

The C-terminal RING and DTC domains of mammalian Deltex proteins catalyze ADP-ribosylation at the C terminus of ubiquitin.
Abstract
Cellular cross-talk between ubiquitination and other posttranslational modifications contributes to the regulation of numerous processes. One example is ADP-ribosylation of the carboxyl terminus of ubiquitin by the E3 DTX3L/ADP-ribosyltransferase PARP9 heterodimer, but the mechanism remains elusive. Here, we show that independently of PARP9, the conserved carboxyl-terminal RING and DTC (Deltex carboxyl-terminal) domains of DTX3L and other human Deltex proteins (DTX1 to DTX4) catalyze ADP-ribosylation of ubiquitin’s Gly76. Structural studies reveal a hitherto unknown function of the DTC domain in binding NAD+. Deltex RING domain recruits E2 thioesterified with ubiquitin and juxtaposes it with NAD+ bound to the DTC domain to facilitate ADP-ribosylation of ubiquitin. This ubiquitin modification prevents its activation but is reversed by the linkage nonspecific deubiquitinases. Our study provides mechanistic insights into ADP-ribosylation of ubiquitin by Deltex E3s and will enable future studies directed at understanding the increasingly complex network of ubiquitin cross-talk.
INTRODUCTION
Reversible posttranslational modifications (PTMs) are used by eukaryotic cells to dynamically regulate signaling events (1). Attachment of PTMs rapidly alters the properties of a protein and, by extension, its functions. While a single PTM is capable of achieving this purpose, many proteins are modified by several PTMs, which exponentially increases the number of possible biological consequences (2). Another layer complexity is achieved when ubiquitin (Ub), itself a PTM, is modified by phosphorylation, deamidation, acetylation, or adenosine 5′-diphosphate (ADP)–ribosylation (3). A number of studies have shown that cross-talk occurs between ubiquitination and poly-ADP-ribosylation (PARylation) whereby PARylation serves as a signal to target substrates for polyubiquitination and subsequent degradation (4–6). In addition, ADP-ribosylation of Ub on Arg42 by Legionella pneumophila SidE family proteins or on Gly76 by the complex comprising E3 Deltex-3-like protein ligase (DTX3L) and protein mono-ADP-ribosyltransferase poly(ADP-ribose) polymerase 9 (PARP9) has been shown to modulate ubiquitination (7–9).
In mammals, three families of proteins are primarily responsible for attaching and removing ADP-ribose moieties during ADP-ribosylation–dependent signaling events. The PARP family of proteins, of which PARP9 is a member, catalyzes the transfer of ADP-ribose to target proteins, whereas ADP-ribosylglycohydrolases and macrodomains remove these signals. PARPs share an evolutionarily conserved catalytic domain essential for binding NAD+ (nicotinamide adenine dinucleotide). These enzymes transfer one or more moieties of ADP-ribose from NAD+ to substrate while releasing nicotinamide, resulting in mono-ADP-ribosylated (MAR) or poly-ADP-ribosylated (PAR) substrates. ADP-ribose can be transferred to side chains with a nucleophilic nitrogen, oxygen, or sulfur atom to generate an N-, O-, or S-glycosidic linkage, respectively (10, 11). Moreover, an O-glycosidic linkage can also form on the C-terminal carboxylate. Although PARP9 is a member of the PARP family, it lacks catalytic activity in auto-ADP-ribosylation assays (12).
PARP9 binds DTX3L (13), which shares a conserved C terminus with Deltex proteins that comprises a RING finger domain and a ~150-residue Deltex C-terminal (DTC) domain of unknown function (14). The presence of a RING finger domain is characteristic of RING-type E3 ligases, which bind E2~Ub thioester intermediate (~ indicates a thioester bond) and facilitate transfer of the C terminus of Ub directly from E2~Ub to substrate (15). The sequential actions of the E1-E2-E3 enzyme cascade are required to achieve ubiquitination (16, 17). E1 uses Mg2+–adenosine 5′-triphosphate (ATP) to activate and then transfer the C terminus of Ub to E2’s catalytic cysteine to form E2~Ub. E3s then catalyze transfer of the C terminus of Ub to the side chain of a lysine residue on substrate (18). Like other PTMs, ubiquitination on a substrate is reversible and is removed by deubiquitinases (DUBs) (19).
We wanted to investigate how the cross-talk between ADP-ribosylation and ubiquitination is mediated by the DTX3L/PARP9 heterodimer. Previously, the formation of ADP-ribosylated–Ub (ADPr-Ub) was reported to depend on both the RING domain of DTX3L and the catalytic domain of PARP9. However, we unexpectedly found that the E3 ligase DTX3L alone was sufficient to catalyze ADP-ribosylation of Ub in the presence of NAD+ and components of the ubiquitination cascade. By truncating DTX3L, we pinpointed that the RING and DTC (hereafter referred to as RD) domains were the minimum subunit required to perform this reaction. Because this region is conserved in the Deltex family, we tested all its members and found that they share this ability to ADP-ribosylate Ub. We hypothesized that the RD domains have to physically interact with both NAD+ and E2~Ub, and using DTX1, we demonstrated that the conserved DTC domain binds NAD+. We found that ADPr-Ub generation by DTX1 relies on NAD+ binding to the DTC domain, E2~Ub recruitment by the RING domain, and the structural arrangement of the two domains. This Ub modification is susceptible to removal by the linkage nonspecific DUB Ub-specific protease 2 (USP2), thereby revealing the dynamic nature of this signal. Overexpression of DTX2 in human embryonic kidney (HEK) 293 cells could overcome signal removal by DUBs and allowed us to detect an ADPr-Ub signal in a cellular environment.
RESULTS
Deltex proteins catalyze ADP-ribosylation of Ub
To elucidate the mechanism of Ub ADP-ribosylation by DTX3L/PARP9, we initially used 32P-NAD+ or biotin-NAD+ to follow the formation of 32P-ADPr-Ub or biotin-ADPr-Ub, respectively, in the presence of E1, the E2 UbcH5B, Mg2+-ATP, and DTX3L/PARP9 complex, DTX3L or PARP9 (Fig. 1A and fig. S1A). As previously reported (9), ADPr-Ub formed with DTX3L/PARP9 complex but not with PARP9 alone; unexpectedly, however, DTX3L alone was sufficient to generate ADPr-Ub. DTX3L has three predicted N-terminal domains (D1, D2, and D3), a RING domain bearing its ligase activity, and a DTC domain of unknown function (Fig. 1B). To map the domain or domains responsible for ADP-ribosylation of Ub, we tracked the formation of ADPr-Ub by truncated DTX3L variants and found that the minimum fragment comprised the RD domains; neither the RING nor DTC domain alone formed 32P-ADPr-Ub (Fig. 1A) or biotin-ADPr-Ub (fig. S1A). To further probe requirements for ADP-ribosylation of Ub by DTX3L, we omitted each reactant individually (E1, E2, Mg2+-ATP, and biotin-NAD+) and tracked biotin-ADPr-Ub formation. Formation of biotin-ADPr-Ub by full-length DTX3L (DTX3L-FL) or DTX3L-RD required the presence of a complete set of ubiquitination cascade components (E1, E2 UbcH5B, Mg2+-ATP, and Ub) and NAD+ (Fig. 1, C and D).
Fig. 1. All Deltex E3s ADP-ribosylate Ub via the RING-DTC domains.
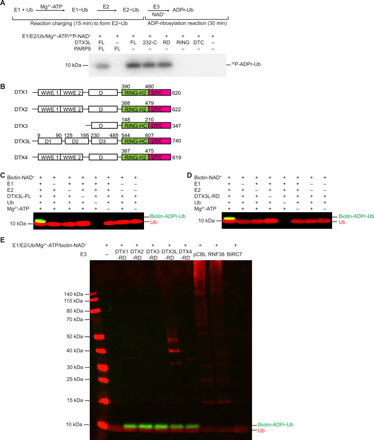
(A) Reduced autoradiogram showing Ub ADP-ribosylation reactions with DTX3L and PARP9 variants in the presence of E1, E2 UbcH5B, Mg2+-ATP, Ub, and 32P-NAD+. The reaction scheme is shown above, where E2~Ub was first generated before the addition of E3 and NAD+ to initiate the reaction. (B) Domain organization of the Deltex family of E3s. All members share adjoining C-terminal RD domains. DTX1, DTX2, and DTX4 have tandem WWE domains at the N terminus and C3H2C3-type RING fingers. DTX3 and DTX3L have C3HC4-type RING fingers. DTX3L has a unique N-terminal region that mediates its interaction with PARP9, while DTX1 to DTX4 contain a proline-rich region in domain D. (C) Western blot of in vitro Ub ADP-ribosylation reactions in which E1, E2 UbcH5B, Ub, Mg2+-ATP, or full-length DTX3L (DTX3L-FL) has been omitted as indicated in the presence of biotin-NAD+ and separated by SDS–polyacrylamide gel electrophoresis (PAGE) in reducing conditions. (D) As in (C), but with DTX3L-RD. (E) Western blot of in vitro reactions with E1, E2 UbcH5B, Mg2+-ATP, Ub, biotin-labeled NAD+, and the indicated Deltex protein RING-DTC domains, pCBL, RNF38, or BIRC7 separated by SDS-PAGE in reducing conditions. For (C) to (E), α-Ub is shown in red, and NeutrAvidin DyLight is shown in green.
Given that the RD domains are the defining characteristic of the Deltex family of E3s (Fig. 1B), we next investigated whether ADP-ribosylation of Ub is a general feature of this family. ADPr-Ub formation was observed in assays with each of the five Deltex RD domains but not with RING domains from other E3s such as pCBL (phosphorylated-Tyr371 Casitas B-lineage lymphoma protein), RNF38 (RING finger protein 38), or BIRC7 (baculoviral inhibitor of apoptosis protein repeat-containing protein 7) (Fig. 1E). In addition, we also confirmed that like DTX3L, DTX1-RD and DTX2-RD depend on a complete set of ubiquitination cascade components and NAD+ to form ADPr-Ub (fig. S1, B and C). Together, our results reveal a previously unknown function of the Deltex proteins in catalyzing ADP-ribosylation of Ub through their RD domains.
The DTC domain of Deltex accommodates NAD+
Building on our finding that ADP-ribosylation of Ub by Deltex RD domains relies on NAD+ and components of the ubiquitination cascade, we hypothesized that the DTX1-RD needs to (i) accommodate NAD+ and (ii) recruit E2~Ub. To investigate whether Deltex RD domains bind NAD+, we purified 15N-DTX1-RD and acquired 1H-15N heteronuclear single-quantum coherence (HSQC) spectra of 15N-DTX1-RD alone and in the presence of NAD+ (Fig. 2A). Chemical shift perturbations (CSPs) induced upon the addition of NAD+ suggest that it binds DTX1-RD.
Fig. 2. DTX1 DTC domain binds NAD+.
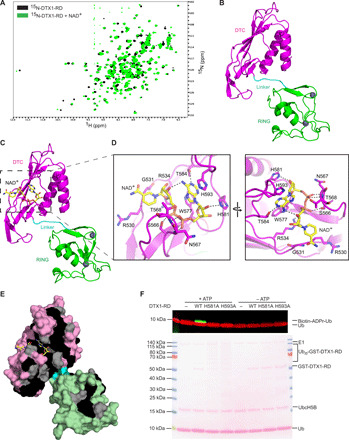
(A) 1H-15N HSQC spectra of 15N-DTX1-RD (black) and after the addition of NAD+ (green). ppm, parts per million. (B) Cartoon representation of the structure of the DTX1-RD. The RING, linker, and DTC domains are colored green, cyan, and magenta, respectively. Zn2+ atoms are depicted as gray spheres. (C) Structure of DTX1-RD bound to NAD+. DTX1-RD is colored as in (B) and in the same orientation. NAD+ is shown in sticks with C atoms colored yellow, O atoms colored red, N atoms colored blue, and P atoms colored orange. (D) Close-up view of NAD+-binding site corresponding to the region outlined in (C) (left). A different close-up view of the NAD+-binding site is shown in the right panel. Key NAD+-binding residues are shown in sticks, and atoms are colored as in (C). Black dashes indicate hydrogen bonds. (E) The DTX1-RD is shown as a surface representation with identical and conserved residues from the sequence alignment in fig. S2C colored black and gray, respectively. The RING, linker, and DTC domains are colored light green, cyan, and light pink, respectively. (F) Western blot (top) and Ponceau stain (bottom) of in vitro ADP-ribosylation reactions with DTX1-RD wild-type (WT) or indicated variants in the presence of E1, UbcH5B, Mg2+-ATP, Ub, and 32P-NAD+. Reactions without ATP were used to show similar E3 loading.
To gain further insight into the RD domains and how they accommodate NAD+, we determined crystal structures of the DTX1-RD alone and in a complex with NAD+ to 1.88 and 1.74 Å, respectively (Fig. 2, B and C, and Table 1). The structure of the RING domain resembles the nuclear magnetic resonance (NMR) structures of mouse [Protein Data Bank (PDB): 1V87] and human (PDB: 6IR0) DTX2 RING domain [root mean square deviation (RMSD) values of 0.938 Å across 71 Cα atoms and 1.237 Å across 55 Cα atoms, respectively], and the DTC domain resembles the crystal structure of human DTX3L DTC domain (PDB: 3PG6; RMSD value of 0.603 Å across 111 Cα atoms). In both the apo and complex structures, the RING domain is connected to the DTC domain by a short five–amino acid linker, and this conformation does not shift upon binding NAD+ (RMSD value of 0.184 Å across 222 Cα atoms). NAD+ binds to a pocket in the DTC domain (Fig. 2D) in which it adopts a compact conformation such that the adenine amino group forms a hydrogen bond with the C3 hydroxyl group of the nicotinamide ribose moiety (Fig. 2D and fig. S2, A and B). The adenine ring is stabilized by π-stacking interactions with aromatic side chains of Trp577 and His593. In addition, hydrogen bonds are observed between Thr584 and N1 of the adenine ring and between His581 and the adenosine ribose. The pyrophosphate forms hydrogen bonds with side chains of Arg534 and Ser566 and backbone amides of Asn567 and Thr568.
Table 1. Data collection and refinement statistics.
| DTX1-RD | DTX1-RD-NAD+ | |
| PDB code | 6Y5N | 6Y5P |
| Data collection | ||
| Space group | P212121 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 67.49, 86.550, 129.830 | 66.85, 85.25, 130.95 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90, 90, 90 |
| Resolution (Å) | 36.01–1.88 (1.88–1.93)* | 66.85–1.74 (1.79–1.74)* |
| Rmerge (%) | 7.3 (117.0) | 5.6 (133.4) |
| I/σI | 14.7 (1.4) | 16.0 (1.0) |
| Completeness (%) | 100.0 (100.0) | 100 (100) |
| Redundancy | 6.6 (6.8) | 6.4 (5.9) |
| Refinement | ||
| Resolution (Å) | 36.01–1.88 (1.88–1.93)* | 65.47–1.74 (1.79–1.74)* |
| No. reflections | 119069 | 148052 |
| Rwork/Rfree (%) | 18.71/21.05 | 18.53/21.31 |
| No. atoms | ||
| Protein | 7128 | 7144 |
| Ions | 4 | 4 |
| Ligand | 140 | |
| Water | 526 | 420 |
| B factors (Å2) | ||
| Protein | 37.26 | 35.96 |
| Ions | 57.68 | 43.01 |
| Ligand | 56.44 | |
| Water | 45.89 | 46.42 |
| RMSDs | ||
| Bond lengths (Å) | 0.011 | 0.010 |
| Bond angles (°) | 1.079 | 1.103 |
*Values in parentheses are for highest-resolution shell.
Sequence alignment of the Deltex RD domains reveals that the aforementioned residues involved in binding NAD+ are identical (Fig. 2E and fig. S2C), except for His593, Asn567, and Thr568. However, differences in these three residues should not affect NAD+ binding. In other Deltex proteins, His593 is conserved as an aromatic residue and can putatively form π-stacking interactions with the adenine ring. Because only the backbone amides of Asn567 and Thr568 play a role in binding NAD+, the side chain properties are likely to be negligible. High sequence conservation in the binding pocket suggests that Deltex proteins are likely to bind NAD+ in a similar manner.
To validate the NAD+-binding motif observed in our structure, we purified DTX1-RD H581A and H593A mutants and tested their ability to form ADPr-Ub. Both variants were defective in promoting ADP-ribosylation of Ub (Fig. 2F) but did not affect E3 ligase activity, as demonstrated by the discharge of Ub from UbcH5B~Ub to l-lysine (fig. S2, D and E). Together, these results show that DTX1-RD binds NAD+ via the DTC domain to prime NAD+ for ADPr-Ub formation.
RING domain of Deltex interacts with E2~Ub
We next scrutinized our second hypothesis that ADP-ribosylation of Ub by RD domains requires E2~Ub recruited in a RING domain–dependent manner. Initially, we tested whether this fragment was capable of promoting ubiquitination. Sequence alignment of the conserved RD region from Deltex family proteins (fig. S2C) reveals the absence of a RING domain “linchpin” arginine that helps promote the closed E2~Ub conformation in which Ub is primed for transfer (18, 20). To assess whether Deltex fragments encompassing the RD domains or RING domain were active in Ub transfer, we purified glutathione S-transferase (GST)–tagged DTX1-RD and DTX1-RING and showed that both are competent in autoubiquitination despite lacking a linchpin arginine (Fig. 3A and fig. S3A).
Fig. 3. DTX1 RD relies on recruitment of E2~Ub for the formation of ADPr-Ub.
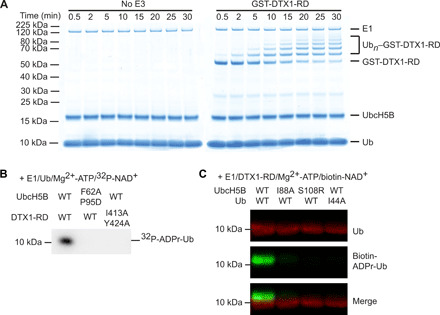
(A) Reduced SDS-PAGE gels following autoubiquitination of GST-DTX1-RD over time. Ubn, Ub chains of length n. (B) Reduced autoradiogram of Ub ADP-ribosylation reactions with DTX1-RD and UbcH5B variants in the presence of E1, Mg2+-ATP, Ub, and 32P-NAD+. (C) Western blot of in vitro reactions with E1, Mg2+-ATP, DTX1-RD, and biotin-labeled NAD+ and the indicated UbcH5B or Ub variant, separated with SDS-PAGE under reducing conditions; α-Ub is shown in red, and NeutrAvidin DyLight is shown in green.
To test our hypothesis that the RING domain is responsible for recruiting E2~Ub in the RD domain–catalyzed reaction, we examined the formation of ADPr-Ub in the presence of 32P-NAD+ with UbcH5B F62A P95D, an E2 variant that cannot bind the RING domain (21), and DTX1-RD I413A Y424A, a RING variant suggested to impair interactions with E2 based on similarities to other RING E3s (fig. S3B). Ile413 in DTX1 is equivalent to Ile440 in MDM2 (murine double minute 2) and Ile26 in BRCA1 (breast cancer type 1 susceptibility protein), and this residue has been demonstrated to be important for binding E2 in both of these RING E3s (22, 23). The UbcH5B and DTX1-RD mutants both showed defects in Ub transfer and abolished ADP-ribosylation of Ub (Fig. 3B and fig. S3, C to F). Thus, the ability of the DTX1 RING domain to recruit E2~Ub is required for ADPr-Ub formation.
Subsequently, we examined the effects of disrupting the primed UbcH5B~Ub conformation required for canonical Ub transfer on the formation of ADPr-Ub. We mutated key residues involved in maintaining the closed conformation, including Ub’s Ile44 to Ala and UbcH5B’s Ile88 to Ala or Ser108 to Arg, which were previously shown to hinder RING E3-mediated Ub transfer (21, 24). These mutants diminished Ub transfer activity in DTX1-RD autoubiquitination assays (fig. S3, G to I) and also abrogated ADP-ribosylation of Ub (Fig. 3C). Collectively, these data suggest that the DTX1 RING domain binds UbcH5B~Ub in the canonical closed conformation to promote the formation of ADPr-Ub.
Ub ADP-ribosylation depends on Deltex RING-DTC conformation
Our data demonstrate that priming of E2~Ub for Ub transfer by the RING domain and recruitment of NAD+ by the DTC domain are indispensable for ADPr-Ub formation. However, whether the arrangement of the two domains relative to one another is critical for this reaction is unclear. In DTX3L, neither the RING nor the DTC domain alone catalyzed ADP-ribosylation of Ub (Fig. 1A). Likewise, in DTX1, neither domain alone was sufficient to promote the formation of ADPr-Ub (Fig. 4A). Furthermore, when we titrated DTX1-RING with excess DTX1-DTC domain in trans, the ability to form ADPr-Ub was not restored (Fig. 4A). Thus, the reaction must occur in cis, suggesting that the conformational arrangement of the RD domains is crucial.
Fig. 4. Flexible arrangement of RING-DTC allows the formation of ADPr-Ub.
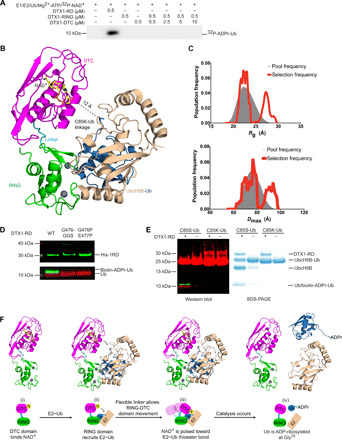
(A) Reduced autoradiogram showing Ub ADP-ribosylation reactions with DTX1-RD or DTX1 RING domain (DTX1-RING) with increasing concentrations of DTX1-DTC domain (DTX1-DTC) in the presence of E1, E2 UbcH5B, Mg2+-ATP, Ub, and+ 32P-NAD+. (B) Model of UbcH5B~Ub complex bound to DTX1-RD obtained by aligning the RING domain of DTX1 to RNF38 bound to E2~Ub (PDB: 4V3L), with RMSD value of 2.072 Å over 76 Cα atoms. UbcH5B~Ub complex is shown as a cartoon representation with UbcH5B in wheat, and Ub in shown blue. DTX1-RD and NAD+ are colored as in Fig. 2C. The stable conjugate of E2~Ub (UbcH5B C85K–Ub) was used in the modeling, and the C85K-Ub isopeptide linkage is shown in sticks. (C) Population frequency versus Rg (top) and Dmax (bottom), respectively, obtained modeling the solution structure of DTX1-RD using an ensemble of conformations. The gray area represents the distribution of the initial pool of generated structures, and the red line shows the two different populations of conformers obtained with this analysis. (D) Western blot of in vitro reactions with E1, UbcH5B, Ub, Mg2+-ATP, biotin-labeled NAD+, and the indicated DTX1-RD linker variant, separated with SDS-PAGE under reducing conditions; α-His (~30 kDa) is shown in green, α-Ub (~10 kDa) is shown in red, and NeutrAvidin DyLight is shown in green. (E) Left: Western blot of the hydrolysis of UbcH5B C85S–Ub or UbcH5B C85K–Ub with biotin-labeled NAD+ in the presence and absence of DTX1-RD; α-Ub is shown in red, and NeutrAvidin DyLight is shown in green. Right: SDS-PAGE gel of reactions in the left panel. (F) Proposed mechanism of ADP-ribosylation of Ub’s C terminus by DTX1-RD.
In all available structures of E2~Ub bound to a RING domain in which Ub is primed for transfer, E2~Ub adopts a closed conformation in which the globular body of Ub is proximal to the E2, and the tail of Ub is locked into E2’s active site groove (18, 24, 25). On the basis of these similarities, we modeled E2~Ub onto our DTX1-RD-NAD+ complex structure (Fig. 4B). In our model, NAD+ is situated ~12 Å away from the UbcH5B~Ub thioester bond. We postulated that the linker between the RD domains might be sufficiently flexible to bridge this gap. To probe its flexibility, we collected small-angle x-ray scattering (SAXS) data on DTX1-RD in solution. The linear Guinier region gave a radius of gyration (Rg) of 24.9 Å, whereas the calculated Rg for the crystal structure was 23.5 Å (fig. S4A). The Dmax calculated from the P(r) from the solution data was 90 Å, whereas it measured 74.7 Å in the crystal structure, suggesting that DTX1-RD can adopt a more extended conformation in solution.
Next, the crystal structure was split into two separate domains [RING (388 to 471) and DTC (479-C)], and the solution structure of DTX1-RD was modeled using an ensemble of conformations (fig. S4B) (26). Two populations best fit the data: One is a compact population with an Rg and Dmax of around 22 and 70 Å, respectively, and the second is a population with an Rg and Dmax of 25 and 96 Å, respectively (Fig. 4C). These data suggest that the two domains have some flexibility in relation to each other in solution (fig. S4C) and that there is scope to accommodate binding to E2~Ub or substrates. We hypothesize that this flexible linker enables the RD domains to sample different positions that bring E2~Ub closer to NAD+.
To assess the importance of linker flexibility, we inserted a Gly-Gly-Ser linker after G476 (G476-GGS) to alter the linker length or mutated G476 and E477 to proline (G476P E477P) to restrict the rotational flexibility. Both mutants exhibited comparable E3 activity in discharging E2~Ub as wild-type DTX1-RD (fig. S4, D and E) but failed to ADP-ribosylate Ub (Fig. 4D). These results suggest that both precise length and flexibility of the linker connecting the two domains are important for ADP-ribosylation of Ub.
Next, we investigated whether ADP-ribosylation of Ub occurs on E2~Ub conjugate or after Ub is discharged by using Ub covalently linked via its C terminus to the active site of UbcH5B (UbcH5B–Ub) with an oxyester linkage (UbcH5B C85S–Ub) or an isopeptide linkage (UbcH5B C85K–Ub) as a substrate in the presence of DTX1-RD and biotin-NAD+. UbcH5B C85K–Ub is not labile, whereas UbcH5B C85S–Ub releases Ub slowly on its own and when in the presence of a RING domain. Covalently bound Ub from both UbcH5B–Ub complexes was not modified. The formation of biotin-ADPr-Ub was only observed when Ub was released from UbcH5B C85S–Ub in the presence of DTX1-RD (Fig. 4E).
On the basis of these results, we propose the following Ub ADP-ribosylation reaction mechanism model by Deltex-RD domains (Fig. 4F): (i) The DTC domain binds NAD+, (ii) the RING domain recruits E2~Ub and promotes the formation of the closed E2~Ub conformation favorable for Ub transfer, (iii) the flexible linker allows the E2~Ub to be brought into close proximity with NAD+, and (iv) this facilitates ADP-ribosylation of Ub upon its release from E2~Ub. In our DTX1-RD-NAD+ model with E2~Ub, conserved residues in the RD domains are localized to the NAD+ and E2~Ub-binding sites (fig. S4F), thus raising the likelihood that our observations in DTX1-RD are extendable to other Deltex proteins.
ADP-ribosylation of Ub at G76 is reversible
To identify the extent and site of ADP-ribosylation on Ub, we generated ADPr-Ub and incubated it with the principal PAR-removing enzyme poly(ADP-ribose) glycohydrolase (PARG), which hydrolyzes the O-glycosidic ribose-ribose 1-2′ bonds between ADP-ribose moieties in PAR but cannot cleave the link between the terminal ADP-ribose and protein (27). In the presence of PARG, ADPr-Ub levels were not noticeably diminished (fig. S5A). In addition, no differences in apparent molecular weight between Ub and purified ADPr-Ub were detected by SDS–polyacrylamide gel electrophoresis (PAGE), and no band smearing such as that observed in PARP1-mediated PARylation reactions was evident (fig. S5B). We next performed the ADP-ribosylation reaction using 15N-Ub, purified ADPr-15N-Ub, and acquired 1H-15N HSQC spectra. Comparison of the spectra of 15N-Ub and ADPr-15N-Ub revealed marked CSPs in Ub’s C-terminal diglycine motif, with the largest CSP observed for Gly76 (Fig. 5, A and B), while no CSPs were observed in control samples without DTX1-RD or NAD+ (fig. S5, C and D). These data suggest that Ub is mono-ADP-ribosylated on Gly76 and are consistent with previous findings by Yang and colleagues (9).
Fig. 5. Ub ADP-ribosylation on Gly76 is reversible by USP2.
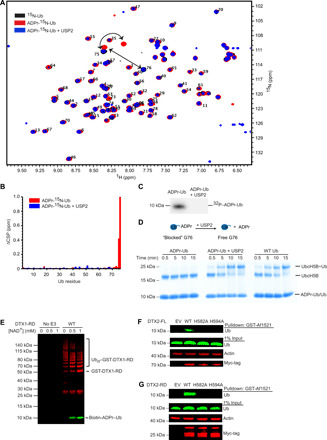
(A) 1H-15N HSQC spectra of wild-type 15N-Ub (black), ADPr-15N-Ub (red), and ADPr-15N-Ub following treatment with USP2 (blue). Upon ADP-ribosylation, peaks from Gly75 and Gly76 undergo distinct shifts. (B) Changes in CSP (ΔCSP) determined by 1H-15N-HQSC for each residue of ADPr-15N-Ub (red) and ADPr-15N-Ub following treatment with USP2 (blue) compared to wild-type 15N-Ub. (C) Autoradiogram of 32P-ADPr-Ub before and after treatment with USP2. (D) Nonreduced SDS-PAGE gel following the formation of UbcH5B~Ub using purified ADPr-Ub, purified ADPr-Ub treated with USP2 (middle), or wild-type Ub (right). (E) Western blot of GST-DTX1-RD autoubiquitination in the presence of biotin-NAD+; α-GST is shown in red, and NeutrAvidin DyLight is shown in green and separated by SDS-PAGE in reducing conditions. (F) Western blot of GST-Af1521 pull-downs on whole-cell lysates of HEK293 cells transfected with empty vector (EV) or full-length Myc-tagged DTX2 variants as indicated. Probed using α-Ub, α-actin, and α-Myc-tag with 1% of input loaded as a loading control. (G) As in (F), but with Myc-tagged DTX2-RD variants.
ADPr-Ub modification can be recycled by as-yet unidentified factor(s) in cell lysates (fig. S5A) (9). Given that DUBs can hydrolyze ester, thioester, and amide linkages at the C terminus of Ub to expose the C-terminal diglycine motif (28), we reasoned that Ub-linkage nonspecific DUBs might hydrolyze ADPr-Ub. We tested a panel of DUBs for their ability to remove ADPr from ADPr-Ub: Ub-specific proteases (USPs; USP2, USP5, USP7, and USP21), ovarian tumor proteases (OTUs; vOTU, OTUB1, OTUD3, and OTULIN), yeast Ub C-terminal hydrolase 1 (YUH1), and associated molecule with the SH3 (Src homology 3) domain of signal transduction adaptor molecule (STAM) (AMSH). Of these, the linkage nonspecific DUBs (USPs, vOTU, and YUH1) reduced or eliminated the signal from ADPr-Ub (fig. S5E). The addition of USP2 (29) to ADPr-15N-Ub reverted the CSPs of Ub’s Gly75 and Gly76 to that observed in 15N-Ub (blue; Fig. 5, A and B) and promoted the release of 32P-ADPr or biotin-labeled ADPr from 32P-ADPr-Ub (Fig. 5C) or biotin-labeled ADPr-Ub (fig. S5A), respectively. Liquid chromatography–mass spectrometry (LC-MS) analysis of ADPr-Ub treated with USP2 revealed a peak consistent with the release of ADP-ribose, confirmed by comparison to an authentic ADP-ribose standard (fig. S6). These results confirm DUBs as factors for removing ADPr from ADPr-Ub to yield free Ub and support the finding that ADP-ribosylation occurs on the C terminus of Ub.
The unmodified C-terminal diglycine motif of Ub is important for its activation and transfer along the E1-E2-E3 cascade (17), and any C-terminal modification would render Ub unusable. In contrast to unmodified Ub, when purified ADPr-Ub was incubated with Uba1 and UbcH5B, no E2~Ub was formed (Fig. 5D). Upon treating ADPr-Ub with USP2, Ub was reactivated and formed UbcH5B~Ub (Fig. 5D). Considering that the Deltex-RD can carry out both ubiquitination and Ub ADP-ribosylation, we wondered what might regulate these two opposing reactions. To probe whether the two reactions occur at the same time, we performed GST-DTX1-RD autoubiquitination assays in the presence of NAD+. With increasing concentrations of NAD+, ADPr-Ub formation increased, and GST-DTX1-RD autoubiquitination decreased (Fig. 5E). These data suggest that NAD+ concentration and the presence of linkage nonspecific DUBs can contribute to regulation of these functionally divergent reactions.
Detecting ADPr-Ub in a cellular environment
Having characterized ADPr-Ub formation by Deltex proteins in vitro, we wanted to determine whether this species exists in a cellular environment. We sought out conditions in which transient expression of Deltex was high to overcome potential difficulties associated with cleavage of the ADPr-Ub linkage by DUBs and other as-yet unidentified proteins. Comparison of DTX1 and DTX2 revealed that DTX2 was better expressed so we opted to use it in our cell-based experiments (fig. S7A). Initially, we confirmed that DTX2-RD is active in autoubiquitination assays and that mutations of residues in the putative NAD+-binding site abolish ADPr-Ub formation (fig. S7, B and C). We used the ADP-ribose binding properties of GST-Af1521 macro domain (30) to pull down ADP-ribosylated protein from cell lysates of HEK293 cells overexpressing DTX2 variants and probed for Ub. ADPr-Ub was detected in pull-downs from cells expressing wild-type full-length DTX2 (DTX2-FL) or DTX2-RD but not the control or the DTX2 mutants expected to be incapable of binding NAD+ (Fig. 5, F and G). Approximately 1% of the Ub pool from the whole-cell input was present as ADPr-Ub.
DISCUSSION
In the past few years, identifying PTMs of Ub itself has expanded our knowledge of the Ub code (3, 31). Recently, Ub was reported to be ADP-ribosylated on its C terminus by the E3 ligase/PARP complex DTX3L/PARP9 (9), yet how PARP9, an inactive ADP-ribosyltransferase (ART), contributed to this reaction was unclear. Here, we show that ADP-ribosylation of Ub’s C terminus is carried out by the RD domains of DTX3L and the other Deltex family members. These domains have dual activity in that they recruit E2~Ub and catalyze Ub transfer in the canonical fashion of RING E3 ligases, but in the presence of NAD+, they also promote the formation of ADPr-Ub. ADPr-Ub cannot be activated by E1 and therefore cannot be used for ubiquitination by the E1-E2-E3 cascade. To form ADPr-Ub, Deltex DTC domain binds NAD+, and the RING domain recruits and primes E2~Ub in the closed conformation for Ub transfer; when the thioester bond of E2~Ub and NAD+ are juxtaposed, ADP-ribosylation of Ub proceeds upon release of Ub from E2. The Deltex RD domains enable E2~Ub and NAD+ to be in close proximity and thereby facilitate ADP-ribosylation of the C-terminal Gly of Ub.
This is the first instance in which a function for the DTC domain has been demonstrated. Previously, Yang and colleagues (9) proposed that the catalytic domain of PARP9 in the DTX3L/PARP9 complex mediates ADP-ribosylation of Ub. However, their observation was made in the context of DTX3L/PARP9 heterodimer, and DTX3L was not tested on its own. We demonstrated that DTX3L catalyzed Ub ADP-ribosylation in the absence of PARP9 and further showed that the RD domains of other Deltex proteins share this function. The role PARP9 plays in modulating the catalytic activity of DTX3L requires further investigation.
At least four families of NAD+-binding proteins have previously been identified in mammals: ARTs, ADP-ribosylcyclases, sirtuins, and oxidoreductases (32). In DTX1-RD-NAD+, the chemical environment of the NAD+-binding site is unique compared to these other families, suggesting that Deltex proteins comprise a fifth family of NAD+-binding proteins. Within these NAD+-binding families, Deltex proteins, ARTs, and sirtuins catalyze NAD+-dependent transfer of ADP-ribose to substrates by breaking the bond between the nicotinamide and ADP-ribose moieties of NAD+ and releasing nicotinamide. ARTs and some sirtuins catalyze ADP-ribose via an SN1-like mechanism in which nicotinamide leaves, thereby generating an oxocarbenium ion intermediate. In other sirtuins, ADP-ribose transfer is proposed to proceed via an SN2-like mechanism. Regardless of mechanism, in both these families, the conformation of NAD+ is constrained to prime the bond between the nicotinamide and nicotinamide-linked ribose for catalysis. Thus, both of these families have conserved residues that function to lock these NAD+ moieties in place; in contrast, there are very few interactions with these moieties in Deltex proteins, suggesting that ADP-ribosylation of Ub Gly76 by Deltex RD domains does not follow known ADP-ribosylation mechanisms.
In our structural model of DTX1-RD domains bound to E2~Ub and NAD+, there is a large distance between the E2~Ub thioester bond and NAD+. For ADP-ribosylation of Ub to occur, the E2~Ub thioester bond and NAD+ must juxtapose. The DTX1-RD structure shows that a five-residue linker separates the RD domains, and the length of this linker is strictly conserved across all Deltex proteins. Our SAXS analyses show that the RD domains can adopt different conformations in solution likely through the flexibility of this linker. When we altered the linker length or restricted its flexibility, Ub ADP-ribosylation was abolished. Thus, linker length and flexibility are crucial in facilitating juxtaposition of RING-bound E2~Ub and DTC-bound NAD+ for Ub ADP-ribosylation. Considering that the Deltex RING domain also catalyzes substrate ubiquitination, the linker flexibility will be essential to bring RING-bound E2~Ub away from the NAD+-binding pocket to allow access by the substrate’s lysine.
ADP-ribosylation and ubiquitination both require a nucleophile to correspondingly attack NAD+ or E2~Ub. ADP-ribosylation of Ub only occurs when Ub is released from E2–Ub as demonstrated by our assays with UbcH5B C85S–Ub and UbcH5B C85K–Ub. The carbonyl carbon is susceptible to nucleophilic attack, including by water, in the oxyester and thioester but not the amide, suggesting that the reactivity of the carbonyl carbon is important. We postulate that ADPr-Ub formation proceeds via one of two scenarios. In the first scenario, the RING-bound E2~Ub is hydrolyzed, thereby releasing the Gly76 carboxylate group, which, in turn, functions as a nucleophile in the ADP-ribosylation reaction. Gly76 is ADP-ribosylated because of its close proximity with NAD+ bound to the DTC domain. In the second scenario, NAD+ is first hydrolyzed into nicotinamide and ADP-ribose, and the ribose O1′ then attacks the thioester bond.
We show that Deltex RD domains have dual activity. DTX1 and DTX2 are active E3 ligases in autoubiquitination assays, and existing literature shows that Deltex proteins are active in ubiquitinating substrates such as MEKK1 (mitogen-activated protein kinase kinase kinase 1) (33) and histones (9, 34, 35). It is therefore intriguing that these RING E3 ligases also ADP-ribosylate Ub on Gly76, thereby preventing ubiquitination. While this dual activity seems counterintuitive, it might serve as an autoinhibitory mechanism to quench ligase activity. Our work and the DTX3L/PARP9 study (9) both show that NAD+ concentrations influence the switch between ubiquitination and ADP-ribosylation of Ub in vitro. However, understanding the full extent of this dual activity and its regulation requires further study.
The extent of any PTM including ADPr-Ub turnover in the cell relies on the forward and reverse reactions (32). Here, we show that while ADP-ribosylation of Ub’s Gly76 blocks activation by E1, it is reversed by the Ub-linkage nonspecific DUB USP2 (29), suggesting a dynamic regulation of this PTM (Fig. 5). This reverse reaction poses a challenge in detecting ADPr-Ub in cells. Moreover, ADP-ribose itself is a labile and highly charged PTM, making it difficult to identify as an intact modification using proteomics methods (36). One strategy used to enrich for ADP-ribosylated proteins is to perform pull-downs with Af1521 macrodomain, which binds ADPr with a Kd (dissociation constant) of 130 nM (37). Given that the total Ub concentration in HEK293 is ~85 μM (38) and that the abundance of ADPr-Ub is limited, we had to drive the forward reaction to overcome removal of the ADP-ribose moiety by DUBs and other unidentified factors and to fulfill the minimum concentration requirements for pull-down with Af1521 macrodomain. We successfully detected ADPr-Ub with GST-Af1521 pull-downs by overexpressing DTX2. Because only a small percentage of Ub (~1%) retains ADP-ribose, it is unlikely that it will stall the general ubiquitination process. The effects of ADPr-Ub generated by Deltex might be regulated by NAD+ concentration or occur in certain compartments that have high Deltex concentrations. Identifying binding motifs that specifically recognize ADPr-Ub will help us to understand the functional relevance of this signal.
In summary, we show that the DTC domain from Deltex family E3s binds NAD+ and is required for ADP-ribosylation of Ub’s C terminus. The conserved Deltex RING-DTC domains enable binding of E2~Ub via the RING domain and NAD+ via the DTC domain. Flexibility of the linker connecting the RD domains facilitates the juxtaposition of the thioester bond and NAD+, where, upon release of Ub, Ub’s Gly76 carboxylate group is ADP-ribosylated. Like other Ub conjugations on Gly76, this signal is dynamic and can be removed by nonspecific DUBs such as USP2. An increasing number of studies have shown that bacterial effectors catalyze ADP-ribosylation of Ub to block activation by E1. Examples include ADP-ribosylation of Ub’s Arg42 and Thr66 by L. pneumophila SidE and Chromobacterium violaceum CteC family proteins, respectively (7, 8, 39). Our work reveals the role of mammalian Deltex family proteins in catalyzing ADP-ribosylation of Ub. These findings should pave the way for future studies on understanding the biology of the Deltex family of E3s and the functions of ADPr-Ub.
MATERIALS AND METHODS
Cloning and plasmids
Constructs were generated using standard polymerase chain reaction techniques and verified by automated sequencing. Codon-optimized G-blocks were purchased from Integrated DNA Technologies and used as templates for subcloning wild-type variants of DTX1, DTX2, DTX3, DTX3L, and PARP9. Mammalian Gene Collection (MGC) sequence-verified complementary DNA (cDNA) (Dharmacon MHS6278-211690540) was used as template to subclone the region of the gene encoding the RD domains of DTX4. RD domains comprise residues 388-C of DTX1, residues 390-C of DTX2, residues 148-C of DTX3, residues 544-C of DTX3L, and residues 387-C of DTX4. Unless otherwise specified, proteins are from human. For bacterial expression, DTX variants were cloned into pABLO tobacco etch virus (TEV), a modified form of pGEX4T-3 (GE Healthcare) containing an N-terminal GST tag, followed by a TEV cleavage site, a second ribosomal binding site, and a second multiple cloning site, or pRSF_Duet TEV, a modified form of pRSF_Duet-1 (Novagen) containing a TEV cleavage site following the hexahistidine tag. To generate DTX3L-FL–PARP9 complex from baculovirus, DTX3L was subcloned into pGEX4T-1 TEV (GE Healthcare), in which a TEV protease cleavage site replaces the thrombin site. Subsequently, GST-DTX3L was subcloned into pACEBAC1 vector. Full-length codon-optimized PARP9 was cloned into pACEBAC1 vector with an N-terminal noncleavable 6× His-tag. A multigene expression cassette containing GST-DTX3L/His-PARP9 was then constructed by using I-Ceu1 and BstX1 restriction sites. EmBacY cells were transformed with the resulting pACEBAC1 GST-DTX3L/His-PARP9 construct to obtain a bacmid for Sf9 insect cell infection and protein expression. For fluorescent labeling, Gly-Gly-Ser-Cys-Ub was cloned into a modified version of pGEX4T-1 (GE Healthcare) in which a noncleavable hexahistidine tag precedes the glutathione tag, and a TEV cleavage site is present instead of thrombin. For mammalian expression, native DTX1 (NM004416) and DTX2 (NM020892) were cloned into pcDNA3 with an N-terminal Myc tag.
Bacterial plasmids for DUBs were obtained from Addgene: USP5 (a gift from C. Arrowsmith; Addgene plasmid no. 25299), USP7 (a gift from T. Sixma; Addgene plasmid no. 63573), USP21 (a gift from C. Arrowsmith; Addgene plasmid no. 25147), vOTU amino acids 1 to 183 (a gift from D. Komander; Addgene plasmid no. 61589), OTUB1 (a gift from C. Wolberger; Addgene plasmid no. 26959), OTUD3 (a gift from D. Komander; Addgene plasmid no. 61411), and OTULIN (a gift from D. Komander; Addgene plasmid no. 61464). USP2 amino acid 260-C was constructed synthetically (codon-optimized G-blocks from Integrated DNA Technologies), and YUH1 and AMSH as described previously (40, 41).
Protein expression and purification
Protocols for generating UbcH5B variants (42), Arabidopsis thaliana Uba1 (24), hexahistidine-tagged human Uba1 (43), Ub variants (24, 44), Tyr371-phosphorylated c-CBL (residues 47 to 435) (42), stably conjugated UbcH5B C85S–Ub (24), UbcH5B C85K–Ub (45), GST-Af1521 (46), USP2 (29), USP7 (47), vOTU (48), OTUB1 (49), OTUD3 (50), OTULIN (51), YUH1 (40), and AMSH (41) have been described previously. PARP1-His purification was as described (52) but without size exclusion chromatography.
Proteins were expressed in Escherichia coli BL21(DE3) Gold cells. Cultures were grown in Luria-Bertani (LB) medium at 37°C to an OD600 (optical density at 600 nm) of 0.6 to 0.8 and induced with 0.2 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at 20°C for 12 to 16 hours, or cultures were grown in LB-based autoinduction medium (Formedium) supplemented with 0.5% glycerol. 15N-labeled DTX1-RD and 15N-labeled Ub were obtained from M9 minimal medium as described previously (40, 53).
Cells were harvested by centrifugation and lysed with a microfluidizer. Cells expressed with a GST-tag were resuspended in 50 mM tris-HCl (pH 8.0), 200 mM NaCl, and 5 mM β-mercaptoethanol (BME). Cells expressed with a hexahistidine or hexahistidine-maltose binding protein (MBP) tag were resuspended in 25 mM tris-HCl (pH 7.6), 200 mM NaCl, 10 mM imidazole, and 5 mM BME. Cell lysates were cleared by high-speed centrifugation. The clarified lysates were applied to glutathione affinity or Ni2+-agarose, depending on the tag system, and incubated for 1 to 2 hours on a rotary shaker at 4°C. The beads were washed in buffers similar to the lysis buffer; GST-tagged proteins were eluted in 50 mM tris-HCl (pH 8.0), 200 mM NaCl, 1 mM dithiothreitol (DTT), and 10 mM glutathione, and His-tagged proteins were eluted in 25 mM tris-HCl (pH 7.6), 200 mM NaCl, 5 mM BME, and 200 mM imidazole. Tagged proteins were buffer exchanged into 150 mM NaCl, 25 mM tris-HCl (pH 7.6), and 1 mM DTT before snap-freezing in liquid nitrogen and storage at −80°C.
For removal of tags, protein samples were dialyzed against 25 mM tris-HCl (pH 7.6), 150 mM NaCl, and 5 mM BME overnight at 4°C in the presence of TEV protease except for Gly-Gly-Ser-Cys-Ub. The proteins of interest were separated from the affinity tags or the remaining uncleaved proteins by applying the dialyzed and cleaved samples onto the same resin and collecting the flowthrough. GGSC-Ub was cleaved with TEV from a glutathione agarose column in 50 mM tris-HCl (pH 8.0), 200 mM NaCl, and 5 mM DTT using a peristaltic pump-driven circular system. Further purification was performed by size exclusion chromatography on a Superdex 75 column (GE Healthcare) and pre-equilibrated in 25 mM tris-HCl (pH 7.6), 150 mM NaCl, and 1 mM DTT. Protein concentrations were determined either by measurement of the absorbance at 280 nm based on molar extinction coefficients calculated from the relevant sequences using Expasy’s ProtParam or by Bradford assay using bovine serum albumin (BSA) as a standard.
For protein expression in Sf9 insect cells, cells were lysed by sonication, and GST-DTX3L/His-PARP9 was purified by Ni-NTA affinity purification, followed by glutathione-sepharose affinity purification in buffers as described above. Recombinant full-length human PARP9 was purchased from Abcam (ab79665).
Crystallization of DTX1-RD
Purified DTX1-RD (~10 mg/ml) was screened by sitting drop vapor diffusion in a range of commercially available screens. Crystals of DTX1-RD were obtained in BCS™ condition 44 (Molecular Dimensions). Crystals of DTX1-RD in complex with NAD+ were obtained by co-crystallizing DTX1-RD and 2 mM NAD+ in JCSG+™ condition 59 (Qiagen).
Data collection, structure determination, and refinement
Data were collected at Diamond Light Source stations I04 and I04-1 and processed by the automatic XIA2 pipeline (54) using AutoPROC (55) for DTX1-RD and DIALS (56) for DTX1-RD-NAD+ complex. The structure of DTX1-RD was determined using the single-wavelength anomalous scattering (SAS) protocol of Auto-Rickshaw, the European Molecular Biology Laboratory (EMBL)–Hamburg automated crystal structure determination platform (57), with the output: “The input diffraction data were prepared and converted for use in Auto-Rickshaw using programs in the CCP4 suite (58). Structure factor amplitudes (FA) values were calculated using the program SHELXC (59). On the basis of an initial analysis of the data, the maximum resolution for substructure determination and initial phase calculation was set to 3.85 Å. All the eight heavy atoms requested were found using the program SHELXD (60). The correct hand for the substructure was determined using the programs ABS (61) and SHELXE (62). Initial phases were calculated after density modification using the program SHELXE (62). The initial phases were improved using density modification and phase extension to 1.98-Å resolution using the program RESOLVE (63). A total of 47.28% of the model was built using the program ARP/wARP (64, 65).” The DTX1-RD-NAD+ complex structure was determined by rigid body positioning of the apo model into the isomorphous dataset. The structures were refined in PHENIX (66) or BUSTER-TNT (version 2.10.3, Global Phasing, UK) and manually inspected and manipulated in Coot (67). Validation of the refined structures was performed using Coot and MolProbity (68). Structural figures were made in PyMOL, and sequence alignments were made in Aline (69).
Generation of fluorescently labeled Ub
A twofold molar excess of IRDye 800CW Maleimide (LI-COR Biosciences) was incubated with GGSC-Ub for 2.5 hours at room temperature in the dark. Excess dye was removed by passing over two Zeba Spin Desalting Columns (Thermo Fisher Scientific) consecutively. The protein concentration of the labeled Ub was estimated on the basis of a 95% recovery rate per Zeba column pass. Labeled Ub was subsequently mixed with a 30-fold molar excess of unlabeled GGSC-Ub to generate a working stock of fluorescently labeled Ub (F-Ub) for assays.
Ub ADP-ribosylation assays
Reactions were performed in 50 mM tris-HCl (pH 7.6), 50 mM NaCl, 5 mM MgCl2, 5 mM ATP, 0.2 μM A. thaliana Uba1, 2 μM UbcH5B, 20 μM Ub, 0.5 μM E3, and 200 μM NAD+ supplemented with 10 μM biotin-NAD+ or 32P-NAD+ (40 nCi/μl). All reaction components except the E3 were assembled and incubated at room temperature for 15 to 20 min to allow E2~Ub formation. Subsequently, E3 was added to the mixture and incubated at 30°C for 30 min. For USP2 treatment, 32P-ADPr-Ub was incubated with USP2 (1 μM) at room temperature for 1 hour. Reactions were quenched with 2× loading dye (LD) containing 500 mM DTT and resolved by SDS-PAGE. Reactions using biotin-NAD+ were transferred to a nitrocellulose membrane, Ponceau staining was performed on the membrane where indicated and completely destained, blocked with 5% BSA, probed with mouse anti-Ub (Santa Cruz Biotechnology, cat. no. sc-8017; 1:1000), and then incubated with DyLight 800 Conjugated NeutrAvidin (Thermo Fisher Scientific, cat. no. 22853; 1:10,000) and goat anti-mouse IRDye 680LT (LI-COR Biosciences, cat. no. 925-68020; 1:20,000). Membranes were scanned on an Odyssey CLx Imaging System (LI-COR Biosciences). For radioactive ADP-ribosylation assays, gels were dried and visualized by autoradiography or exposed to a phosphorimager and scanned using a Typhoon FLA6000. ADP-ribosylation assays with UbcH5B-Ub conjugate were performed in the same buffer (without Uba1, UbcH5B, and Ub) and contained 5 μM DTX1-RD and either 20 μM UbcH5B C85S–Ub or 20 μM UbcH5B C85K–Ub. For all assays (ADP-ribosylation, lysine discharge, and autoubiquitination), concentrations of variants of DTX1-RD, UbcH5B, and Ub were normalized to wild-type protein based on densities of InstantBlue (Expedeon)–stained protein bands following separation by SDS-PAGE using a GS-800 Calibrated Densitometer (Bio-Rad). For UbcH5B–Ub assays, UbcH5B C85S–Ub and UbcH5B C85K–Ub were normalized to each other based on densities as described above.
ADPr-Ub generation and purification
ADPr-Ub or ADPr-15N-Ub were generated by reacting 0.2 μM A. thaliana Uba1, 2 μM UbcH5B, 100 μM Ub or 15N-Ub, 2 μM DTX1-RD, and 1.25 mM NAD+ at 30°C for 16 hours. Control Ub reactions where either DTX1-RD or NAD+ was omitted were prepared in the same manner. For ADPr-Ub or ADPr-15N-Ub, fresh 0.1 μM A. thaliana Uba1 and 1 μM Tyr371-phosphorylated c-CBL Y368F (residues 47 to 435) (42) were added, and the mixture was incubated at 23°C for 1 hour to use up unreacted Ub. ADPr-Ub, ADPr-15N-Ub, and control Ub were separated from other components using a Superdex75 gel filtration column (GE Healthcare) in 25 mM tris-HCl (pH 7.6), 150 mM NaCl, and 1 mM DTT. For USP2-treated ADPr-Ub, 1 μM USP2 was added to 100 μM purified ADPr-Ub, incubated at 23°C for 1 hour, and further purified using a Superdex75 gel filtration column (GE Healthcare) as described above.
Reactivity of ADPr-Ub
To investigate the reactivity of ADPr-Ub, E2 charging assays were performed in 50 mM tris-HCl (pH 7.6), 50 mM NaCl, 5 mM MgCl2, 5 mM ATP, 1 μM A. thaliana Uba1, 10 μM UbcH5B, and either 50 μM ADPr-Ub, 50 μM ADPr-Ub treated with USP2, or 50 μM wild-type Ub. Samples were taken at the indicated time points, quenched with 4× LD, resolved by SDS-PAGE, and stained with InstantBlue (Expedeon). For ADPr-Ub removal by a panel of DUBs, 1 μM DUBs were used.
Lysine discharge assays
UbcH5B (20 μM) was charged with human Uba1 (0.4 μM) and F-Ub (90 μM) for 15 min at 23°C in 50 mM tris-HCl (pH 7.6), 50 mM NaCl, 5 mM MgCl2, and 5 mM ATP. Charging was stopped by incubating the reaction with 45 mM EDTA for 1 to 2 min. Discharge was initiated by the addition of a mixture containing l-lysine (300 mM) and DTX1-RD variant (4 μM). Reactions were quenched at the indicated time points with 4× LD and resolved by SDS-PAGE. Gels were scanned with an Odyssey CLx Imaging System (LI-COR Biosciences). Final concentrations are in parenthesis except for UbcH5B and F-Ub, which were 9 and 45 μM, respectively.
Quantification and statistical analysis
For lysine discharge assays, Image Studio Lite (LI-COR Biosciences) was used to quantify the intensity (I) of each band, and the % UbcH5B~F-Ub remaining was calculated as described previously (53). Data are presented as an average ± SD based on three or five replicates per measurement.
Autoubiquitination assays
Autoubiquitination assays were performed at 23°C in 50 mM tris-HCl (pH 7.6), 50 mM NaCl, 5 mM MgCl2, 5 mM ATP, inorganic pyrophosphatase (0.3 U/ml), creatine kinase (0.3 U/ml) and 5 mM creatine phosphate, human Uba1 (0.4 μM), UbcH5B variants (10 μM), Ub or 2TK-Ub variants (50 μM), and GST-tagged DTX variants (2.7 μM). For Fig. 5E, 0, 0.5, or 1 mM NAD+ was added. Reactions were quenched at the indicated time points with 2× LD containing 500 mM DTT and resolved by SDS-PAGE.
PARP1 autoPARylation assay
PARP1 autoPARylation assay was performed at 30°C in 40 mM Hepes (pH 7.5), 100 mM NaCl, 4 mM MgCl2, 1 mM Tris(2-carboxyethyl)phosphine (TCEP), duplex activating DNA (1 μM), PARP1-His (1 μM), and NAD+ (20 μM). After 30 min, reaction was quenched with 4× LD and resolved by SDS-PAGE.
Solution NMR experiments
All solution NMR data were acquired on a Bruker Avance III 600 MHz spectrometer with a cryogenic triple resonance inverse (TCI) probe. Each sample (15N-Ub or 15N-DTX1-RD) was prepared at a concentration of ~100 μM in NMR buffer [20 mM sodium phosphate (pH 7.0), 100 mM NaCl, and 7.5% D2O]. A temperature series ranging from 278 to 317 K was measured at increments of 3 K on a sample of 15N-Ub to determine the optimal temperature for analysis of samples. Given that Gly75 and Gly76 were easily observed at 278 K, we used 278 K for most of our analysis. The known peak assignment for 15N-Ub at 298 K was traced across the temperature range to assign 1H-15N HSQC spectra for Ub at 278 K. Each 1H-15N HSQC spectrum was recorded with 64 complex points and a sweep width of 36 parts per million (ppm) in the 15N dimension. All spectra were processed with 256 points using Bruker TopSpin version 3.5 patch level 7. All spectra were analyzed using CARA and NMRFAM-SPARKY (70).
CSPs were calculated as follows
Cell culture and pull-down experiments
HEK 293 cells were obtained from the American Type Culture Collection, revived and sent for in-house cell line authentication by short tandem repeat profiling using GenePrint 10 System (Promega), and tested for mycoplasma. HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, penicillin (100 U/ml), streptomycin (100 μg/ml), and gentamicin (10 μg/ml). Transfections were performed using Lipofectamine 2000 reagent. Cells were harvested 40 hours after transfection and lysed in lysis buffer [50 mM tris-HCl (pH 7.6), 150 mM NaCl, 1% IGEPAL, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail]. Whole-cell lysate protein concentration was measured using Bio-Rad reagent, and similar amounts of total protein were incubated with GST-Af1521 (20 μg of GST-Af1521/10 μl of glutathione-agarose beads) for 3 hours at 4°C on an end-to-end mixer. Beads were washed with 5 × 500 μl of lysis buffer and eluted with 1× LD + 250 mM DTT. Eluates were boiled at 95°C for 10 min, spun down, and separated by SDS-PAGE. The samples were then transferred onto a nitrocellulose membrane using the Trans-Blot Turbo Transfer System (Bio-Rad), blocked in 5% BSA in tris-buffered saline at room temperature for 1 hour, and probed with mouse anti-Ub antibody at 4°C overnight as described above. The following day, the membrane was washed, incubated with goat anti-mouse IRDye800CW (LI-COR Biosciences, cat. no. 925-32210; 1:20,000), and scanned using an LI-COR Odyssey CLx Imaging System.
SAXS experiments
Purified protein was shipped to Diamond Light Source, where it was applied onto a Superdex 200 Increase 3.2 column equilibrated in buffer containing 25 mM tris-HCl (pH 7.6), 150 mM NaCl, and 1 mM DTT at 0.16 mg/ml, before being exposed to the x-ray beam as part of the standard setup at station B21. Data were analyzed using ScÅtter version 3.2h (available from station B21, Diamond Light Source). Analysis of the movement of the DTC and RING domains in relation to each other was performed by EOM (ensemble optimization method) (26).
Mass spectrometry
ADPr-Ub (453 μM) was reacted with 11 μM USP2 in a buffer containing 25 mM Hepes (pH 7), 150 mM NaCl, and 1 mM BME for 30 min at room temperature. ADP-ribose standard (400 μM) was prepared in the same buffer. Samples were quenched in extraction solution (methanol, acetonitrile, and water; 5:3:2) at 4°C. Following 15-min incubation at −20°C, the extracts were centrifuged at 16,100g for 10 min at 4°C to remove precipitated protein. LC-MS analysis was carried out on the supernatants as previously described (71). Identification of ADPr peak was confirmed by comparison of the exact mass and the retention time observed using a commercial ADPr standard analyzed under the same LC-MS conditions. The extracted ion chromatograms of ADPr were generated using FreeStyle software (Thermo Fisher Scientific).
Supplementary Material
Acknowledgments
We thank the DLS for access to beamline I04 and I04-1 (proposal mx16258) that contributed to the results presented here, Core Services and Advanced Technologies at the Cancer Research UK Beatson Institute (C596/A17196) with particular thanks to Molecular Technology services, and C. Winchester for her assistance in preparing and critically reviewing this manuscript. Funding: This work was supported by the Cancer Research UK (A23278) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 647849) to D.T.H. Author contributions: C.C., L.B., M.G., M.A.N., S.F.A., G.J.S., and D.T.H. generated constructs and purified proteins. C.C. and L.B. performed all the in vitro assays. M.G. performed protein crystallization and determined the structures. C.C., M.A.N., and B.O.S. performed NMR and analyzed the data. L.B. and D.S. performed mass spectrometry experiment. C.C., L.B., M.G., and D.T.H. wrote the manuscript. All authors commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Coordinates and structure factors for the crystal structures of DTX1-RD apo (PDB: 6Y5N) and DTX1-RD bound to NAD+ (PDB: 6Y5P) have been deposited in the PDB.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/38/eabc0418/DC1
REFERENCES AND NOTES
- 1.Deribe Y. L., Pawson T., Dikic I., Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 17, 666–672 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Hunter T., The age of crosstalk: Phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Song L., Luo Z.-Q., Post-translational regulation of ubiquitin signaling. J. Cell Biol. 218, 1776–1786 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang H. C., Lee Y.-I., Shin J.-H., Andrabi S. A., Chi Z., Gagné J.-P., Lee Y., Ko H. S., Lee B. D., Poirier G. G., Dawson V. L., Dawson T. M., Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proc. Natl. Acad. Sci. U.S.A. 108, 14103–14108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levaot N., Voytyuk O., Dimitriou I., Sircoulomb F., Chandrakumar A., Deckert M., Krzyzanowski P. M., Scotter A., Gu S., Janmohamed S., Cong F., Simoncic P. D., Ueki Y., La Rose J., Rottapel R., Loss of Tankyrase-mediated destruction of 3BP2 is the underlying pathogenic mechanism of cherubism. Cell 147, 1324–1339 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Y., Liu S., Mickanin C., Feng Y., Charlat O., Michaud G. A., Schirle M., Shi X., Hild M., Bauer A., Myer V. E., Finan P. M., Porter J. A., Huang S. M. A., Cong F., RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat. Cell Biol. 13, 623–629 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Qiu J., Sheedlo M. J., Yu K., Tan Y., Nakayasu E. S., Das C., Liu X., Luo Z.-Q., Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 533, 120–124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhogaraju S., Kalayil S., Liu Y., Bonn F., Colby T., Matic I., Dikic I., Phosphoribosylation of ubiquitin promotes serine ubiquitination and impairs conventional ubiquitination. Cell 167, 1636–1649.e13 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Yang C.-S., Jividen K., Spencer A., Dworak N., Ni L., Oostdyk L. T., Chatterjee M., Kuśmider B., Reon B., Parlak M., Gorbunova V., Abbas T., Jeffery E., Sherman N. E., Paschal B. M., Ubiquitin modification by the E3 ligase/ADP-ribosyltransferase Dtx3L/Parp9. Mol. Cell 66, 503–516.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson B. A., Kraus W. L., New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 13, 411–424 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Cohen M. S., Chang P., Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nat. Chem. Biol. 14, 236–243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aguiar R. C. T., Takeyama K., He C., Kreinbrink K., Shipp M. A., B-aggressive lymphoma family proteins have unique domains that modulate transcription and exhibit poly(ADP-ribose) polymerase activity. J. Biol. Chem. 280, 33756–33765 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Takeyama K., Aguiar R. C. T., Gu L., He C., Freeman G. J., Kutok J. L., Aster J. C., Shipp M. A., The BAL-binding protein BBAP and related Deltex family members exhibit ubiquitin-protein isopeptide ligase activity. J. Biol. Chem. 278, 21930–21937 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Obiero J., Walker J. R., Dhe-Paganon S., Fold of the conserved DTC domain in deltex proteins. Proteins 80, 1495–1499 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Deshaies R. J., Joazeiro C. A. P., RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Hershko A., Ciechanover A., Varshavsky A., The ubiquitin system. Nat. Med. 6, 1073–1081 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Dye B. T., Schulman B. A., Structural mechanisms underlying posttranslational modification by ubiquitin-like proteins. Annu. Rev. Biophys. Biomol. Struct. 36, 131–150 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Buetow L., Huang D. T., Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 17, 626–642 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mevissen T. E. T., Komander D., Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 86, 159–192 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Pruneda J. N., Littlefield P. J., Soss S. E., Nordquist K. A., Chazin W. J., Brzovic P. S., Klevit R. E., Structure of an E3:E2~Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol. Cell 47, 933–942 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buetow L., Gabrielsen M., Anthony N. G., Dou H., Patel A., Aitkenhead H., Sibbet G. J., Smith B. O., Huang D. T., Activation of a primed RING E3-E2-ubiquitin complex by non-covalent ubiquitin. Mol. Cell 58, 297–310 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Nomura K., Klejnot M., Kowalczyk D., Hock A. K., Sibbet G. J., Vousden K. H., Huang D. T., Structural analysis of MDM2 RING separates degradation from regulation of p53 transcription activity. Nat. Struct. Mol. Biol. 24, 578–587 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christensen D. E., Brzovic P. S., Klevit R. E., E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat. Struct. Mol. Biol. 14, 941–948 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Dou H., Buetow L., Sibbet G. J., Cameron K., Huang D. T., BIRC7-E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat. Struct. Mol. Biol. 19, 876–883 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plechanovová A., Jaffray E. G., Tatham M. H., Naismith J. H., Hay R. T., Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature 489, 115–120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tria G., Mertens H. D. T., Kachala M., Svergun D. I., Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2, 207–217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barkauskaite E., Jankevicius G., Ladurner A. G., Ahel I., Timinszky G., The recognition and removal of cellular poly(ADP-ribose) signals. FEBS J. 280, 3491–3507 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Amerik A. Y., Hochstrasser M., Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 1695, 189–207 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Renatus M., Parrado S. G., D’Arcy A., Eidhoff U., Gerhartz B., Hassiepen U., Pierrat B., Riedl R., Vinzenz D., Worpenberg S., Kroemer M., Structural basis of ubiquitin recognition by the deubiquitinating protease USP2. Structure 14, 1293–1302 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karras G. I., Kustatscher G., Buhecha H. R., Allen M. D., Pugieux C., Sait F., Bycroft M., Ladurner A. G., The macro domain is an ADP-ribose binding module. EMBO J. 24, 1911–1920 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herhaus L., Dikic I., Expanding the ubiquitin code through post-translational modification. EMBO Rep. 16, 1071–1083 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hottiger M. O., Hassa P. O., Lüscher B., Schüler H., Koch-Nolte F., Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 35, 208–219 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Liu W.-H., Lai M.-Z., Deltex regulates T-cell activation by targeted degradation of active MEKK1. Mol. Cell. Biol. 25, 1367–1378 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y., Mao D., Roswit W. T., Jin X., Patel A. C., Patel D. A., Agapov E., Wang Z., Tidwell R. M., Atkinson J. J., Huang G., McCarthy R., Yu J., Yun N. E., Paessler S., Lawson T. G., Omattage N. S., Brett T. J., Holtzman M. J., PARP9-DTX3L ubiquitin ligase targets host histone H2BJ and viral 3C protease to enhance interferon signaling and control viral infection. Nat. Immunol. 16, 1215–1227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan Q., Dutt S., Xu R., Graves K., Juszczynski P., Manis J. P., Shipp M. A., BBAP monoubiquitylates histone H4 at lysine 91 and selectively modulates the DNA damage response. Mol. Cell 36, 110–120 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daniels C. M., Ong S.-E., Leung A. K. L., The promise of proteomics for the study of ADP-ribosylation. Mol. Cell 58, 911–924 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jungmichel S., Rosenthal F., Altmeyer M., Lukas J., Hottiger M. O., Nielsen M. L., Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol. Cell 52, 272–285 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Kaiser S. E., Riley B. E., Shaler T. A., Trevino R. S., Becker C. H., Schulman H., Kopito R. R., Protein standard absolute quantification (PSAQ) method for the measurement of cellular ubiquitin pools. Nat. Methods 8, 691–696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan F., Huang C., Wang X., Tan J., Cheng S., Wan M., Wang Z., Wang S., Luo S., Li A., Guo X., Feng M., Liu X., Zhu Y., Zhou Y., Threonine ADP-ribosylation of ubiquitin by a bacterial effector family blocks host ubiquitination. Mol. Cell 78, 641–652.e9 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Nakasone M. A., Livnat-Levanon N., Glickman M. H., Cohen R. E., Fushman D., Mixed-linkage ubiquitin chains send mixed messages. Structure 21, 727–740 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCullough J., Clague M. J., Urbé S., AMSH is an endosome-associated ubiquitin isopeptidase. J. Cell Biol. 166, 487–492 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dou H., Buetow L., Hock A., Sibbet G. J., Vousden K. H., Huang D. T., Structural basis for autoinhibition and phosphorylation-dependent activation of c-Cbl. Nat. Struct. Mol. Biol. 19, 184–192 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berndsen C. E., Wolberger C., A spectrophotometric assay for conjugation of ubiquitin and ubiquitin-like proteins. Anal. Biochem. 418, 102–110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pickart C. M., Raasi S., Controlled synthesis of polyubiquitin chains. Methods Enzymol. 399, 21–36 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Dou H., Buetow L., Sibbet G. J., Cameron K., Huang D. T., Essentiality of a non-RING element in priming donor ubiquitin for catalysis by a monomeric E3. Nat. Struct. Mol. Biol. 20, 982–986 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martello R., Leutert M., Jungmichel S., Bilan V., Larsen S. C., Young C., Hottiger M. O., Nielsen M. L., Proteome-wide identification of the endogenous ADP-ribosylome of mammalian cells and tissue. Nat. Commun. 7, 12917 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faesen A. C., Dirac A. M. G., Shanmugham A., Ovaa H., Perrakis A., Sixma T. K., Mechanism of USP7/HAUSP activation by its C-Terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol. Cell 44, 147–159 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Akutsu M., Ye Y., Virdee S., Chin J. W., Komander D., Molecular basis for ubiquitin and ISG15 cross-reactivity in viral ovarian tumor domains. Proc. Natl. Acad. Sci. U.S.A. 108, 2228–2233 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang T., Yin L., Cooper E. M., Lai M.-Y., Dickey S., Pickart C. M., Fushman D., Wilkinson K. D., Cohen R. E., Wolberger C., Evidence for bidentate substrate binding as the basis for the K48 linkage specificity of otubain 1. J. Mol. Biol. 386, 1011–1023 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mevissen T. E. T., Hospenthal M. K., Geurink P. P., Elliott P. R., Akutsu M., Arnaudo N., Ekkebus R., Kulathu Y., Wauer T., El Oualid F., Freund S. M. V., Ovaa H., Komander D., OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 154, 169–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keusekotten K., Elliott P. R., Glockner L., Fiil B. K., Damgaard R. B., Kulathu Y., Wauer T., Hospenthal M. K., Gyrd-Hansen M., Krappmann D., Hofmann K., Komander D., OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 153, 1312–1326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langelier M.-F., Planck J. L., Servent K. M., Pascal J. M., Purification of human PARP-1 and PARP-1 domains from escherichia coli for structural and biochemical analysis. Methods Mol. Biol. 780, 209–226 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Gabrielsen M., Buetow L., Nakasone M. A., Ahmed S. F., Sibbet G. J., Smith B. O., Zhang W., Sidhu S. S., Huang D. T., A general strategy for discovery of inhibitors and activators of RING and U-box E3 ligases with ubiquitin variants. Mol. Cell 68, 456–470.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winter G., xia2: An expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 (2010). [Google Scholar]
- 55.Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., Bricogne G., Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D. Struct. Biol. 67, 293–302 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Winter G., Waterman D. G., Parkhurst J. M., Brewster A. S., Gildea R. J., Gerstel M., Fuentes-Montero L., Vollmar M., Michels-Clark T., Young I. D., Sauter N. K., Evans G., DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. D. Struct. Biol. 74, 85–97 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Panjikar S., Parthasarathy V., Lamzin V. S., Weiss M. S., Tucker P. A., Auto-Rickshaw: An automated crystal structure determination platform as an efficient tool for the validation of an X-ray diffraction experiment. Acta Crystallogr. D. Struct. Biol. 61, 449–457 (2005). [DOI] [PubMed] [Google Scholar]
- 58.Collaborative Computational Project, Number 4 , The CCP4 suite: Programs for protein crystallography. Acta Crystogr. D Biol. Crystallogr. 50, 760–763 (1994). [DOI] [PubMed] [Google Scholar]
- 59.G. M. Sheldrick, H. A. Hauptman, C. M. Weeks, R. Miller, I. Uson, Direct methods, in International Tables for Crystallography, M. Rossmann, E. Arnold, Eds. (Kluwer Academic Publishers, Dordrecht/Boston/London, 2001), vol. F, pp. 333–345. [Google Scholar]
- 60.Schneider T. R., Sheldrick G. M., Substructure solution with SHELXD. Acta Crystallogr. D. Biol. Crystallogr. 58, 1772–1779 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Hao Q., ABS: A program to determine absolute configuration and evaluate anomalous scatterer substructure. J. Appl. Cryst. 37, 498–499 (2004). [Google Scholar]
- 62.Sheldrick G. M., Macromolecular phasing with SHELXE. Z. Kristallogr. 217, 644–650 (2002). [Google Scholar]
- 63.Terwilliger T. C., Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56, 965–972 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perrakis A., Morris R., Lamzin V. S., Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6, 458–463 (1999). [DOI] [PubMed] [Google Scholar]
- 65.Morris R. J., Zwart P. H., Cohen S., Fernandez F. J., Kakaris M., Kirillova O., Vonrhein C., Perrakis A., Lamzin V. S., Breaking good resolutions with ARP/wARP. J. Synchrotron Radiat. 11, 56–59 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crysallogr D Biol Crystallogr 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen V. B., Arendall W. B., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bond C. S., Schüttelkopf A. W., ALINE: A WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr. D Biol. Crystallogr. 65, 510–512 (2009). [DOI] [PubMed] [Google Scholar]
- 70.Lee W., Tonelli M., Markley J. L., NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.G. M. Mackay, L. Zheng, N. J. van den Broek, E. Gottlieb, in Methods Enzymol (Elsevier, 2015), vol. 561, pp. 171–196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/38/eabc0418/DC1