

Vibrational quantum dynamics of conical intersection mediating singlet fission provides mechanistic insight on a structural level.
Abstract
Singlet fission is a spin-allowed exciton multiplication process in organic semiconductors that converts one spin-singlet exciton to two triplet excitons. It offers the potential to enhance solar energy conversion by circumventing the Shockley-Queisser limit on efficiency. We study the primary steps of singlet fission in a pentacene film by using a combination of TG and 2D electronic spectroscopy complemented by quantum chemical and nonadiabatic dynamics calculations. We show that the coherent vibrational dynamics induces the ultrafast transition from the singlet excited electronic state to the triplet-pair state via a degeneracy of potential energy surfaces, i.e., a multidimensional conical intersection. Significant vibronic coupling of the electronic wave packet to a few key intermolecular rocking modes in the low-frequency region connect the excited singlet and triplet-pair states. Along with high-frequency local vibrations acting as tuning modes, they open a new channel for the ultrafast exciton transfer through the resulting conical intersection.
INTRODUCTION
Conversion of sunlight into electrical current with photovoltaic cells is one of the promising technologies to harness renewable energy. Concerted research efforts are directed to obtain cost-effective third-generation photovoltaics with improved power conversion efficiency. Singlet fission (SF) (1) is an exciton multiplication process, which could be used to overcome the fundamental thermodynamic constraint on efficiency, known as the Shockley-Queisser limit (∼30% for an ideal single-junction silicon cell) (2). The SF process has been widely observed in acenes (3), diphenylisobenzofurans (4), carotenoids (5), and other conjugated molecules (6, 7). In the SF process, a photoexcited singlet exciton (S1) is converted to two triplet excitons (2 × T1), which requires another electron in the S0 state of an adjacent molecule to be promoted to an excited electronic state. The generally accepted mechanism of SF involves three steps: (i) primary SF (PSF), which is a spin-conserved step; (ii) spatially separated triplet-pair state (SSP); and (iii) spin decoherence (SD) processes
| (1) |
There has been a persistent debate in the literature about the nature of the intermediates in the SF process (8). Recent consensus is that in PSF, the singlet excited state (S1) is first converted into a spin-correlated triplet pair 1(T1T1), which further forms an SSP 1(T1⋯T1) with an overall singlet spin retained in the process. The final process leading to the generation of free triplet excitons (T1 + T1) from the interacting triplet pair 1(T1⋯T1) happens via spin decoherence (9, 10). If the energy levels in the system are such that 2E(T1) < E(S1), then SF is an overall exothermic process. On the other hand, if 2E(T1) > E(S1), then the SF process is endothermic. The detailed molecular insight of the process holds the key to the realization of synthetic control over the process. Thus, it becomes imperative to reveal the underlying mechanisms of the SF processes in different molecular systems.
Pentacene and its derivatives have shown great promise as potential SF-based materials for photovoltaics. Baldo and co-workers (11, 12) have demonstrated an external quantum efficiency of >126% based on pentacene/C60 in photovoltaics. This level of external quantum efficiency for charge collection has fueled the great interest in pentacene and its derivatives in determining the underlying photophysical mechanisms for the SF process (13). In crystals of pentacene and its derivatives, SF is an exothermic process, which makes SF in pentacene energetically favorable. The process of PSF occurs on the time scale of 100 fs to form the spin-correlated triplet pair 1(T1T1) (13, 14). This 1(T1T1) pair is shown to remain bound for hundreds of picoseconds before spin decoherence forms 2 × T1 (15). There have been numerous studies to unravel the dynamics of the 1(T1T1) formation. In 2012, Zhu and co-workers (16) have reported the direct observation of the 1(T1T1) state using time-resolved two-photon photoemission spectroscopy. They have measured ∼20-fs rise time in the triplet population in both tetracene and pentacene, which they attributed to the formation of the multiexciton state. Motivated by this, Berkelbach et al. (17–19) proposed a microscopic theory to study the coherent dynamics in singlet exciton fission. However, first-principles calculations suggested that, instead of the superexchange between the singlet and the triplet pair, strong electronic couplings to charge transfer states could possibly mediate the evolution of the singlet exciton to the multiexciton state (20). Moreover, a phenomenological model has been constructed to reproduce the time scale of SF in TIPS [6,13-bis(triisopropylsilylethynyl)]–pentacene (21). These results support a scenario where SF is mediated by a coherent superexchange mechanism via an energetically higher-lying charge transfer state. In addition, the role of the charge transfer state in the PSF has been further advocated by studies on intramolecular SF in herringbone-packed SF materials (22–24) and pentacene crystals (25).
In contrast to these ideas, recently, vibronic coherence has been considered as a mediator between the singlet and triplet-pair states. On the basis of a vibrational analysis, Kukura and co-workers (26) have proposed that the ultrafast SF in pentacene and its derivatives is realized by a conical intersection (CI) between the singlet exciton and triplet-pair state. In this work, however, the details of the vibrational dynamics remained elusive. Moreover, two-dimensional (2D) electronic spectroscopy has been applied to study the SF of a pentacene crystal (27). In this study, intramolecular ground-state vibrations are identified, and the associated vibronic coherence has been considered to trigger the PSF from the singlet to triplet-pair states. A vibronic dimer model has been constructed on the basis of intramolecular vibrations to fit the time scale of PSF (28). In addition, this vibronic coherence has also been reported in tetracene (29) and hexacene (3). The dynamics of SF in rubrene has been examined by transient absorption spectroscopy; the results demonstrated that the PSF is induced by a symmetry-breaking mode (30). Apparently, despite these enormous theoretical and experimental efforts, the understanding of the primary step of SF, i.e., the dynamics of the formation of the 1(T1T1) state and the role of intermolecular vibrations in pentacene remained elusive.
To clearly distinguish the different mechanistic aspects of ultrafast SF in pentacene, we examine the process of PSF in a pentacene film using a combination of ultrafast heterodyne-detected transient-grating (TG) and 2D electronic spectroscopy. Because of the near-zero background and the associated higher sensitivity relative to pump-probe methods (31), TG spectroscopy allows us to directly capture the small signal modulations because of the nuclear vibrational coherence that induces the transition from the singlet exciton to the triplet-pair state. On the basis of a vibrational analysis, several key modes are identified in the low- and high-frequency region, which trigger SF, i.e., the transition from the singlet exciton to the triplet-pair state. Moreover, the SF process is further examined by 2D electronic spectroscopy, and the associated correlation analysis excludes any functional role of electronic coherence and reveals the existence of vibronic coherence connecting the singlet exciton and triplet-pair state (32). Quantum chemistry calculations reveal the significant strength of the vibronic coupling of the molecular rocking vibrations along the longitudinal molecular axis in a pentacene dimer, which markedly modulates the electronic coupling between the singlet exciton and triplet-pair state. They play the role of intermolecular vibrations. With the assistance of vibronic couplings to intramolecular vibrational modes, intermolecular vibrations open up new channels, which induce SF in pentacene on an ultrafast time scale.
RESULTS
Thin films of pentacene were prepared on 2-mm-thick quartz substrates using the thermal evaporation method. The details of the sample preparation and characterization are given in Materials and Methods. Figure 1A represents the ground-state absorption spectrum (red circles) of a 200-nm-thin pentacene film at room temperature (296 K). The lowest singlet state for pentacene lies at 677 nm (∼14,800 cm−1) (27). To perform transient spectroscopic measurements on these pentacene thin films, a broadband pulse is generated by a home-built nonlinear optical parametric amplifier (NOPA) and then judiciously tuned-in frequency. The spatial and temporal chirp of the pulse is compensated by the combination of a prism pair and a deformable mirror. The laser pulse spectrum used for the measurements is shown as the blue shaded region in Fig. 1A. Within the probe window (650 to 750 nm), the primary step of SF is monitored by capturing the ground-state bleach (GSB; S0 → S1), stimulated emission, and excited state absorption [ESA; 1(T1T1) → 1(T1T2)] features, which are illustrated in Fig. 1B. The absorption signature of S1 → Sn lies at λ < 645 nm (27), which is out of our probing range.
Fig. 1. SF in a pentacene film: 2D electronic spectroscopy and correlation analysis indicate a vibrationally driven primary step, PSF.
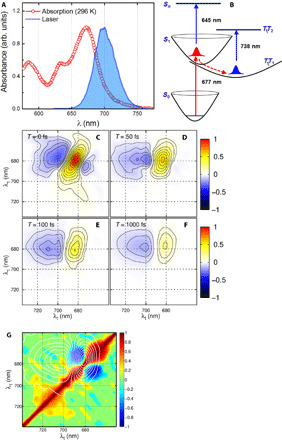
(A) Ground-state absorption spectrum of a pentacene film on quartz substrate (red dots) and laser spectrum is used in this measurement (light-blue shadow). (B) Calculated site energies of the ground, singlet excited states (S0, S1, and Sn), and triplet-pair states [1(T1T1) and 1(T1T2)]. The selected 2D electronic spectra (real part) for selected waiting times are shown from (C to F). Positive diagonal and negative off-diagonal features denote the GSB and ESA, respectively. (G) 2D correlation map obtained from a correlation analysis along the diagonal direction. Two negative peaks are shown in the region of the GSB (the 2D spectrum at T = 500 fs is shown as a white contour), which indicates the vibrational origin of the oscillations.
2D electronic spectroscopy
To examine the coherent dynamics coupled with the primary electronic transition of SF, we measure the 2D electronic spectra of the pentacene film at room temperature (296 K) (for details on the experimental conditions, see Materials and Methods). 2D electronic spectroscopy provides additional resolution along the excitation frequency axis, which helps to decongest the spectra due to different overlapping electronic transitions. In Fig. 1 (C to F), the real parts of the 2D electronic spectra are shown for selected waiting times. The GSB and ESA are separated along the excitation (λτ) and probe (λt) frequencies. At T = 0 fs, the center peak at 680 nm is strongly stretched along the diagonal direction, which illustrates the significant inhomogeneous broadening of the excited electronic transition in the pentacene film. The 2D spectrum at T = 0 fs also carries a small contribution originating from the quartz substrate (33). With increasing waiting times, the magnitude of the central diagonal peak is strongly decreased because of the decay of the stimulated-emission component (see the 2D spectrum at T = 50 fs in Fig. 1D). This decay is primarily due to the population transfer from the singlet state to the triplet-pair state (13). The ESA band in the 2D spectrum at T = 50 fs, appearing as an off-diagonal feature at (λτ, λt) = (680, 700) nm, corresponds to the 1(T1T1) → 1(T1T2) transition. These ESA features allow us to follow the coherent generation of the 1(T1T1) state and subsequent dynamics over time. To further resolve the rates of the underlying electronic transitions, several series of consecutive 2D spectra at different times have been analyzed by the global fitting approach. The obtained time scale of the kinetics and the decay-associated spectra are shown in section S2. The fastest decay component reveals a time scale of ∼100 fs, which is in good agreement with earlier reports on the PSF process in pentacene (26, 27, 34). In addition, information on the coherent dynamics can be obtained by removing the kinetics from the measured data. Here, we first perform the 2D correlation analysis to distinguish the origin of coherence based on the correlation or anticorrelation of peaks along the diagonal direction (see the Supplementary Materials for details) (35). In Fig. 1G, the 2D correlation spectrum is shown together with the overlap of the 2D electronic spectrum at T = 500 fs (white contour lines) to clarify the spectral position of the correlations. In the correlation map, two negative anticorrelation peaks with strong magnitudes are symmetrically located along the diagonal direction, which match the peak of the GSB in the 2D spectrum at T = 500 fs. Earlier theoretical work (35) has demonstrated that the anticorrelation in the 2D correlation spectrum indicates the existence of vibrational coherence, while the positive correlation corresponds to electronic coherence in the respective area of the 2D spectrum. In Fig. 1G, there is no sign of any correlation in the position of the ESA peak, which signifies the absence of electronic quantum coherence mediating the generation of the triplet-pair state. Thus, our measurement does not support the conclusion in (16).
TG spectroscopy
After excluding the role of electronic quantum coherence, we measure the TG response of the pentacene thin film to capture the wave-packet dynamics associated with the PSF transition of the singlet exciton S1 to the triplet-pair state 1(T1T1). The details of the experimental setup and the measurement conditions are described in Materials and Methods. In Fig. 2A, we show the TG spectrum with positive (centered at 685 nm) and negative bands (centered at 705 nm). The TG spectrum evolves considerably in the initial 200 fs and then is largely invariant for the remaining times. The prominent rapid decay of the signal at 685 nm has been assigned in earlier reports to the decay of the stimulated emission from the singlet exciton due to PSF (13). The residual signal at 685 nm at later times corresponds to GSB. The ESA feature centered at 705 nm corresponds to the transition 1(T1T1) → 1(T1T2). Because of the enhanced sensitivity of TG measurements to capture coherences, the oscillations riding on these transient features are clearly visible in Fig. 2A. Traces of the averaged kinetics of the GSB and the ESA regions (marked as black, 685 to 690 nm; green, 700 to 705 nm, dashed lines) are shown in Fig. 2 (B and C), respectively. To retrieve the coherent vibrational modes associated with the transient dynamics, we use the exponential function to fit the TG spectrum and then use a Fourier transform analysis of the residuals after removing the decay components at the probed frequencies (for further description of the global fitting analysis, see the Supplementary Materials). To show the excellent quality of the fits, two averaged traces (red and blue dashed lines) are shown along with the fitted decay curves in Fig. 2 (B and C). The retrieved vibrational frequencies of the GSB and the ESA are shown in Fig. 2 (D and E), respectively. The identified low- and high-frequency modes are marked. Notably, the low-frequency modes at 140 and 460 cm−1 are present in the GSB and the ESA bands. In addition, the high-frequency modes at 1206, 1300, and 1370 cm−1 are shown in the GSB and the ESA bands, which manifest the adjacent vibrational coherence during the transfer of the wave packet from the singlet to the triplet-pair state.
Fig. 2. Vibrational coherence mediating the PSF process is captured by TG measurements.
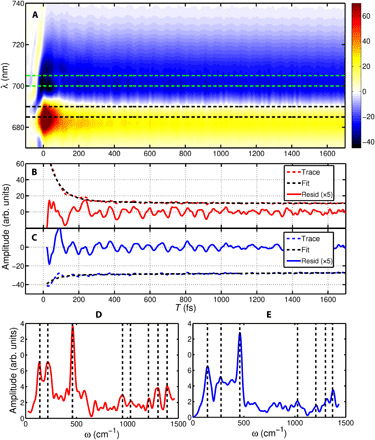
(A) Measured TG spectrum of pentacene film with a time step of 2 fs. The GSB (positive) and the ESA (negative) in the spectrum originate from the corresponding transitions in Fig. 1B, as red (S0 → S1) and blue arrows [1(T1T1) → 1(T1T2)]. The averaged kinetics of the GSB [685 to 690 nm, marked by black dashed lines in (A)] and ESA [700 to 705 nm, marked by green dashed lines in (A)] are shown as red and blue dashed lines in (B) and (C), respectively. The associated exponential fitting curves are presented as black dashed lines. The obtained residuals of the GSB and the ESA bands are magnified and shown as red and blue solid lines in (B) and (C), respectively. A Fourier transform has been performed to examine the vibrational dynamics, the obtained power spectra of GSB and ESA are shown as red and blue lines in (D) and (E), respectively. In (D), the marked dashed lines indicate the vibrational frequencies at 140, 218, 460, 953, 1030, 1206, 1300, and 1370 cm−1, respectively. In addition, the marked dashed lines in ESA show the frequencies of 140, 276, 460, 1027, 1206, 1300, and 1370 cm−1.
To explore the vibrational dynamics, we perform the wavelet analysis on the residuals of the kinetic analysis in the GSB and the ESA regions of the TG spectrum. The details of the wavelet analysis have been described in the Supplementary Materials. In Fig. 3A, the magnified residual of the GSB is shown as a red solid line. The vibrational dynamics in the high- and low-frequency ranges obtained from GSB residuals of the decay fits are presented in Fig. 3 (B and C), respectively. On the basis of the wavelet analysis, we obtain the lifetime of the vibrational coherence corresponding to the high-frequency modes to be 150 fs. However, poor frequency resolution limits our resolution of the modes, which results in a broadband oscillation at ∼1300 cm−1 due to the nonlinear scale of the frequency resolution in the wavelet analysis (marked by the black dashed arrow in Fig. 3B). The low-frequency region of the wavelet-analyzed spectrum shows a splitting of vibrational frequencies, as becomes visible in Fig. 3C. Initially, a positive peak can be seen at the frequency of 170 cm−1, which shows a linear increase in its frequency for up to 250 cm−1 within 500 fs. In addition, one new frequency at 140 cm−1 is gradually generated on an ultrafast time scale of 100 fs, which has been marked by a magenta box in Fig. 3C. This splitting of oscillations is highlighted by two black dashed arrows. The observed evolution of the two new vibrational modes at 140 and 250 cm−1 suggests changes in the vibrational frequencies after the transfer from the potential energy surfaces (PESs) of the singlet exciton state to that of the triplet-pair state. We have also performed the wavelet analysis of the residual of the ESA portion of the TG spectrum shown in Fig. 3D. We obtain, in the high-frequency range, a weak signal of an oscillation lasting for 150 fs, as shown in Fig. 3E. In Fig. 3F, the results of the wavelet analysis in the low-frequency regime of the ESA band show a vibrational mode of 140 cm−1 that lasts for a long time. It is marked by a black dashed arrow. In addition, the comparison of the phases of the vibrational oscillations in the wavelet analysis distinctly reveals opposite phases of the GSB (Fig. 3, B and C), as compared to the ESA (Fig. 3, E and F). This points to the coherent transfer of populations between different electronic states (36). Thus, based on the 2D vibrational analysis, we clearly identify a few key vibrational modes in the low- and high-frequency regime, which induce the coherent generation of the triplet-pair state 1(T1T1).
Fig. 3. Time evolution of vibrational coherences shows the structural evolution by low-frequency modes.
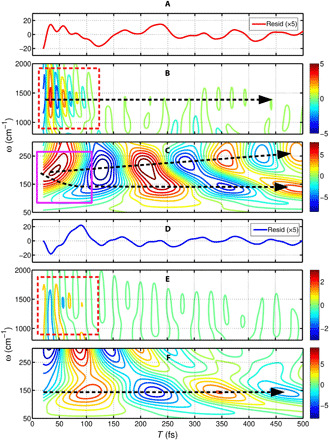
(A) Magnified residuals of the GSB band. The dynamics of each vibrational mode is revealed by the wavelet analysis. The high- and low-frequency ranges are shown in (B) and (C), respectively. In (B), we show the vibrational mode at frequency of ∼1300 cm−1 with a lifetime of 150 fs, which is highlighted by the dashed arrow. In (C), we initially find a strong vibration at 170 cm−1, which reduce its frequency to 140 cm−1 on a time scale of 100 fs (marked in magenta box). Moreover, one new frequency at 250 cm−1 is gradually generated within the initial 500 fs. (D) Magnified residuals of the ESA. The high- and low-frequency ranges of the vibrations are shown in (E) and (F), respectively. In (E), a broadband vibration (∼1300 cm−1) shows weak oscillations decaying on a time scale of 150 fs. In addition, one low-frequency mode at 140 cm−1 shows strong oscillation in the ESA band, which is highlighted by a dashed arrow.
Calculations and modeling
To assign the experimentally observed coherent vibrational dynamics, we have performed quantum chemical calculations on a pentacene pair [see Fig. 4 (A and B)]. The initial geometry of the pentacene pair has been extracted from the crystal structure (37), and the ground state was optimized by a mixed quantum mechanical–molecular mechanical approach on the density functional theory (DFT) level with the general Amber force field for the pentacene dimer inside the crystal structure (see fig. S3). The calculated vibrational frequencies and the associated coupling strengths between the singlet and triplet-pair electronic states are shown in Fig. 4 (E and F) (see section S7). To account for the triplet-pair generation, an ab initio exciton model has been used, which yields information on the molecular vibrations of the pentacene pair and the Huang-Rhys factors (parameters are associated with the displacements of the equilibrium positions of the nuclei on a photoexcitation of molecule) of the singlet excited and triplet-pair states. Using these calculations, vibrational modes with strong Huang-Rhys factors are clearly identified, as shown in Fig. 4 (E and F). Among them, the low-frequency mode of 177 cm−1 corresponds to the intermolecular rocking motion in the pentacene dimer, which, based on our simulations, significantly modulates the electronic couplings between the singlet exciton and triplet-pair state (the details are described in section S9). In addition, a few high-frequency modes (1013, 1196, and 1517 cm−1) with strong Huang-Rhys factors are resolved in Fig. 4F. On the basis of the calculations, these high-frequency modes correspond to the C─C and C═C stretching modes, which strongly modulate the site energies (energy level of the molecular excited states) of the electronic excited state of the pentacene molecule and can be classified as intramolecular vibrations. These key modes have strong vibronic couplings of ∼100 meV to the electronic states, which implies that these high-frequency modes can significantly reduce the effective energy gap between the singlet and triplet-pair states and, thus, make them even nearly degenerate in the vicinity of a CI (38). Moreover, to distinguish the vibrational coherence on each of the S1 and 1(T1T1) PES, we have calculated the vibrational modes of both electronic states. The results are shown in Fig. 4 (C and D). We observe that the mode at 177 cm−1 changes its frequency to 150 cm−1 on the triplet-pair state PES (notice the correlation analysis of the vibrational modes in fig. S6), which is in excellent agreement with the observations revealed in our TG measurements and shown in Fig. 3 (C and F).
Fig. 4. Calculated vibrational modes and PESs.
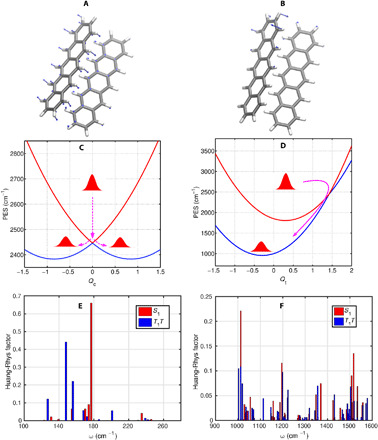
(A and B) Few key modes identified from the calculations. The low-frequency mode (177 cm−1 in A) is associated with the intermolecular rocking motion of two pentacene molecules along the longitudinal molecular axis, which serves as the intermolecular vibration. The calculated high-frequency mode of 1013 cm−1 (in B) corresponds to the intramolecular vibration. Constructed PES along the coupling (C) and the tuning (D) modes, which are based on quantum chemistry calculations for the Huang-Rhys factor between the singlet excited and triplet-pair states. The calculated Huang-Rhys factors of the low- and high-frequency modes are shown in (E) and (F), respectively. Red and blue bars correspond to the modes of the singlet and triplet-pair states.
To illustrate the role of the identified key modes (inter- and intramolecular vibrations) both in theory and experiment, we construct a simple two-state two-mode model to calculate the wave-packet dynamics during the PSF process (more details of the calculation are described in the section S10). We construct the PESs with an energy difference of 850 cm−1 and the electronic coupling of 6 cm−1 (39) between the two electronic states. The reaction coordinates for the transition are selected according to the strong vibronic interaction of the intramolecular vibrations with 1013 cm−1 (although there are a few more high-frequency modes) and the intermolecular vibration with 177 cm−1. The corresponding intra- and intermolecular vibrations can now be identified as tuning (Qt) and coupling (Qc) modes, respectively. On the basis of the computed vibronic coupling strengths, the CI can be established at the reaction coordinate Qt = 1.4, as shown in Fig. 4D. In this figure, the wave packet has the possibility to pass through the PES of the singlet excited state to the PES of the triplet-pair state when vibrational coherence is generated between two vibrational levels of the tuning mode. On the basis of these parameters, we simulate the coherent dynamics of the wave packet on the PESs. For this, we assume the initial wave packet to be in the lowest vibrational level of the singlet excited state and calculate the time evolution of the density matrix associated to the two-state two-mode model. Because of the strong vibronic couplings in the vicinity of the CI, the wave-packet dynamics is calculated by the numerically exact hierarchy equation of motion approach (40) using graphics processing unit (GPU) parallelization (41, 42). The calculated results of the wave-packet dynamics are shown in Fig. 5 on the singlet exciton state and the triplet-pair state, respectively. On the basis of the calculation, we observe that, initially, the wave packet is mainly located on the singlet excited state PES. With on-going time evolution, the wave packet propagates to the right side and reaches Qt = 1.4 where it can decay to the lower excited state through the CI. After that, the wave packet on the lower excited state moves further to the right side to Qt = 2.5 and then returns back to the degenerate point. The wave packet repeats the propagation process again to reach the CI and transitions to the lower excited state. After 100 fs, we observe that the amplitude of the wave packet on the 1(T1T1) state PES gradually reaches its plateau before 400 fs. The obtained time scale of the population transfer for the S1 to 1(T1T1) process is ∼180 fs [the population dynamics of S1 and 1(T1T1) are shown in the Supplementary Materials], which qualitatively agrees with the SF time scale reported by other groups (13). Moreover, the periodic motion of the wave packet is shown to have the period of ∼32 fs, which perfectly matches the vibrational frequency of the tuning mode in the modeling. On the basis of the calculations, we demonstrate the transfer of vibrational coherence of the intramolecular vibration from the singlet state to the triplet-pair state, which agrees with the observations in our 2D vibrational maps obtained from the TG and 2D electronic spectra. The wave-packet dynamics along the coupling mode is shown in Fig. 5 (C and D). The results show that the vibrational coherence of the low-frequency coupling mode does not transfer to the 1(T1T1) state, which is in complete agreement with our experimental observation from TG measurements (Fig. 3, C and F). Thus, based on the kinetic analysis and the coherence transfers, our model of the CI fully reproduces the experimental observations revealed by TG and 2D electronic measurements. However, clearly, due to the simplicity of this model, we are not able to reproduce the frequency upshift of the coupling mode from 170 to 250 cm−1.
Fig. 5. Calculated wave-packet dynamics in the vicinity of the CI.
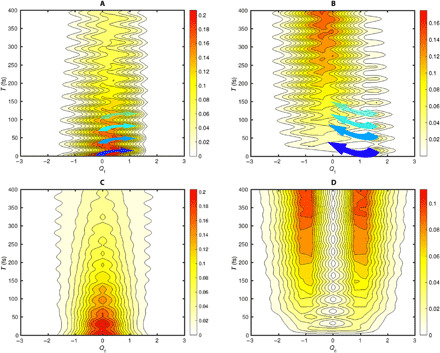
The calculated wave-packet dynamics of S1 and T1T1 along the tuning mode are shown in (A) and (B). The wave-packet dynamics along coupling mode are shown in (C) and (D), respectively.
To further identify the functional role of the intermolecular vibrations, we turn off the vibronic coupling of the coupling mode by setting Λ = 0 cm−1 (the model parameters are described in the Supplementary Materials). We find that in this condition, the transfer of the wave-packet population is significantly slower than observed in the experiment (∼100 fs) because the electronic coupling between the singlet excited state and triplet-pair state is too weak to produce such a fast transfer. It underpins the fact that solely vibronic coupling to the electronic state is not sufficient to reproduce the ultrafast PSF dynamics (29). In addition, the theoretical model presented by Bakulin and co-workers (27) perfectly reproduces the time scale of PSF in pentacene, however, with rather strong electronic couplings between S1 and 1(T1T1). Such a scenario is not supported by the previous quantum chemistry calculations (20). Moreover, to monitor the role of the charge separation (CT) state, the population dynamics of PSF has been determined by other groups. Their calculations show long-lived electronic quantum coherence between the S1, CT, and the 1(T1T1) states originating from the strong electronic couplings between the CT and the S1 and 1(T1T1) states (21). However, our correlation analysis of the measured 2D electronic spectra clearly demonstrates the absence of long-lived electronic coherence. In addition to the intramolecular mode of 1013 cm−1 used in our modeling, there are a few more high-frequency modes that are identified both in the measurements of the TG spectra. With the intermolecular vibration at 177 cm−1, these multivibrational modes generate a CI of higher-dimensional PESs. On the basis of our work, it becomes clear that a few high-frequency modes with strong vibronic couplings further reduce the barrier for the transition from the S1 to 1(T1T1) states, and the low-frequency modes help to bridge between the two PESs by forming the CI, which leads to the SF on an ultrafast time scale (43).
DISCUSSION
We note here that our modeling is limited to a pentacene dimer. In pentacene films, the initially created exciton states are more delocalized, typically over a few (four to five) molecules, but the notion of a coherence length is ill defined. Some estimate can be gained from the decay time of electronic quantum coherence observed in our 2D spectroscopic measurement. The antidiagonal linewidth in Fig. 1C at T = 0 gives a homogeneous linewidth of ∼170 cm−1, as lower estimate given the interference with the ESA (44, 45). This linewidth corresponds to the lifetime of electronic dephasing of ∼61 fs, which is related to the coupling to the bath. The uncorrelated bath fluctuations limit the coherence length to a few pentacene molecules at any given instant. The absence of long-lived electronic coherence and subsequent SF demonstrate that the delocalized exciton rapidly collapses to a localized triplet-pair state, where two electrons occupy adjacent pentacene molecules. Nevertheless, it would be interesting to extend the exciton basis beyond the model pentacene dimer to see what effect this would have on the required vibronic couplings. Such an extended basis is computationally beyond the scope of the present work. We expect that the basic physics would remain unchanged given the expected rapid decoherence and localization of the excitation. The present results illustrate that the ultrafast transition from the singlet to the triplet-pair state, mediated by a strong vibronic coupling of intermolecular vibration, destroys the electronic quantum coherence and the spatial extent of the nascent exciton state (46).
CONCLUSIONS
In summary, we have performed a detailed investigation of the mechanism of the primary step of SF in a pentacene film by using a combination of ultrafast heterodyne-detected TG and 2D electronic spectroscopy. The wave-packet dynamics has been monitored to capture the coherent S1 → 1(T1T1) crossing by tracking the vibrational coherence from the singlet exciton to the spin-correlated triplet-pair state. A time-domain analysis of the vibrational transients reveals the existence of a novel low-frequency mode of 177 cm−1 corresponding to the rocking motion along the longitudinal molecular axis in a pentacene dimer, with high-frequency modes remaining unchanged. These observed key modes show strong vibronic couplings, which bridge the singlet exciton to the triplet-pair state. In addition, based on our calculations, the low-frequency rocking motion strongly modulates the electronic coupling between the two electronic states. The high-frequency vibrational modes, which correspond to the intramolecular vibrations with significant vibronic couplings, reduce the energy gap between the singlet exciton and triplet-pair states and render them nearly degenerate. Our work demonstrates, in detail, experimentally and theoretically that the concept of a CI can be constructed on the basis of the interplay of intermolecular (low frequency) and intramolecular (high frequency) modes coupled to the electronic degrees of freedom. This CI markedly speeds up the process of SF. Hence, our work reveals a novel dynamical mechanism in which low-frequency vibrational modes play a key role in SF. They modulate the couplings of two electronic states and, by this, open a new channel to the desired states by forming a CI between two PESs. This mechanism can be used as a basic design principle for developing new materials to achieve an efficient PSF process in future chemical architectures by exploring the constructive role of vibrational coherences.
MATERIALS AND METHODS
Sample preparation
Quartz substrates were sonicated in a water solution of 2.5% Hellmanex III soap, deionized water, acetone, and isopropyl alcohol, each at 50∘C for 10 min. The cleaned substrates were blow dried with compressed air and then transferred into a UV-Ozone cleaner (UVO-Cleaner, Model 30, Jelight Company Inc.) for 10 min before being loaded into the vacuum chamber (base pressure of approximately 5 ×10−6 mbar; B30 from Oerlikon Leybold Vacuum Dresden GmbH, Germany; with major parts upgraded). This chamber opens up to air, i.e., samples fabricated in this chamber will see air when removed from the chamber for further measurements. Pentacene thin films with a thickness of 200 nm were obtained by thermal evaporation at the rate of 0.5 Å/s. A quartz crystal oscillator was used to monitor the evaporation rate. The evaporated thin films were transferred into a glove box with exposure to air as little as possible. Then, the samples were kept sealed in N2 before they were used for time-resolved optical studies. X-ray diffraction (XRD) thin film measurements of pentacene were performed using a SmartLab x-ray diffractometer (Rigaku Corporation) (the XRD data are shown in the Supplementary Materials). The voltage and current of the instrument were set as 40 kV and 30 mA. The x-ray wavelength is 1.54059 Å. Ultraviolet-visible (UV-vis) measurement of the samples was performed in a LAMBDA 1050 UV/Vis spectrophotometer (PerkinElmer Inc.).
TG and 2D electronic measurements with experimental conditions
Details of the experimental setup have already been described in earlier reports of our group (44). The measurements have been performed on a diffractive optics–based all-reflective 2D spectrometer with a phase stability of λ/160 (47). The laser beam from a home-built NOPA (pumped by a commercial femtosecond PHAROS laser from Light Conversion) is compressed to ∼18 fs using the combination of a deformable mirror (OKO Technologies) and a prism pair. Frequency-resolved optical grating (FROG) measurement is used to characterize the temporal profile of the compressed beam, and the obtained FROG traces are evaluated using a commercial program FROG3 (Femtosecond Technologies). A broadband spectrum obtained carried a linewidth of ∼100 nm (full width at half maximum), centered at 700 nm, which covered the electronic transitions to the first excited state and the ESA in triplet-pair states. Three pulses are focused on the sample with the spot size of ∼100 μm, and the photon echo signal is generated at the phase-matching direction. The photon echo signals are collected using Sciencetech spectrometer model 9055, which is coupled to a charge-coupled device linear array camera (Entwicklungsbüro Stresing). The 2D spectra for each waiting time T were collected by scanning the delay time τ = t1 − t2 in the range of [−128 to 128 fs] with a delay step of 1 fs. At each delay step, 100 spectra were averaged to reduce the noise to signal ratio. The waiting time T = t3 − t2 was linearly scanned in the range of 2.1 ps with steps of 10 fs. For all measurements, the energy of the excitation pulse is attenuated to 10 nJ with 1-kHz repetition rates. Phasing of obtained 2D spectra was performed using an “invariance theorem” (48). To optimize the contribution of ESA, the pentacene film was slightly tilted (5° to 6°) relative to the plane perpendicular to the incident beam.
Calculations
The ab initio exciton model (49) was used to study SF in a pentacene crystal. In this approach, the elements of the exciton model Hamiltonian matrix (namely, the excitation energies and couplings) are calculated in an ab initio manner without empirical parameters from first principles. We have reported the inclusion of CT excited states in the exciton model, and in this work, the multiexcitonic excited states are further involved to properly describe the SF process. The ab initio exciton model provides a more efficient approach as compared to the multiconfigurational methods and minimizes the errors without introducing empirical parameters in the Hamiltonian matrix.
We have used a QM/MM setup where a pentacene dimer is described by the ab initio exciton model, and the surrounding pentacene molecules are modeled by the general Amber force field (for the definition of the QM/MM regions, see the Supplementary Materials) (50). For the QM region, the long-range corrected (LRC) density functional LRC-ω PBEh (51) and the cc-pVDZ basis set (52) were used in DFT and time-dependent DFT calculations, with dispersion correction modeled by Grimme’s dispersion (53) with Becke-Johnson damping (54). Ab initio exciton model calculations were carried out using the GPU-accelerated TeraChem code (55). The exciton model for the pentacene dimer in the QM region was set up to involve two locally excited (LE) states (one on each pentacene molecule), two CT excited states, and one TT (triplet-pair) excited state. We have documented the exciton model in details in our previous work (49, 56). The spin-adapted wave function of the TT state is used to facilitate the evaluation of the couplings. Diagonalization of the exciton model Hamiltonian gives the adiabatic excited states as linear combinations of the diabatic excited state (namely, the LE, CT, and TT states). The gradient of the ab initio exciton model was derived on the basis of the Hellmann-Feynman theorem.
Comparison with the multiconfigurational method suggests that the ab initio exciton model systematically overestimates the vertical excitation energies, while the ordering of the states and the energy gaps are nicely predicted by the model. In particular, the exciton model gives a Davydov splitting of 0.12 eV, which corresponds well with the experimental measurement and the multiconfigurational result (20). We have optimized the geometries and performed a numerical frequency analysis for the adiabatic S1 and S2 states of the QM pentacene dimer, which are of dominant TT and LE character, respectively. The Huang-Rhys factors Si were calculated as , where mi, ωi, and ΔQi are the reduced mass, frequency, and the equilibrium displacement of the i-th vibrational mode, respectively. For the calculation of the coherent dynamics, the population dynamics of the wave packet in the two-state two-mode model is being calculated by the hierarchical equation of motion (40) with GPU parallelization (Tesla K80). To obtain the stable Hamiltonian matrix, 20 (Qt) and 10 (Qc) vibrational levels are used in the tuning and coupling modes, respectively. To monitor the wave-packet dynamics on two reaction coordinates, we perform the projection calculation based on the theoretical work in (57, 58).
Supplementary Material
Acknowledgments
We thank V. I. Prokhorenko for help with the 2D setup and for providing the 2D data analysis software. We thank M. Riede for help in the sample preparation. Funding: This work was supported by the Max Planck Society and the Excellence Cluster “CUI: Advanced Imaging of Matter” of the Deutsche Forschungsgemeinschaft (DFG), EXC 2056, project ID 390715994. P.K.N. acknowledges the support via intramural funds at TIFR Hyderabad from the Department of Atomic Energy (DAE), India. H.Y. acknowledges the China Scholarship Council (CSC) and China Oxford Scholarship Fund (COSF) for support. Work by T.J.M. and X.L. was supported by the AMOS program of the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, and Biosciences Division. Author contributions: H.-G.D. and A.J. conceived the research and planned the experiment. H.-G.D., A.J., and V.T. performed the optical measurements and analyzed the experimental data. H.Y. and P.K.N. prepared the pentacene films and characterized them. X.L., Z.L., X.-L.Z., and T.J.M. performed quantum chemistry calculations. H.-G.D. developed a theoretical model together with M.T. and calculated the wave packet dynamics under the supervision of M.T. Moreover, H.-G.D., A.J., Z.L., M.T., and R.J.D.M. wrote the manuscript with inputs from all the authors. The project was supervised by R.J.D.M. Competing interest: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The Supplementary Materials includes the XRD data of the pentacene film, the description of the global fitting approach and the fitted kinetic traces, calculated vibrational modes and its Huang-Rhys factors, and the modeling of the wave-packet dynamics on the PESs. All relevant experimental data and code for calculations are available from the authors upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/38/eabb0052/DC1
REFERENCES AND NOTES
- 1.Smith M. B., Michl J., Singlet fission. Chem. Rev. 110, 6891–6936 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Tayebjee M. J. Y., McCamey D. R., Schmidt T. W., Beyond shockley-queisser: Molecular approaches to high-efficiency photovoltaics. J. Phys. Chem. Lett. 6, 2367–2378 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Monahan N. R., Sun D., Tamura H., Williams K. W., Xu B., Zhong Y., Kumar B., Nuckolls C., Harutyunyan A. R., Chen G., Dai H.-L., Beljonne D., Rao Y., Zhu X.-Y., Dynamics of the triplet-pair state reveals the likely coexistence of coherent and incoherent singlet fission in crystalline hexacene. Nat. Chem. 9, 341–346 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Smith M. B., Michl J., Recent advances in singlet fission. Annu. Rev. Phys. Chem. 64, 361–386 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Musser A. J., Maiuri M., Brida D., Cerullo G., Friend R. H., Clark J., The nature of singlet exciton fission in carotenoid aggregates. J. Am. Chem. Soc. 137, 5130–5139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Margulies E. A., Miller C. E., Wu Y., Ma L., Schatz G. C., Young R. M., Wasielewski M. R., Enabling singlet fission by controlling intramolecular charge transfer in π-stacked covalent terrylenediimide dimers. Nat. Chem. 8, 1120–1125 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Margulies E. A., Logsdon J. L., Miller C. E., Ma L., Simonoff E., Young R. M., Schatz G. C., Wasielewski M. R., Direct observation of a charge-transfer state preceding high-yield singlet fission in Terrylenediimide thin films. J. Am. Chem. Soc. 139, 663–671 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Miyata K., Conrad-Burton F. S., Geyer F. L., Zhu X.-Y., Triplet pair states in singlet fission. Chem. Rev. 119, 4261–4292 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Breen I., Tempelaar R., Bizimana L. A., Kloss B., Reichman D. R., Turner D. B., Triplet separation drives singlet fission after femtosecond correlated triplet pair production in rubrene. J. Am. Chem. Soc. 139, 11745–11751 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Pensack R. D., Ostroumov E. E., Tilley A. J., Mazza S., Grieco C., Thorley K. J., Asbury J. B., Seferos D. S., Anthony J. E., Scholes G. D., Observation of two triplet-pair intermediates in singlet exciton fission. J. Phys. Chem. Lett. 7, 2370–2375 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Congreve D. N., Lee J., Thompson N. J., Hontz E., Yost S. R., Reusswig P. D., Bahlke M. E., Reineke S., Voorhis T. V., Baldo M. A., External quantum efficiency above 100% in a singlet-exciton-fissionbased organic photovoltaic cell. Science 340, 334–337 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Thompson N. J., Congreve D. N., Goldberg D., Menon V. M., Baldo M. A., Slow light enhanced singlet exciton fission solar cells with a 126% yield of electrons per photon. Appl. Phys. Lett. 103, 263302 (2013). [Google Scholar]
- 13.Wilson M. W. B., Rao A., Ehrler B., Friend R. H., Singlet exciton fission in polycrystalline pentacene: From photophysics toward devices. Acc. Chem. Res. 46, 1330–1338 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Jundt C., Klein G., Sipp B., Le Moigne J., Joucla M., Villaeys A. A., Exciton dynamics in pentacene thin films studied by pump-probe spectroscopy. Chem. Phys. Lett. 241, 84–88 (1995). [Google Scholar]
- 15.Folie B. D., Haber J. B., Refaely-Abramson S., Neaton J. B., Ginsberg N. S., Long-lived correlated triplet pairs in a π-stacked crystalline pentacene derivative. J. Am. Chem. Soc. 140, 2326–2335 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Chan W.-L., Ligges M., Zhu X.-Y., The energy barrier in singlet fission can be overcome through coherent coupling and entropic gain. Nat. Chem. 4, 840–845 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Berkelbach T. C., Hybertsen M. S., Reichman D. R., Microscopic theory of singlet exciton fission. I. General formulation. J. Chem. Phys. 138, 114102 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Berkelbach T. C., Hybertsen M. S., Reichman D. R., Microscopic theory of singlet exciton fission. II. Application to pentacene dimers and the role of superexchange. J. Chem. Phys. 138, 114103 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Berkelbach T. C., Hybertsen M. S., Reichman D. R., Microscopic theory of singlet exciton fission. III. Crystalline pentacene. J. Chem. Phys. 141, 074705 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Zeng T., Hoffmann R., Ananth N., The low-lying electronic states of pentacene and their roles in singlet fission. J. Am. Chem. Soc. 136, 5755–5764 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Tamura H., Huix-Rotllant M., Burghardt I., Olivier Y., Beljonne D., First-principles quantum dynamics of singlet fission: Coherent versus thermally activated mechanisms governed by molecular π stacking. Phys. Rev. Lett. 115, 107401 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Zirzlmeier J., Lehnherr D., Coto P. B., Chernick E. T., Casillas R., Basel B. S., Thoss M., Tykwinski R. R., Guldi D. M., Singlet fission in pentacene dimers. Proc. Natl. Acad. Sci. U.S.A. 112, 5325–5330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanders S. N., Kumarasamy E., Pun A. B., Trinh M. T., Choi B., Xia J., Taffet E. J., Low J. Z., Miller J. R., Roy X., Zhu X.-Y., Steigerwald M. L., Sfeir M. Y., Campos L. M., Quantitative intramolecular singlet fission in bipentacenes. J. Am. Chem. Soc. 137, 8965–8972 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Lukman S., Chen K., Hodgkiss J. M., Turban D. H. P., Hine N. D. M., Dong S., Wu J., Greenham N. C., Musser A. J., Tuning the role of charge-transfer states in intramolecular singlet exciton fission through side-group engineering. Nat. Commun. 7, 13622 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hart S. M., Silva W. R., Frontiera R. R., Femtosecond stimulated Raman evidence for charge-transfer character in pentacene singlet fission. Chem. Sci. 9, 1242–1250 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Musser A. J., Liebel M., Schnedermann C., Wende T., Kehoe T. B., Rao A., Kukura P., Evidence for conical intersection dynamics mediating ultrafast singlet exciton fission. Nat. Phys. 11, 352–357 (2015). [Google Scholar]
- 27.Bakulin A. A., Morgan S. E., Kehoe T. B., Wilson M. W. B., Chin A. W., Zigmantas D., Egorova D., Rao A., Real-time observation of multiexcitonic states in ultrafast singlet fission using coherent 2D electronic spectroscopy. Nat. Chem. 8, 16–23 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Refaely-Abramson S., da Jornada F. H., Louie S. G., Neaton J. B., Origins of singlet fission in solid pentacene from an ab initio Green’s function approach. Phys. Rev. Lett. 119, 267401 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Stern H. L., Cheminal A., Yost S. R., Broch K., Bayliss S. L., Chen K., Tabachnyk M., Thorley K., Greenham N., Hodgkiss J. M., Anthony J., Head-Gordon M., Musser A. J., Rao A., Friend R. H., Vibronically coherent ultrafast triplet-pair formation and subsequent thermally activated dissociation control efficient endothermic singlet fission. Nat. Chem. 9, 1205–1212 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Miyata K., Kurashige Y., Watanabe K., Sugimoto T., Takahashi S., Tanaka S., Takeya J., Yanai T., Matsumoto Y., Coherent singlet fission activated by symmetry breaking. Nat. Chem. 9, 983–989 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Johnson P. J. M., Halpin A., Morizumi T., Prokhorenko V. I., Ernst O. P., Miller R. J. D., Local vibrational coherences drive the primary photochemistry of vision. Nat. Chem. 7, 980–986 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Jonas D. M., Vibrational and nonadiabatic coherence in 2D electronic spectroscopy, the Jahn-Teller effect, and energy transfer. Annu. Rev. Phys. Chem. 69, 327–352 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Jha A., Duan H.-G., Tiwari V., Nayak P. K., Snaith H. J., Thorwart M., Miller R. J. D., Direct observation of ultrafast exciton dissociation in lead iodide perovskite by 2D electronic spectroscopy. ACS Photonics 5, 852–860 (2018). [Google Scholar]
- 34.Rao A., Wilson M. W. B., Albert-Seifried S., Pietro R. D., Friend R. H., Photophysics of pentacene thin flim: The role of exciton fission and heating effects. Phys. Rev. B 84, 195411 (2011). [Google Scholar]
- 35.Butkus V., Zigmantas D., Valkunas L., Abramavicius D., Vibrational vs. electronic coherence in 2D spectrum of molecular systems. Chem. Phys. Lett. 545, 40–43 (2012). [Google Scholar]
- 36.Xian R., Corthey G., Rogers D. M., Morrison C. A., Prokhorenko V. I., Hayes S. A., Dwayne Miller R. J., Coherent ultrafast lattice-directed reaction dynamics of triiodide anion photodissociation. Nat. Chem. 9, 516–522 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Schiefer S., Huth M., Dobrinevski A., Nickel B., Determination of the crystal structure of substrate-induced pentacene polymorphs in fiber structured thin films. J. Am. Chem. Soc. 129, 10316–10317 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Ito S., Nagami T., Nakano M., Density analysis of intra- and intermolecular vibronic couplings toward bath engineering for singlet fission. J. Phys. Chem. Lett. 6, 4972–4977 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Ab initio exciton model calculations show rather weak electronic couplings between the singlet exciton and the triplet-pair state, V = 6 cm−1 due to the quite different electronic configuration of the singlet and triplet-pair states. This value is smaller than the parameters used in (27).
- 40.Ishizaki A., Tanimura Y., Quantum dynamics of system strongly coupled to low-temperature colored noise bath: Reduced hierarchy equations approach. J. Phys. Soc. Jpn. 74, 3131–3134 (2005). [Google Scholar]
- 41.Kreisbeck C., Kramer T., Long-lived electronic coherence in dissipative exciton dynamics of light-harvesting complexes. J. Phys. Chem. Lett. 3, 2828–2833 (2012). [Google Scholar]
- 42.Tsuchimoto M., Tanimura Y., Spins dynamics in a dissipative environment: Hierarchal equations of motion approach using a graphics processing unit (GPU). J. Chem. Theory Comput. 11, 3859–3865 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Based on our two-state two-mode model, the transfer with time ∼180 fs of the PSF is slightly slower than the observation of ∼100 fs. With few high-frequency modes, we expect that a multichannel modeling of a conical intersection speeds up the population transfer. However, the computational costs of the simulations are too large with the numerically exact method.
- 44.Duan H.-G., Prokhorenko V. I., Cogdell R. J., Ashraf K., Stevens A. L., Thorwart M., Miller R. J. D., Nature does not rely on long-lived electronic quantum coherence for photosynthetic energy transfer. Proc. Natl. Acad. Sci. U.S.A. 114, 8493–8498 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Halpin A., Johnson P. J. M., Tempelaar R., Murphy R. S., Knoester J., Jansen T. L. C., Miller R. J. D., Two-dimensional spectroscopy of a molecular dimer unveils the effects of vibronic coupling on exciton coherences. Nat. Chem. 6, 196–201 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Duan H.-G., Nalbach P., Miller R. J. D., Thorwart M., Ultrafast energy transfer in excitonically-coupled molecules induced by a nonlocal Peierls phonon. J. Phys. Chem. Lett. 10, 1206–1211 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Prokhorenko V. I., Picchiotti A., Maneshi S., Miller R. J. D., Broadband electronic two-dimensional spectroscopy in the deep UV. Ultrafast Phenomena XIX 162, 432–435 (2015). [Google Scholar]
- 48.Hybl J. D., Ferro A. A., Jonas D. M., Two-dimensional fourier transform electronic spectroscopy. J. Chem. Phys. 115, 6606 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Li X., Parrish R. M., Liu F., Kokkila Schumacher S. I. L., Martínez T. J., An ab initio exciton model including charge-transfer excited states. J. Chem. Theo. Comput. 13, 3493–3504 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A., Development and testing of a general amber force field. J. Comput. Chem. 15, 1157–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Rohrdanz M. A., Martins K. M., Herbert J. M., A long-range-corrected density functional that performs well for both ground-state properties and time-dependent density functional theory excitation energies, including charge-transfer excited states. J. Chem. Phys. 130, 054112 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Dunning T. H., Jr., Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023 (1989). [Google Scholar]
- 53.Grimme S., Antony J., Ehrlich S., Krieg H., A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010). [DOI] [PubMed] [Google Scholar]
- 54.Grimme S., Ehrlich S., Goerigk L., Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011). [DOI] [PubMed] [Google Scholar]
- 55.Ufimtsev I. S., Martinez T. J., Quantum chemistry on graphical processing units. 2. Direct self-consistent-field implementation. J. Chem. Theory Comput. 5, 1004–1015 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Sisto A., Glowacki D. R., Martinez T. J., Ab initio nonadiabatic dynamics of multichromophore complexes: A scalable graphical-processing-unit-accelerated exciton framework. Acc. Chem. Res. 47, 2857–2866 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Manthe U., Köppel H., New method for calculating wave packet dynamics: Strongly coupled surfaces and the adiabatic basis. J. Chem. Phys. 93, 345 (1990). [Google Scholar]
- 58.Qi D.-L., Duan H.-G., Sun Z.-R., Miller R. J. D., Thorwart M., Tracking an electronic wave packet in the vicinity of a conical intersection. J. Chem. Phys. 147, 074101 (2017). [DOI] [PubMed] [Google Scholar]
- 59.V. I. Prokhorenko, Global analysis of multi-dimensional experimental data. European Photochemistry Association Newletter June 2012, p. 21.
- 60.Milota F., Prokhorenko V. I., Mancal T., von Berlepsch H., Bixner O., Kauffmann H. F., Hauer J., Vibronic and vibrational coherences in two-dimensional electronic spectra of supramolecular J-Aggregates. J. Phys. Chem. A 117, 6007–6014 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cohen A., Kovačevič J., Wavelets: The mathematical background. Proc. IEEE 84, 514–522 (1996). [Google Scholar]
- 62.Harrop J. D., Taraskin S. N., Elliot S. R., Instantaneous frequency and amplitude identification using wavelets: Application to glass structure. Phys. Rev. E Stat. Nonlin. Soft Matter. Phys. 66, 026703 (2002). [DOI] [PubMed] [Google Scholar]
- 63.J. van den Berg, Wavelets in Physics (Cambridge Univ. Press, 2004).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/38/eabb0052/DC1