

Abstract
Pulmonary delivery of lipid-based nanotherapeutics by inhalation presents an advantageous alternative to oral and intravenous routes of administration that avoids enzymatic degradation in gastrointestinal tract and hepatic first pass metabolism and also limits off-target adverse side effects upon heathy tissues. For lung-related indications, inhalation provides localized delivery in order to enhance therapeutic efficacy at the site of action. Optimization of physicochemical properties, selected drug and inhalation format can greatly influence the pharmacokinetic behavior of inhaled nanoparticle systems and their payloads. The present review analyzes a wide range of nanoparticle systems, their formulations and consequent effect on pharmacokinetic distribution of delivered active components after inhalation.
Keywords: Nanomedicine, Liposomes, Nanostructured Lipid Carriers, Dendrimers, Lung Cancer, Inhalation Delivery
1. Introduction
Pulmonary delivery of nanotherapeutics via inhalation techniques has been an area of investigation for several decades and is attracting further prevalence in recent years. The natural characteristics of the lungs including their large absorptive surface area, exposure to high blood flow, thin alveolar epithelial layer and slow cell surface clearance make inhalation a unique approach for both systemic and local delivery of therapeutics. Inhalation administration is widely considered as an appealing noninvasive alternative to conventional invasive techniques given its capacity to localize the delivery of therapeutic agents to the lung with enhanced bioavailability and efficacy of complex formulations at their site of action in addition to systemic distribution. Localized pulmonary delivery by inhalation also provide significant advantages in circumventing non-specific toxicities in other major organs, degradation of active moieties in the gastrointestinal tract and avoiding first pass metabolism by the liver (Figure 1) [1]. In contrast, systemic administration generally promotes adverse effects through high drug exposure to other organs and limits therapeutic effect at the site of action. However, majority of free drugs, native nucleic acids and peptides cannot be delivered in their native form into the lungs by inhalation necessitating a special dosage form or nanotechnology-based delivery system that can be inhaled.
Figure 1.

Advantages and challenges of pulmonary drug delivery. Modified from [1], image credits to iStock/snegok13 with permissions.
To date, there have been many studies were carried out in order to understand the in-vitro and in-vivo efficacy of delivered nanotherapies to the lung. However, few of these investigations have attempted to capture and model the pharmacokinetic profiles of their developed formulation. This present review analyzes several nanotherapeutic approaches for pulmonary delivery of inhalable formulations in previous studies that have collected and modeled the pharmacokinetic properties of the utilized vehicle. The review also summarizes various therapeutic targets of inhalation delivery with an emphasis on lung cancer. Lastly, this manuscript also examines some of the efforts in clinical trials applying nanotherapies for the treatment of lung cancer.
2. Determinant Characteristics of Nano-Formulations
One must consider the key variables that determine a formulation’s efficiency in order to develop a vehicle that targets the specific site of action, avoids degradation and exhibits a robust absorption and elimination profile after inhalation delivery. These considerations primarily include the vehicle’s size, charge, uniformity, final pH, porosity and additional targeting moieties that could influence the formulation’s activity (Figure 2). The ideal nanoparticle would achieve systemic exposure to all parts of the lung including deep lung deposition, un-loading of the drug at the target site of action, high degree of homogeneity, avoidance of any degradation or uptake by macrophages and favorable kinetics that limit saturation. Many studies have been employed to investigate the changes and effects on the manipulation with these characteristics and the findings are summarized herein.
Figure 2.
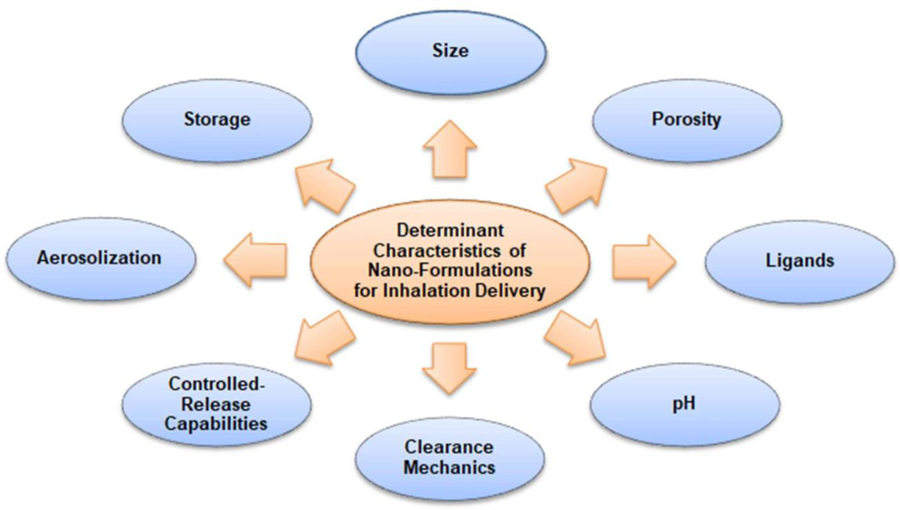
Main key variables that determine a formulation‟s efficiency in order to develop a vehicle that targets the specific site of action, avoids degradation and exhibits a robust absorption and elimination profile after inhalation delivery.
2.1. Size
Medications suitable for inhalation usually are delivered via jet or ultrasonic nebulizer, metered-dose inhaler, or dry powder inhaler in a form of aerosols or dry powders, respectively. Speaking about optimal size distribution for inhalation therapy it is important to distinguish between dry powder and aerosol nebulization. It is equally important to mention that the size of nanoparticles/nanotherapeutics dispersed by inhalation devices is different to the aerosol particle/droplet size. Such differences are discussed later in the review. For inhalation and pulmonary delivery purposes, the aerodynamic diameter of nanoparticles is described as the diameter of a sphere of unit density and optimally captures part of the aerodynamic behavior. Particles of analogous aerodynamic diameters will have equivalent velocities in the air stream regardless of their densities. These geometric diameters can be measured through current techniques such as laser diffraction or image analysis and light scattering, where results can be translated to the aerodynamic diameter by employing a recognized equation (Eq. 1) that relates the aerodynamic diameter with geometric diameter and density [2].
| (Eq. 1) |
In this model, Da represents the aerodynamic diameter, Dg represents the geometric diameter, ρ is the particle density, X is the dynamic shape factor of the particle and ρ0 is the unit particle density. For inhaled particles, there are three key variables that influence the distribution of a given payload through the airways, which are inertial impaction, gravitational sedimentation and diffusion [3, 4]. Precise measurements of aerodynamic diameters and the correlative area of deposition vary in literature but not to a significant degree. Small molecules larger than 5 micrometers (μm) are highly cohesive and have shown poor inhalation performance and flow [5, 6]. It is generally accepted that particles with aerodynamic diameters between 5–10 μm are primarily influenced by inertial impaction and lack the capacity in altering their trajectories resulting in deposition within the primary bronchi or upper airways [7–10]. Particles with an aerodynamic diameter between 1–5 μm deposit in secondary bronchi and particles with a 1–3 μm aerodynamic diameter accumulate in the bronchioles whereby gravitational sedimentation is the leading determinant [10, 11]. Lastly, particles smaller than 1–2 μm in aerodynamic diameter are primarily retained by alveoli and can be phagocytosed by alveolar macrophages. They also are susceptible to mucociliary clearance or exhalation due to their nanoscopic form and propensity to remain in the airstream. The prominent factor for deposition at these size ranges is through diffusive means [10–12]. With respect to aerosolization performance, inhaled formulations are measured and often described in terms of the fine particle fraction (FPF) [13], which is the fraction of emitted particles that are smaller than the upper limit of particle size considered respirable. This limit can be pinpointed at 5 μm as described earlier but can also be defined as 3.5 μm for more stringent purposes. A useful device in describing the overall size of inhalable formulations is the mass median aerodynamic diameter (MMAD), which is defined as the aerodynamic diameter in which half of the particles are smaller [13].
2.2. Porosity
An alternative to nanoparticle inhalation was introduced by Edward et al. with their concept of large porous particles. These formulations are large in geometric size, approximately 10 μm, but behave similar to particles that have aerodynamic diameters smaller than 5 μm due to their low density [14–16]. Due to their larger geometric sizes, these porous particles are able to overcome interparticle forces thus enabling enhanced aerosol performance and greater deposition within deep lung. Further credit to their enlarged size, these porous molecules can efficiently avoid being enveloped by alveolar macrophages [14, 15, 17]. Recent literature has commended the plausibility of utilizing porous particles at the nanoscale to further improve inhalation kinetics. Low-density hollow particles have been previously manufactured through spray-drying emulsions encompassing propellants and phospholipids [14, 15, 17–19]. Tsapsis et al. have generated enlarged porous formulations of their nanoparticles employing specific spray-drying methods without excipients, which can fragment into single nanoparticles upon reconstitution (Figure 3) [20].
Figure 3.

A representative scanning electron microscopy images of large porous nanoparticles (LPNP). (A) Hollow sphere LPNP obtained from the spray drying of a solution of polystyrene nanoparticles (170 nm). (B) Magnified view of the particle surface. (C) LPNPs dissolve readily into the nanoparticles in solution. Redrawn with permission from [20].
In order to form hollow LPNPs by a spray-drying procedure, the ratio between the time required for diffusion of a solute or nanoparticle from the periphery of the droplet to its center and the time required for a droplet to dry (Td). The first time depends on the ration between the ratio of the squared droplet radius (R2) and the solute or NP diffusion coefficient (D). This ratio defines a dimensionless mass transport number (an effective Peclet number) that characterizes the relative importance of diffusion and convection (Eq. 2):
| (Eq. 2) |
Here, Pe is the Peclet number, R is the radius of the droplet, D is the diffusion coefficient and Td is the time needed for the droplet to dry [20]. For inhaled nanoparticle formulations, the optimal Peclet number should be much greater than 1 as this indicates less time for molecules to redistribute to the center of the receding droplet resulting in an accumulation at the air-water interface. Conversely, when the Peclet number is less than 1, molecular constituents accelerate towards the center of the receding droplet by diffusion resulting in comparatively dense dried particles. Continued drying facilitates molecular cohesion through physical forces (i.e. van der Waals forces) or entrenchment in an excipient matrix which forms a shell in the early phases of drying. Increased vapor pressure breaks the cell and continues to leak until the final phase of drying, which generates the porous product [20]. The resulting physical characteristics, such as porosity and morphology, were determinant on a number of factors including the type of excipients used, chemical nature, nanoparticle concentration and size [20–22]. Among these determinants, phospholipid concentration was identified as the dominating influence on the degree of hollowness of generated products [21]. It was also found that the degree of hollowness had a direct correlation with the amount of drug released from the construct [22].
2.3. Ligands and pH
The conjugation of ligands on nanoparticle vehicles can prove to be strategically prudent dependent on the potency of the formulation to yield greater effects at the site of action, shielding from pre-mature degradation, improved targeting and/or controlled release kinetics. Our lab has previously investigated the efficacy of conjugating a modified synthetic analog of luteinizing hormone-releasing hormone (LHRH) as a cancer targeting agent to different nanoparticles [23–27]. LHRH has affinity for receptors that are overexpressed on the plasma membranes of lung (and other) cancer cells and practically not expressed in healthy visceral organs (Figure 4) [27]. It was observed that LHRH-peptide targeted nanoparticles could achieve statistically significant levels of depositing their payload in tumor cells while minimizing delivery to non-targeted healthy cells and organs [24, 25, 27]. Similar approaches have been adopted elsewhere seeking to exploit on the overexpression of specific receptors in lung tumors such as epidermal growth factor (EGF) and folate receptors. Tseng et al. noted increased deposition and retention of cisplatin-loaded gelatin nanoparticles that was biotinylated and EGF-modified [28]. Due to the enhanced delivery to the local site, anti-tumor efficacy was greater in modified nanoparticles compared to the non-modified and control (free cisplatin) counterparts [29]. Inhalable magnetic nanoparticles are another kind of these adaptations that facilitate improved targeting capabilities to allow for tumor ablation to occur at the optimal sites, utilizing an alternating magnetic field that stimulates the superparamagnetic iron oxide payload to heat at cellular lethal temperatures [30]. Frequency of the magnetic field can control the heat generated, which can dissipate over small spaces owing to the high thermal conductivity of water resulting in concentrated heating. Highly conjugated inhalable polyethylene glycol (PEG) dendrimer systems exhibit improved retention in lung and avoid degradation given the greater steric hindrance, which will be explored in a later section [31]. Pre-mature release of encapsulated drugs from nanoparticle vehicles may also lead to non-specific toxicity in healthy lung tissue. It is generally understood that the extracellular microenvironment of tumor tissues exhibits lower pH levels than their healthy counterparts. Sense and respond formulations exploit this divergence enabling a triggered release mechanism through the design of pH-sensitive fusogenic lipid nano-vesicles. The low extracellular pH levels promote nanovesicle fusion with tumor plasma and lysosomal membranes resulting in the targeted delivery of the anticancer payload [32–34].
Figure 4.
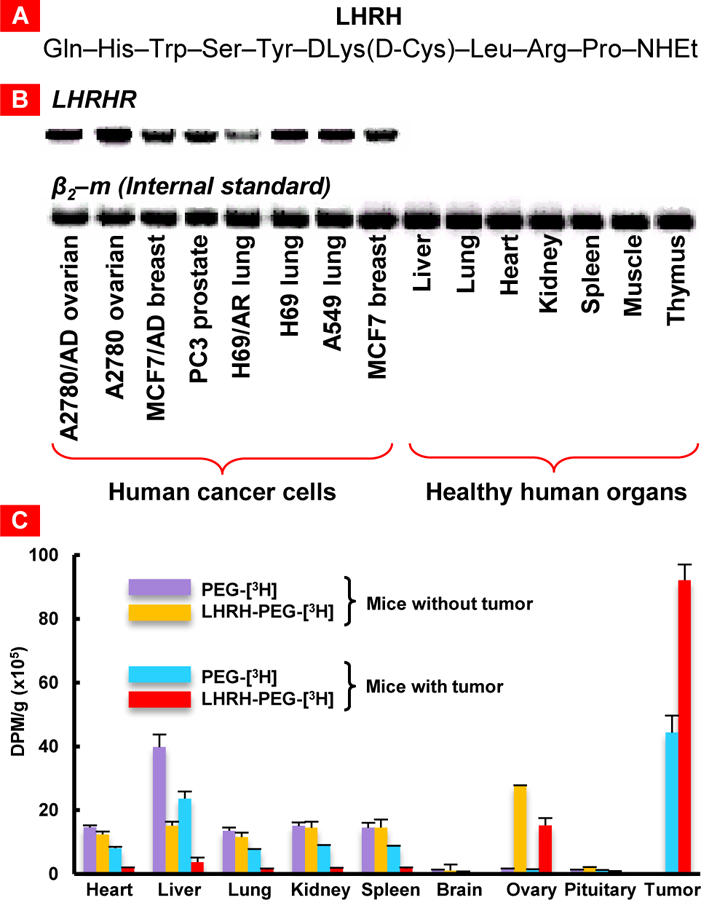
Luteinizing hormone-releasing hormone (LHRH) as a cancer targeting moiety. (A) Sequence of the modified synthetic analog of natural LHRH peptide. (B) Expression of genes encoding LHRH receptor (LHRHR) in different human cancer cells and healthy human organs. β2-microglobulin (β2-m) was used as an internal standard. (C) Distribution of tritium-labeled PEG and LHRH-PEG conjugates in different tissues of mice without tumor and mice bearing xenografts of A2780 human ovarian carcinoma. Radioactivity is expressed in disintegrations per min (dpm) per g of tissue weight. Means ± SD are shown. Redrawn from [27, 35, 36].
2.4. Clearance Mechanics
Particles clear from the lung via three main mechanisms: mucociliary clearance, phagocytosis and systemic uptake. Ciliated columnar epithelium produces mucus, facilitating the trapping of deposited particles in the upper air network. The majority of insoluble particles with a diameter of greater than 5 μm is deposited in the upper airways and eliminated via mucociliary clearance, which is the dominant mechanism in this region [37, 38]. The trapped particles are propelled by the propagation of whipping cilia in a proximal direction resulting in the particles to be swallowed or coughed out. Macrophages are also present in the upper respiratory tract though phagocytosis or endocytosis is less prominent in this area [39, 40]. Particles between 1–5 μm in diameter are most prone to elimination by phagocytosis via alveolar macrophages [41–44]. Physicochemical properties and surface chemistry of the nanoparticle will determine likelihood and kinetics of macrophage uptake. Once digested, the nanoparticle and the encapsulated payload are subjected to lysosomal digestion or removal into the lymph or cleared via mucociliary elimination [45–48]. Phagocytosis is documented to be the dominating clearance mechanism in deep lung. Anything smaller than 200 nanometer (nm) would be un-recognizable to macrophages due to small size or accelerated uptake by lung epithelial cells [49–51]. The rate of absorption into systemic circulation is dependent on the vehicle’s lipophilicity and molecular weight, whereby low molecular weight lipophilic compounds are most readily absorbed [52].
2.5. Controlled-Release Capabilities
By manipulating some of the aforementioned characteristics, nanoparticle formulations have the potential to be optimized for controlled-release capabilities. Ideally, these vehicles will be small enough to warrant deposition in deep lung. Once there, the main clearing mechanism is either systemic or macrophage uptake depending on the size of the nanoparticle. If these nanoparticles are conjugated with external ligands or within the optimal size range, they could be retained for longer periods of time by avoiding receptor-mediated uptake by lung epithelium or otherwise macrophage elimination. These particles will need to maintain their formulation within the surfactant lining layer of the lung [53], which is primarily (~90%) composed of phospholipids and some (~10%) proteins [54]. These alveolar surfactants are naturally amphiphilic and have the capability to dissolve or deplete the lipids on the nanoparticle membranes [55]. By avoiding pre-mature elimination before reaching the site of action and pre-mature degradation in the lining layer of the lung, the nanoparticle formulation could experience an overall improvement in controlled-release kinetics.
2.6. Aerosolization and Storage of Nanoparticles
To optimize delivery, nanoparticle suspensions are aerosolized into droplets with relevant characteristics such as uniformity and appropriate aerodynamic diameters. The most popularized method in converting nanoparticle suspensions into highly inhalable droplets is via a nebulizer device [56]. Nebulizers employ condensed air pressure to transform a suspension of nanoparticles into droplets suitable for inhalation [57]. Our lab has previously demonstrated that one-jet collision nebulizer can generate effective inhalable droplets of a liposome nanoparticle suspension encapsulating anticancer drugs and/or small-interfering RNA (siRNA) without compromising structural integrity or biological activity of the liposome and its constituents [58]. Recent advances in nebulizer technology has made way for more efficient and portable nebulizers such as vibrating mesh nebulizers, which have been previously utilized to aerosolize a nanocapsule suspension encapsulating paclitaxel and demonstrated an FPF greater than 80% without changing the determinant properties of the formulation [59].
An alternative means of generating inhalable droplets can be through pressurized metered dose inhalers (pMDI), which convert the nanoparticle suspension into droplets via a compressed propellant (i.e. hydrofluoroalkane). This route can also maintain the vehicle’s structural integrity as well as the biological activity of its encapsulated drugs, offers a high degree of portability and can be utilized for inhaled delivery. It has been previously established that pMDI can transform a dendrimer-siRNA construct suspension into respirable droplets with an FPF of ~77% where integrity and biological activity remain unchanged even after long-term exposure to the hydrofluoroalkane propellant [60]. Despite these advantages, pDMI technology is constrained by its low efficiency as only ~10% of the aerosol emitted deposit in deep lung [61]. Further limitations are experienced by patients who lack hand-mouth coordination as error has been previously documented [62–64]. pDMI is additionally constrained by its inability to effectively deposit high-dose medications [57].
Whether a nebulizer or pDMI is applied, nanoparticles are often in suspension form before they are delivered. Storing liquid suspensions for long-term periods could lead to physicochemical instabilities like hydrolysis of polymer, drug leakage/degradation and aggregation (especially at smaller sizes, i.e. < 200 nm) [65, 66]. Alternatively, storing nanoparticle formulations as dry powders has shown improved long-term stability when compared with their liquid suspension counterpart. The maintenance of nanoparticle formulations as efficient and reliable dry powders is largely dependent on generating an accurate size. Towards this end, nanoparticles can be dried with or without excipients via freeze-drying, spray freeze-drying and spray-drying to achieve uniformly sized and stable powders [67]. Further complications arise due to the small phenotype and cohesive nature of nanoparticles because constituents smaller than 10 μm reportedly demonstrate poor inhalation and flow capabilities [5, 6, 68, 69]. To improve performance, particles can be formulated as “interactive mixtures”, in which nanoparticles can attach to the surfaces of larger carriers [70, 71]. Co-drying nanoparticles with excipients can produce inhalable nanoparticle aggregates in an excipient matrix [72–74]. L-leucine is a widely recognized force-control mediator that can decrease inter-particle cohesion and enhance the dispersibility of nanoparticles [75, 76]. L-Leucine has been successfully co-freeze-dried with paclitaxel-cisplatin nanoparticles of nano-aggregate sizes between 1–5 μm. This formulation did not indicate any cytotoxic effects up to 5 mg/mL in A549 cells and delivered an FPF of >70% [77]. Variability in the utility of differing excipients has been previously noted as spray-dried formulations of doxorubicin-loaded bovine serum albumin nanoparticles demonstrated more efficient FPF with trehalose when compared with L-leucine, which generated irregularly shaped products [78].
Additional efforts to improve pulmonary delivery of nanoparticle formulations have been introduced through the use of effervescent technology. By co-spray-drying nanoparticles with effervescent excipients, nanoparticles experience more rapid release of their payload upon dissolution of the excipients in aqueous media [79]. This is caused by the effervescent effect and can be achieved through the combination of citric acid with ammonia and sodium bicarbonate. To prevent pre-mature effervescing, the pH of the feed solution is kept low during the drying process or particle formation [79]. Initial studies employing the effervescent co-spray-drying technology showed average MMADs of ~5 μm with no phenotypic change or morbidity to the tested rats upon application, thus confirming the safety and tolerability of the system [80]. It has been successfully showcased that an effervescent carrier co-spray-dried with doxorubicin-loaded nanoparticles can effectively distribute throughout the lung [81]. Further studies confirm longer survival rates of mice receiving doxorubicin-loaded n-butylcyanoacrylate nanoparticles co-spray-freeze-dried with effervescent excipients when compared to control groups receiving free doxorubicin via inhalation or doxorubicin solution via intravenous administration [82].
3. Pharmacokinetics of Dendrimers
3.1. PEGylated Dendrimers
In recent years, PEGylated polylysine dendrimers have exhibited considerable versatility and potential as nanometer-sized drug delivery vehicles due to their inherent structure and biocompatibility [83–85]. Dendrimers exhibit molecular dimensions similar to proteins between 2–20 nm in diameter and typically demonstrate high monodispersity. Their reactive polyfunctional surface make dendrimers favorable subjects for conjugating drugs, proteins, targeting ligands, solubility enhancers and a multitude of other surface functional groups that can govern the dendrimer’s kinetic or physicochemical properties. Dendrimer synthesis can also be regulated to a high standard resulting in well-defined and uniform macromolecules in which by-products of differing molecular weights synthesized by radical mediated polymerization can be avoided [86]. These formulations have been previously studied in vitro and in vivo and noted for their excellent stability, facile enzymatic degradation and ensuing renal elimination of degraded low molecular weight products [83]. Dendrimers based on poly-amino acid constructs are biodegradable and demonstrate reduced risks for immunological stimulation and response. This platform can be prepared as an inhalable therapy via aerosolization utilizing a microsprayer for therapeutic delivery to the lungs and has been previously observed to show controlled release characteristics when designed in this format.
Size and molecular weight of dendrimer vehicles can significantly influence the pharmacokinetic characteristics of varying formulations administered by intratracheal (IT) instillation. Polylysine dendrimers conjugated with surface amino groups of 2300 Dalton (Da) polyethylene glycol polymers (PEG2300) have shown high retention in the lungs up to 1 week but limited availability in systemic circulation after intratracheal delivery [86]. Conversely, the smallest dendrimer conjugated with 200 Da PEG polymers (PEG200) showed relatively good absorption of >20% bioavailability but limited lung retention. At earlier time points (2h), 51.5% of the dose was intact and recovered in bronchoalveolar lavage fluid (BALF) with only 12.4% present in lung homogenate. Whereas PEG2300 had relatively low quantities of intact dendrimer present in BALF and the majority of the dose, approximately 80% after 1 week, was recovered in feces suggesting high clearance via the mucociliary escalator. Relatively, PEG2300 did exhibit higher quantities of the dosage found in lung homogenate when compared with other collected organs and tissues [86]. This is consistent with previous findings that dendrimer retention and stability in lung tissues increases with increasing chain lengths of conjugated PEG products. These observations are also consistent with other macromolecular formulations (nanoparticles, proteins) that the increasing rate of absorption from lungs into systemic circulation is inversely proportional with molecular mass [87–89]. In general, the optimal formulation was found to be the dendrimer of compromising size conjugated with 570 Da PEG polymers (PEG570) and exhibited the more favorable pharmacokinetic (PK) parameters. After 48h, the PEG200 group showed significant levels of degraded 3H-radiolabeled material in the BALF and lungs, indicating significant biodegradation and/or enzymatic cleavage of the intended products occurring in the lung. The PEG570 group showed a 2-fold increase in Cmax, a 5–10-fold increase in Fabs (absolute bioavailability) % and similar values for Tmax when compared with PEG2300 group. On the other hand, both PEG570 and PEG200 had similar Fabs values with PEG570 showing a 6.5-fold increase in Cmax and a 13-fold increase in Tmax (time at which maximum concentration is achieved). Although the dendrimer of smallest size (PEG200) displayed a more immediate and rapid absorption profile, the same group was also subject to greater rates of degradation [86].
Two similar dendrimer formulations were synthesized where both systems were conjugated with 1100 Da linear PEG polymers (PEG1100) and one formulation had an additional surface ε-amino acid group and α-carboxyl OtBu-methotrexate linked via a hexapeptide linker (MTX dendrimer) [28]. These dendrimers were deployed in-vivo in male Sprague-Dawley rats via intravenous and intratracheal administration as a comparative study where all blood samples were collected through the right carotid artery. The systemic concentrations for both PEG1100 and MTX groups were low, where < 3% of the dose was absorbed over 7 days. This could be the result of tight junctions between alveolar epithelial cells that inhibit the paracellular transport of macromolecules. A large proportion of the dose was recovered in lung tissue for both dendrimers (approximately 40% of the nominal dose) with concentrations increasing over a 3-day period. Concentrations of both dendrimers were high in BALF on the initial day of administration and dropped below 5% by the third day. Despite these low figures, the more hydrophobic MTX dendrimer did exhibit a 2-fold increase in pulmonary bioavailability when compared to the standard PEG structure. Confirmed by the size-exclusion chromatography (SEC) profiles of urine samples, a higher concentration of 3H-radiolabled low molecular weight products were identified for the MTX formulation compared to the fully PEGylated construct. The SEC profiles of lung tissue homogenate supernatant also showed increasing concentrations of 3H-radiolabeled low molecular weight products for both dendrimer groups over the course of the study with higher levels observed for the standard PEG group. This is consistent with previous data which showed that polylysine dendrimers with decreased degrees of PEGylation and increased exposure of the scaffold to proteolytic enzymes increases in-vivo instability [90, 91].
Aside from the lung, the remainder of the dosage was traced and distributed to feces, urine or liver for both dendrimers. This suggests that a large portion of the delivered dose was cleared through the mucociliary escalator and absorbed in the gastrointestinal tract. MTX dendrimer recovered in feces was also 2-fold higher than its PEG counterpart, which is indicative of higher mucociliary clearance between the two. However, this is in agreement with previous research which showed that hydrophobic molecules are more readily eliminated from the lungs when compared with hydrophilic formulations. The intravenous (IV) group exhibited high levels of dendrimer in liver and urine with minimal quantities in kidney, spleen or lung [31].
3.1.2. Pharmacokinetics of PEGylated Dendrimers for Lung Cancer Treatment
Clinical studies have shown that the sole administration of cytotoxic drugs against lung cancer requires high local drug concentrations and instigates lung-related toxicities as a result [92–94]. However, on-going development in nanoparticle formulation strategies has been of interest to researchers seeking of improving the delivery of cytotoxic drugs for therapeutic effect with greater degrees of control and precision. Dendrimers also provide a distinct advantage with drug conjugation and cleavage selectivity within the tumor microenvironment that could lend to greater levels of control over the delivery and kinetics of the cytotoxic drug [95]. Dendrimers allow for loading of anticancer drugs either by conjugation to their surface or via encapsulation inside the structure (Figure 5). A 56 kilo-Dalton (kDa) PEGylated polylysine dendrimer conjugated with doxorubicin via an acid labile linker (D-DOX) was explored for its therapeutic utility against lung-resident cancer in a comparative study between inhalation and IV administration as well as delivery of free non-bound doxorubicin alone [85]. In-vivo bioluminescence studies observing the anti-tumor efficacy of the inhaled dendrimer-doxorubicin (D-DOX) formulation showed a reduction of lung tumor burden by >95% when compared to the IV and control forms of D-DOX. Lung tumor regression was seen in five out of the nine rats where one animal showed complete inhibition after 1 week of treatment. Biodistribution data collected from lung tissue and BALF indicate a rapid clearance of D-DOX, approximately 60% of the IT dose, within the first 24h of administration. A large portion of the dose, approximately 20%, was cleared via the muociliary escalator and was recovered in feces. Clearance of D-DOX greatly decelerated after the initial 24h where peak concentrations of D-DOX occurred 3–4 days after administration and approximately 15% of the dose remained in lung tissue and BALF after 1 week. The prolonged residence of inhaled D-DOX in lung is an encouraging development for additional studies on controlled release mechanics and further improvements as t1/2 (half-life) was approximately 49 hours whereas the IV administered D-DOX had a t1/2 of approximately 25 hours.
Figure 5.
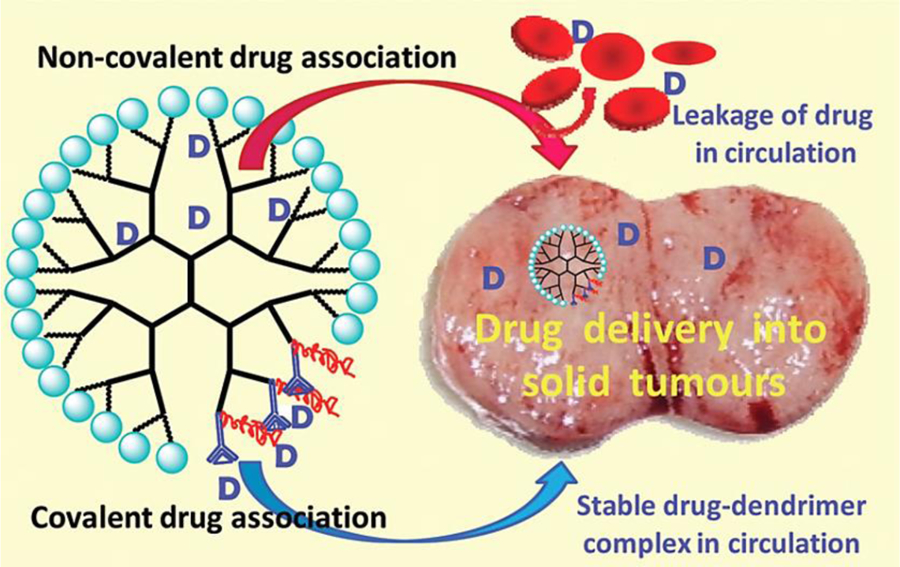
Dendrimers as drug carriers for anticancer drugs. Association of anticancer drugs with dendrimers may be achieved via covalent conjugation to the surface, or by encapsulation of drugs within the structure. Reproduced with permission from [95].
Further exposure of D-DOX to MAT 13762 IIIB cells in-vitro showed that roughly 12% of the doxorubicin associated with D-DOX was internalized by the cell line. Cellular levels of 3H-associated with the dendrimer scaffold were below the limit of quantification. This suggests that the PEGylated dendrimer was not internalized into lung cancer cells and that cellular uptake of doxorubicin liberated from D-DOX occurred only after the drug was released from the dendrimer scaffold. Alexa-flour-405 labelled D-DOX taken from the middle region of a lung tumor lobe showed deep penetration by D-DOX under a glass microscope 24h after IT instillation where concentrations were found to be 100-fold higher when compared to the rats administered IV doxorubicin. The proportion of total D-DOX and free doxorubicin in lung tumors in-vivo could not be determined as tissue homogenization resulted in the liberation of doxorubicin presenting a key limitation and future direction for further pharmacokinetic investigative efforts. Furthermore, IT-administered D-DOX was better tolerated than IT-administered doxorubicin solution as the latter showed greater signs of pulmonary congestion and inflammation. These symptoms manifested through breathing difficulties, rapid loss of body weight, increased number of neutrophils and alveolar macrophages. The inhaled D-DOX formulation was also found to reach an AUC of approximately 266 μg/mL·h, which shows additional potential for tumor penetration from both the ‘air side’ (via the lung) and the ‘blood side’ from the systemic circulation [85].
4. Clinical Application of Chemotherapeutic Formulations Administered via Inhalation
4.1. Doxorubicin via Inhalation
The first study to evaluate the clinical safety of doxorubicin administered via inhalation was a Phase I multicenter dose escalation study in patients with developed cancer affecting the lung [96]. The primary objective was to determine the maximal tolerated dose of inhaled doxorubicin administered every 3 weeks via a high-efficiency nebulizer, the OncoMyst model CDD-2a. In conjunction with this goal, the team also documented toxicity/adverse effects and evaluated initial pharmacokinetic profiles. The results showed that inhaled doxorubicin can be delivered safely however, high dosages do instigate pulmonary toxicities. Significant pulmonary toxicities were observed at the higher dosages of 7.5 and 9.4 mg/m2 where further dose escalation would not be feasible. For most patients subjected to the maximum tolerated dose (7.5 mg/m2) and below, the variability in pulmonary function tests were within the limits established by the study. Only 1 of the 11 patients treated at the advised Phase II dose of 7.5 mg/m2 developed significant pulmonary toxicity that manifested by a considerable drop in forced vital capacity. The generated pharmacokinetic profiles for inhaled doxorubicin were consistent with the minimal systemic toxicity observed during the study. Cmax (maximum concentration) was observed at the first sampling point (as expected) and reached 47.8 ng/mL, which is well below the systemic concentrations observed after IV administration. It has been previously established that the absorption of small molecules via inhalation is primarily determined by lipophilicity rather than size [97]. Being that doxorubicin is a relatively hydrophobic/lipophilic drug, the clinical data is consistent with its initial rapid absorption in the lung within the range of minutes after inhalation [96].
This investigated formulation of inhaled doxorubicin did show a moderate antitumoral response. One partial response was observed with a patient affected by spindle cell sarcoma. The response was initially documented at a dosage level of 1.9 mg/m2 with six cycles of administration. Further escalation of the patient’s dosage form to 6.0 mg/m2 did not result in any additional response. Eight patients observed stabilization of their disease with treatment cycles ranging between 5 and 15. Two patients had soft tissue sarcoma, two patients were affected by bronchoalveolar carcinoma, one had endometrial carcinoma and three were affected with thyroid cancer. Six patients also demonstrated stability of their disease over the duration of 3 courses but were withdrawn due to the protocol and study arrangement [96].
4.2. Combination Study: Doxorubicin via Inhalation and Cisplatin via IV
A subsequent phase II clinical study was initiated by the same group [97] evaluating the efficacy of inhaled doxorubicin given in combination with IV administered cisplatin. Since non-small cell lung cancer (NSCLC) is a considered a systemic disease and the toxicities observed in the phase I study do not readily overlap with platinum-based doublet chemotherapy for NSCLC, the study directors hypothesized that the added IV chemotherapy could bolster therapeutic effects for beneficial responses. This was a multicenter dose-escalation phase II study in patients with metastatic NSCLC who were inexperienced to chemotherapy treatment where the primary objectives were characterizing both the therapeutic potential and toxic effects of the combination therapy. Given their therapeutic efficacy and favorable toxicity profiles, docetaxel and cisplatin were selected as the standard doublet to be combined with the inhalation dosage. The initial dose of inhaled doxorubicin hydrochloride was 6.0 mg/m2 and allowed to escalate to a maximum level of 7.5 mg/m2, where both initial and maximal ranges were previously established from the initial phase I study. Patients were administered doxorubicin via inhalation 1–3 hours prior to receiving IV chemotherapy. Docetaxel and cisplatin were administered in standard practice at 75 mg/m2 with specified dose reductions for non-pulmonary toxicity. This treatment course repeated every 3 weeks provided that patients had recovered from the previous cycle. A total of 43 patients were enrolled in this study where 9 received the maximal dose level of inhaled doxorubicin (7.5 mg/m2) and 34 received the initial phase II dose (6.0mg/m2). After the second patient was subjected to the maximal dose level (7.5 mg/m2) and experienced a drop in the diffusing capacity for carbon monoxide (DLCO) after the first cycle, the data safety monitoring board suggested the phase II portion be conducted at 6 mg/m2 for inhaled doxorubicin. A total of 34 patients received the phase II dose level where 28 underwent at least one cycle of treatment. Of these 34 patients, 7 patients dropped out due to mounting evidence of disease progression in three individuals, adverse events observed in two and two withdrawals with the consent or direction of their physician (one each). Of the 25 registered patients at the phase II level, 24 individuals were evaluable, and 21 patients received at least two cycles of treatment. Among the 24 evaluable individuals treated at the phase II dosage level (6.0 mg/m2), 6 patients experienced partial responses and 1 had a complete response. Among the 17 non-responders, 13 patients had stable disease progression for up to eight courses of treatment and 4 individuals had further progression of their disease states. The most common adverse symptoms related to the IV chemotherapy and were mild in nature, which included (grade 1–2) alopecia, anorexia, diarrhea, nausea and cough. Although the study was able to demonstrate the safety of the combination therapy, the results in overall response rate did not show any statistically significant improvements even when looking only at intrathoracic tumors. Further complications have been noted amongst patients with advanced lung cancers who were habitual smokers or ex-smokers, where many of these individuals were unable to satisfy the pulmonary function test requirements for eligibility in this study [97].
4.3. Clinical Phase I Studies of Liposome Formulations against Lung Cancer
Cisplatin has also been encapsulated in a liposomal formulation and investigated in a clinical Phase I study against metastatic lung cancer given its anti-tumor activity previously reported in-vivo and its capability to circumvent the development of toxic side effects occurring at other major organs or tissues. For intravenous application, the major dose-limiting toxicity is usually associated with nephrotoxicity, peripheral neuropathy, ototoxicity, hypersensitivity reactions and myelosuppression. Initial in-vitro studies utilizing liposomal encapsulated cisplatin against the human lung tumor cell line (NCI-H460) showed no significant alterations to the cytotoxic properties of the nebulized drug [98]. Additional confidence was garnered from the in-vivo studies with Sprague-Dawley rats as the liposomal encapsulated cisplatin displayed sustained release properties and led to much higher ratios of lung/kidney levels when compared to IV-administered cisplatin. This result was indicative of reduced cisplatin concentrations at the kidneys and thus minimizes potential risk for nephrotoxicity typically associated with IV-administered cisplatin. Another study exploring the sustained release lipid inhalation targeting (SLIT) cisplatin also did not show any signs of toxicity or histopathological changes within lung, kidneys or bone marrow [98]. From these in-vivo studies, it was observed that SLIT cisplatin was well-tolerated and provided significant anti-tumor activity with minimal systemic toxicity and exposure. A total of 17 patients were enrolled for the dose-escalation study, which started at 1.5mg/m2, which increased up to 24mg/m2 and up to a maximal deliverable dosage of 48 mg/m2. Inhalation treatments were administered between 1–4 consecutive days in 21-day cycles where nebulization was performed over 20 min and never exceeded 3 nebulizations for any given session with a maximum of two sessions per day. At 48 mg/m2, dose limiting toxicity was still not achieved however inhalation time was the limiting constraint for additional escalation. Other than nausea and vomiting, the primary observed side effects were respiratory related where 11 patients experienced dyspnea, 5 patients experienced productive/irritative cough and 8 patients experienced hoarseness. Patients’ anti-tumor activity was assessed utilizing the Response Evaluation Criteria in Solid Tumors guidelines in which 12 patients achieved a “best response” of stable disease while 4 patients had progressive disease. In general, inhalation therapy remains at a disadvantage given the low deposition efficiency of the drug within the target area. From the PK data, only ~10%−15% of the dose will reach the site of action during jet nebulization. Droplets with MMADs between 1–5 μm have been documented to deposit in the central and peripheral airways by way of gravitational sedimentation and inertial impaction. Smaller particles will be exhaled, and larger particles will experience steric obstruction, thereby remaining in the upper airways. Overall, the study did achieve a significant antitumor response with limited systemic toxicities [98].
Tissue distribution, therapeutic index and the general kinetics of a drug can be significantly altered by formulating active pharmaceutical ingredients (API) into liposomes. Interleukin-2 (IL-2) has been well-documented for its anti-tumor capabilities both in-vivo and in the clinic [99, 100], however, it is generally associated with debilitating adverse effects that include malaise, vascular leak syndrome, eosinophilia, fever, fatigue, chills and sweats [101–104]. Canine studies exploring the utility of aerosolized IL-2 liposomes did demonstrate immune activation, lower systemic toxicities and anti-tumor activity with a 33% complete response rate [105–108]. IL-2 can bind to either a trimeric high-affinity receptor, resulting in proliferation and activation of T-lymphocytes, or bind to a dimeric receptor of lower affinity on NK cells and monocytes [109, 110]. Previous inhalation studies have also noted IL-2’s capability to increase accessory function of alveolar macrophages and increase the number of immunocompetent cells in bronchoalveolar lavage (BAL) [111, 112]. In-depth analysis of nebulized IL-2 liposomes to canine lung showed a significantly improved local effect when compared to freely administered cytokine with increased BAL cell number and functional activity. A phase I dose escalation study exploring the utility of inhaled IL-2 liposomes recruited 9 patients with various cancer types including sarcoma, renal cell and melanoma [113]. Initial inhalation treatment was given on the first day over 4 sessions to account for any adverse effects. Patients then self-administered IL-2 liposomes for one month with three dosages per day. The initial cohort with a starting dosage of 1.5x106 IU/dose showed one episode of upper respiratory symptoms that persisted for 3 days. Outside of this data point, the second cohort of 3x106 IU/dose and third cohort of 6x106 IU/dose were completed without any evidence of toxicity. Of the 9 recruited patients, 7 were evaluable for antitumor response where 3 patients had progressive diseases, 2 patients experienced stable diseases and one patient with complete remission one month after treatment had ended. No significant toxic effects were observed except for the initial respiratory complication and an optimal dosage was determined at 3–6x106 IU 3 times a day. Like the aforementioned cisplatin phase I study, optimal MMAD was identified around 2.0 μm with a mode size of about 1.0 μm. This was further confirmed in a radiolabeled IL-2 liposome study administered via inhalation to canine that demonstrated deposition to all ventilated parts of the lung with a 1:1 ratio between central and peripheral deposition [107]. Overall, the study is a key milestone in exhibiting acceptable tolerability and anti-tumor efficacy.
5. Pharmacokinetics of Liposomes and Nanostructured Lipid Carriers for Lung Cancer
5.1. Nanostructured Lipid Carriers
Nanostructured lipid carriers (NLCs) are widely employed for many drug delivery applications and exhibit specific advantages over other formulations due to their inherent phenotype. In contrast to liposomes that have a bi-layer lipid membrane, NLCs have one-layer membranes with hydrophilic heads exposed to the extracellular environment and hydrophobic tails encapsulating any constituents therein. This characteristic allows for easy integration of lipophilic drugs within the lipid core. A prime illustration was investigated with 9-bromo-noscapine (9-Br-Nos), a lesser known chemotherapeutic platform that has previously demonstrated the capability to arrest cell cycle progression in non-small cell lung cancer cells during the mitosis phase by influencing tubulin polymerization [114, 115]. Despite a favorable therapeutic profile, in-vivo or clinical applications of 9-Br-Nos are limited by its high lipophilicity and poor aqueous solubility that lead to suboptimal therapeutic efficacy at the local site of action [116]. However, NLCs present a feasible solution for a compatible encapsulation of the compound and focused delivery to the site of action. Another complication, as previously established, is the aggregation of nanoparticles with diameters smaller than 200 nm. Although alternative strategies have been studied in varying the principal excipients such as the surfactant or cryoprotectant, recent literature investigating the utility of effervescent excipients in NLCs has shown efficacious rapid dispersion, potent targeting and high dissolution [80, 81]. The rapid release nanostructured lipid particles (NLPs) of 9-Br-Nos (9-Br-Nos-RR-NLPs) given through inhalation was compared with 9-Br-Nos suspension administered intravenously, 9-Br-Nos suspension and 9-Br-Nos-NLP administered via inhalation [114].
Plasma and tissue concentration-time profiles of 9-Br-Nos were developed for all (previously determined) time points where all pharmacokinetic parameters were derived using the linear trapezoidal rule up to the last sampling point with detectable levels and extrapolated to infinity (AUCinf). The analysis was accomplished utilizing WinNonlin software version 4.1® (Pharsight, Mountain View, CA) with one-compartmental method [114]. Other variables calculated included the elimination rate constant (ke, h−1) half-life (t1/2, h), area under the curve at the last sampling point (AUClast, h μg/mL), mean residence time (MRT, h), total clearance rate (CL, L/h), and volume of distribution (Vd, L) [114]. Below is a summary of how the variables were calculated and inter-relate to one another:
| (Eq. 3) |
| (Eq. 4) |
| (Eq. 5) |
| (Eq. 6) |
| (Eq. 7) |
The ke was calculated from the slope of the time points in the final log linear portion of the drug-concentration profile through least square linear regression analysis. AUMC was attained from a plot of product of plasma drug concentration and time (C · t) vs t from zero to infinity [114]. In general, the inhalable 9-Br-Nos-RR-NLPs showed enhanced drug delivery capacity and an improved PK profile in lungs following nose only exposure. The elimination rate constant of RR-NLPs was significantly lower than the individual NLP or dry powder formulation. The half-life of RR-NLPs was also improved to 4.529 hours compared with the 4.029 hours observed in NLPs and 2.585 hours from dry powder. Furthermore, the AUClast of RR-NLPs did indicate enhanced exposure of drug (with a higher end value) in lung when compared with powder or NLPs. However, the MRT of RR-NLPs at 1.749 h−1 was noticeably lower than the 2.92 h−1 of the stand-alone NLP formulation. The better performance in a lower elimination rate constant and enhanced drug exposure can be attributed to the spherical shape of the RR-NLPs and slight negative charge. Alveolar macrophages exhibit a negative charge owing to the sialic acid residue in the membrane [117]. Macrophages also have an innate preference to engulf rod-shaped particles and do so more efficiently compared to nanospheres [118]. Due to the ionic repulsion and increased steric hindrance, NLPs and RR-NLPs are better retained in lungs for longer durations.
Cytotoxicity assays comparing the NLP and RR-NLP formulations against A549 non-small cell lung cancer cell line showed greater apoptotic potency amongst the RR group likely due to their unique rapid drug releasing capability and smaller mean particle size [119]. Confocal microscopy further validated these findings where 9-Br-Nos-RR-NLPs exhibited greater fluorescence intensity in A549 cells indicated an increased cellular uptake when compared to the base NLP formulation. This may be attributed to the rapid internalization of RR-NLPs via endocytosis due to their rough surface morphology as their NLP counterpart exhibit a smooth spherical shape. Previous research has pointed towards the importance of energy-dependent mechanisms for the uptake of nanoparticles and includes caveolae-independent endocytosis, clathrin endocytosis, caveolae-mediated micropinocytosis and clathrin-mediated micropinocytosis [120]. Contrastingly, energy-independent endocytosis mechanisms have also been hypothesized as possible transport mechanism and cellular uptake of nanoparticles with average sizes below 100 nm [121]. The structural and chemical similarities between NPs and the cellular plasma membrane promote uptake by diffusion and facilitate drug delivery to the interior of the cell. For the RR-NLPs formulation and data, it was determined that energy dependent and passive diffusion mechanisms dominated the transport within A549 cells. Sodium azide, an energy depleter, decreased the cellular uptake and showcased a key role in the energy dependent endocytosis mechanism. Clathrin-mediated endocytosis was also observed and is a routine process of all eukaryotic cells to intake nutrients [114]. Further, macropinocytosis also occurred for particles larger than 100 nm and likely transpired as a result of any RR-NLPs that did not effervesce well and aggregated. Some aggregation may have occurred during the storage period due to polymorphic transition from unstable alpha form to the stable beta form [122].
Despite the advantages that NLCs exhibit which include good tolerability, biodegradability and greater stability against shear forces produced during nebulization when compared to liposomes, emulsions and polymeric nanoparticles, there are still limitations [123–126]. Risk of gelation, low drug loading and rug leakage caused by lipid polymorphism during storage have been a few of the primary challenges [127]. Celecoxib (Cxb) is a lipophilic therapeutic drug that has exhibited anticancer capabilities by inhibiting cyclooxygenase-2 (COX-2) enzyme, which is over-expressed in several malignant cancer types and hypothesized to play a key role in the pathogenesis of NSCLC [128, 129]. Prior preclinical data alludes to the essential role that the COX-2/prostaglandin E2 signaling pathway contributes to the malignant characteristics of NSCLC through inhibiting apoptosis, promoting angiogenesis and suppressing the immune response [130]. Cxb has been noted for its ability to modulate the IL-10 production in the lung microenvironment and inhibit the overproduction of prostaglandin E2 in lung cancer patients [130]. Cxb has also demonstrated synergistic anticancer effects when administered in tandem with other anticancer drugs such as docetaxel [128]. To increase the loading capacity of Cxb, an exploratory formulation’s degree of organization was decreased where the NLC was developed to comprise of an inner oil core encapsulated by an exterior solid shell allowing for a high payload of Cxb. The entrapment efficiency of the Cxb-NLC formulation was calculated to be 95.6% and 4% w/w, respectively. In-vitro release studies of the Cxb-NLC formulation also showed controlled release characteristics as 8–10% of Cxb was released after 8h, 34% released after 24h and >80% released after 72h [55].
Pharmacokinetic and tissue distribution studies compared the Cxb-NLC formulation against normal Cxb-Solution (Soln), both of which were delivered via inhalation by nebulization. Following 30 min of nebulizing Cxb-Soln and Cxb-NLC, the degree of Cxb deposition in the lungs was 84.48% and 78.4% of Cxb dose per lung tissue, respectively. Cxb-Soln lung concentrations fell to 4.9% 6h post-nebulization and fell below the limit of quantification at 12h post-dosing. Contrastingly, Cxb-NLC exhibited superior lung residence where Tmax was observed 4.5 h post-inhalation, which was followed by a slow elimination phase and well-above the limit of quantification for up to 24h post-dosing. In general, all calculated PK parameters within plasma and lung displayed improved Cxb concentrations, residence and exposure in the local environment (data summarized in Table 1) [55].
Table 1.
Plasma pharmacokinetic and lung disposition parameters of Celecoxib encapsulated in nanostructured lipid carriers (Cxb-NLC) and in solution (Cxb-Soln) following nebulization and inhalation for 30 min in Balb/c mice*. Mean values (percent coefficient of variation) are shown. For all values, P < 0.05 when compared with tle-o Cxb-Soln. Modified from [55].
| Plasma** | Lung*** | ||||
|---|---|---|---|---|---|
| Parameter | Cxb-NLC | Cxb-soln | Parameter | Cxb-NLC | Cxb-Soln |
| Tmax, h | 4.50 (22.2) | 1.38 (54.5) | Tmax, h | 1.13 (66.7) | 0.5 (0.0) |
| Cmax, μg·mL−1·mg−1 | 0.11 (19.3) | 0.02 (9.6) | Cmax, μg·g−1·mg−1 | 0.21 (12.8) | 0.23 (24.3) |
| AUCt, μg·h·mL−1·mg−1 | 1.1 (4.8) | 0.05 (15.82) | AUCt, μg·h·g−1·mg−1 | 1.26 (14.6) | 0.36 (9.8) |
| AUCinf, μg·h·mL−1·mg−1 | 1.27 (10.5) | 0.07 (15.4) | AUCinf, μg·h·g−1·mg−1 | 1.36 (16.1) | 0.42 (6.6) |
| Cl, L·kg−1·h−1 | 0.93 (4.72) | 20.03 (4.72) | Cl, L·kg−1·h−1 | 0.81 (16.3) | 27.77 (10.3) |
Tmax is the time at which the maximum concentration is observed; Cmax maximum concentration per mg of dose; AUC(t) dose normalized total area under the curve (time zero to last measurable concentration per mg of drug dose), AUCinf/dose dose normalized total area under curve (time zero to infinity per mg of drug dose), Cl clearance.
Blood values are normalized per mL of blood and mg of applied dose.
Lung parameters are normalized per g of lung tissue mass and mg of applied dose.
Cxb-NLC showed improved Cxb plasma concentration levels at all time points, which was attributed to the formulation composition as well as particle size. Modifications of these components exhibited a robust lung residence that avoided rapid clearance and over-saturation, which could lead to complications including inflammation and other negative effects. The exterior shell of the NLC was composed of a lipophilic triglyceride (i.e. Compritol, etc.) in order to increase adsorption to the surface of vascular epithelium. Previous investigations have also showed that nanoparticles smaller than 260 nm can escape macrophage detection and phagocytic uptake [131]. The controlled release behavior of Cxb can be attributed to the presence of Compritol in mixture as the long-chain fatty acid (behenic acid, C22) is known for its slow degradation when compared to shorter chain length fatty acids and exhibits low pulmonary toxicity [131]. Due to the high encapsulation efficiency of Cxb in NLC, the observed AUC/D was 4-fold higher than its Cxb-Soln counterpart following nebulization and inhalation in Balb/c mice [55]. The low molecular weight of the Cxb-NLC complex also promoted absorbance via passive diffusion through the lung epithelial membrane after inhalation. The highest concentration of diffusion is expected to be in the alveoli region as the thin monolayer is made of compact and broad cells over a large surface area [53].
An interest of the biotechnology industry has also been attracted to NLC formulations for highly lipophilic anti-cancer therapies such as paclitaxel. NanOlogy LLC (Lawrence, KS) has patented its NanoPac® technology that processes paclitaxel with compressed antisolvents into uncoated submicron crystal particles in 600–800 nm of size that can be delivered via nebulization [132]. This formulation was intentionally designed large enough to avoid systemic uptake into plasma and be retained in lung as well as malignant sites for a reservoir depot effect. This promotes a slow release effect of paclitaxel into the surrounding fluids and tissues at constant saturation levels. A pharmacokinetic study involving 90 rodent animals investigated the feasibility of inhaled NanoPac at high dosages (IHNP-HD) and low dosage formats (IHNP-LD). Lung tissue and plasma samples were collected at pre-determined time points up to 336 h post-exposure. Initial paclitaxel exposure to the right lobes of the lung was higher in the IHNP-LD and IHNP-HD when compared with the intravenous group where Cmax was 3.5- and 7-fold greater, respectively. Following completion of nebulization, inhaled NanoPac exhibited a much slower clearance rate than the intravenous group and observed a 3-fold increase in T1/2 for both high and low dosage formats. Paclitaxel concentrations were quantifiable up to the 72h time point whereas both inhaled NanoPac arms were above the limit of quantification up to 336 h (2 weeks) post-dosing. Overall paclitaxel deposition and retention was characterized by observing AUClast in lung, which was 5.5- and 18-times higher for IHNP-LD and IHNP-HD groups, respectively, when compared to the intravenous arm. When these numbers are dose normalized, IHNP-LD and IHNP-HD showed 39- and 43-fold increases of paclitaxel exposure per drug unit dose when compared to the intravenous dosing group. 17.75- and 13-fold increases were also documented when comparing paclitaxel plasma concentrations between the IV and the IHNP-LD as well as IHNP-HD, respectively [132].
The extended residence of NanoPac inside lung after inhalation allows the possibility of increased efficacy with the bioavailable concentration of paclitaxel gated by saturation levels in the surrounding environment. This approach could prove beneficial when applied in tandem with conventional therapies to treat diseases such as NSCLC for increased anti-tumor efficacy while also minimizing any substantial systemic toxicity. Although residual NanoPac crystals have been observed in lung lobes for up to 336 h post-administration, histopathological examination of the IHNP-LD/HD were indistinguishable with untreated controls post 336 h time point. Nanoparticle characterization analysis showed that both NanoPac suspensions exhibited MMADs ~2 μm [132]. The sizing aspect could pose further complications when translating to the clinic as human physiology is inherently larger when compared to the rodent model. Further efforts may be necessary to tailor the formulation to clinical applications with appropriate size and API concentrations. Despite these limitations, the study was able to showcase a significantly higher degree of deposition and retention of paclitaxel within the local lung environment as well as uptake into systemic circulation.
5.2. Liposomes
Liposomes have been widely investigated as therapeutic delivery vehicles and recognized as one of the leading lipid-based carriers. Formulations can vary between a single bilayer lipid membrane (unilamellar liposome) and multiple bilayer lipid membranes (multilamellar liposomes). The outer shell is typically comprised of polymers such as PEG to allow for biocompatibility and additional conjugated components that can improve targeting/efficacy. Further manipulation of a liposome’s membrane composition can generate neutral, negatively charged and cationic formulations, which can be utilized to form complexes with negatively charged nucleic acids [1]. Similar to NLCs, liposomal formulations delivered via inhalation also experience rapid clearance, fast absorption or prolonged residence leading to oversaturation. It seems there is an optimal window of residence that augments the controlled release kinetics of nanoparticles while avoiding rapid clearance/absorption as well as oversaturation leading to inflammation.
PEG-phospholipids are amphiphilic polymers with a hydrophobic distearoylphosphatidylethanolamine (DSPE) block and a hydrophilic PEG block. In aqueous conditions, these polymers can self-assemble to form micellar structures that are endogenous to the lungs [133]. The PEGylated micelles and liposomes exhibit improved potential for controlled release kinetics due to the long fatty acyl chains comprising the outer shell, which confer less mobility to the encapsulated drug [133–136]. The same micelles have previously been demonstrated to actively accumulate in the Lewis Lung Carcinoma model in mice after IV administration [137]. The average micelle size was roughly 5 nm with notable uniformity and narrow size distribution (± 0.7 nm). Even among nanoparticles, these micelles are classified as ultra-small vehicles that can avoid phagocytosis by macrophages as they are too small to be detected. They are also more readily incorporated into the “respirable percentage” of aerosolized droplets due to their small size and phenotype as an aqueous colloidal dispersion drug carrier. Following intratracheal deposition, these nanoparticles typically remain in the lung lining fluid until they are dissolved and display prolonged residence within the lungs [138].
Drug release studies were performed in-vitro utilizing a dialysis bag with a molecular weight cut-off of 10 kDa and conditions that mimic the lung environment. These studies showed that only 22% of paclitaxel was released within the first hour of incubation demonstrating a slow release profile, which is contrary to many other sustained release formulations that have displayed an initial rapid burst of drug. Paclitaxel continued to slowly release with 90% of the drug emancipated at 8h after which time, the release profile was constant until the last observed time point of 24h. The PK studies observed 3 distinct formulation groups which included intratracheally administered paclitaxel loaded micelles, IV administered paclitaxel loaded micelles and intratracheally administered Taxol as the control group. The AUC of paclitaxel was significantly higher in the lungs of intratracheally administered PEG-lipid micelles to rats and experienced a ~45-fold increase when compared with the same formulation administered intravenously. Targeting efficiency (Te) was measured and calculated by dividing the AUC0–12 of the target tissue by the sum of all AUC0–12 of non-targeted tissues. Therefore, calculations were feasible for all organ tissues obtained and included lungs, blood, liver, kidney, heart and spleen. Targeting efficiency to the lungs was found to be 132-fold higher in the pulmonary route when compared with the IV counterpart with values of 6.57 and 0.05, respectively. Intratracheal administration of PEG-lipid micelles also resulted in reduced paclitaxel concentrations in plasma as well as the other peripheral organs observed. Despite the higher AUC exhibited in IT administration, Cmax was much higher in the IV route where the majority of the dosage was found in the liver or spleen. This is a good indication as it signifies lower systemic exposure of paclitaxel to other organs when the formulation is applied via IT route and demonstrates improved localization of the chemotherapy. The Taxol group also yielded an 8-fold higher Cmax which has been attributed to the free-state of paclitaxel and thereby, rapid access to systemic circulation. Stability studies of the paclitaxel loaded micelles suspended in water did not show any changes in particle size after 3 months of storage at room temperature, which is an encouraging finding for mass-scale production [138].
In a similar study, paclitaxel was also encapsulated in liposomes with employing dilauroylphosphatidycholine (DLPC) as part of their membrane formulation as opposed to DSPE [139]. The study setup also compared its formulation through two routes of administration: via inhalation through jet nebulization and IV. The measured AUC in the lungs of the aerosol group was 26-fold greater than that of the IV group. In-vivo anti-tumor activity was also evaluated by measuring predefined parameters that included lung weights, number of tumors and median tumor size on the lung surface. The treated mice were compared with untreated control mice and mice that received DLPC aerosol treatment only. The paclitaxel encapsulated DLPC liposome (PTX-DLPC) group showed similar lung weights to tumor-free lung weights. Whereas the DLPC-treated and untreated groups showed an increase in lung weights by 58% and 39%, respectively, when compared with tumor-free lung. Mean size of tumors and mean number of tumor lesions was also reduced in the treated group. A previous study by the same group also demonstrated that the addition of 5% CO2 to the breathing air can improve the deposition efficiency of therapies administered via jet nebulizer as it resulted in a 3-fold increase of drug [140]. In another experiment, the frequency of treatment was altered from 3 times per week to twice weekly for 2.5 weeks. The results still demonstrated a significant reduction in the lung weights of PTX-DLPC mice when compared with the control group. However, the reduction in dosage frequency proved less efficacious than the original 3 times per week treatment [141]. This is consistent with previous studies that showed that in-vitro cytotoxicity of paclitaxel is more dependent on the prolonged duration of exposure rather than increased PTX concentrations [142].
Despite the clinical benefits of inhalable nanotherapies, there is still a lack of understanding on the lung clearance kinetics of nanoparticle drug carriers and the influence lung inflammation can have on their clearance. For example, the accumulation of nanoparticles in the lung over a series of doses can result in “nanoparticle overload” and instigate local inflammatory responses [143–145]. Various inflammatory lung diseases and lung inflammation can compromise the integrity of respiratory function, alveolar epithelium and lung mucus volume as well as its composition. This in turn can affect clearance pathways, lung distribution and general kinetics of inhaled nanomedicines [146–148]. It is also generally understood that pulmonary delivery of nanoparticles or proteins can induce mild inflammatory effects in healthy lungs, which can result in increased pro-inflammatory cytokines or alveolar macrophage presence [145, 149]. A previous study demonstrated that lung clearance kinetics of a non-PEGylated anionic liposome was considerably different from that of a non-PEGylated anionic solid lipid nanoparticle of similar charge and size [150]. Ciprofloxacin is an antibiotic medication that has no intrinsic pro-inflammatory or anti-inflammatory properties and showed high encapsulation efficiency in liposomes. Ciprofloxacin-loaded PEGylated 3H-labelled liposomes were delivered by intratracheal administration to the lungs of healthy rats and rats with bleomycin-induced lung inflammation to characterize the difference lung clearance and kinetics between the two states. Ciprofloxacin and 3H-lablled liposomes were quantified simultaneously utilizing liquid chromatography mass spectroscopy (LC/MS) and liquid scintillation counting, respectively [151].
IV plasma profile after intratracheal administration exhibited a biphasic clearance for both 3H-lablled carrier and ciprofloxacin. The initial clearance of 3H-lablled liposome was rapid where ~2% of the initial dose remained in plasma after 48h. The slower elimination phase that followed showed a terminal half-life of approximately 4 days. On the other hand, ciprofloxacin (IV) cleared more rapidly from plasma than the radiolabeled carrier with a terminal half-life of 2h which indicate its swift liberation from the liposome. Plasma concentrations of 3H-lablled liposomes after pulmonary delivery were steadily low when compared to the IV group. There was an initial rapid absorption phase of 3H-liposomes and followed by a slower absorption rate phase. Healthy lungs did demonstrate an elimination phase whereas the inflamed lung group showed 3H plasma concentrations that plateaued between 4–7 days. Contrastingly, the plasma profile of ciprofloxacin-loaded liposomes after pulmonary administration showed no clear absorption phase as the first blood sample time point was 30 min. By this time point, the bulk of the drug was likely released from the carrier, absorbed into plasma and cleared. However, a distinct elimination phase could be characterized with a slow half-life of 20h, which is considerably longer than the IV counterpart. Despite the initial burst liberation of drug from the liposome, the formulation did promote a reservoir for the steady and prolonged release of drug in the lungs over time [151].
In general, the data suggests that systemic absorption of liposomes is limited after pulmonary intratracheal administration in rats for both healthy and inflamed lung types. Size exclusion chromatography (SEC) profiles of 3H lipids after IV administration indicated that intact liposomes were able to be systemically absorbed up to 24h post-delivery. Though after 72h, the 3H species detected in plasma were primarily products of lipid biodegradation or liberated 3H lipids. Comparatively, SEC profiles after pulmonary administration detected predominantly free 3H lipids and a minor percentage of lipid-bound plasma proteins, suggesting that the systemic absorption of intact liposomes was limited. The marked difference between the healthy and inflamed groups is exhibited in the slower portion of the elimination phase that showed prolonged residence of liposomes in the inflamed lungs. Despite the marked difference in liposomal elimination between IV and IT administrations, this action is not reflected in the pharmacokinetics of ciprofloxacin, which is more frequently examined [151]. Lung inflammation has been documented to increase alveolar macrophage content, induce local edema and change lung mucus viscosity as well as composition [154, 155]. These contributing factors put together may be responsible for the increased liposome retention in lung lining fluid and increased their exposure to alveolar macrophages. Despite the increased macrophage presence, lung inflammation can also decrease macrophage-mediated lung elimination of intratracheally instilled liposomes and impair alveolar macrophage mobility, which are likely by-products associated with the increased viscosity of mucus [156].
5.3. Liposomes vs Nanostructured Lipid Carries
A comparative study exploring the PK differences between liposomal and nanostructured lipid carrier (NLC) systems further reinforced that the speed and degree of absorption of macromolecules from the lungs is sub-par and a rate-limiting consideration [87, 143, 150, 157]. Tight junctions between alveolar epithelial cells are a large contributing factor to the limited paracellular transport of macromolecules. The estimated pore radii of distal capillary endothelium and respiratory epithelium are 6.5–7.5 nm and 0.5–0.9 nm, respectively, effectively serving as a natural physical barrier against transport [158, 159]. This is in good agreement with previous lung retention data, which indicated a prolonged residence of nanomaterials in the lungs of rats via intratracheal instillation after an initial period of rapid clearance where >10% of the dose remained after 2 weeks [150]. Other dog, rodent and human subjects administered with 99mTc-labelled liposomes corroborate these findings [107, 160–162]. More than 51% of the 14C-dipalmitoyl phosphatidylcholine (DPPC) liposome dose remained in the lungs of rats 1 day following pulmonary delivery (intratracheal administration) [163]. A similar formulation of 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC) and DPPC-based liposomes showed ~80% of the inhaled pulmonary dose in the lungs of humans after 1 day [160]. Consensus across multiple studies in rats with varying formulations indicates that residence time can be influenced by controlling structural and chemical characteristics of the carrier or payload. Inhaled levonorgestrel loaded liposomes instilled intratracheally exhibit a Tmax and half-life of 7h and 64h, respectively [164]. Whereas, thymopentin loaded solid lipid nanocarriers (SLNs) exhibited a Tmax and half-life of 1h and 6h, respectively, following pulmonary administration by inhalation [165].
For both NLC and liposome formulations, the comparative study data showed mucociliary clearance to be the dominant mechanism by which 3H-labelled lipids were eliminated from the lungs of rats after intratracheal instillation [150]. Absolute bioavailability was difficult to accurately determine as pulmonary administration of 3H-labelled lipids resulted in large recovery of the dosage (~24%) in feces. This data confirms mucociliary clearance from the lung and liberation of the free 3H-lipid in the GI tract, which contributed to plasma concentrations as lipid micelle or free lipid and skewing the absolute bioavailability. Despite this contribution, both nanomaterials exhibited prolonged residence in the lungs with roughly ~40% of the pulmonary dose remaining after 3 days and approximately ~30% remained after 1 week. This suggests that the majority of the detected 3H-lipid plasma concentrations of both nanomaterials were largely a result of direct absorption from the lungs after intratracheal instillation [150]. Notably however, mucociliary clearance was more prevalent for the NLC group compared to the liposomal formulation, which could be attributed to NLC’s tendency to form aggregates within the lungs, while liposomes are primarily eliminated by alveolar macrophages. Once phagocytosed, alveolar macrophages will either transport towards the lung lymphatics or clear via the mucociliary escalator. The comparative results provide key evidence that alveolar macrophages can distinguish, seize and consume nanosized particles as small as ~170 nm in diameter. This is in direct contrast to the previous studies which determined that particles smaller than ~260 nm were likely to escape phagocytic uptake by macrophages [131, 166]. Despite the lack of prolific in-vivo investigations and data, the comparative study data infers that anionic liposomes are likely to have superior intrinsic mucus penetrating properties and capability to be absorbed by alveolar epithelium when compared to anionic NLCs. Nevertheless, it is generally known that coating polymeric nanoparticles with hydrophilic polymers (such as PEG or pluronic) can improve lung absorbance and reduce mucociliary clearance [167]. In our lab, an original complex multifunctional liposomal drug delivery system containing an anticancer drug, suppressors of multidrug resistance and antiapoptotic defense was developed and tested on an orthotopic model of lung cancer (Figure 6) [152, 153, 168]. Human A549 lung adenocarcinoma epithelial cells (expressing MRP and BCL2 proteins) transfected with luciferase were intratracheally injected into the lungs of nude mice. The liposomal system was delivered by inhalation using a specially designed nose-only exposure chamber equipped by Collison nebulizer. Experimental drug distribution data of inhaled liposomes within treated mice showed preferential accumulation and retention in lungs with tumor. A superior antitumor effect was also observed by our inhaled complex system (Lip-DOX-MRP1-BCL2 ASO) when compared with intravenously injected free, liposomal drug, and even a mixture of liposomal DOX, BCL2 and MRP1 ASOs delivered by inhalation. Inhalation delivery also limited adverse side effects of this toxic system upon healthy organs (Figure 6).
Figure 6.
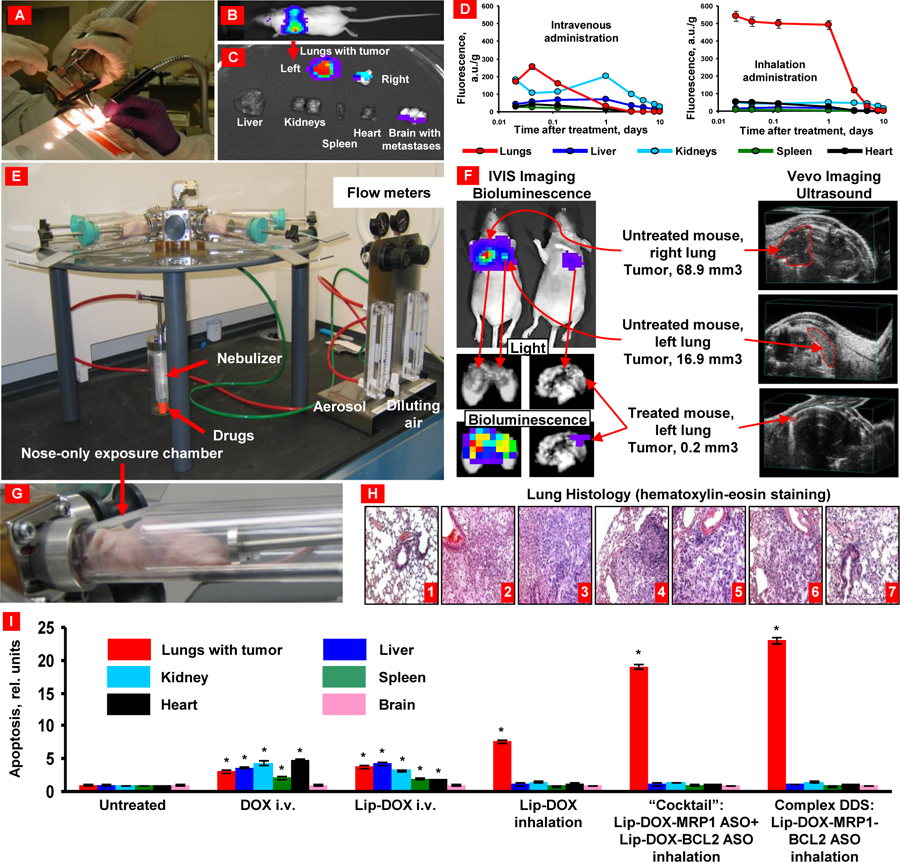
Orthotopic lung tumor model and inhalation treatment of lung cancer. (A) Human lung cancer cells transfected with luciferase were intratracheally injected into the lungs of nude mice. (B) Typical bioluminescent image of a mouse with lung tumor 4 weeks after instillation of cancer cells. Intensity of bioluminescence is expressed by different colors, with blue reflecting the lowest intensity and red indicating the highest intensity. (C) Bioluminescence of excised mouse organs. (D) Inhalation delivery enhances lung exposure to liposomal drug and limits its content in other organs. (E) Installation for inhalation treatment. (F) Inhalation treatment of mice with orthotopic human lung cancer by DOX combined with inhibitors of pump and nonpump cellular resistance significantly decreases tumor size. Representative bioluminescent (IVIS imaging system) and ultrasound (Vevo imaging system) images of mice (untreated and treated within 4 weeks). (G) Mouse inside the nose-only exposure chamber. (H) Lung tissue histology (H&E staining). 1 – control (no tumor); 2 – untreated tumor; 3 – DOX (i.v.); 4 – liposomal-DOX (i.v.); 5 – liposomal-DOX (inhalation); 6 – mixture (“cocktail”): liposomal-DOX-MRP1 ASO + liposomal-DOX-BCL2 ASO (inhalation); 7 – complex DDS: liposomal-DOX-MRP1- BCL2 ASO (inhalation). (I) Inhalation delivery enhances cell death induction in the lungs with tumor and limit adverse side effects in other organs. Means ± SD are shown. *P < 0.05 when compared with untreated animals. Redrawn from: [24, 152, 153].
5.4. Targeted Nanostructured Lipid Carriers
Targeting ligands have been widely employed in nanotechnologies to optimize their pharmacokinetic and pharmacodynamic properties. A diverse range of moieties have been investigated and include aptamers, carbohydrates, vitamins, peptides, proteins and monoclonal antibodies. Continued efforts in innovating these types of vehicles have resulted in the development of hierarchical targeting entities where each layer in the formulation is designed with a different objective. One such example involved a multifunctional NLC nanoparticle that utilized hierarchical targeting in 3-stages that included lung tissue targeting, cancer cell targeting and mitochondrial targeting (final destination of the payload) [169]. This was achieved through a RGDfk-histidine-poly (lactic-co-glycolic acid) copolymers (PLGA) structure where PLGA is a biodegradable and biocompatible material. The size and other physicochemical properties of the NLC formulation were optimized for inhalation administration, which enabled primed lung tissue targeting and is Stage I of the hierarchical design. Stage II involved the utility of RGDfk receptors that recognize the integrin αvβ3 receptor on lung cancer cell membranes and were bound to the surface of the NLC system through chemical grafting. Stage III focused on the intracellular kinetics including mitochondrial targeting and lysosomal escape. Histidine groups were engineered into the NLC system to facilitate the ‘proton sponge’ effect whereby increasing proton influx and resulting in endosomal bursting. The histidine group also promotes a positive charge, aiming to bring the vehicle in proximity to the negatively charged mitochondrial membrane. The active pharmaceutical ingredient (API) employed was yuanhuacine extracted from the Chinese medicine Thymelaeaceae, which has reported for its in vitro anticancer capabilities (especially for lung cancer) [170].
The PK study showed greater plasma AUC, longer mean retention time and lower Cmax for inhaled hierarchical-NLCs when compared with the IV administration. These statistics point to decreased risks of drug toxicity and the potential for a less frequent dosing schedule. The targeting efficiency was also monitored as the relative AUC0-t value of the target tissue to the non-target tissue, providing a ratio and estimation of selectivity. There was a significant increase in the Te of hierarchical-NLCs administered via inhalation when compared with the IV counterpart and was observed consistently across all non-targeted tissues in the study that included liver, spleen, kidney and reproductive organs. Anti-tumor efficacy was examined through in-vitro apoptosis assays utilizing the human lung cancer cell line (A549) and showed ~33.09 % apoptosis rates for the hierarchical-NLC NP while the control group was tracked at ~4.70 %. Traditional NLC formulation (non-hierarchical) encapsulating yuanhuacine was also tested in-vitro and yielded 25.65 % apoptosis rate. Solo treatment of yuanhuacine yielded an apoptosis rate of 18.55 % [169]. Also, observed decreases in mitochondrial membrane potential was an early indicator in the process of cascade apoptosis [171]. Cell apoptosis is irreversible once the membrane potential collapses. Further evidence of the induced activation of caspace (leading to cell death) was detected with increasing concentrations of Cytochrome C, which is an integral component of the mitochondrial respiratory chain and indicative of increasing apoptosis processes [169]. Overall, the data suggests that the hierarchical-NLC systems have demonstrated comparable robustness and therapeutic efficacy to warrant further investigation.
5.5. Targeted Liposomes
Active targeting liposomes typically display enhanced anti-tumor therapeutic efficacy due to their ability to selectively bind to over-expressed receptors of cancer cells. However, accurate selection of efficacious API(s), targeting ligand(s) and physicochemical properties of the liposome system are critical to ensure optimal results. Expression of transferrin receptor (TFR) is low in most healthy cells and is overexpressed (~100-fold) in many tumor types including lung adenocarcinoma, prostate, ovarian, brain and breast [172, 173]. TFRs are transmembrane proteins that regulate the uptake of iron through receptor-mediated endocytosis to help satisfy the metabolic requirements of tumor cells [174, 175]. TFRs are highly expressed in the A549 cancer cell line and further corroborated by an investigation utilizing transferrin-conjugated liposomes loaded with doxorubicin, which did observe higher in vitro uptake in A549 cells [176]. A T7 (HAIYPRH) peptide was identified by phage display and enhanced with a biopanning process [177]. The T7 peptide binds to a small cavity on the TFR surface and is subsequently transferred inside the cell through endocytosis through the assistance of transferrin (TF) [172]. T7 peptides also do not actively compete with TF for binding to TFR and enhances cellular uptake of the TF-conjugated LP system [177, 178]. Quercetin (QR) is a natural flavonoid widely found in nature (i.e. fruits, vegetables, etc.) and has been noted for its anticancer capabilities [179]. QR has been reported for its ability to suppress the overexpression of Aurora-B kinase, inactivate the Akt-1 pathway and promote the MEK-ERK pathway [180, 181]. QR’s utility in lung chemotherapy has been limited due to its low bioavailability, low water solubility and rapid elimination from plasma [182, 183]. A T7-conjugated QR-loaded liposome system (T7-QR-LP) was developed for pulmonary administration (intratracheal aerosol instillation) against lung cancer though and showed only slight improvements in anti-tumor efficiency [184]. Although the T7-QR- P system’s features showed adequate versatility including particle size, polydispersity index, encapsulation efficiency, and stability studies, it only slightly improved the in-vivo therapeutic efficacy of QR. The mean survival time of the control groups were averaging ~40 days while T7-QR-LP group’s mean survival team averaged ~69 days after dosing [184]. Despite the optimization of the platform and targeted nature of the system, therapeutic efficacy was still relatively low.
Other well-known target for cancer-specific delivery of therapeutics is folate receptor which is practically not expressed in healthy cells and overexpressed in tumor cells including NSCLC [185–187]. In particular, it was used for the inhalation delivery of docetaxel by cancer-targeted liposomes [188]. Drug containing liposomes were prepared as inhalable dry powders. The physicochemical properties and PK of the docetaxel (DTX)-loaded folic acid (FA)-conjugated liposomes (LPs-DTX-FA) were tracked and evaluated to assess if PK or anti-tumor efficacy were altered after co-spray drying the inhaled dry powder [188]. The LP-DTX-FA system exhibited an average PDI of ~0.229, zeta potential of −28.6 ± 2.6m V and diameter of 100.1 ± 1.0 nm. Of note, the empty liposomes were smaller (89.2 ± 0.3nm) than the DTX-loaded systems, suggesting that the presence of docetaxel increased the volume of liposomes. In-vitro cellular uptake assays showed that energy-depended endocytosis was primarily responsible for the cellular internalization of such liposomes. It was found that the cellular internalization of LP-DTX-FA suspensions decreased significantly at 4°C when compared with room temperature. The measured concentration of docetaxel in BALF was 128.30 ± 25.89 ng/mL 12h after co-spraying administration via inhalation. The calculated AUC value for docetaxel in lung was 25-fold higher after intratracheal administration of drug-loaded liposomes when compared to IV administration (Figure 7). Furthermore, the highest concentration of docetaxel in the lungs for the IT route was observed at 30 min after liposome exposure and was 45-fold higher than the IV counterpart, implying a potential for a better therapeutic efficacy of local inhalation delivery of liposomal drug formulation. Although both routes of administration exhibited close AUC0–12h for all other measured organs (heart, liver, spleen and kidney), AUC of docetaxel in spleen, kidney, liver and heart were 17-fold, 22-fold, 43-fold and 50-fold lower, respectively, when compared with IT administration creating prerequisites for fewer adverse side effects of the delivered drug. Furthermore, MTT assay was employed for measuring cytotoxicity of the drug loaded liposomes and showed higher cytotoxicity of cancer targeted LP-DTX-FA system when compared with non-targeted one [188].
Figure 7.
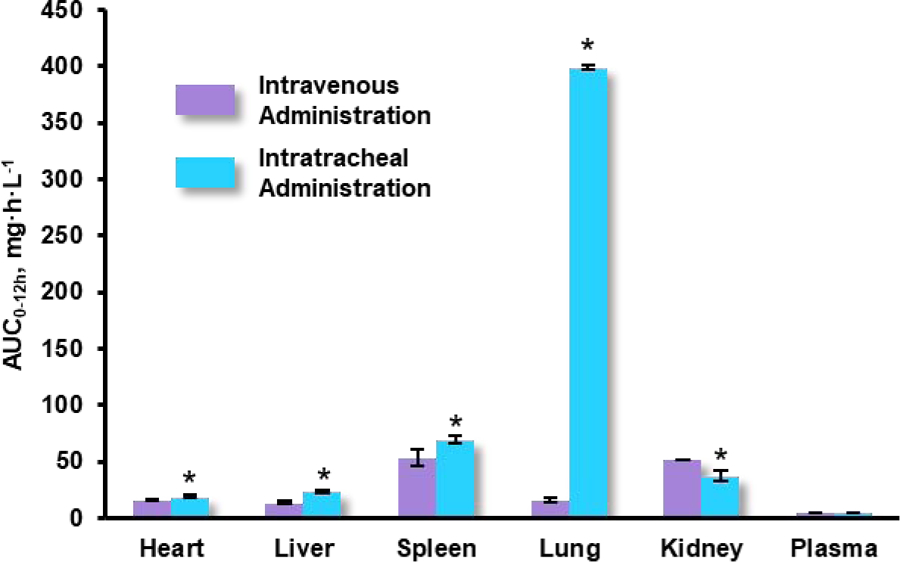
Area under the curve (AUC) for docetaxel delivered by folic acid-conjugated liposomes from t = 0 to t = 12h in heart, liver, spleen, lung, kidney, and plasma following intravenous and intratracheal administration of docetaxel-loaded suspensions (1 mg/kg). Means ± SD (n=6) are shown. *P < 0.05 when compared with intravenous administration. Redrawn based on the data in [188].
The use of targeted liposomal systems in clinical applications is limited by the heterogenous nature of cancer cells that limits efficient binding of cancer-targeted formulations as well as binding-site barriers that prevent deep penetration of liposomes into tumors [190]. To address these complications in a targeting LP system, a dual-ligand delivery platform was developed to enhance the vehicle’s deep penetration and targeted accumulation capabilities [189] (Figure 8A). Tumor microenvironments typically exhibit a relatively low concentration of oxygen due to the poor vasculature and increased consumption of oxygen by cancer cells [191]. The underlying cellular reaction to hypoxia includes the activation of hypoxia-inducible factor (HIF) and results in the over-expression of carbonic anhydrase IX (CA IX) on the cancer cell surface [192]. It has been reported that CA IX expression in healthy tissue is limited and is aberrantly expressed in various types of tumors including lung, breast, kidney and colon [193–199]. The attachment of an anti-CA IX antibody potentially can improve targeting capabilities of drug delivery systems and further limit their accumulation in healthy cells. On the other hand, the hydrophobic feature of cell membranes safeguard against the influx of exogenous materials including bioactive components such as oligonucleotides and proteins [200]. CPP33 is a novel tumor lineage-homing-cell-penetrating peptide which exhibits the capacity to penetrate the cell membrane of A549 cells [201]. A pharmacokinetic study was employed to explore the efficacy of an ant-CA IX antibody and CPP33 dual-ligand liposome system encapsulating triptolide (dl-TPL-LP) delivered via intratracheal aerosol administration [189].
Figure 8.
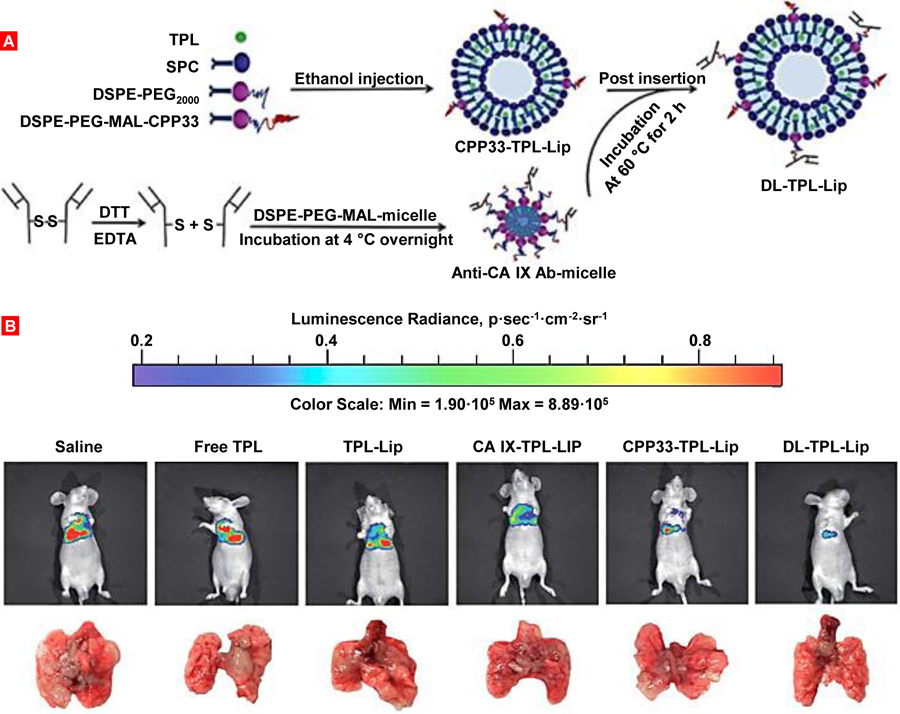
Dual-ligand modified liposomes for local targeted delivery of anticancer drug for treatment of lung cancer. (A) Illustration of the preparation of dual-ligand triptolide (TPL)-loaded liposomes. (B) Representative bioluminescent images and corresponding lung tissue of the mice at day 31 after tumor inoculation. Mice were treated with free triptolide (TPL) and TPL delivered by liposomes (TPL-Lip), anti-carbonic anhydrase IX antibody-modified liposomes (CA IX-TPL-LIP), CPP33-modified liposomes (CPP33-TPL-Lip) or dual-ligand TPL liposomes (DL-TPL-Lip). Mice bearing orthotopic lung tumor were treated every three days four times by intratracheal administration at 0.3 mg/kg of tested substances. Redrawn from [189].
Prior to the PK study, a few in-vitro tests were conducted to verify the tumor penetration, targeting capabilities and anti-tumor efficacy of the tested delivery system. A 3-dimensional tumor spheroid assay showed the dl-TPL-LP could deeply penetrate into different layers of the spheroids when compared to non-modified liposomes, which reflects enhanced penetration of modified liposomes. Cytotoxic damage assays were employed in the 3-dimensional tumor models and showed significantly higher cytotoxicity of dl-TPL-LP when compared to non-modified formulations and the control group. A549 cells were also incubated under hypoxic conditions [202] to mimic the tumor microenvironment and dl-TPL-LP system greatly reduced the migratory activities and proliferation of CA IX-positive A549 cells in this circumstances. After pulmonary administration of the dl-TPL-LP system in rats, the AUC0−∞ remained consistent and a 3-fold decrease in Cmax was observed when compared with the free-TPL formulation also delivered via inhalation. The marked difference was observed through bioluminescence images and mass weights of the dissected lungs of rats treated with free TPL. High tumor proliferation was observed through peak radiance signals and an approximately ~6-fold increase in the weight of the lungs. Comparatively, the bioluminescent images of dl-TPL-LP group showed low radiance signals and approximately ~1-fold increase in lung weight (Figure 8B). Although both free-TPL and dl-TPL-LP groups showed similar AUC0−∞, the anti-tumor efficiencies were radically different and likely attributed to the dual-ligand modification. The reduced Cmax in the blood also shows advantages of targeted system as this indicates a lower distribution of TPL in systemic circulation which creates prerequisites for the limitation of adverse side effects. The dl-TPL-LP also exhibited a ~3-fold increase in half-life when compared to free TPL and is an encouraging feature of the modified liposomal system that creates basis for reduced dosing schedule. Overall, the system showed high encapsulation efficiency, high targeting efficiency, uniformity and robust anti-tumor capabilities after intratracheal aerosol administration [189].
6. Pharmacokinetics of Nanomedicines for other Therapeutic Areas
6.1. Pulmonary Arterial Hypertension
Bosentan is an endothelin receptor antagonist (ERA) and a potent blood vasodilator prescribed to patients with pulmonary arterial hypertension (PAH) due to elevated levels of endothelin (ET1) [203–205]. The oral administration of bosentan has several shortcomings including short duration of action, need for frequent administration, dose-dependent hepatotoxicity, low bioavailability and systemic hypotension. When specifically considering patients with PAH, orally delivered bosentan lacks pulmonary vascular selectivity, which results in peripheral vasodilation and subsequent systemic hypotension which are common side effects for PAH-patients. To address these complications, a biodegradable polymeric nanoparticle colloid composed of PLGA has been developed and investigated for pulmonary administration via respiratory system by intratracheal administration [206]. The respirable controlled release polymeric colloid (RCRPC) exhibits several advantages including biocompatibility, macrophage uptake escape, minimal lung inflammation, controlled release of the API and local delivery to the site of action [207–211]. The release profiles of bosentan from the RCRPC system were characterized as a sustained release pattern (common for colloidal systems in pulmonary delivery) and lacked the initial burst release of API (only about 1.7% of bosentan was released after 0.5h). A wide range of RCRPC systems were developed with different characteristics and tested. The optimal formulation had a particle size of 420 nm (Figure 9A), PDI of 0.39, and an encapsulation efficiency of ~60.5%. The PK studied revealed a ~12.71-fold increase in AUC0-t for intratracheally administered RCRPC when compared with the orally delivered drug suspension (Figure 9B & 9C). The enhanced bioavailability of bosenten achieved after pulmonary administration can be attributed to the RCRPC’s capability to adhere within bronchial/lung tissue and sustain the release of bosenten at the adsorption site. Furthermore, inhalation delivery has the added benefit of avoiding the hepatic first-pass metabolism. It has been reported that PLGA nanoparticles can be retained in alveolar space, type 1 alveolar epithelium cells and basement membrane following IT administration and subsequent absorption by transcellular endocytosis through type 1 alveolar epithelium cells [208]. The congestive effect of IT administered bosentan-loaded RCRPC on pulmonary blood vessels was evident 12h post-dosing and correlates well with the obtained PK data that observed a ~10-fold increase in Cmax when compared with the orally delivered counterpart. By optimizing size, dispersity, physicochemical properties of the carrier and sustained release properties, the bosentan-loaded RCRPC system was able to exhibit improved bioavailability, deep lung deposition, controlled release and sustained vasodilation effect [206]. Therefore, this system potentially can be effective in clinical application where adverse effects of ERA (systemic vasodilation) may be reduced by inhalation delivery.
Figure 9.
Respirable controlled release polymeric colloid (RCRPC) of bosentan for the management of pulmonary hypertension. (A) Representative transmission electron microscope micrograph of the optimized bosentan RCRPC. (B) Bosentan concentration in plasma of rats after the oral administration of bosentan suspension and intratracheal delivery of the optimized bosentan RCRPC. (C) Pharmacokinetics parameters of bosentan in plasma following IT delivery of the optimized RCRPC and oral formulation of bosentan suspension given to rats. Means ± SD are shown. *P < 0.05 when compared with oral administration. Redrawn and replotted based on data presented in [201].
Liposomal encapsulation of PAH therapies is a notable trend as typical medications including ERAs, prostacyclin analogues, nitric oxide (NO) and phosphodiesterase-5 (PDE-5) inhibitors all lack pulmonary specificity, drug stability, ease of administration, safety and efficacy [212]. Systemic administration of these therapies yields many off-target effects including reduced cardiac function and peripheral vasodilation. Furthermore, progression of disease is inadequately inhibited while patient morbidity remains high [213]. Fasudil is an alternative medication to bosentan and has been validated to produce pulmonary vasodilation in both human and animal models of PAH by acting on vascular smooth muscle cells [214]. It is a Rho-kinase inhibitor that is similar to prostacyclins and alleviates the symptoms of PAH by inducing pulmonary vasolidation and downregulating the expression of matrix proteins, cell proliferation markers, growth factors and upregulates the expression of apoptotic markers. For liposomal delivery, fasudil is a weak base, hydrophilic molecule and exhibits low entrapment efficiency in lipid-based systems [215]. For this reason, various passive/active loading methods and drug-to-lipid ratios were analyzed to identify the optimal formulation system. It was found that passive loading methods yielded very low entrapment efficiencies that averaged ~27.4% while active loading methods increased drug entrapment by at least 2.5-fold. 5 sets of formulations (F1 = Passive loading Fasudil; F2 = Active loading pH 5.4; F3 = Active loading pH 3.0; F4 = Active loading pH 7.0; F5 = Active loading pH 8.0) at different pH values were tested where the highest increase in entrapment was observed at pH 3.0 and pH 7.0.
Continued optimization of the drug entrapment method found incubation of the drug/liposome system at 65°C with a 30-minute incubation time yielded maximal drug entrapment performance. Particle size of liposomes ranged between 100 and 250 nm and maintained a PDI of 0.15–0.2 indicating homogenous size distribution. Liposomes were developed using DPPC:Cholesterol (from ovine wool) at a molar ratio of 7:3. F1, F2 and F5 formulations were able to release 50% of their drug content within 8–12h while F3 and F4 released 50% of the drug ~24h showing good promise in delivering fasudil in the distal pulmonary arterioles for a sustained period. The loaded fasudil is initially unprotonated and becomes protonated in the liposome’s rich core and forms neutral complexes with anionic sulfates [216]. It is reasonable to assume that the principal mode of action of drug release is diffusion of the neutral fasudil complex out from the core through the liquefied lipid membranes as phase transition temperature for DPPC is 41°C and near human physiological temperature. This assumption is consistent with a previous report that analyzed temperature-dependent phase transition of DPPC liposome systems of >100nm undergo phase transition in the range of 37–39°C. Stability studies show that storage at 4°C observed a 10–15% reduction in drug entrapment over a period of 28 days while storage at room temperature (25°C) resulted in ~30–35% drug loss in the same time period [215].
IV administration of fasudil yielded a quick rise and rapid decline in drug plasma concentrations with a calculated Cmax of 298 ng/mL and t1/2 of 0.39h. Comparatively, pulmonary administration via intratracheal instillation of plain fasudil yielded a modest rise and sustained elimination profile in drug plasma concentrations with a calculated Cmax of 66.2 ng/mL and t1/2 of 1.17h. Utility of the liposomal formulations yielded fasudil concentrations above therapeutic levels for at least 15-hours, which can be extrapolated into a once or twice-a day dosing schedule. F3 group showed a t1/2 of 4.71h and a Cmax of 89.4 ng/mL at 8h followed by a sharp decline with no detectable drug at 18h. F4 group produced a t1/2 of 3.44 h and a Cmax of 86.9 ng/mL at 2 h. The controlled release characteristics in liposomes can be attributed to multiple factors. When administered via inhalation, liposomes can act as reservoirs that remain submerged in BALF and release drug continuously. Fasudil released from the liposome system will travel via the air-blood airway barrier and enter arterioles through the adventitial side to facilitate its vasodilatory effects. Intact liposomes can also enter systemic circulation via the air-barrier and release their payload in plasma. PD efficacy was analyzed through measuring mean pulmonary arterial pressure (MPAP) and mean systemic arterial pressure (MSAP). IV plain fasudil yielded a 45.6% reduction in MPAP, which quickly diminished 60–80 minutes post-dosing. IT administration of plain fasudil yielded a 38.3% reduction in MPAP where the effect could be extended up to 200 minutes. IT administration of liposomal fasudil observed a maximal reduction of 37.6% in MPAP with its vasodilatory effect observed beyond 3.5 h post instillation where a 20% reduction in MPAP was measured at 200 minutes. On the other hand, MSAP of IV and IT plain fasudil yielded 45.6% and 38.1% decreases in MSAP, respectively, while IT liposomal fasudil produced a 26.4% reduction in MSAP [215]. This data suggests the liposomal formulation delivered via IT route is capable of pulmonary selectivity while reducing its systemic exposure to off-target vasculature and thereby minimizing adverse effects in other parts of the body.
6.2. Liposomal Systems for Other Pathological Conditions
Lung transplantation (LT) has improved in the last decade. However, long-term survival after LT remains inadequate by the on-set of bronchiolitis obliterans syndrome and is regarded as a manifestation of chronic allograft rejection, which occurs in 30–50% of patients 2 years post-operation [218, 219]. Tacrolimus is a primary immunosuppressant and has been found to be more potent, effective, safe and superior than its competitor, cyclosporine, showing favorable results in decreasing the risk of obliterative bronchiolitis, acute rejection and lower toxicity in LT [220–223]. Clinical application of tacrolimus is limited due to a narrow therapeutic index, patient PK variability, potential drug interactions and dose-related efficacy and toxicity issues when considering available oral or parenteral formulations [224]. Encapsulation of tacrolimus in liposomal carriers has its advantages including biocompatibility for most lipophilic drugs, intracellular delivery, aqueous compatibility and the potential for a sustained release profile [225]. Among the options of pulmonary delivery systems, the dry powder inhaler formulations exhibit key features over the alternatives including increased potency, uniform deposition, propellant free, high encapsulation efficiency, reduced toxicity and stability [226–232]. The tacrolimus encapsulated liposomal systems were also tested in tandem with a co-spray drying ingredient to assess their effect on the formulation’s drug release capabilities. Nanoparticles (Figure 10) were spray dried with lactose, trehalose or sucrose and compared with spray dried plain tacrolimus with lactose. 90% drug release was observed from the spray dried plain tacrolimus with lactose formulation and was the most rapid of the four groups. The trehalose formulation produced the most pronounced sustained release capabilities with 90% of the drug released at 18h, followed by the lactose and sucrose [217].
Figure 10.

Spray dried nanoliposomes with trehalose for the inhalation delivery of tacrolimus. (A) A representative scanning electron microphotograph. (B) Surface texture analysis of trehalose based spray dried formulation. Reproduced with permission from [217].
Consistent with other liposomal-based formulations, the in-vivo PK profile of the LP-encapsulated tacrolimus system delivered via IT administration in albino rats showed prolonged residence of up to 24 hours and t1/2 of 16h. Meanwhile, plain tacrolimus administered via IT exhibited a much shorter residence with a t1/2 of 3.9 hours. Controlled release kinetics were also apparent for the LP formulation as 35.47% of the tacrolimus dosage was present in BAL at 4h and 9.47% observed at 12h. Contrastingly, plain tacrolimus was below the level of quantification 4 h post-IT instillation. AUC0–24h of the LP group was 1.83-fold higher than the plain tacrolimus group suggesting greater exposure and therapeutic potential with the lipid-based system [217].
Contraceptive administration experience similar obstacles as oral delivery often interferes with the production and action of endogenously generated steroid hormones [233]. Previous reports have indicated changes in oestradiol metabolism following administration of exogenous hormones [234]. Since oral contraceptives also incur the hepatic first-pass effect, hepatic enzymes and liver microsomal cytochromes (P450) are common in high concentration [235]. Continued stress and interference with liver function is also consistent with reports of oral contraceptive users developing fatty livers as an unwarranted effect of long-term treatment [236]. However, there is rising interest in the exploitation of the lung for systemic delivery of challenging molecules such as proteins, peptides, vaccines and analgesic agents. The large surface area and permeability of the lung tissue similar to other mucosal surfaces provides a promising space for rapid absorption of these delivery-challenged therapies. Levonorgestrel (LN) is a widely employed oral contraceptive where its utility has been regularly in low dosages in the progestogen only pill (POP) and in combination with estrogen in combined oral contraception (COC) formulations [237]. PK analysis of LN delivered orally (LO) against the LN-encapsulated LP (LNLP) formulation revealed similar AUC drug plasma concentrations though vastly different absorption profiles. LO produced a Cmax of 14.4 ± 0.6 ng/mL and t1/2 of 16.9 h while LNLP observed a Cmax of 4.4 ± 0.6 ng/mL and t1/2 of 64.4 h. Solubilization and diffusion of the drug into alveolar fluid prior to absorption into systemic circulation via transcellular transport may be accountable for the controlled release kinetics and zero-order absorption of LN. Pulmonary intratracheal administration and liposomal encapsulation of LN also limit exposure within the therapeutic window of 4–6 ng/mL for a longer period of time (almost 2-fold longer) rather than over-exposure of LN for a shorter duration, which is often attributed to the oral route [164] (Table 2). The data show that the liposomal encapsulated LN’s demonstrates ability to reduce dose-dependent progestronic side effects that are typically associated with orally administered LN.
Table 2.
PK of different formulations after oral and intratracheal pulmonary delivery of levonorgestrel in rats. LP1 - plain drug formulation, LP2 - physical mixture (plain drug along with constituents of liposomes), LP3 - liposomal formulation of levonorgestrel. Means ± SD are shown. Based on data from [164].
| Formulation | Route | AUC* (ng-h/mL) | Percentage relative bioavailability | Tmax (hours) | Cmax (ng/mL) | T1/2 (hours) |
|---|---|---|---|---|---|---|
| LP1 | Oral | 261.41 ± 12.36 | - | 2.1 ± 0.2 | 14.4 ± 0.6 | 16.9 ± 0.2 |
| LP1 | Pulmonary | 255.16 ± 9.87 | 97.6 ± 1.2 | 6 ± 0.2 | 4.4 ± 0.4 | 61.2 ± 0.2 |
| LP2 | Pulmonary | 257.63 ± 10.15 | 98.6 ± 1.4 | 7 ± 0.2 | 4.2 ± 0.5 | 61.4 ± 0.2 |
| LP3 | Pulmonary | 287.24 ± 11.29 | 109.9 ± 1.4 | 6.8 ± 0.2 | 4.4 ± 0.6 | 64.4 ± 0.2 |
AUC indicates the area under the blood levonorgestrel concentration time curve
7. Future Directions
Therapeutic efficacy of NLC and LP formulations are highly dependent upon their API, physicochemical properties and delivery format. Consistent across most pulmonary administered lipid nanoparticle systems is their ability to deliver therapeutic levels of their payload for sustained periods without over-exposing other major organs. A common trend to be weary of is optimization of key characteristics that govern mucociliary clearance, drug release kinetics, deep lung deposition and ability to maintain a reservoir of drug in BAL. Given the importance of controlled release kinetics in NLC/LP therapies, it is imperative that a balanced formulation is developed to ensure optimal saturation of API is stored in BAL whereas over- or under-saturation can lead to inflammatory risks and/or insufficient drug exposure. PK studies have been employed across a variety of diseases. However, PD data seems lacking in most investigations and require further attention. Meanwhile, understanding PD characteristics of various inhaled nanotechnology-based formulations may help in developing novel treatment strategies. Except few studies of PD of inhaled insulin [238], corticosteroids [239, 240] and some other drugs [241–245], PD of inhaled drugs is under investigated in most cases. Also, many studies collate one or two data sets which may include plasma, lung tissue or BAL drug concentrations. However, one could contend that a comprehensive approach and understanding of nanoparticle PK behavior in all 3 data fields is advantageous when optimizing the platform for additional therapeutic efficacy. Clinical applications should also be kept in mind as many variables and physicochemical characteristics are tailored for in-vivo rat/mice studies. Comprehensive approaches for translation of developed inhalation delivery systems into clinical applications should also be investigated. Tumor targeting efficiency can also be improved by the careful selection of the type of ligands for conjugation and/or innovative formulations (e.g. hierarchical liposomal system). Several complex and often stimuli-responsive structures (often defined as hierarchical) such as vesicles incorporating gas bubbles or nanoparticles in the lumen or membrane were developed in the last decade [246–249]. These carriers defy easy classification and require a separate more detail discussion. Overall, the pharmacokinetic profiles of LP, NLC and other nanosized vehicles show promise of localized application, enhancing therapeutic efficacy and warrant further investigation.
Acknowledgements
Funding: This work was supported in part by the National Institutes of Health [grant numbers: CA111766, HL118312, CA209818, CA238871].
Abbreviations
- 9-Br-Nos
9-bromo-noscapine
- 9-Br-Nos-RR-NLP
9-bromo-noscapine, rapid release, nano-lipid particle
- API
Active pharmaceutical ingredient
- AUC
Area under the curve
- AUC0–12
Area under the curve from t=0h to t=12h
- AUC0–t
Area under the curve from t=0 to t=t
- AUCinf
Area under the curve from t=0h extrapolated to t=infinity
- AUClast
Last collated time point for the measured area under the curve
- BAL
Bronchoalveolar lavage
- BALF
Bronchoalveolar lavage fluid
- CA IX
Carbonic anhydrase IX
- CL
Clearance
- Cmax
Highest reached concentration in time-concentration drug profile
- Cxb
Celecoxib
- D-DOX
56 kDa PEGylated polylysine dendrimer conjugated with doxorubicin via an acid labile linker
- DLCO
Diffusing capacity for carbon monoxide
- DLPC
1,2-dilauroyl-sn-glycero-3-phosphocholine
- dl-TPL-LP
Dual-ligand triptolide encapsulated liposome
- DPPC
14C-dipalmitoyl phosphatidylcholine
- DSPE
Distearoylphosphatidylethanolamine
- DTX
Docetaxel
- EGF
Epidermal growth factor
- ERA
Endothelin receptor antagonist
- FA
Folic acid
- Fabs
Absolute bioavailability
- FPF
Fine particle fraction
- HIF
hypoxia inducible factor
- IC50
A concentration that inhibits cell grows on 50%
- IHNP-HD
Inhaled NanoPac at high dosages
- IHNP-LD
Inhaled NanoPac at low dosages
- IT
Intratracheal
- IV
Intravenous
- ke
Elimination rate constant
- LC/MS
Liquid chromatography mass spectroscopy
- LHRH
Luteinizing hormone-releasing hormone
- LN
Levonorgestral
- LP
Liposome/Liposomal
- LT
Lung transplantation
- MMAD
Mass median aerodynamic diameter
- MPAP
Mean pulmonary arterial pressure
- MRT
Mean residence time
- MSAP
Mean systemic arterial pressure
- MTX
Methotrexate
- NLC
Nanostructured lipid carrier
- NLP
Nanostructured lipid particle
- NSCLC
Non-small cell lung cancer
- PAH
Pulmonary arterial hypertension
- PEG
Polyethylene glycol
- PEGxxxx
Polyethylene glycol polymer with molecular weight xxxx Da
- PK
Pharmacokinetic
- PLGA
Poly(lactic-co-glycolic acid)
- pMDI
Pressurized metered dose inhalers
- PTX
Paclitaxel
- QR
Quercetin
- RCRPC
Respirable controlled release polymeric colloid
- SEC
Size exclusion chromatography
- siRNA
Short-interfering RNA
- SLIT
Sustained release lipid inhalation targeting
- SLN
Solid lipid nanocarrier
- Soln
Solution
- t1/2
Half-life
- Te
Targeting efficiency
- TFR
Transferrin receptor
- Tmax
Time at which the highest drug concentration is reached
- TPL
triptolide
- Vd
Volume of distribution
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: None
References
- [1].Kuzmov A, Minko T, Nanotechnology approaches for inhalation treatment of lung diseases, J Control Release, 219 (2015) 500–518, 10.1016/j.jconrel.2015.07.024. [DOI] [PubMed] [Google Scholar]
- [2].Telko MJ, Hickey AJ, Dry powder inhaler formulation, Respir Care, 50 (2005) 1209–1227, [PubMed] [Google Scholar]
- [3].Martonen TB, Katz I, Deposition patterns of polydisperse aerosols within human lungs, Journal of Aerosol Medicine, 6 (1993) 251–274, 10.1089/jam.1993.6.251. [DOI] [Google Scholar]
- [4].Davies CN, Muir DC, Deposition of inhaled particles in human lungs, Nature, 211 (1966) 90–91, 10.1038/211090a0. [DOI] [PubMed] [Google Scholar]
- [5].Schulze D, Powders and bulk solids: Behavior, characterization, storage and flow., Springer, 2010, [Google Scholar]
- [6].Zhou QT, Armstrong B, Larson I, Stewart PJ, Morton DA, Understanding the influence of powder flowability, fluidization and de-agglomeration characteristics on the aerosolization of pharmaceutical model powders, Eur J Pharm Sci, 40 (2010) 412–421, 10.1016/j.ejps.2010.04.012. [DOI] [PubMed] [Google Scholar]
- [7].Stahlhofen W, Gebhart J, Heyder J, Biological variability of regional deposition of aerosol particles in the human respiratory tract, Am Ind Hyg Assoc J, 42 (1981) 348–352, 10.1080/15298668191419866. [DOI] [PubMed] [Google Scholar]
- [8].Sturm R, Hofmann W, A theoretical approach to the deposition and clearance of fibers with variable size in the human respiratory tract, J Hazard Mater, 170 (2009) 210–218, 10.1016/j.jhazmat.2009.04.107. [DOI] [PubMed] [Google Scholar]
- [9].Yeh HC, Schum GM, Models of human lung airways and their application to inhaled particle deposition, Bull Math Biol, 42 (1980) 461–480, 10.1007/bf02460796. [DOI] [PubMed] [Google Scholar]
- [10].Yang W, Peters JI, Williams RO 3rd, Inhaled nanoparticles--a current review, Int J Pharm, 356 (2008) 239–247, 10.1016/j.ijpharm.2008.02.011. [DOI] [PubMed] [Google Scholar]
- [11].Heyder J, Rudolf G, Mathematical models of particle deposition in the human respiratory tract, Journal of Aerosol Science, 15 (1984) 697–707, 10.1016/0021-8502(84)90007-7. [DOI] [Google Scholar]
- [12].Heyder J, Gebhart J, Rudolf G, Schiller CF, Stahlhofen W, Deposition of particles in the human respiratory tract in the size range 0.005–15 μm, Journal of Aerosol Science, 17 (1986) 811–825, 10.1016/0021-8502(86)90035-2. [DOI] [Google Scholar]
- [13].Lin YW, Wong J, Qu L, Chan HK, Zhou QT, Powder production and particle engineering for dry powder inhaler formulations, Curr Pharm Des, 21 (2015) 3902–3916, 10.2174/1381612821666150820111134. [DOI] [PubMed] [Google Scholar]
- [14].Edwards DA, Ben-Jebria A, Langer R, Recent advances in pulmonary drug delivery using large, porous inhaled particles, J Appl Physiol (1985), 85 (1998) 379–385, 10.1152/jappl.1998.85.2.379. [DOI] [PubMed] [Google Scholar]
- [15].Edwards DA, Hanes J, Caponetti G, Hrkach J, Ben-Jebria A, Eskew ML, Mintzes J, Deaver D, Lotan N, Langer R, Large porous particles for pulmonary drug delivery, Science, 276 (1997) 1868–1871, 10.1126/science.276.5320.1868. [DOI] [PubMed] [Google Scholar]
- [16].Sung JC, Pulliam BL, Edwards DA, Nanoparticles for drug delivery to the lungs, Trends Biotechnol, 25 (2007) 563–570, 10.1016/j.tibtech.2007.09.005. [DOI] [PubMed] [Google Scholar]
- [17].Vanbever R, Mintzes JD, Wang J, Nice J, Chen D, Batycky R, Langer R, Edwards DA, Formulation and physical characterization of large porous particles for inhalation, Pharm Res, 16 (1999) 1735–1742, 10.1023/a:1018910200420. [DOI] [PubMed] [Google Scholar]
- [18].Steckel H, Brandes HG, A novel spray-drying technique to produce low density particles for pulmonary delivery, Int J Pharm, 278 (2004) 187–195, 10.1016/j.ijpharm.2004.03.010. [DOI] [PubMed] [Google Scholar]
- [19].Dellamary LA, Tarara TE, Smith DJ, Woelk CH, Adractas A, Costello ML, Gill H, Weers JG, Hollow porous particles in metered dose inhalers, Pharm Res, 17 (2000) 168–174, 10.1023/a:1007513213292. [DOI] [PubMed] [Google Scholar]
- [20].Tsapis N, Bennett D, Jackson B, Weitz DA, Edwards DA, Trojan particles: Large porous carriers of nanoparticles for drug delivery, Proc Natl Acad Sci U S A, 99 (2002) 12001–12005, 10.1073/pnas.182233999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hadinoto K, Phanapavudhikul P, Kewu Z, Tan RB, Dry powder aerosol delivery of large hollow nanoparticulate aggregates as prospective carriers of nanoparticulate drugs: Effects of phospholipids, Int J Pharm, 333 (2007) 187–198, 10.1016/j.ijpharm.2006.10.009. [DOI] [PubMed] [Google Scholar]
- [22].Hadinoto K, Zhu K, Tan RB, Drug release study of large hollow nanoparticulate aggregates carrier particles for pulmonary delivery, Int J Pharm, 341 (2007) 195–206, 10.1016/j.ijpharm.2007.03.035. [DOI] [PubMed] [Google Scholar]
- [23].Taratula O, Garbuzenko OB, Chen AM, Minko T, Innovative strategy for treatment of lung cancer: Targeted nanotechnology-based inhalation co-delivery of anticancer drugs and sirna, J Drug Target, 19 (2011) 900–914, 10.3109/1061186X.2011.622404. [DOI] [PubMed] [Google Scholar]
- [24].Shah V, Taratula O, Garbuzenko OB, Taratula OR, Rodriguez-Rodriguez L, Minko T, Targeted nanomedicine for suppression of cd44 and simultaneous cell death induction in ovarian cancer: An optimal delivery of sirna and anticancer drug, Clin Cancer Res, 19 (2013) 6193–6204, 10.1158/1078-0432.CCR-13-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dharap SS, Minko T, Targeted proapoptotic lhrh-bh3 peptide, Pharm Res, 20 (2003) 889–896, 10.1023/a:1023839319950. [DOI] [PubMed] [Google Scholar]
- [26].Dharap SS, Qiu B, Williams GC, Sinko P, Stein S, Minko T, Molecular targeting of drug delivery systems to ovarian cancer by bh3 and lhrh peptides, J Control Release, 91 (2003) 61–73, 10.1016/s0168-3659(03)00209-8. [DOI] [PubMed] [Google Scholar]
- [27].Dharap SS, Wang Y, Chandna P, Khandare JJ, Qiu B, Gunaseelan S, Sinko PJ, Stein S, Farmanfarmaian A, Minko T, Tumor-specific targeting of an anticancer drug delivery system by lhrh peptide, Proc Natl Acad Sci U S A, 102 (2005) 12962–12967, 10.1073/pnas.0504274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tseng CL, Wang TW, Dong GC, Yueh-Hsiu Wu S, Young TH, Shieh MJ, Lou PJ, Lin FH, Development of gelatin nanoparticles with biotinylated egf conjugation for lung cancer targeting, Biomaterials, 28 (2007) 3996–4005, 10.1016/j.biomaterials.2007.05.006. [DOI] [PubMed] [Google Scholar]
- [29].Tseng CL, Wu SY, Wang WH, Peng CL, Lin FH, Lin CC, Young TH, Shieh MJ, Targeting efficiency and biodistribution of biotinylated-egf-conjugated gelatin nanoparticles administered via aerosol delivery in nude mice with lung cancer, Biomaterials, 29 (2008) 3014–3022, 10.1016/j.biomaterials.2008.03.033. [DOI] [PubMed] [Google Scholar]
- [30].Sadhukha T, Wiedmann TS, Panyam J, Inhalable magnetic nanoparticles for targeted hyperthermia in lung cancer therapy, Biomaterials, 34 (2013) 5163–5171, 10.1016/j.biomaterials.2013.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Haque S, McLeod VM, Jones S, Fung S, Whittaker M, McIntosh M, Pouton C, Owen DJ, Porter CJH, Kaminskas LM, Effect of increased surface hydrophobicity via drug conjugation on the clearance of inhaled pegylated polylysine dendrimers, Eur J Pharm Biopharm, 119 (2017) 408–418, 10.1016/j.ejpb.2017.07.005. [DOI] [PubMed] [Google Scholar]
- [32].Hafez IM, Cullis PR, Roles of lipid polymorphism in intracellular delivery, Advanced Drug Delivery Reviews, 47 (2001) 139–148, 10.1016/s0169-409x(01)00103-x. [DOI] [PubMed] [Google Scholar]
- [33].Cevc G, Richardsen H, Lipid vesicles and membrane fusion, Advanced Drug Delivery Reviews, 38 (1999) 207–232, 10.1016/s0169-409x(99)00030-7. [DOI] [PubMed] [Google Scholar]
- [34].Andresen TL, Jensen SS, Jorgensen K, Advanced strategies in liposomal cancer therapy: Problems and prospects of active and tumor specific drug release, Prog Lipid Res, 44 (2005) 68–97, 10.1016/j.plipres.2004.12.001. [DOI] [PubMed] [Google Scholar]
- [35].Saad M, Garbuzenko OB, Ber E, Chandna P, Khandare JJ, Pozharov VP, Minko T, Receptor targeted polymers, dendrimers, liposomes: Which nanocarrier is the most efficient for tumor-specific treatment and imaging?, J Control Release, 130 (2008) 107–114, 10.1016/j.jconrel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Saad M, Garbuzenko OB, Minko T, Co-delivery of sirna and an anticancer drug for treatment of multidrug-resistant cancer, Nanomedicine (Lond), 3 (2008) 761–776, 10.2217/17435889.3.6.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Moller W, Haussinger K, Winkler-Heil R, Stahlhofen W, Meyer T, Hofmann W, Heyder J, Mucociliary and long-term particle clearance in the airways of healthy nonsmoker subjects, J Appl Physiol (1985), 97 (2004) 2200–2206, 10.1152/japplphysiol.00970.2003. [DOI] [PubMed] [Google Scholar]
- [38].Kreyling WG, Semmler-Behnke M, Moller W, Ultrafine particle-lung interactions: Does size matter?, J Aerosol Med, 19 (2006) 74–83, 10.1089/jam.2006.19.74. [DOI] [PubMed] [Google Scholar]
- [39].Zhang J, Wu L, Chan HK, Watanabe W, Formation, characterization, and fate of inhaled drug nanoparticles, Adv Drug Deliv Rev, 63 (2011) 441–455, 10.1016/j.addr.2010.11.002. [DOI] [PubMed] [Google Scholar]
- [40].Bur M, Henning A, Hein S, Schneider M, Lehr CM, Inhalative nanomedicine-- opportunities and challenges, Inhal Toxicol, 21 Suppl 1 (2009) 137–143, 10.1080/08958370902962283. [DOI] [PubMed] [Google Scholar]
- [41].Tabata Y, Ikada Y, Macrophage phagocytosis of biodegradable microspheres composed of l-lactic acid/glycolic acid homo- and copolymers, J Biomed Mater Res, 22 (1988) 837–858, 10.1002/jbm.820221002. [DOI] [PubMed] [Google Scholar]
- [42].Rudt S, Wesemeyer H, Müller RH, In vitro phagocytosis assay of nano- and microparticles by chemiluminescence. Iv. Effect of surface modification by coating of particles with poloxamine and antarox co on the phagocytic uptake, Journal of Controlled Release, 25 (1993) 123–132, 10.1016/0168-3659(93)90101-a. [DOI] [Google Scholar]
- [43].Rudt S, Müller RH, In vitro phagocytosis assay of nano- and microparticles by chemiluminescence. I. Effect of analytical parameters, particle size and particle concentration, Journal of Controlled Release, 22 (1992) 263–271, 10.1016/0168-3659(92)90101-v. [DOI] [Google Scholar]
- [44].Kawaguchi H, Koiwai N, Ohtsuka Y, Miyamoto M, Sasakawa S, Phagocytosis of latex particles by leucocytes. I. Dependence of phagocytosis on the size and surface potential of particles, Biomaterials, 7 (1986) 61–66, 10.1016/0142-9612(86)90091-8. [DOI] [PubMed] [Google Scholar]
- [45].Lippmann M, Yeates DB, Albert RE, Deposition, retention, and clearance of inhaled particles, Br J Ind Med, 37 (1980) 337–362, 10.1136/oem.37.4.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Langenback EG, Bergofsky EH, Halpern JG, Foster WM, Supramicron-sized particle clearance from alveoli: Route and kinetics, J Appl Physiol (1985), 69 (1990) 1302–1308, 10.1152/jappl.1990.69.4.1302. [DOI] [PubMed] [Google Scholar]
- [47].Geiser M, Morphological aspects of particle uptake by lung phagocytes, Microsc Res Tech, 57 (2002) 512–522, 10.1002/jemt.10105. [DOI] [PubMed] [Google Scholar]
- [48].Ferin J, Pulmonary alveolar pores and alveolar macrophage-mediated particle clearance, Anat Rec, 203 (1982) 265–272, 10.1002/ar.1092030208. [DOI] [PubMed] [Google Scholar]
- [49].Schmid O, Moller W, Semmler-Behnke M, Ferron GA, Karg E, Lipka J, Schulz H, Kreyling WG, Stoeger T, Dosimetry and toxicology of inhaled ultrafine particles, Biomarkers, 14 Suppl 1 (2009) 67–73, 10.1080/13547500902965617. [DOI] [PubMed] [Google Scholar]
- [50].Borm P, Klaessig FC, Landry TD, Moudgil B, Pauluhn J, Thomas K, Trottier R, Wood S, Research strategies for safety evaluation of nanomaterials, part v: Role of dissolution in biological fate and effects of nanoscale particles, Toxicol Sci, 90 (2006) 23–32, 10.1093/toxsci/kfj084. [DOI] [PubMed] [Google Scholar]
- [51].Madl AK, Pinkerton KE, Health effects of inhaled engineered and incidental nanoparticles, Crit Rev Toxicol, 39 (2009) 629–658, 10.1080/10408440903133788. [DOI] [PubMed] [Google Scholar]
- [52].Patton JS, Byron PR, Inhaling medicines: Delivering drugs to the body through the lungs, Nat Rev Drug Discov, 6 (2007) 67–74, 10.1038/nrd2153. [DOI] [PubMed] [Google Scholar]
- [53].Geiser M, Schurch S, Gehr P, Influence of surface chemistry and topography of particles on their immersion into the lung’s surface-lining layer, J Appl Physiol (1985), 94 (2003) 1793–1801, 10.1152/japplphysiol.00514.2002. [DOI] [PubMed] [Google Scholar]
- [54].Goerke J, Pulmonary surfactant: Functions and molecular composition, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, 1408 (1998) 79–89, 10.1016/s0925-4439(98)00060-x. [DOI] [PubMed] [Google Scholar]
- [55].Patlolla RR, Chougule M, Patel AR, Jackson T, Tata PN, Singh M, Formulation, characterization and pulmonary deposition of nebulized celecoxib encapsulated nanostructured lipid carriers, J Control Release, 144 (2010) 233–241, 10.1016/j.jconrel.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rogueda PG, Traini D, The nanoscale in pulmonary delivery. Part 2: Formulation platforms, Expert Opin Drug Deliv, 4 (2007) 607–620, 10.1517/17425247.4.6.607. [DOI] [PubMed] [Google Scholar]
- [57].Zhou QT, Tang P, Leung SS, Chan JG, Chan HK, Emerging inhalation aerosol devices and strategies: Where are we headed?, Adv Drug Deliv Rev, 75 (2014) 3–17, 10.1016/j.addr.2014.03.006. [DOI] [PubMed] [Google Scholar]
- [58].Mainelis G, Seshadri S, Garbuzenko OB, Han T, Wang Z, Minko T, Characterization and application of a nose-only exposure chamber for inhalation delivery of liposomal drugs and nucleic acids to mice, J Aerosol Med Pulm Drug Deliv, 26 (2013) 345–354, 10.1089/jamp.2011-0966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hureaux J, Lagarce F, Gagnadoux F, Vecellio L, Clavreul A, Roger E, Kempf M, Racineux JL, Diot P, Benoit JP, Urban T, Lipid nanocapsules: Ready-to-use nanovectors for the aerosol delivery of paclitaxel, Eur J Pharm Biopharm, 73 (2009) 239–246, 10.1016/j.ejpb.2009.06.013. [DOI] [PubMed] [Google Scholar]
- [60].Conti DS, Brewer D, Grashik J, Avasarala S, da Rocha SR, Poly(amidoamine) dendrimer nanocarriers and their aerosol formulations for sirna delivery to the lung epithelium, Mol Pharm, 11 (2014) 1808–1822, 10.1021/mp4006358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Newman SP, Therapeutic inhalation agents and devices. Effectiveness in asthma and bronchitis, Postgrad Med, 76 (1984) 194–203, 206–197, 10.1080/00325481.1984.11698763. [DOI] [PubMed] [Google Scholar]
- [62].Bennett WD, Smaldone GC, Human variation in the peripheral air-space deposition of inhaled particles, J Appl Physiol (1985), 62 (1987) 1603–1610, 10.1152/jappl.1987.62.4.1603. [DOI] [PubMed] [Google Scholar]
- [63].Crompton GK, Problems patients have using pressurized aerosol inhalers, Eur J Respir Dis Suppl, 119 (1982) 101–104, [PubMed] [Google Scholar]
- [64].Newman SP, Pavia D, Moren F, Sheahan NF, Clarke SW, Deposition of pressurised aerosols in the human respiratory tract, Thorax, 36 (1981) 52–55, 10.1136/thx.36.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chacón M, Molpeceres J, Berges L, Guzmán M, Aberturas MR, Stability and freeze-drying of cyclosporine loaded poly(d,l lactide–glycolide) carriers, European Journal of Pharmaceutical Sciences, 8 (1999) 99–107, 10.1016/s0928-0987(98)00066-9. [DOI] [PubMed] [Google Scholar]
- [66].Abdelwahed W, Degobert G, Stainmesse S, Fessi H, Freeze-drying of nanoparticles: Formulation, process and storage considerations, Adv Drug Deliv Rev, 58 (2006) 1688–1713, 10.1016/j.addr.2006.09.017. [DOI] [PubMed] [Google Scholar]
- [67].Mangal S, Gao W, Li T, Zhou QT, Pulmonary delivery of nanoparticle chemotherapy for the treatment of lung cancers: Challenges and opportunities, Acta Pharmacol Sin, 38 (2017) 782–797, 10.1038/aps.2017.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Desai N, Challenges in development of nanoparticle-based therapeutics, AAPS J, 14 (2012) 282–295, 10.1208/s12248-012-9339-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Todoroff J, Vanbever R, Fate of nanomedicines in the lungs, Current Opinion in Colloid & Interface Science, 16 (2011) 246–254, 10.1016/j.cocis.2011.03.001. [DOI] [Google Scholar]
- [70].Saint-Lorant G, Leterme P, Gayot A, Flament MP, Influence of carrier on the performance of dry powder inhalers, Int J Pharm, 334 (2007) 85–91, 10.1016/j.ijpharm.2006.10.028. [DOI] [PubMed] [Google Scholar]
- [71].Grasmeijer F, Grasmeijer N, Hagedoorn P, Frijlink HW, Haaije de Boer A, Recent advances in the fundamental understanding of adhesive mixtures for inhalation, Curr Pharm Des, 21 (2015) 5900–5914, 10.2174/1381612821666151008124622. [DOI] [PubMed] [Google Scholar]
- [72].Bohr A, Water J, Beck-Broichsitter M, Yang M, Nanoembedded microparticles for stabilization and delivery of drug-loaded nanoparticles, Curr Pharm Des, 21 (2015) 5829–5844, 10.2174/1381612821666151008124322. [DOI] [PubMed] [Google Scholar]
- [73].Yamasaki K, Kwok PC, Fukushige K, Prud’homme RK, Chan HK, Enhanced dissolution of inhalable cyclosporine nano-matrix particles with mannitol as matrix former, Int J Pharm, 420 (2011) 34–42, 10.1016/j.ijpharm.2011.08.010. [DOI] [PubMed] [Google Scholar]
- [74].Kwok PC, Tunsirikongkon A, Glover W, Chan HK, Formation of protein nano-matrix particles with controlled surface architecture for respiratory drug delivery, Pharm Res, 28 (2011) 788–796, 10.1007/s11095-010-0332-2. [DOI] [PubMed] [Google Scholar]
- [75].Mangal S, Meiser F, Tan G, Gengenbach T, Denman J, Rowles MR, Larson I, Morton DA, Relationship between surface concentration of l-leucine and bulk powder properties in spray dried formulations, Eur J Pharm Biopharm, 94 (2015) 160–169, 10.1016/j.ejpb.2015.04.035. [DOI] [PubMed] [Google Scholar]
- [76].Mangal S, Nie H, Xu R, Guo R, Cavallaro A, Zemlyanov D, Zhou QT, Physico-chemical properties, aerosolization and dissolution of co-spray dried azithromycin particles with l-leucine for inhalation, Pharm Res, 35 (2018) 28, 10.1007/s11095-017-2334-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].El-Gendy N, Berkland C, Combination chemotherapeutic dry powder aerosols via controlled nanoparticle agglomeration, Pharm Res, 26 (2009) 1752–1763, 10.1007/s11095-009-9886-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Varshosaz J, Hassanzadeh F, Mardani A, Rostami M, Feasibility of haloperidol-anchored albumin nanoparticles loaded with doxorubicin as dry powder inhaler for pulmonary delivery, Pharm Dev Technol, 20 (2015) 183–196, 10.3109/10837450.2013.852576. [DOI] [PubMed] [Google Scholar]
- [79].Ely L, Roa W, Finlay WH, Lobenberg R, Effervescent dry powder for respiratory drug delivery, Eur J Pharm Biopharm, 65 (2007) 346–353, 10.1016/j.ejpb.2006.10.021. [DOI] [PubMed] [Google Scholar]
- [80].Azarmi S, Lobenberg R, Roa WH, Tai S, Finlay WH, Formulation and in vivo evaluation of effervescent inhalable carrier particles for pulmonary delivery of nanoparticles, Drug Dev Ind Pharm, 34 (2008) 943–947, 10.1080/03639040802149079. [DOI] [PubMed] [Google Scholar]
- [81].Al-Hallak MH, Sarfraz MK, Azarmi S, Roa WH, Finlay WH, Rouleau C, Lobenberg R, Distribution of effervescent inhalable nanoparticles after pulmonary delivery: An in vivo study, Ther Deliv, 3 (2012) 725–734, 10.4155/tde.12.42. [DOI] [PubMed] [Google Scholar]
- [82].Roa WH, Azarmi S, Al-Hallak MH, Finlay WH, Magliocco AM, Lobenberg R, Inhalable nanoparticles, a non-invasive approach to treat lung cancer in a mouse model, J Control Release, 150 (2011) 49–55, 10.1016/j.jconrel.2010.10.035. [DOI] [PubMed] [Google Scholar]
- [83].Kaminskas LM, Kelly BD, McLeod VM, Sberna G, Owen DJ, Boyd BJ, Porter CJ, Characterisation and tumour targeting of pegylated polylysine dendrimers bearing doxorubicin via a ph labile linker, J Control Release, 152 (2011) 241–248, 10.1016/j.jconrel.2011.02.005. [DOI] [PubMed] [Google Scholar]
- [84].Kaminskas LM, McLeod VM, Ascher DB, Ryan GM, Jones S, Haynes JM, Trevaskis NL, Chan LJ, Sloan EK, Finnin BA, Williamson M, Velkov T, Williams ED, Kelly BD, Owen DJ, Porter CJ, Methotrexate-conjugated pegylated dendrimers show differential patterns of deposition and activity in tumor-burdened lymph nodes after intravenous and subcutaneous administration in rats, Mol Pharm, 12 (2015) 432–443, 10.1021/mp500531e. [DOI] [PubMed] [Google Scholar]
- [85].Kaminskas LM, McLeod VM, Ryan GM, Kelly BD, Haynes JM, Williamson M, Thienthong N, Owen DJ, Porter CJ, Pulmonary administration of a doxorubicin-conjugated dendrimer enhances drug exposure to lung metastases and improves cancer therapy, J Control Release, 183 (2014) 18–26, 10.1016/j.jconrel.2014.03.012. [DOI] [PubMed] [Google Scholar]
- [86].Ryan GM, Kaminskas LM, Kelly BD, Owen DJ, McIntosh MP, Porter CJ, Pulmonary administration of pegylated polylysine dendrimers: Absorption from the lung versus retention within the lung is highly size-dependent, Mol Pharm, 10 (2013) 2986–2995, 10.1021/mp400091n. [DOI] [PubMed] [Google Scholar]
- [87].Patton JS, Fishburn CS, Weers JG, The lungs as a portal of entry for systemic drug delivery, Proc Am Thorac Soc, 1 (2004) 338–344, 10.1513/pats.200409-049TA. [DOI] [PubMed] [Google Scholar]
- [88].Bailey MM, Berkland CJ, Nanoparticle formulations in pulmonary drug delivery, Med Res Rev, 29 (2009) 196–212, 10.1002/med.20140. [DOI] [PubMed] [Google Scholar]
- [89].Folkesson HG, Westrom BR, Dahlback M, Lundin S, Karlsson BW, Passage of aerosolized bsa and the nona-peptide ddavp via the respiratory tract in young and adult rats, Exp Lung Res, 18 (1992) 595–614, 10.3109/01902149209031697. [DOI] [PubMed] [Google Scholar]
- [90].Kaminskas LM, Boyd BJ, Karellas P, Krippner GY, Lessene R, Kelly B, Porter CJ, The impact of molecular weight and peg chain length on the systemic pharmacokinetics of pegylated poly l-lysine dendrimers, Mol Pharm, 5 (2008) 449–463, 10.1021/mp7001208. [DOI] [PubMed] [Google Scholar]
- [91].Lee KC, Chae SY, Kim TH, Lee S, Lee ES, Youn YS, Intrapulmonary potential of polyethylene glycol-modified glucagon-like peptide-1s as a type 2 anti-diabetic agent, Regul Pept, 152 (2009) 101–107, 10.1016/j.regpep.2008.09.008. [DOI] [PubMed] [Google Scholar]
- [92].Gagnadoux F, Hureaux J, Vecellio L, Urban T, Le Pape A, Valo I, Montharu J, Leblond V, Boisdron-Celle M, Lerondel S, Majoral C, Diot P, Racineux JL, Lemarie E, Aerosolized chemotherapy, Journal of Aerosol Medicine, 0 (2008) 080207080519480–080207080519489, 10.1089/jam.2007.0656. [DOI] [PubMed] [Google Scholar]
- [93].Zarogoulidis P, Eleftheriadou E, Sapardanis I, Zarogoulidou V, Lithoxopoulou H, Kontakiotis T, Karamanos N, Zachariadis G, Mabroudi M, Zisimopoulos A, Zarogoulidis K, Feasibility and effectiveness of inhaled carboplatin in nsclc patients, Invest New Drugs, 30 (2012) 1628–1640, 10.1007/s10637-011-9714-5. [DOI] [PubMed] [Google Scholar]
- [94].Zarogoulidis P, Giraleli C, Karamanos NK, Inhaled chemotherapy in lung cancer: Safety concerns of nanocomplexes delivered, Ther Deliv, 3 (2012) 1021–1023, 10.4155/tde.12.77. [DOI] [PubMed] [Google Scholar]
- [95].Kaminskas LM, McLeod VM, Porter CJ, Boyd BJ, Association of chemotherapeutic drugs with dendrimer nanocarriers: An assessment of the merits of covalent conjugation compared to noncovalent encapsulation, Mol Pharm, 9 (2012) 355–373, 10.1021/mp2005966. [DOI] [PubMed] [Google Scholar]
- [96].Otterson GA, Villalona-Calero MA, Sharma S, Kris MG, Imondi A, Gerber M, White DA, Ratain MJ, Schiller JH, Sandler A, Kraut M, Mani S, Murren JR, Phase i study of inhaled doxorubicin for patients with metastatic tumors to the lungs, Clin Cancer Res, 13 (2007) 1246–1252, 10.1158/1078-0432.CCR-06-1096. [DOI] [PubMed] [Google Scholar]
- [97].Otterson GA, Villalona-Calero MA, Hicks W, Pan X, Ellerton JA, Gettinger SN, Murren JR, Phase i/ii study of inhaled doxorubicin combined with platinum-based therapy for advanced non-small cell lung cancer, Clin Cancer Res, 16 (2010) 2466–2473, 10.1158/1078-0432.CCR-09-3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Wittgen BP, Kunst PW, van der Born K, van Wijk AW, Perkins W, Pilkiewicz FG, Perez-Soler R, Nicholson S, Peters GJ, Postmus PE, Phase i study of aerosolized slit cisplatin in the treatment of patients with carcinoma of the lung, Clin Cancer Res, 13 (2007) 2414–2421, 10.1158/1078-0432.CCR-06-1480. [DOI] [PubMed] [Google Scholar]
- [99].Dutcher JP, Gaynor ER, Boldt DH, Doroshow JH, Bar MH, Sznol M, Mier J, Sparano J, Fisher RI, Weiss G, et al. , A phase ii study of high-dose continuous infusion interleukin-2 with lymphokine-activated killer cells in patients with metastatic melanoma, J Clin Oncol, 9 (1991) 641–648, 10.1200/JCO.1991.9.4.641. [DOI] [PubMed] [Google Scholar]
- [100].Eberlein TJ, Schoof DD, The role of interleukin-2 in cancer immunotherapy, Compr Ther, 17 (1991) 49–56, [PubMed] [Google Scholar]
- [101].Christiansen NP, Kennedy BJ, Ochoa AC, Skubitz KM, Bach FH, Continuous infusion of recombinant interleukin-2 and lymphokine-activated killer cells in refractory malignancies, Med Pediatr Oncol, 17 (1989) 455–458, 10.1002/mpo.2950170521. [DOI] [PubMed] [Google Scholar]
- [102].Siegel JP, Puri RK, Interleukin-2 toxicity, J Clin Oncol, 9 (1991) 694–704, 10.1200/JCO.1991.9.4.694. [DOI] [PubMed] [Google Scholar]
- [103].Whittington R, Faulds D, Interleukin-2. A review of its pharmacological properties and therapeutic use in patients with cancer, Drugs, 46 (1993) 446–514, 10.2165/00003495-199346030-00009. [DOI] [PubMed] [Google Scholar]
- [104].Christiansen NP, Skubitz KM, Nath K, Ochoa A, Kennedy BJ, Nephrotoxicity of continuous intravenous infusion of recombinant interleukin-2, The American Journal of Medicine, 84 (1988) 1072–1075, 10.1016/0002-9343(88)90314-2. [DOI] [PubMed] [Google Scholar]
- [105].Khanna C, Hasz DE, Klausner JS, Anderson PM, Aerosol delivery of interleukin 2 liposomes is nontoxic and biologically effective: Canine studies, Clin Cancer Res, 2 (1996) 721–734, [PubMed] [Google Scholar]
- [106].Khanna C, Anderson PM, Hasz DE, Katsanis E, Neville M, Klausner JS, Interleukin-2 liposome inhalation therapy is safe and effective for dogs with spontaneous pulmonary metastases, Cancer, 79 (1997) 1409–1421, 10.1002/(sici)1097-0142(19970401)79:7<1409::aid-cncr19>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- [107].Khanna C, Waldrep JC, Anderson PM, Weischelbaum RW, Hasz DE, Katsanis E, Klausner JS, Nebulized interleukin 2 liposomes: Aerosol characteristics and biodistribution, J Pharm Pharmacol, 49 (1997) 960–971, 10.1111/j.2042-7158.1997.tb06024.x. [DOI] [PubMed] [Google Scholar]
- [108].Anderson PM, Hasz D, Dickrell L, Sencer S, Interleukin-2 in liposomes: Increased intravenous potency and less pulmonary toxicity in the rat, Drug Development Research, 27 (1992) 15–31, 10.1002/ddr.430270103. [DOI] [Google Scholar]
- [109].Anderson PM, Sorenson MA, Effects of route and formulation on clinical pharmacokinetics of interleukin-2, Clin Pharmacokinet, 27 (1994) 19–31, 10.2165/00003088-199427010-00003. [DOI] [PubMed] [Google Scholar]
- [110].Smith KA, Lowest dose interleukin-2 immunotherapy [see comments], Blood, 81 (1993) 1414–1423, 10.1182/blood.V81.6.1414.bloodjournal8161414. [DOI] [PubMed] [Google Scholar]
- [111].Lorenz J, Wilhelm K, Kessler M, Peschel C, Schwulera U, Lissner R, Struff WG, Huland E, Huber C, Aulitzky WE, Phase i trial of inhaled natural interleukin 2 for treatment of pulmonary malignancy: Toxicity, pharmacokinetics, and biological effects, Clin Cancer Res, 2 (1996) 1115–1122, [PubMed] [Google Scholar]
- [112].Zissel G, Aulitzky WE, Lorenz J, Huber C, Muller-Quernheim J, Induction of accessory cell function of human alveolar macrophages by inhalation of human natural interleukin-2, Cancer Immunol Immunother, 42 (1996) 122–126, 10.1007/s002620050261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Skubitz KM, Anderson PM, Inhalational interleukin-2 liposomes for pulmonary metastases: A phase i clinical trial, Anticancer Drugs, 11 (2000) 555–563, 10.1097/00001813-200008000-00006. [DOI] [PubMed] [Google Scholar]
- [114].Jyoti K, Kaur K, Pandey RS, Jain UK, Chandra R, Madan J, Inhalable nanostructured lipid particles of 9-bromo-noscapine, a tubulin-binding cytotoxic agent: In vitro and in vivo studies, J Colloid Interface Sci, 445 (2015) 219–230, 10.1016/j.jcis.2014.12.092. [DOI] [PubMed] [Google Scholar]
- [115].Zhou J, Gupta K, Aggarwal S, Aneja R, Chandra R, Panda D, Joshi HC, Brominated derivatives of noscapine are potent microtubule-interfering agents that perturb mitosis and inhibit cell proliferation, Mol Pharmacol, 63 (2003) 799–807, 10.1124/mol.63.4.799. [DOI] [PubMed] [Google Scholar]
- [116].Madan J, Baruah B, Nagaraju M, Abdalla MO, Yates C, Turner T, Rangari V, Hamelberg D, Aneja R, Molecular cycloencapsulation augments solubility and improves therapeutic index of brominated noscapine in prostate cancer cells, Mol Pharm, 9 (2012) 1470–1480, 10.1021/mp300063v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Gallagher JE, George G, Brody AR, Sialic acid mediates the initial binding of positively charged inorganic particles to alveolar macrophage membranes, Am Rev Respir Dis, 135 (1987) 1345–1352, 10.1164/arrd.1987.135.6.1345. [DOI] [PubMed] [Google Scholar]
- [118].Bartneck M, Keul HA, Singh S, Czaja K, Bornemann J, Bockstaller M, Moeller M, Zwadlo-Klarwasser G, Groll J, Rapid uptake of gold nanorods by primary human blood phagocytes and immunomodulatory effects of surface chemistry, ACS Nano, 4 (2010) 3073–3086, 10.1021/nn100262h. [DOI] [PubMed] [Google Scholar]
- [119].Azarmi S, Roa WH, Lobenberg R, Targeted delivery of nanoparticles for the treatment of lung diseases, Adv Drug Deliv Rev, 60 (2008) 863–875, 10.1016/j.addr.2007.11.006. [DOI] [PubMed] [Google Scholar]
- [120].Liu J, Shapiro JI, Endocytosis and signal transduction: Basic science update, Biol Res Nurs, 5 (2003) 117–128, 10.1177/1099800403256860. [DOI] [PubMed] [Google Scholar]
- [121].Rivolta I, Panariti A, Lettiero B, Sesana S, Gasco P, Gasco MR, Masserini M, Miserocchi G, Cellular uptake of coumarin-6 as a model drug loaded in solid lipid nanoparticles, J Physiol Pharmacol, 62 (2011) 45–53, [PubMed] [Google Scholar]
- [122].Yang Y, Corona A 3rd, Henson MA, Experimental investigation and population balance equation modeling of solid lipid nanoparticle aggregation dynamics, J Colloid Interface Sci, 374 (2012) 297–307, 10.1016/j.jcis.2012.02.024. [DOI] [PubMed] [Google Scholar]
- [123].Hitzman CJ, Elmquist WF, Wattenberg LW, Wiedmann TS, Development of a respirable, sustained release microcarrier for 5-fluorouracil i: In vitro assessment of liposomes, microspheres, and lipid coated nanoparticles, J Pharm Sci, 95 (2006) 1114–1126, 10.1002/jps.20591. [DOI] [PubMed] [Google Scholar]
- [124].Chougule MB, Padhi BK, Misra A, Nano-liposomal dry powder inhaler of amiloride hydrochloride, J Nanosci Nanotechnol, 6 (2006) 3001–3009, 10.1166/jnn.2006.405. [DOI] [PubMed] [Google Scholar]
- [125].Videira MA, Botelho MF, Santos AC, Gouveia LF, de Lima JJ, Almeida AJ, Lymphatic uptake of pulmonary delivered radiolabelled solid lipid nanoparticles, J Drug Target, 10 (2002) 607–613, 10.1080/1061186021000054933. [DOI] [PubMed] [Google Scholar]
- [126].Reddy PR, Venkateswarlu V, Pharmacokinetics and tissue distribution of etoposide delivered in long circulating parenteral emulsion, J Drug Target, 13 (2005) 543–553, 10.1080/10611860500403156. [DOI] [PubMed] [Google Scholar]
- [127].Muller RH, Radtke M, Wissing SA, Solid lipid nanoparticles (sln) and nanostructured lipid carriers (nlc) in cosmetic and dermatological preparations, Adv Drug Deliv Rev, 54 Suppl 1 (2002) S131–155, 10.1016/s0169-409x(02)00118-7. [DOI] [PubMed] [Google Scholar]
- [128].Hida T, Kozaki K, Muramatsu H, Masuda A, Shimizu S, Mitsudomi T, Sugiura T, Ogawa M, Takahashi T, Cyclooxygenase-2 inhibitor induces apoptosis and enhances cytotoxicity of various anticancer agents in non-small cell lung cancer cell lines, Clin Cancer Res, 6 (2000) 2006–2011, [PubMed] [Google Scholar]
- [129].Shishodia S, Koul D, Aggarwal BB, Cyclooxygenase (cox)-2 inhibitor celecoxib abrogates tnf-induced nf-kappa b activation through inhibition of activation of i kappa b alpha kinase and akt in human non-small cell lung carcinoma: Correlation with suppression of cox-2 synthesis, J Immunol, 173 (2004) 2011–2022, 10.4049/jimmunol.173.3.2011. [DOI] [PubMed] [Google Scholar]
- [130].Mao JT, Roth MD, Serio KJ, Baratelli F, Zhu L, Holmes EC, Strieter RM, Dubinett SM, Celecoxib modulates the capacity for prostaglandin e2 and interleukin-10 production in alveolar macrophages from active smokers, Clin Cancer Res, 9 (2003) 5835–5841, [PubMed] [Google Scholar]
- [131].Lauweryns JM, Baert JH, Alveolar clearance and the role of the pulmonary lymphatics, Am Rev Respir Dis, 115 (1977) 625–683, 10.1164/arrd.1977.115.4.625. [DOI] [PubMed] [Google Scholar]
- [132].Verco J, Johnston W, Frost M, Baltezor M, Kuehl PJ, Lopez A, Gigliotti A, Belinsky SA, Wolff R, diZerega G, Inhaled submicron particle paclitaxel (nanopac) induces tumor regression and immune cell infiltration in an orthotopic athymic nude rat model of non-small cell lung cancer, J Aerosol Med Pulm Drug Deliv, 32 (2019) 266–277, 10.1089/jamp.2018.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Torchilin VP, Lipid-core micelles for targeted drug delivery, Curr Drug Deliv, 2 (2005) 319–327, 10.2174/156720105774370221. [DOI] [PubMed] [Google Scholar]
- [134].Croy SR, Kwon GS, Polymeric micelles for drug delivery, Curr Pharm Des, 12 (2006) 4669–4684, 10.2174/138161206779026245. [DOI] [PubMed] [Google Scholar]
- [135].Dabholkar RD, Sawant RM, Mongayt DA, Devarajan PV, Torchilin VP, Polyethylene glycol-phosphatidylethanolamine conjugate (peg-pe)-based mixed micelles: Some properties, loading with paclitaxel, and modulation of p-glycoprotein-mediated efflux, Int J Pharm, 315 (2006) 148–157, 10.1016/j.ijpharm.2006.02.018. [DOI] [PubMed] [Google Scholar]
- [136].Zang X, Lee JB, Deshpande K, Garbuzenko OB, Minko T, Kagan L, Prevention of paclitaxel-induced neuropathy by formulation approach, J Control Release, 303 (2019) 109–116, 10.1016/j.jconrel.2019.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Lukyanov AN, Gao Z, Torchilin VP, Micelles from polyethylene glycol/phosphatidylethanolamine conjugates for tumor drug delivery, J Control Release, 91 (2003) 97–102, 10.1016/s0168-3659(03)00217-7. [DOI] [PubMed] [Google Scholar]
- [138].Gill KK, Nazzal S, Kaddoumi A, Paclitaxel loaded peg(5000)-dspe micelles as pulmonary delivery platform: Formulation characterization, tissue distribution, plasma pharmacokinetics, and toxicological evaluation, Eur J Pharm Biopharm, 79 (2011) 276–284, 10.1016/j.ejpb.2011.04.017. [DOI] [PubMed] [Google Scholar]
- [139].Koshkina NV, Waldrep JC, Roberts LE, Golunski E, Melton S, Knight V, Paclitaxel liposome aerosol treatment induces inhibition of pulmonary metastases in murine renal carcinoma model, Clin Cancer Res, 7 (2001) 3258–3262, [PubMed] [Google Scholar]
- [140].Koshkina NV, Knight V, Gilbert BE, Golunski E, Roberts L, Waldrep JC, Improved respiratory delivery of the anticancer drugs, camptothecin and paclitaxel, with 5% co2-enriched air: Pharmacokinetic studies, Cancer Chemother Pharmacol, 47 (2001) 451–456, 10.1007/s002800000230. [DOI] [PubMed] [Google Scholar]
- [141].Fujimoto S, Chikazawa H, Schedule-dependent and -independent antitumor activity of paclitaxel-based combination chemotherapy against m-109 murine lung carcinoma in vivo, Jpn J Cancer Res, 89 (1998) 1343–1351, 10.1111/j.1349-7006.1998.tb00532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Liebmann JE, Cook JA, Lipschultz C, Teague D, Fisher J, Mitchell JB, Cytotoxic studies of paclitaxel (taxol) in human tumour cell lines, Br J Cancer, 68 (1993) 1104–1109, 10.1038/bjc.1993.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Patton JS, Mechanisms of macromolecule absorption by the lungs, Adv Drug Deliv Rev, 19 (1996) 3–36, [Google Scholar]
- [144].Oberdorster G, Lung particle overload: Implications for occupational exposures to particles, Regul Toxicol Pharmacol, 21 (1995) 123–135, 10.1006/rtph.1995.1017. [DOI] [PubMed] [Google Scholar]
- [145].Forbes B, O’Lone R, Allen PP, Cahn A, Clarke C, Collinge M, Dailey LA, Donnelly LE, Dybowski J, Hassall D, Hildebrand D, Jones R, Kilgour J, Klapwijk J, Maier CC, McGovern T, Nikula K, Parry JD, Reed MD, Robinson I, Tomlinson L, Wolfreys A, Challenges for inhaled drug discovery and development: Induced alveolar macrophage responses, Adv Drug Deliv Rev, 71 (2014) 15–33, 10.1016/j.addr.2014.02.001. [DOI] [PubMed] [Google Scholar]
- [146].Barlow PG, Brown DM, Donaldson K, MacCallum J, Stone V, Reduced alveolar macrophage migration induced by acute ambient particle (pm10) exposure, Cell Biol Toxicol, 24 (2008) 243–252, 10.1007/s10565-007-9033-y. [DOI] [PubMed] [Google Scholar]
- [147].Moller W, Hofer T, Ziesenis A, Karg E, Heyder J, Ultrafine particles cause cytoskeletal dysfunctions in macrophages, Toxicol Appl Pharmacol, 182 (2002) 197–207, 10.1006/taap.2002.9430. [DOI] [PubMed] [Google Scholar]
- [148].Johnson LN, Koval M, Cross-talk between pulmonary injury, oxidant stress, and gap junctional communication, Antioxid Redox Signal, 11 (2009) 355–367, 10.1089/ars.2008.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Jones RM, Neef N, Interpretation and prediction of inhaled drug particle accumulation in the lung and its associated toxicity, Xenobiotica, 42 (2012) 86–93, 10.3109/00498254.2011.632827. [DOI] [PubMed] [Google Scholar]
- [150].Haque S, Whittaker M, McIntosh MP, Pouton CW, Phipps S, Kaminskas LM, A comparison of the lung clearance kinetics of solid lipid nanoparticles and liposomes by following the (3)h-labelled structural lipids after pulmonary delivery in rats, Eur J Pharm Biopharm, 125 (2018) 1–12, 10.1016/j.ejpb.2018.01.001. [DOI] [PubMed] [Google Scholar]
- [151].Haque S, Feeney O, Meeusen E, Boyd BJ, McIntosh MP, Pouton CW, Whittaker M, Kaminskas LM, Local inflammation alters the lung disposition of a drug loaded pegylated liposome after pulmonary dosing to rats, J Control Release, 307 (2019) 32–43, 10.1016/j.jconrel.2019.05.043. [DOI] [PubMed] [Google Scholar]
- [152].Garbuzenko OB, Saad M, Pozharov VP, Reuhl KR, Mainelis G, Minko T, Inhibition of lung tumor growth by complex pulmonary delivery of drugs with oligonucleotides as suppressors of cellular resistance, Proc Natl Acad Sci U S A, 107 (2010) 10737–10742, 10.1073/pnas.1004604107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Garbuzenko OB, Kuzmov A, Taratula O, Pine SR, Minko T, Strategy to enhance lung cancer treatment by five essential elements: Inhalation delivery, nanotechnology, tumor-receptor targeting, chemo- and gene therapy, Theranostics, 9 (2019) 8362–8376, 10.7150/thno.39816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Lai SK, Wang YY, Wirtz D, Hanes J, Micro- and macrorheology of mucus, Adv Drug Deliv Rev, 61 (2009) 86–100, 10.1016/j.addr.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Rubin BK, Mucus structure and properties in cystic fibrosis, Paediatr Respir Rev, 8 (2007) 4–7, 10.1016/j.prrv.2007.02.004. [DOI] [PubMed] [Google Scholar]
- [156].Renwick LC, Brown D, Clouter A, Donaldson K, Increased inflammation and altered macrophage chemotactic responses caused by two ultrafine particle types, Occup Environ Med, 61 (2004) 442–447, 10.1136/oem.2003.008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Miller MR, Raftis JB, Langrish JP, McLean SG, Samutrtai P, Connell SP, Wilson S, Vesey AT, Fokkens PHB, Boere AJF, Krystek P, Campbell CJ, Hadoke PWF, Donaldson K, Cassee FR, Newby DE, Duffin R, Mills NL, Correction to”inhaled nanoparticles accumulate at sites of vascular disease”, ACS Nano, 11 (2017) 10623–10624, 10.1021/acsnano.7b06327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Folkesson HG, Matthay MA, Westrom BR, Kim KJ, Karlsson BW, Hastings RH, Alveolar epithelial clearance of protein, J Appl Physiol (1985), 80 (1996) 1431–1445, 10.1152/jappl.1996.80.5.1431. [DOI] [PubMed] [Google Scholar]
- [159].Taylor AE, Gaar KA Jr., Estimation of equivalent pore radii of pulmonary capillary and alveolar membranes, Am J Physiol, 218 (1970) 1133–1140, 10.1152/ajplegacy.1970.218.4.1133. [DOI] [PubMed] [Google Scholar]
- [160].Saari SM, Vidgren MT, Koskinen MO, Turjanmaa VM, Waldrep JC, Nieminen MM, Regional lung deposition and clearance of 99mtc-labeled beclomethasone-dlpc liposomes in mild and severe asthma, Chest, 113 (1998) 1573–1579, 10.1378/chest.113.6.1573. [DOI] [PubMed] [Google Scholar]
- [161].Barker SA, Taylor KMG, Short MD, The deposition and clearance of liposome entrapped 99mtc-dtpa in the human respiratory tract, International Journal of Pharmaceutics, 102 (1994) 159–165, 10.1016/0378-5173(94)90051-5. [DOI] [Google Scholar]
- [162].Saari M, Vidgren MT, Koskinen MO, Turjanmaa VMH, Nieminen MM, Pulmonary distribution and clearance of two beclomethasone liposome formulations in healthy volunteers, International Journal of Pharmaceutics, 181 (1999) 1–9, 10.1016/s0378-5173(98)00398-6. [DOI] [PubMed] [Google Scholar]
- [163].Morimoto Y, Adachi Y, Pulmonary uptake of liposomal phosphatidylcholine upon intratracheal administration to rats, Chem Pharm Bull (Tokyo), 30 (1982) 2248–2251, 10.1248/cpb.30.2248. [DOI] [PubMed] [Google Scholar]
- [164].Shahiwala A, Misra A, Pulmonary absorption of liposomal levonorgestrel, AAPS PharmSciTech, 5 (2004) E13, 10.1208/pt050113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Li YZ, Sun X, Gong T, Liu J, Zuo J, Zhang ZR, Inhalable microparticles as carriers for pulmonary delivery of thymopentin-loaded solid lipid nanoparticles, Pharm Res, 27 (2010) 1977–1986, 10.1007/s11095-010-0201-z. [DOI] [PubMed] [Google Scholar]
- [166].Geiser M, Casaulta M, Kupferschmid B, Schulz H, Semmler-Behnke M, Kreyling W, The role of macrophages in the clearance of inhaled ultrafine titanium dioxide particles, Am J Respir Cell Mol Biol, 38 (2008) 371–376, 10.1165/rcmb.2007-0138OC. [DOI] [PubMed] [Google Scholar]
- [167].Lai SK, Wang YY, Hanes J, Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues, Adv Drug Deliv Rev, 61 (2009) 158–171, 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Garbuzenko OB, Mainelis G, Taratula O, Minko T, Inhalation treatment of lung cancer: The influence of composition, size and shape of nanocarriers on their lung accumulation and retention, Cancer Biol Med, 11 (2014) 44–55, 10.7497/j.issn.2095-3941.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Chen R, Xu L, Fan Q, Li M, Wang J, Wu L, Li W, Duan J, Chen Z, Hierarchical pulmonary target nanoparticles via inhaled administration for anticancer drug delivery, Drug Deliv, 24 (2017) 1191–1203, 10.1080/10717544.2017.1365395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Hong JY, Nam JW, Seo EK, Lee SK, Daphnane diterpene esters with anti-proliferative activities against human lung cancer cells from daphne genkwa, Chem Pharm Bull (Tokyo), 58 (2010) 234–237, 10.1248/cpb.58.234. [DOI] [PubMed] [Google Scholar]
- [171].Ly JD, Grubb DR, Lawen A, The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update, Apoptosis, 8 (2003) 115–128, 10.1023/a:1022945107762. [DOI] [PubMed] [Google Scholar]
- [172].Han L, Huang R, Liu S, Huang S, Jiang C, Peptide-conjugated pamam for targeted doxorubicin delivery to transferrin receptor overexpressed tumors, Mol Pharm, 7 (2010) 2156–2165, 10.1021/mp100185f. [DOI] [PubMed] [Google Scholar]
- [173].Singh M, Transferrin as a targeting ligand for liposomes and anticancer drugs, Curr Pharm Des, 5 (1999) 443–451, [PubMed] [Google Scholar]
- [174].Deshpande PP, Biswas S, Torchilin VP, Current trends in the use of liposomes for tumor targeting, Nanomedicine (Lond), 8 (2013) 1509–1528, 10.2217/nnm.13.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Daniels TR, Bernabeu E, Rodriguez JA, Patel S, Kozman M, Chiappetta DA, Holler E, Ljubimova JY, Helguera G, Penichet ML, The transferrin receptor and the targeted delivery of therapeutic agents against cancer, Biochim Biophys Acta, 1820 (2012) 291–317, 10.1016/j.bbagen.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Anabousi S, Bakowsky U, Schneider M, Huwer H, Lehr CM, Ehrhardt C, In vitro assessment of transferrin-conjugated liposomes as drug delivery systems for inhalation therapy of lung cancer, Eur J Pharm Sci, 29 (2006) 367–374, 10.1016/j.ejps.2006.07.004. [DOI] [PubMed] [Google Scholar]
- [177].Lee JH, Engler JA, Collawn JF, Moore BA, Receptor mediated uptake of peptides that bind the human transferrin receptor, Eur J Biochem, 268 (2001) 2004–2012, 10.1046/j.1432-1327.2001.02073.x. [DOI] [PubMed] [Google Scholar]
- [178].Wang Z, Zhao Y, Jiang Y, Lv W, Wu L, Wang B, Lv L, Xu Q, Xin H, Enhanced anti-ischemic stroke of zl006 by t7-conjugated pegylated liposomes drug delivery system, Sci Rep, 5 (2015) 12651, 10.1038/srep12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Srivastava S, Somasagara RR, Hegde M, Nishana M, Tadi SK, Srivastava M, Choudhary B, Raghavan SC, Quercetin, a natural flavonoid interacts with DNA, arrests cell cycle and causes tumor regression by activating mitochondrial pathway of apoptosis, Sci Rep, 6 (2016) 24049, 10.1038/srep24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Nguyen TT, Tran E, Nguyen TH, Do PT, Huynh TH, Huynh H, The role of activated mek-erk pathway in quercetin-induced growth inhibition and apoptosis in a549 lung cancer cells, Carcinogenesis, 25 (2004) 647–659, 10.1093/carcin/bgh052. [DOI] [PubMed] [Google Scholar]
- [181].Xingyu Z, Peijie M, Dan P, Youg W, Daojun W, Xinzheng C, Xijun Z, Yangrong S, Quercetin suppresses lung cancer growth by targeting aurora b kinase, Cancer Med, 5 (2016) 3156–3165, 10.1002/cam4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Suntres ZE, Liposomal antioxidants for protection against oxidant-induced damage, J Toxicol, 2011 (2011) 152474, 10.1155/2011/152474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Lamson DW, Brignall MS, Antioxidants and cancer, part 3: Quercetin, Altern Med Rev, 5 (2000) 196–208, [PubMed] [Google Scholar]
- [184].Riaz MK, Zhang X, Wong KH, Chen H, Liu Q, Chen X, Zhang G, Lu A, Yang Z, Pulmonary delivery of transferrin receptors targeting peptide surface-functionalized liposomes augments the chemotherapeutic effect of quercetin in lung cancer therapy, Int J Nanomedicine, 14 (2019) 2879–2902, 10.2147/IJN.S192219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Yoo HS, Park TG, Folate-receptor-targeted delivery of doxorubicin nano-aggregates stabilized by doxorubicin-peg-folate conjugate, J Control Release, 100 (2004) 247–256, 10.1016/j.jconrel.2004.08.017. [DOI] [PubMed] [Google Scholar]
- [186].Yuan Z, Chen D, Zhang S, Zheng Z, Preparation, characterization and evaluation of docetaxel-loaded, folate-conjugated peg-liposomes, Yakugaku Zasshi, 130 (2010) 1353–1359, 10.1248/yakushi.130.1353. [DOI] [PubMed] [Google Scholar]
- [187].Zhai G, Wu J, Xiang G, Mao W, Yu B, Li H, Piao L, Lee LJ, Lee RJ, Preparation, characterization and pharmacokinetics of folate receptor-targeted liposomes for docetaxel delivery, J Nanosci Nanotechnol, 9 (2009) 2155–2161, 10.1166/jnn.2009.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Zhu X, Kong Y, Liu Q, Lu Y, Xing H, Lu X, Yang Y, Xu J, Li N, Zhao D, Chen X, Lu Y, Inhalable dry powder prepared from folic acid-conjugated docetaxel liposomes alters pharmacodynamic and pharmacokinetic properties relevant to lung cancer chemotherapy, Pulm Pharmacol Ther, 55 (2019) 50–61, 10.1016/j.pupt.2019.02.001. [DOI] [PubMed] [Google Scholar]
- [189].Lin C, Zhang X, Chen H, Bian Z, Zhang G, Riaz MK, Tyagi D, Lin G, Zhang Y, Wang J, Lu A, Yang Z, Dual-ligand modified liposomes provide effective local targeted delivery of lung-cancer drug by antibody and tumor lineage-homing cell-penetrating peptide, Drug Deliv, 25 (2018) 256–266, 10.1080/10717544.2018.1425777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Cai D, Gao W, He B, Dai W, Zhang H, Wang X, Wang J, Zhang X, Zhang Q, Hydrophobic penetrating peptide pfvyli-modified stealth liposomes for doxorubicin delivery in breast cancer therapy, Biomaterials, 35 (2014) 2283–2294, 10.1016/j.biomaterials.2013.11.088. [DOI] [PubMed] [Google Scholar]
- [191].Raghunand N, Gatenby RA, Gillies RJ, Microenvironmental and cellular consequences of altered blood flow in tumours, Br J Radiol, 76 Spec No 1 (2003) S11–22, 10.1259/bjr/12913493. [DOI] [PubMed] [Google Scholar]
- [192].Pastorek J, Pastorekova S, Hypoxia-induced carbonic anhydrase ix as a target for cancer therapy: From biology to clinical use, Semin Cancer Biol, 31 (2015) 52–64, 10.1016/j.semcancer.2014.08.002. [DOI] [PubMed] [Google Scholar]
- [193].McDonald PC, Winum JY, Supuran CT, Dedhar S, Recent developments in targeting carbonic anhydrase ix for cancer therapeutics, Oncotarget, 3 (2012) 84–97, 10.18632/oncotarget.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Vermylen P, Roufosse C, Burny A, Verhest A, Bosschaerts T, Pastorekova S, Ninane V, Sculier JP, Carbonic anhydrase ix antigen differentiates between preneoplastic malignant lesions in non-small cell lung carcinoma, Eur Respir J, 14 (1999) 806–811, 10.1034/j.1399-3003.1999.14d14.x. [DOI] [PubMed] [Google Scholar]
- [195].Swinson DE, Jones JL, Richardson D, Wykoff C, Turley H, Pastorek J, Taub N, Harris AL, O’Byrne KJ, Carbonic anhydrase ix expression, a novel surrogate marker of tumor hypoxia, is associated with a poor prognosis in non-small-cell lung cancer, J Clin Oncol, 21 (2003) 473–482, 10.1200/JCO.2003.11.132. [DOI] [PubMed] [Google Scholar]
- [196].Le QT, Chen E, Salim A, Cao H, Kong CS, Whyte R, Donington J, Cannon W, Wakelee H, Tibshirani R, Mitchell JD, Richardson D, O’Byrne KJ, Koong AC, Giaccia AJ, An evaluation of tumor oxygenation and gene expression in patients with early stage non-small cell lung cancers, Clin Cancer Res, 12 (2006) 1507–1514, 10.1158/1078-0432.CCR-05-2049. [DOI] [PubMed] [Google Scholar]
- [197].Liao SY, Aurelio ON, Jan K, Zavada J, Stanbridge EJ, Identification of the mn/ca9 protein as a reliable diagnostic biomarker of clear cell carcinoma of the kidney, Cancer Res, 57 (1997) 2827–2831, [PubMed] [Google Scholar]
- [198].Saarnio J, Parkkila S, Parkkila AK, Haukipuro K, Pastorekova S, Pastorek J, Kairaluoma MI, Karttunen TJ, Immunohistochemical study of colorectal tumors for expression of a novel transmembrane carbonic anhydrase, mn/ca ix, with potential value as a marker of cell proliferation, Am J Pathol, 153 (1998) 279–285, 10.1016/S0002-9440(10)65569-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Wykoff CC, Beasley N, Watson PH, Campo L, Chia SK, English R, Pastorek J, Sly WS, Ratcliffe P, Harris AL, Expression of the hypoxia-inducible and tumor-associated carbonic anhydrases in ductal carcinoma in situ of the breast, Am J Pathol, 158 (2001) 1011–1019, 10.1016/S0002-9440(10)64048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Wang F, Wang Y, Zhang X, Zhang W, Guo S, Jin F, Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery, J Control Release, 174 (2014) 126–136, 10.1016/j.jconrel.2013.11.020. [DOI] [PubMed] [Google Scholar]
- [201].Kondo E, Saito K, Tashiro Y, Kamide K, Uno S, Furuya T, Mashita M, Nakajima K, Tsumuraya T, Kobayashi N, Nishibori M, Tanimoto M, Matsushita M, Tumour lineage-homing cell-penetrating peptides as anticancer molecular delivery systems, Nat Commun, 3 (2012) 951, 10.1038/ncomms1952. [DOI] [PubMed] [Google Scholar]
- [202].Wong BC, Zhang H, Qin L, Chen H, Fang C, Lu A, Yang Z, Carbonic anhydrase ix-directed immunoliposomes for targeted drug delivery to human lung cancer cells in vitro, Drug Des Devel Ther, 8 (2014) 993–1001, 10.2147/DDDT.S63235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Williamson DJ, Wallman LL, Jones R, Keogh AM, Scroope F, Penny R, Weber C, Macdonald PS, Hemodynamic effects of bosentan, an endothelin receptor antagonist, in patients with pulmonary hypertension, Circulation, 102 (2000) 411–418, 10.1161/01.cir.102.4.411. [DOI] [PubMed] [Google Scholar]
- [204].Clozel M, Breu V, Gray GA, Kalina B, Loffler BM, Burri K, Cassal JM, Hirth G, Muller M, Neidhart W, et al. , Pharmacological characterization of bosentan, a new potent orally active nonpeptide endothelin receptor antagonist, J Pharmacol Exp Ther, 270 (1994) 228–235, [PubMed] [Google Scholar]
- [205].Roux S, Breu V, Ertel SI, Clozel M, Endothelin antagonism with bosentan: A review of potential applications, J Mol Med (Berl), 77 (1999) 364–376, 10.1007/s001090050363. [DOI] [PubMed] [Google Scholar]
- [206].Hanna LA, Basalious EB, ON EL, Respirable controlled release polymeric colloid (rcrpc) of bosentan for the management of pulmonary hypertension: In vitro aerosolization, histological examination and in vivo pulmonary absorption, Drug Deliv, 24 (2016) 188–198, 10.1080/10717544.2016.1239661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Nahar K, Absar S, Patel B, Ahsan F, Starch-coated magnetic liposomes as an inhalable carrier for accumulation of fasudil in the pulmonary vasculature, Int J Pharm, 464 (2014) 185–195, 10.1016/j.ijpharm.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Hara K, Tsujimoto H, Tsukada Y, Huang CC, Kawashima Y, Tsutsumi M, Histological examination of plga nanospheres for intratracheal drug administration, Int J Pharm, 356 (2008) 267–273, 10.1016/j.ijpharm.2007.12.041. [DOI] [PubMed] [Google Scholar]
- [209].Beck-Broichsitter M, Gauss J, Packhaeuser CB, Lahnstein K, Schmehl T, Seeger W, Kissel T, Gessler T, Pulmonary drug delivery with aerosolizable nanoparticles in an ex vivo lung model, Int J Pharm, 367 (2009) 169–178, 10.1016/j.ijpharm.2008.09.017. [DOI] [PubMed] [Google Scholar]
- [210].Beck-Broichsitter M, Kleimann P, Gessler T, Seeger W, Kissel T, Schmehl T, Nebulization performance of biodegradable sildenafil-loaded nanoparticles using the aeroneb pro: Formulation aspects and nanoparticle stability to nebulization, Int J Pharm, 422 (2012) 398–408, 10.1016/j.ijpharm.2011.10.012. [DOI] [PubMed] [Google Scholar]
- [211].Beck-Broichsitter M, Schmehl T, Gessler T, Seeger W, Kissel T, Development of a biodegradable nanoparticle platform for sildenafil: Formulation optimization by factorial design analysis combined with application of charge-modified branched polyesters, J Control Release, 157 (2012) 469–477, 10.1016/j.jconrel.2011.09.058. [DOI] [PubMed] [Google Scholar]
- [212].Corris PA, Alternatives to lung transplantation: Treatment of pulmonary arterial hypertension, Clin Chest Med, 32 (2011) 399–410, 10.1016/j.ccm.2011.02.015. [DOI] [PubMed] [Google Scholar]
- [213].Vachiery JL, Prostacyclins in pulmonary arterial hypertension: The need for earlier therapy, Adv Ther, 28 (2011) 251–269, 10.1007/s12325-011-0005-5. [DOI] [PubMed] [Google Scholar]
- [214].Oka M, Fagan KA, Jones PL, McMurtry IF, Therapeutic potential of rhoa/rho kinase inhibitors in pulmonary hypertension, Br J Pharmacol, 155 (2008) 444–454, 10.1038/bjp.2008.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Gupta V, Gupta N, Shaik IH, Mehvar R, McMurtry IF, Oka M, Nozik-Grayck E, Komatsu M, Ahsan F, Liposomal fasudil, a rho-kinase inhibitor, for prolonged pulmonary preferential vasodilation in pulmonary arterial hypertension, J Control Release, 167 (2013) 189–199, 10.1016/j.jconrel.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Ishida T, Takanashi Y, Kiwada H, Safe and efficient drug delivery system with liposomes for intrathecal application of an antivasospastic drug, fasudil, Biol Pharm Bull, 29 (2006) 397–402, 10.1248/bpb.29.397. [DOI] [PubMed] [Google Scholar]
- [217].Chougule M, Padhi B, Misra A, Nano-liposomal dry powder inhaler of tacrolimus: Preparation, characterization, and pulmonary pharmacokinetics, Int J Nanomedicine, 2 (2007) 675–688, [PMC free article] [PubMed] [Google Scholar]
- [218].Boehler A, Estenne M, Obliterative bronchiolitis after lung transplantation, Curr Opin Pulm Med, 6 (2000) 133–139, 10.1097/00063198-200003000-00009. [DOI] [PubMed] [Google Scholar]
- [219].Hertz MI, Taylor DO, Trulock EP, Boucek MM, Mohacsi PJ, Edwards LB, Keck BM , The registry of the international society for heart and lung transplantation: Nineteenth official report-2002, J Heart Lung Transplant, 21 (2002) 950–970, 10.1016/s1053-2498(02)00498-9. [DOI] [PubMed] [Google Scholar]
- [220].Griffith BP, Bando K, Hardesty RL, Armitage JM, Keenan RJ, Pham SM, Paradis IL, Yousem SA, Komatsu K, Konishi H, et al. , A prospective randomized trial of fk506 versus cyclosporine after human pulmonary transplantation, Transplantation, 57 (1994) 848–851, 10.1097/00007890-199403270-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Onsager DR, Canver CC, Jahania MS, Welter D, Michalski M, Hoffman AM, Mentzer RM Jr., Love RB, Efficacy of tacrolimus in the treatment of refractory rejection in heart and lung transplant recipients, J Heart Lung Transplant, 18 (1999) 448–455, 10.1016/s1053–2498(99)00016–9. [DOI] [PubMed] [Google Scholar]
- [222].Treede H, Klepetko W, Reichenspurner H, Zuckermann A, Meiser B, Birsan T, Wisser W, Reichert B, Tacrolimus versus cyclosporine after lung transplantation: A prospective, open, randomized two-center trial comparing two different immunosuppressive protocols, The Journal of Heart and Lung Transplantation, 20 (2001) 511–517, 10.1016/s1053-2498(01)00244-3. [DOI] [PubMed] [Google Scholar]
- [223].Knoop C, Thiry P, Saint-Marcoux F, Rousseau A, Marquet P, Estenne M, Tacrolimus pharmacokinetics and dose monitoring after lung transplantation for cystic fibrosis and other conditions, Am J Transplant, 5 (2005) 1477–1482, 10.1111/j.1600-6143.2005.00870.x. [DOI] [PubMed] [Google Scholar]
- [224].Ihara H, Shinkuma D, Ichikawa Y, Nojima M, Nagano S, Ikoma F, Intra- and interindividual variation in the pharmacokinetics of tacrolimus (fk506) in kidney transplant recipients--importance of trough level as a practical indicator, Int J Urol, 2 (1995) 151–155, 10.1111/j.1442-2042.1995.tb00444.x. [DOI] [PubMed] [Google Scholar]
- [225].Waldrep JC, New aerosol drug delivery systems for the treatment of immune-mediated pulmonary diseases, Drugs Today (Barc), 34 (1998) 549–561, 10.1358/dot.1998.34.6.485253. [DOI] [PubMed] [Google Scholar]
- [226].Letsou GV, Safi HJ, Reardon MJ, Ergenoglu M, Li Z, Klonaris CN, Baldwin JC, Gilbert BE, Waldrep JC, Pharmacokinetics of liposomal aerosolized cyclosporine a for pulmonary immunosuppression, The Annals of Thoracic Surgery, 68 (1999) 2044–2048, 10.1016/s0003-4975(99)01183-2. [DOI] [PubMed] [Google Scholar]
- [227].Joshi M, Misra AN, Pulmonary disposition of budesonide from liposomal dry powder inhaler, Methods Find Exp Clin Pharmacol, 23 (2001) 531–536, 10.1358/mf.2001.23.10.677118. [DOI] [PubMed] [Google Scholar]
- [228].Joshi M, Misra A, Disposition kinetics of ketotifen from liposomal dry powder for inhalation in rat lung, Clin Exp Pharmacol Physiol, 30 (2003) 153–156, 10.1046/j.1440-1681.2003.03813.x. [DOI] [PubMed] [Google Scholar]
- [229].Shah SP, Misra A, Development of liposomal amphotericin b dry powder inhaler formulation, Drug Deliv, 11 (2004) 247–253, 10.1080/10717540490467375. [DOI] [PubMed] [Google Scholar]
- [230].Lo YL, Tsai JC, Kuo JH, Liposomes and disaccharides as carriers in spray-dried powder formulations of superoxide dismutase, J Control Release, 94 (2004) 259–272, 10.1016/j.jconrel.2003.09.019. [DOI] [PubMed] [Google Scholar]
- [231].Lu D, Hickey AJ, Liposomal dry powders as aerosols for pulmonary delivery of proteins, AAPS PharmSciTech, 6 (2005) E641–648, 10.1208/pt060480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Vermehren C, Frokjaer S, Aurstad T, Hansen J, Lung surfactant as a drug delivery system, Int J Pharm, 307 (2006) 89–92, 10.1016/j.ijpharm.2005.10.029. [DOI] [PubMed] [Google Scholar]
- [233].Gruber CJ, Tschugguel W, Schneeberger C, Huber JC, Production and actions of estrogens, N Engl J Med, 346 (2002) 340–352, 10.1056/NEJMra000471. [DOI] [PubMed] [Google Scholar]
- [234].Mueck AO, Seeger H, Graser T, Oettel M, Lippert TH, The effects of postmenopausal hormone replacement therapy and oral contraceptives on the endogenous estradiol metabolism, Horm Metab Res, 33 (2001) 744–747, 10.1055/s-2001-19139. [DOI] [PubMed] [Google Scholar]
- [235].Kent UM, Mills DE, Rajnarayanan RV, Alworth WL, Hollenberg PF, Effect of 17-alpha-ethynylestradiol on activities of cytochrome p450 2b (p450 2b) enzymes: Characterization of inactivation of p450s 2b1 and 2b6 and identification of metabolites, J Pharmacol Exp Ther, 300 (2002) 549–558, 10.1124/jpet.300.2.549. [DOI] [PubMed] [Google Scholar]
- [236].Rosing J, Middeldorp S, Curvers J, Thomassen MCLG, Nicolaes GAF, Meijers JCM, Bouma BN, Büller HR, Prins MH, Tans G, Low-dose oral contraceptives and acquired resistance to activated protein c: A randomised cross-over study, The Lancet, 354 (1999) 2036–2040, 10.1016/s0140-6736(99)06092-4. [DOI] [PubMed] [Google Scholar]
- [237].McCann MF, Potter LS, Progestin-only oral contraception: A comprehensive review, Contraception, 50 (1994) S1–195, [PubMed] [Google Scholar]
- [238].Patton JS, Bukar JG, Eldon MA, Clinical pharmacokinetics and pharmacodynamics of inhaled insulin, Clinical Pharmacokinetics, 43 (2004) 781–801, 10.2165/00003088-200443120-00002. [DOI] [PubMed] [Google Scholar]
- [239].Derendorf H, Nave R, Drollmann A, Cerasoli F, Wurst W, Relevance of pharmacokinetics and pharmacodynamics of inhaled corticosteroids to asthma, Eur Respir J, 28 (2006) 1042–1050, 10.1183/09031936.00074905. [DOI] [PubMed] [Google Scholar]
- [240].Winkler J, Hochhaus G, Derendorf H, How the lung handles drugs, Proceedings of the American Thoracic Society, 1 (2004) 356–363, 10.1513/pats.200403-025MS. [DOI] [PubMed] [Google Scholar]
- [241].Dershwitz MMDPD, Walsh John L.M.D., Morishige Richard J.R.R.T., Connors Patricia M.R.N.B.S.N., Rubsamen Reid M.M.D., Shafer Steven L.M.D., Rosow Carl E.M.D.P.D., Pharmacokinetics and pharmacodynamics of inhaled versus intravenous morphine in healthy volunteers Anesthesiology: The Journal of the American Society of Anesthesiologists, 93 (2000) 619–628, [DOI] [PubMed] [Google Scholar]
- [242].Hubner M, Hochhaus G, Derendorf H, Comparative pharmacology, bioavailability, pharmacokinetics, and pharmacodynamics of inhaled glucocorticosteroids, Immunol Allergy Clin North Am, 25 (2005) 469–488, 10.1016/j.iac.2005.05.004. [DOI] [PubMed] [Google Scholar]
- [243].Ahrens RC, Harris JB, Milavetz G, Annis L, Ries R, Use of bronchial provocation with histamine to compare the pharmacodynamics of inhaled albuterol and metaproterenol in patients with asthma, Journal of Allergy and Clinical Immunology, 79 (1987) 876–882, 10.1016/0091-6749(87)90235-1. [DOI] [PubMed] [Google Scholar]
- [244].Marino MT, Costello D, Baughman R, Boss A, Cassidy J, Damico C, van Marle S, van Vliet A, Richardson PC, Pharmacokinetics and pharmacodynamics of inhaled glp-1 (mkc253): Proof-of-concept studies in healthy normal volunteers and in patients with type 2 diabetes, Clinical Pharmacology & Therapeutics, 88 (2010) 243–250, 10.1038/clpt.2010.85. [DOI] [PubMed] [Google Scholar]
- [245].Boyd SD, Hadigan C, McManus M, Chairez C, Nieman LK, Pau AK, Alfaro RM, Kovacs JA, Calderon MM, Penzak SR, Influence of low-dose ritonavir with and without darunavir on the pharmacokinetics and pharmacodynamics of inhaled beclomethasone, Journal of acquired immune deficiency syndromes (1999), 63 (2013) 355–361, 10.1097/QAI.0b013e31829260d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Kauscher U, Holme MN, Bjornmalm M, Stevens MM, Physical stimuli-responsive vesicles in drug delivery: Beyond liposomes and polymersomes, Adv Drug Deliv Rev, 138 (2019) 259–275, 10.1016/j.addr.2018.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Li Y, Feng X, Wang A, Yang Y, Fei J, Sun B, Jia Y, Li J, Supramolecularly assembled nanocomposites as biomimetic chloroplasts for enhancement of photophosphorylation, Angew Chem Int Ed Engl, 58 (2019) 796–800, 10.1002/anie.201812582. [DOI] [PubMed] [Google Scholar]
- [248].Zhang J, Yang C, Pan S, Shi M, Li J, Hu H, Qiao M, Chen D, Zhao X, Eph a10-modified ph-sensitive liposomes loaded with novel triphenylphosphine-docetaxel conjugate possess hierarchical targetability and sufficient antitumor effect both in vitro and in vivo, Drug Deliv, 25 (2018) 723–737, 10.1080/10717544.2018.1446475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Zhang X, Zong W, Bi H, Zhao K, Fuhs T, Hu Y, Cheng W, Han X, Hierarchical drug release of ph-sensitive liposomes encapsulating aqueous two phase system, Eur J Pharm Biopharm, 127 (2018) 177–182, 10.1016/j.ejpb.2018.02.021. [DOI] [PubMed] [Google Scholar]