

This review provides an overview of recent advances in understanding the interactions and signaling of PD-1 and its ligands.
Abstract
Programmed Death-1 (PD-1; CD279) is an inhibitory receptor induced in activated T cells. PD-1 engagement by its ligands, PD-L1 and PD-L2, maintains peripheral tolerance but also compromises anti-tumor immunity. Blocking antibodies against PD-1 or its ligands have revolutionized cancer immunotherapy. However, only a fraction of patients develop durable antitumor responses. Clinical outcomes have reached a plateau without substantial advances by combinatorial approaches. Thus, great interest has recently emerged to investigate, in depth, the mechanisms by which the PD-1 pathway transmits inhibitory signals with the goal to identify molecular targets for improvement of the therapeutic success. These efforts have revealed unpredictable dimensions of the pathway and uncovered novel mechanisms involved in PD-1 and PD-L1 regulation and function. Here, we provide an overview of the recent advances on the mechanistic aspects of the PD-1 pathway and discuss the implications of these new discoveries and the gaps that remain to be filled.
INTRODUCTION
Programmed death–1 (PD-1) was discovered in 1992 by T. Honjo and colleagues in Kyoto University as an apoptosis-associated gene. However, PD-1 overexpression was not required for apoptosis (1). Later studies from the same group identified that PD-1 expression was induced through signaling by antigen receptors of T and B lymphocytes (2) and was involved in the inhibition of immune responses, as PD-1–deficient mice developed autoimmune phenotypes (3–6). The discovery that the PD-1 ligands, PD-L1 (CD274; also known as B7-H1) and PD-L2 (CD273; also known as B7-DC), are expressed in cancer cells (7) and antigen-presenting cells (APCs) of the tumor microenvironment (TME) (8, 9), and the role of PD-1 as T cell inhibitory receptor of PD-L1 (10), led to the targeting of PD-1 and its ligands for induction of antitumor T cell responses. The development and usage of such blocking antibodies brought a revolution in cancer immunotherapy (11–13).
Preclinical studies and clinical trials generated with anti–CTLA-4 (cytotoxic T lymphocyte–associated protein 4) blocking compounds had a significant impact on the acceleration of clinical trials and U.S. Food and Drug Administration approval of compounds blocking PD-1 and its ligands. Antibodies blocking PD-1 and its ligands have reduced toxicity compared to CTLA-4 blocking compounds and efficacy in a broader spectrum of cancer types (14). These aspects combined provided an excellent opportunity for the administration of PD-1–based immunotherapy in the outpatient setting. In addition to being used as monotherapy, PD-1/PD-L1 blocking immunotherapy is currently administered in combination with other treatments such as blockade of checkpoint inhibitors (mainly CTLA-4), chemotherapy, targeted therapy with small-molecule inhibitors, cancer vaccines, or agonist antibodies. It should be noted that after monotherapy with PD-1 blocking compounds, only a fraction of patients develop durable clinical responses while most patients develop only transient responses or no responses at all.
However, although several different antibodies blocking PD-1 and its ligands are currently in clinical use, this small fraction of durable responses have been achieved with all of them in various types of cancer. This outcome indicates that targeting of this pathway has an inherently high potential to induce antitumor immunity. However, critical information is missing on how to assess the involvement of the PD-1 pathway in cancer-mediated immunosuppression and design its appropriate therapeutic exploitation required in each individual patient. These issues have sparked great interest during the past 5 years to investigate in depth the mechanisms by which the PD-1:PD-L1 pathway transmits inhibitory signals with the goal to identify new targets to intervene properly and enhance the therapeutic success.
EXPRESSION OF PD-1 AND ITS LIGANDS
PD-1 and its ligands are expressed in several cell types of the innate and adaptive immune system, and their expression pattern has been extensively studied [reviewed in (15) and (16)]. Although initial work focused on the transcriptional mechanisms that induce the expression of PD-1 and its ligands, recent studies determined that PD-1 and PD-L1 expression is also regulated at the posttranslational level (Fig. 1). CRISPR-based screening identified Fut8, a core fucosylase of PD-1–N-linked oligosaccharides at positions N49 and N74, to regulate cell-surface expression of PD-1 (Fig. 1A), and T cells treated with a cellular fucosylation inhibitor had stronger antitumor reaction in vivo (17). However, the precise biochemical mechanisms of how core fucosylation affects PD-1 structure and function remain to be elucidated. PD-1 expression may also be regulated by E3 ligase FBXO38, which mediates K48-linked polyubiquitination at the PD-1 K233 site and subsequent proteasome degradation (Fig. 1B) (18).
Fig. 1. Posttranslational modifications regulate PD-1 expression.
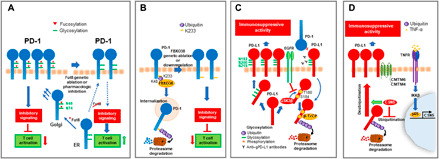
(A) In the endoplasmic reticulum (ER), PD-1 is glycosylated at residues N49 and N74, which are subsequently fucosylated (red triangle) in the Golgi apparatus, resulting in sustained expression of PD-1 at the cell membrane and transmission of inhibitory signals. Genetic depletion or pharmacologic inhibition of Fut8 fucosyltransferase decreases PD-1 fucosylation, expression, and inhibitory signaling, resulting in increased T cell activation. (B) FBXO38 ubiquitin ligase mediates a K48-linked ubiquitination of PD-1 at K233, resulting in PD-1 internalization and proteasomal degradation. Genetic ablation or down-regulation of FBXO38 results in increased PD-1 expression, leading to enhanced inhibitory signaling and T cell suppression. (C) Expression of PD-L1 in tumor cells is stabilized by glycosylation. This is antagonized by GSK3β, which binds to the nonglycosylated form of PD-L1, leading to phosphorylation at T180 and S184, and β-TrCP–mediated PD-L1 ubiquitination and proteasomal degradation. EGFR-mediated signals inhibit GSK3β-mediated PD-L1 phosphorylation and degradation and promote PD-L1 stabilization and immunosuppressive function. Antibodies targeting glycosylated PD-L1 (anti–gPD-L1) block interaction with PD-1 and induce PD-L1 internalization and degradation. (D) In cancer cells, TNFR (TNF receptor)–mediated signaling results in IKKβ (inhibitor of nuclear factor κB kinase β)–mediated p65 activation and nuclear translocation, leading to transcription of CSN5, which stabilizes PD-L1 by direct deubiquitination or by inhibiting PD-L1 ubiquitination, resulting in enhanced immunosuppressive activity. CMTM4/6 associates with PD-L1 at the cell surface, reducing its ubiquitination and increasing the half-life of PD-L1 protein.
PD-L1 expression and stability is also regulated at the posttranslational level by glycosylation and ubiquitination (19, 20). Only nonglycosylated PD-L1 was found to interact with glycogen synthase kinase 3β (GSK3β), leading to phosphorylation-dependent proteasome degradation of PD-L1 by β-transducin repeat-containing protein (β-TrCP) (Fig. 1C) (19). Conversely, COP9 signalosome 5 (CSN5), induced by nuclear factor κB p65, is required for tumor necrosis factor–α (TNF-α)–mediated PD-L1 stabilization in cancer cells either by direct deubiquitination or by inhibiting ubiquitination and degradation of PD-L1 (Fig. 1D) (20).
PD-L1 glycosylation is also essential for PD-1/PD-L1 interaction. A monoclonal antibody targeting glycosylated PD-L1 blocked interaction with PD-1, promoted PD-L1 internalization and degradation in triple-negative breast cancer (TNBC) cells in vitro, and eradicated TNBC cells in tumor-bearing mice (21). This observation is exciting because the efficacy of checkpoint blockade is only limited in patients with TNBC and suggests that targeting PD-L1 glycosylation might be a promising strategy to increase the success of PD-1 checkpoint immunotherapy. Glycosylation is essential even for soluble PD-L1 to maintain binding capacity to PD-1 (22). In contrast, PD-1 glycosylation is not essential for PD-1/PD-L1 interaction. Structure studies revealed that PD-1 glycosylation sites are located away from PD-1/PD-L1–binding interface, suggesting that modifications of PD-1 glycosylation would not have direct influence on this interaction (23). Consistently, binding of the therapeutic blocking antibodies nivolumab and pembrolizumab to PD-1 was independent of PD-1 glycosylation (24, 25). These recent findings link glycosylation and ubiquitination pathways to the stringent regulation of PD-1 and PD-L1 and open new potential therapeutic strategies to enhance the efficacy of cancer immunotherapy.
Regulators of PD-L1 protein on the cell surface were revealed recently by two independent genome-wide CRISPR-Cas9 screens in pancreatic cancer cells and human haploid HAP1 cells (26, 27). In both studies, CMTM6 was identified as a critical stabilizer of PD-L1 in a broad range of cancer cells, including melanoma, pancreatic cancer, thyroid cancer, breast cancer, non–small cell lung cancer (NSCLC), and colorectal cancer. CMTM6 was found to bind and colocalize with PD-L1 on the plasma membrane and in recycling endosomes, which prevented PD-L1 from lysosome-dependent degradation during recycling (Fig. 1D) (26). In the absence of CMTM6, tumor cells had reduced PD-L1 protein expression, which could be partially reverted by deletion of the STUB1 E3 ubiquitin ligase, identified as a ubiquitin ligase responsible for PD-L1 degradation and down-regulation of PD-L1 expression (27). In a modified genetic screen in CMTM6-deficient haploid cells, CMTM4, but not other CMTM family members, was found as a complemental regulator of PD-L1 expression (27). Notably, interference with CMTM6 expression impaired constitutive and interferon-γ (IFN-γ)–induced protein expression of PD-L1 on tumor cells, without affecting the PD-L1 transcriptional level (26, 27). By destabilization of PD-L1 on tumor cells, CMTM6 depletion significantly mitigated the suppression of tumor-specific T cell activity both in vitro and in vivo (26, 27). These findings highlight CMTM6 and its regulatory role on PD-L1 protein as a potential therapeutic target to overcome immune suppression in the TME.
Besides membrane-bound PD-L1 and PD-1, soluble forms also exist. A soluble monomeric form of PD-L1 can be produced in vitro by cancer cell lines that express PD-L1 and by activated monocyte-derived dendritic cells (DCs) that also express high levels of PD-L1 (28, 29). It has been reported that soluble PD-L1 retains its inhibitory function (28) and is generated by matrix metalloproteases, which cleave it from the surface of cells (28, 30). High levels of soluble PD-L1 in sera of patients with melanoma before immune checkpoint therapy were associated with increased likelihood of progressive disease after treatment with CTLA-4–blocking antibodies (31). Soluble PD-L1 was also detected in plasma samples from patients with NSCLC correlating with poor prognosis and reduced survival (22, 32) and in plasma samples from patients with pancreatic cancer (33). Recently, a new secreted PD-L1 splice variant has been identified with a unique 18–amino acid tail containing a cysteine that allows it to homodimerize and more effectively inhibit lymphocyte function than monomeric soluble PD-L1 (34).
STRUCTURE AND INTERACTION OF PD-1 AND ITS LIGANDS
Structure
PD-1 and its ligands are members of the B7/CD28/CTLA-4 family of receptors, which are type I transmembrane glycoproteins sharing a basic structural pattern consisting of an immunoglobulin (Ig) variable–type (IgV) extracellular domain, a transmembrane region, and a cytoplasmic tail that serves as a docking site for signaling or scaffolding proteins (35). So far, structural studies of PD-1 and its ligands have focused mainly on the extracellular domains and have revealed the very details of interaction interfaces (35–42). PD-1 contains a front β sheet face comprising the CC′FG strands and a back β sheet face comprising the AA′BDE strands. The binding of PD-1 to its ligands involves the front β sheet “faces” of the interacting molecules, with additional contributions of the FG loops. Differences in the interfaces formed between PD-1 and each of its ligands explain the higher affinity that PD-L2 exhibits toward PD-1, which is attributed to its unique sequence characteristics. In particular, tryptophan W110 in PD-L2, which is conserved among species, forms the largest numbers of contacts with PD-1 residues but is replaced with alanine in PD-L1. Deletion or mutagenesis of W110 in PD-L2 to alanine results in reduced binding affinity with PD-1 (38). Although other members of the family such as CD28, CTLA-4, and ICOS form disulfide-based covalent homodimers (43), PD-1 lacks the analogous cysteine residue and does not form such dimers. Moreover, although B7-1 and B7-2 can form noncovalent dimers (44, 45), PD-1, PD-L1, and PD-L2 are mainly known to exist and interact as monomers (35, 37–41).
Progress in the structural characterization of the human PD-1/PD-L1 interaction (41) has enabled the development of small-molecule modulators that can directly bind to PD-L1 and effectively inhibit PD-1/PD-L1 interaction in vitro by inducing PD-L1 dimerization through the PD-1 interacting surface (46, 47). These small molecules were capable of restoring the activity of T cells in vitro (48); however, the in vivo efficacy of these compounds remains to be determined. Notably, the PD-1/PD-L1 complex was shown to form an interface of high structural similarity with the interphase of antigen-binding Fv domains of antibodies and T cell receptors (TCRs) raising the possibility for the synergistic formation of a binding site for a third molecule (37). So far, no such third player in this interaction has been identified and the biological significance of this observation remains to be determined.
Interaction between PD-1 and its ligands
According to the canonical PD-1/PD-L1 pathway, binding between PD-1 and PD-L1 in trans, when these surface receptors are expressed on T cells and APCs (or cancer cells), respectively, triggers inhibitory signaling to attenuate T cell responses, and these inhibitory signals are blocked by antibodies against PD-1 and its ligands (Fig. 2A). However, a recent study showed that PD-1/PD-L1 interaction can also occur in cis on certain cancer cells and tumor-infiltrating APCs when these cells coexpress both PD-1 and PD-L1 (49). By pioneering in vitro reconstitution of lipid bilayer systems and cell culture assays, the study demonstrated that PD-1 and PD-L1 coexpressed on the same APC or cancer cell interact with each other on the same cell surface in cis, resulting in the decreased ability of PD-L1 to bind PD-1 on T cells in trans. This in cis interaction prevents canonical PD-1/PD-L1 inhibitory signals (Fig. 2B) (49). This mode of in cis PD-1/PD-L1 interaction might explain why, in certain cases, PD-1 blockade fails to enhance T cell responses since PD-1 antibody binding on tumor-expressed PD-1 might release PD-L1 and make it available to interact with PD-1 expressed on T cells to inhibit T cell signaling and cytotoxicity (Fig. 2B). This new dimension of PD-1 regulation might have therapeutic implications in tumor immunotherapy, as it might guide the need for combined PD-1 and PD-L1 blocking strategies to overcome such outcomes.
Fig. 2. Interaction modes of PD-1 and PD-L1.
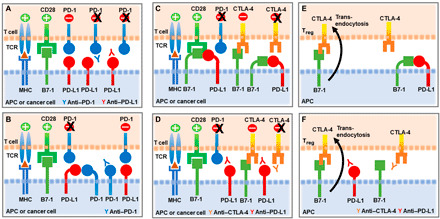
(A) According to the canonical PD-1/PD-L1 interaction, PD-L1 expressed on APCs or tumor cells interacts with PD-1 expressed on T cells in trans to attenuate activation mediated by TCR/MHC (major histocompatibility complex) and CD28/B7-1 interactions. Blocking antibodies against PD-1 or PD-L1 alleviate T cell inhibition by preventing trans PD-1/PD-L1 interaction. (B) When PD-1 and PD-L1 are coexpressed on APCs or tumor cells, PD-1 binds to PD-L1 in cis, diminishing the ability of PD-L1 to bind PD-1 expressed on T cells in trans. (C) PD-L1/B7-1 interaction in cis on APCs or tumor cells disrupts PD-1/PD-L1 binding in trans, resulting in diminished PD-1–mediated T cell inhibition. Binding of PD-L1/B7-1 in cis does not disrupt the binding of B7-1 to CD28, and costimulatory effects of B7-1/CD28 interaction are preserved. In contrast, binding of PD-L1/B7-1 in cis disrupts the B7-1/CTLA-4 axis. (D) Adding a blocking anti–PD-L1 antibody to disrupt PD-1/PD-L1 interaction can also disrupt PD-L1/B7-1 interaction and allow released B7-1 to bind to CTLA-4 and deliver inhibitory signals. In this case, a blocking anti–CTLA-4 antibody might be beneficial by preventing CTLA-4–mediated T cell inhibition. (E) PD-L1/B7-1 interaction in cis on APCs prevents regulatory T cell (Treg) CTLA-4–mediated trans-endocytosis of B7-1 that leads to B7-1 depletion from APC surface. (F) Anti–PD-L1 antibody can have a negative impact on immunotherapy by disrupting PD-L1/B7-1 interaction and allowing B7-1 binding to CTLA-4 on Tregs, CTLA-4–mediated trans-endocytosis of B7-1, and diminished B7-1–mediated costimulation. Such negative effect of anti–PD-L1 antibody might be alleviated by an anti–CTLA-4 antibody.
Besides interacting with PD-1, PD-L1 also interacts with B7-1 (CD80) and was initially thought that this in trans PD-L1/B7-1 interaction can bidirectionally mediate inhibitory signals to T cells (36, 50). A subsequent study reported that B7-1 coexpression with PD-L1 on tumor cells could overcome PD-L1–mediated inhibition but did not provide a mechanistic explanation (51). This mystery remained unsolved until the preference of cis versus trans interaction between PD-L1 and B7-1 was recently examined (52). By using transfected cell lines, cell-to-cell binding assays, NanoBiT proximity assays, enzyme-linked immunosorbent assay, and flow cytometry, these studies concluded that PD-L1/B7-1 interaction occurred exclusively in cis between PD-L1 and B7-1 molecules expressed on the same cell but not in trans between PD-L1 and B7-1 expressed on two separate cells (52). Shortly thereafter, a different study examined the functional implications of the in cis PD-L1/B7-1 interaction and determined that in cis PD-L1/B7-1 interaction on APCs disrupts PD-1/PD-L1 binding in trans (Fig. 2C). Through this mechanism, APCs expressing substantial amounts of B7-1 mediate diminished T cell inhibition via PD-1 (53).
Almost in parallel, another study also reported that in cis PD-L1/B7-1 interaction on APCs disrupts PD-1/PD-L1 binding in trans (54) and determined that in cis PD-L1/B7-1 interaction does not disrupt binding of B7-1 to CD28, and costimulatory effects of B7-1/CD28 interaction are preserved (Fig. 2C). Instead, in cis PD-L1/B7-1 interaction prevents B7-1 from binding to CTLA-4 and disrupts the CTLA-4 trans-functional axis (Fig. 2C) (54). In this setting, adding an anti–PD-L1 blocking antibody to disrupt PD-1/PD-L1 interaction can also result in disruption of PD-L1/B7-1 interaction and allow released B7-1 to bind to CTLA-4 and deliver inhibitory signals through the B7-1/CTLA-4 axis (Fig. 2D). In this case, combining anti–PD-L1 with a blocking anti–CTLA-4 antibody is beneficial since it prevents inhibitory signals mediated from B7-1/CTLA-4 interaction (Fig. 2D). Furthermore, in cis PD-L1/B7-1 interaction on APCs could prevent CTLA-4–mediated trans-endocytosis of B7-1 that leads to B7-1 depletion from APC surface (Fig. 2E) (55, 56). In this scenario, anti–PD-L1 antibody may exert a negative effect on cancer immunotherapy by disrupting in cis PD-L1/B7-1 interaction and allowing the released B7-1 on APCs to interact with CTLA-4 on regulatory T cells (Tregs), resulting in enhanced CTLA-4–mediated B7-1 depletion from APCs and decreased B7-1–mediated costimulation (Fig. 2F). This effect might be avoided by combining an anti–CTLA-4 antibody that disrupts B7-1/CTLA-4 interaction (Fig. 2F) (54). These findings suggest that the levels of B7-1 and PD-L1 expression on APCs and tumor cells might serve as guiding biomarkers for the proper selection of combination therapies targeting PD-1, PD-L1, and CTLA-4.
PD-L2 interactions may also be complex. The role of PD-L2 in T cell responses remains controversial because both coinhibitory and costimulatory functions have been reported (7, 57). Besides interacting with PD-1, PD-L2 interacts with repulsive guidance molecule b (RGMb), a coreceptor for bone morphogenetic proteins, which is expressed on the surface of naive T cells, macrophages, neutrophils, and DCs and regulates respiratory tolerance (58). A mutant PD-L2 (K113S), which loses interaction with PD-1, binds RGMb with similar affinity to wild-type PD-L2 and costimulates CD4+ T cell responses to promote T helper 1 (TH1) polarization (59), while suppressing TH2-mediated responses in an experimental mouse model of asthma (59). Thus, the precise role of PD-L2 in regulating T cell differentiation and function in vivo remains to be determined.
MECHANISMS AND TARGETS OF PD-1 SIGNALING
PD-1:SHP-2 interaction and SHP-2 activation
Although the extracellular structure and interactions of PD-1 are extensively studied, little information is known about its intracellular structure and signaling mechanisms. The cytoplasmic tail of PD-1 contains two tyrosine-based structural motifs, an immunoreceptor tyrosine-based inhibitory motif (ITIM) (V/L/I/XpYXX/L/V), and an immunoreceptor tyrosine-based switch motif (ITSM) (TXpYXXV/I) (60). Mutational studies have shown that PD-1 inhibitory function is dependent on the ITSM phosphotyrosine, which preferentially recruits Src homology region 2 domain-containing phosphatase-2 (SHP-2), resulting in dephosphorylation and down-regulation of downstream signaling pathways (61–63). On the basis of these findings, an antibody specific for phosphorylated PD-1-ITSM Y248 (thereafter named pY-ITSM) was generated and tested for its ability to detect PD-1–mediated inhibitory signaling (64). These studies showed that phosphorylation of PD-1 at the ITSM Y248 was up-regulated by simultaneous TCR/PD-1 coligation. Detection of PD-1–ITSM Y248 phosphorylation on T cells from human peripheral blood correlated with markers of impaired effector function. PD-1–ITSM Y248 phosphorylation was detected on T cells in tumor-draining lymph nodes of tumor-bearing mice. pY-ITSM–positive cells were also detected in biopsies of patients with glioblastoma. These results suggest that detection of PD-1 pY-ITSM in T cells might serve as a more biologically relevant biomarker than PD-1 expression, identifying T cells that are actively undergoing immunosuppression through PD-1 ligation, which depends on phosphorylation of ITSM Y248 and recruitment of SHP-2 (64).
Although it is widely accepted that SHP-2 is a key mediator of PD-1 inhibitory function, the precise mechanism of how PD-1 engagement leads to SHP-2 enzymatic activation has remained puzzling. SHP-2 has two tandem SH2 domains, N-terminal (N-SH2) and C-terminal SH2 (C-SH2), followed by a single phosphatase [protein tyrosine phosphatase (PTP)] domain, and a C-terminal hydrophobic tail with two tyrosine phosphorylation sites. At the basal state, the N-SH2 domain of SHP-2 folds into an autoinhibitory closed conformation to directly block the active PTP site. Interaction of the N-SH2 domain with phosphotyrosine peptide disrupts this interaction and activates the enzyme. Binding of both SH2 domains is required for full SHP-2 enzymatic activation, with the C-SH2 domain contributing to binding energy and specificity and N-SH2 having a direct role in enzymatic activation (65–67). For example, binding of both SH2 domains of SHP-2 in tandem with the tyrosines of a biphosphorylated peptide from insulin receptor substrate 1 can induce potent SHP-2 catalytic activity (65). A similar type of interaction and SHP-2 activation mechanism has been recently hypothesized to occur between SHP-2 and PD-1 (68). In support of this hypothesis, structural characterization and biochemical analysis of SHP-2 activity in vitro provided evidence of a two-step binding model, according to which PD-1 phosphotyrosine at the ITSM motif (pY-ITSM) binds to C-SH2 with strong affinity, recruiting PD-1 to SHP-2, while phosphotyrosine at the ITIM motif (pY-ITIM) binds to N-SH2, displacing it from the PTP site and activating the phosphatase (Fig. 3A) (69). In contrast to previous observations (61–63), the latter study found that besides ITSM, ITIM also contributed to PD-1 inhibitory function, although to a lesser extent, supporting a potential model requiring both domains ITIM and ITSM for activating SHP-2 phosphatase (69).
Fig. 3. PD-1/SHP-2 interaction modes.
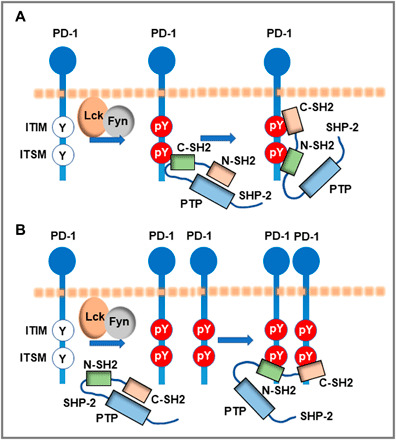
(A) Two-step binding model, according to which SHP-2 C-SH2 binds to PD-1 pY-ITSM with strong affinity, recruiting PD-1 to SHP-2, while PD-1 pY-ITIM binds to N-SH2, displacing it from the PTP site to activate the phosphatase. (B) Dimerization model, according to which SHP-2 bridges two pY-ITSM residues on two PD-1 molecules via its N-SH2 and C-SH2 domains forming a PD-1:PD-1 dimer and inducing SHP-2 activation.
Almost in parallel, it was determined that besides binding of the two SHP-2 SH2 domains in tandem with the two phosphotyrosines of the PD-1 cytoplasmic tail, SHP-2 and PD-1 have the biophysical properties to interact via engagement of both SHP-2 SH2 domains with phosphorylated PD-1–ITSM Y248 from two different PD-1 molecules (70). By combining biochemical and biophysical methods as well as confocal microscopy, this study revealed that this alternative mode of PD-1:SHP-2 interaction preferentially occurred in live cells, where, after PD-1 phosphorylation, SHP-2, via its N-SH2 and C-SH2 domains, could bridge two phosphorylated pY-ITSM residues on two PD-1 molecules localized at the plasma membrane, forming a PD-1:PD-1 dimer (Fig. 3B) (70). SHP-2 interaction with two tandemly connected ITSM phosphopeptides induced robust SHP-2 enzymatic activation, suggesting that a combination of strong binding ITSM motifs can form an SHP-2–dependent PD-1 dimer and simultaneously activate the phosphatase. The discovery of this alternative binding mode that leads to the formation of an SHP-2–dependent PD-1 dimer explains how PD-1/SHP-2 interaction can occur by the involvement of only one PD-1 phosphotyrosine (61–63) and resolves a long-standing conundrum of how a single docking site within the PD-1 cytoplasmic tail can activate SHP-2 and mediate PD-1 inhibitory function (70). Development of novel techniques to monitor the activity of SHP-2 in live cells will shed light on the detailed interplay between these PD-1:SHP-2 interaction modes in live cells.
Signaling pathways targeted by PD-1
Multiple studies have investigated the effects of PD-1 on key signaling pathways activated by TCR and costimulatory receptors to determine how PD-1 ligation inhibits the expansion and effector differentiation of activated T cells and suppresses cytokine production. There has been a long debate about which signaling pathway is the primary target of PD-1. Most studies support a model in which PD-1 primarily targets the TCR and TCR downstream cascades (61–63, 71–74). However, it has also been suggested that the coreceptor CD28 is preferred over the TCR as the PD-1 primary target (75), whereas other studies have reported that both the TCR and CD28 are equally targeted by PD-1 (76). Despite the debate about the PD-1 primary target, there is an agreement that SHP-2 is currently the only identified direct partner of PD-1 in normal T cells responsible for mediating PD-1 inhibitory signals. For this reason, it was unexpected that mice with T cell–specific SHP-2 deletion could be benefitted by PD-1 blockade in the context of cancer, suggesting the potential existence of additional PD-1 signaling partners that can mediate PD-1 inhibitory function in the absence of SHP-2 (77). An explanation to this finding came from the detailed molecular characterization of human PD-1 signalosome by quantitative interactomics, which demonstrated the ability of SHP-1 phosphatase to replace SHP-2 and compensate for PD-1 inhibitory function when SHP-2 is absent (76).
SHP-1 and SHP-2 phosphatases not only are involved in PD-1 signaling but also play critical and opposing roles in TCR-mediated activation. Although SHP-1 mediates inhibitory signals (78), SHP-2 is considered a positive regulator of T cell activation (79–81) and may act by dephosphorylating inhibitory sites of positive regulators. Both phosphatases are involved in the formation of the TCR signalosome (82), and their phosphatase activities are regulated by phosphorylation/dephosphorylation as well as by oxidation/reduction reactions (83–85). Since PD-1 expression is induced only after activation and participates in TCR signaling microclusters upon binding to PD-L1 (63), it is reasonable to speculate that PD-1 molecules are clustered and stabilized by PD-L1 interactions around TCR molecules, serving as “switches” of positive signalosome conformations to negative ones. Some proteins recently identified as molecular partners in the PD-1 signalosome such as Grb2, Lck, and ZAP-70 (76) are also components of the TCR signalosome. Further studies are required to elucidate the complex interplay between TCR and PD-1 signalosomes, determine their shared molecular partners and signaling mechanisms involving SHP-1 and/or SHP-2, and identify their direct substrates.
Although PD-1–mediated signaling and functional effects are mainly directed toward T cells, two studies raised the question whether the PD-1:PD-L1 pathway might signal in cancer cells. It was reported that subpopulations of established human and murine melanoma cell lines and subpopulations of malignant cells in melanomas from patients’ biopsies express PD-1 (86, 87). Unlike T cells, in which PD-1 ligation causes inhibition of PI3K/Akt, mitogen-activated protein kinase (MAPK), and mammalian target of rapamycin (mTOR) pathways, PD-1 ligation in melanoma cells was found to activate these pathways and promote tumor growth. These studies suggested that cancer cells might use the PD-1:PD-L1 pathway to support their growth by triggering mTOR signaling in trans in neighboring tumor cells (86).
PD-1 cross-talk with costimulatory receptors
PD-1–targeted tumor immunotherapies enhance T cell responses but show limited efficacy in many cases. A better understanding of how PD-1 cross-talks with other costimulatory receptors will benefit not only antibody-based but also cell-based immunotherapies. It has been shown that PD-1 signaling can be overcome by interleukin-2 (IL-2) and that only cytokines that activate signal transducer and activator of transcription 5 (STAT5) can rescue PD-1 inhibition (88). Under optimal conditions of stimulation, CD28 costimulation can overcome PD-1–mediated inhibition by augmenting IL-2 production (88). However, ICOS-mediated costimulation, which leads to lower amount of IL-2 production, cannot overcome PD-1–mediated inhibition. Thus, costimulatory signals mediated by ICOS are more susceptible to PD-1–mediated negative regulation than those provided by CD28 costimulation (89). In a recent study, overexpression of c-Jun, an AP-1 family transcription factor that drives IL-2 transcription associated with productive T cell activation, prevented CAR (chimeric antigen receptor) T cell exhaustion, increased functional capacity, diminished terminal differentiation, and improved antitumor potency in multiple in vivo models (90), presumably by effectively competing with inhibitory signals through PD-1 and other checkpoint inhibitory receptors. It is remarkable that, albeit inhibitory, PD-1 is highly expressed in T follicular helper (Tfh) cells, particularly those localized inside the germinal center (GC) territory (91). However, instead of being inhibited, Tfh cells are highly functional and sensitive to antigen presented by cognate B cells. Tfh cells also highly express ICOS, and although ICOS costimulation is insufficient to fully restore proliferation upon PD-1 engagement (89), in Tfh cells, ICOS is indispensable for bypassing PD-1–mediated inhibition of PI3K downstream of CXCR5, thereby allowing Tfh to migrate to the GC follicle (91). Paradoxically, PD-1 was also found to have a positive effect on Tfh and to be required for IL-21 production and for setting a sufficiently stringent threshold for the GC B cell competition. Thus, by controlling both proper positioning and helper functions of Tfh cells, the inhibitory receptor PD-1 acts in concert with ICOS and plays an essential role in the GC response. Further understanding of how the signaling machinery of PD-1 competes and cooperates with signaling of ICOS and other costimulatory receptors may allow for efficient therapeutic interventions in immunotherapies.
MECHANISMS AND OUTCOMES OF PD-L1 SIGNALING
Expression of PD-L1 in cancer cells (7, 8) was initially considered a major mechanism of cancer-mediated T cell immunosuppression and exhaustion. Subsequently, it became apparent that PD-L1 expressed in antigen-presenting myeloid cells in the TME is equally important for mediating immunosuppressive properties to tumor-specific T cells (9). PD-L1–expressing DCs and macrophages might have a dominant role in mediating T cell immunosuppression, as PD-L1 expression on these two APC types of the TME can predict the efficacy and therapeutic outcome of PD-L1/PD-1 blockade (92). Responses to therapeutic blockade can also be developed as a consequence to blocking T cell immunosuppression mediated by PD-L1–expressing APCs in non-tumor sites (93). Robust antitumor T cell responses are induced in tumor-draining lymph nodes by blocking PD-L1–mediated inhibitory effects of host APCs in extratumoral sites.
PD-L1–mediated signaling was first studied in cancer cells. PD-L1 is considered an immunological shield that transmits anti-apoptotic signals in cancer cells after ligation by PD-1 expressed in T cells, thereby inducing resistance against T cell–mediated killing and protecting cancer cells (94). Subsequent studies suggested that PD-L1 can activate cancer cell–intrinsic signals in a PD-1–independent manner and enhance cancer cell proliferation and survival through the inhibition of autophagy and mTOR activation (95). No obvious signaling sequences related to signal transduction have been predicted or identified for PD-L1 cytoplasmic tail, suggesting that PD-L1 is using nonconventional signaling motifs. Recently, three conserved sequences in PD-L1 cytoplasmic tail were identified, which were termed “RMLDVEKC,” “DTSSK,” and “QFEET” motifs (96). Notably, lysines 271 and 280 within RMLDVEKC and DTSSK motifs are putative targets for ubiquitination, leading to PD-L1 destabilization and down-regulation (20). The conserved RMLDVEKC motif is required to counteract IFN-β toxicity mediated by PD-L1 engagement, while the DTSSK motif acts as a negative regulator of PD-L1 function to transduce signals that counteract IFN signal transduction and its toxicity in cancer cells (Fig. 4) (96). These findings mechanistically extended the early observations that PD-L1 serves as a direct defense for cancer cells (94) and revealed that abrogation of PD-L1 expression or antibody-mediated PD-L1 blockade sensitizes cancer cells to IFN cytotoxicity through a STAT3/caspase-7–dependent pathway (Fig. 4). In human carcinomas, somatic mutations within these novel nonconventional PD-L1 motifs resulted in enhanced cytotoxic activities from type I and type II IFN, providing evidence for the significant biological relevance of these PD-L1 cytoplasmic regions (96). In Hodgkin’s lymphoma (HL) cell lines, engagement of PD-L1 by an agonistic antibody can increase cell survival and proliferation. Moreover, in patients with HL, serum levels of soluble PD-1 were significantly higher than healthy controls. Both membrane-bound and soluble forms of PD-1 were able to induce PD-L1–mediated signaling in HL cell lines, which was associated with the activation of the MAPK pathway and increased mitochondrial oxygen consumption, events that were reversed by PD-1 blockade (97). Further studies are needed to determine which binding partners of the cytoplasmic domain of PD-L1 mediate these functions.
Fig. 4. Signaling through PD-L1 protects tumor cells from IFN-mediated cytotoxicity.
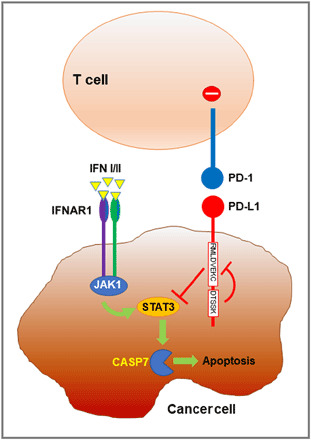
PD-L1 expressed on tumor cells engages PD-1 to deliver inhibitory signals to T cells. PD-L1 may also deliver inhibitory signals to tumor cells to attenuate IFN-mediated cytotoxicity through a STAT3/caspase-7–dependent pathway. The conserved RMLDVEKC motif of PD-L1 is required to counteract IFN toxicity, while the DTSSK motif prevents this function. Thus, PD-L1 provides tumor cells with a dual escape mechanism from T cell–dependent cytotoxicity. IFNAR1, interferon alpha/beta receptor alpha chain.
PD-L1 is expressed in T cells after activation, but its ability to transmit signals to T cells has been controversial. Initially, a specific and significant bidirectional interaction between B7-1 and PD-L1 that inhibits T cell responses was demonstrated (36). These effects were thought to be mediated by PD-L1 expressed in T cells that could transmit inhibitory signals after being ligated by B7-1 in trans. This conclusion was based on studies using anti–CD3/B7-1 Ig-coated beads that inhibited the proliferation of CD28/CTLA-4–deficient T cells in vitro (36). However, later studies with in vivo models of T cell activation and tolerance identified that it was not PD-L1 but B7-1 expressed on antigen-specific T cells that was responsible for transmitting coinhibitory signals (50). Using an agonist anti–PD-L1 antibody, a different study determined that PD-L1 can mediate inhibitory signaling to T cells by inducing increased phosphorylation of p38, leading to apoptosis (98). Similarly, in a mouse model of pancreatic ductal adenocarcinoma (PDA) and in biopsies of human PDA, up-regulation of PD-L1 on T cells in response to antigen presentation and inflammatory signals induced intracellular signaling equally suppressive to that of PD-1 (99). PD-L1 engagement on T cells induced STAT3-dependent inhibitory signaling in CD4+ T cells, which reduced TH1 (T-bet and IFN-γ) and TH2 (GATA3) polarization but favored a partial TH17 differentiation. In addition, PD-L1 expressed on T cells inhibited neighbor T cells not only by engaging PD-1 but also by promoting an M2 macrophage polarization in the TME, thereby suppressing neighbor effector T cells (99).
Contrary to the studies indicating that B7-1 mediates inhibitory signals to T cells (50), in trans PD-L1/B7-1 interaction was reported to induce activation of alloreactive T cells and graft versus leukemia effector function (100). In light of the recent findings that PD-L1/B7-1 interaction occurs only in cis (52–54), while PD-1/PD-L1 interaction can occur both in cis and in trans (49), caution is required in the interpretation of these findings. It is also possible that various receptor:ligand pairs transmitting distinct signals might be formed in the immune microenvironment, leading to combined effects and functional outcomes.
These new developments regarding the signaling interplay among PD-1, PD-1 ligands, and B7-1 not only shed light on previously unexplained observations but also call for careful attention in the interpretation of experimental outcomes. At this point, it is unclear whether cell-specific expression of these receptors in the TME, or other sites with critical role in mounting antitumor immune responses, might have implications on patient stratification, therapeutic responses to PD-1 blocking immunotherapy, or guidance for combinatorial therapies.
PD-1 DETERMINES T CELL DIFFERENTIATION AND FUNCTION BY REGULATING METABOLIC REPROGRAMMING
In T cells, metabolism not only supports growth and proliferation but also plays a critical role in driving differentiation (101). Oxidative phosphorylation is the main source of energy for naive T cells, but upon activation, naïve T cells undergo metabolic reprogramming to aerobic glycolysis, also known as the Warburg effect, while simultaneously augmenting glutamine uptake and catabolism to maintain effector T cell fitness and drive differentiation of memory T cells (102). PD-1 signaling does not shut down T cell metabolism globally but diverts metabolic reprogramming by favoring the utilization of fatty acids in β-oxidation (FAO) while impairing glycolysis, glutaminolysis, and metabolism of branched-chain amino acids (103, 104). Pharmacologic inhibition of glycolysis blocks differentiation of TH17 cells while promoting development of Tregs (105, 106). Thus, together with decreasing the threshold to transforming growth factor–β (TGF-β)–mediated signals (72, 107), PD-1 also favors the development of Tregs by reprogramming T cell metabolism.
Regulation of the cell redox state is an important component of cellular metabolism. In T cells, reactive oxygen species (ROS) are required for cellular and signaling processes, leading to T cell activation (108). However, excessive ROS production can have detrimental effects in T cells (109). The fine balance between mechanisms of ROS generation and detoxification is critical for cell proliferation, differentiation, and survival. Very limited information is available regarding the effects of PD-1 on T cell oxidative state. T cells receiving PD-1 signals maintain metabolically active mitochondria, higher FAO, more pronounced decrease in the levels of reduced glutathione (GSH), and higher levels of GSH-cysteine disulfide compared to T cells activated without PD-1 ligation, potentially indicating a more oxidative environment (103).
A key mediator of oxidative detoxification is the peroxisome proliferator–activated receptor γ (PPARγ) coactivator-1a (PGC-1a) (110, 111), which also promotes mitochondrial biogenesis, and its overexpression can reinvigorate T cells in the TME, resulting in enhanced antitumor efficacy even in the presence of high expression of PD-1 and other checkpoint inhibitors (112). Thus, it is possible that the combination of mitochondrial biogenesis, increase of mitogenic ROS, and the right amount of ROS detoxifiers to prevent excessive ROS-mediated damage can bypass signals imposed by inhibitory receptors. Toward this direction, a recent study determined that ROS generation by the use of ROS precursors or mitochondrial uncouplers synergized with the tumoricidal activity of PD-1 blockade, leading to the expansion of effector/memory CTLs in the TME (113). These CTLs activated both mTOR and adenosine monophosphate–activated protein kinase (AMPK) and their downstream transcription factors such as PGC-1a and its cofactor NRF2, which is a major transcriptional inducer of antioxidant genes. Under these conditions, PPARs that regulate fatty acid metabolism, and T-bet that controls IFN-γ transcription and CTL effector function, were also increased. Furthermore, mTOR, AMPK, or PGC-1a activators synergized with PD-1 blockade and improved antitumor efficacy (113).
The highly oxidative TME not only alters the function of T effector cells directly but also mediates a dominant detrimental effect on Tregs, which succumb to oxidative stress and undergo apoptosis due to their weak NRF2-associated antioxidant system, resulting in the release of large amounts of adenosine triphosphate and its conversion to adenosine via CD39 and CD73, leading to enhanced immunosuppression via the adenosine and A2A pathways (114). Thus, in addition to suppressing T effector cell function directly, PD-1 also compromises antitumor immunity by decreasing the threshold of TGF-β–mediating signaling and altering metabolic reprogramming (72, 103, 107), thereby favoring the generation of Tregs, which are highly vulnerable to ROS-dependent cell death in the TME and produce immunosuppressive intermediates that prevent the responses of T effector cells to PD-1 blockade. Thus, the oxidative pathway could be considered as a metabolic checkpoint, potentially amenable to modulation for enhancing the efficacy of therapeutics targeting PD-1.
SIGNALING AND FUNCTION OF THE PD-1 PATHWAY IN INNATE IMMUNE CELLS
As PD-1 expression and function have been originally assigned to activated lymphocytes T and B cells (2), attention has been paid to the expression and function of PD-1 in T cells, whereas in cells of the innate immune compartment, attention has been paid to the expression of PD-L1. In the context of cancer, it is widely accepted that checkpoint blockade reverts the PD-1–induced exhausted T cell state and unleashes reinvigorated T cells to attack cancer. However, PD-1 is also expressed in cells of the innate immune system (115–118). In cancer mouse models, activated natural killer (NK) cells express PD-1, which may mediate suppressive signals. Under these conditions, NK cell activation can be induced by PD-1/PD-L1 blockade (119). PD-1 is expressed in innate lymphoid cells (ILCs), particularly in the ILC2 subset (116, 120). ILC2s are important regulators of immune responses and tissue homeostasis. In adipose tissues, ILC2s maintain homeostasis by promoting beiging of white adipocytes and supporting a TH2 environment enriched in eosinophils and M2 macrophages. In the context of obesity, ILC2 function is impaired as a consequence of signals mediated by IL-33 that is increased under the control of adipocyte-produced TNF. This correlates with PD-1 expression and imbalance of metabolic homeostasis (120). PD-1 signaling in ILC2 is also involved in antitumor responses (121). In a mouse model of PDA, ILC2s infiltrate tumors and activate tissue-specific CD8+-mediated tumor immunity. Tumor-infiltrated ILC2s express PD-1, and PD-1 blockade in combination with IL-33 can induce ILC2 activation and enhance antitumor responses. PD-1+ ILC2s are present in biopsies of patients with PDA (121).
PD-1 expression on myeloid cells is increasingly being appreciated. PD-1 is up-regulated in several myeloid cell populations and regulates their differentiation and function. For instance, TLR signaling induces PD-1 expression in macrophages, which negatively correlates with M1 polarization (122). In the context of infection, PD-1 expression in macrophages plays a pathologic role by suppressing the innate inflammatory response to sepsis (115) and inhibiting Mycobacterium tuberculosis phagocytosis (117). Similarly, in the context of cancer, PD-1 expression inversely correlates with M1 polarization and phagocytic potency of tumor-associated macrophages against tumor (123, 124). Myeloid cells of the TME are derived from myeloid progenitors of the bone marrow bone, specifically common myeloid progenitors (CMPs) and granulocyte/macrophage progenitors (GMPs), which expand during cancer-mediated emergency myelopoiesis. Terminally differentiated myeloid cells are essential innate immune cells required for the activation of adaptive immunity. Strong activation signals mediated by pathogen-associated molecular pattern or danger-associated molecular pattern molecules lead to a transient expansion and subsequent differentiation of myeloid progenitors to mature monocytes and granulocytes to protect the host. In contrast, during emergency myelopoiesis, driven by continuous low-level stimulation mediated by cancer-derived factors and cytokines, CMPs but predominantly GMPs undergo modest expansion with hindered differentiation, leading to the accumulation of myeloid cells with immunosuppressive and tumor-promoting properties, named myeloid-derived suppressor cells (MDSCs) (125).
Recent studies revealed that the PD-1:PD-L1 axis is activated in CMP and GMP myeloid progenitors that accumulate during cancer-driven emergency myelopoiesis (126). This is an early event that occurs during the growth of cancer cells and occurs even before the tumor obtains a detectable size. PD-L1 is constitutively expressed on CMPs and GMPs, whereas PD-1 expression displayed a notable increase on GMPs that arose during tumor-driven emergency myelopoiesis. By generating mice with myeloid-specific or T cell–specific ablation of the Pdcd1 gene, it was determined that myeloid-specific but not T cell–specific PD-1 ablation prevented the accumulation of GMPs and immunosuppressor MDSCs while inducing systemic output of effector myeloid cells and TEM (T effector memory) cells with improved functionality and eliminated tumor growth despite preserved PD-1 expression in T cells (Fig. 5). At a biochemical level, PD-1 may directly inhibit signaling in myeloid cells, as previously shown for T cells. Growth factors driving emergency myelopoiesis mediated enhanced activation of ERK1/2s (extracellular signal–regulated kinases 1/2) and mTOR1 kinase complex in PD-1–deficient myeloid progenitors. In response to these factors, PD-1–deficient myeloid progenitors displayed metabolic reprogramming characterized by increased intermediates of glycolysis, pentose phosphate pathway, and tricarboxylic acid cycle but, most prominently, elevated cholesterol. As cholesterol is required for differentiation of inflammatory macrophages and DC and promotes antigen-presenting function (127), these findings indicate that metabolic reprogramming of emergency myelopoiesis and differentiation of effector myeloid cells might be a key mechanism of antitumor immunity mediated by PD-1 blockade. Thus, antitumor T cell responses are guided by the consequences of PD-1 signaling in myeloid cells, and PD-1 ablation in T cells, alone, might not be sufficient to promote sustained antitumor function. Instead, it might rather work against antitumor immunity by promoting the accumulation of terminally differentiated T effector cells that promote the generation of MDSCs (126). Consistent with these findings, triggering PD-1 on monocytes from patients with chronic lymphocytic leukemia hampers glycolysis, phagocytosis, and Bruton’s tyrosine kinase signaling, whereas disrupting PD-1/PD-L1 signaling reverses these immune metabolic dysfunctions (124).
Fig. 5. PD-1 regulates the differentiation and lineage fate commitment of myeloid progenitors during cancer-mediated emergency myelopoiesis and determines the efficiency of T cell antitumor responses.
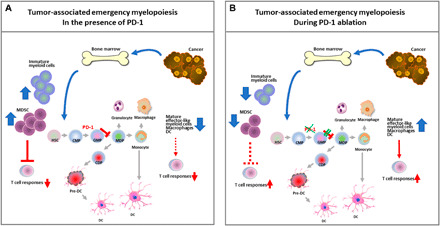
(A) During cancer-driven emergency myelopoiesis, PD-1 is up-regulated on CMPs but mostly in GMPs and inhibits signaling and metabolic reprogramming mediated by growth factors driving emergency myelopoiesis, resulting in accumulation of immature myeloid cells and immunosuppressor MDSCs, and decreased systemic output of effector myeloid cells. (B) PD-1 ablation in myeloid cells promotes signaling and metabolic reprogramming mediated by growth factors of emergency myelopoiesis and leads to the output of effector myeloid cells with improved antigen-presenting function that drive T effector memory cell responses and antitumor protection. HSC, hematopoietic stem cell; CMP, common myeloid progenitor; GMP, granulocyte/monocyte progenitor; MDP, monocyte/dendritic cell progenitor; CDP, common dendritic cell progenitor; DC, dendritic cell; CSF, cancer-produced soluble factor.
To date, very few studies have investigated the impact of metabolic dysfunction on patients’ resistance to checkpoint immunotherapy. Growing evidence suggests that hyperglycemia and cholesterolemia skew hematopoietic stem cells to enhance myelopoiesis and production of proinflammatory myeloid cells (128, 129). Such enhanced myelopoiesis propagates inflammation from the bone marrow to the adipose tissue and the vasculature and contributes to the increased production of TNF-α, IL-6, IL-1, and C-reactive protein, leading to insulin resistance (130, 131). Further, low-grade inflammation, persistent myelopoiesis, and MDSC expansion have been proposed as potent inducers of immunosenescence in age-related immune deficiency (132). Therefore, PD-1 blockade in patients with metabolic comorbidities, and in the elderly, might exacerbate persistent myelopoiesis and systemic inflammation. This emphasizes the need for patient stratification and metabolic monitoring in immunotherapy recipients. In addition, combining checkpoint immunotherapy with immunometabolic targets might be increasingly required, while confronting the worldwide pandemic of metabolic syndrome and the growing number of aging individuals treated with checkpoint immunotherapy. Novel biomarkers that allow the clinical team to choose the therapy that matches the biology of each cancer as well as the host’s metabolic status and immune system are required for personalized precision cancer therapy.
CONCLUDING REMARKS
The PD-1 pathway is a promising target for immunotherapies against cancer and chronic infections. The current paradigm dictates the use of blocking antibodies against the components of this pathway to enhance T cell responses against tumors or chronic infections. However, recently identified novel mechanisms that regulate the expression and function of PD-1 and PD-L1 suggest new approaches to manipulate this pathway therapeutically (Table 1). (i) PD-1 and its ligands are expressed not only on T cells but also on cells of the myeloid compartment and affect immune responses systemically. Thus, preclinical studies targeting this pathway should evaluate functional responses, differentiation profiles, and metabolic programs of both innate and adaptive immune cells. Identification of the precise role of each immune subset in such systemic responses may generate new potential targets for combinatorial therapies with checkpoint inhibitors. (ii) PD-1 and its ligands interact not only in trans between neighboring cells but also in cis on the same cell surface, highlighting the complexity of competitive interactions among costimulatory and coinhibitory molecules. These observations can enable the identification of better biomarkers to stratify patients and guide new strategies for achieving either induction or suppression of immune responses. (iii) Besides the current norm of developing blocking antibodies, progress on the structural characterization of PD-1/PD-L1 interaction has allowed for the development of small-molecule modulators to target and disrupt their extracellular interaction interfaces and posttranslational modifications. Further advancements in characterizing the cytoplasmic structures, biochemical events, and interactions of the components of the PD-1 pathway will enable the development of small-molecule modulators to target this pathway intracellularly. Such approaches might result in treatments that can be used in combination with antibody-based immunotherapies to improve therapeutic success.
Table 1. Potential new interventions for immunotherapy by targeting the PD-1 pathway.
Candidate therapeutic targets (together with key relevant references), mechanism of action, and outcome of each targeted therapy on T cell responses.
| Treatment/targets | Mechanism of action | Outcome in T cells |
| Inhibition of PD-1 core fucosylation (17) |
Decreased surface expression of PD-1 on T cells |
Increased T cell function |
| Enhancing expression of ubiquitin ligase FBXO38 (18) |
Increased PD-1 degradation in T cells |
Increased T cell function |
| EGFR inhibitors + PD-1 blockade (19) |
Decreased PD-L1 expression |
Increased T cell function |
| CSN5 inhibitors (or curcumin) + CTLA-4 blockade (20) |
Decreased PD-L1 expression |
Increased T cell function |
| CMTM4/6 degradation (26, 27) |
Decreased PD-L1 expression |
Increased T cell function |
| Antibodies against glycosylated PD-L1 (21) |
PD-L1 internalization and degradation |
Increased T cell function |
Acknowledgments
Funding: This work was supported by NIH grants R01CA238263, R01CA212605, and R01CA229784 (V.A.B.) and by NIH grant R21AR073494 (N.P.). Author contributions: N.P. generated the main body of the manuscript and prepared figures. Q.W. and L.S. generated sections of the manuscript. V.A.B. generated sections of the manuscript, prepared figures, guided the coauthors, and was responsible for the organization of the document. Competing interests: V.A.B. has patents (U.S. application serial no. 09/644,934; date of patent, 30 August 2005; and serial no. 10/002,775; date of patent, May 2006) on the PD-1 pathway licensed by Bristol-Myers Squibb, Roche, Merck, EMD-Serono, Boehringer Ingelheim, AstraZeneca, Novartis, and Dako. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the authors.
REFERENCES AND NOTES
- 1.Ishida Y., Agata Y., Shibahara K., Honjo T., Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11, 3887–3895 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agata Y., Kawasaki A., Nishimura H., Ishida Y., Tsubata T., Yagita H., Honjo T., Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 8, 765–772 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Nishimura H., Nose M., Hiai H., Minato N., Honjo T., Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11, 141–151 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Nishimura H., Okazaki T., Tanaka Y., Nakatani K., Hara M., Matsumori A., Sasayama S., Mizoguchi A., Hiai H., Minato N., Honjo T., Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291, 319–322 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Wang J., Yoshida T., Nakaki F., Hiai H., Okazaki T., Honjo T., Establishment of NOD-Pdcd1−/− mice as an efficient animal model of type I diabetes. Proc. Natl. Acad. Sci. U.S.A. 102, 11823–11828 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J., Okazaki I.-m., Yoshida T., Chikuma S., Kato Y., Nakaki F., Hiai H., Honjo T., Okazaki T., PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int. Immunol. 22, 443–452 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Latchman Y., Wood C. R., Chernova T., Chaudhary D., Borde M., Chernova I., Iwai Y., Long A. J., Brown J. A., Nunes R., Greenfield E. A., Bourque K., Boussiotis V. A., Carter L. L., Carreno B. M., Malenkovich N., Nishimura H., Okazaki T., Honjo T., Sharpe A. H., Freeman G. J., PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2, 261–268 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Dong H., Strome S. E., Salomao D. R., Tamura H., Hirano F., Flies D. B., Roche P. C., Lu J., Zhu G., Tamada K., Lennon V. A., Celis E., Chen L., Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 8, 793–800 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Curiel T. J., Wei S., Dong H., Alvarez X., Cheng P., Mottram P., Krzysiek R., Knutson K. L., Daniel B., Zimmermann M. C., David O., Burow M., Gordon A., Dhurandhar N., Myers L., Berggren R., Hemminki A., Alvarez R. D., Emilie D., Curiel D. T., Chen L., Zou W., Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 9, 562–567 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Freeman G. J., Long A. J., Iwai Y., Bourque K., Chernova T., Nishimura H., Fitz L. J., Malenkovich N., Okazaki T., Byrne M. C., Horton H. F., Fouser L., Carter L., Ling V., Bowman M. R., Carreno B. M., Collins M., Wood C. R., Honjo T., Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brahmer J. R., Tykodi S. S., Chow L. Q. M., Hwu W.-J., Topalian S. L., Hwu P., Drake C. G., Camacho L. H., Kauh J., Odunsi K., Pitot H. C., Hamid O., Bhatia S., Martins R., Eaton K., Chen S., Salay T. M., Alaparthy S., Grosso J. F., Korman A. J., Parker S. M., Agrawal S., Goldberg S. M., Pardoll D. M., Gupta A., Wigginton J. M., Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garon E. B., Rizvi N. A., Hui R., Leighl N., Balmanoukian A. S., Eder J. P., Patnaik A., Aggarwal C., Gubens M., Horn L., Carcereny E., Ahn M.-J., Felip E., Lee J.-S., Hellmann M. D., Hamid O., Goldman J. W., Soria J.-C., Dolled-Filhart M., Rutledge R. Z., Zhang J., Lunceford J. K., Rangwala R., Lubiniecki G. M., Roach C., Emancipator K., Gandhi L.; KEYNOTE-001 Investigators , Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 372, 2018–2028 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Ansell S. M., Lesokhin A. M., Borrello I., Halwani A., Scott E. C., Gutierrez M., Schuster S. J., Millenson M. M., Cattry D., Freeman G. J., Rodig S. J., Chapuy B., Ligon A. H., Zhu L., Grosso J. F., Kim S. Y., Timmerman J. M., Shipp M. A., Armand P., PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 372, 311–319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das S., Johnson D. B., Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 7, 306 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Francisco L. M., Sage P. T., Sharpe A. H., The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 236, 219–242 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okazaki T., Chikuma S., Iwai Y., Fagarasan S., Honjo T., A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 14, 1212–1218 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Okada M., Chikuma S., Kondo T., Hibino S., Machiyama H., Yokosuka T., Nakano M., Yoshimura A., Blockage of core fucosylation reduces cell-surface expression of PD-1 and promotes anti-tumor immune responses of T cells. Cell Rep. 20, 1017–1028 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Meng X., Liu X., Guo X., Jiang S., Chen T., Hu Z., Liu H., Bai Y., Xue M., Hu R., Sun S.-c., Liu X., Zhou P., Huang X., Wei L., Yang W., Xu C., FBXO38 mediates PD-1 ubiquitination and regulates anti-tumour immunity of T cells. Nature 564, 130–135 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Li C.-W., Lim S.-O., Xia W., Lee H.-H., Chan L.-C., Kuo C.-W., Khoo K.-H., Chang S.-S., Cha J.-H., Kim T., Hsu J. L., Wu Y., Hsu J.-M., Yamaguchi H., Ding Q., Wang Y., Yao J., Lee C.-C., Wu H.-J., Sahin A. A., Allison J. P., Yu D., Hortobagyi G. N., Hung M.-C., Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 7, 12632 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim S.-O., Li C.-W., Xia W., Cha J.-H., Chan L.-C., Wu Y., Chang S.-S., Lin W.-C., Hsu J.-M., Hsu Y.-H., Kim T., Chang W.-C., Hsu J. L., Yamaguchi H., Ding Q., Wang Y., Yang Y., Chen C.-H., Sahin A. A., Yu D., Hortobagyi G. N., Hung M.-C., Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 30, 925–939 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li C.-W., Lim S.-O., Chung E. M., Kim Y.-S., Park A. H., Yao J., Cha J.-H., Xia W., Chan L.-C., Kim T., Chang S.-S., Lee H.-H., Chou C.-K., Liu Y.-L., Yeh H.-C., Perillo E. P., Dunn A. K., Kuo C.-W., Khoo K.-H., Hsu J. L., Wu Y., Hsu J.-M., Yamaguchi H., Huang T.-H., Sahin A. A., Hortobagyi G. N., Yoo S. S., Hung M.-C., Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell 33, 187–201.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takeuchi M., Doi T., Obayashi K., Hirai A., Yoneda K., Tanaka F., Iwai Y., Soluble PD-L1 with PD-1-binding capacity exists in the plasma of patients with non-small cell lung cancer. Immunol. Lett. 196, 155–160 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Chen D., Tan S., Zhang H., Wang H., He W., Shi R., Tong Z., Zhu J., Cheng H., Gao S., Chai Y., Qi J., Xiao M., Yan J., Gao G. F., The FG loop of PD-1 serves as a “Hotspot” for therapeutic monoclonal antibodies in tumor immune checkpoint therapy. iScience 14, 113–124 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tan S., Zhang H., Chai Y., Song H., Tong Z., Wang Q., Qi J., Wong G., Zhu X., Liu W. J., Gao S., Wang Z., Shi Y., Yang F., Gao G. F., Yan J., An unexpected N-terminal loop in PD-1 dominates binding by nivolumab. Nat. Commun. 8, 14369 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Na Z., Yeo S. P., Bharath S. R., Bowler M. W., Balıkçı E., Wang C.-I., Song H., Structural basis for blocking PD-1-mediated immune suppression by therapeutic antibody pembrolizumab. Cell Res. 27, 147–150 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burr M. L., Sparbier C. E., Chan Y.-C., Williamson J. C., Woods K., Beavis P. A., Lam E. Y. N., Henderson M. A., Bell C. C., Stolzenburg S., Gilan O., Bloor S., Noori T., Morgens D. W., Bassik M. C., Neeson P. J., Behren A., Darcy P. K., Dawson S.-J., Voskoboinik I., Trapani J. A., Cebon J., Lehner P. J., Dawson M. A., CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mezzadra R., Sun C., Jae L. T., Gomez-Eerland R., de Vries E., Wu W., Logtenberg M. E. W., Slagter M., Rozeman E. A., Hofland I., Broeks A., Horlings H. M., Wessels L. F. A., Blank C. U., Xiao Y., Heck A. J. R., Borst J., Brummelkamp T. R., Schumacher T. N. M., Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549, 106–110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frigola X., Inman B. A., Lohse C. M., Krco C. J., Cheville J. C., Thompson R. H., Leibovich B., Blute M. L., Dong H., Kwon E. D., Identification of a soluble form of B7-H1 that retains immunosuppressive activity and is associated with aggressive renal cell carcinoma. Clin. Cancer Res. 17, 1915–1923 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frigola X., Inman B. A., Krco C. J., Liu X., Harrington S. M., Bulur P. A., Dietz A. B., Dong H., Kwon E. D., Soluble B7-H1: Differences in production between dendritic cells and T cells. Immunol. Lett. 142, 78–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y., Wang Q., Shi B., Xu P., Hu Z., Bai L., Zhang X., Development of a sandwich ELISA for evaluating soluble PD-L1 (CD274) in human sera of different ages as well as supernatants of PD-L1+ cell lines. Cytokine 56, 231–238 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Zhou J., Mahoney K. M., Giobbie-Hurder A., Zhao F., Lee S., Liao X., Rodig S., Li J., Wu X., Butterfield L. H., Piesche M., Manos M. P., Eastman L. M., Dranoff G., Freeman G. J., Hodi F. S., Soluble PD-L1 as a biomarker in malignant melanoma treated with checkpoint blockade. Cancer Immunol. Res. 5, 480–492 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okuma Y., Hosomi Y., Nakahara Y., Watanabe K., Sagawa Y., Homma S., High plasma levels of soluble programmed cell death ligand 1 are prognostic for reduced survival in advanced lung cancer. Lung Cancer 104, 1–6 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Kruger S., Legenstein M.-L., Rösgen V., Haas M., Modest D. P., Westphalen C. B., Ormanns S., Kirchner T., Heinemann V., Holdenrieder S., Boeck S., Serum levels of soluble programmed death protein 1 (sPD-1) and soluble programmed death ligand 1 (sPD-L1) in advanced pancreatic cancer. Oncoimmunology 6, e1310358 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahoney K. M., Shukla S. A., Patsoukis N., Chaudhri A., Browne E. P., Arazi A., Eisenhaure T. M., Pendergraft W. F. III, Hua P., Pham H. C., Bu X., Zhu B., Hacohen N., Fritsch E. F., Boussiotis V. A., Wu C. J., Freeman G. J., A secreted PD-L1 splice variant that covalently dimerizes and mediates immunosuppression. Cancer Immunol. Immunother. 68, 421–432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X., Schwartz J.-C. D., Guo X., Bhatia S., Cao E., Lorenz M., Cammer M., Chen L., Zhang Z.-Y., Edidin M. A., Nathenson S. G., Almo S. C., Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 20, 337–347 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Butte M. J., Keir M. E., Phamduy T. B., Sharpe A. H., Freeman G. J., Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 27, 111–122 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin D. Y.-W., Tanaka Y., Iwasaki M., Gittis A. G., Su H.-P., Mikami B., Okazaki T., Honjo T., Minato N., Garboczi D. N., The PD-1/PD-L1 complex resembles the antigen-binding Fv domains of antibodies and T cell receptors. Proc. Natl. Acad. Sci. U.S.A. 105, 3011–3016 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lázár-Molnár E., Yan Q., Cao E., Ramagopal U., Nathenson S. G., Almo S. C., Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc. Natl. Acad. Sci. U.S.A. 105, 10483–10488 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Y., Liu P., Gao F., Cheng H., Qi J., Gao G. F., A dimeric structure of PD-L1: Functional units or evolutionary relics? Protein Cell 1, 153–160 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng X., Veverka V., Radhakrishnan A., Waters L. C., Muskett F. W., Morgan S. H., Huo J., Yu C., Evans E. J., Leslie A. J., Griffiths M., Stubberfield C., Griffin R., Henry A. J., Jansson A., Ladbury J. E., Ikemizu S., Carr M. D., Davis S. J., Structure and interactions of the human programmed cell death 1 receptor. J. Biol. Chem. 288, 11771–11785 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zak K. M., Kitel R., Przetocka S., Golik P., Guzik K., Musielak B., Dömling A., Dubin G., Holak T. A., Structure of the complex of human programmed death 1, PD-1, and its ligand PD-L1. Structure 23, 2341–2348 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lázár-Molnár E., Scandiuzzi L., Basu I., Quinn T., Sylvestre E., Palmieri E., Ramagopal U. A., Nathenson S. G., Guha C., Almo S. C., Structure-guided development of a high-affinity human Programmed Cell Death-1: Implications for tumor immunotherapy. EBioMedicine 17, 30–44 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwartz J.-C. D., Zhang X., Nathenson S. G., Almo S. C., Structural mechanisms of costimulation. Nat. Immunol. 3, 427–434 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Ikemizu S., Gilbert R. J. C., Fennelly J. A., Collins A. V., Harlos K., Jones E. Y., Stuart D. I., Davis S. J., Structure and dimerization of a soluble form of B7-1. Immunity 12, 51–60 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Zhang X., Schwartz J.-C. D., Almo S. C., Nathenson S. G., Crystal structure of the receptor-binding domain of human B7-2: Insights into organization and signaling. Proc. Natl. Acad. Sci. U.S.A. 100, 2586–2591 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zak K. M., Grudnik P., Guzik K., Zieba B. J., Musielak B., Dömling A., Dubin G., Holak T. A., Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 7, 30323–30335 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guzik K., Zak K. M., Grudnik P., Magiera K., Musielak B., Törner R., Skalniak L., Dömling A., Dubin G., Holak T. A., Small-molecule inhibitors of the programmed cell death-1/programmed death-ligand 1 (PD-1/PD-L1) interaction via transiently induced protein states and dimerization of PD-L1. J. Med. Chem. 60, 5857–5867 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Skalniak L., Zak K. M., Guzik K., Magiera K., Musielak B., Pachota M., Szelazek B., Kocik J., Grudnik P., Tomala M., Krzanik S., Pyrc K., Dömling A., Dubin G., Holak T. A., Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 8, 72167–72181 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao Y., Harrison D. L., Song Y., Ji J., Huang J., Hui E., Antigen-presenting cell-intrinsic PD-1 neutralizes PD-L1 in cis to attenuate PD-1 signaling in T cells. Cell Rep. 24, 379–390.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park J.-J., Omiya R., Matsumura Y., Sakoda Y., Kuramasu A., Augustine M. M., Yao S., Tsushima F., Narazaki H., Anand S., Liu Y., Strome S. E., Chen L., Tamada K., B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood 116, 1291–1298 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haile S. T., Bosch J. J., Agu N. I., Zeender A. M., Somasundaram P., Srivastava M. K., Britting S., Wolf J. B., Ksander B. R., Ostrand-Rosenberg S., Tumor cell programmed death ligand 1-mediated T cell suppression is overcome by coexpression of CD80. J. Immunol. 186, 6822–6829 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaudhri A., Xiao Y., Klee A. N., Wang X., Zhu B., Freeman G. J., PD-L1 binds to B7-1 only in cis on the same cell surface. Cancer Immunol. Res. 6, 921–929 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sugiura D., Maruhashi T., Okazaki I.-m., Shimizu K., Maeda T. K., Takemoto T., Okazaki T., Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 364, 558–566 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Zhao Y., Lee C. K., Lin C.-H., Gassen R. B., Xu X., Huang Z., Xiao C., Bonorino C., Lu L.-F., Bui J. D., Hui E., PD-L1:CD80 Cis-heterodimer triggers the co-stimulatory receptor CD28 while repressing the inhibitory PD-1 and CTLA-4 pathways. Immunity 51, 1059–1073.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wing K., Onishi Y., Prieto-Martin P., Yamaguchi T., Miyara M., Fehervari Z., Nomura T., Sakaguchi S., CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Hou T. Z., Qureshi O. S., Wang C. J., Baker J., Young S. P., Walker L. S. K., Sansom D. M., A transendocytosis model of CTLA-4 function predicts its suppressive behavior on regulatory T cells. J. Immunol. 194, 2148–2159 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tseng S.-Y., Otsuji M., Gorski K., Huang X., Slansky J. E., Pai S. I., Shalabi A., Shin T., Pardoll D. M., Tsuchiya H., B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 193, 839–846 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiao Y., Yu S., Zhu B., Bedoret D., Bu X., Francisco L. M., Hua P., Duke-Cohan J. S., Umetsu D. T., Sharpe A. H., DeKruyff R. H., Freeman G. J., RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J. Exp. Med. 211, 943–959 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nie X., Chen W., Zhu Y., Huang B., Yu W., Wu Z., Guo S., Zhu Y., Luo L., Wang S., Chen L., B7-DC (PD-L2) costimulation of CD4+ T-helper 1 response via RGMb. Cell. Mol. Immunol. 15, 888–897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shinohara T., Taniwaki M., Ishida Y., Kawaichi M., Honjo T., Structure and chromosomal localization of the human PD-1 gene (PDCD1). Genomics 23, 704–706 (1994). [DOI] [PubMed] [Google Scholar]
- 61.Okazaki T., Maeda A., Nishimura H., Kurosaki T., Honjo T., PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl. Acad. Sci. U.S.A. 98, 13866–13871 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chemnitz J. M., Parry R. V., Nichols K. E., June C. H., Riley J. L., SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J. Immunol. 173, 945–954 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Yokosuka T., Takamatsu M., Kobayashi-Imanishi W., Hashimoto-Tane A., Azuma M., Saito T., Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 209, 1201–1217 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bardhan K., Aksoylar H.-I., Le Bourgeois T., Strauss L., Weaver J. D., Delcuze B., Charest A., Patsoukis N., Boussiotis V. A., Phosphorylation of PD-1-Y248 is a marker of PD-1-mediated inhibitory function in human T cells. Sci. Rep. 9, 17252 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pluskey S., Wandless T. J., Walsh C. T., Shoelson S. E., Potent stimulation of SH-PTP2 phosphatase activity by simultaneous occupancy of both SH2 domains. J. Biol. Chem. 270, 2897–2900 (1995). [DOI] [PubMed] [Google Scholar]
- 66.Hof P., Pluskey S., Dhe-Paganon S., Eck M. J., Shoelson S. E., Crystal structure of the tyrosine phosphatase SHP-2. Cell 92, 441–450 (1998). [DOI] [PubMed] [Google Scholar]
- 67.Neel B. G., Gu H., Pao L., The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28, 284–293 (2003). [DOI] [PubMed] [Google Scholar]
- 68.Peled M., Tocheva A. S., Sandigursky S., Nayak S., Philips E. A., Nichols K. E., Strazza M., Azoulay-Alfaguter I., Askenazi M., Neel B. G., Pelzek A. J., Ueberheide B., Mor A., Affinity purification mass spectrometry analysis of PD-1 uncovers SAP as a new checkpoint inhibitor. Proc. Natl. Acad. Sci. U.S.A. 115, E468–E477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marasco M., Berteotti A., Weyershaeuser J., Thorausch N., Sikorska J., Krausze J., Brandt H. J., Kirkpatrick J., Rios P., Schamel W. W., Köhn M., Carlomagno T., Molecular mechanism of SHP2 activation by PD-1 stimulation. Sci. Adv. 6, eaay4458 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patsoukis N., Duke-Cohan J. S., Chaudhri A., Aksoylar H.-I., Wang Q., Council A., Berg A., Freeman G. J., Boussiotis V. A., Interaction of SHP-2 SH2 domains with PD-1 ITSM induces PD-1 dimerization and SHP-2 activation. Commun. Biol. 3, 128 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sheppard K. A., Fitz L. J., Lee J. M., Benander C., George J. A., Wooters J., Qiu Y., Jussif J. M., Carter L. L., Wood C. R., Chaudhary D., PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 574, 37–41 (2004). [DOI] [PubMed] [Google Scholar]
- 72.Patsoukis N., Brown J., Petkova V., Liu F., Li L., Boussiotis V. A., Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 5, ra46 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Patsoukis N., Li L., Sari D., Petkova V., Boussiotis V. A., PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol. Cell. Biol. 33, 3091–3098 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mizuno R., Sugiura D., Shimizu K., Maruhashi T., Watada M., Okazaki I.-m., Okazaki T., PD-1 primarily targets TCR signal in the inhibition of functional T cell activation. Front. Immunol. 10, 630 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hui E., Cheung J., Zhu J., Su X., Taylor M. J., Wallweber H. A., Sasmal D. K., Huang J., Kim J. M., Mellman I., Vale R. D., T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science 355, 1428–1433 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Celis-Gutierrez J., Blattmann P., Zhai Y., Jarmuzynski N., Ruminski K., Grégoire C., Ounoughene Y., Fiore F., Aebersold R., Roncagalli R., Gstaiger M., Malissen B., Quantitative interactomics in primary T cells provides a rationale for concomitant PD-1 and BTLA coinhibitor blockade in cancer immunotherapy. Cell Rep. 27, 3315–3330.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rota G., Niogret C., Dang A. T., Barros C. R., Fonta N. P., Alfei F., Morgado L., Zehn D., Birchmeier W., Vivier E., Guarda G., Shp-2 is dispensable for establishing T cell exhaustion and for PD-1 signaling in vivo. Cell Rep. 23, 39–49 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Zhang J., Somani A.-K., Siminovitch K. A., Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin. Immunol. 12, 361–378 (2000). [DOI] [PubMed] [Google Scholar]
- 79.Frearson J. A., Alexander D. R., The phosphotyrosine phosphatase SHP-2 participates in a multimeric signaling complex and regulates T cell receptor (TCR) coupling to the Ras/mitogen-activated protein kinase (MAPK) pathway in Jurkat T cells. J. Exp. Med. 187, 1417–1426 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tailor P., Jascur T., Williams S., von Willebrand M., Couture C., Mustelin T., Involvement of Src-homology-2-domain-containing protein-tyrosine phosphatase 2 in T cell activation. Eur. J. Biochem. 237, 736–742 (1996). [DOI] [PubMed] [Google Scholar]
- 81.Nguyen T. V., Ke Y., Zhang E. E., Feng G.-S., Conditional deletion of Shp2 tyrosine phosphatase in thymocytes suppresses both pre-TCR and TCR signals. J. Immunol. 177, 5990–5996 (2006). [DOI] [PubMed] [Google Scholar]
- 82.Presotto D., Erdes E., Duong M. N., Allard M., Regamey P.-O., Quadroni M., Doucey M.-A., Rufer N., Hebeisen M., Fine-tuning of optimal TCR signaling in tumor-redirected CD8 T cells by distinct TCR affinity-mediated mechanisms. Front. Immunol. 8, 1564 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Meng T.-C., Fukada T., Tonks N. K., Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell 9, 387–399 (2002). [DOI] [PubMed] [Google Scholar]
- 84.Kwon J., Qu C.-K., Maeng J.-S., Falahati R., Lee C., Williams M. S., Receptor-stimulated oxidation of SHP-2 promotes T-cell adhesion through SLP-76-ADAP. EMBO J. 24, 2331–2341 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Choi S., Warzecha C., Zvezdova E., Lee J., Argenty J., Lesourne R., Aravind L., Love P. E., THEMIS enhances TCR signaling and enables positive selection by selective inhibition of the phosphatase SHP-1. Nat. Immunol. 18, 433–441 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kleffel S., Posch C., Barthel S. R., Mueller H., Schlapbach C., Guenova E., Elco C. P., Lee N., Juneja V. R., Zhan Q., Lian C. G., Thomi R., Hoetzenecker W., Cozzio A., Dummer R., Mihm M. C. Jr., Flaherty K. T., Frank M. H., Murphy G. F., Sharpe A. H., Kupper T. S., Schatton T., Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell 162, 1242–1256 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schatton T., Murphy G. F., Frank N. Y., Yamaura K., Waaga-Gasser A. M., Gasser M., Zhan Q., Jordan S., Duncan L. M., Weishaupt C., Fuhlbrigge R. C., Kupper T. S., Sayegh M. H., Frank M. H., Identification of cells initiating human melanomas. Nature 451, 345–349 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carter L., Fouser L. A., Jussif J., Fitz L., Deng B., Wood C. R., Collins M., Honjo T., Freeman G. J., Carreno B. M., PD-1:PD-L inhibitory pathway affects both CD4+ and CD8+ T cells and is overcome by IL-2. Eur. J. Immunol. 32, 634–643 (2002). [DOI] [PubMed] [Google Scholar]
- 89.Bennett F., Luxenberg D., Ling V., Wang I.-M., Marquette K., Lowe D., Khan N., Veldman G., Jacobs K. A., Valge-Archer V. E., Collins M., Carreno B. M., Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: Attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J. Immunol. 170, 711–718 (2003). [DOI] [PubMed] [Google Scholar]
- 90.Lynn R. C., Weber E. W., Sotillo E., Gennert D., Xu P., Good Z., Anbunathan H., Lattin J., Jones R., Tieu V., Nagaraja S., Granja J., de Bourcy C. F. A., Majzner R., Satpathy A. T., Quake S. R., Monje M., Chang H. Y., Mackall C. L., c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 576, 293–300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shi J., Hou S., Fang Q., Liu X., Liu X., Qi H., PD-1 controls follicular T helper cell positioning and function. Immunity 49, 264–274.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lin H., Wei S., Hurt E. M., Green M. D., Zhao L., Vatan L., Szeliga W., Herbst R., Harms P. W., Fecher L. A., Vats P., Chinnaiyan A. M., Lao C. D., Lawrence T. S., Wicha M., Hamanishi J., Mandai M., Kryczek I., Zou W., Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Invest. 128, 805–815 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tang H., Liang Y., Anders R. A., Taube J. M., Qiu X., Mulgaonkar A., Liu X., Harrington S. M., Guo J., Xin Y., Xiong Y., Nham K., Silvers W., Hao G., Sun X., Chen M., Hannan R., Qiao J., Dong H., Peng H., Fu Y.-X., PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J. Clin. Invest. 128, 580–588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Azuma T., Yao S., Zhu G., Flies A. S., Flies S. J., Chen L., B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 111, 3635–3643 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Clark C. A., Gupta H. B., Sareddy G., Pandeswara S., Lao S., Yuan B., Drerup J. M., Padron A., Conejo-Garcia J., Murthy K., Liu Y., Turk M. J., Thedieck K., Hurez V., Li R., Vadlamudi R., Curiel T. J., Tumor-intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res. 76, 6964–6974 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gato-Cañas M., Zuazo M., Arasanz H., Ibañez-Vea M., Lorenzo L., Fernandez-Hinojal G., Vera R., Smerdou C., Martisova E., Arozarena I., Wellbrock C., Llopiz D., Ruiz M., Sarobe P., Breckpot K., Kochan G., Escors D., PDL1 signals through conserved sequence motifs to overcome interferon-mediated cytotoxicity. Cell Rep. 20, 1818–1829 (2017). [DOI] [PubMed] [Google Scholar]
- 97.Jalali S., Price-Troska T., Bothun C., Villasboas J., Kim H.-J., Yang Z.-Z., Novak A. J., Dong H., Ansell S. M., Reverse signaling via PD-L1 supports malignant cell growth and survival in classical Hodgkin lymphoma. Blood Cancer J. 9, 22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu X., Wu X., Cao S., Harrington S. M., Yin P., Mansfield A. S., Dong H., B7-H1 antibodies lose antitumor activity due to activation of p38 MAPK that leads to apoptosis of tumor-reactive CD8+ T cells. Sci. Rep. 6, 36722 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Diskin B., Adam S., Cassini M. F., Sanchez G., Liria M., Aykut B., Buttar C., Li E., Sundberg B., Salas R. D., Chen R., Wang J., Kim M., Farooq M. S., Nguy S., Fedele C., Tang K. H., Chen T., Wang W., Hundeyin M., Rossi J. A. K., Kurz E., Haq M. I. U., Karlen J., Kruger E., Sekendiz Z., Wu D., Shadaloey S. A. A., Baptiste G., Werba G., Selvaraj S., Loomis C., Wong K.-K., Leinwand J., Miller G., PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat. Immunol. 21, 442–454 (2020). [DOI] [PubMed] [Google Scholar]
- 100.Ni X., Song Q., Cassady K., Deng R., Jin H., Zhang M., Dong H., Forman S., Martin P. J., Chen Y.-Z., Wang J., Zeng D., PD-L1 interacts with CD80 to regulate graft-versus-leukemia activity of donor CD8+ T cells. J. Clin. Invest. 127, 1960–1977 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.MacIver N. J., Michalek R. D., Rathmell J. C., Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 31, 259–283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang R., Dillon C. P., Shi L. Z., Milasta S., Carter R., Finkelstein D., McCormick L. L., Fitzgerald P., Chi H., Munger J., Green D. R., The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Patsoukis N., Bardhan K., Chatterjee P., Sari D., Liu B., Bell L. N., Karoly E. D., Freeman G. J., Petkova V., Seth P., Li L., Boussiotis V. A., PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 6, 6692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Parry R. V., Chemnitz J. M., Frauwirth K. A., Lanfranco A. R., Braunstein I., Kobayashi S. V., Linsley P. S., Thompson C. B., Riley J. L., CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 25, 9543–9553 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Michalek R. D., Gerriets V. A., Jacobs S. R., Macintyre A. N., Maciver N. J., Mason E. F., Sullivan S. A., Nichols A. G., Rathmell J. C., Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shi L. Z., Wang R., Huang G., Vogel P., Neale G., Green D. R., Chi H., HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 208, 1367–1376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Francisco L. M., Salinas V. H., Brown K. E., Vanguri V. K., Freeman G. J., Kuchroo V. K., Sharpe A. H., PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 206, 3015–3029 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sena L. A., Li S., Jairaman A., Prakriya M., Ezponda T., Hildeman D. A., Wang C.-R., Schumacker P. T., Licht J. D., Perlman H., Bryce P. J., Chandel N. S., Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Belikov A. V., Schraven B., Simeoni L., T cells and reactive oxygen species. J. Biomed. Sci. 22, 85 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bhalla K., Hwang B. J., Dewi R. E., Ou L., Twaddel W., Fang H.-b., Vafai S. B., Vazquez F., Puigserver P., Boros L., Girnun G. D., PGC1α promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 71, 6888–6898 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.St-Pierre J., Drori S., Uldry M., Silvaggi J. M., Rhee J., Jäger S., Handschin C., Zheng K., Lin J., Yang W., Simon D. K., Bachoo R., Spiegelman B. M., Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127, 397–408 (2006). [DOI] [PubMed] [Google Scholar]
- 112.Scharping N. E., Menk A. V., Moreci R. S., Whetstone R. D., Dadey R. E., Watkins S. C., Ferris R. L., Delgoffe G. M., The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45, 374–388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chamoto K., Chowdhury P. S., Kumar A., Sonomura K., Matsuda F., Fagarasan S., Honjo T., Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc. Natl. Acad. Sci. U.S.A. 114, E761–E770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Maj T., Wang W., Crespo J., Zhang H., Wang W., Wei S., Zhao L., Vatan L., Shao I., Szeliga W., Lyssiotis C., Liu J. R., Kryczek I., Zou W., Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 18, 1332–1341 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Huang X., Venet F., Wang Y. L., Lepape A., Yuan Z., Chen Y., Swan R., Kherouf H., Monneret G., Chung C.-S., Ayala A., PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl. Acad. Sci. U.S.A. 106, 6303–6308 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yu Y., Tsang J. C. H., Wang C., Clare S., Wang J., Chen X., Brandt C., Kane L., Campos L. S., Lu L., Belz G. T., McKenzie A. N. J., Teichmann S. A., Dougan G., Liu P., Single-cell RNA-seq identifies a PD-1hi ILC progenitor and defines its development pathway. Nature 539, 102–106 (2016). [DOI] [PubMed] [Google Scholar]
- 117.Shen L., Gao Y., Liu Y., Zhang B., Liu Q., Wu J., Fan L., Ou Q., Zhang W., Shao L., PD-1/PD-L pathway inhibits M.tb-specific CD4+ T-cell functions and phagocytosis of macrophages in active tuberculosis. Sci. Rep. 6, 38362 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pesce S., Greppi M., Tabellini G., Rampinelli F., Parolini S., Olive D., Moretta L., Moretta A., Marcenaro E., Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 139, 335–346.e3 (2017). [DOI] [PubMed] [Google Scholar]
- 119.Hsu J., Hodgins J. J., Marathe M., Nicolai C. J., Bourgeois-Daigneault M.-C., Trevino T. N., Azimi C. S., Scheer A. K., Randolph H. E., Thompson T. W., Zhang L., Iannello A., Mathur N., Jardine K. E., Kirn G. A., Bell J. C., McBurney M. W., Raulet D. H., Ardolino M., Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Invest. 128, 4654–4668 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Oldenhove G., Boucquey E., Taquin A., Acolty V., Bonetti L., Ryffel B., Le Bert M., Englebert K., Boon L., Moser M., PD-1 is involved in the dysregulation of type 2 innate lymphoid cells in a murine model of obesity. Cell Rep. 25, 2053–2060.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 121.Moral J. A., Leung J., Rojas L. A., Ruan J., Zhao J., Sethna Z., Ramnarain A., Gasmi B., Gururajan M., Redmond D., Askan G., Bhanot U., Elyada E., Park Y., Tuveson D. A., Gönen M., Leach S. D., Wolchok J. D., DeMatteo R. P., Merghoub T., Balachandran V. P., ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature 579, 130–135 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen W., Wang J., Jia L., Liu J., Tian Y., Attenuation of the programmed cell death-1 pathway increases the M1 polarization of macrophages induced by zymosan. Cell Death Dis. 7, e2115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gordon S. R., Maute R. L., Dulken B. W., Hutter G., George B. M., McCracken M. N., Gupta R., Tsai J. M., Sinha R., Corey D., Ring A. M., Connolly A. J., Weissman I. L., PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Qorraj M., Bruns H., Böttcher M., Weigand L., Saul D., Mackensen A., Jitschin R., Mougiakakos D., The PD-1/PD-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia 31, 470–478 (2017). [DOI] [PubMed] [Google Scholar]
- 125.Gabrilovich D. I., Ostrand-Rosenberg S., Bronte V., Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 12, 253–268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Strauss L., Mahmoud M. A. A., Weaver J. D., Tijaro-Ovalle N. M., Christofides A., Wang Q., Pal R., Yuan M., Asara J., Patsoukis N., Boussiotis V. A., Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 5, eaay1863 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tall A. R., Yvan-Charvet L., Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 15, 104–116 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nagareddy P. R., Murphy A. J., Stirzaker R. A., Hu Y., Yu S., Miller R. G., Ramkhelawon B., Distel E., Westerterp M., Huang L.-S., Schmidt A. M., Orchard T. J., Fisher E. A., Tall A. R., Goldberg I. J., Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 17, 695–708 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nagareddy P. R., Kraakman M., Masters S. L., Stirzaker R. A., Gorman D. J., Grant R. W., Dragoljevic D., Hong E. S., Abdel-Latif A., Smyth S. S., Choi S. H., Korner J., Bornfeldt K. E., Fisher E. A., Dixit V. D., Tall A. R., Goldberg I. J., Murphy A. J., Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 19, 821–835 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Makki K., Froguel P., Wolowczuk I., Adipose tissue in obesity-related inflammation and insulin resistance: Cells, cytokines, and chemokines. ISRN Inflamm. 2013, 139239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sharma M., Boytard L., Hadi T., Koelwyn G., Simon R., Ouimet M., Seifert L., Spiro W., Yan B., Hutchison S., Fisher E. A., Ramasamy R., Ramkhelawon B., Moore K. J., Enhanced glycolysis and HIF-1α activation in adipose tissue macrophages sustains local and systemic interleukin-1β production in obesity. Sci. Rep. 10, 5555 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Salminen A., Kaarniranta K., Kauppinen A., Immunosenescence: The potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell. Mol. Life Sci. 76, 1901–1918 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]