

A generalizable strategy enables the expansion of the bioink palette and bioprinting capabilities.
Abstract
A major challenge in three-dimensional (3D) bioprinting is the limited number of bioinks that fulfill the physicochemical requirements of printing while also providing a desirable environment for encapsulated cells. Here, we address this limitation by temporarily stabilizing bioinks with a complementary thermo-reversible gelatin network. This strategy enables the effective printing of biomaterials that would typically not meet printing requirements, with instrument parameters and structural output largely independent of the base biomaterial. This approach is demonstrated across a library of photocrosslinkable bioinks derived from natural and synthetic polymers, including gelatin, hyaluronic acid, chondroitin sulfate, dextran, alginate, chitosan, heparin, and poly(ethylene glycol). A range of complex and heterogeneous structures are printed, including soft hydrogel constructs supporting the 3D culture of astrocytes. This highly generalizable methodology expands the palette of available bioinks, allowing the biofabrication of constructs optimized to meet the biological requirements of cell culture and tissue engineering.
INTRODUCTION
The emergence of three-dimensional (3D) bioprinting has opened up new avenues for engineering in vitro living systems that can be used in tissue regeneration, biological modeling, and cell-based diagnostics (1, 2). 3D bioprinting is based on the free-form fabrication of biomaterials into customized geometries informed by digital design. The most common modality for tissue fabrication is extrusion bioprinting, in which living constructs are additively manufactured via layer-by-layer deposition of cellularized bioinks (1, 3). The design of bioinks is a central topic in this field-formulations rarely have both the physicochemical properties required for the 3D printing process and the physicochemical cues to meet the biological needs of the encapsulated cells. Moreover, these characteristics are often mutually exclusive. For example, high-concentration bioinks generally have a high viscosity that aids filament extrusion and maintains the structural stability during sequential layering (4); however, the resulting high-density polymer networks can hinder essential cellular activities (e.g., spreading, migration, and proliferation) (5, 6) and present an environmental stiffness that is incompatible with cells of soft-tissue origin. This paradox has resulted in a compressed “biofabrication window” containing a select group of feasible bioinks, with other biomaterial systems requiring major compromises in either printability or biocompatibility (7, 8).
In recent years, this challenge has been addressed through the development of advanced printing modalities (9, 10). For instance, 3D printers can be adapted with transparent nozzles, allowing bioinks to be partially photocrosslinked using ultraviolet (UV) light irradiation immediately before extrusion (10). This in situ crosslinking approach can aid the printability of certain photocrosslinkable bioinks with low initial viscosity; however, this approach is still limited by the mechanical stiffness of the extruded material, which needs to support the weight of each additive layer. The recent developments in suspension-based bioprinting could potentially address this issue (11); however, such a strategy would require the careful design of a suspending biomaterial that is able to support, but not deform, soft bioinks. While highly specific bioink formulations (12) or customized printing methodologies (13) can address the printability of certain biomaterial systems, these do not provide a standardized method that can be applied across different 3D printing scenarios. This consideration highlights the motivation for a more generalizable strategy that extends the biofabrication window across a broad palette of printable, cytocompatible bioinks.
In this work, we address this unmet need through the use of complementary network bioinks, a broadly applicable strategy that enables the effective 3D printing of a range of hydrogels at different polymer concentrations (Table 1). These bioinks comprise two miscible polymer networks with complementary gelation mechanisms that regulate different stages of the fabrication process. A thermo-responsive gelatin network provides excellent extrusion and structural stability during 3D printing, while a photocrosslinkable network allows the printed structure to be stabilized by covalent crosslinking. The combination of these two polymers leads to a complementary network hydrogel where dissociation of the thermo-reversible network does not affect the photocrosslinked network (Fig. 1A). We show that a tight range of printing parameters can be broadly applied across different bioinks, with a conserved outcome across 12 different polymers and 20 different formulations. The consistent printability of complementary network bioinks eliminates the need to tune the physicochemical properties of the bioink for sequential layering. Thus, we propose an alternative approach to bioprinting, in which the biological performance of different bioinks are pre-screened to select an optimal formulation for the bioprinting application. We demonstrate this approach using two targeted applications: screening hydrogel stiffness to support the 3D culture of printed astrocytes, and identifying optimal biopolymer formulations for mineralized tissue engineering. These applications demonstrate how a standardized bioprinting process with an extended biofabrication window can be used to quantitatively compare and optimize different biomaterial formulations to meet the biological needs of printed cell cultures and engineered tissues.
Table 1. Bioink library.
Polymer backbones and photocrosslinkable side groups used in this study. Dithiothreitol was used as crosslinker in the step-growth mechanism.
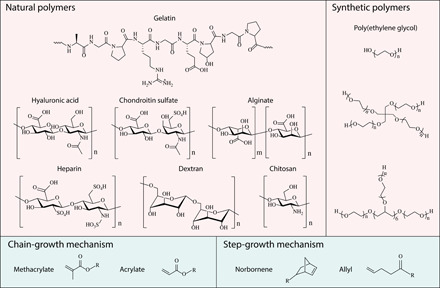
Fig. 1. Complementary network bioink 3D printing process.
(A) (i) Schematic of the direct 3D printing of nonviscous, photocrosslinkable bioinks, which form droplets during extrusion and undergo spreading once deposited, even with light irradiation during the printing process. (ii) Schematic of the 3D printing of complementary network bioinks in three core steps: cooling-induced deposition mediated by the gelation of gelatin, photocrosslinking of the bioink, and thermal liberation of gelatin during incubation. (B) Oscillatory temperature sweep (between 37°C and 7°C at a rate of 5°C min−1; closed markers indicate cooling and open markers indicate heating) showing reversible cooling-induced gelation of HAMA+, while undoped HAMA exhibits a low viscosity across the monitored temperature range. (C) The injection of HAMA+ through a needle generates stable filaments in both air and PBS, while undoped HAMA does not (both formulations supplemented with dye for visualization). (D) Storage (G′) and loss (G″) moduli during an oscillatory time sweep of the 2.5 wt % HAMA+ bioink during stages of cooling (sharp temperature change from 37°C to 15°C), UV light exposure (10 mW cm−2), and heating back to 37°C. (E) Representative images of 3D printed tubular and lattice constructs using 2.5 wt % HAMA and HAMA+ (containing 5 wt % Fluorescein-gelatin). A frequency of 1.5 Hz and a strain of 1% were used for oscillatory tests. Scale bars, 5 mm (C and E, left) and 1 mm (E, right). Credit for all photographs in this figure: Liliang Ouyang, Imperial College London.
RESULTS
Demonstrating the principle of complementary network bioinks
We used methacrylated hyaluronic acid (HAMA) as a representative bioink to illustrate the challenges of current printing methods and the advantages of complementary network bioinks. HAMA is a semisynthetic photocrosslinkable hydrogel that is widely used in tissue engineering due to its excellent biocompatibility and ease of crosslinking (14). However, non-crosslinked HAMA has rheological properties that are poorly suited to filament extrusion and layer-by-layer assembly, a situation that is exacerbated when using low concentration formulations. We demonstrated this limitation by measuring the rheological properties of a 2.5 weight % (wt %) non-crosslinked HAMA solution (temperature sweep between 37°C and 7°C). Under these conditions, the polymer solution presented a complex viscosity of ~15 mPa·s and a temperature-independent complex modulus (G) below 0.3 Pa (Fig. 1B). Moreover, in a strain sweep test, the storage modulus (G′) was consistently lower than the loss modulus (G″), which indicated that the non-crosslinked HAMA remained in a liquid state across this strain range (10−2 to 104%) (fig. S1A). These rheological properties make 2.5 wt % HAMA unsuitable for extrusion printing, as evidenced by the solution droplet jetting when passed through a standard needle (25-gauge) and rapidly diffusing when injected into phosphate-buffered saline (PBS) (Fig. 1C). A filament deformation experiment, based on methods by Therriault et al. (15), showed that the filaments produced by 2.5 wt % HAMA were unable to bridge small gaps (≥0.5 mm) (fig. S1B).
However, the rheological properties of 2.5 wt % HAMA could be tuned for filament extrusion simply by adding gelatin, a common biopolymer that undergoes a thermo-reversible sol-gel transition through the entanglement of a helical biopolymer network. A composite formulation of 2.5 wt % HAMA and 5 wt % gelatin (henceforth referred to as HAMA+) exhibited thermal gelation at 17.2°C (Fig. 1B) and shear-thinning behavior at 25°C (fig. S1A). HAMA+ could thus be extruded into continuous hydrogel filaments, which maintained their structural integrity in PBS (Fig. 1C) and could bridge gaps as large as 4 mm (fig. S1B). After extrusion, printed complementary network bioinks were designed to undergo three processes: thermosetting, photocrosslinking, and thermal release of gelatin (Fig. 1A). We thus designed and implemented a multistep rheological test sequence. HAMA+ exhibited low shear modulus when the temperature was maintained at 37°C (G′ < 0.5 Pa) but underwent a rapid sol-gel transition upon cooling the stage (G′ ~ 750 Pa and G″ ~ 20 Pa after 20 min at 15°C). At this point, in situ UV light irradiation was used to trigger the covalent crosslinking of HAMA via free radical polymerization of the methacrylate groups, which initiated an immediate and rapid increase in shear modulus (G′ ~ 10 kPa and G″ ~ 70 Pa after 5-min exposure to 365-nm light at 10 mW cm−2). Last, a temperature ramp was used to liquefy the gelatin component by thermal disassembly of the gelatin network, which was evidenced by a slow decrease in shear modulus (G′ ~ 3.5 kPa and G″ ~ 24 Pa after 30 min at 37°C) (Fig. 1D).
These rheological tests indicated that HAMA+ should be compatible with a 3D printing process using the following parameters: extrusion from a heated nozzle (25°C to 26°C), deposition onto a cool print bed to initiate thermosetting (15°C), free radical photocrosslinking to stabilize the structure (UV light irradiation), and thermal release of gelatin (37°C) (Fig. 1A). Using these parameters, we successfully 3D printed 2.5 wt % HAMA+ into standard tubular and lattice constructs, with the latter structure exhibiting the well-defined square pores that are characteristic of printable bioink formulations (Fig. 1E). This result was in stark contrast to HAMA (2.5 wt %) without gelatin, which rapidly coalesced upon deposition and formed a single homogeneous droplet instead of a hollow tube. The loss of integrity could be partially remedied by using UV light exposure at the print bed to photocrosslink HAMA during deposition; however, even under these conditions, the filaments spread laterally and layers fused axially to form structures with very poorly defined features (Fig. 1E). These observations are consistent with literature showing the poor printability of HAMA, even at concentrations as high as 20 wt % (4).
Bioink library expansion
We next sought to generalize the principles of complementary network bioinks across a broad library of polymers, including matrix-derived proteins (gelatin), matrix-derived glycosaminoglycans (hyaluronic acid, chondroitin sulfate, and heparin), other natural polysaccharides (dextran, chitosan, and alginate), and synthetic derivatives [poly(ethylene glycol) (PEG)] (Table 1). These polymers were functionalized with different photocrosslinkable moieties supporting chain-growth polymerization (methacryloyl and acryloyl groups) and step-growth polymerization (norbornene and allyl groups). Overall, we used a library of 12 photocrosslinkable polymers: HAMA, norbornene-functionalized hyaluronic acid (HANB), gelatin methacryloyl (GelMA), allylated gelatin (GelAGE), methacrylated chondroitin sulfate (CSMA), methacrylated dextran (DexMA), methacrylated alginate (AlgMA), methacrylated chitosan (ChiMA), methacrylated heparin (HepMA), PEG diacrylate (PEGDA), eight-arm PEG acrylate (PEGA), and norbornene-functionalized four-arm PEG (PEGNB). Mixing these photocrosslinkable polymers with 5 wt % gelatin formed the following composite bioinks that we shall subsequently refer to as HAMA+, HANB+, GelMA+, GelAGE+, CSMA+, DexMA+, AlgMA+, ChiMA+, HepMA+, PEGDA+, PEGA+, and PEGNB+.
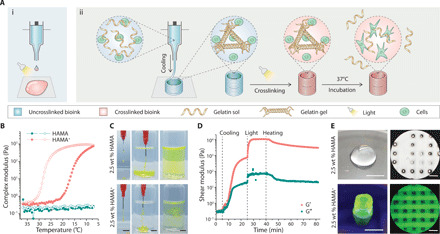
We optimized the 3D printing parameters for each of these complementary network bioinks at different concentrations (table S1); however, it was clear that printability was conserved across the library. Moreover, each of the complementary network bioinks were printed using a nozzle temperature between 23°C and 27°C, a pneumatic pressure between 0.5 and 1.1 bar, and a printing speed between 1.5 and 3 mm s−1. This consistency is highly advantageous, particularly for multimaterial 3D printing, and confirms that the gelatin component was principally dictating the bioink printability. We successfully 3D printed each of the complementary network bioinks into standard tubular structures, with consistent and accurate replication of the digital design (Fig. 2A). The dimensions of the printed tubes were highly consistent across the bioink library, with an outer diameter between 5.1 and 5.5 mm and a height between 6.3 and 6.7 mm (Fig. 2B). In all cases, the measured dimensions matched the input values (diameter, 5 mm; height, 6 mm), with the marginal expansion attributed to the width of the filament building blocks. Nevertheless, it was clear that the complementary network bioinks were highly compatible with extrusion printing, and thus, we explored the 3D printing of complex geometries encompassing challenging features (overhangs, thin walls, and branches) that are highly desirable in applications such as tissue engineering. Taking PEGA+ as an example, we readily printed different geometric assemblies (e.g., multilayer lattices and pyramidal structures), anatomical shapes (e.g., ear and brain), and even unsupported hollow constructs (e.g., trifurcated tube) (Fig. 2C). We observed excellent extrusion consistency and structural stability throughout the 3D printing process, which was evident by the high lateral and axial uniformity observed across the multilayer lattice (Fig. 2D).
Fig. 2. 3D printing a library of complementary network bioinks.
(A) Representative images and (B) measured outer diameters and heights of 3D printed tubular constructs (upper row, oblique view; bottom row, top view) using various complementary network bioinks, all containing 5 wt % Fluorescein-gelatin. (C) Representative images (top row, top view; bottom row, side view) of 3D complex 3D printed structures (from left to right: lattice, ear-shape, brain-shape, pyramid, and trifurcated tubular constructs) using 2.5 wt % PEGA+ bioink (containing 5 wt % Rhod-gelatin). (D) Side (left) and top (right) views of a printed lattice structure (2.5 wt % PEGA+) with the intensity profile showing structural uniformity. (E) Printed heterogeneous tubular and (F) tracheal-esophageal models before (0 day) and after incubation (1 and 4 days) at 37°C. HAMA+ (2.5 wt %; clear phase) and GelMA+ (5 wt %; green phase) were printed alternately along the longitudinal or transverse directions. Scale bars, 5 mm. Credit for all photographs in this figure: Liliang Ouyang, Imperial College London.
We next explored the long-term stability of the printed structures for each of the complementary network bioinks. Tubular structures were printed, photocrosslinked, and then incubated in PBS at 37°C for 1 week, with the hypothesis that hydration would lead to changes in the swelling of the photocrosslinked hydrogels. The tubular structure was maintained across this period for all of the printed structures (fig. S2, A and B), with the majority of constructs remaining within 10% of their preincubation dimensions (those printed using 2.5 wt % HAMA+, 5 wt % PEGA+, 2.5 wt % CSMA+, 5 wt % DexMA+, 1 wt % AlgMA+, 1 wt % ChiMA+, and 10 wt % HepMA+). We observed deswelling in the photocrosslinkable gelatin constructs (5 wt % GelMA+ and 5 wt % GelAGE+) and swelling in the norbornene-based constructs (5 wt % PEGNB+ and 2.5 wt % HANB+), changes that occurred predominantly within the first 24 hours (fig. S2, C and D). It should be noted that swelling and deswelling are intrinsic material characteristics related to the concentration, crosslinking, and hydrophilicity of the network, and thus largely independent of the printing process.
We next explored whether different complementary network bioinks could be sequentially printed to form heterogeneous 3D structures. HAMA+ (2.5 wt %) and GelMA+ (5 wt %) were selected, because these bioinks exhibit compatible photocrosslinking chemistry (methacryloyl groups) to enable covalent crosslinking and tight adhesion between printed layers. These bioinks were extruded via separate nozzles into two different print designs: tubular constructs with alternating layers to test axial fusion and a tracheal-esophageal model to test lateral fusion (Fig. 2, E and F). These structures were successfully printed, and structural integrity was well maintained during long-term incubation (21 days) (fig. S3A), with distinct boundaries retained throughout this period (fig. S3, B and C). This highlights another major advantage of complementary network bioinks; the thermal gelation removes the need for photocrosslinking during printing, thus allowing multiple bioinks to be sequentially printed and then collectively photocrosslinked in a single postprinting irradiation step.
On the basis of the previous rheological testing, we anticipated that the elevated temperature during incubation (37°C) would lead to liquification and release of the gelatin from the photocrosslinked printed structures. We visualized this process by using fluorescein-labeled gelatin and observed a gradual decrease in the fluorescence of 2.5 wt % HAMA+ printed tubes over a 20-day incubation in PBS (fig. S4A). This observation was supported by measuring the fluorescence intensity of the supernatant throughout this period. A burst release was observed from 2.5 wt % HAMA+, with 73% of the gelatin liberated after 1 day, a value that rose to 94% after 15 days (fig. S4B). Similar profiles and values were recorded for the other polymers: 2.5 wt % HANB+ (65 and 84%), 5 wt % GelMA+ (76 and 93%), 5 wt % GelAGE+ (78 and 89%), 2.5 wt % CSMA+ (73 and 86%), 5 wt % DexMA+ (71 and 84%), 1 wt % AlgMA+ (68 and 85%), 1 wt % ChiMA+ (88 and 92%), 5 wt % PEGA+ (57 and 69%), and 5 wt % PEGNB+ (62 and 83%) (fig. S4C), with a marked reduction in gelatin release observed at room temperature (fig. S4D). The concentration of the photocrosslinkable hydrogel also affected the rate of release: 93, 85, and 83% of the gelatin was measured in the supernatant after 3 days for 1, 2.5, and 5 wt % HAMA+, respectively. Similar trends were observed for HANB+ and GelMA+, suggesting that increasing the density of the photocrosslinked network slows the mass transfer and release of gelatin (fig. S4E). Nevertheless, these data demonstrate that the majority of gelatin is thermally released from the complementary network hydrogels without the need for any specific washing procedures.
Soft hydrogel printing
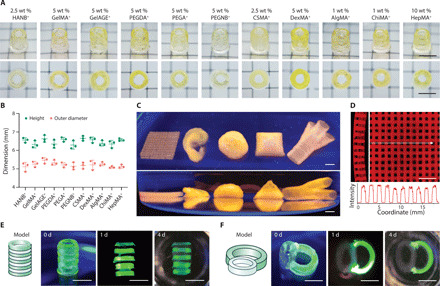
Our primary motivation for developing complementary network bioinks was to enable the 3D printing of soft hydrogel constructs that are generally considered unprintable. We first compared the mechanical properties of constructs generated by three complementary network bioinks (HANB+, GelMA+, and GelAGE+) to equivalent structures produced from their gelatin-free counterparts. Initially, the gelatin component contributed to higher observed compressive moduli (E) for the complementary network structures; however, these values decreased to the level of the gelatin-free counterparts after the thermal release of gelatin (1 day) (fig. S5A). This was most clearly demonstrated with 5 wt % GelMA+, which had a relatively high initial stiffness (E = 7.7 ± 0.9 kPa) that was reduced after incubation (E = 2.2 ± 0.3 kPa) to a value similar to undoped 5 wt % GelMA (E = 1.5 ± 0.9 kPa). This demonstrated that the long-term mechanical properties of the printed structures were minimally affected by the inclusion of gelatin. Moreover, scanning electron microscopy (SEM) revealed similar microstructures and microporosity for the freeze-dried hydrogels with or without gelatin after 1-day incubation (fig. S5B). These results suggested that soft printed structures could be attained simply by using 5 wt % gelatin and a low concentration of photocrosslinkable hydrogel. We explored this possibility by 3D printing HAMA+ into tubular structures across a range of concentrations (0.5 to 10.0 wt %, referring to the HAMA component) (Fig. 3A). Excellent printability was observed across this range; even the low concentrations of 0.5 and 1 wt % HAMA+ retained their printed form after immersion and incubation (1 day) in PBS (Fig. 3B). The structural integrity of the 0.5 wt % HAMA+ after gelatin release was particularly notable considering that HAMA at this concentration cannot support itself in air, either after gelatin release (Fig. 3B) or when cast as a gelatin-free hydrogel (fig. S6A). Mechanical testing was used to compare the hydrogels formed using HAMA+ and HAMA across the same concentration range (0.5 to 10.0 wt %). Minimal differences were observed between the complementary network hydrogels and their gelatin-free counterparts after 1 day of incubation in PBS at 37°C. As a result, we were able to generate structures with highly tunable stiffnesses, with this polymer range yielding a post-incubation compressive modulus from ~2 to ~200 kPa (Fig. 3C). We also observed excellent printability and structural integrity for other low-concentration complementary network bioinks (1.5 wt % CSMA+ and 1.5 wt % PEGA+) (Fig. 3D), structures with the minimum polymer concentration required to support self-standing CSMA and PEGA hydrogels (fig. S6B).
Fig. 3. 3D printing soft hydrogel constructs.
(A) Representative images of 3D printed tubular structures using HAMA+ bioinks across various concentrations of HAMA. (B) Comparison of 3D printed tubes before (0 day) and after incubation (1 day in air or PBS) using either 0.5 or 1 wt % HAMA+. (C) The compressive moduli of cylinders cast from HAMA formulations (with and without 5 wt % gelatin) across various concentrations before (0 day) and after (1 day) incubation. Two-tailed Mann-Whitney test, *P < 0.05; n.s., not significant (n ≥ 3). (D) Comparison of 3D printed tubes before (0 day) and after incubation (1 day in air or PBS) using 1.5 wt % CSMA+ and 1.5 wt % PEGA+. Fluorescein-gelatin was used for visualization. Scale bars, 5 mm. Credit for all photographs in this figure: Liliang Ouyang, Imperial College London.
3D printing soft hydrogels is motivated by the need to generate structures that can effectively support the culture of cells originating from soft tissues. Given the extremely low stiffness of brain tissue (0.1 to 1 kPa), we sought to examine whether complementary network bioinks could be used to support the 3D printing and culture of neural cells. We selected astrocytes, which require a 3D culture environment to avoid the emergence of a reactive inflammatory phenotype (16). We encapsulated primary astrocytes in mold-cast GelMA+ across a wide concentration range (2.5 to 10.0 wt %, referring to the GelMA component). We observed a clear stiffness-dependent behavior over a 7-day culture period, with only low-concentration GelMA+ (2.5 and 3.0 wt %) supporting the formation of elongated processes that are characteristic of healthy astrocytes (Fig. 4A). These visual observations were supported by an alamarBlue assay, which revealed a similar stiffness-dependent effect on cellular metabolism after 1 day, and evidence of astrocyte proliferation only in the low-concentration bioinks (2.5 and 3.0 wt %) after 3 days of culture (Fig. 4B). We used this screening to select the optimal GelMA+ concentration (2.5 wt %) and used this to successfully print astrocyte-laden 3D multilayer lattices (Fig. 4C). LIVE/DEAD staining performed after 1 day revealed a highly viable population of astrocytes (93 ± 3%) (Fig. 4D), with an elongated cell morphology (Fig. 4E). The cell viability was well maintained, with no significant difference after 3 days (89 ± 3%), 5 days (90 ± 3%), and 7 days (88 ± 3%). The cultured astrocytes were homogeneously distributed throughout the construct (fig. S7) and exhibited a characteristic stellar morphology as shown by volumetric confocal fluorescence microscopy (Fig. 4F).
Fig. 4. Bioprinting and culture of constructs containing astrocytes.
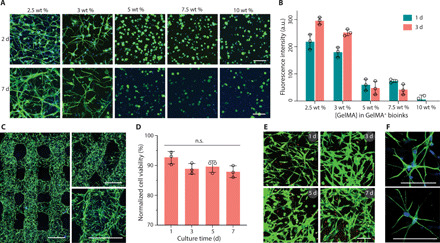
(A) Fluorescence microscopy images (green, phalloidin; blue, DAPI) and (B) metabolic activity of astrocytes (2 × 106 cells ml−1) encapsulated in GelMA+ hydrogels with varied GelMA concentrations (2.5 to 10 wt %). a.u., arbitrary unit. (C) Confocal laser scanning microscopy images (green, phalloidin; blue, DAPI) of printed multilayer lattices with 90° or 60° angles between adjacent layers (2 days). (D) Quantified cell viability and (E) LIVE/DEAD-stained astrocytes (green, calcein AM; red, ethidium homodimer-1) after bioprinting and culture for up to 7 days. Two-tailed Mann-Whitney test (n = 3). (F) High-magnification fluorescence microscopy images of astrocytes (green, phalloidin; blue, DAPI) in printed constructs (2 days). GelMA+ (2.5 wt %) and astrocytes (2 × 106 ml−1) were used for bioprinting in (C) to (F). Scale bars, 100 μm (A, E, and F) and 500 μm (C).
Tissue engineering optimization
In addition to screening hydrogel concentrations for cell viability and morphology, we used the complementary network bioinks to screen different photocrosslinkable polymers before their use in tissue engineering. We chose to compare three formulations (2.5 wt % HAMA+, 5 wt % GelMA+, and 2.5 wt % CSMA+) that had similar storage moduli after incubation (~1 kPa) to enable a fair comparison between polymers. We first investigated the possibility that cells might be lost into the cell medium during the release of gelatin, a process that could affect their utility in tissue engineering. We performed a PicoGreen assay on cellularized hydrogels (HAMA, GelMA, CSMA, HAMA+, GelMA+, and CSMA+) to assess the DNA content before and after gelatin release (0 and 5 hours). These measurements indicated that the initial DNA content was consistent between the different complementary network hydrogels (fig. S8A) and was not reduced after a 5-hour incubation period (fig. S8B). This suggests that the photocrosslinked polymer network is effective in preventing cell loss during gelatin release. Continuing DNA measurements over a 24-hour period revealed cell proliferation for the HAMA- and GelMA-based hydrogels, with significant increases when gelatin was included in the initial formulation.
We next assessed cartilage formation using the 3D culture of juvenile bovine articular chondrocytes within the complementary network bioinks. It should be noted that the use of juvenile chondrocytes has been shown to offer benefits over their adult counterparts for cartilage tissue engineering (17). Chondrocytes were immobilized in each of the formulations (2.5 × 107 cells ml−1) and then cultured for 42 days in chondrogenic medium. The tissues were harvested and sectioned for analysis, with safranin O staining revealing an increasing density of sulfated glycosaminoglycans for CSMA+, HAMA+, and GelMA+, respectively (fig. S8C). The cartilage constructs were further assessed by performing a dimethylmethylene blue (DMMB) assay to quantify the amount of sulfated glycosaminoglycan. This analysis revealed a significantly higher dry mass fraction of sulfated glycosaminoglycan in the cartilage tissues engineered using HAMA+ (32 ± 2%) and GelMA+ (35 ± 14%) when compared to CSMA+ (13 ± 6%) (fig. S8D). The inclusion of gelatin appeared to have no detrimental effect on the quantity or distribution of sulfated glycosaminoglycans (fig. S8, C and D).
A similar screen was performed for bone tissue engineering, with an osteogenic sarcoma cell line (Saos-2) encapsulated in the same three formulations (7.5 × 106 cells ml−1) and cultured for 14 days in osteogenic medium. The tissues were harvested for analysis, with Alizarin Red S staining revealing a much higher density of calcium for GelMA+ when compared to CSMA+ and HAMA+ (fig. S8E). An alkaline phosphatase (ALP) assay further suggested a higher mineralizing enzyme activity for GelMA+ (84 ± 7 nmol min−1 μg−1 of DNA) than for CSMA+ (72 ± 10 nmol min−1 μg−1 of DNA) or HAMA+ (28 ± 3 nmol min−1 μg−1 of DNA) (fig. S8F). As with the cartilage tissue engineering example, the inclusion of gelatin generally had no effect upon matrix production, as illustrated with calcium deposition (fig. S8E) and mineralizing enzyme activity (fig. S8F), although a small decrease in ALP activity was observed for GelMA+ when compared to GelMA.
On the basis of this screen, we selected 5 wt % GelMA+ as the optimized complementary network bioink for 3D printing mineralized tissue constructs. We successfully printed a centimeter-sized porous cylinder (diameter, 2 cm; height, 1 cm) containing highly uniform lattices to enable effective nutrient access (Fig. 5A). Over a 14-day culture in osteogenic medium, the cellularized construct transitioned from a clear hydrogel into an opaque structure. A lateral section stained with hematoxylin and eosin indicated homogeneous tissue formation across a maintained lattice structure (Fig. 5B). Meanwhile, Alizarin Red S staining of sections taken from the top and bottom of the constructs demonstrated uniformity in calcium deposition throughout the construct (Fig. 5C). We next fabricated larger and more complex structures, including a ~4-cm-long, ~2-cm-wide, and ~1-cm-high bone-like geometry (fig. S8G) and a ~3-cm-high and ~1.5-cm-wide trifurcated tube with a hollow interior and overhanging walls (Fig. 5D). Both tissue constructs turned opaque throughout and maintained their printed geometries after 14 days of osteogenic culture. The uniformity of mineralization was assessed by taking six cross-sectional segments along the 3-cm longitudinal axis of the trifurcated tube. An ALP assay revealed mineralizing enzyme activity across the construct, with no significant differences observed in the different regions of the tissue (Fig. 5E). This uniformity was confirmed with Alizarin Red S staining, with sections taken from the bottom and top segments showing a similar high density of calcium deposition (Fig. 5F).
Fig. 5. Exploring complementary network bioinks for tissue engineering.
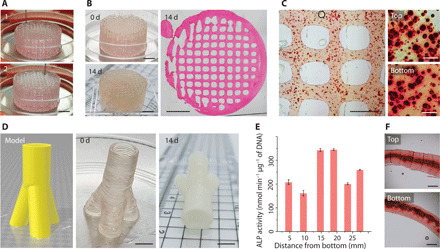
(A) Bioprinting a porous cylinder construct (diameter, 2 cm; height, 1 cm) with Saos-2 cells in the bioink. Numbers on the images indicate the printing process. (B) Images of a printed cylinder before (0 day) and after culture (14 days). The microscopy image (hematoxylin and eosin staining) of the cylinder cross section indicates the maintenance of a uniform pattern during tissue formation. (C) Alizarin Red S staining of printed samples (14 days). The higher-magnification images indicate the top and bottom layers along the height of the cylinder. (D) Trifurcated tubular bioprinted constructs that replicate their digital design. (E) Normalized ALP activity and (F) Alizarin Red S staining of samples at different positions along the length of the trifurcated tube after 14 days of culture. GelMA+ (5 wt %), Saos-2 (7.5 × 106 ml−1), and osteogenic medium were used throughout. Scale bars, 5 mm (A, B, and D), 1 mm (C, left, and F), and 100 μm (C, right). Credit for all photographs in this figure: Liliang Ouyang, Imperial College London.
DISCUSSION
A major bottleneck in 3D bioprinting is the small number of effective bioinks. A particular issue is that many biomaterial formulations are effective in 3D cell culture and tissue engineering but widely considered unprintable. Here, we addressed this challenge by introducing a library of 12 complementary network bioinks comprising photocrosslinkable hydrogels (various forms of gelatin, hyaluronic acid, chondroitin sulfate, dextran, alginate, chitosan, heparin, and PEG) supplemented with 5 wt % gelatin. These complementary network bioinks were highly effective for 3D printing using thermal gelation and photocrosslinking, unlike their gelatin-free counterparts, which were either unprintable or only printable over a certain concentration threshold. We demonstrated excellent printability using hydrogels that have not previously been used as the base material in 3D bioprinting (e.g., HepMA, PEGNB, CSMA, and DexMA) and polymer concentrations that are considered unprintable [e.g., 0.5 wt % HAMA (10), 5 wt % GelAGE (18), and 2.5 and 3.0 wt % GelMA (19)]. The different complementary network bioinks were printed using a tight range of process parameters, thus providing a generalized 3D printing methodology with broadened scope. Unlike the use of viscosity modifiers [e.g., alginate (20) and cellulose (21, 22)] that are retained in the bioprinted structure, the complementary network bioinks naturally release the majority of gelatin at 37°C. Critically, the inclusion of gelatin did not notably affect the mechanical properties of the hydrogels after thermal release, nor the ability of encapsulated chondrocytes and osteosarcoma cells to produce cartilaginous or mineralized tissue, respectively.
These results lay the basis of a generalizable approach to 3D printing, in which the concentration or type of base polymer can be tuned with minimal concern over printability. In particular, this ability enabled us to address the enduring challenge of soft hydrogel printing. Biomaterial stiffness is a critical factor in 3D cell culture and tissue engineering; however, soft hydrogels are generally formed from low-concentration polymer solutions with viscosities that are too low for effective 3D printing. Here, we show that complementary network bioinks could be readily used for 3D printing hydrogels at the structural integrity limit of the photocrosslinkable hydrogel (1.5 wt % CSMA+, 1.5 wt % PEGA+, and 1 wt % HAMA+). We even used 0.5 wt % HAMA+, which could not support its weight in air, to 3D print super-soft hydrogels with maintained structure during incubation in PBS. We exploited soft hydrogel bioprinting to fabricate 3D constructs that were stiffness-optimized for astrocyte culture. Our screening process illustrated the common dichotomy of 3D printing and cell culture; GelMA at printable concentrations (≥5 wt %) was incompatible with astrocyte culture, and conversely, the concentrations that supported proliferative astrocytes with stellar processes (2.5 and 3 wt %) are widely considered to be unprintable (10, 19–21, 23). However, by using complementary network bioinks, we were able to 3D print structures with 2.5 wt % GelMA, i.e., without needing to compromise either the printing process or the cell culture.
Aside from soft hydrogel printing, we also used complementary network bioinks to systematically compare how different biopolymers supported in vitro cartilage and bone tissue engineering. This screening process identified GelMA+ as the preferred biomaterial, and thus, we used it as a bioink to 3D print complex geometries for bone tissue engineering. The ability to fabricate a stable lattice geometry within 3D printed centimeter-sized constructs was used to provide a porous network for nutrient diffusion and ensure consistent mineralization throughout the engineered construct, a key factor that should allow the scale-up of complementary network bioinks for printing even larger tissue geometries. Nevertheless, a challenge of scaling up this methodology would be to maintain cell viability for prolonged fabrication periods (24). In our case, the largest, centimeter-scale structures were fabricated from a single print lasting approximately 2 hours. It should also be noted that the inclusion of cells can affect the rheological properties and printability of a bioink (13, 25, 26). While we achieved good printability using complementary network hydrogels with cell densities of up to 2.5 × 107 cells ml−1, it is possible that higher cell densities could affect the printing process. Another important and enduring challenge of hydrogel-based musculoskeletal tissue engineering is replicating the mechanical properties of the native tissues. The mechanical properties of the tissue constructs could potentially be improved by adjusting the bioink (e.g., through the use of double-network bioinks), coprinting solid polymeric scaffolds (e.g., by melt electrowriting), incorporating mechanical stimulation into the tissue culture setup, or extending the maturation period.
In summary, we introduce the concept of complementary network bioinks as a generalized methodology for 3D bioprinting. This approach offers previously unidentified bioprinting capabilities wherein the polymer type, photocrosslinking kinetics, and bioink concentration can all be selected in the absence of any constraints imposed by the printability of the formulation. In turn, this offers a more effective methodological approach to 3D printing living tissue constructs, whereby the bioink can be readily tuned to meet the biological requirements of the encapsulated cells. Our demonstration of soft hydrogel printing for astrocyte culture indicates the suitability of complementary network bioinks for the biofabrication of living constructs of soft-tissue origins (e.g., other neural cells, adipose, kidney, pancreas, and lung). While we have presented an extensive library of complementary network bioinks comprising 12 photocrosslinkable polymers, this approach should be generalizable to other hydrogel networks with alternative crosslinking mechanisms, such as those triggered by the addition of chemical crosslinkers, changes in pH, enzyme catalysis, or exposure to ultrasound (7, 27, 28). The only classes of biomaterials that should pose difficulties are those that cannot be made miscible with gelatin and those that rely on an opposing thermal gelation (e.g., poloxamer 407). We have also used a very standard thermal extrusion–based approach that is compatible with existing 3D bioprinting methods (11, 23, 29). Together, these design factors should enable complementary network bioinks to find broad applicability across different biomaterial systems and 3D printing protocols, and provide new opportunities for biofabrication and tissue engineering.
MATERIALS AND METHODS
Materials
Unless otherwise stated, all the chemicals were purchased from Sigma-Aldrich, and deionized (DI) water was used throughout. Apart from GelAGE (70% substitution, 300 g Bloom, Sigma-Aldrich), PEGDA (6 kDa, Sigma-Aldrich), and PEGNB (20 kDa, JenKem Technology), all the other photocrosslinkable polymers were synthesized as described within Materials and Methods.
HAMA was prepared using an established protocol (30). Methacrylic anhydride (3 equiv.) was added dropwise to an aqueous 1 wt % solution of hyaluronic acid (75 kDa, Lifecore) on ice for 6 to 8 hours while maintaining the pH ~8. The mixture was then allowed to react overnight at 4°C and then neutralized to pH 7 to 7.5 and dialyzed [molecular weight cutoff (MWCO), 6 to 8 kDa] against water for 1 week. 1H nuclear magnetic resonance (NMR) was used to determine the degree of functionalization assuming substitution at the 6-position previously reported in the literature (30). Because the integrals of a (acetyl-CH3) and b (methacrylate-CH3) overlap, we make the assumption that when c + d = 2 (methacrylic vinyl protons), b = 3 and then a = 12.23. In the case of 100% degree of functionalization, the ratio of (c + d):a = 2:3; therefore, the true integration of c + d = 0.49, and the degree of functionalization is (0.49/2) × 100% = ~25% (fig. S9A).
HANB was prepared using an established protocol (31). Hyaluronic acid tetrabutylammonium salt was dissolved in dimethyl sulfoxide (DMSO; 0.5 wt %), before adding 5-norbornene-2-methylamine (0.32 equiv.) and benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate (0.3 equiv.). The reaction was performed at room temperature for 2 hours, followed by precipitation against acetone and then dialysis (MWCO, 6 to 8 kDa) against water for 1 week. 1H NMR was used to determine the degree of functionalization using the norbornene vinyl protons, b and c, by reference to the acetyl-CH3 protons, a = 3. The degree of functionalization is therefore (0.46/2) × 100% = ~23% (fig. S9B).
GelMA was prepared using an established protocol (32). Methacrylic anhydride (0.6 g of methacrylic anhydride per 1 g of gelatin) was added dropwise to an aqueous 10 wt % solution of gelatin (300 g Bloom, type A, from porcine skin) while stirring at 50°C. After 3-hour reaction, the solution was centrifuged at 3500g for 3 min and the supernatants were collected and diluted with four volumes of water. This solution was dialyzed (MWCO, 12 to 14 kDa) against water at 40°C for 1 week, followed by pH adjustment to 7.4 using 1 M NaHCO3. A Fluoraldehyde assay (32) showed a quantitative degree of functionalization of ~80% (fig. S9C).
PEGA was prepared using a protocol adapted from the literature (33). Eight-arm PEG (40 kDa, JenKem Technology) was dissolved in anhydrous dichloromethane at a concentration of 10 wt %, followed by adding potassium carbonate (12.8 equiv., previously dried at 120°C under vacuum overnight) portion-wise under a nitrogen blanket, vigorously stirred at room temperature. Acryloyl chloride (6.38 equiv., previously distilled over hydroquinone) was added dropwise, and the reaction was stirred under nitrogen at room temperature in the dark for 3 days. Methanol (0.15 volume equiv.) was added to the reaction mixture to quench unreacted acryloyl chloride and allowed to stir for 30 min. The reaction was filtered, and the solvent was removed using a rotary evaporator. The crude polymer was dissolved in a minimum amount of tetrahydrofuran and precipitated dropwise into cold diethyl ether (10 volume equiv.). The precipitate was filtered and dried under vacuum. 1H NMR analysis showed a quantitative degree of functionalization (>95%) by integration of the acrylate vinyl protons, a, b, and c, against the terminal-CH2-CH2-O- protons, d (fig. S9D).
CSMA was prepared using an established protocol (34). Methacrylic anhydride (37 equiv.) was added dropwise to 2 wt % aqueous solution of chondroitin sulfate (type A, from bovine trachea) while stirring, followed by pH adjustment to 8.0 using 5 N NaOH. After 24 hours of stirring at room temperature, the solution was precipitated in ethanol (8 volume equiv.) and the isolated precipitate was redissolved in water (2 volume equiv.). The solution was dialyzed (MWCO, 3.5 kDa) against water for 1 week. 1H NMR was used to determine the degree of functionalization assuming substitution at the 6-position previously reported in the literature. Because the integrals of a (acetyl-CH3) and b (methacrylate-CH3) overlap, we make the assumption that when c + d = 2 (methacrylic vinyl protons), b = 3 and then a = 4.64. In the case of 100% degree of functionalization, the ratio of (c + d):a = 2:3; therefore, the true integration of c + d = 1.29, and the degree of functionalization is (1.29/2) × 100% = ~65% (fig. S9E).
DexMA was prepared using an established protocol (35). Dextran (70 kDa, TCI Chemicals) was dissolved in 10 wt % LiCl/DMF (N,N′-dimethylformamide) solvent while stirring at 90°C to form a 1 wt % solution. After complete dissolution, the solution was cooled to 60°C and triethylamine (5 equiv.) was added as a catalyst. Methacrylic anhydride (2 equiv.) was then added to the reaction dropwise and stirred for 10-hour reaction at room temperature. The mixture was precipitated in cold isopropyl alcohol, washed several times with isopropyl alcohol, and then redissolved in water. The solution was dialyzed (MWCO, 12 to 14 kDa) against water for 1 week. 1H NMR analysis showed a degree of functionalization of ~10% by integration of the methacrylate protons, b, c, and d, against the dextran H1 proton, a (fig. S9F).
AlgMA was synthesized using alginate from brown algae. Alginate was dissolved in 0.1 M MES [2-(N-morpholino)ethanesulfonic acid] buffer (pH 6.5) at the concentration of 0.6 wt %. After complete dissolution, N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC; 1.3 equiv.) and N-hydroxysuccinimide (NHS; 1 equiv.) were added dropwise and stirred for 20-min reaction at room temperature. N-(3-aminopropyl) methacrylamide hydrochloride was dissolved in a minimal amount of MES buffer and added dropwise to the mixture for an overnight reaction at room temperature. The product was purified by precipitation, filtration, washing with acetone, and drying in vacuum overnight. The degree of functionalization was determined using 1H NMR, following a previously reported method (36). On the basis of 1H NMR assignments of alginate by Jensen and co-workers (37), the protons from approximately 4.30 to 3.45 ppm (parts per million) can be assigned to 2α, 3α, and 4α of α-l-guluronic acid and 2β, 3β, 4β, and 5β of β-d-mannuric acid. Protons 1α, 5α, and 1β overlap with the residual water peak and were not used. Assuming 100% degree of functionalization, the ratio of methacrylamide protons to alginate protons should be 10:7. If we let the alginate protons = 7, we expect a total of 10 methacrylamide protons; however, we only obtain 0.34 + 0.36 + 1.21 = 1.91 protons. The degree of functionalization, therefore, is (1.91/10) × 100% = ~19% (fig. S9G).
ChiMA was prepared using an established protocol (38). Glycol chitosan (Sigma-Aldrich, G7753) was purified by filtering the 1.3% aqueous solution through a 0.45-μm filter to remove insoluble impurities. The solution was freeze-dried, and the dried powder was then dissolved in water at a concentration of 2 wt % while stirring. The solution was adjusted to pH 9.0, and methacrylic anhydride (0.7 equiv.) was added dropwise for an overnight reaction at room temperature. The solution was dialyzed (MWCO, 1 kDa) against water for 1 week. The degree of functionalization is equivalent to 20 to 25% according to the adapted protocol (38).
HepMA was synthesized from heparin (porcine sodium salt). EDC (5 equiv.) and NHS (5 equiv.) were added to a 1 wt % aqueous solution of heparin while stirring. After 15 min, predissolved 2-aminoethylmethacrylamide hydrochloride (2 equiv.) was added dropwise and the mixture was stirred overnight at room temperature. The solution was dialyzed (MWCO, 3.5 kDa) against water for 4 days. 1H NMR analysis was used to determine the degree of functionalization. Heparin is composed of repeating disaccharide unit, m, of α-l-idopyranosyluronic acid 2-sulfate and 2-deoxy-2-sulfamino-α-d-glucopyranose 6-sulfate, β-d-glucuronic acid, n, and 2-acetamido-2-deoxy-α-d-glucose, o (39). We use the acetyl group of n as our reference for the degree of functionalization, which is determined to be 17% and falls within the expected range for heparin (39). If we make the assumption that the heparin we used is the same, we can set the reference to the integral of the acetyl group from N-acetylated glucosamine (known from heparin). In the case of 100% incorporation, we would expect a ratio of acetyl to methacrylamide protons of 3:2. When we set the acetyl group to 3, we get a value of 8.06 for both methacrylamide protons. We know that only 17% of these acetyl groups are in heparin; therefore, 0.17 × 8.06 = 1.37 gives us the true integration of the methacrylamide protons. We know that the ratio of methacrylamide protons to H2 protons is 2:1; however, we get a degree of functionalization of (1.37/2) × 100% = ~69% (fig. S9H).
Fluorescein-conjugated gelatin (Fluorescein-gelatin) and rhodamine-conjugated gelatin (Rhod-gelatin) were synthesized by reacting gelatin with NHS-Fluorescein and NHS-Rhodamine (Thermo Fisher Scientific), respectively. Gelatin was fully dissolved in phosphate buffer (pH ~8.1) at 50°C at a concentration of 10 wt %, followed by the addition of NHS-Fluorescein or NHS-Rhodamine (30 mg per 1 g of gelatin). The reaction was performed for 3 to 8 hours at 50°C in the dark and subsequently quenched by adding water (4 volume equiv.). The product was then dialyzed (MWCO, 5 kDa) against water at 40°C for 1 week. All dialyzed solutions above were freeze-dried and stored at −20°C before use.
Cell culture and isolation
Primary rat astrocytes were obtained from P2 neonatal rat pups as previously described (40). The culture of primary astrocytes was performed in accordance with the UK Animals (Scientific Procedures) Act (1986)/European Directive (2010/63/EU) and approved by the University College London (UCL) Animal Welfare and Ethics Review Board. Briefly, cerebral cortices were dissected out and finely chopped. Following a series of tissue digestion, cell isolation, and purification steps, the resulting cell suspension was collected and centrifuged at 400g. The pellet was resuspended in high-glucose Dulbecco’s modified Eagle’s medium (HG-DMEM) supplemented with 10 volume % fetal bovine serum (FBS) and 1 volume % penicillin-streptomycin (P/S). Cells were cultured in poly-d-lysine (PDL)–coated T-175 flasks and cultured for 14 days at 37°C, 5% CO2 until 100% confluent. Loosely adherent microglial cell populations were detached by shaking on an orbital shaker overnight at 37°C, 5% CO2 and subsequently removed. Astrocytes were detached from flasks by incubation with 0.25% trypsin-EDTA solution for 7 to 10 min at 37°C. Cell stocks were frozen down in HG-DMEM with 10% DMSO at −140°C for later use. Cells were thawed and cultured in PDL-coated flasks and expanded until 80 to 90% confluency before use. Astrocytes were used immediately and not passaged further.
Human bone osteosarcoma cells (Saos-2, American Type Culture Collection) were cultured in α-modified Eagle’s medium (α-MEM) supplemented with 10 volume % FBS and 1 volume % P/S at 37°C, 5% CO2. Primary bovine articular chondrocytes were obtained from 4-week-old calf hind legs, sourced from a local abattoir. Articular cartilage, dissected from the femoral condyle under sterile conditions, was enzymatically digested for 15 hours at 37°C in serum-free low-glucose DMEM supplemented with 1 volume % P/S and 0.2 wt % type II collagenase. The tissue digest was passed through a 100-μm cell strainer to remove undigested cartilage. The resulting suspension was then centrifuged at 700g for 12 min, resuspended in fresh DMEM, centrifuged at 600g for 8 min, resuspended in fresh DMEM, and then passed through a 40-μm cell strainer to obtain a single-cell suspension for use. Chondrocytes were used immediately and not passaged further.
Bioink preparation
Complementary network bioinks were composed of a photocrosslinkable component, gelatin, photo-initiator [lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP)], and cells. A master list of bioink formulations can be found in table S1. In all cases, the gelatin and LAP concentrations were 5 wt % and 2 mM, respectively. For physical visualization and release studies, Fluorescein-gelatin or Rhod-gelatin was mixed with unmodified gelatin at a mass ratio of 1:24. In the cases of thiol-ene reaction bioinks (GelAGE, HANB, and PEGNB), dithiothreitol (DTT) was included as a crosslinker. For PEG-based bioink preparation, a minimal amount of acetic acid was introduced to avoid phase separation when mixing with 5 wt % gelatin, a protocol adapted from the literature (41). At neutral pH, aqueous mixtures of gelatin and PEG will form a liquid-liquid phase-separated system when the critical concentration of either gelatin or PEG is exceeded. The addition of acetic acid enables the formation of a miscible single phase by protonating basic amino acids present in gelatin, which reduces gelatin-gelatin attraction through incorporating electrostatic charge repulsion (42). According to the optimization results (fig. S10), we added 0.05 and 0.01 volume % acetic acid in formulations containing 5 and 2.5 wt % PEG, respectively, regardless of the macromer used. For sterile culture studies, gelatin solutions were heated to 70°C for 30 min for three cycles, while LAP and DTT solutions were filter-sterilized by 0.22-μm filter. Photocrosslinkable polymers were sterilized either by sterile filtration or by 30-min UV treatment for solutions or powders, respectively.
Rheological measurements
To determine the thermal responsivity of the hydrogels, oscillatory temperature sweeps were performed using a rheometer (AR2000, TA Instruments) using a cone geometry (2°; diameter, 40 mm). The temperature was set for a ramp change between 37° and 7°C at a change rate of 5°C min−1. To measure the dynamic response of the formulations to temperature and light, we used a rheometer (MR 302, Anton Paar) equipped with a temperature-controlled photorheology plate for in situ photo-polymerization. In oscillatory time sweeps, the temperature was initially set at 37°C and then markedly decreased to 15°C for 20 min, after which UV light (365 nm, 10 mW cm−2, OmniCure S1500) was introduced for 5 min. A frequency of 1.5 Hz and a strain of 1% were used throughout. Oscillatory strain sweeps (10−2 to 104%) were performed at room temperature (25°C) using a frequency of 1.5 Hz.
Mechanical testing
Unconfined compression testing was performed with hydrogel cylinders (diameter, 4.5 mm; height, 3 mm) using a uniaxial mechanical tester (Electroforce 3200, TA Instruments) equipped with a 250g force sensor. Ramp compression at a speed ratio of 0.01 mm s−1 was applied to obtain stress-strain curves. The compressive modulus was calculated within the strain range of 0.1 to 0.2 mm mm−1.
Gelatin release study
A hydrogel cylinder model (diameter, 3.5 mm; height, 2 mm) was used to test the release of the gelatin from photocrosslinked complementary network bioinks. Typically, 20 μl of Fluorescein-gelatin–containing bioinks was cast in a disposable truncated syringe (0.5 ml, BD), followed by photocrosslinking for 5 min (365 nm, 10 mW cm−2, Cambridge UVTEC). The generated hydrogels were transferred to separate 1.5-ml Eppendorf tubes filled with 1 ml of PBS for incubation at 37°C or room temperature. At given time intervals, 500 μl of supernatant was collected and replaced with 500 μl of fresh PBS. After the final collection, the remaining solution together with the hydrogel cylinder was homogenized for 10 min using a TissueLyser II (QIAGEN) with a frequency of 22 Hz. The fluorescence intensity from the lysed solution and all the harvested solutions were obtained at 525 nm (excitation, 490 nm) using a plate reader (SpectraMax M5). The percentage release at each time point was calculated by normalization to the total release value.
Bioink screening
For astrocyte culture optimization, GelMA+ bioinks with varied GelMA concentrations (2.5 to 10 wt %) were used to resuspend astrocytes at a cell density of 2 × 106 cells ml−1. Cell-laden bioinks (25 μl) were cast in disposable syringe molds (1 ml, BD), followed by 5-min light irradiation (365 nm, 10 mW cm−2, Cambridge UVTEC). Astrocyte growth medium (HG-DMEM supplemented with 10 volume % FBS and 1 volume % P/S) was used for the culture of mold-cast and bioprinted astrocyte-laden constructs.
For chondrocyte and Saos-2 cell culture optimization, 5 wt % GelMA, 2.5 wt % HAMA, 5 wt % CSMA, and their complementary network counterparts were used to resuspend chondrocytes and Saos-2 cells at a cell density of 2.5 × 107 and 7.5 × 106 cells ml−1, respectively. Cell-laden bioinks (50 μl) were cast in disposable syringe molds (1 ml, BD), followed by 5-min light irradiation (365 nm, 10 mW cm−2, Cambridge UVTEC). Chondrogenic medium [HG-DMEM supplemented with 1× Insulin-Transferrin-Selenium (ITS), proline (50 μg ml−1), 1% nonessential amino acids, 1 volume % P/S, ascorbic acid (50 μg ml−1), 100 nM dexamethasone, transforming growth factor–β3 (TGF-β3) (10 ng ml−1)] was used for the culture of chondrocyte-laden constructs. Osteogenic medium (α-MEM supplemented with 10% FBS, 1% P/S, 100 μM ascorbic acid, 10 nM dexamethasone, and 5 mM β-glycerophosphate) was used for the culture of mold-cast and bioprinted Saos-2–laden constructs.
3D bioprinting
The 3D bioprinting process was performed in a temperature-controlled manner (13). Briefly, bioinks were gently mixed and incubated at 37°C for 10 min and then loaded into a print cartridge equipped with 25-gauge disposable needle. The cartridge was loaded to the printer (3D-Bioplotter, EnvisionTEC) with the nozzle set at a temperature around 23° to 27°C. The bioinks were allowed to equilibrate for 10 to 15 min before printing on a precooled plate (15°C). The pneumatic pressure and nozzle moving speed were coordinated to deliver continuous filaments. The detailed printing parameters for individual bioinks can be found in table S1. For the printing of centimeter-sized tissue constructs, the printing speed was altered to 8 to 10 mm s−1. After printing, the printed constructs were treated with light (365 nm, 10 mW cm−2, Cambridge UVTEC) for 150 s. To avoid oxygen inhibition, another 150-s light treatment was applied after covering the constructs with a 2 mM LAP solution, as previously suggested (4). After washing with PBS, the cell-laden constructs were incubated at 37°C and 5% CO2. The corresponding medium was changed after 1 hour, 24 hours, and then every 2 days. No further washing steps were applied.
Shape fidelity characterization
To assess filament extrusion, bioinks were incubated at 15°C for 10 min and then extruded in air or PBS through a 25-gauge needle. A further setup adapted from literature (15) was used to assess the formation or deformation of self-supporting filaments. HAMA and HAMA+ bioinks were deposited continuously onto a custom plate with gaps of varied size (0.5 to 5 mm) using the corresponding bioprinting parameters. The greatest width gap that a bioink could successfully bridge was used as a measure of the shape fidelity at the filament level.
Metabolic activity assay
An alamarBlue assay (Thermo Fisher Scientific) was used to determine the metabolic activity of cells embedded in hydrogels. Briefly, alamarBlue reagent was mixed with culture medium at a volume ratio of 1:9 to form the working solution. At defined time points, cell-laden constructs were washed with PBS and incubated with the working solution for 3 hours. The supernatant was transferred to a black, clear-bottom 96-well plate for fluorescence reading at 590 nm (excitation wavelength of 560 nm) using a multimode plate reader (EnVision).
Microscopy
Astrocytes in hydrogels were stained with Alexa Fluor 488 phalloidin (5 U ml−1 for 1 hour, Thermo Fisher Scientific) and 4′,6-diamidino-2-phenylindole (DAPI; 5 μg ml−1 for 15 min, Thermo Fisher Scientific) to visualize the F-actin fibers and overall cell morphology. To assess cell viability, LIVE/DEAD staining was conducted by immersing the cell-laden hydrogel into calcein AM/ethidium homodimer-1 (Invitrogen) solution (each at 1 × 10−6 M) for 20 min, followed by gentle washing with PBS. All optical microscope images were taken using a widefield fluorescence microscope (Olympus BX51) or a confocal laser scanning microscope (Leica SP5). SEM was used to observe the microstructure of different hydrogels. Hydrogels were prepared as indicated previously and incubated for 24 hours, followed by freeze-drying. The freeze-dried samples were then sputter-coated with gold (thickness, 10 to 15 nm) and imaged with a Zeiss Auriga SEM system at 5 kV. It should be noted that this approach introduces defects to the hydrogel during freeze-drying and does not represent the structure in the aqueous swollen state.
Histology
Tissue constructs were harvested and washed in PBS, fixed with 4% paraformaldehyde for 2 hours, and washed 70 volume % ethanol for paraffin embedding. Sections (10 μm) on SuperFrost Plus slides (Thermo Scientific, UK) were deparaffinized by immersing in histology cleaning agent (Histo-Clear, National Diagnostics) for 5 min, followed by rehydration in 100 volume % ethanol, 70 volume % ethanol, and then water subsequently for 2 min for each step. Cartilage slides were stained with 0.02 wt % fast green FCF for 4 min and then dipped thrice in 1 volume % acetic acid. This was followed by staining with safranin O for 6 min, 10 dips in 95 volume % ethanol, 20 dips in 100 volume % ethanol, and a 1-min immersion in 100 volume % ethanol. Bone slides were stained with hematoxylin and eosin for 5 min and Alizarin Red S (2 wt %, pH 4.2) for 2 min on consecutive sections. The stained slides were dehydrated in 70 and 100 volume % ethanol sequentially, followed by drying and mounting in Histomount (National Diagnostics). A widefield microscope (Olympus BX51) was used for imaging.
DMMB assay
Engineered cartilage was digested and analyzed using methods described in the literature (43). Freeze-dried tissue constructs were weighed and then digested by adding one volume of digestion solution, stirring at 37°C overnight, adding a second volume of digestion solution, and stirring at 65°C for a further 2 hours. The digestion solution was trypsin (2 mg/ml; N-tosyl-l-phenylalanine chloromethyl ketone treated), 1 mM iodoacetamide, 1 mM EDTA (ethylenediaminetetraacetic acid), and pepstatin A (10 μg ml−1), dissolved in water, and buffered with tris-HCl (6.6 mg ml−1) at pH 7.4. The digestion was terminated by heating the solution to 100°C for 15 min. After centrifugation at 5000g, the supernatant containing the glycosaminoglycan was carefully collected and stored at 4°C until use. Diluted samples and chondroitin sulfate standards were added to a 96-well plate (20 μl per well), before adding an aqueous solution of 1,9-dimethyl-methylene blue (16 μg ml−1), glycine (3.04 mg ml−1), and sodium chloride (2.37 mg ml−1; 200 μl per well). The absorbance was measured immediately at 525 nm (SpectraMax M5). The quantity of sulfated glycosaminoglycan was extrapolated from the standard curve and normalized by taking into account the dilution factor and the dry mass of the tissue constructs.
ALP activity assay
Engineered bone-like tissue constructs were harvested and immersed in ALP lysis buffer [1 mM MgCl2, 20 mM ZnCl2, and 0.1% octyl-β-glucopyranoside in 10 mM tris(hydroxymethyl)aminomethane buffer at pH 7.4], followed by homogenizing (22 Hz for 10 min) using a TissueLyser II (QIAGEN). The samples were then incubated with p-nitrophenol phosphate buffer at 37°C for 30 min, together with a standard curve with 0 to 800 μM 4-nitrophenol solutions. The reaction was terminated by adding an equal volume of 1 N NaOH and measured with absorbance at 405 nm using a plate reader (SpectraMax M5). A PicoGreen dsDNA assay kit was used for DNA quantification. Briefly, the PicoGreen working solution was added to an equivalent volume of samples and DNA standards in a 96-well plate. After 5-min incubation, the fluorescence emission was measured at 520 nm with an excitation wavelength of 480 nm (SpectraMax M5). The ALP activity was determined by normalizing to DNA quantity.
Statistical analyses
All statistical analyses were performed using GraphPad Prism X9. Unless otherwise noted, all data were presented as means ± SD and all statistical comparisons were made using a two-tailed Mann-Whitney test or paired Wilcoxon test.
Supplementary Material
Acknowledgments
We acknowledge L. Li for help with the schematic drawing in Fig. 1, S. C. Skaalure for help with materials preparation, S. Treumuth for help with cell isolation and culture, T. Benge for help with ALP assay, G. Miklosic and A. Moore for help with mechanical data analysis, and J. Tang for discussion in SEM imaging. C.L-R. acknowledges her secondary supervisor J. Phillips (UCL). We also acknowledge J. Phillips for the gift of astrocytes. M.M.S. is also affiliated to the Department of Medical Biochemistry and Biophysics, Karolinska Institute, Sweden. Funding: L.O. and M.M.S. acknowledge the financial support from Engineering and Physical Sciences Research Council (EPSRC) Programme Grant “Engineering Growth Factor Microenvironments—A New Therapeutic Paradigm for Regenerative Medicine” (EP/P001114/1). J.P.K.A. acknowledges support from the Medical Research Council (MRC) (MR/S00551X/1). J.P.W. and M.M.S. acknowledge the financial support from the UK Regenerative Medicine Platform “Acellular / Smart Materials—3D Architecture” (MR/R015651/1). M.M.S. and Y.L. acknowledge support from the Wellcome Trust Senior Investigator Award (098411/Z/12/Z). C.L-R. acknowledges funding from EPSRC studentship “Development of New Biomaterials for Regenerative Medicine” (no. 1975740). D.H. acknowledges funding from the European Union’s Horizon 2020 research and innovation programme under Marie Skłodowska-Curie grant agreement no. 839111. Author contributions: L.O. designed the study and performed the experiments. M.M.S. supervised the study. L.O. and J.P.K.A. wrote the manuscript with input from other authors. J.P.K.A., Y.L., and J.A.B. contributed to the scientific discussion. Y.L., J.P.W., D.H., and J.A.B. contributed to the distribution of polymers. J.P.K.A. and C.L-R. contributed to the isolation and distribution of primary cells. K.Z. contributed to the SEM imaging. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Raw research data are available online at DOI: 10.5281/zenodo.3932099.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/38/eabc5529/DC1
REFERENCES AND NOTES
- 1.Ouyang L., Armstrong J. P. K., Salmeron-Sanchez M., Stevens M. M., Assembling living building blocks to engineer complex tissues. Adv. Funct. Mater. 30, 1909009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moroni L., Burdick J. A., Highley C., Lee S. J., Morimoto Y., Takeuchi S., Yoo J. J., Biofabrication strategies for 3D in vitro models and regenerative medicine. Nat. Rev. Mater. 3, 21–37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levato R., Jungst T., Scheuring R. G., Blunk T., Groll J., Malda J., From shape to function: The next step in bioprinting. Adv. Mater. 32, 1906423 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ouyang L., Highley C. B., Rodell C. B., Sun W., Burdick J. A., 3D printing of shear-thinning hyaluronic acid hydrogels with secondary cross-linking. ACS Biomater Sci. Eng. 2, 1743–1751 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Zaman M. H., Trapani L. M., Sieminski A. L., MacKellar D., Gong H., Kamm R. D., Wells A., Lauffenburger D. A., Matsudaira P., Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc. Natl. Acad. Sci. U.S.A. 103, 10889–10894 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaudhuri O., Gu L., Klumpers D., Darnell M., Bencheriff S. A., Weaver J. C., Huebsch N., Lee H.-P., Lippens E., Duda G. N., Mooney D. J., Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat. Mater. 15, 326–334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malda J., Visser J., Melchels F. P., Jüngst T., Hennink W. E., Dhert W. J. A., Groll J., Hutmacher D. W., 25th anniversary article: Engineering hydrogels for biofabrication. Adv. Mater. 25, 5011–5028 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Chimene D., Lennox K. K., Kaunas R. R., Gaharwar A. K., Advanced bioinks for 3D printing: A materials science perspective. Ann. Biomed. Eng. 44, 2090–2102 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Colosi C., Shin S. R., Manoharan V., Massa S., Costantini M., Barbetta A., Dokmeci M. R., Dentini M., Khademhosseini A., Microfluidic bioprinting of heterogeneous 3D tissue constructs using low-viscosity bioink. Adv. Mater. 28, 677–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ouyang L., Highley C. B., Sun W., Burdick J. A., A generalizable strategy for the 3D bioprinting of hydrogels from nonviscous photo-crosslinkable inks. Adv. Mater. 29, 1604983 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Lee A., Hudson A. R., Shiwarski D. J., Tashman J. W., Hinton T. J., Yerneni S., Bliley J. M., Campbell P. G., Feinberg A. W., 3D bioprinting of collagen to rebuild components of the human heart. Science 365, 482–487 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Armstrong J. P. K., Burke M., Carter B. M., Davis S. A., Perriman A. W., 3D bioprinting using a templated porous bioink. Adv. Healthc. Mater. 5, 1724–1730 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Ouyang L., Yao R., Zhao Y., Sun W., Effect of bioink properties on printability and cell viability for 3D bioplotting of embryonic stem cells. Biofabrication 8, 035020 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Burdick J. A., Prestwich G. D., Hyaluronic acid hydrogels for biomedical applications. Adv. Mater. 23, H41–H56 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Therriault D., White S. R., Lewis J. A., Rheological behavior of fugitive organic inks for direct-write assembly. Appl. Rheol. 17, 10112-1–10112-8 (2007). [Google Scholar]
- 16.Watson P. M. D., Kavanagh E., Allenby G., Vassey M., Bioengineered 3D glial cell culture systems and applications for neurodegeneration and neuroinflammation. SLAS Discov. 22, 583–601 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Smeriglio P., Lai J. H., Dhulipala L., Behn A. W., Goodman S. B., Smith R. L., Maloney W. J., Yang F., Bhutani N., Comparative potential of juvenile and adult human articular chondrocytes for cartilage tissue formation in three-dimensional biomimetic hydrogels. Tissue Eng. Part A 21, 147–155 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Bertlein S., Brown G., Lim K. S., Jungst T., Boeck T., Blunk T., Tessmar J., Hooper G. J., Woodfield T. B. F., Groll J., Thiol-ene clickable gelatin: A platform bioink for multiple 3D biofabrication technologies. Adv. Mater. 29, 1703404 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Yin J., Yan M., Wang Y., Fu J., Suo H., 3D Bioprinting of low-concentration cell-laden gelatin methacrylate (GelMA) bioinks with a two-step cross-linking strategy. ACS Appl. Mater. Interfaces 10, 6849–6857 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Zhu K., Chen N., Liu X., Mu X., Zhang W., Wang C., Zhang Y. S., A general strategy for extrusion bioprinting of bio-macromolecular bioinks through alginate-templated dual-stage crosslinking. Macromol. Biosci. 18, 1800127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shin S., Park S. S., Park M., Jeong E., Na K., Youn H. J., Hyun J., Cellulose nanofibers for the enhancement of printability of low viscosity gelatin derivatives. BioResources 12, 2941–2954 (2017). [Google Scholar]
- 22.Xu W., Molino B. Z., Cheng F., Molino P. J., Yue Z., Su D., Wang X., Willför S., Xu C., Wallace G. G., On low-concentration inks formulated by nanocellulose assisted with gelatin methacrylate (GelMA) for 3D printing toward wound healing application. ACS Appl. Mater. Interfaces 11, 8838–8848 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ouyang L., Armstrong J. P. K., Chen Q., Lin Y., Stevens M. M., Void-free 3D bioprinting for in situ endothelialization and microfluidic perfusion. Adv. Funct. Mater. 30, 1908349 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy S. V., De Coppi P., Atala A., Opportunities and challenges of translational 3D bioprinting. Nat. Biomed. Eng. 4, 370–380 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Billiet T., Gevaert E., De Schryver T., Cornelissen M., Dubruel P., The 3D printing of gelatin methacrylamide cell-laden tissue-engineered constructs with high cell viability. Biomaterials 35, 49–62 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Diamantides N., Dugopolski C., Blahut E., Kennedy S., Bonassar L. J., High density cell seeding affects the rheology and printability of collagen bioinks. Biofabrication 11, 045016 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Nele V., Schutt C. E., Wojciechowski J. P., Kit-Anan W., Doutch J. J., Armstrong J. P. K., Stevens M. M., Ultrasound-triggered enzymatic gelation. Adv. Mater. 32, 1905914 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo C., Li C., Mu X., Kaplan D. L., Engineering silk materials: From natural spinning to artificial processing. Appl. Phys. Rev. 7, 011313 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Ruijter M., Ribeiro A., Dokter I., Castilho M., Malda J., Simultaneous micropatterning of fibrous meshes and bioinks for the fabrication of living tissue constructs. Adv. Healthc. Mater. 8, 1800418 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khetan S., Guvendiren M., Legant W. R., Cohen D. M., Chen C. S., Burdick J. A., Degradation-mediated cellular traction directs stem cell fate in covalently crosslinked three-dimensional hydrogels. Nat. Mater. 12, 458–465 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vega S. L., Kwon M. Y., Song K. H., Wang C., Mauck R. L., Han L., Burdick J. A., Combinatorial hydrogels with biochemical gradients for screening 3D cellular microenvironments. Nat. Commun. 9, 614 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loessner D., Meinert C., Kaemmerer E., Martine L. C., Yue K., Levett P. A., Klein T. J., Melchels F. P. W., Khademhosseini A., Hutmacher D. W., Functionalization, preparation and use of cell-laden gelatin methacryloyl-based hydrogels as modular tissue culture platforms. Nat. Protoc. 11, 727–746 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Zhang Z., Loebus A., de Vicente G., Ren F., Arafeh M., Ouyang Z., Lensen M. C., Synthesis of poly(ethylene glycol)-based hydrogels via amine-michael type addition with tunable stiffness and postgelation chemical functionality. Chem. Mater. 26, 3624–3630 (2014). [Google Scholar]
- 34.Wang L.-F., Shen S.-S., Lu S.-C., Synthesis and characterization of chondroitin sulfate–methacrylate hydrogels. Carbohyd. Polym. 52, 389–396 (2003). [Google Scholar]
- 35.Kim S.-H., Chu C.-C., Synthesis and characterization of dextran-methacrylate hydrogels and structural study by SEM. J. Biomed. Mater. Res. 49, 517–527 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Bencherif S. A., Sands R. W., Bhatta D., Arany P., Verbeke C. S., Edwards D. A., Mooney D. J., Injectable preformed scaffolds with shape-memory properties. Proc. Natl. Acad. Sci. U.S.A. 109, 19590–19595 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jensen H. M., Larsen F. H., Engelsen S. B., Characterization of alginates by nuclear magnetic resonance (NMR) and vibrational spectroscopy (IR, NIR, Raman) in combination with chemometrics. Methods Mol. Biol. 1308, 347–363 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Amsden B. G., Sukarto A., Knight D. K., Shapka S. N., Methacrylated glycol chitosan as a photopolymerizable biomaterial. Biomacromolecules 8, 3758–3766 (2007). [DOI] [PubMed] [Google Scholar]
- 39.Gatti G., Casu B., Hamer G. K., Perlin A. S., Studies on the conformation of heparin by 1H and 13C NMR spectroscopy. Macromolecules 12, 1001–1007 (1979). [Google Scholar]
- 40.East E., Golding J. P., Phillips J. B., A versatile 3D culture model facilitates monitoring of astrocytes undergoing reactive gliosis. J. Tissue Eng. Regen. Med. 3, 634–646 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang J., Guo Z., Timmerman A., Grijpma D., Poot A., Enhanced mechanical and cell adhesive properties of photo-crosslinked PEG hydrogels by incorporation of gelatin in the networks. Biomed. Mater. 14, 024102 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Thompson R. W. Jr., Latypov R. F., Wang Y., Lomakin A., Meyer J. A., Vunnum S., Benedek G. B., Evaluation of effects of pH and ionic strength on colloidal stability of IgG solutions by PEG-induced liquid-liquid phase separation. J. Chem. Phys. 145, 185101 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Armstrong J. P. K., Shakur R., Horne J. P., Dickinson S. C., Armstrong C. T., Lau K., Kadiwala J., Lowe R., Seddon A., Mann S., Anderson J. L. R., Ferriman A. W., Hollander A. P., Artificial membrane-binding proteins stimulate oxygenation of stem cells during engineering of large cartilage tissue. Nat. Commun. 6, 7405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/38/eabc5529/DC1