

Abstract
The capsular polysaccharides (CPS) of Campylobacter jejuni contain multiple heptose residues with variable stereochemical arrangements at C3, C4, C5, and C6. The immediate precursor to all of these possible variations is currently believed to be GDP-d-glycero-α-d-manno-heptose. Oxidation of this substrate at C4 enables subsequent epimerization reactions at C3, C4, and C5 that can be coupled to the dehydration/reduction at C5/C6. However, the enzyme responsible for the critical oxidation of C4 from GDP-d-glycero-α-d-manno-heptose has remained elusive. The enzyme Cj1427 from C. jejuni NCTC 11168 was shown to catalyze the oxidation of GDP-d-glycero-α-d-manno-heptose to GDP-d-glycero-4-keto-α-d-lyxo-heptose in the presence of α-ketoglutarate using mass spectrometry and NMR spectroscopy. At pH 7.4 the apparent kcat is 0.6 s−1, with a value of kcat/Km of 1.0 × 104 M−1 s−1 for GDP-d-glycero-α-d-manno-heptose. α-Ketoglutarate is required to recycle the tightly bound NADH nucleotide in the active site of Cj1427, which does not dissociate from the enzyme during catalysis.
Graphical Abstract
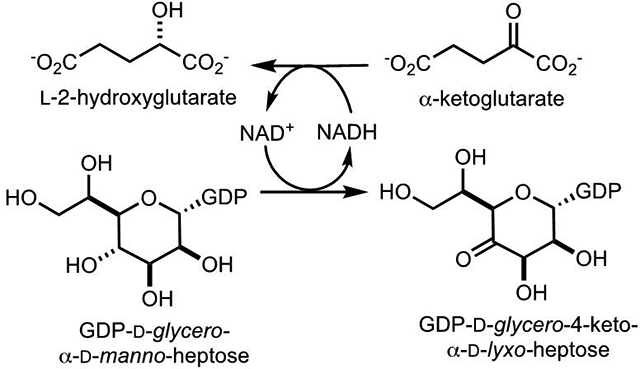
Introduction
Heptose units are common among the many different capsular polysaccharides (CPS) from the various strains of the human pathogen Campylobacter jejuni (1–3). As of 2019, there are 12 known CPS structures and 9 of these contain heptose moieties with 13 different stereochemical variations at C3 through C6 (3, 4). The CPS of C. jejuni NCTC 11168 (HS:2) contains a single heptose moiety identified as d-glycero-l-gluco-heptose (4, 5). Three enzymes have been implicated in the transformation of GDP-d-glycero-α-d-manno-heptose (1) to GDP-d-glycero-β-l-gluco-heptose (4) and the proposed biosynthetic pathway is presented in Scheme 1 (6). These enzymes include Cj1427 (Uniprot: Q0P8I7, Cj1428 (Uniprot: Q0P8I6), and Cj1430 (Uniprot: Q0P8I4). However, there remains uncertainty about the specific enzyme required for the initial oxidation of C4 from GDP-d-glycero-α-d-manno-heptose (6).
Scheme 1:
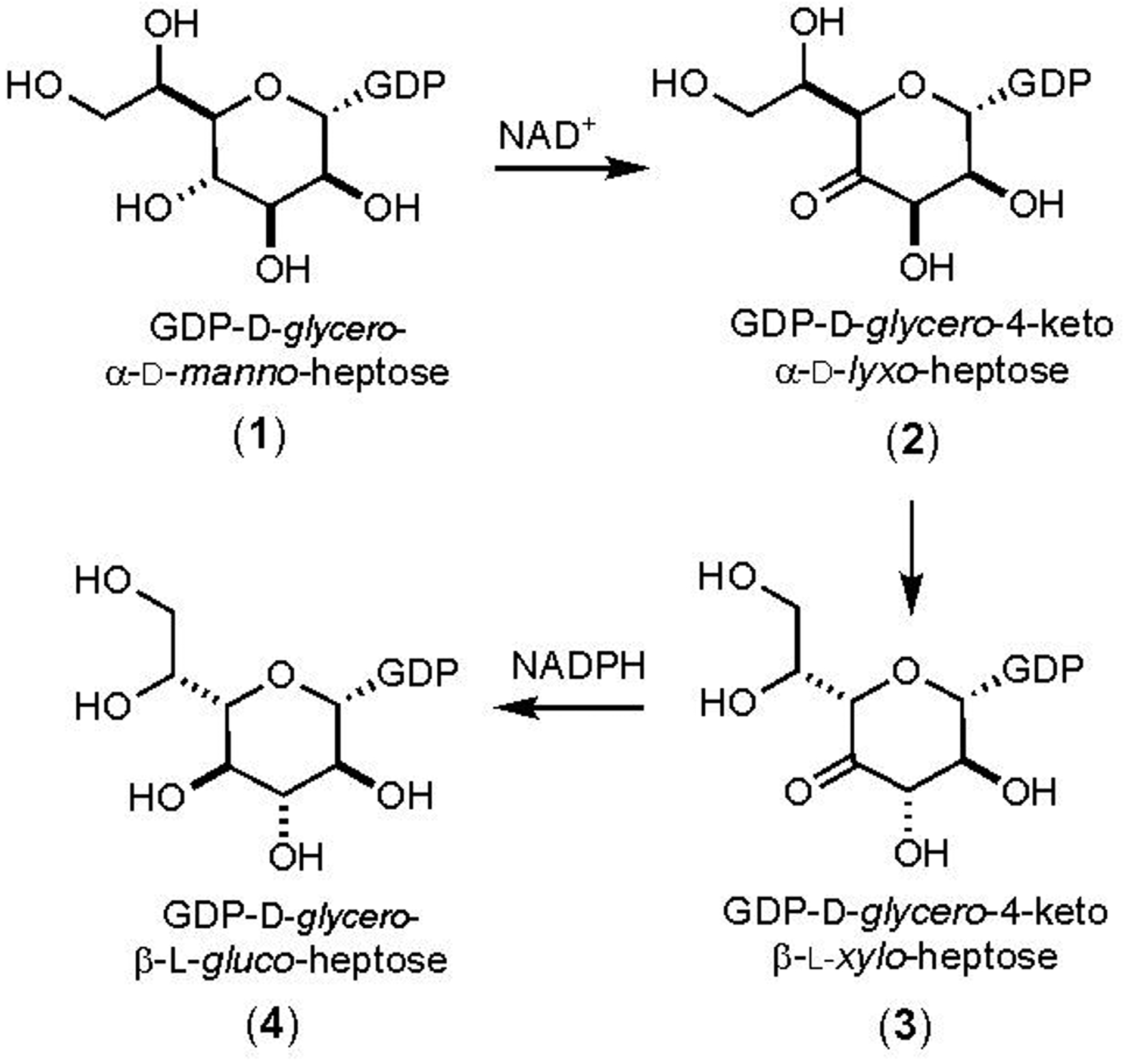
Proposed biosynthetic pathway for formation of GDP-d-glycero-β-l-gluco-heptose starting from GDP-d-glycero-α-d-manno-heptose (6).
The Crueznet laboratory has tested Cj1427 as a potential catalyst for the oxidation of GDP-d-glycero-α-d-manno-heptose (1) but was unable to demonstrate any activity with this substrate and have thus concluded that this enzyme is not part of the pathway for the biosynthesis of GDP-d-glycero-β-l-gluco-heptose (6). They have, however, used the enzyme DdahA, from C. jejuni strain 81–176, to catalyze the NAD+/NADH dependent oxidation/dehydration/reduction of GDP-d-glycero-α-d-manno-heptose (1) to GDP-6-deoxy-4-keto-α-d-lyxo-heptose (5) as illustrated in Scheme 2 (6). They have subsequently shown that this product is a suitable substrate for Cj1430 where C3 and C5 are epimerized to form GDP-6-deoxy-4-keto-β-l-xylo-heptose (6) and that Cj1428 will catalyze the reduction of this product to GDP-6-deoxy-β-l-gluco-heptose (7). This set of experiments has thus identified the probable catalytic activities of Cj1430 and Cj1428 with the surrogate 6-deoxy substrate and thus the specific enzyme responsible for the initial oxidation of GDP-d-glycero-α-d-manno-heptose has remained elusive.
Scheme 2:
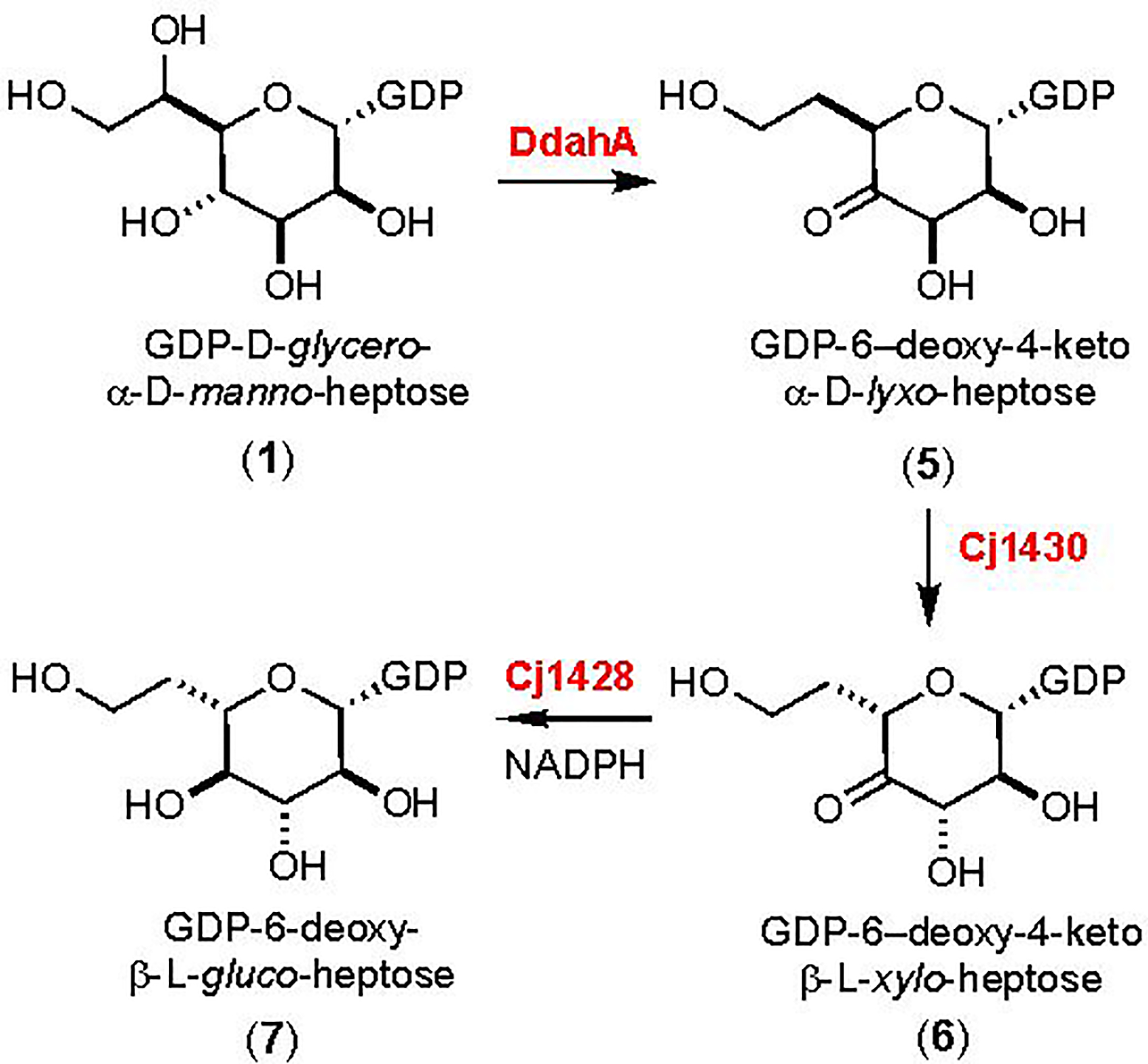
Catalytic activities of DdahA, Cj1430, and Cj1428 (6).
Curiously, it was found that Cj1427 could catalyze the NADH-dependent reduction of GDP-6-deoxy-4-keto-α-d-lyxo-heptose (5) to GDP-6-deoxy-α-d-manno-heptose (8) as shown in Scheme 3. It is not obvious why Cj1427 could catalyze the reduction of compound 5 but not the oxidation of compound 1. However, we have shown that Cj1427 co-purifies with a tightly bound NADH cofactor and that this enzyme requires the addition of α-ketoglutarate to re-oxidize the bound nicotinamide cofactor to NAD+ (7). We have also demonstrated that Cj1427 is capable of binding GDP-d-glycero-α-d-manno-heptose (1) in the active site of this enzyme with C4 positioned for direct hydride transfer to the nicotinamide cofactor (7).
Scheme 3:
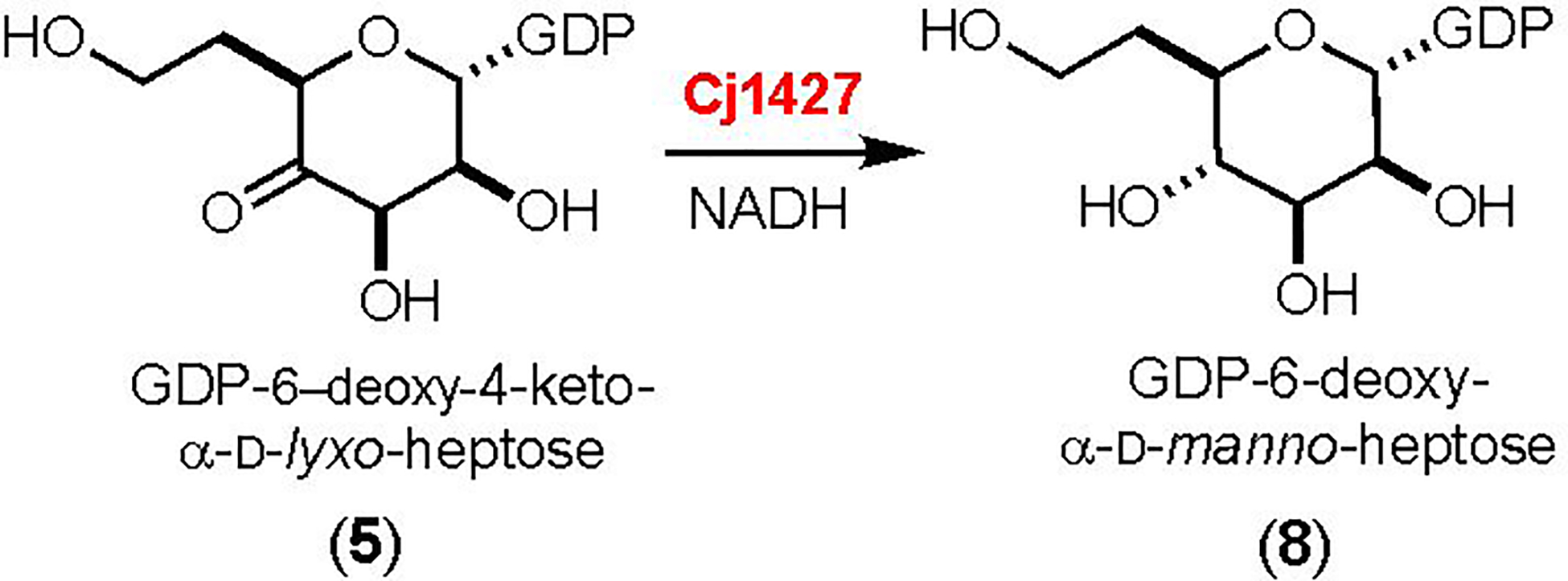
Reaction catalyzed by Cj1427 with the surrogate substrate 5 (6).
In this paper, we demonstrate that Cj1427 and its close functional homologues are fully conserved among C. jejuni strains that possess a heptose moiety with a hydroxyl group at C6 within their CPS. We further prove that Cj1427 catalyzes the oxidation of C4 of GDP-d-glycero-α-d-manno-heptose (1) to form GDP-d-glycero-4-keto-α-d-lyxo-heptose (2) in the presence of α-ketoglutarate by NMR spectroscopy and mass spectrometry. We have measured the steady-state kinetic constants for Cj1427 using a coupled enzyme assay with Cj1430 and Cj1428, and have characterized the single turnover oxidation of Cj1427 with α-ketoglutarate using stopped-flow kinetic measurements.
Materials and Methods
Materials and Equipment.
All materials used in this study were obtained from Sigma-Aldrich, Carbosynth, or GE Healthcare Bio-Sciences, unless stated otherwise. E. coli strains XL1 Blue and BL21-Gold (DE3) were obtained from New England Biolabs. NMR spectra were collected on a Bruker Avance 500 MHz system equipped with a triple resonance cyroprobe. Mass spectrometry samples were collected on an MDS-Sciex 4000 Qtrap system or a Thermo Scientific Q Exactive Focus system run in the negative ion mode. UV spectra were collected with a SpectraMax340 UV-visible plate reader using 96-well NucC plates. α-Ketoglutarate was purchased from AK scientific (Union City, CA). Single turnover reactions with Cj1427 were monitored using a Kintek AutoSF-120 stopped-flow fluorimeter (Kintek Corp, Austin, TX) equipped with a Semrock Brightline 438 ± 24 nm cutoff filter. The slit width of the monochromator and the light filter were set at 0.6 mm. Data collection was conducted using Kintek stopped-flow software Ver. 1.0.2359, and initial equation-based fitting of data and fit by simulation were conducted using KinTek Global Kinetic Explorer Pro, Ver. 6.3.180116.
Bioinformatic Analysis of Cj1427 and DdahA from C. jejuni.
The protein sequences of 484 C. jejuni strains were obtained from the NCBI assembly database (8). These genomes were subsequently compiled into a custom C. jejuni database using the command-line applications provided by NCBI (9). The protein sequences for Cj1427 and DdahA were used as BLAST search queries against the custom database containing all protein FASTA sequences from the 484 C. jejuni strains. Sequence similarity networks (SSN) were generated by directly submitting the FASTA sequences from the closest homolog from each of the 484 C. jejuni strains to the EFI-EST webtool (10). All network layouts were created and visualized using Cytoscape 3.4 (11).
Cloning of the Genes for Cj1427, Cj1428 and Cj1430.
The method for cloning the gene for Cj1427 was described previously (7). The genes for Cj1428 (Uniprot ID: Q0P8I6), and Cj1430 (Uniprot ID: Q0P8I4) were amplified from the genomic DNA of C. jejuni strain NCTC 11168 (ATCC-700819D-5). The gene for Cj1428 was amplified using the primer pair:
5’- GATTG GGATCC ATGCAAACAAATTCAAAAATATATATAGCG −3’
5’- GGCAG CTCGAG TCAATTTTGTGTTTTATACCATTCATAC −3’
The gene for Cj1430 was amplified using the primer pair:
5’- GATTG GGATCC ATGGCAATAGAATTTGATATACAAGAATC −3’
5’- GGCAG CTCGAG TTATCCTTTATTTTTAGTTGCAAGAATATC −3’
For each pair of primers, restriction sites for BamHI and XhoI (underlined) were introduced into the forward and reverse primers, respectively. These sites allow for the addition of an N-terminal 6x-His-tag in the pET30a+ expression vector for all three enzymes. Procedures for gene amplification, restriction digest, ligation, and plasmid isolation were followed as previously reported (12). Plasmids were fully sequenced to confirm correctly cloned genes in the appropriate vector.
Purification of Cj1427, Cj1428 and Cj1430.
Recombinant plasmids containing the genes for Cj1427, Cj1428, or Cj1430 were used to transform E. coli BL21 (DE3) cells using the method of heat-shock (13). Conditions for expression and purification were followed as previously described (7, 12). Briefly, a 20-mL starter culture was used to inoculate two 1-L flasks of lysogeny broth (LB) medium. Growth continued at 37 °C until OD600 = 0.6, followed by the addition of 1.0 mM isopropyl β-thiogalactoside (IPTG) to induce protein expression for 18 h at 22 °C. Growth and expression of Cj1427, Cj1428, and Cj1430 proceeded with no other additions to the growth medium. The cells were harvested by centrifugation and stored at −80 °C. For purification, frozen cell pellets were resuspended in 50 mM HEPES/K+, pH 8.5, with 250 mM KCl and 10 mM imidazole, and lysed by sonication. The enzymes were purified using a HisTrap HP column, buffer exchanged into 50 mM HEPES/K+, pH 8.5, with 250 mM KCl, and concentrated as described previously (7, 12). Final enzyme concentrations were determined from the absorbance at 280 nm and the Bradford assay (Bio-Rad, Hercules CA) for Cj1427, Cj1428, and Cj1430 (14). The extinction coefficients used for Cj1427 and Cj1428 were ε280 = 24,500 M−1 cm−1 and ε280 = 34,595, respectively (15). These values were estimated based on the protein sequences including the His-tag and linkers (ε280 = 28,100 M−1 cm−1 for 1427 and ε280 = 38,195 M−1 cm−1 for Cj1428) and corrected for bound NADH and NADPH, which has an extinction coefficient determined to be ε280 = 3,600 M−1 cm−1 calculated from free NADH and NADPH. The extinction coefficient used for Cj1430 was ε280 = 41,495 M−1 cm−1. Yields of purified protein were 50–75 mg for Cj1427, 150–200 mg for Cj1428, and 75–100 mg for Cj1430 from 2.0 L of cell culture.
Synthesis of GDP-d-glycero-α-d-manno-heptose.
GDP-d-glycero-α-d-manno-heptose (1) was synthesized using Cj1423, Cj1424, Cj1425, GmhB, and TktA in a single reaction vessel starting from d-ribose-5-P and hydroxypyruvate following methodology described previously (3). GDP-d-glycero-α-d-manno-heptose was purified by DEAE anion exchange chromatography as outlined previously (3, 12, 16). GDP-d-glycero-α-d-manno-heptose was precipitated from a solution of ethanol/water (60/40) at −20 °C. The white precipitate was collected using gravity paper filtration and dried under vacuum to remove any remaining ethanol. A fine white powder was collected and stored in a desiccator at −20 °C until further use. The identity of GDP-d-glycero-α-d-manno-heptose was confirmed by mass spectrometry and 1H NMR spectroscopy.
Synthesis of [2-2H]-d,l-2-Hydroxyglutarate.
α-Ketoglutarate (84 mg) was dissolved into 5.0 mL of H2O. A second solution was made containing 1.5 eq (38 mg) of sodium borodeuteride (NaBD4) in H2O (3.0 mL). To the stirring solution of α-ketoglutarate, the solution of NaBD4 was added dropwise over 2–3 h. The final mixture was lyophilized to dryness to obtain a white solid of the disodium salt of [2-2H]-d,l-2-hydroxyglutarate. The structure was confirmed by 13C and 1H NMR spectroscopy.
Enzymatic Oxidation of GDP-d-glycero-α-d-manno-heptose.
Cj1427 was exchanged into 50 mM ammonium bicarbonate buffer (pH 8.0) using VivaSpin 3K spin concentrators. For oxidation of GDP-d-glycero-α-d-manno-heptose (1), Cj1427 (20 μM) was mixed with 2.0 mM GDP-d-glycero-α-d-manno-heptose and 20 mM α-ketoglutarate in 50 mM ammonium bicarbonate buffer, pH 8.0. For measurement of hydrogen/deuterium exchange at C4 of the substrate, Cj1427 (20 μM) was mixed with 2.0 mM GDP-d-glycero-α-d-manno-heptose, 0.5 mM α-ketoglutarate, and 20 mM [2-2H]-d,l-2-hydroxyglutarate in 50 mM ammonium bicarbonate buffer, pH 8.0. Mass spectra were collected in negative ion mode after 1–2 h of incubation at 25 °C.
For measurement of the oxidation of C4 by NMR spectroscopy, Cj1427 (20 μM) was mixed with 2.0 mM GDP-d-glycero-α-d-manno-heptose and 10 mM α-ketoglutarate in 50 mM potassium phosphate/D2O buffer, pH 8.0. The oxidation of GDP-d-glycero-α-d-manno-heptose was followed over time by 1H NMR spectroscopy. For deuterium exchange at C4, Cj1427 (20 μM) was mixed with 2.0 mM GDP-d-glycero-α-d-manno-heptose, 0.5 mM α-ketoglutarate, and 20 mM [2-2H]-d,l-2-hydroxyglutarate in potassium phosphate/D2O buffer, pH 8.0. Exchange of the hydrogen at C4 with deuterium was monitored for 1 h by 1H NMR spectroscopy with spectra collected every 1 min.
Determination of Kinetic Constants for Cj1427.
The steady-state kinetic constants for Cj1427 were determined using a coupled assay with Cj1430 and Cj1428 at 30 °C. A solution of Cj1427 (0.5 μM) was made in 50 mM HEPES/KCl buffer, pH 7.4 with an excess of Cj1430 (10 μM) and Cj1428 (10 μM). To this solution, 300 μM NADPH was added. Cj1428 oxidizes exogenous NADPH to NADP+ to catalyze the reduction of the substrate to product. The oxidation of NADPH to NADP+ was monitored using UV spectroscopy at 340 nm. The enzyme solution was added to the wells containing varying concentrations of GDP-d-glycero-α-d-manno-heptose (0–400 μM) with a fixed amount of α-ketoglutarate (1.0 mM) or varying concentrations of α-ketoglutarate (0–2000 μM) with a fixed amount of GDP-d-glycero-α-d-manno-heptose (500 μM). The apparent values of kcat and kcat/Km were determined by fitting the initial velocity data to eq. 1 using GraFit 5, where v is the initial velocity of the reaction, E is the enzyme concentration, kcat is the turnover number, and Km is the Michaelis constant.
| (1) |
Stopped-Flow Kinetics of NADH Oxidation Using α-Ketoglutarate.
Cj1427 was diluted into 50 mM HEPES/KCl buffer, pH 7.4, to a final concentration of 30 μM. A stock solution of 1.0 mM α-ketoglutarate was made in 50 mM HEPES/KCl buffer, pH 7.4. Various concentrations of α-ketoglutarate (5–600 μM) were made from the stock solution. Cj1427 and substrate were mixed in a 1:1 ratio in the stopped-flow apparatus and changes in NADH fluorescence were collected for 0.5 s. The instrument was set to collect 2000 total points with 1000 points in the first 50 ms and 1000 points over the remaining 450 ms. Collected traces represent an average of at least seven reactions at each substrate concentration. Data were exported from the KinTek stopped-flow instrument and imported into Kintek Global Kinetic Explorer. In Kintek Global Kinetic Explorer, the first 100 ms of the stopped-flow fluorescence traces were fit to a single exponential function (eq 2).
| (2) |
Subsequently, the observed rate constants (λ1) were plotted versus the concentration of α-ketoglutarate. The concentration dependence of the observed rate constant (obtained from the single exponential fit) was fit by Grafit to a hyperbola (eq 3).
| (3) |
The concentration dependence of kobs versus the concentration of α-ketoglutarate was consistent with a model for a two-step reaction where K1 represents the equilibrium constant for the binding of S to E (k1/k2), k3 represents the rate constant of the second step in the forward direction, k4 represents the rate constant of the second step in the reverse direction (Scheme 4)(18, 19).
Scheme 4:
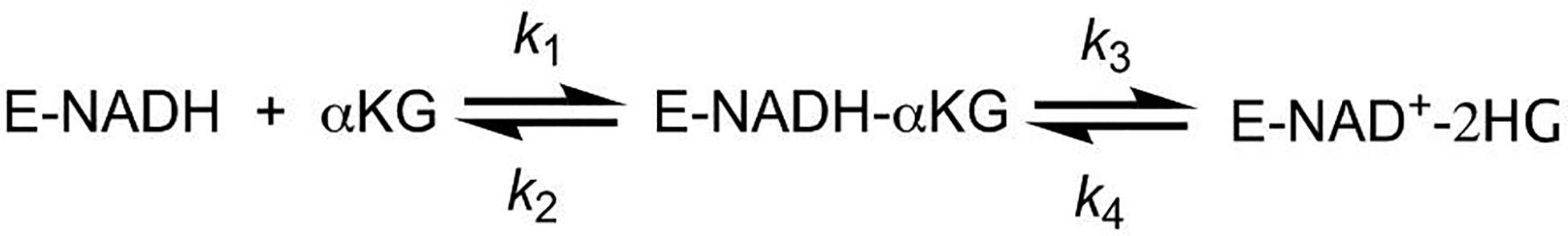
Minimal kinetic model for the oxidation of E•NADH by α-ketoglutarate.
Fitting of Stopped-Flow Data by Simulation.
The conventional analysis of the stopped-flow data and resulting minimal kinetic model were used as initial guides to fit the data by simulation using Kintek Global Kinetic Explorer (19–21). During the process of global optimization, the data collected over a series of concentrations within a given experiment were scaled using a correction factor (less than 3%) for each trace to correct for lamp drift between traces.
Results
Bioinformatic Analysis of Cj1427 and DdahA Homologs in C. jejuni.
Cj1427 shares 97% sequence identity to WcaG from C. jejuni 81–176 (HS:23/36) (6). Both enzymes have been shown to catalyze the NADH-dependent reduction of GDP-6-deoxy-4-keto-α-d-lyxo-heptose (5) to GDP-6-deoxy-α-d-manno-heptose (8) (6, 22). However, these two enzymes were also reported to be unable to catalyze the oxidation of GDP-d-glycero-α-d-manno-heptose (1) to GDP-d-glycero-α−4-keto-d-lyxo-heptose (2). It seemed curious to us that these enzymes should be able to catalyze the reduction of 5 but unable to catalyze the oxidation of 1 (7). As this latter transformation is essential for the biosynthesis of all CPS structures that contain a 6-hydroxy heptose moiety, a bioinformatic analysis of these enzymes was conducted to better understand the sequence diversity and prevalence of Cj1427 and its homologs among other strains of C. jejuni. A similar analysis was conducted with the dehydratase, DdahA, which produces the key 6-deoxy-4-keto intermediate (22, 23).
Homologs to Cj1427 were found in 208 out of the 484 (43%) C. jejuni genomes queried. A sequence similarity comparison among the Cj1427 homologs shows >97% sequence conservation among the group (Figure 1a). In the 276 strains that do not have a homolog to Cj1427, the closest protein shares <27% sequence identity to Cj1427. For DdahA, homologs were found in 265 out of 484 (55%) C. jejuni genomes. A sequence comparison among these homologs, shows >91% sequence conservation (Figure 1b). In the 219 genomes that do not contain a DdahA homolog, no protein shares more than 29% sequence identity to DdahA.
Figure 1.
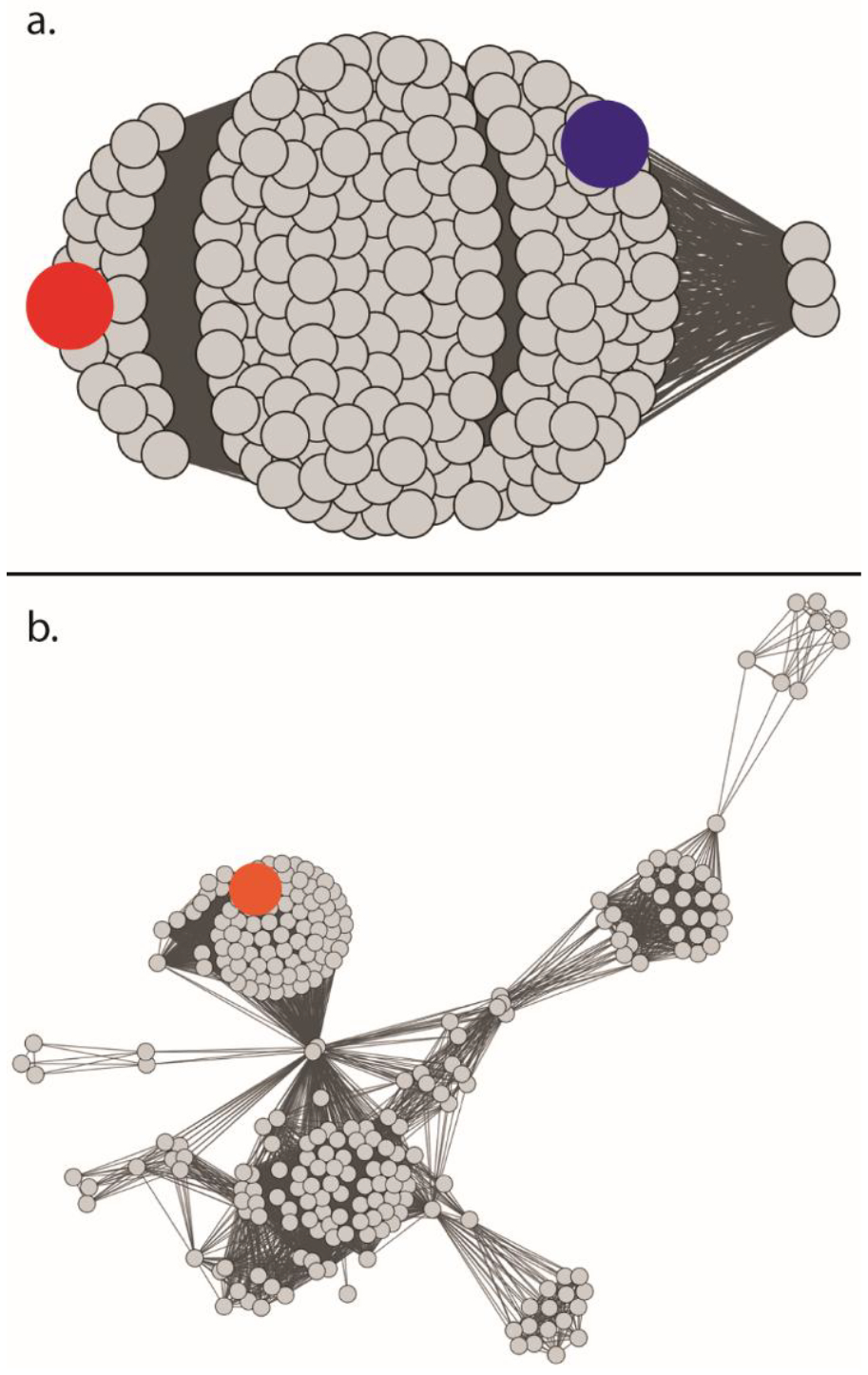
Sequence similarity networks of homologs to Cj1427 and DdahA. (a) Sequence similarity network of the 208 homologs of Cj1427 and WcaG. The alignment value cutoff is set to 98% identity. The sequences for Cj1427 and WcaG are shown as the blue and red nodes, respectively. (b) Sequence similarity network of the 265 homologues of DdahA. The alignment value cutoff was set to 96% identity. The sequence for DdahA is shown as the orange node.
Interestingly, 92 out of the 484 genomes (19%) contain homologs to both Cj1427 and DdahA. A total of 24% of the genomes (116 out of 484) only contain homologs to Cj1427 and 36% of the genomes (173 out of 484) only contain homologs to DdahA (Figure 2). A pattern emerges when the known structures of the heptose residues are mapped to the presence of Cj1427 and DdahA homologs. Strains of C. jejuni that contain CPS structures that possess both 6-deoxy- and 6-hydroxy-heptose moieties have homologs to both Cj1427 and DdahA (Figure 2, purple). Strains of C. jejuni that utilize only the 6-deoxy-heptose moiety contain only homologs to DdahA (Figure 2, blue). There is a single known CPS structure that contains only the 6-hydroxy-heptose residue from C. jejuni NCTC 11168 (HS:2) with Cj1427 (Figure 2, red). However, from this analysis, there are 115 genomes that contain only a homologue to Cj1427 whose CPS structures are currently unknown, but are expected to contain only 6-hydroxy-heptose residues in their CPS structures as they lack a homolog to DdahA in their genome.
Figure 2:
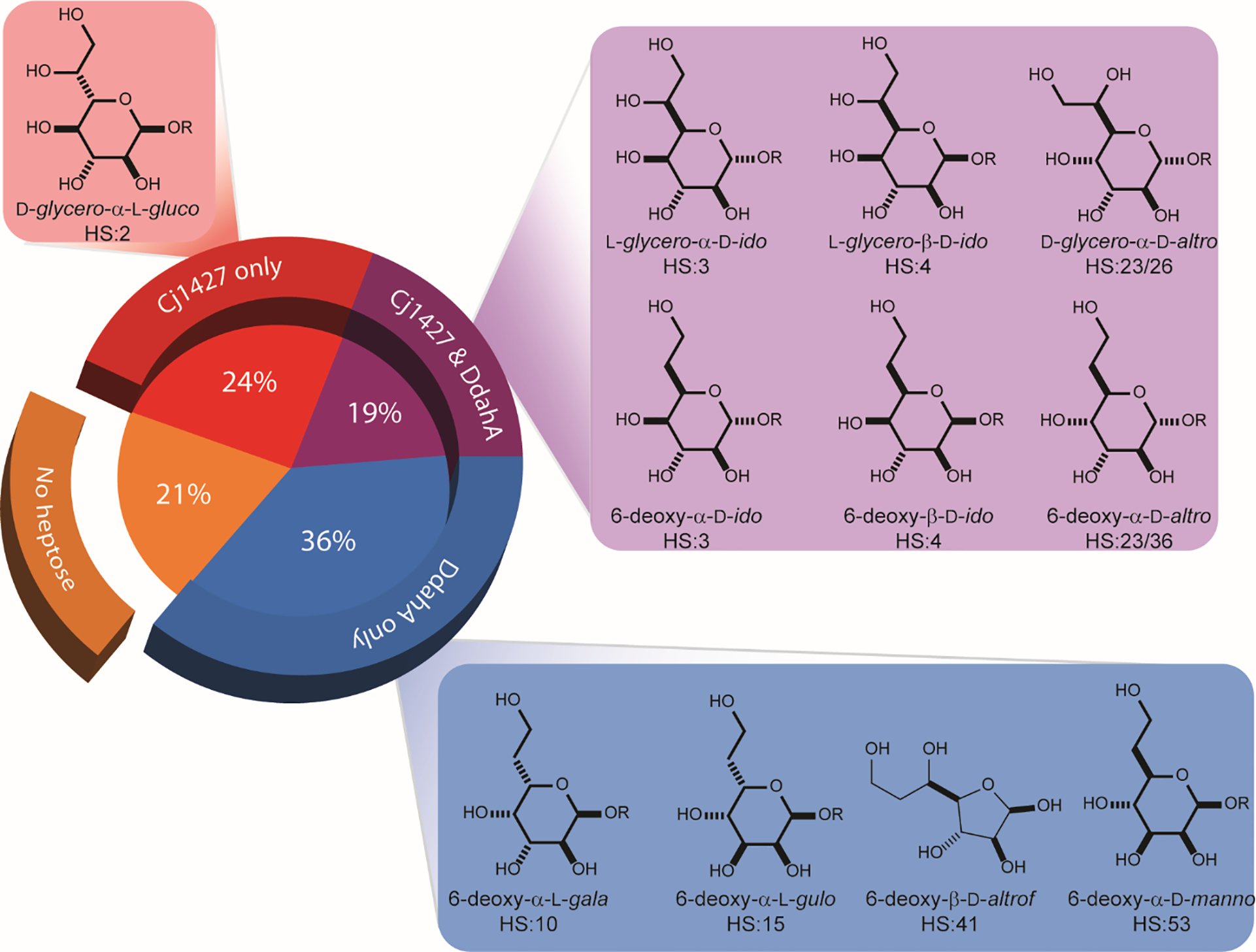
Distribution of heptose structures versus Cj1427 and DdahA homologs among 484 C. jejuni genomes.
Cj1427 Catalyzed Oxidation of GDP-d-glycero-α-d-manno-heptose.
α-Ketoglutarate is required to oxidize the tightly bound NADH of Cj1427 to NAD+ and L-2-hydroxyglutarate (7). The reactivity of GDP-d-glycero-α-d-manno-heptose (1) with Cj1427 was initially addressed by mass spectrometry (Figure 3). Cj1427 was first incubated with GDP-d-glycero-α-d-manno-heptose in the presence of a 10-fold excess of α-ketoglutarate. The mass spectrum of the reaction mixture shows two sets of peaks at an m/z of 634.08 and 632.07 Da. One set is identical to the control of GDP-d-glycero-α-d-manno-heptose alone, with a peak at 634.08 Da for M–H+ and observed isotopic peaks at 635.09 Da and 636.09 Da (Figure 3a). The other set represents the oxidized GDP-d-glycero-α-d-lyxo-heptose product, indicated by the loss of 2 Da yielding a peak at 632.07 Da for M–H+ and a single observable isotopic peak at 633.07 Da (Figure 3b).
Figure 3.
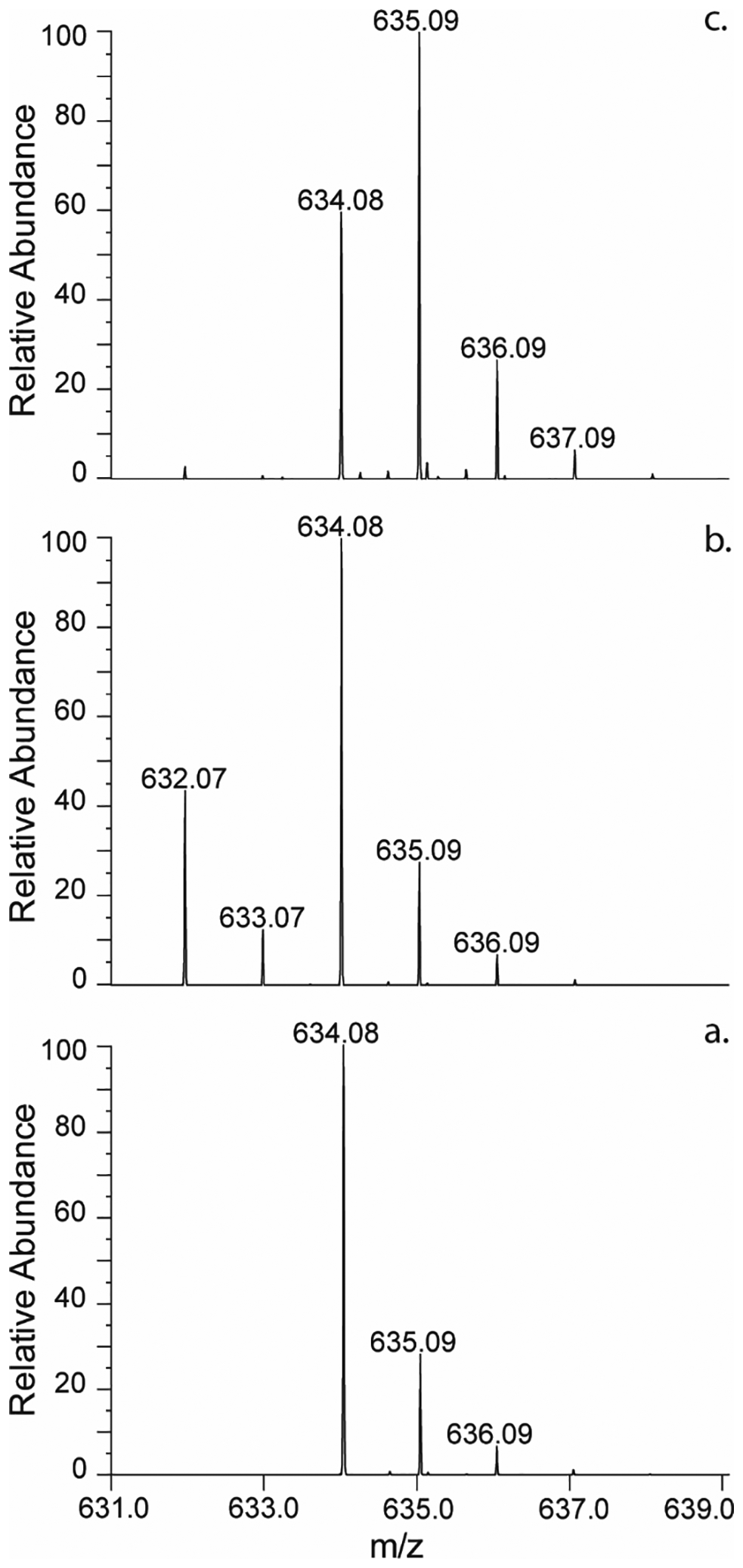
Negative ESI mass spectra of GDP-d-glycero-α-d-manno-heptose (1) with Cj1427. (a) Control mass spectrum of GDP-d-glycero-α-d-manno-heptose (m/z = 634.08 Da for M–H+). (b) Mass spectrum of the reaction mixture containing Cj1427 (20 μM), α-ketoglutarate (20 mM), and GDP-d-glycero-α-d-manno-heptose (2.0 mM) in 50 ammonium bicarbonate buffer, pH 8.0. The identified ions correspond to unreacted GDP-d-glycero-α-d-manno-heptose (m/z = 634.08 Da for M–H+) and the 4-keto product, GDP-d-glycero-4-keto-α-d-lyxo-heptose (m/z = 632.07 Da for M–H+). (c) Mass spectrum of the reaction containing Cj1427 (20 μM), α-ketoglutarate (0.5 mM), [2-2H]-d,l-2-hydroxyglutarate (20 mM) and GDP-d-glycero-α-d-manno-heptose (2.0 mM) in 50 mM ammonium bicarbonate buffer, pH 8.0. The identified ions correspond to unreacted GDP-d-glycero-α-d-manno-heptose (m/z = 634.08 Da for M–H+) and [2H]-GDP-d-glycero-α-d-manno-heptose (m/z = 635.09 Da for M–H+).
In the second experiment, Cj1427 was incubated with GDP-d-glycero-α-d-manno-heptose (1), α-ketoglutarate and a 10-fold excess of [2-2H]-d,l-2-hydroxyglutarate. The mass spectrum of the reaction products shows a peak for unreacted GDP-d-glycero-α-d-manno-heptose at 634.08 Da for M–H+ and a second peak for [2H]-GDP-d-glycero-α-d-manno-heptose at 635.09 Da for M–H+. The corresponding isotopic peaks appear at 636.09 Da and 637.09 Da. These results are indicative of deuterium incorporation into GDP-d-glycero-α-d-manno-heptose obtained via the transfer of deuterium from [2-2H]-d,l-2-hydroxyglutarate to NAD+ forming deuterated NAD(D) and subsequent reduction of the oxidized GDP-manno-heptose product.
The mass spectrometric analysis demonstrates that Cj1427 catalyzes the oxidation of GDP-d-glycero-α-d-manno-heptose (1) in the presence of α-ketoglutarate and can exchange a single hydrogen with deuterium from [2-2H]-d,l-2-hydroxyglutarate. To help quantify the oxidation reaction and identify the specific proton exchanged with deuterium, the same reactions were monitored by 1H NMR spectroscopy. The portion of the 1H NMR spectrum showing the anomeric hydrogens at C1 of the ribose and heptose portions of GDP-d-glycero-α-d-manno-heptose is presented in Figure 4a (22, 24, 25). When Cj1427 is added to GDP-d-glycero-α-d-manno-heptose in the presence of a 10-fold excess of α-ketoglutarate, a broad doublet peak appears with a chemical shift of 5.54 ppm (Figure 4b). After one hour the area of this new resonance is 0.3 compared to the anomeric proton from the ribose ring (5.86 ppm). There is an equivalent reduction to 0.7 for the anomeric proton of the heptose ring of the unreacted starting material (5.42 ppm).
Figure 4.
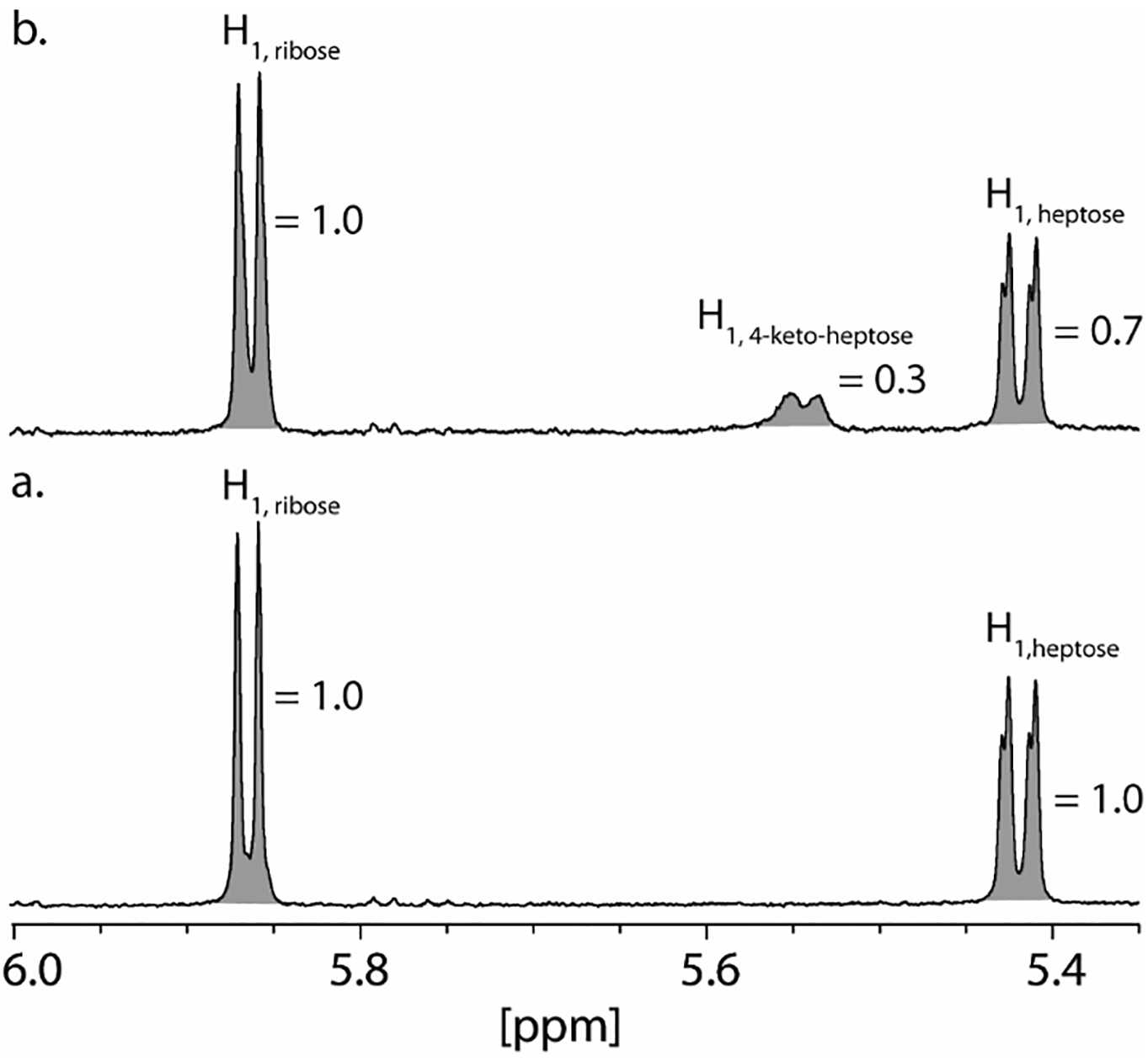
Portion of the 1H NMR spectrum of GDP-d-glycero-α-d-manno-heptose (1) and GDP-d-glycero-4-keto-α-d-lyxo-heptose (2). (a) Control 1H NMR spectrum of GDP-d-glycero-α-d-manno-heptose. (b) 1H NMR spectrum of GDP-d-glycero-α-d-manno-heptose (2.0 mM) after the addition of Cj1427 (20 μM) for 1 h in the presence of 20 mM α-ketoglutarate in 50 mM potassium phosphate buffer, pH 8.0. Peak assignments were made from previously publications (22, 24, 25). Integrations are shown in shaded area for each resonance.
In a parallel experiment, Cj1427 was mixed with GDP-d-glycero-α-d-manno-heptose (1) in the presence of α-ketoglutarate and a 10-fold excess of [2-2H]-d,l-2-hydroxyglutarate (Figure 5). The 1H NMR spectrum of GDP-d-glycero-α-d-manno-heptose is illustrated in Figure 5a where the sum of the integrals for H2 and H6 are collectively assigned a value of 2.0 at 3.91 to 3.96 ppm. Between 3.64 and 3.72 ppm are the mixed resonances for the 2 protons from C7 and the single proton from C4 with a relative integral sum of 2.9. After Cj1427 is added to an excess of [2-2H]-d,l-2-hydroxyglutarate in the presence of α-ketoglutarate, the resonance corresponding to hydrogen at C4 of GDP-d-glycero-α-d-manno-heptose is diminished significantly and replaced with deuterium from [2-2H]-l-2-hydroxyglutarate (Figure 5b). The integral, relative to that observed for the sum of H2 and H6 is 1.7. In addition, the resonances for the protons corresponding to H3 and H5 (3.80 to 3.85 ppm) of GDP-d-glycero-α-d-manno-heptose show the loss of spin coupling with the hydrogen that was originally at C4. Collectively, these experiments demonstrate unequivocally that Cj1427 catalyzes the oxidation of GDP-d-glycero-α-d-manno-heptose at C4 in the presence of added α-ketoglutarate.
Figure 5.
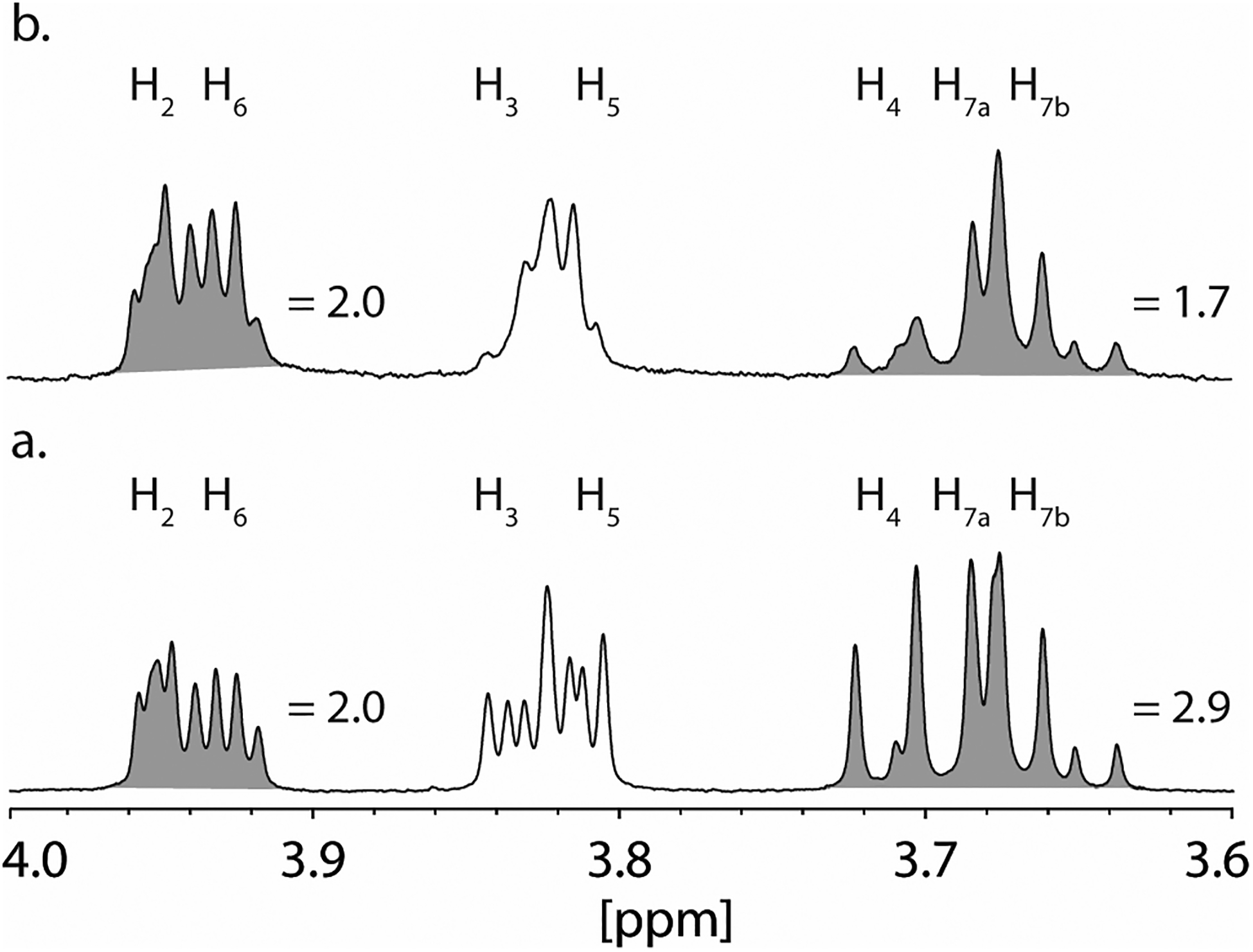
1H NMR spectra of GDP-d-glycero-α-d-manno-heptose and [4-2H]-GDP-d-glycero-α-d-manno-heptose. (a) 1H NMR spectrum of GDP-d-glycero-α-d-manno-heptose. Resonance assignments were made from previously publications (22, 24, 25). (b) 1H NMR spectrum of GDP-d-glycero-α-d-manno-heptose (2.0 mM) after the addition of Cj1427 (20 μM) in the presence of 0.5 mM α-ketoglutarate, and 20 mM [2-2H]-d,l-2-hydroxyglutarate in 50 mM potassium phosphate buffer, pH 8.0 for 50 min. Integrations are shown in shaded area under the resonances.
Determination of Steady-State Kinetic Constants for Cj1427.
The steady-state kinetic constants for Cj1427 were determined using a coupled assay with Cj1430 and Cj1428 in the presence of α-ketoglutarate. Cj1430 was previously shown to catalyze the epimerization of C3 and C5 in the transformation of GDP-6-deoxy-4-keto-α-d-lyxo-heptose (5) to GDP-6-deoxy-4-keto-β-l-xylo-heptose (6) (6). Cj1428 was also demonstrated to catalyze the reduction of this product to GDP-6-deoxy-β-l-gluco-heptose (7) (6). Cj1428 behaves differently than Cj1427, utilizing exogenous NADPH to catalyze the reduction reaction during formation of the final product (Scheme 2). Using these enzymes, the kinetic constants of Cj1427 were determined by following the conversion of NADPH to NADP+ with an excess of Cj1430 and Cj1428 at 340 nm. The steady-state kinetic constants for Cj1427 are summarized in Table 1.
Table 1.
Steady-state kinetic parameters for Cj1427a
| Enzyme | Substrate | kcat (s−1) | Km (μM) | kcat/Km (M−1 s−1) |
|---|---|---|---|---|
| Cj1427 | GDP-d-glycero-α-d-manno-heptoseb | 0.64 ± 0.02 | 63 ± 5 | (10.2 ± 0.8) × 103 |
| Cj1427 | α-ketoglutaratec | 0.55 ± 0.01 | 130 ± 8 | (4.3 ± 0.3) × 103 |
Errors were calculated from the standard deviation of the fitting results. Reactions were monitored by UV coupled assay at 340 nm in 50 mM HEPES/KCl, pH 7.4 at 30 °C in the presence of 300 μM NADPH and 10 μM Cj1430 and Cj1428.
The concentration of GDP-d-glycero-α-d-manno-heptose was varied with a fixed concentration of α-ketoglutarate (1.0 mM).
Concentration of α-ketoglutarate was varied with a fixed concentration GDP-d-glycero-d-manno-heptose (0.5 mM).
The coupled enzyme assay demonstrated that the addition of Cj1430 is required for observation of the catalytic activity of Cj1428, agreeing with previously published observations (6). Cj1427 has an apparent kcat of between 0.55 and 0.66 s−1. The observed Km for GDP-d-glycero-α-d-manno-heptose (1) is about 2-fold less than that for α-ketoglutarate, yielding a slightly higher kcat/Km value of (10.2 ± 0.8) × 103 M−1 s−1 compared to (4.3 ± 0.3) × 103 M−1 s−1
Stopped-Flow Kinetics of E-NADH Oxidation by α-Ketoglutarate.
To better understand the rate-limiting step for the reaction catalyzed by Cj1427, a single turnover experiment was conducted by measuring the oxidation of the NADH that was tightly bound within the active site of Cj1427 to NAD+ after the binding of α-ketoglutarate. The reaction was followed during the transient kinetic time scale by monitoring the changes in fluorescence over time. After mixing Cj1427•NADH with α-ketoglutarate, there was a rapid loss of fluorescence that reaches a final plateau in ~0.5 s. Due to this rapid change in fluorescence, the instrument was set to collect equal points (1000 points each) during the first 50 ms and the last 450 ms. The first 50 ms were best approximated by a single exponential as illustrated in Figure 6a. The concentration dependence of the observed rate constant is hyperbolic (Figure 6b).
Figure 6.
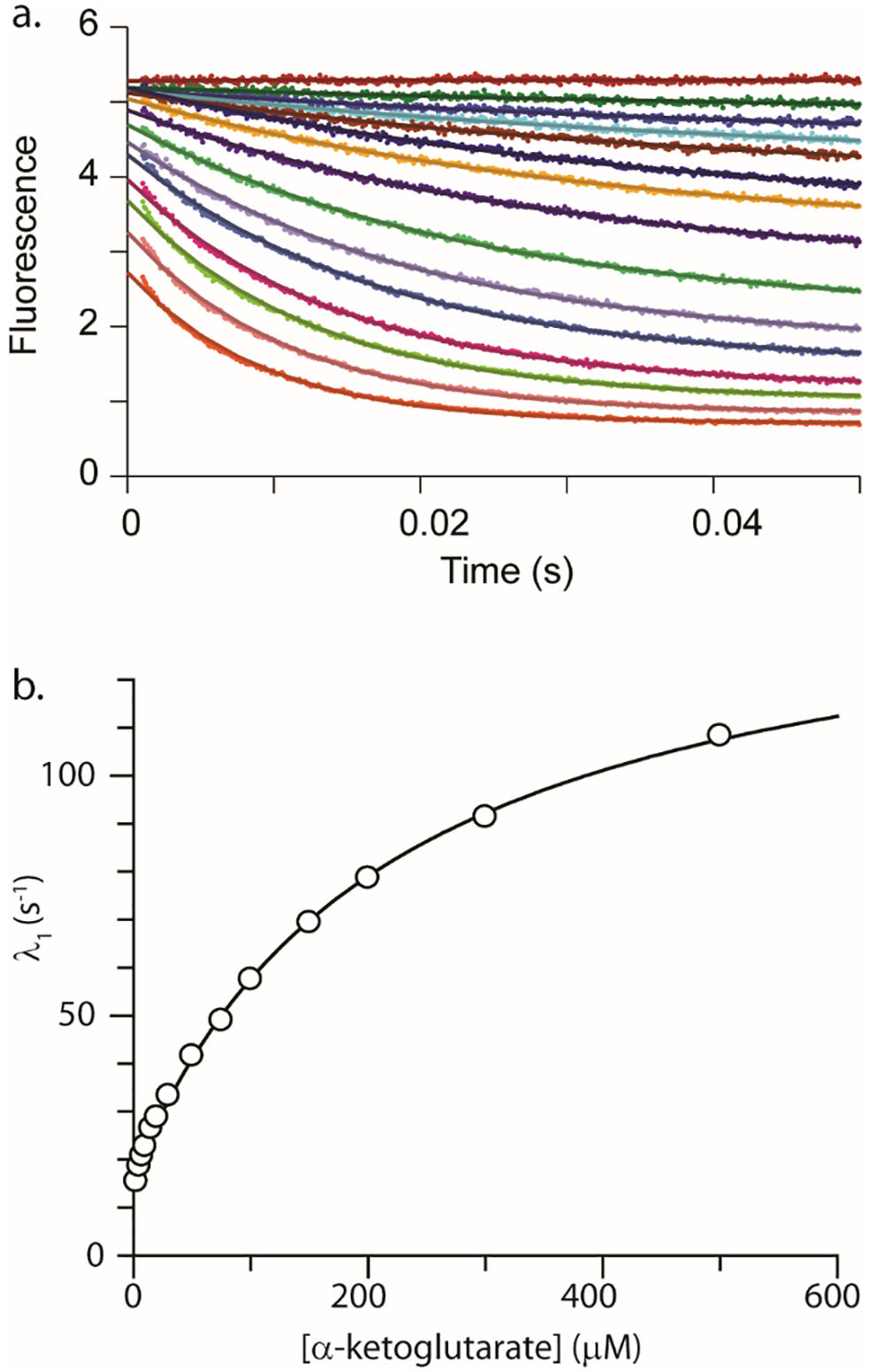
Stopped-flow fluorescence of Cj1427-NADH after the addition of α-ketoglutarate. (a) Stopped-flow fluorescence traces (0.05 s) showing Cj1427•NADH conversion to Cj1427•NAD+ with various amount of α-ketoglutarate. The data were fit to a single exponential (equation 2) shown as solid lines. Data were collected using 15 μM Cj1427 (final concentration) with various concentrations of α-ketoglutarate in 50 mM HEPES/KCl buffer, pH 7.4. The concentrations of α-ketoglutarate are 0, 2.5, 5, 7.5, 10, 15, 20, 30, 50, 75, 100, 150, 200, 300, 500 μM. (b) Observed rate constant (λ1) from the 0.05 s stopped-flow data for Cj1427 reacting with α-ketoglutarate plotted versus the concentration of α-ketoglutarate. The solid line is the fit of the data to equation 3.
This relationship suggests a minimal two-step model (Scheme 4) with a rapid equilibrium step (K1 = k1/k2) followed by a rate-limiting step governed by a forward (k3) and reverse step (k4)(18, 19). Analysis of the concentration dependence of the observed rate constant yields an apparent K1 = .0046 μM (Kd = k2/k1 = 217 μM) and a maximum rate of change in fluorescence of ~146 s−1 (k3 + k4). The well-defined intercept yields an estimate for k4 = 16 s−1 and thus k3 = 130 s−1.
Kinetic Model for Fitting by Simulation.
The kinetic model used for fitting by simulation was developed using the conventional analysis of the data. Initially, the data were fit to a single exponential, yielding a minimal two-step model with estimates for K1, k3, and k4. Initial attempts at fitting by simulation using the model provided by Scheme 4 could not account for the data in its entirety. Simply increasing the individual rate constants resulted in changes to the overall equilibrium defined by the spacing of the traces or changes to observed rate of fluorescence decay. The initial model failed to account for the very rapid loss of fluorescence signal in the first 10 ms, most of which occurs in the dead time of the instrument for the higher concentrations of α-ketoglutarate, resulting in a lack of data to define this phase. Attempts at fitting to higher order exponential equations did not improve these results.
To remedy this problem, the original kinetic model was expanded to include a change in the fluorescence of NADH upon binding α-ketoglutarate, where the fluorescence (F) of the tertiary complex Cj1427•NADH•αKG is different from the binary complex Cj1427•NADH (Fo). The UV spectrum for NADH bound to Cj1427 is red-shifted, leading to a maximum absorbance of 350 nm, instead of 340 nm as found for free NADH (7). Therefore, it is reasonable to allow for the fluorescence of the Cj1427•NADH•αKG ternary complex to be different than the Cj1427•NADH complex, as changes occur to the active site structure upon binding α-ketoglutarate.
One additional constraint was introduced to yield the final fit. As indicated by the conventional analysis, there is insufficient data to define the individual rate constants for k1 and k2. To account for the rapid equilibrium in the fit by simulation, the value for k1 was held fixed at the lower limit required by the data to simulate the loss of fluorescence signal within the dead-time of the instrument, a value equal to 5.5 × 106 M−1 s−1. This value is only limited by diffusion (~1 × 108 M−1 s−1) with the corresponding adjusted values of k2. As a result, the optimal values for k2 are defined with respect to the set value of k1, therefore only actually represent a value of Kd, which is constrained by the data.
Fitting the data to the slightly modified model shows an improved fit to the data than the single exponential equation, especially during the early phase of the course (<10 ms) and yields well-constrained values for the dissociation constant (Kd) for α-ketoglutarate, the individual rate constants k3 and k4, and the change in fluorescence (ΔF) for the Cj1427•NADH•α-KG tertiary complex (Figure 7). The best values for the individual rate constants are summarized in Table 2. Error ranges were provided by FitSpace confidence contour analysis and represent the upper and lower limits of a 10% threshold on the summation of the squared error (Figure 8) (26).
Figure 7.
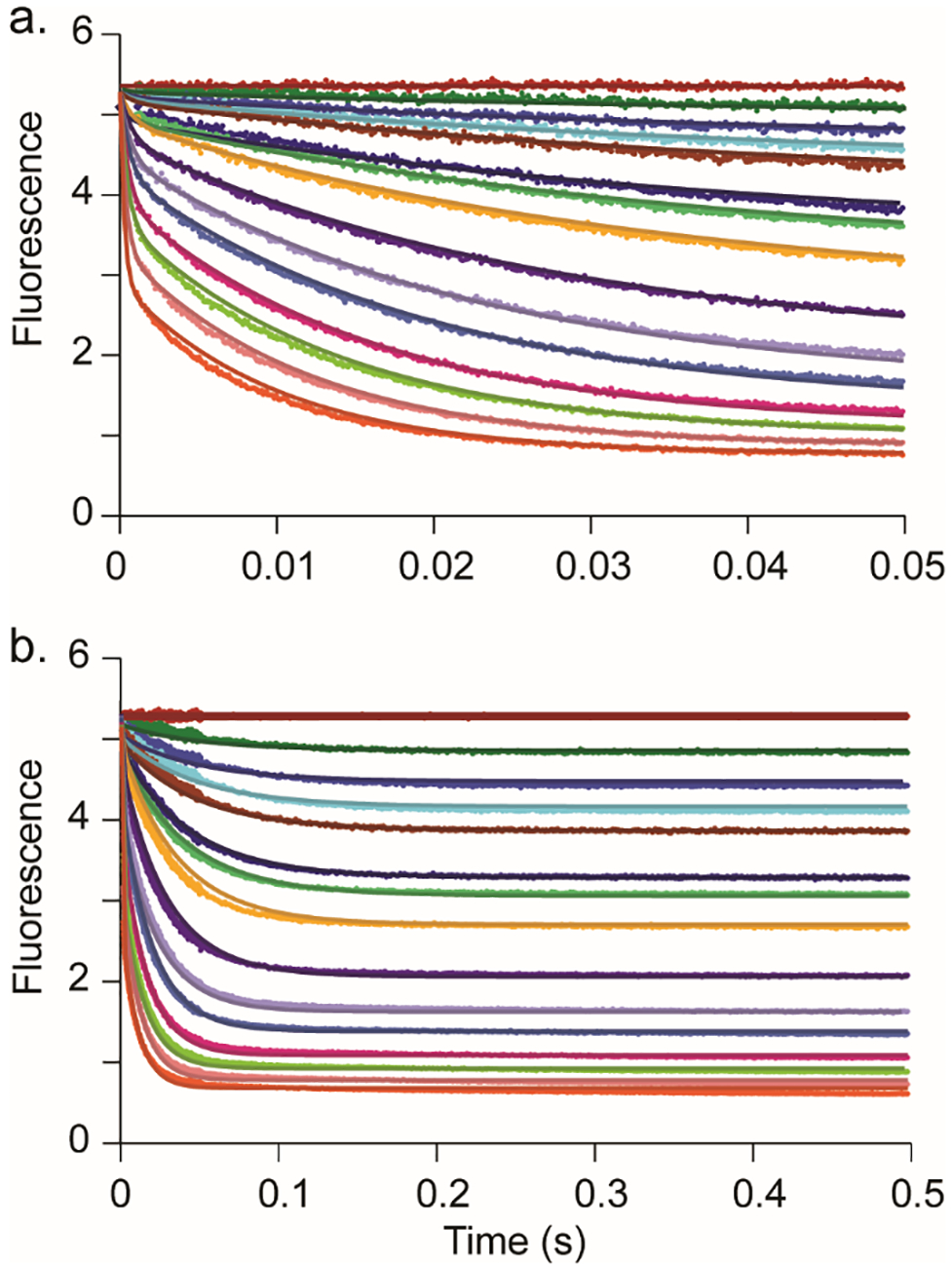
Fit of stopped-flow traces by simulation. (a) Stopped-flow fluorescence traces (0.05 s) monitoring the changes in fluorescence of Cj1427 bound NADH conversion to NAD+ with various α-ketoglutarate concentrations. (b) Stopped-flow fluorescence traces (0.5 s) monitoring the changes in fluorescence of Cj1427 bound NADH conversion to NAD+ with various α-ketoglutarate concentrations. The solid lines show the simultaneous fit by simulation to the model shown in Scheme 4. The concentrations of α-ketoglutarate are 0, 2.5, 5, 7.5, 10, 15, 20, 30, 50, 75, 100, 150, 200, 300, 500 μM. Data were collected using 15 μM Cj1427 (final concentration) with α-ketoglutarate in 50 mM HEPES/KCl buffer, pH 7.4. Colors are maintained in both panels.
Table 2.
Individual Rate Constants and Errorsa
| Lower limitb | Upper Limitb | % rangec | Best fit | |
|---|---|---|---|---|
| f/f0d | 0.23 | 0.349 | 21% | 0.29 |
| Kde (μM) | 140 | 200 | 18% | 165 |
| k3 (s−1) | 97.2 | 140 | 18% | 116 |
| k4 (s−1) | 11.5 | 14.2 | 11% | 12.8 |
The data are fit globally to the mechanism shown in Scheme 4.
The upper and lower limits reflect a threshold of a 10% deviation from the minimal SSE in the confidence contours. FitSpace error confidence contours are shown in Figure 8 (19).
The percentage range was calculated by dividing the mean of the range by the best fit value as (upper − lower)/(2 × best fit). This reflects the allowable variation of each best fit value as a percentage.
The fluorescence scaling factor is shown as the fractional change in enzyme fluorescence (see the text).
The value for k1 (α-ketoglutarate binding) was assumed to be fast and held fixed at a lower limit of 5.5 × 106 M−1 s−1, so fitting to derive k2 defined only Kd.
Figure 8.
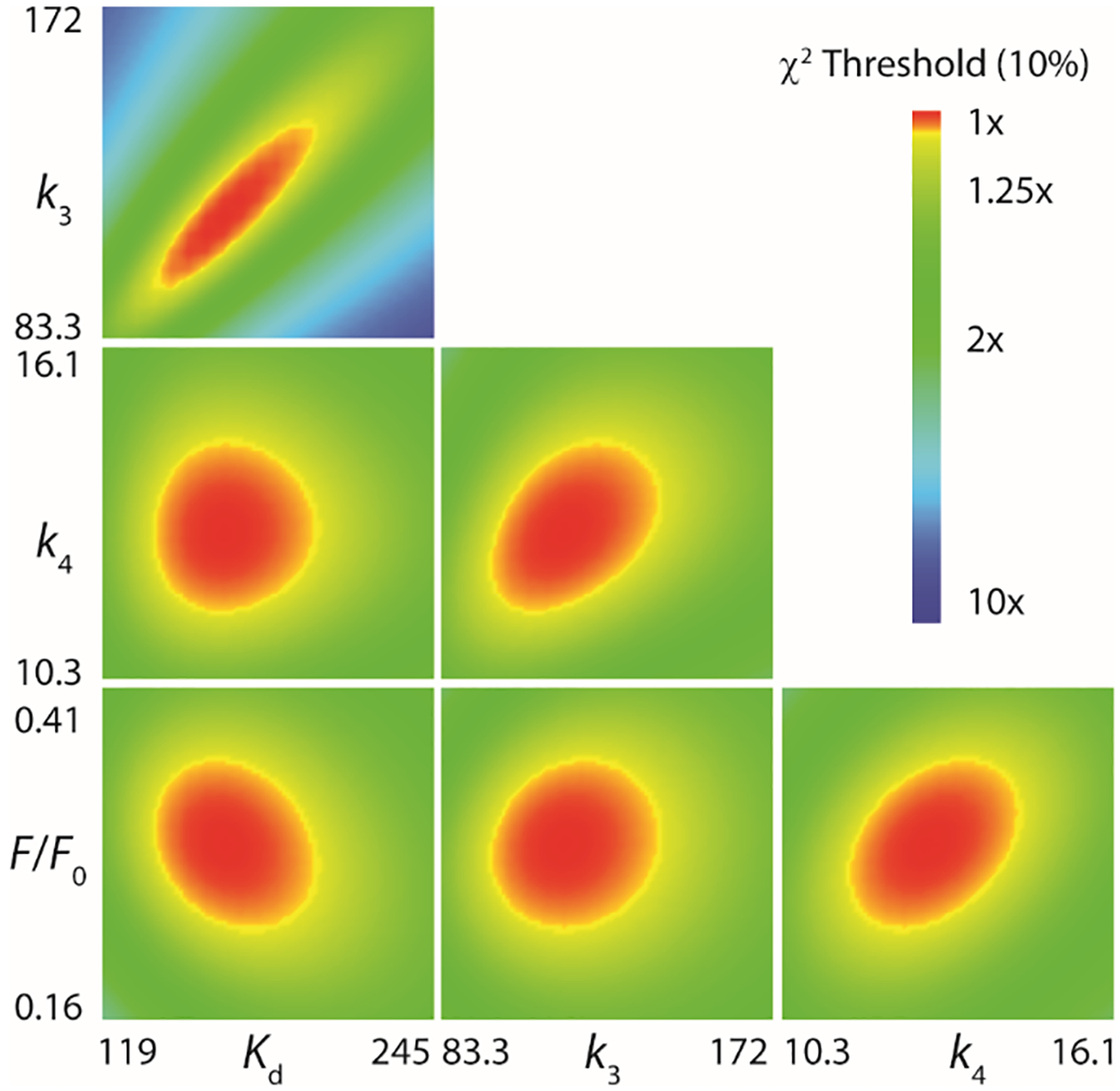
FitSpace confidence contours from the fit by simulation for the reaction of Cj1427•NADH with α-ketoglutarate (26). FitSpace confidence contours (10% χ2) for the fit of the kinetic data shown in the Figure 8 and Scheme 4. Included is the observed change in the fluorescence for the species Cj1427•NADH•αKG, relative to that for Cj1427•NADH. Optimal values and error ranges are provided in Table 2.
Discussion
Currently, there are 13 known heptoses contained within the various C. jejuni CPS structures (3, 4). Twelve of these heptoses can be derived directly from GDP-d-glycero-α-d-manno-heptose (1) by modifying the stereochemistry at C3, C4, C5 or C6. The biosynthetic pathways to generate the 6-deoxy heptose derivatives are known based on the characterization of the pathway for GDP-6-deoxy-α-d-altro-heptose (22). However, the biosynthesis of the heptose moieties with a hydroxylated C6 remain elusive due to an apparent lack of an enzyme that is known to catalyze the formation of the critical GDP-d-glycero-4-keto-d-lyxo-heptose (2) intermediate as shown in Scheme 1 (6, 7). The most likely candidate enzyme is Cj1427 since this enzyme was previously shown to catalyze the reduction of GDP-6-deoxy-4-keto-α-d-lyxo-heptose (5) to GDP-6-deoxy-α-d-manno-heptose (8) (6, 22). We have shown that Cj1427 co-purifies with tightly bound NADH enabling it to catalyze a reduction reaction, but not an oxidation reaction (7). We also showed that α-ketoglutarate can convert the bound NADH cofactor to NAD+, which can subsequently be used to catalyze the oxidation of l-2-hydroxyglutarate. Moreover, a crystal structure showed that Cj1427 is capable of binding GDP-d-glycero-α-d-manno-heptose and places it in a prime position to catalyze the oxidation of C4 due to its location near the nicotinamide cofactor (7).
Here we report an in-depth bioinformatic analysis of Cj1427 and the dehydratase, DdahA. This analysis shows that Cj1427 homologs are found in 208 out of 484 sequenced C. jejuni genomes and share a very high sequence conservation, >97%. Furthermore, Cj1427 and its homologs are exclusively found in C. jejuni species known to yield a heptose moiety with an intact hydroxyl group at C6 in the final CPS structure. Species that only have a 6-deoxy heptose contain only homologs to the dehydratase, DdahA. Many of the known CPS structures utilize 6-deoxy and 6-hydroxyl heptoses and contain homologs to both Cj1427 and DdahA in their gene clusters. This includes the well-characterized strain C. jejuni 81–176 (HS:23/36) in which the biosynthesis of GDP-6-deoxy-α-d-altro-heptose is well described, but the pathway for the formation of GDP-d-glycero-α-d-altro-heptose has remained elusive (22).
In the presence of α-ketoglutarate, we show that Cj1427 catalyzes the oxidation of GDP-d-glycero-α-d-manno-heptose by mass spectrometry and NMR spectroscopy. There is a loss of 2 Da in the mass spectrum and a new resonance corresponding to the anomeric proton on the heptose ring of GDP-d-glycero-4-keto-α-d-lyxo-heptose. We further show that Cj1427 catalyzes the exchange of a single proton with deuterium from [2-2H]-d,l-2-hydroxyglutarate and confirm the proton is at C4 by 1H NMR spectroscopy. Using a coupled kinetic assay with Cj1430 and Cj1428, we were able to measure the maximum turnover rate for Cj1427 to be ~0.6 s−1 and a kcat/Km for GDP-d-glycero-α-d-manno-heptose to be 1 × 104 M−1 s−1. Compared to the other measured steady-state kinetic constants for enzymes involved in the biosynthesis of GDP-d-glycero-α-d-manno-heptose (i.e. Cj1424, Cj1425, Cj1152, and Cj1423), Cj1427 with GDP-d-glycero-α-d-manno-heptose has the highest specificity constant and the second fastest kcat (3). We have shown that Cj1427 utilizes a ping-pong mechanism for the oxidation of GDP-d-glycero-α-d-manno-heptose followed by reduction of α-ketoglutarate to recycle the bound NADH cofactor.
Stopped-flow studies with Cj1427 were used to further investigate the kinetics of the recycling mechanism with α-ketoglutarate. Fitting the data with both conventional and simulation methods show that the oxidation of NADH to NAD+ by α-ketoglutarate is very fast (>100 s−1). This rate constant is more than 2 orders of magnitude greater than the overall kcat for Cj1427. This suggests that a step associated with the oxidation of GDP-d-glycero-α-d-manno-heptose is the rate-limiting step of the mechanism, while the recycling of the cofactor back to NAD+ occurs quite rapidly. The stopped-flow data also suggest a rapid equilibrium for the formation of the Cj1427•NADH•α-KG ternary complex. These results clearly indicate that Cj1427 is the “previously missing” enzyme for the oxidation of GDP-d-glycero-α-d-manno-heptose (1) to the GDP-d-glycero-4-keto-α-d-lyxo-heptose (2). This is believed to be the essential first step to enable the stereochemical modifications to occur subsequently at C3, C4, and C5. The complete depiction of the reaction catalyzed by Cj1427 is presented in Scheme 6.
Scheme 6:
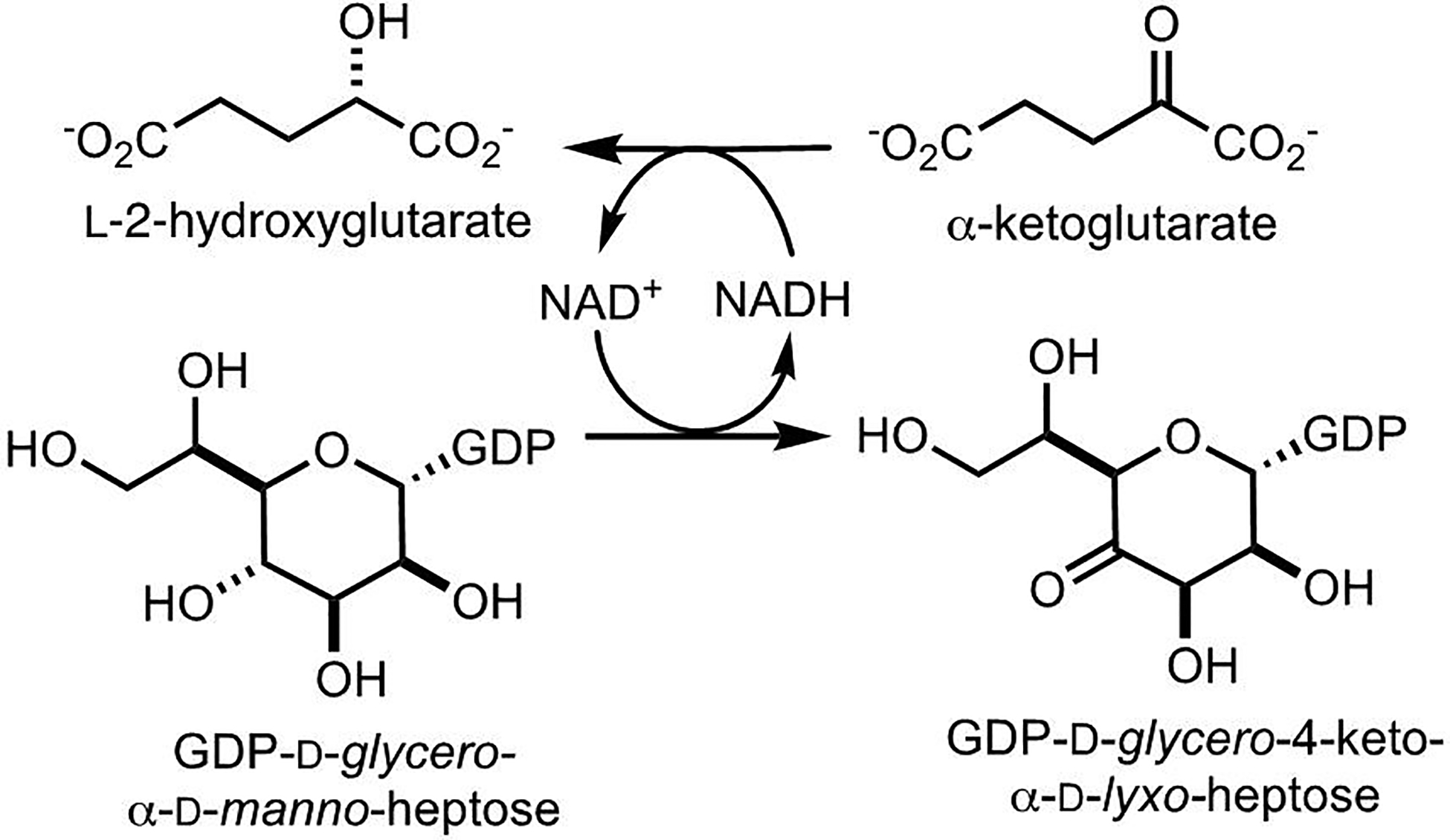
Mechanism for the oxidation of GDP-d-glycero-α-d-manno-heptose to GDP-d-glycero-4-keto-α-d-lyxo-heptose by α-ketoglutarate via tightly bound NADH.
Compared to other enzymes that are known to recycle a bound NADH cofactor with α-ketoglutarate, Cj1427 shares very little sequence identity to PdxB, SerA, or WlbA (7). For these enzymes the α-ketoglutarate co-substrate requirement for their catalytic activity was not obvious but for the fact that subsequent enzymes produced α-ketoglutarate as part of an aminotransferase reaction via l-glutamate (17, 28–30). It is highly likely that there are other “inactive” dehydrogenase activities that function similarly with no obvious signature for a required co-substrate. In conclusion, we have shown that Cj1427 is responsible for catalyzing the oxidation at C4 of GDP-d-glycero-α-d-manno-heptose (1). Cj1427 and its homologs are required for the biosynthesis of heptose residues which contain a 6-hydroxyl group within the capsular polysaccharides of C. jejuni.
CONCLUSIONS
The enzyme Cj1427 from Campylobacter jejuni NCTC11168 catalyzes the oxidation of GDP-d-glycero-α-d-manno-heptose to GDP-d-glycero-4-keto-α-d-lyxo-heptose in the presence of added α-ketoglutarate. This result demonstrates for the first time how GDP-d-glycero-4-keto-α-d-lyxo-heptose is made as a precursor for subsequent modifications to C6, C5, C4, and C3 of the heptose moiety in the capsular polysaccharides within the various strains of C. jejuni. α-Ketoglutarate is required to recycle tightly bound NADH after each catalytic cycle and thus this enzyme functions as a rare NAD+/NADH-dependent dehydrogenase with a ping pong reaction mechanism.
Acknowledgements
We thank Professor Thomas D. Meek for the use of his stopped-flow spectrophotometer.
Funding
This work was supported in parts by grants from the Robert A. Welch Foundation (A-840) and the National Institutes of Health (GM122825).
Footnotes
Accession Codes
Cj1427 Q0P8I7
The authors declare no competing financial interest.
REFERENCES
- (1).García-Sánchez L, Melero B, and Rovira J (2018) Campylobacter in the Food Chain, In Advances in Food and Nutrition Research (Rodríguez-Lázaro D, Ed.), pp 215–252, Academic Press. [DOI] [PubMed] [Google Scholar]
- (2).Burnham PM, and Hendrixson DR (2018) Campylobacter jejuni: collective components promoting a successful enteric lifestyle, Nat. Rev. Microbiol, 16, 551–565. [DOI] [PubMed] [Google Scholar]
- (3).Huddleston JP, and Raushel FM (2019) Biosynthesis of GDP-d-glycero-α-d-manno-heptose for the Capsular Polysaccharide of Campylobacter jejuni, Biochemistry, 58, 3893–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Monteiro MA, Noll A, Laird RM, Pequegnat B, Ma Z, Bertolo L, DePass C, Omari E, Gabryelski P, Redkyna O, Jiao Y, Borrelli S, Poly F, and Guerry P (2018) Campylobacter jejuni capsule polysaccharide conjugate vaccine, In Carbohydrate-based vaccines: from concept to clinic, pp 249–271, American Chemical Society. [Google Scholar]
- (5).Michael FS, Szymanski CM, Li J, Chan KH, Khieu NH, Larocque S, Wakarchuk WW, Brisson J-R, and Monteiro MA (2002) The structures of the lipooligosaccharide and capsule polysaccharide of Campylobacter jejuni genome sequenced strain NCTC 11168, Eur. J. Biochem, 269, 5119–5136. [DOI] [PubMed] [Google Scholar]
- (6).McCallum M, Shaw GS, and Creuzenet C (2013) Comparison of predicted epimerases and reductases of the Campylobacter jejuni D-altro- and l-gluco-heptose synthesis pathways, J. Biol. Chem, 288, 19569–19580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Huddleston JP, Anderson TK, Spencer KD, Thoden JB, Raushel FM, and Holden HM (2019) Structural analysis of Cj1427, an Essential NAD-dependent Dehydrogenase for the Biosynthesis of the Heptose Residues in the Capsular Polysaccharides of Campylobacter jejuni, Biochemistry, 59, 0000–0000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kitts PA, Church DM, Thibaud-Nissen F, Choi J, Hem V, Sapojnikov V, Smith RG, Tatusova T, Xiang C, Zherikov A, DiCuccio M, Murphy TD, Pruitt KD, and Kimchi A (2016) Assembly: a resource for assembled genomes at NCBI, Nucleic Acids Res, 44, D73–D80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL (2009) BLAST+: architecture and applications, BMC Bioinform, 10, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, and Whalen KL (2015) Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks, Biochim. Biophys. Acta, Proteins Proteomics, 1854, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks, Genome. Res, 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Huddleston JP, Thoden JB, Dopkins BJ, Narindoshvili T, Fose BJ, Holden HM, and Raushel FM (2019) Structural and functional characterization of YdjI, an aldolase of unknown specificity in Escherichia coli K12, Biochemistry, 58, 3340–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Inoue H, Nojima H, and Okayama H (1990) High efficiency transformation of Escherichia coli with plasmids, Gene, 96, 23–28. [DOI] [PubMed] [Google Scholar]
- (14).Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- (15).Gasteiger E, Hoogland C, Gattiker A, Duvaud S. e., Wilkins MR, Appel RD, and Bairoch A (2005) Protein identification and analysis tools on the ExPASy server, In The Proteomics Protocols Handbook (Walker JM, Ed.), pp 571–607, Humana Press, Totowa, NJ. [Google Scholar]
- (16).Huddleston JP, and Raushel FM (2019) Functional characterization of YdjH, a sugar kinase of unknown specificity in Escherichia coli K12, Biochemistry, 58, 3354–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Thoden JB, and Holden HM (2010) Structural and functional studies of WlbA: A dehydrogenase involved in the biosynthesis of 2,3-diacetamido-2,3-dideoxy-D-mannuronic acid, Biochemistry, 49, 7939–7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Johnson KA (1992) Transient-State Kinetic Analysis of Enzyme Reaction Pathways, In The Enzymes (Sigman DS, Ed.), pp 1–61, Academic Press. [Google Scholar]
- (19).Huddleston JP, Schroeder GK, Johnson KA, and Whitman CP (2012) A Pre-Steady State Kinetic Analysis of the αY60W Mutant of trans-3-Chloroacrylic Acid Dehalogenase: Implications for the Mechanism of the Wild-Type Enzyme, Biochemistry, 51, 9420–9435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Huddleston JP, Wang SC, Johnson KA, and Whitman CP (2017) Resolution of the uncertainty in the kinetic mechanism for the trans-3-Chloroacrylic acid dehalogenase-catalyzed reaction, Archives of Biochemistry and Biophysics, 623–624, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Johnson KA, Simpson ZB, and Blom T (2009) Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data, Anal. Biochem, 387, 20–29. [DOI] [PubMed] [Google Scholar]
- (22).McCallum M, Shaw SD, Shaw GS, and Creuzenet C (2012) Complete 6-Deoxy-d-altro-heptose biosynthesis pathway from Campylobacter jejuni, J. Biol. Chem, 287, 29776–29788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).McCallum M, Shaw GS, and Creuzenet C (2011) Characterization of the dehydratase WcbK and the reductase WcaG involved in GDP-6-deoxy-d-manno-heptose biosynthesis in Campylobacter jejuni, Biochem. J, 439, 235–248. [DOI] [PubMed] [Google Scholar]
- (24).Butty FD, Aucoin M, Morrison L, Ho N, Shaw G, and Creuzenet C (2009) Elucidating the formation of 6-deoxyheptose: biochemical characterization of the GDP-d-glycero-d-manno-heptose C6 dehydratase, DmhA, and its associated C4 reductase, DmhB, Biochemistry, 48, 7764–7775. [DOI] [PubMed] [Google Scholar]
- (25).Kneidinger B, Graninger M, Puchberger M, Kosma P, and Messner P (2001) Biosynthesis of nucleotide-activated d-glycero-d-manno-heptose, J. Biol. Chem, 276, 20935–20944. [DOI] [PubMed] [Google Scholar]
- (26).Johnson KA, Simpson ZB, and Blom T (2009) FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data, Anal. Biochem, 387, 30–41. [DOI] [PubMed] [Google Scholar]
- (27).Mukherjee K, Huddleston JP, Narindoshvili T, Nemmara VV, and Raushel FM (2019) Functional Characterization of the ycjQRS Gene Cluster from Escherichia coli: A Novel Pathway for the Transformation of D-Gulosides to D-Glucosides, Biochemistry, 58, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Rudolph J, Kim J, and Copley SD (2010) Multiple turnovers of the nicotino-enzyme PdxB require alpha-keto acids as cosubstrates, Biochemistry, 49, 9249–9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhao G, and Winkler ME (1996) A novel α-ketoglutarate reductase activity of the serA-encoded 3-phosphoglycerate dehydrogenase of Escherichia coli K-12 and its possible implications for human 2-hydroxyglutaric aciduria, J. Bacteriol, 178, 232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Larkin A, and Imperiali B (2009) Biosynthesis of UDP-GlcNAc(3NAc)A by WbpB, WbpE, and WbpD: enzymes in the Wbp pathway responsible for O-antigen assembly in Pseudomonas aeruginosa PAO1, Biochemistry, 48, 5446–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]