometries for in situ bone regeneration.
Keywords: 3D printing, mesenchymal stem cells, osteogenic differentiation, stem cell-derived extracellular matrix, hydrogels
Graphical Abstract
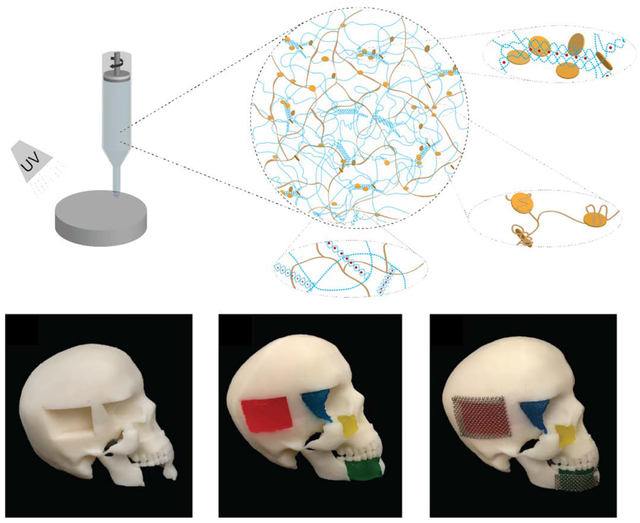
3D printed osteogenic scaffolds is designed for bone bioprinting using nanoengineered ionic-covalent entanglement ink (NICE). Osteoinductive characteristics is imparted by bioconditioning of 3D printed scaffolds via deposition of anabolic bone extracellular matrix (ECM) secreted by induced pluripotent stem cell (iP-hMSCs) derived human mesenchymal stem cells (hMSCs).
1. INTRODUCTION
Approximately 200,000 craniomaxillofacial (CMF) injuries occur annually due to trauma, congenital malformations, neoplasia and neurosurgical interventions[1]. Some of the complex CMF cases require repair by cranioplasty to protect intracranial contents, re-establish anatomical boundaries between intra and extra-cranial structures, restore aesthetic craniofacial contour, and support craniofacial soft tissues[2]. Although the material choice and practices regarding cranioplasty can be largely dependent on the nature of the defect and surgeon preferences[3,4], autologous bone grafts are considered the gold standard reconstructive material for CMF repair[5,6]. However, this technique relies on a limited source of donor tissue, incurs donor-site morbidity, and the complex geometries of bone within the CMF region cannot be easily recapitulated [2,3]. Alternatively, common alloplastic materials used for CMF repair include polymethyl methacrylate (PMMA)[7], hydroxyapatite cement[8], polyether ether ketone (PEEK)[9], and titanium[10]. However, these materials are more susceptible to infection, implant extrusion and exposure[2], tissue necrosis, and stress shielding[11,12] compared to autologous grafts. Despite these limitations, custom-shaped alloplastic implants, specifically those made from PEEK[9] or titanium[10], have been successful in in restoring craniofacial contours. Given that PEEK and titanium are non-resorbable, these materials cannot be fully incorporated and replaced by newly formed bone. Thus, there is a need to develop approaches to designing patient-specific biodegradable bone grafts to stimulate bone tissue regeneration.
Human mesenchymal stem cells (hMSCs) from bone marrow represent an attractive candidate for bone repair due to their ability to stimulate bone healing. However, poor retention of hMSCs at the implantation site limits their clinical efficacy [13]. To address this limitation, we have utilized cell derived extracellular matrix that has been shown to enhance hMSC retention and accelerate bone repair in rodent calvarial, femoral, and spinal defect models[13–15]. Briefly, hMSCs treated with 2-chloro-5-nitrobenzanilide (GW9662), a PPAR-γ inhibitor, secrete a collagenous extracellular matrix (ECM) that resembles the composition of anabolic bone, including high levels of Collagen VI and XII[13]. A significant drawback to using hMSCs to produce ECM and other complex biological products is their limited proliferation potential and variability in cell behavior between donors. Further, when cell banks become exhausted, new donor sources must be identified and re-validated to establish similar efficacy. To address these limitations, we have generated hMSCs from human induced pluripotent stem cells (iP-hMSCs)[16]. Unlike donor-derived hMSCs, undifferentiated iPSCs divide without senescence until they are differentiated into iP-hMSCs, providing a theoretically limitless supply of reproducible biomaterial for the production of the anabolic bone matrix described in this work. This provides a potential strategy to produce large quantities of ECM generated from a genetically identical source of iP-hMSCs in a reproducible manner, effectively dismissing the need for variable donor-derived biomaterial. While this ECM can mimic a healing microenvironment, a lack of structural form limits its practicality for bone repair.
Recent developments in additive manufacturing provide an opportunity to design patient-specific scaffolds with tunable architectures[17]. A range of polymeric biomaterials can be 3D printed for tissue engineering and regenerative medicine. However, a common limitation of these 3D printed structures when applied to bone tissue engineering is limited osteoinductive characteristics. The osteoinductive properties of polymeric scaffolds can be improved by incorporating inorganic biomaterials meant to mimic or substitute for bone mineral, such as calcium phosphates[18], specifically hydroxyapatites[19,20] or tricalcium phosphates[21], calcium sulfates[22], and bioactive glass[23]. Polycaprolactone (PCL), a thermoplastic polymer commonly used as a biomaterial ink due to its relatively low melting temperature (60 °C) and biocompatibility[24], has recently been blended with hydroxyapatite[25] and decellularized trabecular bone particles[26] for 3D printing of osteogenic scaffolds for craniofacial regeneration. Calcium phosphate bone cement (CPC) paste has successfully been printed under mild temperature conditions (<37 °C) [27] and can be used to print drug loaded scaffolds[18]. However, seeded hMSCs form a dense layer on the surface of the scaffolds and have difficulties migrating into the scaffold material[27].
While hydrogel-based inks can address some of these issues with improved biocompatibility, superior cell-adhesion, osteoinductivity, and biodegradability, this typically comes at the expense of print fidelity and mechanical strength[28]. Strategies to improve printability include the use of various reinforcement techniques, such as dual crosslinking, interpenetrating polymer networks (IPNs), nanocomposites reinforcement and co-printing hydrogel inks with thermoplastic [24,29]. We have recently introduce a new class of ink, Nanoengineered Ionic-Covalent Entanglement (NICE), capable of printing high aspect ratio structures with high fidelity thanks a dual reinforcement strategy utilizing IPNs with nanoparticles[28]. Ionic-Covalent Entanglement (ICE), a type of IPN, is engineered by combining covalently crosslinked gelatin methacryloyl (GelMA) with ionically crosslinked kappa-Carrageenan (κCA). GelMA facilitates cell adhesion due to the presence of cell-binding domains and can be enzymatically degraded to assist cell-induced matrix remodeling[28,30]. In contrast, κCA, the second polymeric network, provides mechanical rigidity through its cation-crosslinked network[31–33]. In addition, nSi form reversible non-covalent bonds with both polymers due to their discotic surface charge and high surface area, further strengthening the ICE network[34]. Our recent data demonstrate that addition of nSi to GelMA/κCA hydrogels results in superior printability and with the ability to support the fabrication of large anatomical-size constructs [28]. Our previous studies also demonstrate the inherent osteoinductive ability of nSi[35,36]. Specifically, nSi can stimulate both osteogenic and chondrogenic differentiation of stem cells in absence of inductive agents [37]. While the addition of nSi to polymeric network imparts osteoinductive characteristics, it fails to mimic the complex microenvironment of healing bone.
Here, we augment the osteoinductive ability of 3D printed NICE scaffolds by deposition an anabolic bone ECM scaffold surface. First, we characterized ECM deposition and decellularization on NICE scaffolds. To mimic the use of bone marrow derived cellular material in bone grafting, we assessed the capacity of bNICE to enhance in vitro osteogenic differentiation of newly seeded bone marrow hMSCs. Finally, we demonstrated the ability of NICE to print anatomically complex geometries, especially for craniomaxillofacial injuries as they present a unique challenge in aesthetically recapitulating craniofacial contours.
2. RESULTS & DISCUSSION
2.1. Fabrication and Characterization of Bioconditioned NICE Scaffolds
Effective 3D bioprinting via extrusion-based methods requires ink to exhibit shear-thinning properties, allowing the bioink to freely flow from the printing nozzle in a way that reduces the shear stresses experienced by cells during extrusion. The ink should also quickly recover its viscosity after extrusion in order to minimize further deformation until a more permanent crosslinking method can be utilized for long term stability. Furthermore, 3D printing hydrogel-based scaffolds in the z-direction can be challenging as it requires the biomaterial ink to be strong enough to withstand the weight of subsequent layers. Laponite, the nSi used in this study, has shown promise as a material for bioprinting due to its excellent biocompatibility, ease in forming gels, and shear thinning properties[38]. Laponite has been added to inks of varying crosslinking mechanisms, e.g. poly(ethylene glycol) (PEGDA), alginate, and gelatin[39,40], to modulate and improve rheological properties for 3D printing. Furthermore, the high surface area and charge of Laponite can also be exploited for drug-delivery purposes, promoting sustained release of protein therapeutics sequestered to 3D printed scaffolds[41,42].
In this study, the NICE ink platform utilizes nSi and ICE to modulate the rheological properties of GelMA-based ink for suitable 3D printing. κCA was chosen for the ionic cross-linking polymer since it has been used for its thickening properties in the past[43]. For this work, we utilized 2% w/v of κCA, below the toxicity levels reported for both oral consumption and intravenous consumption[44]. Characterization of both printability and mechanical reinforcement has been demonstrated previously[28]. Most notably, shear stresses generated during extrusion (on the order of 100 Pa) result in an apparent viscosity of less than 10 Pa·s that allows the ink to exhibit liquid-like behavior during extrusion, while removal of shear stress after deposition results in re-solidification to retain shape after extrusion. While NICE bioink has been utilized to 3D print large free-standing tubular structures and bioprint encapsulated cell[28], here we have printed a simple disc with a 30 mm diameter and 0.6mm thick for investigating cell-scaffold interactions.
Augmenting bioactivity (e.g. osteoinductivity) and promoting cellular attachment to synthetic scaffolds for bone repair is a common strategy. While functionalizing synthetic scaffolds/hydrogels with singular peptide sequences can improve cellular attachment [45] and even bone healing response[46], peptide instability remains an issue[46]. In addition, the use of single peptide sequence fails to mimic the complex array of signals stemming from an osteogenic niche. While the use multiple peptide sequences to target specific cell lines or a specific phase of bone healing is an ongoing area of interest meant to address these limitations[46], this presents its own challenges. Specifically, designing and controlling the ratios of multiple peptide motifs can be challenging task that one can expect to increase in difficulty as the number of sequences increases[47].
We have demonstrated that utilizing the ECM secreted by hMSCs treated with GW9662 enhanced cell retention and accelerated bone repair in a calvaria murine lesion model[13]. However, the translation to clinical use of hMSC-derived matrices could be hindered by donor variance. We hypothesized that coating the surface of 3D-printed NICE scaffolds with the osteo-regenerative ECM derived from iP-hMSCs would result in a scaffold biofabrication methodology with customizable geometries and promising bone healing capabilities. This was achieved by culturing iP-hMSCs on surface of 3D-printed NICE scaffolds for 10 days in the presence of GW9662 followed by a modified decellularization consisting of a series of detergent, enzyme, and solvent washes (Figure 1). Decellularization by chemical and enzymatic means has been used to facilitate the removal of residual cellular and nuclear material from tissues[48]. Although proteases, such as trypsin, can be effective in removing attached cells, this was avoided in the decellularization of bNICE scaffolds to avoid proteolysis of the deposited ECM[49]. Ideally, the decellularization process would remove enough cellular and nuclear debris to avoid adverse host response when implanted while conserving the structure and bioactivity of the anabolic bone matrix to elicit a robust regenerative response. Thus, decellularization was achieved through enzymatic and chemical means. Hoechst staining revealed no discernable nuclear material on the surface of the bNICE scaffolds (Figure 2A). The amended decellularization protocol reduced the DNA content to less than 50 ng per mg of scaffold dry weight, a threshold demonstrating successful decellularization[53]. Scanning electron microscopy (SEM) was used to characterize the deposition of ECM before and after decellularization. (Figure 2B). A smooth surface was observed on the freshly printed NICE. In contrast, a dense cell sheet and an underlying fibrillar ECM can be seen on the bioconditioned scaffolds before decellularization. After decellularization, the fibrillar structure of the ECM remains intact, while no cell-like structures can be seen. The choice of decellularization method ultimately decides whether the potential beneficial effects of cell-derived ECM can be realized. To validate the ECM derived from iP-hMSCs was of similar composition to that derived from hMSCs, we immunostained for Collagen VI and XII. Similar to previous work done on hMSCs cultured in both 2D [13] and 3D cultures[14], iP-hMSCs treated with GW9662 successfully deposited the anabolic bone ECM on 3D printed NICE scaffolds. As shown on Figure 2C, there was an increase in deposition of Collagens VI and XII on the bNICE scaffolds compared to NICE scaffolds. Based on our previous observations that nSi can sequester various types of proteins for prolong duration[41,50–52], we expected that deposited ECM will be adsorbed strongly on NICE structure due to presence of nSi. To verify the nSi did not chelate the detergent used in the lysis buffer, we acquired the FTIR spectra of NICE scaffolds at various stages of the decellularization process (Figure 2D), revealing the removal of the detergent.
Figure 1.
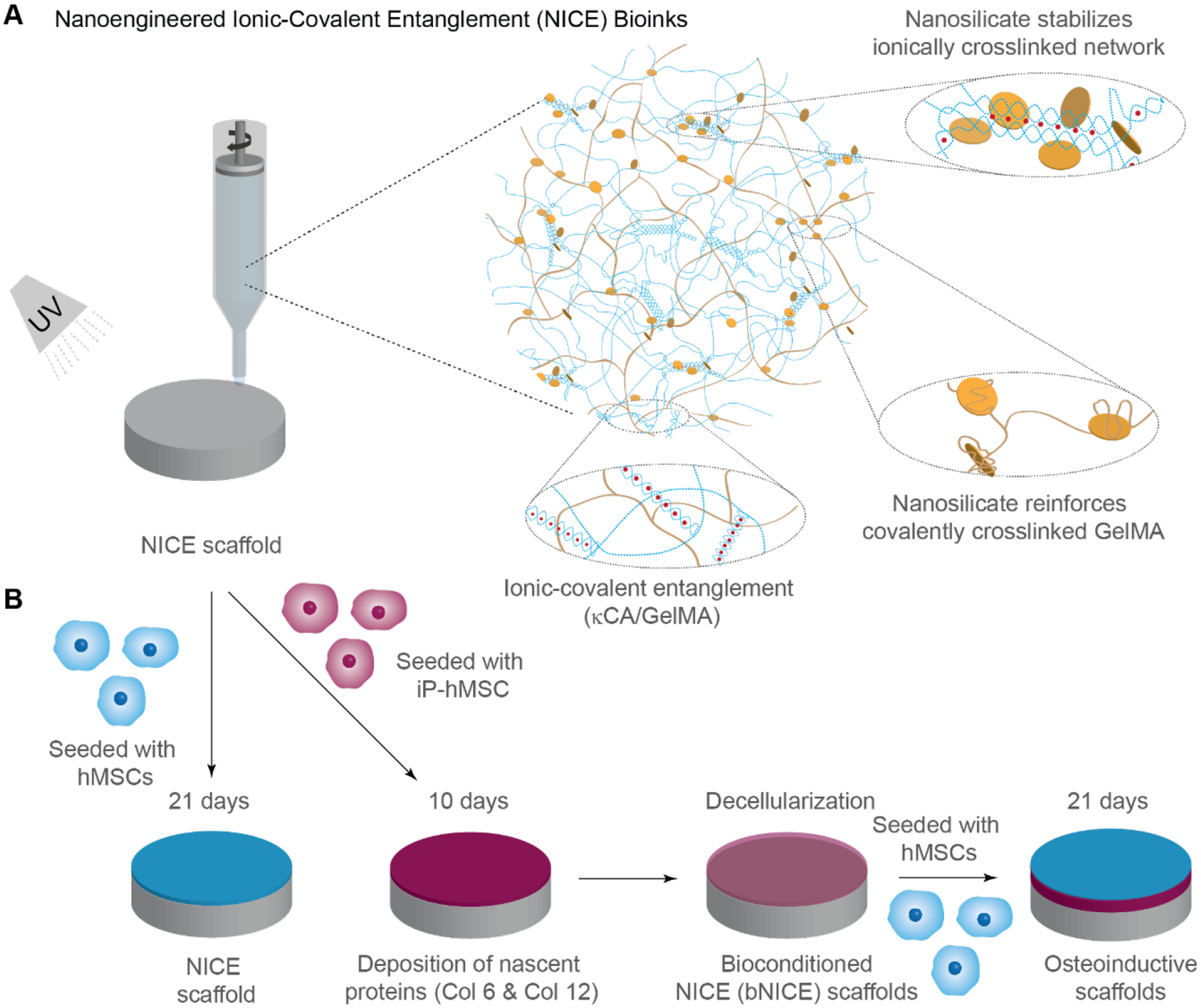
Schematic illustrating the development of NICE and bioconditioned NICE (bNICE) scaffolds. (A) Nanocomposite reinforcement between nSil and crosslinked GelMA and the ionic-covalent entanglement of the independent polymeric networks of κCA and GelMA allows for NICE ink to be both elastic and highly printable. (B) iP-hMSCs were seeded on NICE scaffolds and cultured in the presence of GW9662 for 10 days followed by decellularization. The scaffolds modified with iP-hMSC-derived ECM, or bNICE, were seeded with hMSCs and evaluated in vitro for osteogenic differentiation after 8 and 21 days of culture.
Figure 2. Characterization of NICE and bNICE scaffolds.
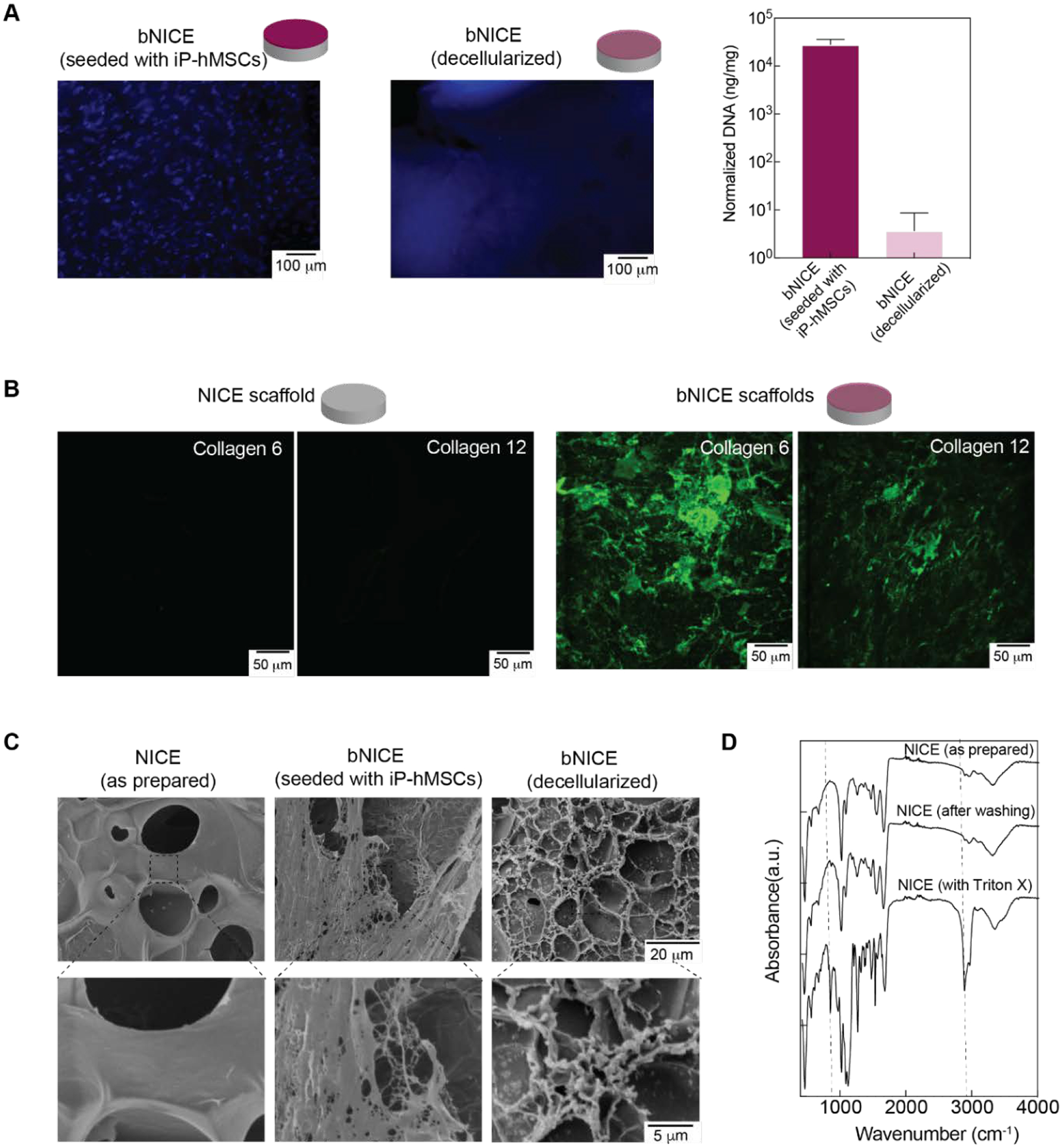
(A) Effectiveness of the decellularization process in removing DNA while preserving anabolic collagens was evaluated. The degree of DNA removal was evaluated by both qualitative and quantitative means. Hoechst staining revealed well-defined nuclei prior to decellularization are no longer present after the process. Scale bar 100 μm. Quantification of DNA via Cyquant showed five orders of magnitude decrease after decellularization. (B) SEM images of acellular NICE and bNICE scaffolds before and after decellularization. A smooth surface can be seen on the acellular bare scaffolds. Both cells and the underlying ECM are visible prior to decellularization while the fibrillar ECM remains after decellularization. (C) Immunostaining of Coll VI and XII showed intact ECM deposition in the bNICE scaffolds after decellularization that was not observed on bare NICE scaffolds. Scale bar represents 50 μm. (D) FTIR shows the removal of lysis buffer.
Reduction of DNA content and conservation of the composition and ultrastructure of the ECM are recognized as the benchmarks in successful decellularization[48], which were effectively met. While repeated freeze-thawing has been successfully implemented as an inexpensive and easily accessible method for decellularizing thermoplastic-based 3D-printed scaffolds[54], this approach was deemed unsuitable for hydrogel-based scaffolds due to its high water content and the risk of negatively impacting the scaffold structure. Given the detrimental effects common decellularization methods can have on ECM, exploiting programmed cell death has been proposed as a novel decellularization method[55]. Cell death could be induced through mitochondrial apoptosis by exposure to “lethal environmental conditions”, e.g. nitric oxide, or delivery death receptor ligands to activate extrinsic apoptotic pathway, or by genetically engineering cell lines to under apoptosis in response to caspase 9[55]. The efficiency of this method has been already demonstrated in hMSC[56], and presents an attractive avenue for modifying our iP-hMSCs.
2.2. Mechanical Characterization
In addition to maintaining structural integrity and determining shape fidelity, the mechanical properties of 3D-printed scaffolds also affects cell functions such as proliferation and differentiation. Although we have previously demonstrated that the NICE ink system can produce highly elastic and mechanically stiff scaffolds, we assessed the effects of bioconditioning and decellularization on material properties. Biopsy punches (diameter of 6 mm diameter, 600 μm thick) taken from the printed scaffolds were used for mechanical characterization. A uniaxial cyclic compression test revealed freshly printed NICE scaffolds to have a compressive modulus of approximately 78±17 kPa, which is 2-fold higher than the observed values for the bNICE scaffolds (38±7 kPa) (Figure 3A). Although both NICE and bNICE scaffolds could undergo high levels of strain without failure, the NICE scaffolds were shown to be tougher than bNICE, storing approximately 40 kJ/m3 compared to 25 kJ/m3. Future work will investigate the individual contributions of the 10-day matrix deposition process and the decellularization step on scaffold mechanical properties and thus optimize strategies to minimize these detrimental effects.
Figure 3.
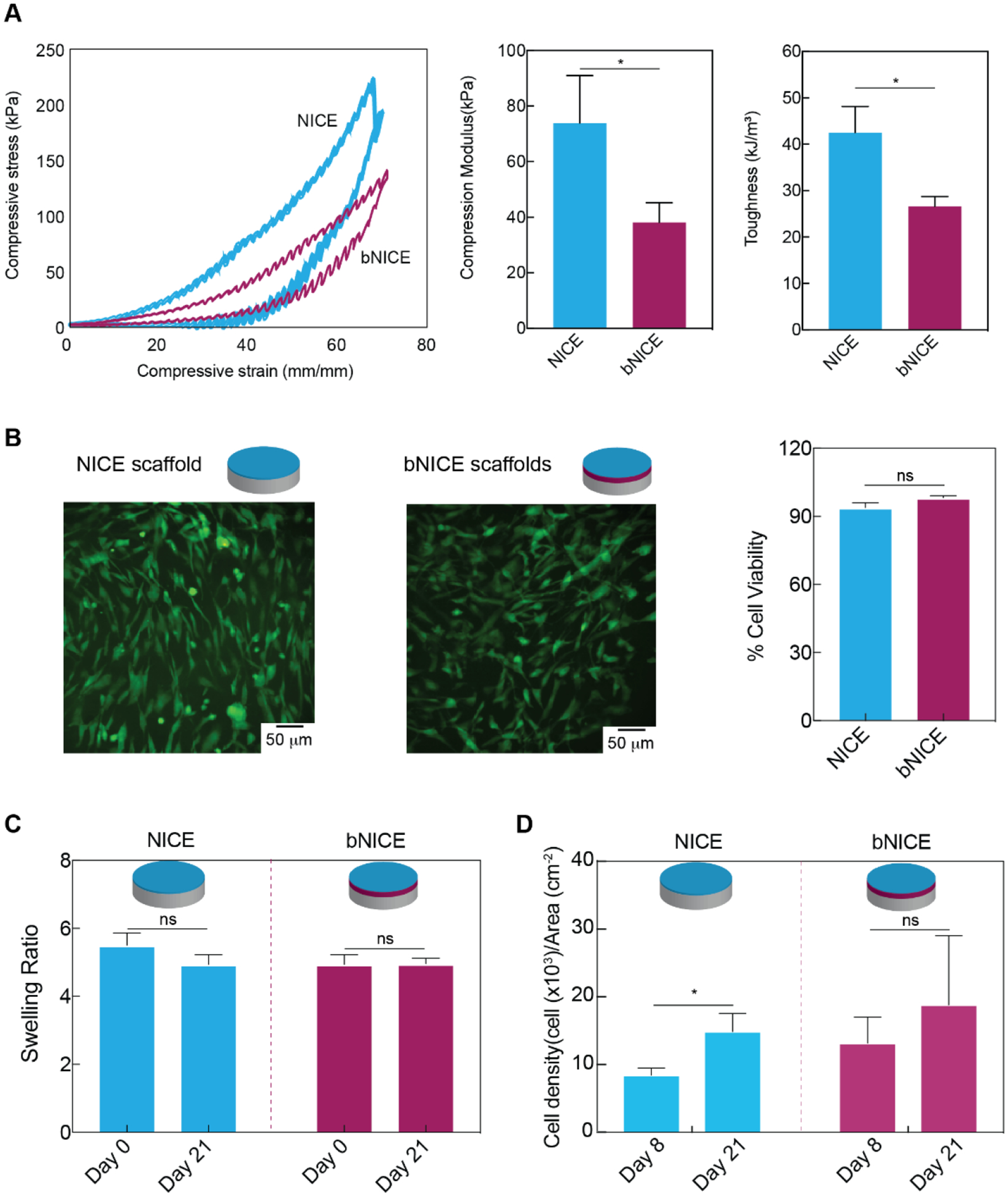
Effects of decellularization process on the mechanical properties of NICE hydrogels and hMSCs viability on both NICE and bNICE scaffolds. (A) Representative stress–strain response of scaffolds undergoing cyclic compression to 70% strain. Analysis on the average compressive modulus and toughness (n=3, student’s t-test, p<0.05) demonstrated a decrease in response to the decellularization process. (B) Live/dead staining of hMSCs present on scaffolds demonstrated an abundance of live cells and sparsely distributed dead cells. Cell viability 48 hr post-seeding was 93.9% for bare scaffolds, while the bNICE scaffolds demonstrated cell viability of 98.2%. A two-tailed student’s t-test analysis established that the difference in viability was statistically significant (n=3, student’s t-test p<0.05). (C) No significant effect on swelling was observed due to the bioconditioning or decellularization process. (D) A significant increase in cell density was observed between days 8 and 21 on NICE scaffolds (n=3, student’s t-test p<0.05). No significant difference was observed on the bNICE scaffolds.
2.3. hMSCs Viability and iP-hMSCs Invasion into NICE and Swelling Degree of NICE and bNICE
We anticipated that bone marrow aspirate is currently the most clinically relevant source of osteoprogenitor cell source, hence we used bone marrow derived hMSCs to assess cell behavior on our iP-hMSC-derived ECM. To gauge hMSCs viability and verify a desired confluency was achieved before GW9662 stimulation, hMSCs were cultured on NICE and bNICE scaffolds for 2 days in CCM before performing live/dead staining. Representative images demonstrate the hMSCs have a spindle-shaped morphology on both bare and bioconditioned scaffolds (Figure 3B). Summarized results from multiple experiments indicate similarly high levels of cell viability on both NICE and bNICE. No significant changes in swelling of NICE and bNICE scaffolds were observed over a 21-day culture (Figure 3C). Comparable swelling ratios for a GelMA/nSi nanocomposite have been reported elsewhere[57]. Cell density assessed via GAPDH expression over the course of the culture period revealed a significant increase between days 8 and 21 on the NICE scaffolds. While the cell density between days 8 and 21 on the bNICE also increased, the trend was not significant (Figure 3D). As would be expected, cell density on day 8 on both scaffolds were higher than the seeding density (5000 cells/cm2).
To assess NICE scaffolds propensity to be remodeled and allow for cell infiltration, we seeded iP-hMSCs onto the surface of the 3D printed NICE scaffolds and imaged after 21 days of culture. Confocal images of actin-stained iP-hMSCs cultured on NICE scaffolds for 21 day showed iP-hMSCs were able to migrate into the scaffolds (Figure 4). Orthogonal projections of the z-stack images revealed that despite the 100% infill density used, iP-hMSCs were able to migrate over 100 microns into the scaffold, suggesting that the cells are able to remodel the scaffold in order to invade. Exploring the degree at which GW-stimulated iP-hMSC can deposit the anabolic matrix in 3D culture rather than on the surface of the scaffold is of future interest.
Figure 4.
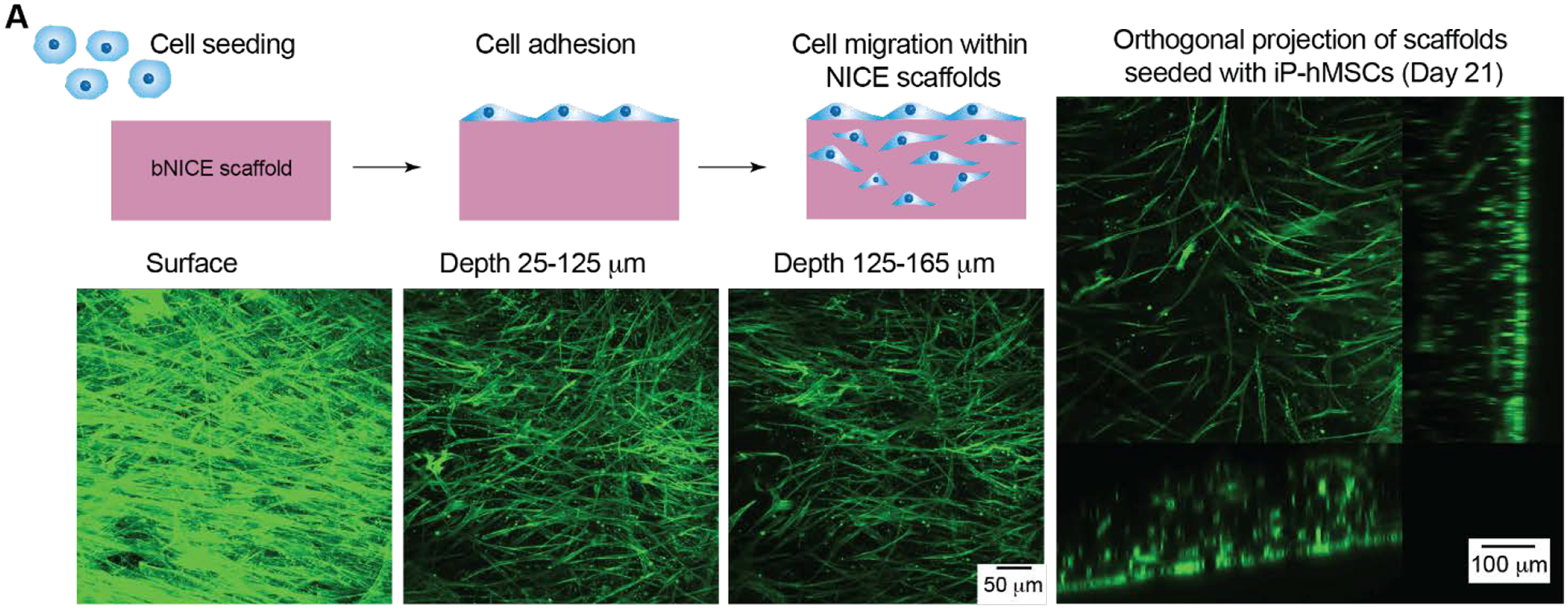
iP-hMSCs adhere and migrate in NICE scaffolds over 21 day-cultivation. (A) Maximum projections at various depths. (B) Orthogonal projections show a dense layer on the surface of the scaffold and migration into the scaffold.
2.4. hMSCs Gene Expression on NICE and bNICE
We have been previously demonstrated hMSCs secrete Collagen VI and XII in response to GW9662 treatment[13] and a similar response was observed with iP-hMSCs via immunostaining (cf. Figure 2C). We next determined if hMSCs could be further stimulated to express additional collagens VI and XII on bNICE. Given that deposition of collagens precedes ossifications and is considered early indication of osteogenic differentiation, expression of these early collagens was analyzed after 8 days of culture. Consistent with our previous findings, Collagens VI and XII were each significantly upregulated in response to GW9662 treatment in hMSCs cultured on bare NICE scaffolds (Figure 5A). In contrast, expression of these matrix proteins was low in GW9662-treated hMSCs cultured on bNICE, suggesting a negative feedback regulates the deposition of Collagens VI and XII.
Figure 5.
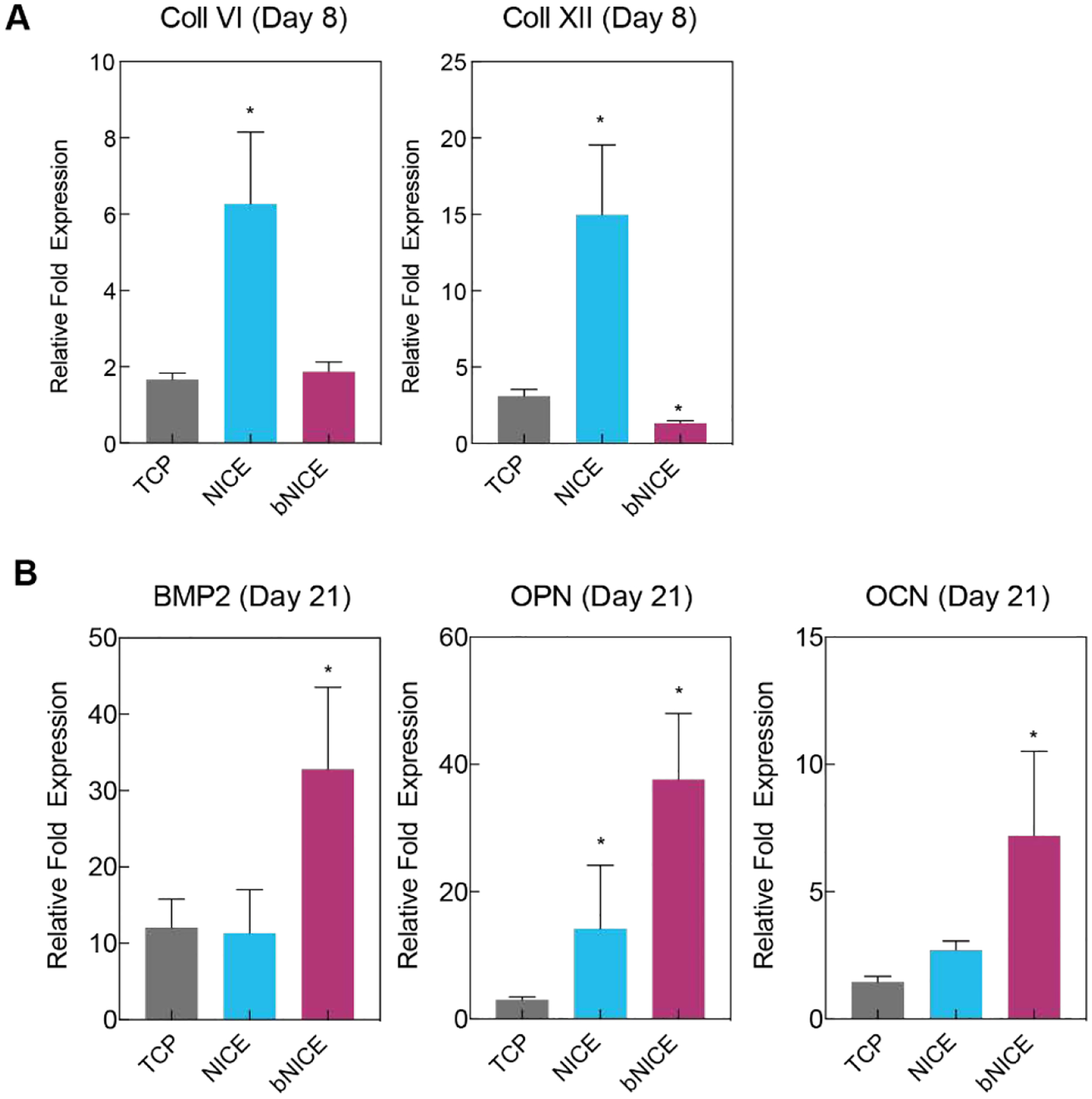
Bioconditioned scaffolds upregulate expression of key osteogenic markers. (A) Quantitative RT-PCR analysis showed an upregulation of Coll VI and Coll XII in response to GW9662 treatment, but not to bioconditioned scaffolds after 8 days of culture. (B) An increase at the transcription level of BMP2, osteocalcin, and osteopontin was observed after 21 days of culture on bioconditioned scaffolds. Fold changes were normalized to the expression levels of uninduced hMSCs after 8 or 21 days of 2D culture. A one-way ANOVA, followed by a Tukey’s multiple comparison test was performed on ΔΔCT values (* p<0.05).
In addition to improving cellular retention at an injury site, the anabolic bone ECM has been shown to actively induce the secretion of various osteogenic factors, e.g. BMP2[14]. Thus, we investigated the effects of iP-hMSC-derived ECM on the expression of late osteogenic markers osteocalcin (OCN), osteopontin (OPN), and BMP2 of hMSCs after 21 days of culture on NICE and bNICE scaffolds (Figure 5B). The bioconditioning treatment significantly increased the expression of all three markers relative to bare NICE scaffolds. Thus, surface modification of NICE with anabolic bone ECM causes a switch in hMSC phenotype from matrix secretion to an osteogenic phenotype. Future work will investigate the mechanism of this putative negative feedback mechanism in which the anabolic bone ECM negatively regulates the further expression of ECM components.
Although not as highly upregulated as those in bNICE scaffolds, it is important to note the osteogenic markers were still highly expressed by hMSCs cultured on bare NICE scaffolds, with OPN expression even being higher than in hMSCs that were osteogenically induced in tissue culture polystyrene (TCP). We attributed the inherent osteoinductive properties of the bare NICE scaffolds to both the presence of nSi and GW9662 treatment. The addition of nSil to a 3D hydrogel matrix has been shown to facilitate osteogenic differentiation of encapsulated hMSCs in a concentration dependent manner[30]. While the hMSCs were cultured on the surface of NICE and bNICE scaffolds, we hypothesized that the dissolution of nSi into its ionic components occurs during the matrix remodeling promotes osteogenic differentiation of hMSCs.
2.4. In-vitro Mineralization
A hallmark of late osteogenic differentiation is the mineralization of the deposited matrix. Therefore, we assessed the calcium content of NICE and bNICE scaffolds that were cultured with hMCSs for 21 days. In addition, the inherent mineral content of freshly printed NICE and of freshly decellularized bNICE scaffolds were also examined, designating both as “Day 0” time points. Though not significantly different, slightly higher levels of calcium were measured in freshly decellularized bNICE scaffolds compared to freshly 3D-printed NICE, indicating that the bioconditioned matrix is partially mineralized prior to the seeding of hMSCs (Figure 6A). Culturing hMSCs on both NICE and bNICE for an additional 21 days substantially increased calcium content, with the amount significantly higher on the bNICE scaffolds (Figure 6B). Given the crucial roles osteopontin[58,59] and BMP2[60] play in regulating biomineralization, these results are consistent with the gene expression trends observed in the bNICE scaffolds (cf. Figure 5B), Although the initial quantity of calcium in the bNICE scaffolds started was somewhat higher than bare scaffolds, further inspection revealed the bioconditioning process contributed a small percentage of the final calcium content (~3%) of the bNICE scaffolds cultured for 21 days. In addition, the low levels of calcium detected on freshly printed NICE indicate that the divalent cations present in nSil did not interfere with the o-cresolphthalein based assay. To further characterize the newly mineralized matrix, we acquired FTIR spectra of cultured NICE and bNICE scaffolds and of freshly printed NICE. When compared to the spectra of freshly printed NICE, two unique bands became apparent in the cultured samples. Upon further examination, these bands were attributed to the deposited collagen (2930 cm−1) and of phosphates (1020 cm−1) in the apatite structure[61]. The phosphate signal from the mineralized matrix was robust enough to obscure the double peaks at 1000 and 1050 cm−1 seen in freshly printed NICE scaffolds, which are consistent with an interaction between the nanosilicate and the GelMA network of the NICE ink[36].
Figure 6.
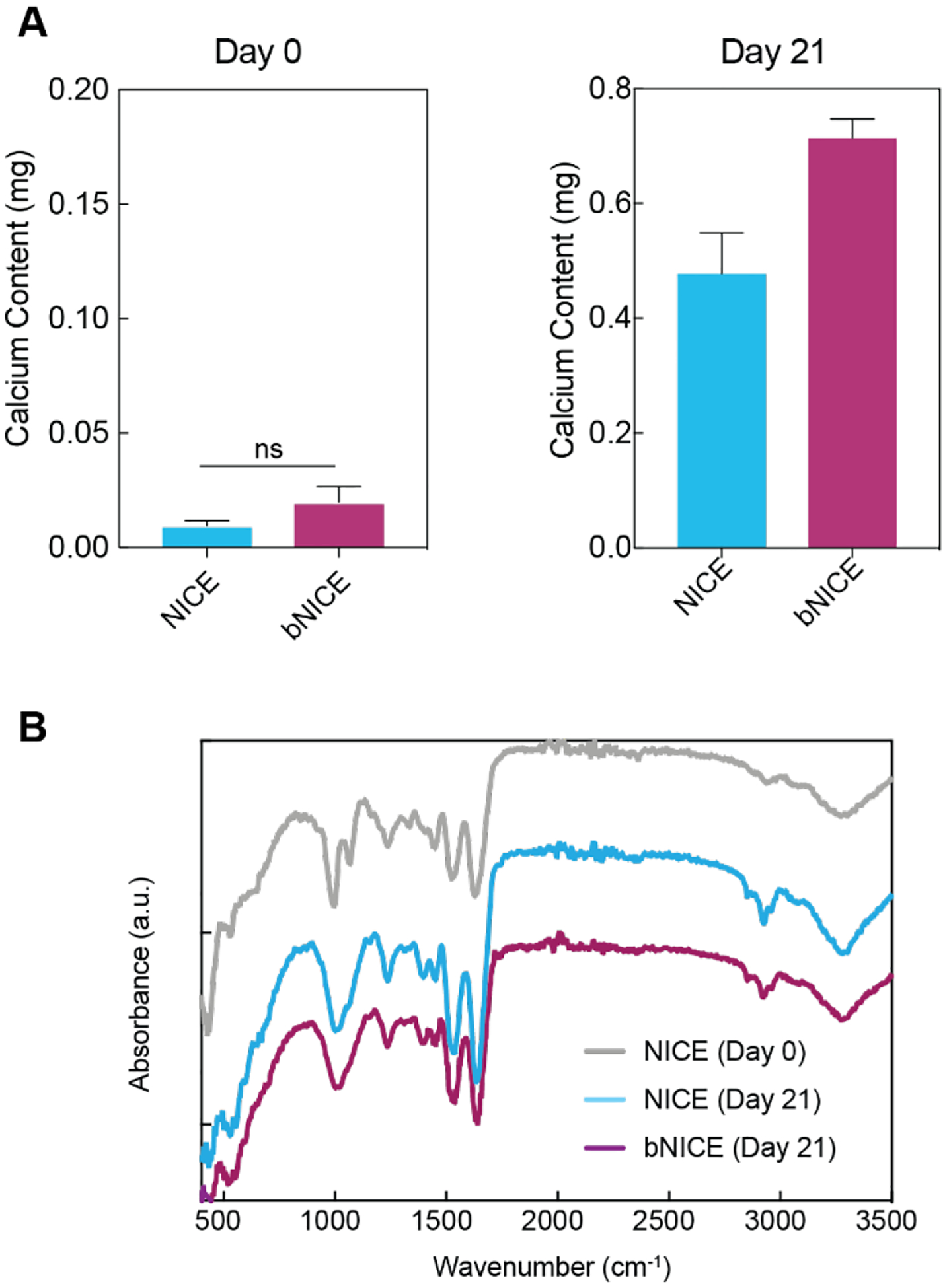
Calcium content on bare and bioconditioned scaffolds. (A) Calcium content of freshly printed NICE and recently decellularized bNICE scaffolds were low relative to the cultured scaffolds. After 21 days of culture, calcium content from cultured NICE and bNICE scaffolds were significantly different, with higher mineralization detected on bioconditioned scaffolds. Statistical analysis was performed using student’s t test (* p<0.05). (B) FTIR spectra of the scaffolds revealed bands corresponding to both collagen deposition (2930 cm−1) and phosphates (1020 cm−1) in cultured samples. The double peaks at 1000 and 1050 cm−1 on day 0 NICE samples are indicative of the silicate/GelMA interaction.
2.5. 3D printing NICE scaffolds for CMF defects
To explore potential applications of an osteogenically enhanced 3D-printed scaffold system, we assessed the ability of NICE to be printed into anatomically complex geometries. Craniomaxillofacial injuries are unique in that it requires scaffolds to aesthetically recapitulate craniofacial contours, and thus would benefit greatly from customizable, 3D-printed scaffolds. We previously demonstrated that large, free-standing tubular structures could be printed with NICE ink[28]. Here, idealized bone defects were modeled in Solidworks, and 3D-printed (Figure 7A). The printed NICE scaffolds were easily handled with forceps and press-fitted into the complementary skull defects with minimal effort (Figure 7B). A stainless-steel mesh was added and secured with miniature screws to emulate how these constructs could be used in a surgical setting. In-situ forming scaffolds based on injectable materials have also been explored for the treatment of CMF defects due to their promise as a minimally invasive method to deliver cells and bioactive factors to irregular-shaped defects[62]. These, however, can lack the mechanical strength or fail to mimic key structural parameters (e.g. pore size and interconnectivity), as is the case in hydrogel-based systems and in-situ forming ceramics[62,63]. Thermoresponsive shape memory polymers, whose shape can change upon exposure to heat, have been explored as a press-to-fit treatment option for CMF defects[63,64]. While these scaffolds can be fabricated with tunable pore size/interconnectivity, mechanical properties[64], and degradation rates[65], poor osteoinductive properties presents as an obstacle for bone regeneration.
Figure 7.
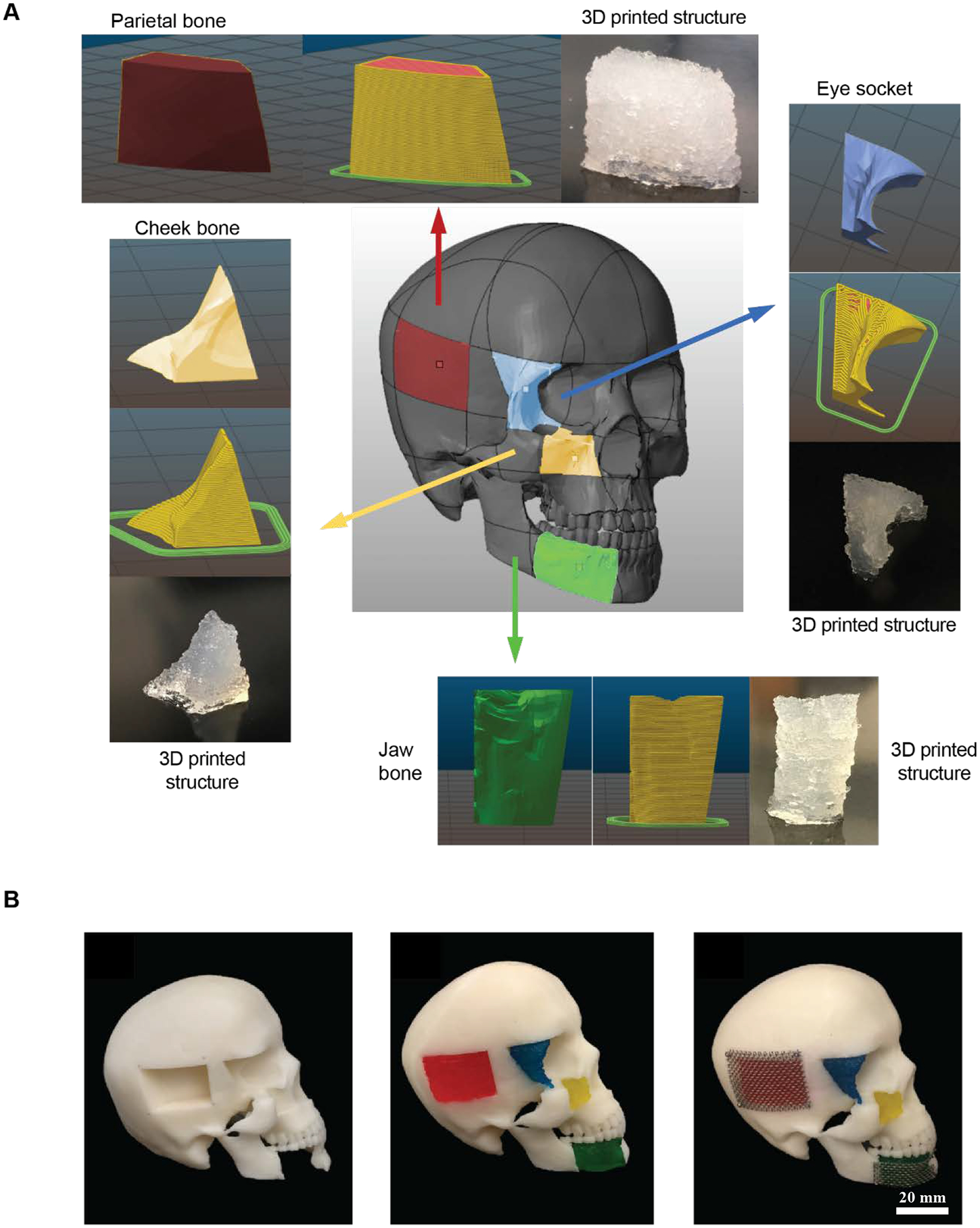
NICE ink reproduces various anatomical defects with high print fidelity. (A) NICE ink reproduce various anatomical defects with high print fidelity. NICE ink can be used to print human craniomaxillofacial defects. Various defects–jaw bone, eye socket, cheek bone, and parietal bone were modeled and printed with a custom-built RepRap Prusa i3-style 3D printer. (B) Proposed clinical implementation of 3D printed NICE scaffolds as a means of providing patient specific craniomaxillofacial defects. 3D printed NICE scaffolds were inserted into a 3D printed human skull with idealized bone defects. A surgical stainless steel mesh was screwed into the skull to help keep the parietal and jaw bones scaffolds in place.
While this study demonstrates a strategy to generating an engineered bone graft designed to mimic autograft, modifications to the steps taken should be considered for future studies. ECM proteins and their binding domains have been used to enhance the bone repair capabilities of scaffolds.[14,54,66–68]. Methods to functionalize scaffolds to present these functional domains include: adsorption from protein stock solutions[67], covalent tethering[69], and direct cell-culture deposition[54,66,68]. Decellularized extracellular matrices derived from tissue culture plastic can be harvested and purified for direct binding to the scaffold via passive adsorption or covalent tethering. In addition, the composition of the decellularized matrix can be altered by varying the culture duration, which can influence the behavior of newly seeded progenitor cells. For example, murine MSCs cultured on more mature (i.e. more mineralized) decellularized matrices were observed to have higher alkaline phosphatase activity, but also reduced cellular proliferation [68]. Alternatively, direct deposition of ECM onto established scaffolds allows for the fabrication of scaffolds with desired microstructures and composition, e.g. titanium meshes [70,71]. Unlike synthetic inks, NICE is proteolytically-degradable[28] and can thus be remodeled in vivo. A 100% infill density was used for the cell culture work in order to avoid confounding issues with nutrient and oxygen transport. Future work will focus on coupling scaffolds with reduced infill density with a perfusion bioreactor system. This would allow for improved cell infiltration and ECM deposition. Furthermore, due to the angiogenic properties of the anabolic bone ECM[14], the resulting mesh/channels from a reduced infill density will likely facilitate vascularization that could be stimulated by the angiogenic factors produced by hMSCs in response to attachment to the anabolic ECM
3. CONCLUSION
We have successfully demonstrated the feasibility of generating 3D printed NICE scaffolds for patient specific craniomaxillofacial defects with high print fidelity. Printed scaffolds are resilient and tough, permitting manual manipulation. They are also slightly compressible, allowing a press-fit into the defect. The scaffolds that were modified with a stem cell-derived extracellular matrix exhibited an increase in osteogenic gene expression and mineralization. The decellularized 3D printed scaffolds described here may be utilized as an alternative to autologous grafts for orthopedic procedures. Due to the promising biological and physical properties of NICE grafts created with 3D printing and bioconditioning, this system will be evaluated for bone regeneration in vivo for spinal fusion in the near future.
4. Experimental Section
4.1. NICE Ink:
Gelatin methacryloyl (GelMA) with 80% methacrylation was synthesized using a previously reported method [72]. Kappa-carrageenan (κCA) was purchased from TCI America (USA), while the nSi (Laponite XLG) were purchased from BYK Additives Inc. NICE ink was synthesized using our published protocol[28]. Briefly, NICE ink was obtained by prepared (10% w/v GelMA, 1% w/v κCA, 2% w/v nanosilicates, and 0.25% w/v Irgacure 2959) by mixing a solution of 20% w/v GelMA, 2% w/v κCA, and 0.5% w/v Irgacure 2959 with a solution of 4% w/v nanosilicates at a 1:1 ratio. The solution was vortexed and sonicated for 2 minutes to ensure homogenous dispersion of components. The ink was then stored overnight at 40°C.
4.2. Printing of NICE Scaffolds:
Printed scaffolds were designed in SolidWorks and exported as STL files. STL files were loaded into Slic3r to customize printing options and converted into G-code printer instructions. Relevant printing parameters include the infill density (100%), layer height (200 μm), extrusion width (500 μm), and print speed (20 mm s−1) for printing and travel moves. The open source user interface, or “host”, Pronterface (https://github.com/kliment/Printrun) was utilized to control the 3D printer. The ink was stored at 37 °C and loaded into an extrusion tube with a 400 μm nozzle tip and extrusion printed through a RepRap Prusa i3-style printer. For the in-vitro work, 30 mm diameter × 0.6mm disks were printed under a biosafety cabinet. The disk scaffolds were covalently crosslinked via exposure to 25 mW cm−2 365nm UV light for 80 seconds. Scaffolds were disinfected in 70% ethanol for 30 minutes. Ionic crosslinking of the scaffolds was completed by submersion in sterile-filtered 5% potassium chloride (KCl) for 30 minutes and were then stored at 4 °C until needed.
4.3. Bioconditioning of 3D printed scaffolds:
iP-hMSCs were generated as previously described[16]. Prior to cell-seeding, scaffolds were rinsed three times with warm PBS followed by a 30-minute incubation at 37 °C in Complete Culture Media (CCM), consisting of α-MEM (Invitrogen), 20% FBS (Atlanta Biologicals), 4mM L-glutamine (Invitrogen), 100 units ml−1 penicillin, and 100 μg ml−1 streptomycin (Hyclone). Excess CCM was removed from the scaffolds prior to iP-hMSCs seeding. Briefly, iP-hMSCs were incubated with the NICE scaffolds at density of 5,000 cells cm−2 in 5 mL of CCM in low-adherence 6 well-plates. Cells were allowed to adhere overnight with orbital shaking at 20 rpm. After 2 days, the type of culture media was changed to Osteogenic Basal Media (OBM)—CCM with 5mM β-glycerophosphate (Calibiochem) and 50 μg ml−1 ascorbic acid (Sigma-Aldrich)—supplemented with 10 μM GW9662 (Sigma-Aldrich) and changed every 2 days thereafter for 10 days. NICE scaffolds were then processed following a decellularization protocol previously described with slight variation[14]. In short, the NICE-iP-hMSC scaffolds were washed in excess PBS and decellularized by lysis in buffer consisting of PBS containing 0.1% (v/v) Triton X-100 (Sigma-Aldrich), 1mM MgCl2, and 1U ml−1 DNAse I for 12 hours at 37°C with orbital shaking at 60 rpm. The decellularized scaffolds were washed in dH2O, acetone, and dH2O. Lastly, the scaffolds were swelled in PBS until further use. The bioconditioned NICE scaffolds were hereafter referred to as bNICE whereas bare scaffolds lacking the iP-hMSCs laid matrix as simply NICE.
4.4. Evaluation of the Degree of Decellularization:
To validate the decellularization process, DNA content before and after decellularization was quantified using CyQuant GR dye (Invitrogen). Briefly, scaffolds were gently washed three times with warm PBS before being transferred to conical tubes. At which point, lysis buffer consisting of PBS with 1 mM MgCl2 and 0.1% Triton X-100 was added. The scaffolds were lysed by vortexing for 2 minutes. Lysates were collected after brief centrifugation while the scaffolds were washed and lyophilized in order to measure their dry weights. Diluted aliquots (100 μL) of the lysates were added to a black Micro-Well 96-well plate (Nunc, Thermo Fisher). An equal volume of the lysis buffer with 2X Cyquant GR dye was added. A DNA standard curve was made following the manufacturer’s instructions, using the lysis buffer described above as the serial diluent. Total DNA content was normalized by scaffold dry weight. For qualitative assessment, scaffolds were stained with 5 μM Hoechst to confirm the absence of nuclear material after decellularization. The FTIR spectra of NICE scaffolds at varying stages of the decellularization process were evaluated to verify the detergent utilized during the decellularization process was removed during washing steps. Briefly, 1 mm thick NICE scaffolds hand casted in 15 mm silicone molds were subjected to the decellularization process to varying degrees, resulting in unwashed NICE scaffolds with lysis buffer, washed NICE scaffolds, and NICE scaffolds before the lysis treatment. The scaffolds were flash frozen with liquid nitrogen and were stored at −80°C overnight before being lyophilized. Prior to analysis on an Alpha Bruker Spectrometer, the dried samples were flattened into thin disks using a mortar and pestle.
4.5. Bone Marrow Derived hMSCs Culture:
Bone marrow derived hMSCs were acquired from the Texas A&M Health Science Center adult stem cell distribution facility in accordance with their institutionally approved protocols. The hMSCs were cultured in both bare and bioconditioned NICE scaffolds in a similar fashion to iP-hMSCs—seeded at density of 5,000 cells cm−2 in CCM in low-adherence 6 well plates for 2 days. The hMSCs and scaffolds were then cultured for an additional 8 or 21 days in OBM supplemented with 10 μM of GW9662, with media changes occurring every 2–3 days.
4.6. ECM Characterization via Immunohistochemistry and SEM:
The deposited ECM was characterized via SEM imaging. Freshly printed NICE and bNICE scaffolds before and after the decellularization process were flash frozen with liquid nitrogen and were then stored at −80°C overnight. Frozen samples were then lyophilized, gold sputter coated, and imaged on a JCM 5000. Immunostaining for Coll VI and XII was performed to confirm the deposition of the distinct, anabolic-like ECM on the bNICE scaffolds. Both NICE and bNICE scaffolds were blocked with 5% goat serum (MP Biomedicals) and 0.3% Triton X-100 (Sigma-Aldrich) for 1 hr at room temperature. Scaffolds were then incubated overnight at 4°C in either rabbit antihuman type VI collagen (Novus Biologicals, Littleton, CO) or rabbit antihuman type XII collagen antibody (Novus) diluted in the blocking buffer at a 1:200 ratio. Samples were washed with PBS before the addition of fluorescein-conjugated goat antirabbit antibody (1:500, Millipore) for 2 hours at room temperature. Finally, samples were washed in PBS and imaged on an upright confocal microscope (Nikon D Eclipse C1).
4.7. Live/dead and actin staining:
hMSCs were cultured on bare and bioconditioned NICE scaffolds for 48 hours in CCM. Samples were then treated with PBS containing 0.1% BSA (Sigma-Aldrich), 1 μM calcein AM (AnaSpec), and 5 μM propidium iodide (Sigma-Aldrich) for 45 min at 37°C. The scaffolds were then washed with PBS and images were collected using an upright confocal microscope (Nikon D Eclipse C1). In order to gauge the ability of the iP-hMSCs to penetrate into the scaffolds from the surface, iP-hMSCs were cultured for 21 days on bare NICE scaffolds in the absence of osteoinductive factors. Cultured scaffolds were washed with PBS before and after being fixed with 4% formalin for 1 hour at room temperature. Samples were then permeabilized with 0.1% Triton-X in PBS,. Sample were then stained with Alexa-Fluor 488 Phalloidin (Life Technologies) according to the manufacturer’s instructions. Z-stack images were taken from the surface, towards a depth of 250 μm, using 10 μm slices. The relative fluorescent intensity units for z-stacked images were evaluated as a function of depth using ImageJ (NIH Image).
4.8. Gene expression analysis using qRT-PCR:
After 8 and 21 days of culture, total RNA was extracted from the hydrogels via a modified protocol using a total RNA isolation kit (High Pure, Roche). Cells were separated from the hydrogels by brief trypsinization and centrifugation before treatment with RNA extraction buffer. RNA was extracted from hMCS cultured on TCP following the manufacturer’s instructions for cells grown in monolayer. The purity (A260/A280~2.0) and concentration of isolated RNA was quantified before being used for cDNA synthesis in 21 μL reaction (Superscript II kit, Invitrogen). Approximately 8 ng of cDNA was amplified in a 20 μL reaction with Brilliant III Ultra-Fast SYBR Green QPCR Master Mix with Low ROX (Agilent) on an Agilent Aria Mx Real-Time PCR System. Primer sequences used can be found in Table 1. Relative gene expression was calculated via ΔΔCT method, using uninduced hMSCs cultured for either 8 or 21 days in CCM on TCP as a control. Induced hMSCs cultured for either 8 or 21 days in OBM on TCP were used as a positive control. In addition, cell number normalized to scaffold area was quantified after 8 and 21 days of culture on the scaffolds via GAPDH expression, using known cell number standards. RNA was extracted from the cell standard in the presence of NICE scaffolds to mimic the RNA extraction efficiency from the cultured samples.
Table 1.
Primers used in this study.
| Target | Sequence |
|---|---|
| human GAPDH | Forward: ctctctgctcctcctgttcgac Reverse: tgagcgatgtggctcggct |
| human Collagen VI | Forward: ccatcgtgcgcagcc Reverse: tgcgccgactcgtgc |
| human Collagen XII | Forward: cttccattgaggcagaagtt Reverse: agacacaagagcagcaatga |
| human BMP2 | Forward: cccagcgtgaaaagagagac Reverse: gagaccgcagtccgtctaag |
| human OCN | Forward: tcacactcctcgccctattg Reverse: ctcttcactacctcgctgcc |
| human OPN | Forward: catcacctgtgccataccagtt Reverse: ttggaagggtctgtggggcta |
4.9. Uniaxial Compression Testing:
Recently printed NICE and recently decellularized bNICE scaffolds were cut into cylinders using a 6 mm biopsy punch. The diameter and thickness (~600 μm) of the samples were verified using digital calipers and the ADMET MTEST Quattro eXpert 7600, respectively. Samples then underwent a single cycle unconfined compression test, strained up to 70% and returned to its starting position strain at a rate of 1 mm min−1. Raw data was analyzed using an Excel macro for compressive modulus (calculated from 0 to 20% strain region), and maximum stress at 70% strain.
4.10. Scaffold Swelling Test:
The equilibrium swelling ratio of NICE and bNICE scaffolds were calculated before and after undergoing 21 days of hMSCs culture. The swelling ratio was calculated as:
Here, Wwet and Wdry represent the weight of the scaffolds equilibrated in DI water for 1 hour and the weight of the scaffolds after lyophilization. To measure the swelling ratio, 6 mm biopsies were taken from recently printed NICE (day 0), recently decellularized bNICE (day 0), and cultured NICE and bNICE scaffolds. The scaffolds were washed with PBS before being incubated with DI water for1 hours at 37°C. After recording the wet weights, scaffolds were lyophilized overnight and weighed again.
4.11. Mineralization Characterization:
The calcium content was measured using an o-Cresolphthalein-Calcium reaction assay (Cayman Chemicals) after 21 days of culture on bare and bioconditioned NICE scaffolds. Briefly, 6 mm biopsies were taken from the cultured scaffolds and were fixed overnight at 4⁰ C in 4% formaldehyde. Samples were then washed three times with DI water before being dried overnight in a desiccator. Calcium was extracted from dried samples by undergoing an overnight acidic digestion at 4⁰ C using 0.5 M HCl on a tube rotator. Samples were centrifuged prior to collecting the supernatant for downstream use. Fourier-transform Infrared (FTIR) spectroscopy was utilized to further characterize the mineralization that occurred on the bare and bNICE scaffolds. Samples were collected via biopsy and dried as described above. Prior to analysis on an Alpha Bruker Spectrometer, the biopsied samples were flattened into thin disks using a mortar and pestle.
4.12. Statistical analysis:
Statistical tests and data plotting were performed with GraphPad Prism version 8.2.1 for Windows. All experiments were done in triplicate. A two-tailed t-test was used to determine statistical significance, assuming unequal sample variance, when comparing between two groups. For multiple variable experiments, statistical analysis was performed using one-way ANOVA, followed by a Tukey’s multiple comparison test.
Acknowledgements
CS and EM contributed equally. AKG would like to acknowledge financial support from the National Institute of Biomedical Imaging and Bioengineering (NIBIB) of the National Institutes of Health (NIH) Director’s New Innovator Award (DP2 EB026265) and the National Science Foundation (NSF) Award (CBET 1705852). RK and CAG would like to acknowledge financial support from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01 AR066033 and R21 AR072292) and the National Science Foundation (NSF) Award (CBET 1264848 and 1264832). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agency.
Contributor Information
Candice Sears, Department of Biomedical Engineering, College of Engineering, Texas A&M University, College Station, Texas 77843, USA.
Eli Mondragon, Department of Biomedical Engineering, College of Engineering, Texas A&M University, College Station, Texas 77843, USA.
Zachary Richards, Department of Biomedical Engineering, College of Engineering, Texas A&M University, College Station, Texas 77843, USA.
Nick Sears, Department of Biomedical Engineering, College of Engineering, Texas A&M University, College Station, Texas 77843, USA.
David Chimene, Department of Biomedical Engineering, College of Engineering, Texas A&M University, College Station, Texas 77843, USA.
Eoin McNeill, Department of Molecular and Cellular Medicine, Texas A&M Health Science Center, College Station, TX, United States.
Carl Gregory, Department of Molecular and Cellular Medicine, Texas A&M Health Science Center, College Station, TX, United States.
Akhilesh K. Gaharwar, Department of Biomedical Engineering, College of Engineering, Texas A&M University, College Station, Texas 77843, USA Department of Material Science and Engineering, College of Engineering, Texas A&M University, College Station, Texas 77843, USA; Center for Remote Health Technologies and Systems, Texas A&M University, College Station, Texas 77843, USA.
Roland Kaunas, Department of Biomedical Engineering, College of Engineering, Texas A&M University, College Station, Texas 77843, USA; Department of Molecular and Cellular Medicine, Texas A&M Health Science Center, College Station, TX, United States.
REFERENCES
- [1].Desai BM, Am. J. Orthop. (Belle Mead. NJ) 2007, 36, 8. [PubMed] [Google Scholar]
- [2].Reddy S, Khalifian S, Flores JM, Bellamy J, Manson PN, Rodriguez ED, Dorafshar AH, Plast. Reconstr. Surg 2014, 133, 864. [DOI] [PubMed] [Google Scholar]
- [3].Yadla S, Campbell PG, Chitale R, Maltenfort MG, Jabbour P, Sharan AD, Neurosurgery 2011, 68, 1124. [DOI] [PubMed] [Google Scholar]
- [4].Sahoo N, Roy ID, Desai AP, Gupta V, J. Craniofac. Surg 2010, 21, 79. [DOI] [PubMed] [Google Scholar]
- [5].Bhumiratana S, Bernhard JC, Alfi DM, Yeager K, Eton RE, Bova J, Shah F, Gimble JM, Lopez MJ, Eisig SB, et al. , Sci. Transl. Med 2016, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Arnaoutakis D, Bahrami A, Cohn JE, Smith JE, JAMA Facial Plast. Surg 2018, 20, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dean D, Topham NS, Rimnac C, Mikos AG, Goldberg DP, Jepsen K, Redtfeldt R, Liu Q, Pennington D, Ratcheson R, Plast. Reconstr. Surg 1999, 104, 705. [DOI] [PubMed] [Google Scholar]
- [8].Matic DB, Manson PN, J. Craniofac. Surg 2004, 15, 415. [DOI] [PubMed] [Google Scholar]
- [9].Bin Lai J, Sittitavornwong S, Waite PD, Oral Maxillofac J. Surg 2011, 69, 1175. [DOI] [PubMed] [Google Scholar]
- [10].Cabraja M, Klein M, Lehmann TN, Neurosurg. Focus 2009, 26, E10. [DOI] [PubMed] [Google Scholar]
- [11].Berman AT, Reid JS, Yanicko DR, Sih GC, Zimmerman MR, Clin. Orthop. Relat. Res 1984, 186, 284. [PubMed] [Google Scholar]
- [12].Wu S, Liu X, Yeung KWK, Liu C, Yang X, Mater. Sci. Eng. R Reports 2014, 80, 1. [Google Scholar]
- [13].Zeitouni S, Krause U, Clough BH, Halderman H, Falster A, Blalock DT, Chaput CD, Sampson HW, Gregory C. a, Sci. Transl. Med 2012, 4, 132ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Clough BH, McCarley MR, Krause U, Zeitouni S, Froese JJ, McNeill EP, Chaput CD, Sampson HW, Gregory CA, J. Bone Miner. Res 2015, 30, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Clough BH, McNeill EP, Palmer D, Krause U, Bartosh TJ, Chaput CD, Gregory CA, Spine J 2017, 17, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhao Q, Gregory CA, Lee H, Reger RL, Qin L, Hai B, Park MS, Yoon N, Clough B, Mcneill E, et al. , Proc. Natl. Acad. Sci. U. S. A 2015, 112, 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Seol YJ, Kang TY, Cho DW, Soft Matter 2012, 8, 1730. [Google Scholar]
- [18].Trombetta R, Inzana JA, Schwarz EM, Kates SL, Awad HA, Ann. Biomed. Eng 2017, 45, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Thakur T, Xavier JR, Cross L, Jaiswal MK, Mondragon E, Kaunas R, Gaharwar AK, J. Biomed. Mater. Res. A 2015, 104, 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gaharwar AK, Dammu SA, Canter JM, Wu CJ, Schmidt G, Biomacromolecules 2011, 12, 1641. [DOI] [PubMed] [Google Scholar]
- [21].Shim JH, Huh JB, Park JY, Jeon YC, Kang SS, Kim JY, Rhie JW, Cho DW, Tissue Eng. - Part A 2013, 19, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang W, Yeung KWK, Bioact. Mater 2017, 2, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gao G, Schilling AF, Yonezawa T, Wang J, Dai G, Cui X, Biotechnol. J 2014, 9, 1304. [DOI] [PubMed] [Google Scholar]
- [24].Chimene D, Kaunas R, Gaharwar AK, Adv. Mater 2019, 32, 1902026. [DOI] [PubMed] [Google Scholar]
- [25].Jakus AE, Rutz AL, Jordan SW, Kannan A, Mitchell SM, Yun C, Koube KD, Yoo SC, Whiteley HE, Richter CP, et al. , Sci. Transl. Med 2016, 8, 358ra127. [DOI] [PubMed] [Google Scholar]
- [26].Hung BP, Naved BA, Nyberg EL, Dias M, Holmes CA, Elisseeff JH, Dorafshar AH, Grayson WL, ACS Biomater. Sci. Eng 2016, 2, 1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lode A, Meissner K, Luo Y, Sonntag F, Glorius S, Nies B, Vater C, Despang F, Hanke T, Gelinsky M, Tissue Eng J. Regen. Med 2014, 8, 682. [DOI] [PubMed] [Google Scholar]
- [28].Chimene D, Peak CW, Gentry JL, Carrow JK, Cross LM, Mondragon E, Cardoso GB, Kaunas R, Gaharwar AK, ACS Appl. Mater. Interfaces 2018, 10, 9957. [DOI] [PubMed] [Google Scholar]
- [29].Chimene D, Lennox KK, Kaunas RR, Gaharwar AK, Ann. Biomed. Eng 2016, 44, 2090. [DOI] [PubMed] [Google Scholar]
- [30].Paul A, Manoharan V, Krafft D, Assmann A, Uquillas JA, Shin SR, Hasan A, Hussain MA, Memic A, Gaharwar AK, et al. , J. Mater. Chem. B 2016, 4, 3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wilson SA, Cross LM, Peak CW, Gaharwar AK, ACS Appl. Mater. Interfaces 2017, 9, 43449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bakarich SE, In Het Panhuis M, Beirne S, Wallace GG, Spinks GM, J. Mater. Chem. B 2013, 1, 4939. [DOI] [PubMed] [Google Scholar]
- [33].Bakarich SE, Gorkin R, In Het Panhuis M, Spinks GM, Macromol. Rapid Commun 2015, 36, 1211. [DOI] [PubMed] [Google Scholar]
- [34].Gaharwar AK, Cross LM, Peak CW, Gold K, Carrow JK, Brokesh A, Singh KA, Adv. Mater 2019, 31, 1900332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gaharwar AK, Mihaila SM, Swami A, Patel A, Sant S, Reis RL, Marques AP, Gomes ME, Khademhosseini A, Adv. Mater 2013, 25, 3329. [DOI] [PubMed] [Google Scholar]
- [36].Xavier JR, Thakur T, Desai P, Jaiswal MK, Sears N, Cosgriff-Hernandez E, Kaunas R, Gaharwar AK, ACS Nano 2015, 9, 3109. [DOI] [PubMed] [Google Scholar]
- [37].Carrow JK, Cross LM, Reese RW, Jaiswal MK, Gregory CA, Kaunas R, Singh I, Gaharwar AK, Proc. Natl. Acad. Sci 2018, 115, E3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Afewerki S, Magalhães LSSM, Silva ADR, Stocco TD, Silva Filho EC, Marciano FR, Lobo AO, Adv. Healthc. Mater 2019, 8, 1900158. [DOI] [PubMed] [Google Scholar]
- [39].Jin Y, Liu C, Chai W, Compaan A, Huang Y, ACS Appl. Mater. Interfaces 2017, 9, 17456. [DOI] [PubMed] [Google Scholar]
- [40].Jin Y, Zhao D, Huang Y, Bio-Design Manuf 2018, 1, 123. [Google Scholar]
- [41].Peak CW, Singh KA, Adlouni M, Chen J, Gaharwar AK, Adv. Healthc. Mater 2019, 8, 1801553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ahlfeld T, Cidonio G, Kilian D, Duin S, Akkineni AR, Dawson JI, Yang S, Lode A, Oreffo ROC, Gelinsky M, Biofabrication 2017, 9, 034103. [DOI] [PubMed] [Google Scholar]
- [43].Sason G, Nussinovitch A, Food Hydrocoll 2018, 79, 136. [Google Scholar]
- [44].Necas J, Bartosikova L, Vet. Med. (Praha) 2013, 58, 187. [Google Scholar]
- [45].Shekaran A, García AJ, J. Biomed. Mater. Res. Part A 2011, 96A, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Pountos I, Panteli M, Lampropoulos A, Jones E, Calori GM, Giannoudis PV, BMC Med 2016, 14, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hoyos-Nogués M, Falgueras-Batlle E, Ginebra M-P, Manero J, Gil J, Mas-Moruno C, Int. J. Mol. Sci 2019, 20, 1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kim AGM, Seon Yu, Majid Marjan, Melchiorri Anthony J., Bioeng. Transl. Med 2018, 4, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Crapo PM, Gilbert TW, Badylak SF, Biomaterials 2011, 32, 3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cross LM, Carrow JK, Ding X, Singh KA, Gaharwar AK, ACS Appl. Mater. Interfaces 2019, 11, 6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Howell DW, Peak CW, Bayless KJ, Gaharwar AK, Adv. Biosyst 2018, 2, 1800092. [Google Scholar]
- [52].Lokhande G, Carrow JK, Thakur T, Xavier JR, Parani M, Bayless KJ, Gaharwar AK, Acta Biomater 2018, 70, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pati F, Jang J, Ha DH, Won Kim S, Rhie JW, Shim JH, Kim DH, Cho DW, Nat. Commun 2014, 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pati F, Song T-H, Rijal G, Jang J, Kim SW, Cho D-W, Biomaterials 2015, 37, 230. [DOI] [PubMed] [Google Scholar]
- [55].Bourgine PE, Pippenger BE, Todorov A, Tchang L, Martin I, Biomaterials 2013, 34, 6099. [DOI] [PubMed] [Google Scholar]
- [56].Ramos CA, Asgari Z, Liu E, Yvon E, Heslop HE, Rooney CM, Brenner MK, Dotti G, Stem Cells 2010, 28, 1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gao Q, Niu X, Shao L, Zhou L, Lin Z, Sun A, Fu J, Chen Z, Hu J, Liu Y, et al. , Biofabrication 2019, 11, 035006. [DOI] [PubMed] [Google Scholar]
- [58].Gericke A, Qin C, Spevak L, Fujimoto Y, Butler WT, Sørensen ES, Boskey AL, Calcif Tissue Int 2005, 77, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zurick KM, Qin C, Bernards MT, J. Biomed. Mater. Res. - Part A 2013, 101 A, 1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yang W, Guo D, Harris MA, Cui Y, Gluhak-Heinrich J, Wu J, Chen X-D, Skinner C, Nyman JS, Edwards JR, et al. , J. Cell Sci 2013, 126, 4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Figueiredo MM, Gamelas JAF, Martins AG, Characterization of Bone and Bone-Based Graft Materials Using FTIR Spectroscopy, 2012.
- [62].Kretlow JD, Young S, Klouda L, Wong M, Mikos AG, Adv. Mater 2009, 21, 3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhang D, George OJ, Petersen KM, Jimenez-Vergara AC, Hahn MS, Grunlan MA, Acta Biomater 2014, 10, 4597. [DOI] [PubMed] [Google Scholar]
- [64].Woodard LN, Page VM, Kmetz KT, Grunlan MA, Macromol. Rapid Commun 2016, 37, 1972. [DOI] [PubMed] [Google Scholar]
- [65].Woodard LN, Grunlan MA, ACS Biomater. Sci. Eng 2019, 5, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Decaris ML, Binder BY, Soicher MA, Bhat A, Leach JK, Tissue Eng. - Part A 2012, 18, 2148. [DOI] [PubMed] [Google Scholar]
- [67].Decaris ML, Mojadedi A, Bhat A, Leach JK, Acta Biomater 2012, 8, 744. [DOI] [PubMed] [Google Scholar]
- [68].Liao J, Guo X, Nelson D, Kasper FK, Mikos AG, Acta Biomater 2010, 6, 2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Liu Q, Limthongkul W, Sidhu G, Zhang J, Vaccaro A, Shenck R, Hickok N, Shapiro I, Freeman T, J. Orthop. Res 2012, 30, 1626. [DOI] [PubMed] [Google Scholar]
- [70].Pham QP, Kasper FK, Mistry AS, Sharma U, Yasko AW, Jansen JA, Mikos AG, J. Biomed. Mater. Res. Part A 2009, 88, 295. [DOI] [PubMed] [Google Scholar]
- [71].Pham QP, Kurtis Kasper F, Scott Baggett L, Raphael RM, Jansen JA, Mikos AG, Biomaterials 2008, 29, 2729. [DOI] [PubMed] [Google Scholar]
- [72].Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A, Biomaterials 2010, 31, 5536. [DOI] [PMC free article] [PubMed] [Google Scholar]