

USP15 deubiquitinase inactivates TET2 to suppress tumor immunity and is a potential therapeutic target of immunotherapy.
Abstract
TET2 DNA dioxygenase is frequently mutated in human hematopoietic malignancies and functionally inactivated in many solid tumors through a nonmutational mechanism. We recently found that TET2 mediates the interferon-JAK-STAT pathway to stimulate chemokine expression and tumor infiltration of lymphocytes (TILs). TET2 is monoubiquitylated at K1299, which promotes its activity. Here, we report that USP15 is a TET2 deubiquitinase and inhibitor. USP15 catalyzes the removal of K1299-linked monoubiquitin and negatively regulates TET2 activity. Gene expression profiling demonstrates that TET2 and USP15 oppositely regulate genes involved in multiple inflammatory pathways, and TET2 is a major target of USP15 function. Deletion of Usp15 in melanoma stimulates chemokine expression and TILs in a TET2-dependent manner, leading to increased response to immunotherapy and extended life span of tumor-bearing mice. These results reveal a previously unknown regulator of TET2 activity and suggest USP15 as a potential therapeutic target for immunotherapy of solid tumors.
INTRODUCTION
Deregulation of DNA methylation is now recognized as a critical feature of most, if not all, cancer types and is an emerging target of cancer therapy (1). DNA methylation is reversibly catalyzed by DNA methyltransferase (DNMT) and the ten-eleven translocation (TET) family of α-ketoglutarate (α-KG)– and Fe(II)-dependent DNA dioxygenases (2, 3). TET enzymes catalyze three steps of iterative oxidation—first converting 5-methylcytosine to 5-hydroxymethyl cytosine (5hmC), then 5hmC to 5-formyl cytosine (5fC), and, lastly, 5fC to 5-carboxy cytosine (5caC). 5caC can be removed by DNA glycosylase TDG, resulting in 5-unmodified cytosine and completing DNA demethylation (4, 5). Human cells express three TET proteins—TET1, TET2, and TET3—which share the same catalytic mechanism, a conserved C-terminal cysteine-rich dioxygenase (CD) domain, and more divergent N-terminal sequences. Loss-of-function mutations in TET2 occur frequently in hematopoietic malignancies of both myeloid, in particular acute myeloid leukemia (AML; ~8 to 15%), and lymphoid lineages, such as angioimmunoblastic T cell lymphoma (~30 to 40%) and diffused large B cell lymphoma (~6%) (6, 7). In a subset of AML with wild-type (WT) TET2 gene, TET2 enzyme is catalytically inactivated by D-2-hydroxyglutarate (D-2-HG), an oncometabolite produced by mutated isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) (8, 9), or functionally inactivated by the mutations in WT1 transcription factor, which binds and recruits TET2 to regulate target genes (10, 11). Mutations targeting TET2, IDH1, IDH2, and WT1 occur in a mutually exclusive manner in about a third of AMLs (12). Deletion of Tet2 in mice resulted in increased hematopoietic stem cell self-renewal and myeloid malignancy (13–15) and accelerated leukemogenesis and lymphomagenesis when combined with other oncogenic mutations (16–19). These genetic studies indicate the critical importance of maintaining TET2 function in suppressing hematopoietic malignancies.
TET genes are rarely mutated or significantly reduced in expression in solid tumors (20). However, the activity of TET enzymes, as determined by its enzymatic product, 5hmC, is significantly decreased in a wide range of solid tumors lacking mutation targeting TET genes (21–28). These findings suggest a broad tumor suppression function by the TET gene beyond hematopoietic malignancies. We recently found that loss of TET activity in melanoma and colon cancer diminished the expression of T helper 1 (TH1)–type chemokines and tumor infiltration of T cells, leading to decreased antitumor immunity and resistance to anti–PD-L1 immunotherapy (20). Functional inactivation of TET enzyme activity through a nonmutational mechanism(s) raise(s) an intriguing possibility that unlike other genomically mutated tumor suppressor genes, restoration of TET function could potentially be exploited to inhibit tumor growth. This possibility was supported by recent finding that ascorbate/vitamin C, a cofactor for α-KG/Fe(II)–dependent dioxygenases, stimulates DNA hypomethylation, reverses aberrant hematopoietic stem and progenitor cell self-renewal, and suppresses leukemia progression (29, 30). We found, along with others, that stimulation of TET activity by systemic injection of vitamin C increased TET activity, chemokine expression, and tumor infiltration of lymphocytes, resulting in enhanced antitumor immunity and increased efficacy of anti–PD-L1 immunotherapy (20, 31, 32).
While the catalytic mechanisms of TET dioxygenases are relatively well established, how TET enzymes are regulated remains poorly understood. The levels of TET mRNA change very little between matched tumor and normal samples across different cancer types (20). Instead, the availability of cofactors, including vitamin C (33, 34), α-KG (35, 36), and oxygen (28), and posttranslation modifications, such as phosphorylation (37), acetylation (38), and ubiquitylation (39), are emerging to play an important role in the regulation of TET activity. We previously found that the VprBP/DCAF1-DDB1-CUL4-ROC1 E3 ubiquitin ligase (CRL4DCAF1) catalyzes monoubiquitylation at a lysine residue that is conserved in all three TET proteins from different species (K1299 in human TET2; Fig. 1A) (39). K1299 monoubiquitylation promotes TET2 binding to DNA and chromatin. K1299 and several additional residues in TET2 involved in binding with DCAF1 are recurrently mutated in AML, leading to functional inactivation of TET2. Unlike K48- and K63-linked polyubiquitylation, which usually leads to irreversible protein degradation, monoubiquitylation regulates the function of substrate via a nonproteolytic mechanism and is often reversibly regulated by a deubiquitinase (DUB) (40). A DUB catalyzing the removal of TET monoubiquitylation is predicted to negatively regulate the activity of TET enzymes and, when disrupted, would stimulate TET activity and the response to immunotherapy. This study was aimed at searching for and functionally characterizing the TET DUB.
Fig. 1. USP15 is a TET2 deubiquitinase.
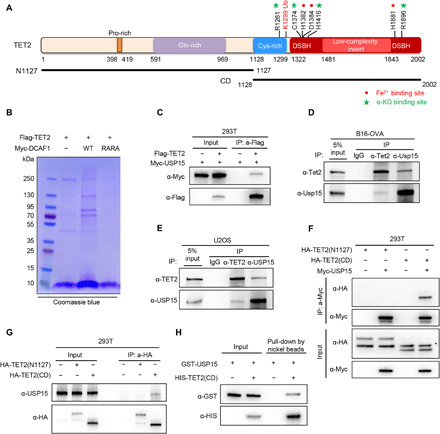
(A) Domain structure, critical residues, and expression vectors of human TET2. (B) Identification of TET2 binding proteins. HEK293T cells were singularly transfected with Flag-TET2 or cotransfected with Flag-TET2 and Myc-DCAF1 or Myc-DCAF1(RARA) mutant. Flag complexes were immunopurified from each set of transfected cells, stained with Coomassie blue, and subjected to mass spectrometric analyses. Proteins identified by the mass spectrometry that were hit by more than two unique peptides are shown in fig. S1A. (C) Ectopically expressed TET2 binds to USP15. HEK293T cells were transiently transfected with FLAG-TET2 and Myc-USP15, and cell lysates were subjected to IP with a Flag antibody, followed by Western blot analysis. (D and E) Endogenous TET2-USP15 associations were examined by reciprocal immunoprecipitation (IP)–Western analyses using indicated antibodies in mouse B16 ovalbumin (OVA) melanoma (D) and human U2OS osteosarcoma (E) cells. (F) Ectopically expressed USP15 binds to the TET2 CD domain. HEK293T cells were transiently transfected with indicated plasmids, cell lysates were subjected to IP with a Myc antibody, followed by Western blot analysis. * indicates nonspecific band. (G) USP15 binds to the TET2 CD domain. HEK293T cells were transiently transfected with the N-terminal or CD domain of TET2. Cell lysates were subjected to IP with HA antibody, followed by WB with indicated antibodies. (H) Purified recombinant GST-USP15 and His-Flag-TET2(CD) were incubated in vitro, pulled down by nickel beads overnight, then eluted with imidazole, and subjected to SDS–polyacrylamide gel electrophoresis (SDS-PAGE). USP15-TET2(CD) binding was examined by Western blot.
RESULTS
USP15 binds to TET2
To identify the DUB of TET2, we established human embryonic kidney (HEK) 293T cells stably expressing a FLAG-tagged TET2 and then ectopically expressed WT DCAF1 to increase TET2 monoubiquitylation which, we reasoned, would increase the interaction of the DUB with TET2. As a control, we ectopically expressed a mutant DCAF1, R1247A/R1283A, which disrupts its binding to DDB1 and thus blocks the ability to ubiquitylate TET2. We then performed immunoprecipitation (IP) followed by mass spectrometric analyses of three Flag immune complexes to identify TET2-interacting proteins. A large number of potential TET2-interacting proteins were identified (Fig. 1B and fig. S1A), including OGT, which was previously identified as a TET2 interacting protein (41, 42). Among these potential TET2 binding proteins, one DUB, USP15, was identified. To confirm the interaction of TET2 with USP15, we examined the binding of their association in cells when both proteins were ectopically expressed by IP–Western blot analysis. Ectopically expressed Myc-USP15 was readily detected in Flag-TET2 immune complex (Fig. 1C). Using antibodies that recognize TET2 or USP15, we detected endogenous interaction between USP15 and TET2 by reciprocal IP-Western assay in both human U2OS cells and mouse B16–ovalbumin (OVA) cells (Fig. 1, D and E). Next, we mapped the USP15 binding site to the C-terminal CD domain of TET2 (Fig. 1, F and G). An in vitro pull-down assay using purified recombinant proteins demonstrated a direct interaction between human USP15 and the CD domain of human TET2 proteins (Fig. 1H and fig. S1B). Collectively, these data demonstrate that USP15 and TET2 physically interact with each other, and the CD domain of TET2 is responsible for their association.
USP15 deubiquitylates TET2 at K1299
To determine whether USP15 catalyzes TET2 deubiquitination, we first ectopically expressed WT or catalytic dead mutant C269A USP15 (43) together with Flag-TET2 and performed in vivo ubiquitylation assay. Overexpression of WT USP15, but not the catalytically inactive mutant, decreased the monoubiquitylation of TET2 (Fig. 2A). Next, we transfected WT or catalytic inactive USP15 into U2OS cells that stably express hemagglutinin (HA)–ubiquitin, followed by IP-Western analysis of endogenous TET2. This experiment showed that monoubiquitylation of endogenous TET2 was markedly reduced when USP15 was overexpressed (Fig. 2B). Conversely, depletion of USP15 in both 293T and U2OS cells significantly increased monoubiquitylation of both ectopically expressed and endogenous TET2 (Fig. 2, C and D). Together, these results demonstrate that USP15 is a TET2 DUB.
Fig. 2. USP15 deubiquitylates TET2 at K1299.
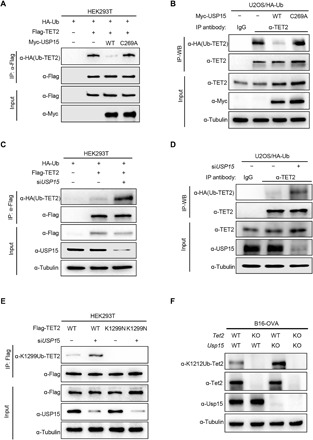
(A) USP15 deubiquitylates TET2. HEK293T cells were transfected with plasmid expressing WT and catalytic mutant USP15 as indicated. Cells were lysed and subjected to IP with FLAG antibody under denaturing conditions (0.1% SDS), and then the precipitates were separated by SDS-PAGE and blotted with indicated antibodies. (B) USP15 decreased TET2 endogenous monoubiquitylation. U2OS cells stably expressing HA-ubiquitin (HA-Ub) were transfected with indicated plasmids for 40 hours. Cells were lysed, and lysates were subjected to IP with TET2 antibody in a denaturing buffer containing 0.1% SDS. TET2 ubiquitylation was detected by α-HA immunoblotting. (C) Knocking down USP15 increased TET2 monoubiquitylation. HEK293T cells were transfected with HA-ubiquitin along with small interfering RNA (siRNA) targeting USP15 for 24 hours and then transfected with Flag-TET2 for another 48 hours. The efficiency of knockdown was verified by immunoblotting. TET2 ubiquitylation was determined by IP–Western blot analysis as described in (A). (D) Knocking down USP15 increased endogenous TET2 monoubiquitylation. U2OS cells stably expressing HA-ubiquitin (HA-Ub) were transfected with siRNA against USP15. TET2 ubiquitylation was determined by IP–Western blot analysis as described in (B). (E) The effect of USP15 on the ubiquitylation of WT or mutant TET2 was determined in HEK293T cells after transfection with indicated plasmids and siUSP15 oligo. Cells were lysed in 0.1% SDS buffer and subjected to IP with FLAG beads under denaturing condition. Precipitates were blotted with FLAG and UbTET2 (K1299) antibodies. (F) Knocking out Usp15 increased monoubiquitylation at K1212 of mouse Tet2 (equivalent to K1299 in human TET2). The indicated cells were lysed in 0.1% SDS buffer and then subjected to Western blot with indicated antibodies.
We previously found that all three TET proteins are monoubiquitylated at a conserved lysine residue—K1589, K1299, and K859 in human TET1, TET2, and TET3, respectively (39). To determine whether USP15 specifically deubiquitylates TET2 at K1299, we transfected Flag-tagged WT or K1299N mutant TET2 with USP15, followed by an in vivo ubiquitylation assay. While USP15 decreased WT TET2 monoubiquitylation levels, overexpression of USP15 could not further decrease K1299 monoubiquitylation (fig. S2A), suggesting that USP15 specifically deubiquitylates TET2 at K1299. Conversely, we demonstrated that depletion of USP15 increases the ubiquitylation of WT, but not K1299N mutant TET2 (Fig. 2E). In Usp15–knockout (KO) and Usp15/Tet2 double-knockout (DKO) B16-OVA cells (fig. S2, B and C), we found that deletion of Usp15 significantly increased monoubiquitylation of Tet2 at K1212 (K1299 in human; Fig. 2F). Together, these results demonstrate that K1299 is the major site of TET2 monoubiquitylation, and USP15 is the principle K1299 DUB of TET2.
USP15 inactivates TET2 and inhibits TET2 binding to substrate DNA
Monoubiquitylation of TET2 is important for its activity and is required for TET2 binding to chromatin (39). To determine the functional significance of USP15-TET2 interaction, we first performed a dot blot experiment to determine 5hmC levels in cells ectopically expressing USP15. We found that overexpression of WT, but not C269A catalytic inactive mutant USP15, reduced TET activity by 67% (Fig. 3A). Knocking down USP15 with two different small interfering RNAs (siRNAs) increased 5hmC levels by 41 and 74%, respectively (Fig. 3B). While knocking out Tet2 in B16-OVA cells significantly decreased 5hmC levels by 75%, deletion of Usp15 increased 5hmC level by 63% in a Tet2-dependent manner (Fig. 3C). Next, we used a more quantitative colorimetric assay to determine the 5hmC in the genome and obtained the same result that depletion of Usp15 increased global 5hmC in a manner that is dependent on Tet2 (Fig. 3D). Furthermore, quantitative fluorescence-activated cell sorting (FACS) analyses also showed that KO of Usp15 increased TET activity in B16-OVA cells expressing Tet2, but not when Tet2 is deleted (Fig. 3E). These results demonstrate that USP15 negatively regulates TET2 activity in cells.
Fig. 3. USP15 inactivates TET2 and impairs TET2 binding to DNA.
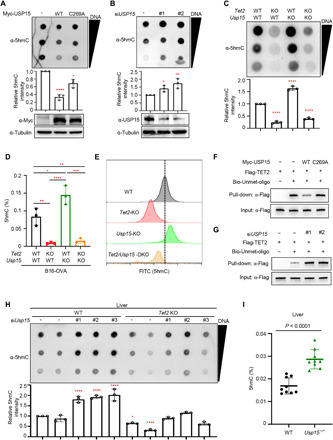
(A and B) U2OS cells were transfected with plasmid expressing indicated WT and mutant USP15 (A) or siRNA targeting USP15 (B). Global 5hmC was determined by dot-blot. (C and D) Total genomic DNA was extracted from indicated cell lines, and 5hmC level was determined by dot blot (C) and an antibody-dependent colorimetric assay using the MethylFlash Hydroxymethylated DNA Quantification Kit (Epigentek) (D), respectively. (E) 5hmC levels by flow cytometry. Four B16-OVA cell lines were immune-stained with an anti-5hmC primary antibody and a fluorescein isothiocyanate (FITC)–labeled secondary antibody. (F and G) Flag-TET2 was transfected in HEK293T cells expressed WT or catalytic mutant USP15 (F), or HEK293T cells with USP15 knocking down by siRNA (G); the expression of individual proteins is shown in fig. S3 (A and B). TET2 was purified and incubated with biotinylated DNA oligonucleotides containing a single unmethylated CpG. The binding of TET2 with DNA was determined by Western blot after pull-down using streptavidin-coated beads. (H) WT or Tet2-KO mice were tail vein injected by three different siRNA targeting Usp15 mixed with Invivofectamine 3.0. After 72 hours, genomic DNA was isolated from mice livers, and the 5hmC levels were determined by dot-blotting with anti-5hmC antibody. The knockdown efficiency was determined by quantitative polymerase chain reaction (qPCR) shown in fig. S3C. (I) Deletion of Usp15 in mice increases TET activity. Total genomic DNA was extracted from the liver of WT and Usp15-KO mice (n = 3 each), and 5hmC levels were assessed by antibody-dependent colorimetric assay. All data in this figure represent means ± SD for triplicate experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Next, we examined whether USP15 affects TET2 binding to its substrate DNA. TET2 was coexpressed with USP15, immunopurified, and then incubated with biotinylated double-stranded DNA oligonucleotides containing a single unmethylated CpG, followed by pull-down using streptavidin-coupled beads. The result showed that coexpression of TET2 with WT, but not the catalytic inactive C269A mutant USP15, reduced TET2 DNA binding (Fig. 3F and fig. S3A). Conversely, silencing USP15 enhanced TET2 DNA binding (Fig. 3G and fig. S3B), demonstrating that USP15 impairs TET2 binding to DNA in vitro.
To support the function of USP15 as an inhibitor of TET in vivo, we delivered three different siUsp15 oligos into WT and Tet2 KO mice by hydrodynamic tail vein injection and determined the 5hmC level in liver genomic DNA. We found that knockdown of Usp15 increased 5hmC levels in mouse liver (Fig. 3H and fig. S3C). Furthermore, we isolated liver and bone marrow from WT or Usp15 KO mice and determined the 5hmC levels by antibody-dependent colorimetric assay. We found that KO of Usp15 increased TET activity in both liver (Fig. 3I) and bone marrow (fig. S3D). Together, these results demonstrate that USP15 inhibits TET binding to DNA and TET activity in vivo.
Usp15 and Tet2 oppositely regulate genes involved in multiple inflammatory process
We next determined whether USP15 can change the expression of TET2 target genes. We previously reported the function of TET2 in mediating interferon-γ (IFN-γ) signaling (20). We therefore determined gene expression in WT, Tet2, and Usp15 KO B16 melanoma cells treated with IFN-γ or untreated (table S1). A total of 1197 genes were induced by 20% or more following IFN-γ treatment. Of these 1197 genes, 394 (33%) were down-regulated by Tet2 KO (by 20% or more), reinforcing the role of TET2 in mediating IFN signaling. Five hundred forty-three (45%), on the other hand, were further up-regulated by Usp15 KO, supporting the important role of USP15 in IFN signaling (44, 45). Notably, 181 IFN-γ–induced genes were oppositely regulated by Usp15 and Tet2 KO (Fig. 4A), which accounts for 33 and 46% of Usp15 inhibited or Tet2-activated genes, respectively. These results suggest that Usp15 is a major inhibitor of Tet2, and Tet2 is a major target of Usp15 in the regulation of IFN-γ signaling. The KEGG (Kyoto Encyclopedia of Genes and Genomes) signaling pathway analysis of these 181 genes revealed that they were enriched in multiple inflammatory signaling pathways, including tumor necrosis factor (TNF), chemokine, Toll-like receptor, and Janus kinase (JAK)–signal transducer and activator of transcription (STAT) signaling pathways (Fig. 4B). Genes involved in the 15 pathways whose expression is most significantly affected by either Tet2 or Usp15 deletion are shown in Fig. 4C. Consistent with our previous discovery (20), Cxcl9, Cxcl10, and Cxcl11 were also down-regulated by Tet2 KO, but their expression was up-regulated by Usp15 KO in B16 cells (Fig. 4, C and D). In addition, several genes whose expression was down-regulated by Tet2 deletion but up-regulated by Usp15 deletion are also linked to the IFN-JAK-STAT signaling pathway, including Oas3, Jak2, Jak3, Stat3, and Stat2, raising an intriguing possibility that, besides chemokine genes, additional genes on the IFN-JAK-STAT pathway may also be regulated by TET2. Together, these results demonstrate that Usp15 and Tet2 oppositely regulate the expression of genes involved in multiple inflammatory signaling pathways.
Fig. 4. Usp15 and Tet2 oppositely regulate genes involved in multiple inflammatory processes.
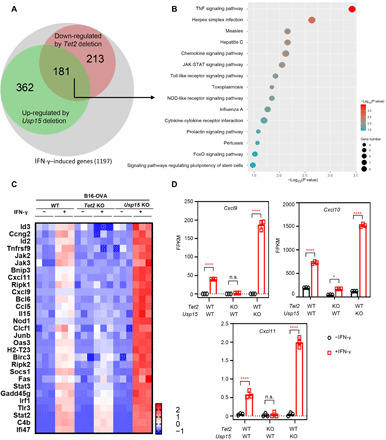
(A) Overlap between IFN-γ–induced genes down-regulated by Tet2 deletion and up-regulated by Usp15 deletion in B16 cells is displayed by a Venn diagram (P < 10−10). (B) The genes regulated by both Usp15 and Tet2 are enriched in multiple inflammatory processes. The 181 genes up- or down-regulated by Usp15 and Tet2 deletion identified in (A )were analyzed by the DAVID gene functional classification tool, and the top 15 enriched pathways (based on the Kyoto Encyclopedia of Genes and Genomes database) are shown and ranked by P value. (C) Heatmap presentation of genes involved in the 15 pathways in (B) whose expressions are most significantly affected by Tet2 and Usp15 deletion. (D) Transcription of chemokine genes Cxcl9, Cxcl10, and Cxcl11 was down-regulated by Tet2 KO and up-regulated by Usp15 KO during IFN-γ treatment. FPKM (fragments per kilobase million) values from RNA sequencing were analyzed and shown. Data represent means ± SD. TNF, tumor necrosis factor. JAK-STAT, Janus kinase–signal transducer and activator of transcription; two-tailed Student’s t-test: *P < 0.05; ****P < 0.0001. n.s., not significant.
Deletion of Usp15 promotes IFN-γ–induced chemokine expression
To confirm that Usp15 negatively regulates IFN-γ–induced chemokine expression, we treated B16-OVA cells of different genotypes with IFN-γ and examined transcription and promoter methylation. We found that deletion of Usp15 substantially increased the mRNA levels of three TH1-type chemokine genes, Cxcl9, Cxcl10, and Cxcl11, and these increases were substantially reduced when Tet2 is deleted (Fig. 5A), suggesting a Usp15-mediated repression of these genes through Tet2. We confirmed this finding by showing that Usp15 deletion increased protein levels of Cxcl9 and Cxcl10 in response to IFN-γ treatment by enzyme-linked immunosorbent assay (ELISA) analysis in a Tet2-dependent manner (Fig. 5B). We then conducted Transwell assay that measures the capacity of cell motility to assess the function of Usp15 to regulate T cell migration in response to the levels of these chemokines via the process of chemotaxis. We found that migration of CD8+ T cells toward conditional media (CM) derived from IFN-γ–treated Usp15 KO culture was accelerated compared with that observed in WT B16-OVA–derived CM (Fig. 5C). This increased T cell migration was largely abolished when the CM was derived from IFN-γ–treated Usp15/Tet2 DKO cell culture, indicating a functional dependency of Usp15 in regulating T cell migration on Tet2. Antibody-mediated blockade of CXCR3 inhibited T cell migration toward WT and Usp15 KO B16-OVA–derived CM. Furthermore, the addition of recombinant murine CXCL10 to CM from Tet2-KO and Tet2/Usp15-DKO cell cultures restored T cell migration. These results demonstrate that Usp15 deletion increased IFN-γ–induced chemokine production through Tet2, which directly leads to the increased migration of CD8+ T cells. Furthermore, we found that ectopic expression of WT TET2, but not the K1299N mutant, largely restored the induction of Cxcl9, Cxcl10, and Cxcl11 by IFN-γ (Fig. 5D and fig. S4). These results suggest that USP15-mediated K1299 deubiquitination of TET2 is important for IFN-γ–induced chemokine expression.
Fig. 5. Deletion of Usp15 promotes IFN-γ–induced chemokine expression.
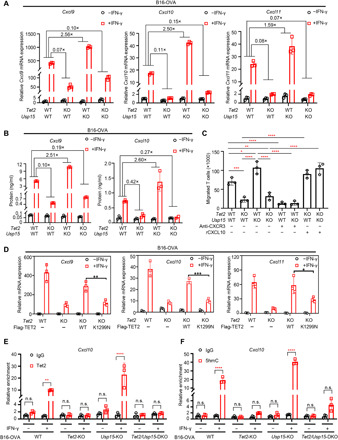
(A) Deletion of Usp15 gene in B16-OVA melanoma cells enhances IFN-γ–induced chemokines gene expression. WT, Tet2-KO, Usp15-KO, or Tet2/Usp15-DKO B16 cells were treated with IFN-γ for 20 hours, and then total RNA was extracted. The relative mRNA levels of chemokines were determined by qPCR. (B) Deletion of Usp15 gene in B16-OVA melanoma cells decreased IFN-γ–induced Cxcl9 and Cxcl10 protein levels. The indicated four cell lines were treated with IFN-γ for 72 hours, and then medium were collected and subjected for ELISA analysis. (C) Deletion of Usp15 impaired T cell attracting by Transwell assay. The indicated four cell lines were treated with IFN-γ for 48 hours, and CM were collected. Triplicate independent experiments were performed for each group. (D) TET2 WT and K1299N mutant were overexpressed in Tet2 KO B16-OVA cells, then cells were treated with IFN-γ for 20 hours, and total RNA was extracted. The TET2 expression levels were determined by Western blot (fig. S4), and the relative mRNA levels of Cxcl9, Cxcl10, and Cxcl11 were determined by qPCR. (E) WT, Tet2-KO, Usp15-KO, or Tet2/Usp15-DKO B16-OVA cells were treated with or without IFN-γ, and Tet2 binding to Cxcl10 was determined by TET2 ChIP-qPCR. (F) Hydroxymethylated DNA immunoprecipitation (hMeDIP) assays were performed in the indicated four cell lines treated with IFN-γ or untreated. 5hmC levels on the Cxcl10 promoter were determined by 5hmC ChIP-qPCR. All error bars shown in this figure represent ±SD for triplicate experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Next, we performed chromatin IP (ChIP)–quantitative polymerase chain reaction (qPCR) analysis to determine whether Usp15 affects Tet2’s binding to the promoter of Cxcl10. We confirmed that IFN-γ stimulated Tet2’s binding to the promoter of Cxcl10 in B16-OVA cells and found that deletion of Usp15 increased Tet2’s binding to the Cxcl10 promoter (Fig. 5E). 5hmC levels of the Cxcl10 promoters were increased by 17-fold in Tet2-WT B16-OVA cells after IFN-γ treatment, and deletion of Usp15 further increased 5hmC at the Cxcl10 promoter 29-fold (Fig. 5F). Deletion of Tet2 nearly completely abolished the effects of Usp15 deletion in stimulating 5hmC. Together, these results demonstrate that Usp15 deletion increases Tet2 binding to chemokine genes’ promoter, leading to increased demethylation and expression of chemokine genes.
Deletion of Usp15 enhances tumor-infiltrating lymphocytes and antitumor immunotherapy
We recently reported that TET activity is important for tumor immunity and response to immunotherapy (20). Negative regulation of Tet activity by Usp15 led us to determine whether deletion of Usp15 could enhance antitumor immunity and immunotherapy efficacy. Tet2-KO B16-OVA cells showed similar rates of proliferation compared with the parental WT B16-OVA cells (fig. S5A), and Usp15 KO increased cell proliferation, but this increase is independent of Tet2. This result indicates that Tet2 does not play a significant intrinsic role in B16-OVA cell proliferation and that Usp15 plays an important role in cell proliferation in vitro through regulating additional substrate(s). Next, we subcutaneously transplanted equal numbers of WT, Tet2-KO, Usp15-KO, or Tet2/Usp15 DKO B16-OVA melanoma cells into C57BL/6J mice, followed by intravenous injection of OT-I cells that recognize the OVA antigen expressed by the B16-OVA cells (46). Consistent with what we previously reported, deletion of Tet2 significantly (P = 0.0009) reduced mean life span of mice transplanted with WT B16-OVA cells from 28 to 20 days [a 28% decrease, group 1 (G1) versus G2; Fig. 6A]. In contrast, deletion of Usp15 significantly (P = 0.0031) extended mean life span of mice transplanted with WT B16-OVA cells from 28 days to more than 35 days (a 25% increase, G1 versus group 3 (G3); Fig. 6A). Comparing mice transplanted with Tet2-KO and Tet2/Usp15 DKO tumor cells, Usp15 deletion still displayed significant (P = 0.029) benefit and extended the mean life span from 20 to 23 days (a 15% increase, G2 versus G4; Fig. 6A), presumably due to the stimulation of TET1 and/or TET3 activity or additional Usp15 substrate(s) that might be involved in tumor immunity.
Fig. 6. Deletion of Usp15 enhances tumor-infiltrating lymphocytes and immunotherapy.
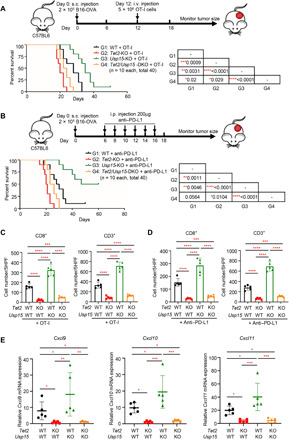
(A) Kaplan-Meier survival curves for mice injected with WT, Tet2-KO, Usp15-KO, or Tet2/Usp15-DKO B16-OVA cells and treated with adoptive T cell immunotherapy are shown. WT or Tet2 KO B16-OVA (2 × 105) cells were subcutaneously (s.c.) injected to C57BL/6J mice at day 0, and 5 × 106 OT-I cells were intravenously (i.v.) injected at day 12. Kaplan-Meier survival curves for these mice are shown (n = 10 mice for each group). The P value was determined using a log-rank (Mantel-Cox) test, comparing each two groups, and is shown in the table of the figure. (B) Kaplan-Meier survival curves for mice injected with WT, Tet2-KO, Usp15-KO, or Tet2-Usp15-DKO B16-OVA cells and treated with anti–PD-L1 therapy are shown. WT or Tet2 KO B16-OVA (2 × 105) cells and anti–PD-L1 antibody were subcutaneously and intraperitoneally injected to C57BL/6J mice at indicated time points, respectively. Kaplan-Meier survival curves for these mice are shown (n = 10 mice for each group). The P value was determined using log-rank (Mantel-Cox) test, comparing each two group, and is shown in the table of the figure. (C and D) Quantification of CD8+ and CD3+ T cells from fig. S5B (C) and fig. S5C (D), respectively. Average cell number per high-power field (HPF) is shown, with five HPFs calculated from each group. Error bars represent ±SD. (E) Deletion of Usp15 in allograft tumors increased chemokine expression in a Tet2-dependent manner. Total mRNA was extracted from the indicated tumor (n = 5 for each group), and mRNA levels of genes were determined by qPCR. Data represent means ± SD. *P < 0.05; **P < 0.01; ***P < 0.001;****P < 0.0001.
Next, we determined the effect of Usp15 deletion for anti–PD-L1 immunotherapy. The mean life span of mice transplanted with Usp15-KO B16-OVA cells (>54 days) was significantly (P = 0.0046) longer by 86.2% than that of mice transplanted with WT cells (29 days, G1 versus G3; Fig. 6B). Mice transplanted with Tet2/Usp15 DKO cells (24.5 days) also had a 16.3% increased mean life span compared with mice transplanted with Tet2-KO cells (20.5 days, G2 versus G4), but the benefits on the life span conferred by Usp15 deletion were much more pronounced in the presence of Tet2 than without it. Associated with these life-span extensions are the increases in tumor infiltrating CD8+ and CD3+ T cells by the Usp15 deletion, and their increases are dependent on the expression of Tet2 in both models, either with injection of OT-I cells (Fig. 6C and fig. S5B) or anti–PD-L1 antibody (Fig. 6D and fig. S5C). Last, we examined the intratumoral chemokine expression and found that while Cxcl9, Cxcl10, and Cxcl11 expressions were significantly decreased in Tet2-KO B16-OVA tumors, their expressions were increased in Usp15-KO tumors in a Tet2-dependent manner (Fig. 6E). Together, these results demonstrate that deletion of Usp15 in tumor stimulates chemokine expression and lymphocyte infiltration and enhances the effect of antitumor immunity and efficacy of anti–PD-L1 immunotherapy.
DISCUSSION
While the catalytic mechanism and genetic function of TET enzymes are relatively well characterized, how TET enzymes are regulated remains poorly understood. The levels of TET mRNA change very little between matched tumor and normal samples across different cancer types (20). Instead, the availability of cofactors (28, 33–36) and posttranslation modifications (37–39) are emerging as significant factors in the regulation of TET activity (see Introduction). One modification is CRL4DCAF1-catalyzed monoubiquitylation, which promotes TET binding to chromatin and TET activity (39). Four lines of evidence presented in this study support USP15 as a major TET DUB and a previously unidentified TET regulator. First, USP15 directly interacts with TET2 (Fig. 1) and catalyzes the TET2 deubiquitylation. We showed that overexpression reduces and depletion of USP15 increases TET2 monoubiquitylation, respectively, and this activity is specific to K1299-linked ubiquitin (Fig. 2). Second, deletion or depletion of Usp15 resulted in increased 5hmC in cultured B16 cells and in mouse liver in a manner that is dependent on Tet2 (Fig. 3). Third, Usp15 and Tet2 oppositely regulate the expression of as many as 181 IFN-γ–induced genes, which counts for 33 and 46% of Usp15-inhibited or Tet2-activated genes, respectively (Fig. 4). This result suggests that Usp15 is a major inhibitor of Tet2 and Tet2 is a major target of Usp15 in the regulation of IFN-γ signaling. Last, deletion of Usp15 in melanoma cells stimulated chemokine expression and tumor infiltration of lymphocytes, resulting in increased response to T cell and immunotherapy and extended life span of tumor-bearing mice. Each of these phenotypes was previously found to be reduced by TET2 deletion in tumor cells or stimulated by the systematic injection of ascorbic acid/vitamin C, a cofactor of TET enzymes (20). Collectively, these results identify USP15 as a bona fide regulator of TET enzymes. TET2, and possibly other TET proteins, is also regulated by p300-mediated acetylation, which promotes TET2 catalytic activity, protein stability, and binding with other partners and is counted by HDCA1/2-mediated deacetylation (38). TET2 was also found to be phosphorylated by AMP (adenosine 5′-monophosphate)–activated protein kinase (AMPK), which promotes TET2 binding to 14-3-3 and TET2 protein stability and is counted by protein phosphatase 2A (PP2A) (37, 47, 48). Our findings provide further evidence supporting the regulation of TET enzyme by reversible posttranslational modifications. Disruptions of these three modifications were linked to various human pathological conditions such as oxidative stress (38), diabetes (37), and cancer (37, 39). It will be important to determine what physiological conditions regulate each of these modifications and whether they cross-talk to integrate different cellular conditions.
An oncogenic activity of USP15 was previously suggested by its function to deubiquitylate and stabilize the transforming growth factor–β receptor and MDM2 and by its amplification and association with poor prognosis in glioblastoma (49). Analysis of a pan-cancer project showed that USP15 was amplified in about 4% of human cancers of different types (glioblastoma, breast, lung, kidney, liver). The mechanism underlying the oncogenic activity of USP15 is not clear. Several studies have previously linked the function of USP15 to IFN signaling during T cell activation (50, 51), viral infection, and neuroinflammation (44). Several cellular substrates of USP15 have been reported that are directly involved in immune response and IFN signaling, including IκBα and TRIM25 E3 ligase, both of which play critical roles in innate immune and inflammation response (52–54), and MDM2 E3 ligase, which promotes the degradation of NFATc2 during T cell activation (50). We found that the expression of 545 of 1197 IFN-γ–induced genes (45%) was up-regulated by Usp15 KO (Fig. 4A), providing further support for the function of USP15 in IFN signaling. Among these IFN-γ–induced genes whose expression was also down-regulated by Tet2 KO are Cxcl9, Cxcl10, and Cxcl11, which are often referred to as TH1-type chemokines and are consistent with our previous discovery (20). In addition to DNA demethylation, several other epigenetic modification enzymes have also been previously linked to the regulation of TH1-type chemokine expression and tumor immunity, including PRC2 complex (e.g., EZH2), DNMTs, and SWI/SNF (switching defective/sucrose non-fermenting) complex (e.g., ARID1A) (55, 56). Together, these findings raise an interesting possibility that these immune response genes, which are not needed during normal growth or homeostasis, are stably suppressed by epigenetic modifications. Our findings also provide a previously unidentified mechanism for the negative regulation of IFN signaling by USP15 by placing USP15 directly onto the JAK-STAT-TET2 axis in mediating the IFN signaling pathway.
The findings presented here also bear important therapeutic implications. Deletion of Usp15 in tumor cells promotes intratumoral chemokine production and tumor infiltration of T cells (Fig. 5), and enhanced response to T cell immunity and anti–PD-L1 therapy, resulting in extended life span of tumor-bearing mice (Fig. 6). These findings present USP15, an enzyme, as a potential therapeutic target for enhancing antitumor immunity and immunotherapy efficacy for solid tumors. DUBs are a newly emerging class of drug targets, and potent and selective DUB inhibitors are being developed (57). Mice lacking Usp15 develop normally and have no obvious tumor phenotype. Furthermore, Usp15 deficiency promotes T cell activation and enhances T cell responses to tumor challenge in vivo. Compared with WT mice, Usp15−/− mice had significantly reduced tumor size and extended life span in the B16 syngeneic tumor model (50). Together with the findings presented here, we suggest that USP15 inhibitors, if developed, could be specific and effective in promoting immunotherapy through enhancing tumor-intrinsic TET activity and chemokine production and T cell–intrinsic activity and IFN-γ production, and with limited toxicity.
MATERIALS AND METHODS
Plasmids
Expression constructs for TET2 and ubiquitin were previously described (39). USP15 was cloned to pcDNA3-3Myc vector. Point mutations for USP15 were generated by site-directed mutagenesis.
Cell cultures and cell transfection
HEK293T and B16-OVA (B16F10 cells expressing OVA) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; CORNING) supplemented with 10% fetal bovine serum (FBS) (CORNING) and 1% penicillin/streptomycin antibiotics (CORNING). U2OS cells were maintained in McCoy’s 5A medium (CORNING) containing 10% FBS and 1% penicillin/streptomycin antibiotics. Cell transfection was carried out by Lipofectamine 2000 according to the manufacturer’s protocol (Life Technologies).
Cell lysis, IP, immunoblotting, and antibodies
Cells were washed with ice-cold phosphate-buffered saline and lysed in NP-40 lysis buffer at 4°C for 30 min. Cell lysates were incubated with anti-Flag beads (Sigma-Aldrich) or protein A/G-agarose (Thermo Fisher Scientific) and antibodies (indicated in the figures) for 3 hours at 4°C. The beads were washed three times with NP-40 buffer and then subjected to SDS–polyacrylamide gel electrophoresis (SDS-PAGE) or eluted by Flag peptides for TET2-DNA binding assay. Western blotting was performed according to standard protocol.
Antibodies to Flag (Clone M2, Sigma-Aldrich), HA (Clone 3F10, Roche), Myc (Clone 9E10, Roche), TET2 (Millipore, catalog no. MABE462; Cell Signaling, 18950), USP15 (Abcam, catalog no. ab71713), STAT1 (Santa Cruz, catalog no. sc-345), and tubulin (Santa Cruz, catalog no. sc-23948) were purchased commercially.
siRNA transfection and RNA interference
All siRNA oligonucleotides were synthesized with 3′ dTdT overhangs by Sigma-Aldrich in a purified and annealed duplex form. The target gene used two effective sequences as follows: human USP15-1, 5′-GGUUGGAAUAAACUUGUCA-3′; USP15-2, 5′-GCACCUUGGAAGUUUACUU-3′. For transfection of siRNA, OPTI-MEM medium (250 μl) was mixed with 10 μl of Lipofectamine 2000 for 5 min and then incubated with another 250 μl of OPTI-MEM medium containing 10 μl of siRNA (20 mM) for 20 min at room temperature. The mixtures were added to cells cultured on a 60-mm plate at 30 to 40% confluence. The knocking down efficiency was determined by Western blot 72 hours after transfection.
Gene deletion by CRISPR-Cas9 system
Tet2 and Usp15 KO B16-OVA cells were generated through the CRISPR (clustered regularly interspaced short palindromic repeats)–Cas9 system by transient CRISPR strategy (58). Cells were transiently transfected with a Cas9 and single-guide RNA (sgRNA) plasmid with enhanced green fluorescent protein (EGFP) expression (PX458; Addgene plasmid no. 48138). The gRNA sequence used for targeting Tet2 was AAAGTGCCAACAGATATCC, and the gRNA sequence used for targeting Usp15 was TGGCGACGCGCAGTCACTT. Following transfection for 2 days, single cells were sorted by FACS based on EGFP expression into 96-well plates. KO clones were validated by Western blot and DNA sequencing.
In vivo ubiquitylation assay
For the in vivo ubiquitylation assay in cells, HEK293T cells or U2OS cells were transfected with plasmid DNA or siRNA as indicated in the figures. Lysates were prepared with NP-40 lysis buffer containing 0.1% SDS. FLAG-TET2 or endogenous TET2 was immunoprecipitated with anti-FLAG (M2) antibody or anti-TET2 antibody, respectively. Then, immunoprecipitates were subsequently subjected to SDS-PAGE. Ubiquitylated TET2 was detected with HA antibody or K1299 site specific antibody.
In vitro pull-down assay
Biotin-labeled single-stranded oligonucleotides used for pull-down assay were synthesized at Sigma-Aldrich as follows: unmethylated CpG probe, 5′-GTATGCCTCATGCCGGACTTAACTGCAGTG-3′ (forward) and 3′-CATACGGAGTACGGCCTGAATTGACGTCAC-5′ (reverse). DNA annealing was performed as described before (39). Two nanomoles of complementary single-stranded oligonucleotides was mixed in annealing buffer [10 mM tris (pH 8.0), 50 mM NaCl, 1 mM EDTA], boiled for 5 min, transferred to a water preheated to 90°C, and gradually cooled overnight.
Flag-TET2 was singly or cotransfected with other plasmids indicated in the figures and immunopurified from HEK293T cells with anti-FLAG M2 agarose beads for 3 hours in a NP-40 lysis buffer [0.3% NP-40, 50 mM tris (pH 7.5), 150 mM NaCl]. Immobilized Flag-tagged proteins were washed three times in the same lysis buffer and eluted with an excess of Flag peptide (Sigma-Aldrich). The annealed DNA was immobilized to streptavidin beads by incubation together at 4°C for 2 hours and washed three times. Flag-TET2 was incubated with immobilized DNA at 4°C for 3 hours and washed three times. The binding efficiency of TET2-DNA was determined by Western blot.
In vivo tumor progression and immunotherapy models
All animal studies were approved by the UNC-CH Institutional Animal Care and Use Committee (IACUC). B16-OVA cells (2 × 105) were subcutaneously transplanted into the back flanks of 5- to 6-week-old C57BL/6J mice (the Jackson laboratory) or nude mice. Tumor size was measured with a caliper every 2 to 3 days, and tumor volume was calculated by width2 × length × 0.523. Mice were euthanized when tumors reached maximum allowed size (20 mm in diameter).
For the adoptive T cell immunotherapy model, OT-I cells were isolated from 6- to 8-week-old C57BL/6-Tg (TcraTcrb)1100Mjb/J mice (the Jackson laboratory, stock no. 003831) using CD8a microbeads (Miltenyi Biotec) according to the manual. OT-I CD8+ T cells (5 × 106) were intravenously transfused into tumor-bearing mice at day 12. For anti–PD-L1 immunotherapy model, mice were intraperitoneally injected with 200 μg of anti–PD-L1 (clone 10F.9G2, BP0101, Bio X Cell) three times per week for 2 weeks after tumor implantation. Mice were monitored for tumor growth every 3 days and euthanized when tumors reached 20 mm in diameter.
ChIP-qPCR assay
ChIP assay was performed as described previously (59). DNA was sheared by sonication using a Covaris sonicator for 12 min at 4°C. ChIP-enriched DNA was analyzed by qPCR with SYBR Green Master Mix. Cxcl10 Primers used for qPCR analysis were as follows: 5′-CGCCGAGTCAGAGCTGCG-3′ (forward) and 5′- GCTTTCTGAGAAACGAAAGCTC-3′ (reverse).
Statistical analyses
Data analysis was performed using GraphPad Prism software. Normally distributed data were analyzed using an unpaired, two-tailed Student’s t test; multiple comparisons used one-way analysis of variance (ANOVA) and corrected by Tukey multiple-comparisons test or Dunnett’s multiple-comparisons test. A log-rank (Mantel-Cox) test was used for the mouse survival assay. Statistical significance was defined as a P value of less than 0.05. Levels of significance were indicated as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplementary Material
Acknowledgments
We thank N. Mailand for providing HA-ubiquitin stably expressed cell lines, and X. Tan for helping with statistical analyses. Funding: This study was supported by a Samuel Waxman Research Foundation Investigator Award and NIH grants CA163834 and R35GM128855 to Y.X. This study was also supported by Shanghai Natural Science Foundation, China (General Program, No. 20ZR1461900) and Fundamental Research Funds for the Central Universities of China (No. 22120200112) to Y.-P. X. Author contributions: Y.-P. X. performed the experimental work including the animal experiments, biochemical and cellular studies, and analyzed the data. L.-L.C. analyzed the RNA-seq data and conducted revision experiments. M.D.S. assisted with animal work. L.L. performed TET2-DNA binding and ChIP experiments. T.N. did the IP–mass spectrometry analysis. Z.L. helped with the flow cytometry. S.-C.S. provided the Usp15 KO mice. N.G.B. participated in the project discussion and manuscript preparation. Y.-P. X. and Y. X. designed the experiments, conceived and supervised the study, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/38/eabc9730/DC1
REFERENCES AND NOTES
- 1.Topper M. J., Vaz M., Marrone K. A., Brahmer J. R., Baylin S. B., The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 17, 75–90 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenberg M. V. C., Bourc'his D., The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 20, 590–607 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Wu X., Zhang Y., TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 18, 517–534 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Tahiliani M., Koh K. P., Shen Y., Pastor W. A., Bandukwala H., Brudno Y., Agarwal S., Iyer L. M., Liu D. R., Aravind L., Rao A., Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ito S., Shen L., Dai Q., Wu S. C., Collins L. B., Swenberg J. A., He C., Zhang Y., Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delhommeau F., Dupont S., Valle V. D., James C., Trannoy S., Massé A., Kosmider O., Le Couedic J.-P., Robert F., Alberdi A., Lécluse Y., Plo I., Dreyfus F. J., Marzac C., Casadevall N., Lacombe C., Romana S. P., Dessen P., Soulier J., Viguié F., Fontenay M., Vainchenker W., Bernard O. A., Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 360, 2289–2301 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Reddy A., Zhang J., Davis N. S., Moffitt A. B., Love C. L., Waldrop A., Leppa S., Pasanen A., Meriranta L., Karjalainen-Lindsberg M.-L., Nørgaard P., Pedersen M., Gang A. O., Høgdall E., Heavican T. B., Lone W., Iqbal J., Qin Q., Li G., Kim S. Y., Healy J., Richards K. L., Fedoriw Y., Bernal-Mizrachi L., Koff J. L., Staton A. D., Flowers C. R., Paltiel O., Goldschmidt N., Calaminici M., Clear A., Gribben J., Nguyen E., Czader M. B., Ondrejka S. L., Collie A., Hsi E. D., Tse E., Au-Yeung R. K. H., Kwong Y.-L., Srivastava G., Choi W. W. L., Evens A. M., Pilichowska M., Sengar M., Reddy N., Li S., Chadburn A., Gordon L. I., Jaffe E. S., Levy S., Rempel R., Tzeng T., Happ L. E., Dave T., Rajagopalan D., Datta J., Dunson D. B., Dave S. S., Genetic and functional drivers of diffuse large B cell lymphoma. Cell 171, 481–494.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chowdhury R., Yeoh K. K., Tian Y.-M., Hillringhaus L., Bagg E. A., Rose N. R., Leung I. K. H., Li X. S., Woon E. C. Y., Yang M., McDonough M. A., King O. N., Clifton I. J., Klose R. J., Claridge T. D. W., Ratcliffe P. J., Schofield C. J., Kawamura A., The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 12, 463–469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S.-H., Ito S., Yang C., Wang P., Xiao M.-T., Liu L.-x., Jiang W.-q., Liu J., Zhang J.-y., Wang B., Frye S., Zhang Y., Xu Y.-h., Lei Q.-y., Guan K. L., Zhao S.-m., Xiong Y., Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-Ketoglutarate-Dependent dioxygenases. Cancer Cell 19, 17–30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y., Xiao M., Chen X., Chen L., Xu Y., Lv L., Wang P., Yang H., Ma S., Lin H., Jiao B., Ren R., Ye D., Guan K.-L., Xiong Y., WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol. Cell 57, 662–673 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rampal R., Alkalin A., Madzo J., Vasanthakumar A., Pronier E., Patel J., Li Y., Ahn J., Abdel-Wahab O., Shih A., Lu C., Ward P. S., Tsai J. J., Hricik T., Tosello V., Tallman J. E., Zhao X., Daniels D., Dai Q., Ciminio L., Aifantis I., He C., Fuks F., Tallman M. S., Ferrando A., Nimer S., Paietta E., Thompson C. B., Licht J. D., Mason C. E., Godley L. A., Melnick A., Figueroa M. E., Levine R. L., DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 9, 1841–1855 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Figueroa M. E., Abdel-Wahab O., Lu C., Ward P. S., Patel J., Shih A., Li Y., Bhagwat N., Vasanthakumar A., Fernandez H. F., Tallman M. S., Sun Z., Wolniak K., Peeters J. K., Liu W., Choe S. E., Fantin V. R., Paietta E., Löwenberg B., Licht J. D., Godley L. A., Delwel R., Valk P. J. M., Thompson C. B., Levine R. L., Melnick A., Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moran-Crusio K., Reavie L., Shih A., Abdel-Wahab O., Ndiaye-Lobry D., Lobry C., Figueroa M. E., Vasanthakumar A., Patel J., Zhao X., Perna F., Pandey S., Madzo J., Song C., Dai Q., He C., Ibrahim S., Beran M., Zavadil J., Nimer S. D., Melnick A., Godley L. A., Aifantis I., Levine R. L., Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20, 11–24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z., Cai X., Cai C.-L., Wang J., Zhang W., Petersen B. E., Yang F.-C., Xu M., Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118, 4509–4518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ko M., Bandukwala H. S., An J., Lamperti E. D., Thompson E. C., Hastie R., Tsangaratou A., Rajewsky K., Koralov S. B., Rao A., Ten-Eleven-Translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc. Natl. Acad. Sci. U.S.A. 108, 14566–14571 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen E., Schneider R. K., Breyfogle L. J., Rosen E. A., Poveromo L., Elf S., Ko A., Brumme K., Levine R., Ebert B. L., Mullally A., Distinct effects of concomitant Jak2V617F expression and Tet2 loss in mice promote disease progression in myeloproliferative neoplasms. Blood 125, 327–335 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shih A. H., Jiang Y., Meydan C., Shank K., Pandey S., Barreyro L., Antony-Debre I., Viale A., Socci N., Sun Y., Robertson A., Cavatore M., de Stanchina E., Hricik T., Rapaport F., Woods B., Wei C., Hatlen M., Baljevic M., Nimer S. D., Tallman M., Paietta E., Cimmino L., Aifantis I., Steidl U., Mason C., Melnick A., Levine R. L., Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell 27, 502–515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rasmussen K. D., Jia G., Johansen J. V., Pedersen M. T., Rapin N., Bagger F. O., Porse B. T., Bernard O. A., Christensen J., Helin K., Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 29, 910–922 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zang S., Li J., Yang H., Zeng H., Han W., Zhang J., Lee M., Moczygemba M., Isgandarova S., Yang Y., Zhou Y., Rao A., You M. J., Sun D., Huang Y., Mutations in 5-methylcytosine oxidase TET2 and RhoA cooperatively disrupt T cell homeostasis. J. Clin. Invest. 127, 2998–3012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y.-p., Lv L., Liu Y., Smith M. D., Li W.-C., Tan X.-m., Cheng M., Li Z., Bovino M., Aubé J., Xiong Y., Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Invest. 129, 4316–4331 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang H., Liu Y., Bai F., Zhang J.-Y., Ma S.-H., Liu J., Xu Z.-D., Zhu H.-G., Ling Z.-Q., Ye D., Guan K.-L., Xiong Y., Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 32, 663–669 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gambichler T., Sand M., Skrygan M., Loss of 5-hydroxymethylcytosine and ten-eleven translocation 2 protein expression in malignant melanoma. Melanoma Res. 23, 218–220 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Jäwert F., Hasséus B., Kjeller G., Magnusson B., Sand L., Larsson L., Loss of 5-hydroxymethylcytosine and TET2 in oral squamous cell carcinoma. Anticancer Res. 33, 4325–4328 (2013). [PubMed] [Google Scholar]
- 24.Haffner M. C., Chaux A., Meeker A. K., Esopi D. M., Gerber J., Pellakuru L. G., Toubaji A., Argani P., Iacobuzio-Donahue C., Nelson W. G., Netto G. J., de Marzo A. M., Yegnasubramanian S., Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2, 627–637 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kudo Y., Tateishi K., Yamamoto K., Yamamoto S., Asaoka Y., Ijichi H., Nagae G., Yoshida H., Aburatani H., Koike K., Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 103, 670–676 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Müller T., Gessi M., Waha A., Isselstein L. J., Luxen D., Freihoff D., Freihoff J., Becker A., Simon M., Hammes J., Denkhaus D., zur Mühlen A., Pietsch T., Waha A., Nuclear exclusion of TET1 is associated with loss of 5-hydroxymethylcytosine in IDH1 wild-type gliomas. Am. J. Pathol. 181, 675–683 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Lian C. G., Xu Y., Ceol C., Wu F., Larson A., Dresser K., Xu W., Tan L., Hu Y., Zhan Q., Lee C.-w., Hu D., Lian B. Q., Kleffel S., Yang Y., Neiswender J., Khorasani A. J., Fang R., Lezcano C., Duncan L. M., Scolyer R. A., Thompson J. F., Kakavand H., Houvras Y., Zon L. I., Mihm M. C. Jr., Kaiser U. B., Schatton T., Woda B. A., Murphy G. F., Shi Y. G., Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 150, 1135–1146 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thienpont B., Steinbacher J., Zhao H., D’Anna F., Kuchnio A., Ploumakis A., Ghesquière B., Van Dyck L., Boeckx B., Schoonjans L., Hermans E., Amant F., Kristensen V. N., Koh K. P., Mazzone M., Coleman M. L., Carell T., Carmeliet P., Lambrechts D., Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 537, 63–68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cimmino L., Dolgalev I., Wang Y., Yoshimi A., Martin G. H., Wang J., Ng V., Xia B., Witkowski M. T., Mitchell-Flack M., Grillo I., Bakogianni S., Ndiaye-Lobry D., Martín M. T., Guillamot M., Banh R. S., Xu M., Figueroa M. E., Dickins R. A., Abdel-Wahab O., Park C. Y., Tsirigos A., Neel B. G., Aifantis I., Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 170, 1079–1095.e20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agathocleous M., Meacham C. E., Burgess R. J., Piskounova E., Zhao Z., Crane G. M., Cowin B. L., Bruner E., Murphy M. M., Chen W., Spangrude G. J., Hu Z., DeBerardinis R. J., Morrison S. J., Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magri A., Germano G., Lorenzato A., Lamba S., Chilà R., Montone M., Amodio V., Ceruti T., Sassi F., Arena S., Abrignani S., D’Incalci M., Zucchetti M., Di Nicolantonio F., Bardelli A., High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 12, eaay8707 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Luchtel R. A., Bhagat T., Pradhan K., Jacobs W. R. Jr., Levine M., Verma A., Shenoy N., High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc. Natl. Acad. Sci. U.S.A. 117, 1666–1677 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minor E. A., Court B. L., Young J. I., Wang G., Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem. 288, 13669–13674 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blaschke K., Ebata K. T., Karimi M. M., Zepeda-Martínez J. A., Goyal P., Mahapatra S., Tam A., Laird D. J., Hirst M., Rao A., Lorincz M. C., Ramalho-Santos M., Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 500, 222–226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang H., Lin H., Xu H., Zhang L., Cheng L., Wen B., Shou J., Guan K., Xiong Y., Ye D., TET-catalyzed 5-methylcytosine hydroxylation is dynamically regulated by metabolites. Cell Res. 24, 1017–1020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris J. P. IV, Yashinskie J. J., Koche R., Chandwani R., Tian S., Chen C.-C., Baslan T., Marinkovic Z. S., Sánchez-Rivera F. J., Leach S. D., Carmona-Fontaine C., Thompson C. B., Finley L. W. S., Lowe S. W., α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573, 595–599 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu D., Hu D., Chen H., Shi G., Fetahu I. S., Wu F., Rabidou K., Fang R., Tan L., Xu S., Liu H., Argueta C., Zhang L., Mao F., Yan G., Chen J., Dong Z., Lv R., Xu Y., Wang M., Ye Y., Zhang S., Duquette D., Geng S., Yin C., Lian C. G., Murphy G. F., Adler G. K., Garg R., Lynch L., Yang P., Li Y., Lan F., Fan J., Shi Y., Shi Y. G., Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 559, 637–641 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y. W., Wang Z., Xie W., Cai Y., Xia L., Easwaran H., Luo J., Yen R.-W. C., Li Y., Baylin S. B., Acetylation enhances TET2 function in protecting against abnormal DNA methylation during oxidative stress. Mol. Cell 65, 323–335 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakagawa T., Lv L., Nakagawa M., Yu Y., Yu C., D’Alessio A. C., Nakayama K., Fan H.-Y., Chen X., Xiong Y., CRL4VprBP E3 ligase promotes monoubiquitylation and chromatin binding of TET dioxygenases. Mol. Cell 57, 247–260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oh E., Akopian D., Rape M., Principles of ubiquitin-dependent signaling. Annu. Rev. Cell Dev. Biol. 34, 137–162 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Chen Q., Chen Y., Bian C., Fujiki R., Yu X., TET2 promotes histone O-GlcNAcylation during gene transcription. Nature 493, 561–564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deplus R., Delatte B., Schwinn M. K., Defrance M., Méndez J., Murphy N., Dawson M. A., Volkmar M., Putmans P., Calonne E., Shih A. H., Levine R. L., Bernard O., Mercher T., Solary E., Urh M., Daniels D. L., Fuks F., TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 32, 645–655 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inui M., Manfrin A., Mamidi A., Martello G., Morsut L., Soligo S., Enzo E., Moro S., Polo S., Dupont S., Cordenonsi M., Piccolo S., USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat. Cell Biol. 13, 1368–1375 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Torre S., Polyak M. J., Langlais D., Fodil N., Kennedy J. M., Radovanovic I., Berghout J., Leiva-Torres G. A., Krawczyk C. M., Ilangumaran S., Mossman K., Liang C., Knobeloch K.-P., Healy L. M., Antel J., Arbour N., Prat A., Majewski J., Lathrop M., Vidal S. M., Gros P., Erratum: USP15 regulates type I interferon response and is required for pathogenesis of neuroinflammation. Nat. Immunol. 17, 1479 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Zhang H., Wang D., Zhong H., Luo R., Shang M., Liu D., Chen H., Fang L., Xiao S., Ubiquitin-specific protease 15 negatively regulates virus-induced type I interferon signaling via catalytically-dependent and -independent mechanisms. Sci. Rep. 5, 11220 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clarke S. R., Barnden M., Kurts C., Carbone F. R., Miller J. F., Heath W. R., Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol. Cell Biol. 78, 110–117 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Chen H., Yu D., Fang R., Rabidou K., Wu D., Hu D., Jia P., Zhao Z., Wu Z., Peng J., Shi Y., Shi Y. G., TET2 stabilization by 14-3-3 binding to the phosphorylated Serine 99 is deregulated by mutations in cancer. Cell Res. 29, 248–250 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kundu A., Shelar S., Ghosh A. P., Ballestas M., Kirkman R., Nam H., Brinkley G. J., Karki S., Mobley J. A., Bae S., Varambally S., Sudarshan S., 14-3-3 proteins protect AMPK-phosphorylated ten-eleven translocation-2 (TET2) from PP2A-mediated dephosphorylation. J. Biol. Chem. 295, 1754–1766 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eichhorn P. J. A., Rodón L., Gonzàlez-Juncà A., Dirac A., Gili M., Martínez-Sáez E., Aura C., Barba I., Peg V., Prat A., Cuartas I., Jimenez J., García-Dorado D., Sahuquillo J., Bernards R., Baselga J., Seoane J., USP15 stabilizes TGF-β receptor I and promotes oncogenesis through the activation of TGF-β signaling in glioblastoma. Nat. Med. 18, 429–435 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Zou Q., Jin J., Hu H., Li H. S., Romano S., Xiao Y., Nakaya M., Zhou X., Cheng X., Yang P., Lozano G., Zhu C., Watowich S. S., Ullrich S. E., Sun S.-C., USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat. Immunol. 15, 562–570 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zou Q., Jin J., Xiao Y., Zhou X., Hu H., Cheng X., Kazimi N., Ullrich S. E., Sun S.-C., T cell intrinsic USP15 deficiency promotes excessive IFN-γ production and an immunosuppressive tumor microenvironment in MCA-induced fibrosarcoma. Cell Rep. 13, 2470–2479 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schweitzer K., Bozko P. M., Dubiel W., Naumann M., CSN controls NF-κB by deubiquitinylation of IκBα. EMBO J. 26, 1532–1541 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Torre S., Polyak M. J., Langlais D., Fodil N., Kennedy J. M., Radovanovic I., Berghout J., Leiva-Torres G. A., Krawczyk C. M., Ilangumaran S., Mossman K., Liang C., Knobeloch K.-P., Healy L. M., Antel J., Arbour N., Prat A., Majewski J., Lathrop M., Vidal S. M., Gros P., USP15 regulates type I interferon response and is required for pathogenesis of neuroinflammation. Nat. Immunol. 18, 54–63 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Pauli E.-K., Chan Y. K., Davis M. E., Gableske S., Wang M. K., Feister K. F., Gack M. U., The ubiquitin-specific protease USP15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci. Signal. 7, ra3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peng D., Kryczek I., Nagarsheth N., Zhao L., Wei S., Wang W., Sun Y., Zhao E., Vatan L., Szeliga W., Kotarski J., Tarkowski R., Dou Y., Cho K., Hensley-Alford S., Munkarah A., Liu R., Zou W., Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li J., Wang W., Zhang Y., Cieślik M., Guo J., Tan M., Green M. D., Wang W., Lin H., Li W., Wei S., Zhou J., Li G., Jing X., Vatan L., Zhao L., Bitler B., Zhang R., Cho K. R., Dou Y., Kryczek I., Chan T. A., Huntsman D., Chinnaiyan A. M., Zou W., Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J. Clin. Invest. 130, 2712–2726 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harrigan J. A., Jacq X., Martin N. M., Jackson S. P., Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 17, 57–78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lv L., Wang Q., Xu Y., Tsao L.-C., Nakagawa T., Guo H., Su L., Xiong Y., Vpr targets TET2 for degradation by CRL4VprBP E3 ligase to sustain IL-6 expression and enhance HIV-1 replication. Mol. Cell 70, 961–970.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/38/eabc9730/DC1