

Abstract
Zika virus (ZIKV) belongs to the Flaviviridae family and is related to other viruses that cause human diseases. Unlike other flaviviruses, ZIKV infection can cause congenital neurologic disorders and replicates efficiently in reproductive tissues1–3,. Here, we show that ZIKV envelope (E) protein is K63-linked polyubiquitinated by the E3-ubiquitin ligase TRIM7. Accordingly, ZIKV replicates less efficiently in brain and reproductive tissues of Trim7−/− mice. Ubiquitinated E is present on infectious Zika virions when released from specific cell types and enhances virus attachment and entry into cells. Specifically, K63-linked polyubiquitin chains directly interact with the Tim-1 (HAVCR1) receptor, enhancing virus replication in cells and in vivo in brain tissue. Recombinant ZIKV mutants lacking ubiquitination are attenuated in human cells and in a mouse model, but not in live mosquitoes. Monoclonal antibodies against K63-linked polyubiquitin specifically neutralize ZIKV and reduce viremia in mice. Collectively, the results demonstrate that ubiquitination of ZIKV E is an important determinant of virus entry, tropism and pathogenesis.
MAIN
Zika virus (ZIKV) is transmitted primarily by peridomestic Aedes mosquitoes, but also can be acquired through sexual, vertical, and blood transfusion routes1,2. ZIKV infection causes congenital abnormalities in fetuses of infected pregnant women3. Although ZIKV is closely related to other flaviviruses that cause human diseases, including dengue (DENV), West Nile (WNV), and yellow fever (YFV), the mechanism of how ZIKV causes neurologic disorders or replicates in reproductive tissues remains unclear.
Ubiquitination of proteins is a post-translation modification process with many cellular functions, including regulation of virus replication4. There is previous evidence that flaviviruses utilize the host Ub system for replication5–7, however whether flaviviruses carry Ub in the infectious virion or whether the Ub machinery is involved in determining virus tropism and pathogenesis has not been explored. Tripartite Motif (TRIM) proteins are a large family of E3-Ub ligases that mediate transfer of Ub to target proteins and many are known to inhibit viral replication4,8,9. However, very few examples exist of TRIM proteins being exploited by viruses to promote virus replication9,10. Here, we report that ZIKV envelope (E) protein is ubiquitinated by the E3-Ub ligase TRIM7, and this modification is a determinant of tissue tropism. A proportion of virions contain ubiquitinated E protein, which promotes more efficient attachment and entry into host cells.
Flavivirus envelope protein is ubiquitinated
Studies have shown that proteasome inhibitors reduce DENV replication7,11–13. Consistent with this, placenta-derived JEG-3 cells pretreated with proteasome inhibitor MG132 are more resistant to ZIKV infection (Extended Data Fig. 1a). To examine whether ubiquitination of viral proteins has a role in flavivirus biology, we performed mass spectrometry (MS) analysis of samples from cells infected with WNV, DENV-2, or ZIKV. This analysis identified ubiquitination on the K38 residue of WNV and DENV E, which is conserved among flaviviruses (Extended Data Fig. 1b). Another ubiquitination site on K281 at the hinge region (“kl loop”) of ZIKV-E was identified; however, K281 is not conserved in flaviviruses (Extended Data Fig. 1b, and14). We focused our studies on E because of its essential function in virus entry15. Co-immunoprecipitation assays (coIP) with Huh7 infected with DENV or ZIKV confirmed that E was ubiquitinated (Extended Data Fig. 1c). Examination of the Ub linkage type revealed that ubiquitinated ZIKV E was mostly associated with K63-linked poly-Ub chains (Extended Data Fig. 1d). We also found that proteasome inhibition significantly reduced viral RNA replication at later time points, but had no effects on virus entry and/or uncoating (Extended Data Fig.1e), as previously proposed for DENV5,6. Since E is critical in mediating virus entry and proteasome inhibition does not have an effect early during infection, we focused our studies on the role of K63-linked polyubiquitination of E independent of the proteasome at early steps of the viral infection cycle.
Ubiquitination of ZIKV E on K38 and K281 during infection is important for replication in a cell-type specific manner
To test whether ZIKV is ubiquitinated on the K38 residue and further confirm ubiquitination on K281, we performed coIP assays of HA-Ub in the presence of wild-type E (E-WT) or K-to-R mutants on residues K38 and K281 (E-K38R and E-K281R). We found that ubiquitination of E was significantly reduced on E-K38R and E-K281R mutants, confirming that E is ubiquitinated on both residues (Fig. 1a). Based on the molecular weight of Ub (~8.5 kDa) and E (~48 kDa), a proportion of ubiquitinated E appears to be in the form of mono or di-ubiquitinated E, or conjugated to a mix of larger polyUb chains (a smear of over 50 kDa, Fig. 1a). To examine the functional significance of ubiquitination in the context of infectious ZIKV, we generated recombinant viruses that lack ubiquitination on E (ZIKV E-K38R, ZIKV E-K281R, or double mutant K38/281R). CoIP assays confirmed reduced ubiquitination on E-K38R and E-K281R (Extended Data Fig. 2a; coIP normalized to equal levels of input E in infected cells as shown in Fig. 1b). Compared with ZIKV E-WT, both ZIKV E-K38R and E-K281R were highly attenuated in two placenta-derived cells, JEG-3 and HTR-8 (Fig. 1c and Extended Data Fig. 2b–c). However, the replication level of ZIKV E-K38R, but not the E-K281R, was also significantly reduced in testis (15P-1) and liver (Huh-7; Extended Data Fig. 2d–e). The input dose of WT and mutant viruses was equal (Extended Data Fig. 2f). In contrast, WT and mutant viruses replicated to similar levels in mosquito C6/36 cells (Fig. 1d). The ZIKV double mutant E-K38/281R did not show an additive effect and replicated similar to the E-K38R (Fig. 1c–d). Therefore, ubiquitination of E plays an important role in viral replication in the human host, but not in the mosquito host.
Fig. 1: ZIKV-E ubiquitination on K38 and K281 promotes virus replication in cells and in vivo.
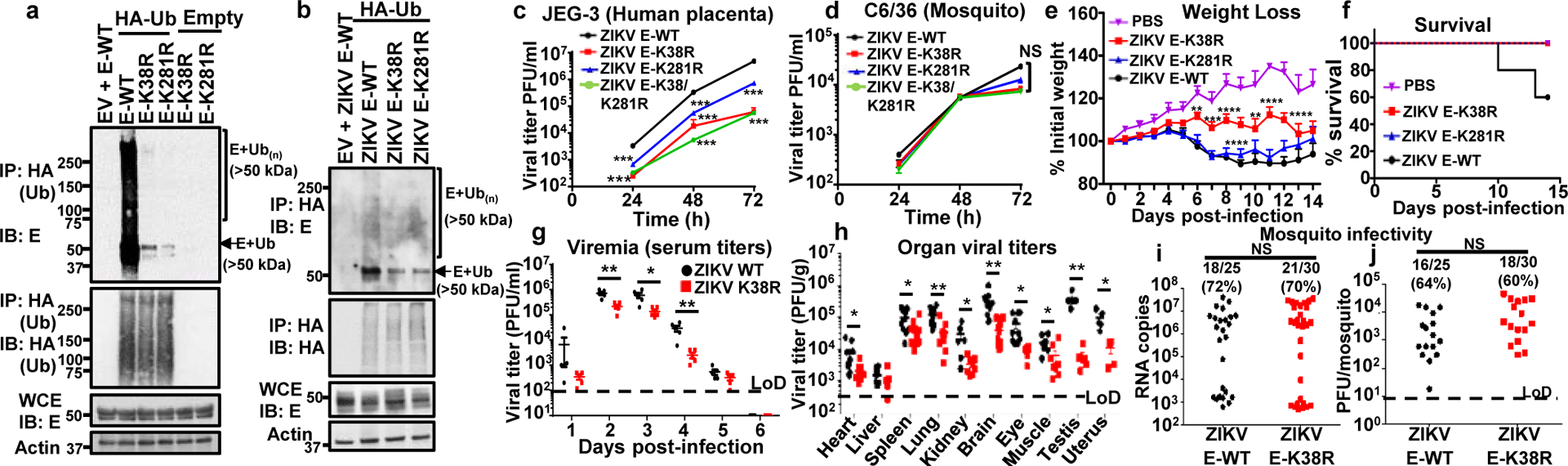
a, Whole cell extracts (WCE) from HEK293T cells transfected with empty vector (EV), E-WT or mutants and HA-Ub were used for IP with anti-HA beads. b, JEG-3 cells stably expressing HA-Ub were infected with ZIKV E-WT, or ZIKV mutants followed by AHA IP. Since the mutant viruses are attenuated, the input E was normalized for immunoprecipitation. c-d, Virus titers in supernatants from infected JEG-3 cells (c); or mosquito C6/36 (d), at MOI 0.5. Representatives from 2 independent experiments (n=3 technical replicates, mean +/−SE, ***P < 0.001). e-h, A129 mice (5 mock) or infected with ZIKV mutants (1×104 PFU, 9 mice/group, from 2 independent experiments). e, Body weight (Two-way ANOVA, Tukey’s test). f, Survival. Virus titers shown in Extended Data Fig 3c–d. g, Same experiment as in e repeated with 5 males, 5 females (Viremia), and h, virus titers (day 6 p.i.). Unpaired two-sided t-test, *p<0.05, **p<0.01. i-j, Mosquito infectivity. Aedes aegypti mosquitoes were fed with bloodmeal (106 PFU/ml) of ZIKV. At day 10, individual mosquitoes were quantified for viral RNA (qPCR, i) and virus (plaque assay, j). LoD, limits of detection. Unpaired, two-sided t-test, NS (Not significant, p>0.05).
Lack of E ubiquitination attenuates ZIKV in mammalian but not in mosquito host in vivo
Since ubiquitination of E-WT is detected in mouse brains and testis (Extended Data Fig. 3a–b), two major sites of ZIKV replication during in vivo infection16, we examined whether lack of ubiquitination on E would lead to altered tissue tropism and, consequently, pathogenesis, using an established mouse model for ZIKV infection (Ifnar1−/−, A129 mice)16. In agreement with the cell culture results, ZIKV E-K38R was significantly attenuated in vivo (Fig. 1e–f and Extended Data Fig. 3c–d). Infection with ZIKV E-WT resulted in weight loss and 40% death. In contrast, mice infected with ZIKV E-K38R showed significantly less weight loss (Fig. 1e) with no major signs of disease or death (Fig. 1f). Accordingly, ZIKV E-K38R viral titers in serum (day 2) and in brain and testis (day 8) were significantly lower than ZIKV E-WT (Extended Data Fig. 3 c–d). Although infection with ZIKV E-K281R did not show significant differences overall as compared to WT virus, it caused slightly less weight loss resulting in 100% survival. Intriguingly, the difference in virus titers between WT and mutant viruses in the eye was marginal compared to other tissues, even though the eye is another target of ZIKV infection17. To explore the possibility of differential tropism between WT and ZIKV E-K38R, we repeated the experiment and measured viral titers in additional tissues. Consistent with the data described above, ZIKV E-K38R viremia was significantly lower than ZIKV E-WT (Fig. 1g). The largest reduction in E-K38R viral titers vs ZIKV E-WT (~1 to ~2 log) was observed in brain and reproductive tissues (uterus and testis), whereas smaller differences (~2–5 fold) were found in heart, liver, lung, kidney, eye, and muscle tissues (Fig. 1h). In contrast, comparable infection rates between the WT and K38R ZIKVs were detected using quantitative RT-PCR (Fig. 1i) and plaque assay (Fig. 1j) in Aedes aegypti mosquitos at day 10 post-bloodmeal feeding. Together, these data suggest that lack of ubiquitination specifically on E-K38 reduces viral pathogenesis and virus replication in a tissue-specific manner in the mammalian host but not in the mosquito host and ubiquitination of E may play a role in tissue and species tropism.
The E3-Ubiquitin ligase TRIM7 ubiquitinates E and promotes ZIKV replication
Since our data indicate that ubiquitination of ZIKV E promotes viral replication and that ubiquitination of E may be a conserved feature in flaviviruses, we searched the literature for E3-Ub ligases that had been previously reported to promote flavivirus replication in genome-wide siRNA knockdown studies. TRIM7 (also called GNIP18), a member of the TRIM family of E3-ligases4,9, was identified as a potential pro-viral factor for YFV [supplementary Table 1 of19]. Expression of the full-length TRIM7 isoform can be detected in the known sites of ZIKV replication16, including placenta20, brain, and testis, although it is also expressed at different levels in other tissues21 (Extended Data Fig. 4a). When TRIM7 expression was knocked down via siRNA, ZIKV replication was significantly reduced in placental JEG-3 and brain-derived HTB-15 cells (Extended Data Fig. 4b–c). These pro-viral effects of TRIM7 require an intact K38 residue on E, because while ZIKV E-WT replicates to lower levels in TRIM7 JEG-3 CRISPR Knockout (TRIM7 KO) cells, no additional difference was observed upon infection with the ZIKV E-K38R mutant between WT and TRIM7 KO cells (Fig. 2a). In contrast to ZIKV, deletion of TRIM7 did not affect DENV replication in a lung A549 TRIM7 KO cell line (Extended Data Fig 4d). Although TRIM7 has been proposed to play antiviral roles against norovirus22 and potentially via induction of IFN23, and indeed TRIM7 KO cells have reduced IFNβ induction upon ZIKV infection or stimulation with double stranded RNA mimic poly I:C (Extended Data Fig. 4e–f), the data suggest that the pro-viral roles of TRIM7 are dominant over its potential IFN-mediated antiviral roles. Furthermore, ectopically expressed TRIM7 increased K63-linked polyubiquitination of WT but not the E mutants (Extended Data Fig. 4g), and correlated with increased virus titers when overexpressed in the liver cell line Huh7 (Extended Data Fig. 4h), in which ZIKV does not normally replicate to optimal levels. Consistent with these data, full-length TRIM7 as well as its short isoform, which lacks the RING-BBOX domains but retains the B30.2 domain, interact with E WT and mutants (Fig. 2b). Endogenous TRIM7 also co-immunoprecipitated with E in ZIKV infected cells (Extended Data Fig. 4i), confirming TRIM7–E interaction. Finally, TRIM7 together with the E2 conjugating enzyme UbcH5a, which was previously identified to interact and promote TRIM7-mediated K63-linked ubiquitination24,25, directly ubiquitinated recombinant ZIKV-E on both K38 and K281 in an in vitro ubiquitination assay (Fig 2c, quantification shown in 2d). Intriguingly, in this in vitro system, although ubiquitination by TRIM7 is reduced in the E-K38R mutant, it appears that some compensation on other residues can occur in these conditions (Fig 2c–d, compare lane 10, 11 with 4, 5).
Fig. 2. TRIM7 ubiquitinates ZIKV-E and promotes virus replication.
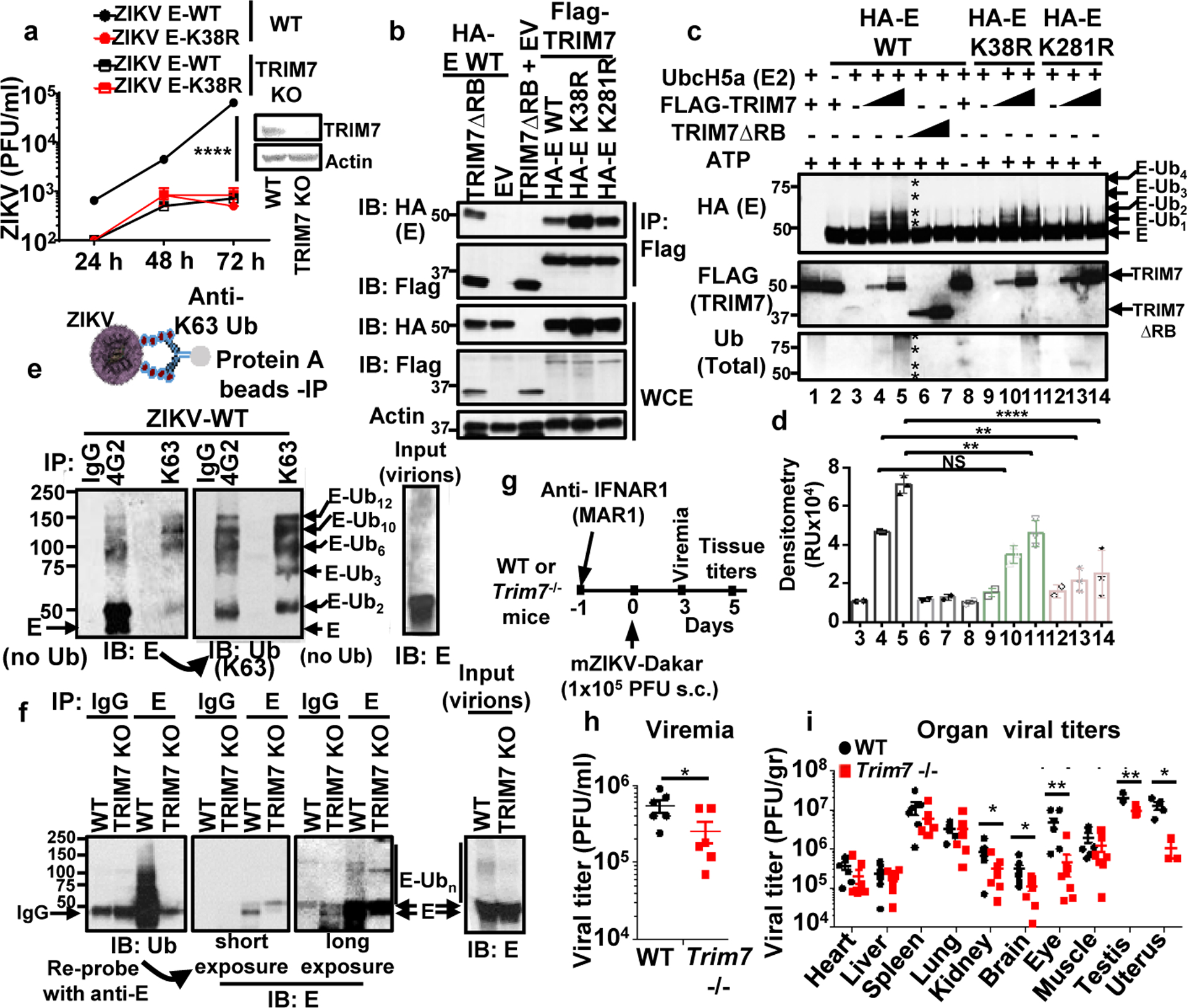
a, Virus titers from TRIM7 JEG-3 Knockout (TRIM7 KO) infected with ZIKVs (MOI 0.5). (n=3, technical replicates, mean +/−SE, multiple t-test, Holm-Sidak correction, ****p<0.0001). b, HEK293Ts transfected with TRIM7 or a short isoform (TRIM7 ΔRB), WT and mutant E, followed by IP. Representative of 2 independent experiments. c, TRIM7 ubiquitinates recombinant ZIKV-E on both K38 and K281 in an in vitro ubiquitination assay. Representative of four independent experiments. Densitometry shown in d (N=3, mean +/− SE, One-way Anova, Tukey’s multiple comparison, **p<0.01, ****p<0.0001, NSp>0.5).). e, K63-linked polyubiquitinated E was detected on ZIKV particles concentrated from supernatants from Vero after IP with anti-K63-linked or anti-E 4G2 antibodies. After immunoblot (IB) with anti-E, the blots were re-probed with anti-K63-Ub. f, IP with anti-E (4G2) of virus stocks from WT or TRIM7 KO JEG-3 cells. Representative from 2 independent experiments. g-i, Trim7−/− and Trim7+/+ littermates from 3 separate CRISPR KO lines (4–5 weeks old) were treated i.p. with anti-IFNAR1 (MAR1–5A3). Next day infections performed with mouse-adapted ZIKV Dakar (footpad, 105 PFU, n=7 Trim7+/+, 8 Trim7−/− mice). Serum (h) and tissue titers (i). Unpaired, t-test, two-sided *p<0.05, **p<0.01.
Since TRIM7 was previously suggested to localize in the Golgi18, we hypothesized that TRIM7 may be recruited to intracellular membranes during exocytosis of progeny virus where it could ubiquitinate E. In mock cells, TRIM7 showed weak diffuse staining with some apparent localization in cytoplasmic structures, as previously reported8,18. In addition, a low proportion of TRIM7 co-localized with WGA, a lectin dye that labels glycoconjugates enriched in Golgi26 (Extended Data Fig. 5a–b, middle panels). However, upon ZIKV infection, there was a striking reorganization of intracellular membranes, as previously reported during flavivirus infection27. Furthermore, ZIKV infection re-localized TRIM7 to these membranes where a small proportion co-localized with E (Extended Data Fig. 5a–b, top panels). Cell fractionation also showed both TRIM7 and its E2-conjugating enzyme UbcH5a co-fractioned with the reticulum marker calnexin in infected cells (Extended Data Fig. 5c).
Infectious ZIKV particles contain a proportion of Ubiquitinated E
We next tested whether mature ZIKV particles released from infected cells contained ubiquitinated E. Supernatants collected from ZIKV-infected JEG-3 cells showed detectable levels of ubiquitinated E (Extended Data Fig. 6a). Moreover, K63-linked polyubiquitinated E was detected on ZIKV particles concentrated from supernatants from Vero cells after IP with an anti-K63-linked specific antibody, and was also able to detect ubiquitinated E when virions were isolated using an anti-E (4G2) antibody (Fig. 2e). Based on molecular weight, potentially up to 12 Ub molecules covalently attached to E could be detected (Fig 2e). In addition, ubiquitinated E was also detected from virus stocks grown and concentrated from WT JEG-3 but was strongly reduced in virus grown in TRIM7 KO JEG-3 cells (Fig. 2f). While deletion of TRIM7 in A549 cells also reduced the infectivity of progeny virus (Extended Data Fig. 6b), it did not appear to affect viral RNA replication or virion release (Extended Data Fig. 6c–d). K63-linked polyubiquitinated E was also detected after IP of E from supernatants containing WT-ZIKV, and was reduced in ZIKV-K38R and K281R mutants (Extended Data Fig. 6e). Interestingly, ubiquitinated E was also detected after IP of ZIKV grown in mosquito C6/36 cells (Extended Data Fig. 6e, lane 7), although it appeared more in shorter K63-linked polyUb chains as compared to the longer Ub chains found on ZIKV E-WT grown in JEG-3 (Extended Data Fig. 6e). Importantly, DENV particles also contained ubiquitinated E (Extended Data Fig. 6f). Together, these data indicate that flaviviruses released from cells contain a proportion of ubiquitinated E.
In an effort to quantify the proportion of ubiquitinated ZIKV particles, we performed IPs with an antibody against Ub-K63 and ZIKV supernatants obtained from different cells, and measured the proportion of viral RNA copies from input virus (Extended Data Fig. 7a). Approximately 5–6% of ZIKV particles grown in placenta JEG-3 cells could be detected with the anti-K63-Ub, which is significantly higher than the proportion of ubiquitinated E of ZIKV grown in Vero cells, and ZIKV grown in JEG-3 TRIM7 KO cells contained near to background levels of ubiquitinated E as compared to an IgG control (Extended Data Fig 7a). Additional evidence that the intact virus particle contains a proportion of ubiquitinated E comes from cryo-electron microscopy (cryoEM) studies. Ubiquitinated Zika virions were labeled using anti-K63-Ub and a gold-anti-IgG secondary antibody. After purification of virus-antibody complexes by sucrose gradient, approximately 15% of virus particles showed at least one gold particles close to the virus (mostly between 50–150 nm away from the viral particle) (Extended Data Fig. 7b). In contrast, no gold particles were found close to the ZIKV-K38/281R double mutant in the same conditions.
TRIM7 is a determinant of ZIKV tissue tropism in vivo
To test the role of TRIM7 in ZIKV replication in vivo, we generated Trim7−/− mice (mutants sequences shown in Supplementary Figure 2) and infections were performed using a previously established protocol28 (Fig. 2g–i). Strikingly, viral titers in serum (Fig 2h) and in kidney, eye, brain, and reproductive tissues (uterus and testis) of Trim7−/− were significantly lower as compared to WT mice (Fig. 2i). In contrast, ZIKV replicated to similar levels in tissues from WT and Trim7−/− including heart, liver, lung and muscle (Fig. 2i), indicating that TRIM7 indeed promotes virus replication in a tissue specific manner. These data suggest TRIM7 may be a determinant of tissue tropism.
Ubiquitination of E is important in virus entry
Since E mediates virus attachment to host cells and induces virus-endosome membrane fusion15,29,30, we examined whether ubiquitination contributes to virus entry. Endosome-virus membrane fusion was analyzed using a lipophilic dye (DiOC18) to label WT and ZIKV mutants. The ability of ZIKV-E-K38R to promote virus-endosome fusion was significantly decreased as compared to ZIKV-E-WT in both JEG-3 and A549 cells (Fig. 3a, and Extended Data Figs. 8 and 9). Ammonium chloride (NH4Cl) treatment, which blocks acidification of the endosome and subsequent fusion, served as a control.
Fig. 3. Ubiquitination of ZIKV-E promotes virus attachment and virus-endosome membrane fusion.
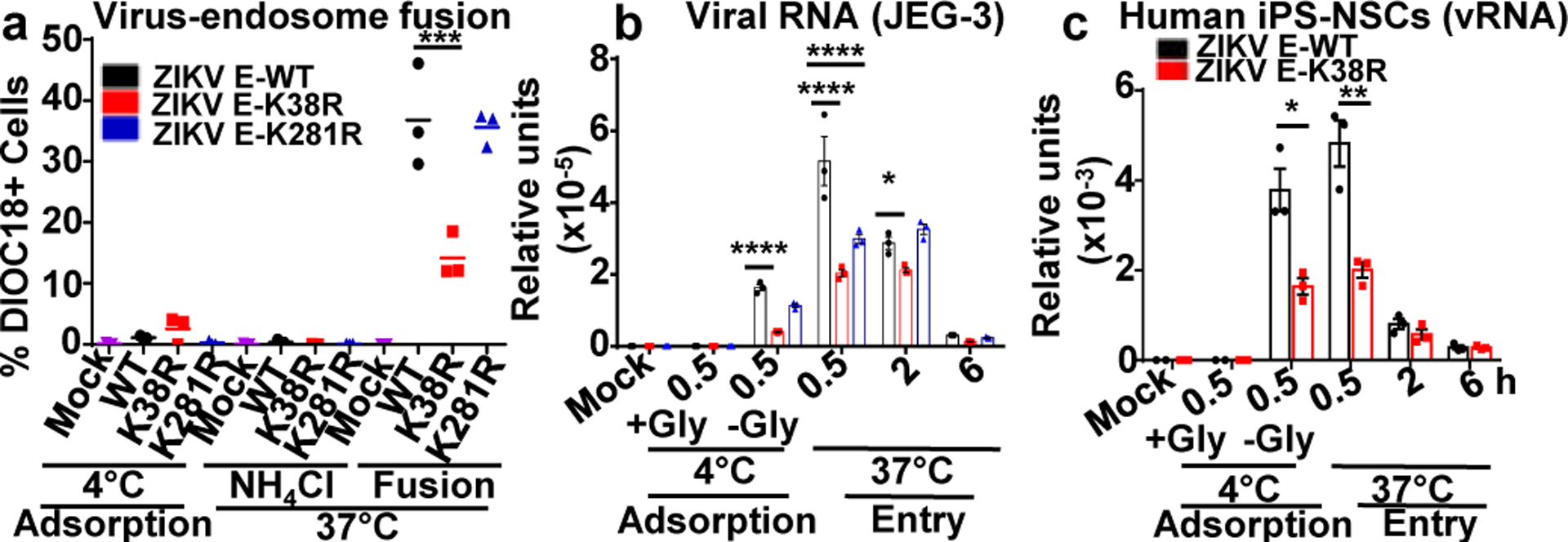
a, ZIKVs were labeled with DiOC18. After filtration, viruses were incubated at 4°C with JEG-3 at MOI 2. After 30 min were washed and collected as adsorbed viruses. Additional samples were then incubated at 37°C for 1h, +/−NH4Cl to block acidification (as control), washed, fixed and quantification by FACS. b-c, Ubiquitination of E promotes virus attachment. JEG-3 (b) or human primary induced-pluripotent neural stem cells (hiPS-NSCs, c), were incubated with viruses as described in a. Viral RNA (qPCR). Incubation at 4°C without Glycine (-Gly) represents attached viruses. Each are representative of 2 independent experiments (N=3, technical replicates, mean +/−SE. Unpaired t-test, two-sided, *p<0.05, **p<0.01 ***p<0.001, ****p<0.0001.
Although the ZIKV-E-K281R did not significantly attenuate fusion in JEG-3 cells, it slightly reduced fusion in A549 cells (Extended Data Figs. 9b–c), suggesting that ubiquitination of E on K281 may affect virus entry/fusion in a cell-type specific manner. Further evidence acquired by measuring viral RNA of adsorbed viruses to cells indicates that the specific ubiquitination on E-K38, and not on E-K281, is responsible for efficient virus attachment to the host cell (Fig. 3b, adsorption at 4°C). Reduced levels of ZIKV E-K38R attachment as compared to ZIKV E-WT were also observed in human primary induced-pluripotent neural stem cells (hiPS-NSCs, Fig. 3c), brain microvascular endothelial (hBMECs), and astrocytes (Extended Data Fig. 10a–b), which also correlated with reduced virus replication by plaque assay (Extended Data Fig. 10c–e). These effects are not due to reduced glycosylation of E because N-glycosidase F can cleave both ZIKV-E mutants (Extended Data Fig. 10f). Indeed, ZIKV E-K38R grown in WT JEG-3 cells has reduced capacity to attach to cells as compared to WT ZIKV, and this is comparable to the reduced ability of ZIKV grown in TRIM7 KO cells to attach to these cells (Extended Data Fig. 10g). To further rule out that reduced attachment of ZIKV E-K38R is due to any minor structural changes caused by the K-to-R mutation, and confirm that ubiquitination enhances entry and replication, WT ZIKV was treated with the deubiquitinase (DUB) ovarian tumor (OTU) of Crimean Congo hemorrhagic fever virus (CCHFV)31, which can cleave Ub chains (Extended Data Fig. 10h). Ubiquitin removal with the OTU reduces ZIKV attachment as compared to an OTU (2A) mutant with reduced activity (Extended Data Fig. 10i). Treatment with this DUB also reduced WT ZIKV replication (Extended Data Fig. 10j).
Antibody mediated neutralization of ZIKV infection
We examined whether the same monoclonal antibody against K63-linked polyUb, used in the IP experiments described above, could neutralize ZIKV infection (Fig. 4a–c). Pre-treatment with this antibody significantly decreased infection of ZIKV WT and ZIKV-E-K281, but not ZIKV-E-K38R, in a dose dependent manner as compared to an IgG control (Fig. 4a). This neutralizing effect was specific for the K63-Ub-linkage because antibodies against K48-linked polyUb did not have major effects (Fig. 4b). As an additional control, we used pan-flavivirus anti-E (4G2) monoclonal antibody, which neutralized WT and ZIKV mutants, especially at higher antibody concentrations (Fig. 4c). The specificity of the anti-K63 polyUb antibody in neutralizing ubiquitinated E could be confirmed in a competition assay in which addition of purified K63-linked polyUb chains reduced the neutralizing activity of anti-K63-Ub, but not of anti-E (4G2) (Fig. 4d). Furthermore, administration of this anti-K63-Ub antibody in mice one day prior to ZIKV infection significantly reduced virus titers as compared to an IgG or PBS control in vivo (Fig. 4e). Importantly, ZIKV produced in mosquito cells was less sensitive to neutralization by the anti-K63-Ub antibody treatment (Fig. 4f), while the anti-E 4G2 antibody inhibited at high concentrations (Fig. 4g).
Fig. 4. Specific anti-K63-linked polyUb antibody neutralizes ZIKV in vitro and in vivo.
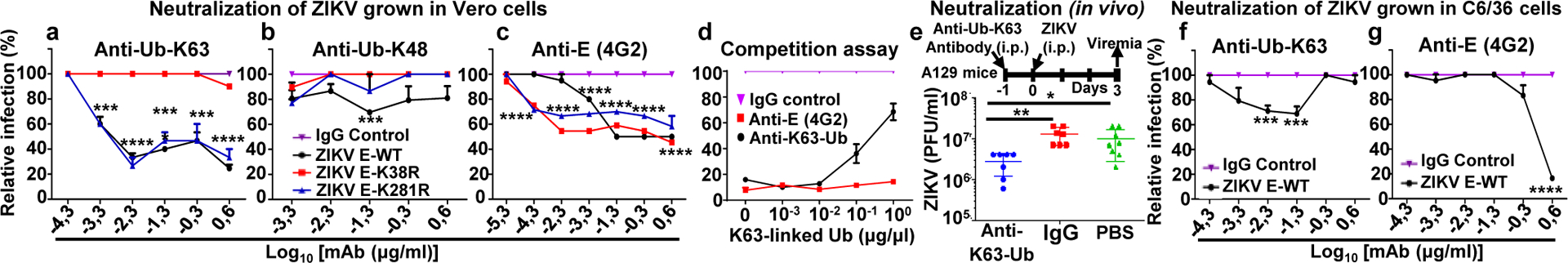
a-d, WT and mutant viruses grown in Vero cells or mosquito C6/36 cells (f-g) were incubated at 37°C with dilutions of indicated antibodies for 1h, followed by plaque assay. d, Purified K63-Ub chains were incubated together with ZIKV WT and anti-K63-Ub antibody, IgG control or 4G2. Relative infection was calculated as a percentage of antibody effects on each virus relative to its own IgG control (n=3, technical replicates, mean +/−SE, Two-Way ANOVA, with Tukey correction.). e, In vivo neutralization assay, A129 mice were inoculated i.p. with anti-K63-Ub or an IgG control. Next day mice were infected with ZIKV (1×104 PFU). Viremia (day 3). N=7, mean +/−SE, Unpaired t-test, two-sided, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Ubiquitination of ZIKV envelope protein promotes binding to the Tim-1 receptor
Although still unclear, multiple receptors have been proposed to mediate ZIKV attachment in specific cell types, including DC-SIGN, AXL, Tyro3, and Tim-132. We tested whether ubiquitination of E may enhance affinity for one of the proposed receptors, Tim-1 (HAVCR1). coIP assays revealed that recombinant Tim-1 interacts with ectopically expressed ZIKV-E (Fig. 5a), as well as with infectious ZIKV E-WT but only minimally with ZIKV-E K38R viral particles (Fig. 5b), suggesting that ubiquitination on K38 residue is responsible for this interaction. In support of this, recombinant purified K63-, but not K48-linked polyUb chains, interact with Tim-1 in the absence of E (Fig 5c). Furthermore, addition of K63-linked polyUb chains compete with ZIKV particles for interaction with Tim-1, while K48-linked polyUb chains do not (Fig. 5d). Ubiquitinated E is also likely to mediate virus attachment to cells, at least in part via Tim-1, because knockdown of Tim-1 in JEG-3 cells significantly reduced the levels of WT ZIKV attachment, whereas the ZIKV E-K38R virus attachment was not further reduced in Tim-1 knockdown cells as compared to controls (Fig 5e). Finally, infection of Tim-1−/− mice (C57BL/6, Ifnar1−/− background) with WT ZIKV exhibited a small (~2.5 fold) but statistically significant reduction in virus titers in the brain as compared to Tim-1+/+ controls (Fig. 5f). In contrast, while replication of ZIKV E-K38R was strongly reduced as compared to ZIKV E-WT in the brain, no difference was observed between WT and Tim-1−/− mice. In addition, no differences were observed in other tissues (e.g. spleen, lung, kidney) between WT and K38R viruses and between Tim-1+/+ and Tim-1−/− mice (Fig 5f). The data suggest that although Tim-1 is not the only receptor that mediates ZIKV entry, it may play a role in specific cell types/tissue like the brain. Taken together, K63-linked ubiquitination on the ZIKV E-K38 residue promotes efficient virus attachment to host receptors, and it is at least in part mediated by Tim-1.
Fig. 5. Ubiquitination of ZIKV E promotes binding to the Tim-1 receptor.
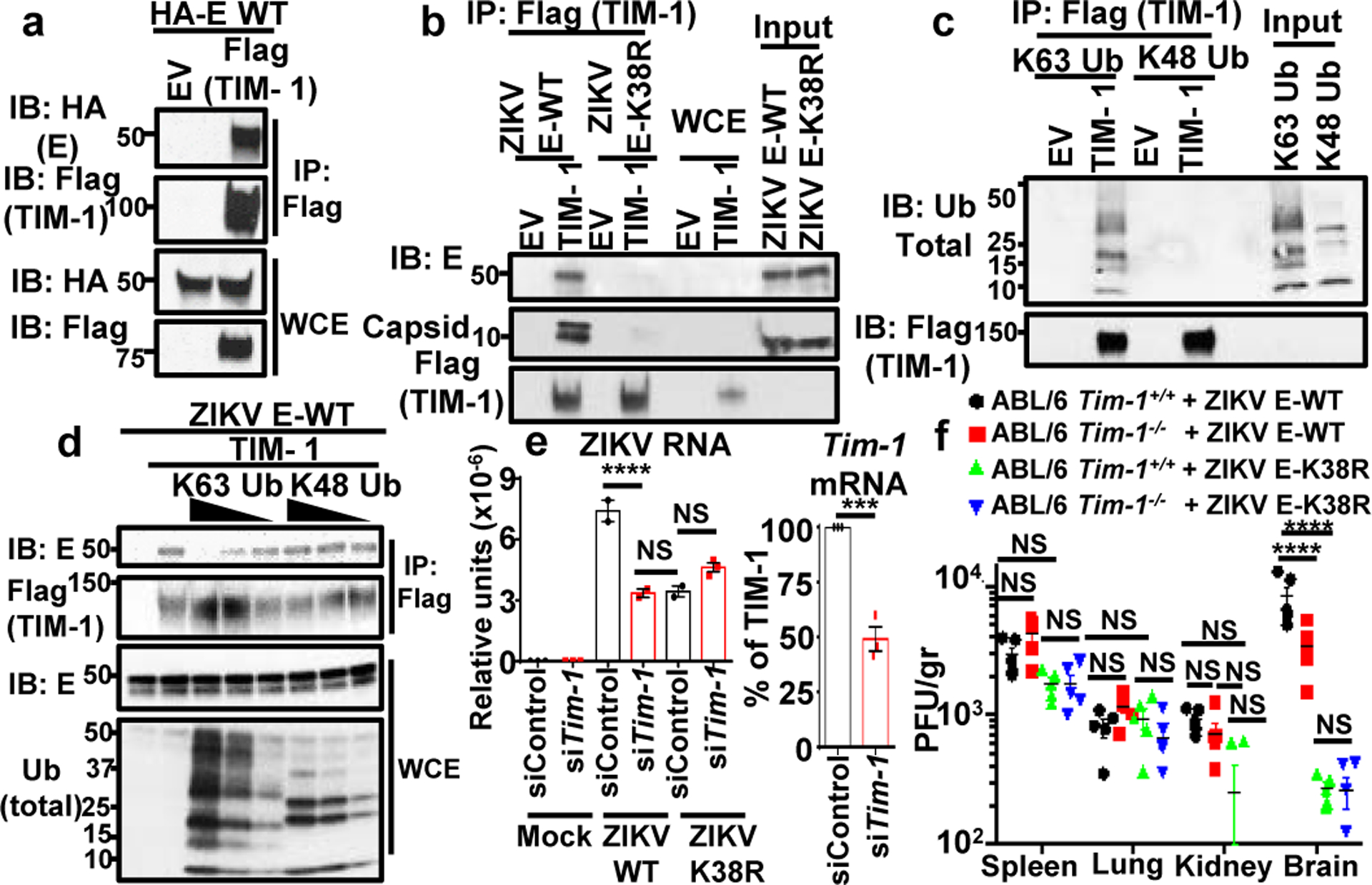
a, WCE from HEK293Ts transfected with empty vector (EV), HA-E WT and Flag-TIM-1 were used for IP with anti-Flag beads. b, Interaction of ZIKV E-WT but not ZIKV-E K38R viral particles with TIM-1. Ectopically expressed TIM-1 was isolated using anti-Flag beads. After washes, viruses were incubated with the TIM-1. c, Interaction of K63-, but not K48-linked polyUb, with TIM-1 in the absence of E, coIP assay. d, WT was bound to TIM-1 containing beads (as in b). Increasing amounts of purified K63-Ub or K48-Ub chains were added. Representatives of 2 independent experiments (a-d). e, Virus attachment (viral RNA, qPCR) after Tim-1 knockdown in JEG-3 (n=2, mean +/−SE, One-Way ANOVA, multiple comparison, Tukey correction, ****p<0.0001, *p<0.05, NS p>0.05). f, Titers in infected Tim-1−/− mice, n=5/group, mean +/−SE, Two-Way ANOVA multiple comparison, Tukey correction. ****p<0.0001, NS p>0.05.
Discussion
We have shown that ubiquitinated E present in infectious ZIKV virions promotes efficient virus entry, however is not a requirement for virus replication, as both ZIKV mutants are attenuated but still able to replicate. Our data support a model in which ubiquitination on E-K38 enhances viral attachment to the cell receptors, thereby increasing the efficiency of virus replication. This occurs in a tissue specific manner and could partially be explained by the expression levels of TRIM7 in combination with other factors including expression of the E2-conjugase UbcH5a or cellular receptors, which may also contribute to the characteristic ZIKV tropism. Our data showing ubiquitination on residue K38, which is conserved in members of the Flaviviridae family33, and that dengue virions also contain K63-linked polyubiquitinated E, raises the possibility that ubiquitination on K38 may be used as a general mechanism in flavivirus entry. An additional ubiquitination site on K281 of ZIKV-E was identified, which is not present in other flaviviruses. The combined ubiquitination on both residues could also contribute to the differential tropism between ZIKV and other flaviviruses.
Despite existing literature on the crucial role of E in binding to host cell receptors or neutralizing antibodies, and on structural studies including ZIKV or E from other flaviviruses14,34,35, to our knowledge, no studies have detected ubiquitination on E of flaviviruses. One possible explanation is that many structural studies have used ZIKV prepared from mosquito cells, which according to our data may produce virions with reduced ubiquitination levels. The presence of only a small proportion of ubiquitinated E on the viral particle, as suggested by our cryoEM, may also contribute to previous observations of imperfect virion symmetry36. Finally, our studies indicate that the anti-K63-linked Ub antibody has neutralizing activity in vivo and could provide a novel therapeutic approach against ZIKV.
Extended Data
Extended Data Fig. 1: Ubiquitination of flavivirus E protein.
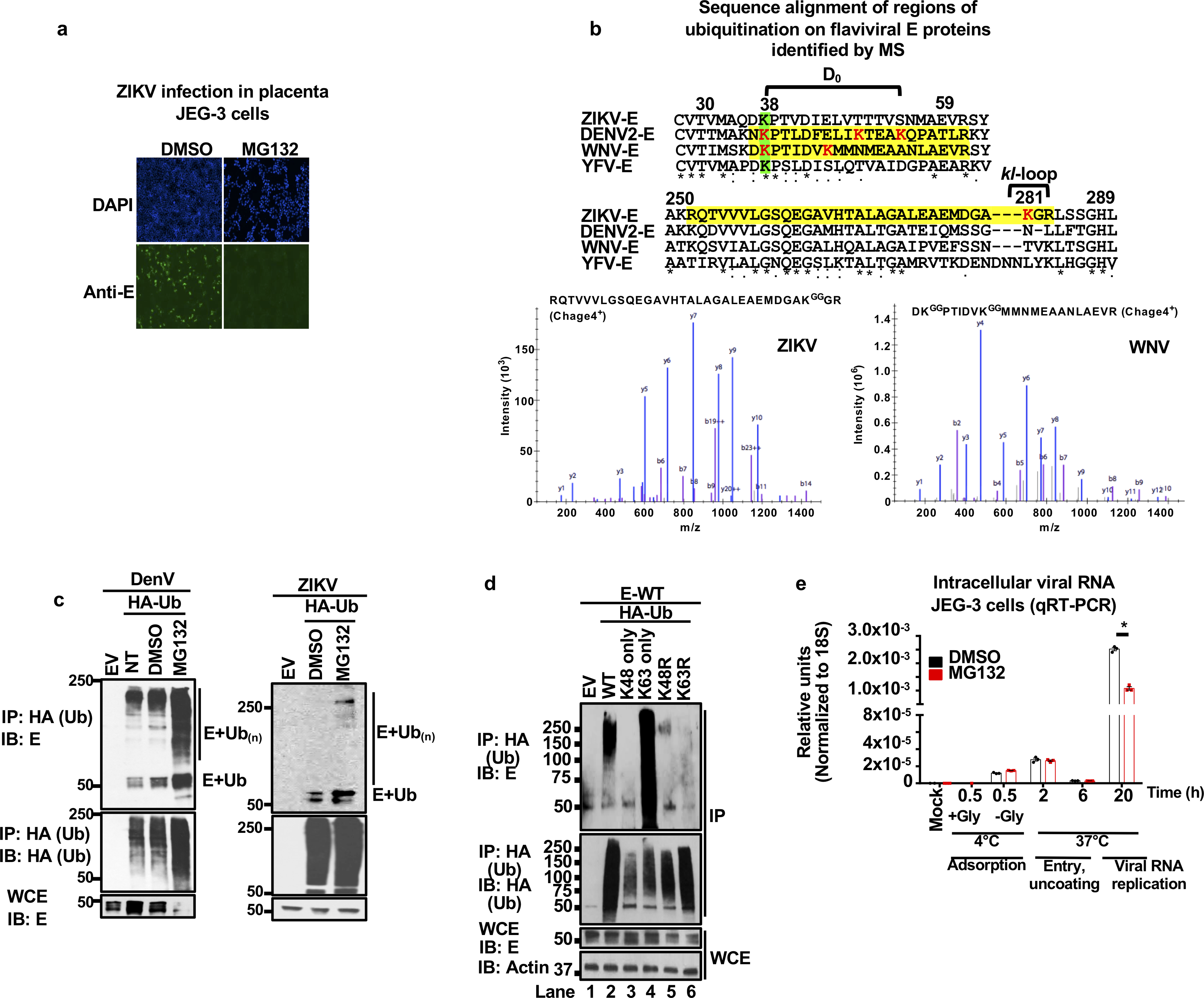
a, Proteasome inhibition blocks ZIKV replication. JEG-3 were pre-treated with DMSO or MG132 (2 h) followed by ZIKV infection (MOI 2, 24 h, visualized by immunofluorescence with anti-E 4G2). b, Ubiquitinated peptides from flavivirus-infected cells identified by mass spectrometry (peptides highlighted in yellow, diglycine residues indicating ubiquitination in red, conserved residues in green). Sequences for strains ZIKV FSS13025 (GenBank: KU955593.1), DENV-2 Y98P (JF327392.1), WNV NY99 (DQ211652) and YFV (ANC33490.1). JEG-3 cells were used for ZIKV infections, Huh7 for DENV, and A549 for WNV (repeated in U2OS cells with identical results, two independent experiments). Representative Mass Spectra for ubiquitinated peptides found for WNV are shown. b and y ions are indicated in blue and red, respectively. c, Whole cell extracts (WCE) from DENV or ZIKV HuH7 transfected with HA-Ub and infected (MOI 2, 20h), followed by DMSO or MG132 treatment (6 h) were used for HA immunoprecipitation (IP). Immunoblots (IB) are shown. NT: non-treated d, WCE from cells transfected with vectors expressing E and Ub-WT or all K-to-R mutants except for K63 or K48 (only), or K48R and K63R, followed by IP. e, JEG-3 pre-treated with MG132 or DMSO. Cells were incubated with ZIKV at 4°C for 30min, followed by wash with or without glycine to test virus adsorption. Additional samples were then switched to 37°C to allow virus internalization. Viral RNA detection by qRT-PCR. Representative of 2 independent experiments, N=3 technical replicates, mean +/− SE, Unpaired t-test, two-sided, * (p < 0.05). NS (Not significant). All experiments are representatives from 2 independent experiments, with similar results.
Extended Data Fig. 2: Differences in Zika virus cell tropism are associated with ZIKV E-ubiquitination.
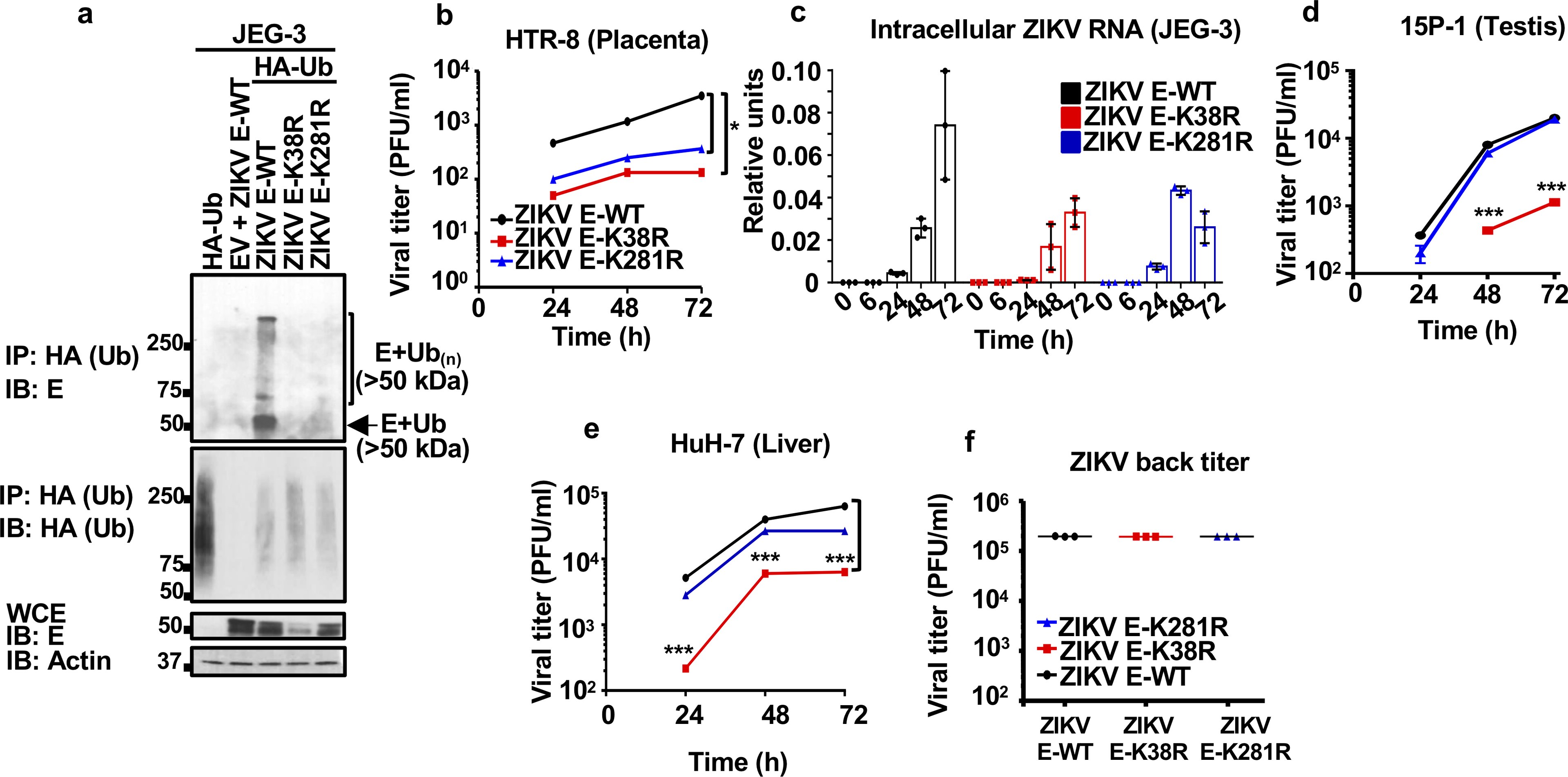
a, JEG-3 cells stably expressing HA-Ub were infected with ZIKV E-WT, or recombinant infectious ZIKV E-K38R and ZIKV E-K281R mutants. WCE were used for IP with HA beads (same experiment as shown in Figure 1b, but without normalizing the input for the IP). Reduced ZIKV E-K38R replication can be seen represented by the levels of E in the WCE. Representative of two independent experiments. b-e, Different cell types were infected with either ZIKV E-WT, E-K38R, or E-K281R (MOI 0.5). Cells were lysed for RNA extraction and virus quantification by qRT-PCR in c, and supernatants were collected for plaque assays at different time-points, for placenta HTR-8 in b, testis 15P-1 in d, liver HuH7 in e. f, Back titration for the virus used on these experiments, and for Fig 1e and 1f. Representatives from 2 independent experiments (n=3 technical replicates, mean +/− SE, multiple t-test, Holm-Sidak correction, *p < 0.05, ***p < 0.001)
Extended Data Fig. 3: Ubiquitination of E in tissues from infected mice.
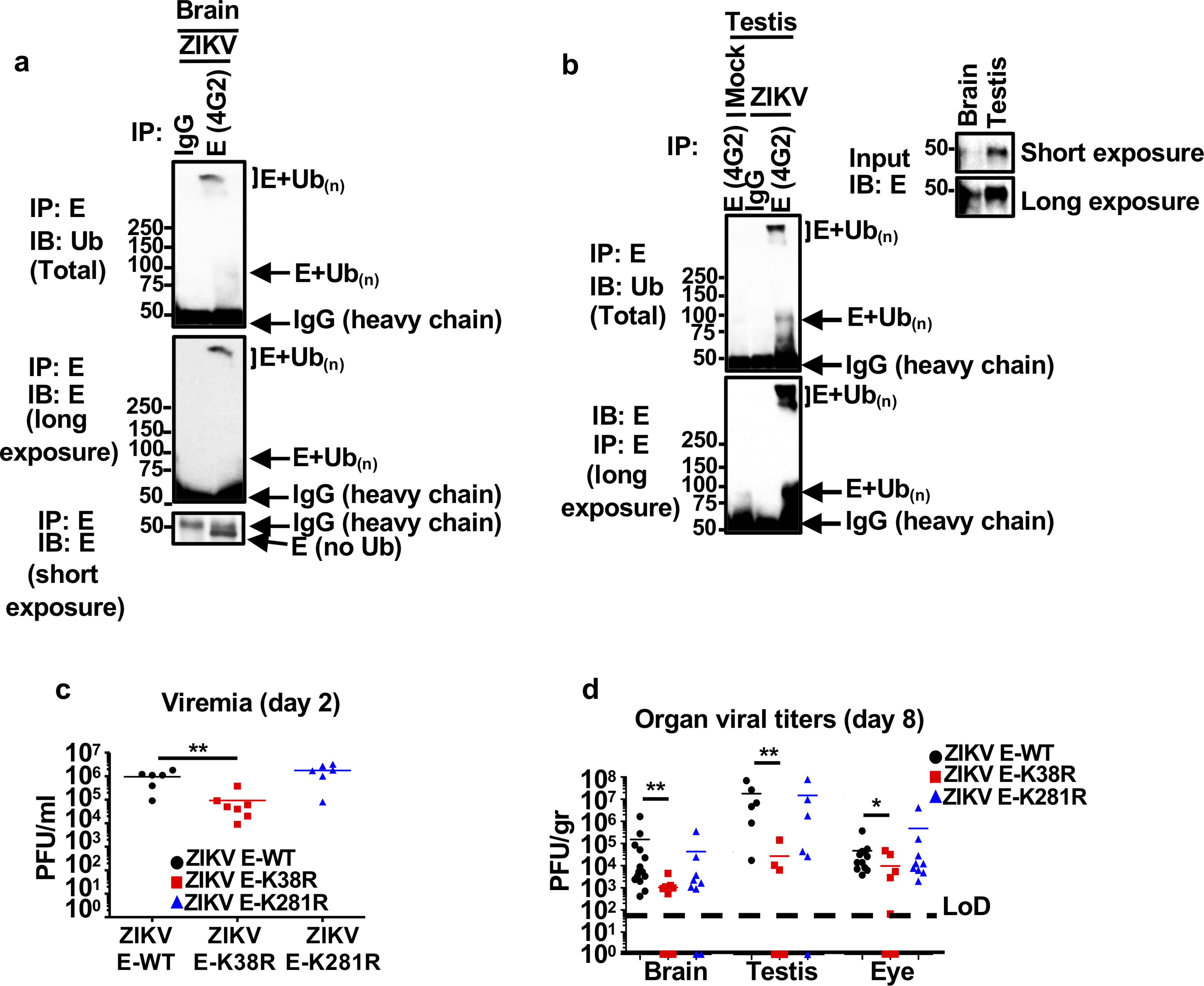
Tissues from testis (a), and brain (b) from mock or ZIKV-WT infected A129 mice were collected at day 8 post-infection. Tissues were homogenized and 200 μg of total input protein was used for immunoprecipitation (IP) of E using 4G2 antibody or an IgG control. Ubiquitination of WT-E was detected with anti-Ub antibody by immunoblot (IB). IPs shown are from mixed tissue lysates from 3 different mice. c-d, A129 mice (male and females) were mock treated (5 mice) or infected with ZIKV E-WT, ZIKV E-K38R or ZIKV E-K281R (1×104 PFU, 9 mice per group, combined from 2 independent experiments). Weight loss and survival is shown in Fig 1e–f. c, serum titers (viremia), were determined at day 2 p.i. by plaque assay, after blood collection from 6 mice for ZIKV E-WT and K38R, and 7 mice for ZIKV E-K38R. d, virus titers (at day 8 p.i.) in brain (14 mice for ZIKV E- WT, and 9 mice for ZIKV E- K38R and ZIKV E- K281R), testis (6 mice/each gorup), and eye (14 mice for ZIKV E-WT, and 9 mice for ZIKV E-K38R and E-K281R). Unpaired, t-test, two-sided, *p < 0.05, **p < 0.01.
Extended Data Fig. 4: TRIM7 interacts with and ubiquitinates ZIKV-E and promotes virus replication.
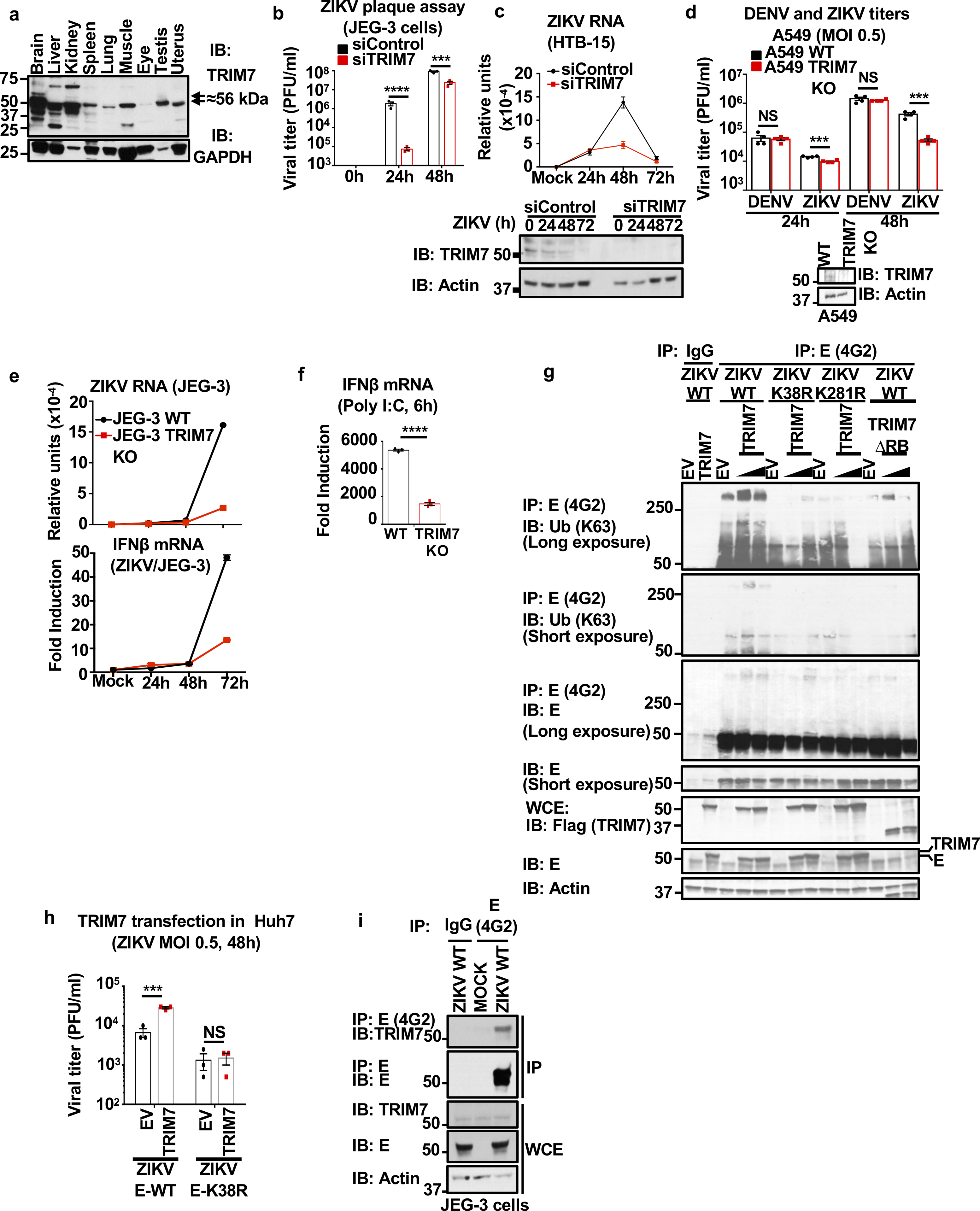
a, Differential expression of TRIM7 in mouse tissues by immunoblot (IB). Predicted molecular weight of full-length TRIM7 is 56 kDa. b-c, TRIM7 knockdown (24h) in JEG-3 (b) or brain HTB-15 cells (c) followed by infection with ZIKV (MOI 1). c, Viral RNA levels were determined by qRT-PCR at different time points (upper panel). TRIM7 knockdown efficiency was confirmed by western blot (lower panel). d, TRIM7 Crispr Knockout A549 and WT parental cells were used for infections with ZIKV or DENV at an MOI of 0.5. Bottom panel shows immunoblot of TRIM7. Plaque assays from supernatants collected at different time-points is shown. e-f, Infections of WT and TRIM7 CRISPR KO JEG-3 cells with ZIKV (MOI 0.5) or poly I:C stimulation (f, transfection of 10 μg/ml with lipofectamine 2000). Quantification of ZIKV RNA (e, top) and IFNβ mRNA expression (e, bottom and f) by qPCR. g, Overexpression of TRIM7 enhances K63-linked polyubiquitination of E-WT but not E-K38R or E-K281R. HEK293T cells were transfected with vectors expressing E-WT, E-K38R, or E-K281R and different amounts of HA-TRIM7 (350ng, 700ng). Thirty hours post-transfection, cells lysates were used for immunoprecipitation (IP) with anti-E 4G2 or isotype control. Immunoblots (IB) with indicated antibodies. h, Transfection of Huh7 cells with empty vector (EV) or vector expressing TRIM7. After 48h cells were infected with ZIKV E-WT or E-K38R. N=3, technical replicates, mean +/−SE. Multiple t-test, two-sided, ***p<0.001, ****p<0.0001, NS (Not significant, p>0.05). i, Endogenous TRIM7 interacts with E in ZIKV placental JEG-3 infected cells. Cells were infected with ZIKV-WT (MOI 2). Thirty hours p.i. cells were lysed and whole cell extracts (WCE) were used for IP with anti-E (4G2) or isotype control. Representatives of 2 independent experiments
Extended Data Fig. 5: TRIM7 co-localizes with E in the Golgi and ZIKV-E is detected in supernatants from infected cells.
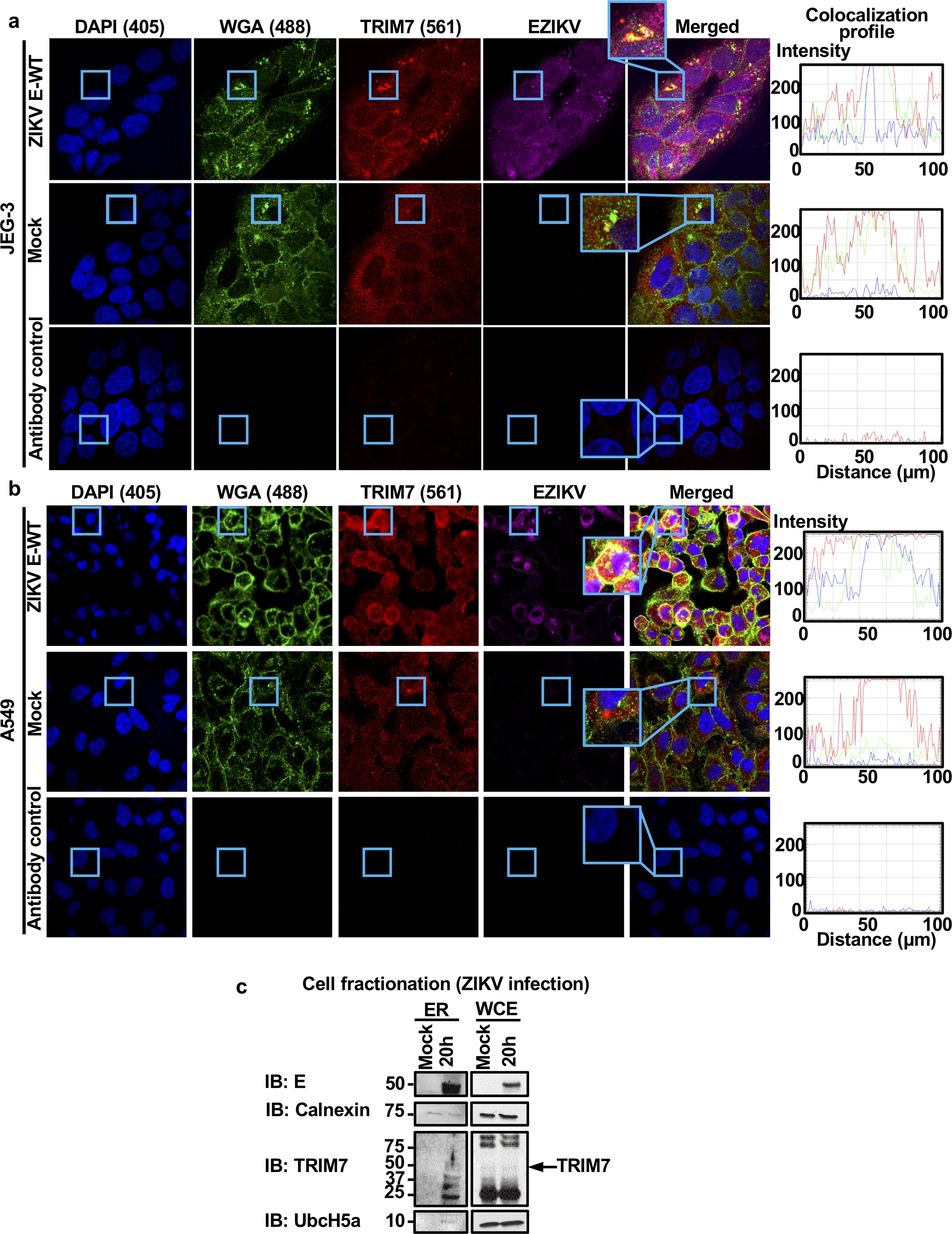
a-b, JEG-3 (a) or A549 (b) cells were mock treated or infected with ZIKV (MOI 2). Twenty-four hours post-infection, cells were fixed and stained for endogenous TRIM7 (red), Golgi (WGA-FITC, green), and E (4G2, purple) for confocal microscopy. Colocalization is shown in rectangles, and RGB profile graphs are on the right. All images were processed identically using the same conditions with ZEN 2.5.75.0 (Zeiss) and RGB profiles were obtained using ImageJ (NIH) v1.52e. c, cell fractionation of infected JEG-3 cells (20h, MOI 2) for ER was performed following manufacturer instructions (Sigma). Representative of two independent experiments.
Extended Data Fig. 6. Zika and dengue virions contain ubiquitinated E, and TRIM7 is important for ZIKV but not DENV replication.
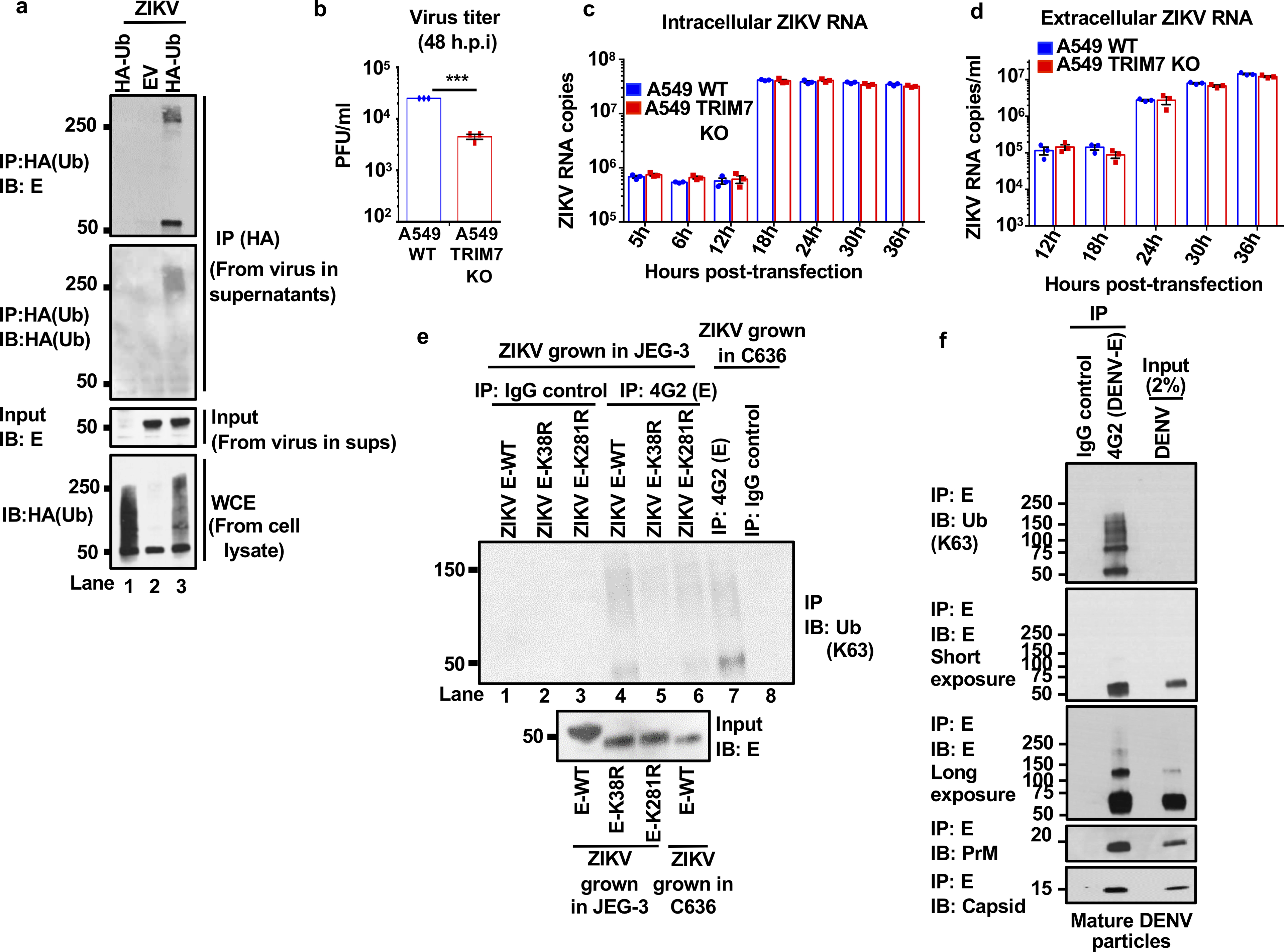
a, Supernatants collected from JEG-3 ZIKV-infected cells stably expressing HA-Ub showed detectable levels of ubiquitinated E. Viruses were immunoprecipitated with anti-HA beads. Immunoblot (IB) for E and HA-Ub. The lower panel shows the total HA-Ub levels expressed in these HA-Ub stable cell lines. b, Infection of A549 WT or TRIM7 KO cells with ZIKV. c-d, Transfection of A549 WT or TRIM7 KO cells with in vitro transcribed ZIKV RNA. Viral RNA was quantified by qPCR from cell lysates (intracellular, c) or from supernatants (extracellular, d). Representatives of 2 independent experiments. N=3, technical replicates, mean +/− SE. e, K63-linked polyubiquitinated E was detected on ZIKV E-WT particles but reduced in ZIKV E-K38R and K281R, after IP anti-E 4G2 antibody. Although reduced, ubiquitination on E from ZIKV grown in mosquito C6/36 can also be detected. f, DENV virus from supernatants from infected BHK-21 was immunoprecipitated with anti-E antibody (4G2) or an IgG control. Immunoblot (IB) for K63-linked polyUb and each viral protein are shown. Representatives of 2 independent experiments.
Extended Data Fig 7. Proportion of ubiquitinated E in Zika virions and CryoElectron microscopy of ubiquitinated ZIKV.
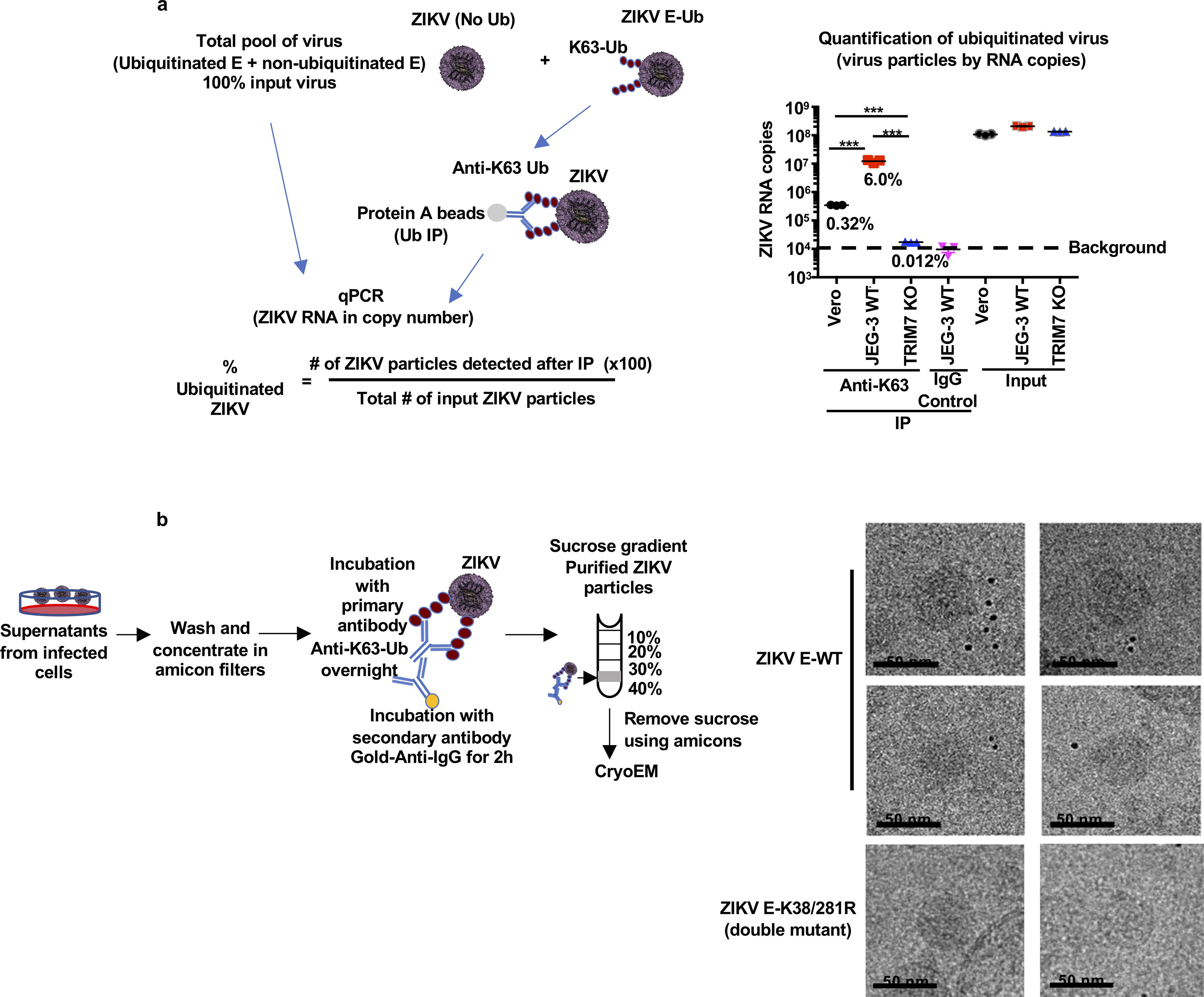
a-b, ZIKV stocks were grown in Vero cells, JEG-3 WT or TRIM7 KO cells were used for IP using an anti-K63-Ub antibody, or an IgG control to set the background levels. The immunoprecipitated virus as well as a sample of input viruses were lysed in Trizol for virus RNA quantification by qPCR (a). The virus RNA copy number was determined using a standard of purified ZIKV RNA and its known molecular weight. The proportion of ubiquitinated virus was calculated taking as 100% the input virus. N=3 technical replicates, mean, unpaired two-sided t-test, ***p<0.001. b, CryoElectron microscopy of ubiquitinated ZIKV. Experimental approach. Supernatants from Vero cells infected with WT or ZIKV E-K38/281R double mutant were washed and concentrated in Amicon filters followed by labelling with a primary antibody against K63-Ub and secondary nano-gold-labelled antibody. Virus-antibody complexes were then purified by sucrose gradient. A visible band containing these complexes was recovered and passed through Amicon filters to remove sucrose and concentrate the complexes. Samples were flash-frozen in liquid ethane cooled to liquid nitrogen temperatures on holey carbon grids and images were recorded in movie mode at 40K magnification using a 200 KV JEOL 2200FS transmission electron microscope. To facilitate visualization of virus particles, frames were further binned 3X to yield a pixel size of 4.398 Å/pixel. These binned micrographs were manually examined using EMAN2. To identify potentially gold-labelled ubiquitinated particles, we looked for spherical particles corresponding to the known ~500 Å (50nm) size of mature ZIKV and which were within 200 Å of the easily recognizable nano-gold clusters. Approximately 15% of visible ~500 Å ZIKV E-WT particles satisfied these criteria. None of the ZIKV E-K38/281R double mutant viruses were found labelled with gold particles (b). The cryoEM experiments with Gold particle labeling were performed only one time due to the large amount of virus needed.
Extended Data Fig. 8: Ubiquitination of ZIKV-E promotes virus-endosome membrane fusion. In JEG-3 cells.
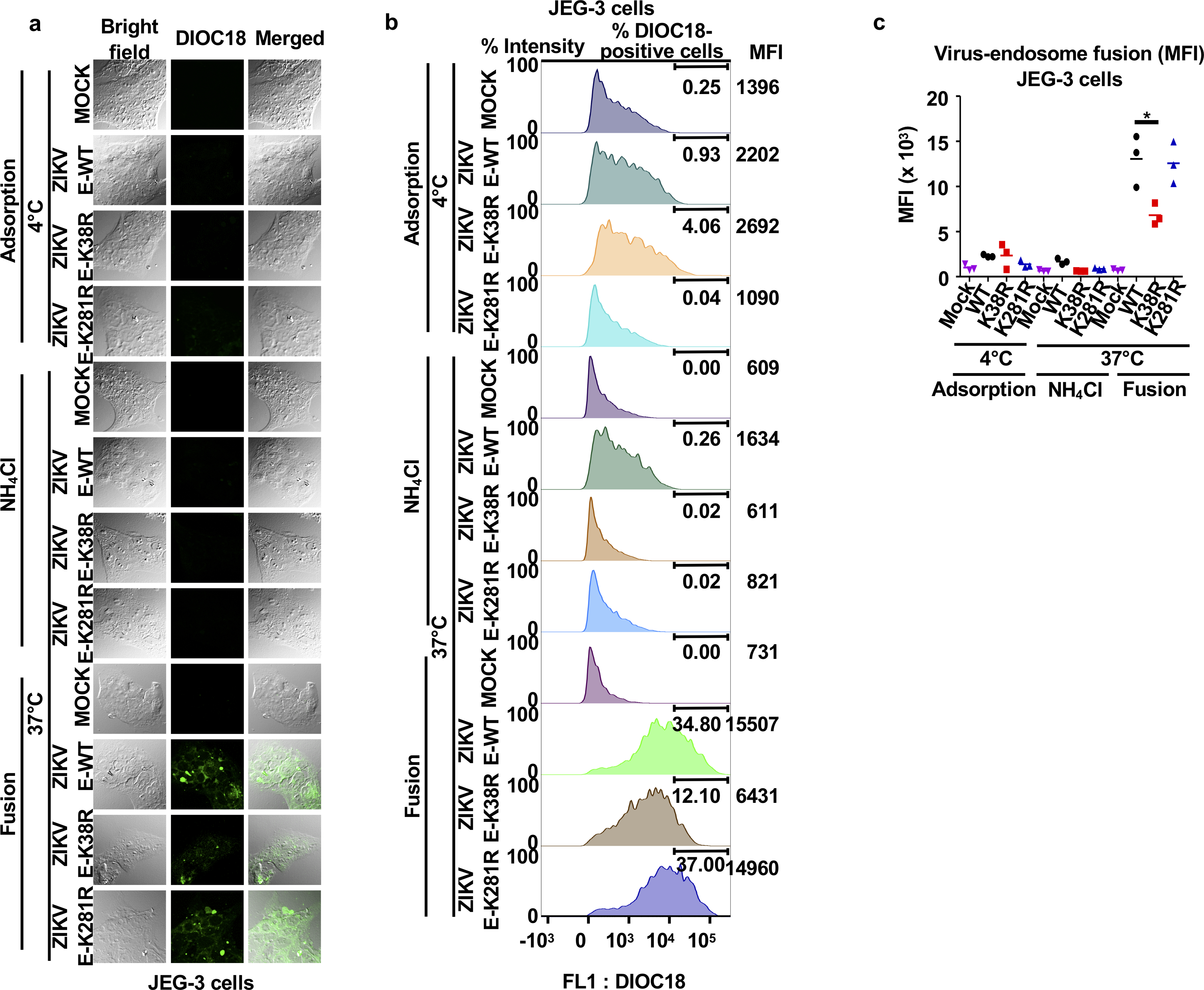
a-c, ZIKV E-WT, E-K38R and E-K281R were labeled with DiOC18. After filtration, viruses were incubated at 4°C with JEG-3 cells at MOI 2 and after 30 min were washed and collected for controls as adsorbed viruses. Additional samples were then incubated at 37°C for 1h, in the presence or absence of NH4Cl to block acidification (as control), washed, fixed and visualized with a confocal microscope (a). The same experiment was repeated for quantification by FACS (mean fluorescence intensity, MFI, shown in b, % of cells infected is shown in main Figure 3a, and c, Representative histograms). N=3, technical replicates, mean, unpaired two-sided t-test , *p < 0.05. Representatives of 2 independent experiments.
Extended Data Fig. 9: Ubiquitination of ZIKV-E promotes virus-endosome membrane fusion in A549 cells.
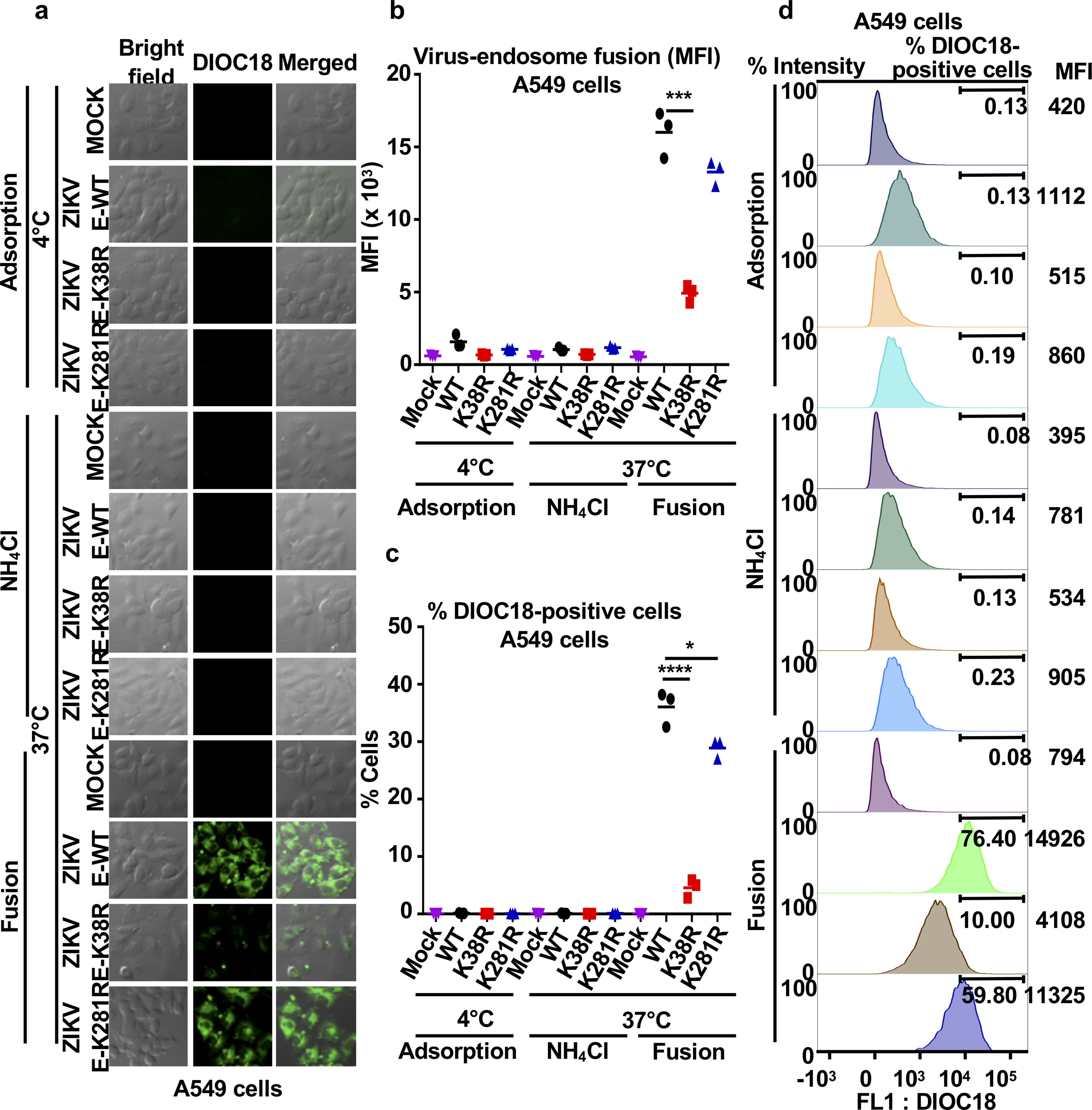
a-d, ZIKV E-WT, E-K38R, and E-K281R were labeled with DiOC18. After filtration, viruses were incubated at 4°C with A549 cells at MOI 2 and after 30min were washed and collected for controls as adsorbed viruses. Additional samples were then incubated at 37°C for 1h, and NH4Cl was used as control to block acidification of the endosome. Cells were washed, fixed, and visualized in a confocal microscope in a. Quantification by FACS is shown (mean fluorescence intensity, MFI) in b-c. d, Representative histograms showing green florescence (FL1: DiOC18) during ZIKV-endosome fusion. N=3, technical replicates, mean, unpaired two-sided t-test, *P < 0.05, ***P < 0.05. Representatives of 2 independent experiments.
Extended data Fig. 10. Ubiquitination of E on K38 promotes ZIKV attachment and enhanced replication in relevant human cells.
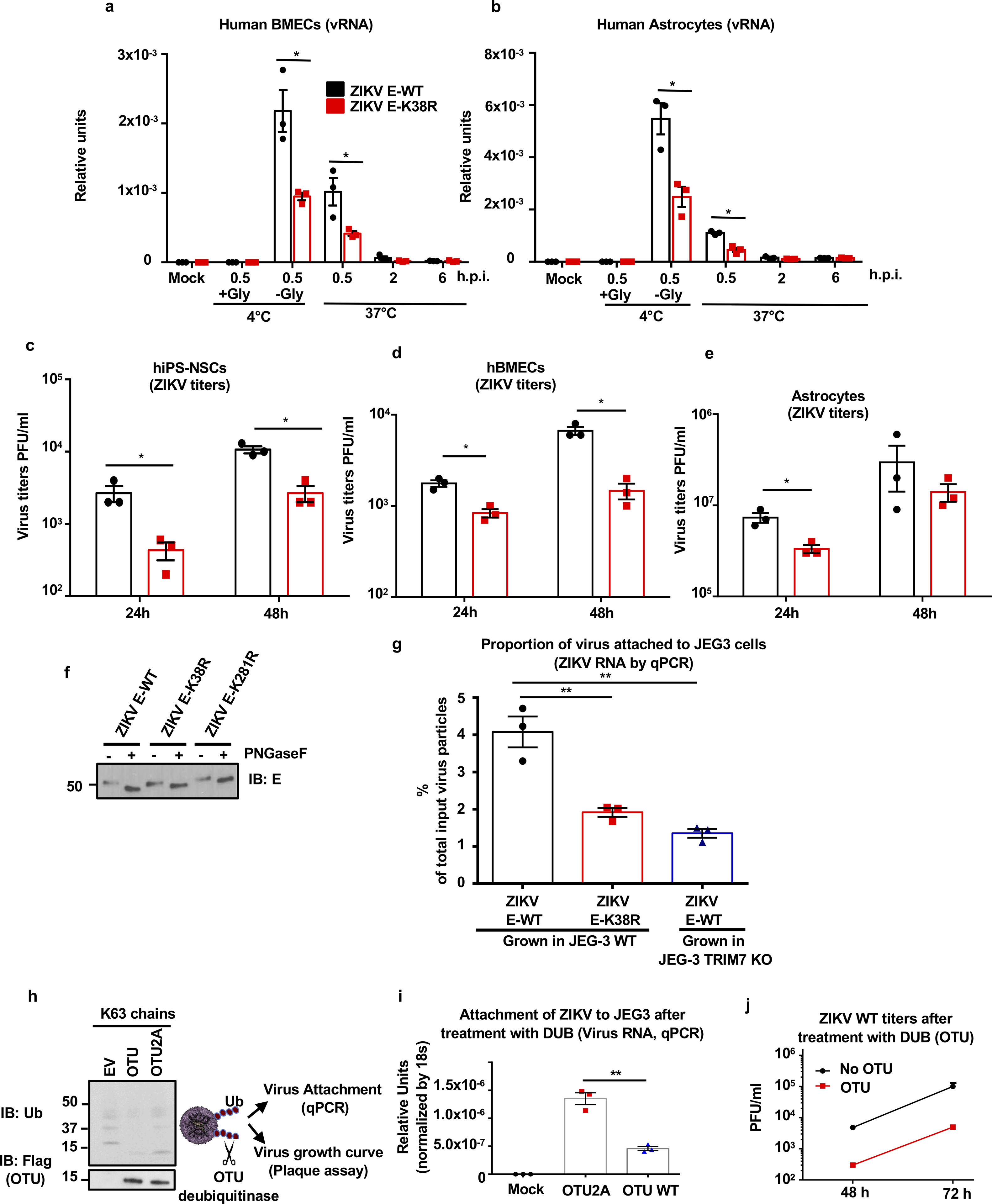
a-e, Human brain microvascular endothelial cells (hBMECs, a, d), human astrocytes (b, e), Human primary induced-pluripotent neural stem cells (hiPS-NSCs, c), were infected with ZIKV E-WT or E- K38R (MOI 2) as described in Fig 3b–c. Viral RNA was quantified by qPCR (a-b) and Virus titers by plaque assay (c-e). One experiment, N=3, technical replicates, mean +/− SE. Unpaired two-sided t-test, *p < 0.05. f, Endoglycosidase analyses of E. Proteins from ZIKV E-WT and E- K38R and E-K281R viruses were analyzed by western blot. Viruses were treated with PNGase F for 1h at 37°C. g, ZIKV E-WT or E-K38R mutant grown in JEG-3 WT or TRIM7 KO were used for attachment assays. Viruses were incubated at 4°C for 30min with JEG-3 cells and attachment was determined by measuring virus RNA by qPCR. The percentage of virus attachment was calculated by taking the input virus as 100%. h-j, The deubiquitinase (DUB) domain of the ovarian tumor (OTU) of the Crimean Congo Hemorrhagic fever (CCHF), which can cleave polyubiquitin chains (shown in h as control for activity), and a mutant (2A) with reduced activity were used to cleave ubiquitinated E of ZIKV. After incubation of ZIKV with purified recombinant OTU, the ability of the deubiquitinated virus to attach to cells and to replicate was tested by incubation with JEG-3 cells at 4°C for 30min and viral RNA quantified by qPCR (i), and replication by plaque assay (j). Representatives of 2 independent experiments, N=3 technical replicates, mean +/−SE, unpaired two-sided t-test, **p < 0.01.
Supplementary Material
Acknowledgments:
The authors wish to thank Drs. Garcia-Blanco, Shelton Bradrick and Ruben Soto (UTMB) for their generosity in sharing reagents and advice, Yuejin Liang (UTMB) for his technical advice on flow cytometry and Melina Fan for providing the lentivirus for establishing stable cell lines expressing HA-Ub (pLenti puro HA-Ub) through Addgene. We thank Dr. Andrea Gamarnik (Instituto Leloir, Buenos Aires, Argentina), Dr. Robin Stephens (UTMB) and Dr. Vineet Menachery (UTMB) for suggestions and helpful discussions, and Dr Tian Wang (UTMB) for providing some control mice. We also thank Dr. Linsey Yeager for editing.
Funding: Dr Rajsbaum’s lab was supported in part by the John Sealy Memorial Endowment Fund for Biomedical Research (UTMB), a research career development award (K12HD052023: BIRCWH program, from NIH ORWH/NICHD), and NIH/NIAID grants R21 AI132479-01, R21 AI126012–01A1 and R01 AI134907-01. T32-AI060549 to S.V.T., and T32 AI060549 to A.H. from NIH/NIAID. P.-Y.S. was supported by NIH grants AI142759, AI127744, and AI136126, and awards from the Kleberg Foundation, John S. Dunn Foundation, Amon G. Carter Foundation, Gilson Longenbaugh Foundation, and Summerfield Robert Foundation. S.M.B. lab was supported in part by the Division of Intramural Research of the NIH/NIAID. J.R.J. and N.J.K. were supported by NIH/NIAID grant U19 AI118610.
Footnotes
Competing interests: Authors declare no competing interests
Methods
Detailed methods and statistics are provided in the Supplementary information
Data and materials availability: All data is available in the main text or the supplementary materials. Mutant viruses may be available upon request after respective material transfer agreements are completed.
References
- 1.Musso D et al. Potential sexual transmission of Zika virus. Emerg Infect Dis 21, 359–361, doi:10.3201/eid2102.141363 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hills Susan L., Morgan Hennessey KR, Williams Charnetta, Oster Alexandra M., Fischer Marc, Mead Paul. Transmission of Zika Virus Through Sexual Contact with Travelers to Areas of Ongoing Transmission — Continental United States, 2016. CDC MMWR Morb Mortal Wkly Rep 65, 215–216 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Driggers RW et al. Zika Virus Infection with Prolonged Maternal Viremia and Fetal Brain Abnormalities. N Engl J Med 374, 2142–2151, doi:10.1056/NEJMoa1601824 (2016). [DOI] [PubMed] [Google Scholar]
- 4.van Tol S, Hage A, Giraldo MI, Bharaj P & Rajsbaum R The TRIMendous Role of TRIMs in Virus-Host Interactions. Vaccines (Basel) 5, doi:10.3390/vaccines5030023 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byk LA et al. Dengue Virus Genome Uncoating Requires Ubiquitination. MBio 7, doi:10.1128/mBio.00804–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandez-Garcia MD et al. Appraising the roles of CBLL1 and the ubiquitin/proteasome system for flavivirus entry and replication. J Virol 85, 2980–2989, doi:10.1128/JVI.02483–10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choy MM et al. Proteasome Inhibition Suppresses Dengue Virus Egress in Antibody Dependent Infection. PLoS Negl Trop Dis 9, e0004058, doi:10.1371/journal.pntd.0004058 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Versteeg GA et al. The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 38, 384–398, doi:10.1016/j.immuni.2012.11.013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hage A & Rajsbaum R To TRIM or not to TRIM: the balance of host-virus interactions mediated by the ubiquitin system. J Gen Virol 100, 1641–1662, doi:10.1099/jgv.0.001341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bharaj P et al. The Host E3-Ubiquitin Ligase TRIM6 Ubiquitinates the Ebola Virus VP35 Protein and Promotes Virus Replication. J Virol 91, doi:10.1128/JVI.00833–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fink J et al. Host gene expression profiling of dengue virus infection in cell lines and patients. PLoS Negl Trop Dis 1, e86, doi:10.1371/journal.pntd.0000086 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Padilla SL, Rodriguez A, Gonzales MM, Gallego GJ & Castano OJ Inhibitory effects of curcumin on dengue virus type 2-infected cells in vitro. Arch Virol 159, 573–579, doi:10.1007/s00705–013-1849–6 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Choy MM, Sessions OM, Gubler DJ & Ooi EE Production of Infectious Dengue Virus in Aedes aegypti Is Dependent on the Ubiquitin Proteasome Pathway. PLoS Negl Trop Dis 9, e0004227, doi:10.1371/journal.pntd.0004227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kostyuchenko VA et al. Structure of the thermally stable Zika virus. Nature 533, 425–428, doi:10.1038/nature17994 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Pierson TC & Kielian M Flaviviruses: braking the entering. Curr Opin Virol 3, 3–12, doi:10.1016/j.coviro.2012.12.001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rossi SL et al. Characterization of a Novel Murine Model to Study Zika Virus. Am J Trop Med Hyg 94, 1362–1369, doi:10.4269/ajtmh.16–0111 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Z et al. Viral Retinopathy in Experimental Models of Zika Infection. Invest Ophthalmol Vis Sci 58, 4355–4365, doi:10.1167/iovs.17–22016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Montori-Grau M et al. GNIP1 E3 ubiquitin ligase is a novel player in regulating glycogen metabolism in skeletal muscle. Metabolism 83, 177–187, doi:10.1016/j.metabol.2018.02.005 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Le Sommer C, Barrows NJ, Bradrick SS, Pearson JL & Garcia-Blanco MA G protein-coupled receptor kinase 2 promotes flaviviridae entry and replication. PLoS Negl Trop Dis 6, e1820, doi:10.1371/journal.pntd.0001820 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skurat AV, Dietrich AD, Zhai L & Roach PJ GNIP, a novel protein that binds and activates glycogenin, the self-glucosylating initiator of glycogen biosynthesis. J Biol Chem 277, 19331–19338, doi:10.1074/jbc.M201190200 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Zhai L, Dietrich A, Skurat AV & Roach PJ Structure-function analysis of GNIP, the glycogenin-interacting protein. Arch Biochem Biophys 421, 236–242 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Orchard RC et al. Identification of Antinorovirus Genes in Human Cells Using Genome-Wide CRISPR Activation Screening. J Virol 93, doi:10.1128/JVI.01324–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu M et al. E3 ubiquitin ligase tripartite motif 7 positively regulates the TLR4-mediated immune response via its E3 ligase domain in macrophages. Mol Immunol 109, 126–133, doi:10.1016/j.molimm.2019.01.015 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Chakraborty A, Diefenbacher ME, Mylona A, Kassel O & Behrens A The E3 ubiquitin ligase Trim7 mediates c-Jun/AP-1 activation by Ras signalling. Nat Commun 6, 6782, doi:10.1038/ncomms7782 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Napolitano LM, Jaffray EG, Hay RT & Meroni G Functional interactions between ubiquitin E2 enzymes and TRIM proteins. Biochem J 434, 309–319, doi:10.1042/BJ20101487 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Chazotte B Labeling membrane glycoproteins or glycolipids with fluorescent wheat germ agglutinin. Cold Spring Harb Protoc 2011, pdb prot5623, doi:10.1101/pdb.prot5623 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Rossignol ED, Peters KN, Connor JH & Bullitt E Zika virus induced cellular remodelling. Cell Microbiol 19, doi:10.1111/cmi.12740 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorman MJ et al. An Immunocompetent Mouse Model of Zika Virus Infection. Cell Host Microbe 23, 672–685 e676, doi:10.1016/j.chom.2018.04.003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fontes-Garfias CR et al. Functional Analysis of Glycosylation of Zika Virus Envelope Protein. Cell Rep 21, 1180–1190, doi:10.1016/j.celrep.2017.10.016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Modis Y, Ogata S, Clements D & Harrison SC Structure of the dengue virus envelope protein after membrane fusion. Nature 427, 313–319, doi:10.1038/nature02165 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Frias-Staheli N et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe 2, 404–416, doi:10.1016/j.chom.2007.09.014 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamel R et al. Biology of Zika Virus Infection in Human Skin Cells. J Virol 89, 8880–8896, doi:10.1128/JVI.00354–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dai L et al. Structures of the Zika Virus Envelope Protein and Its Complex with a Flavivirus Broadly Protective Antibody. Cell Host Microbe 19, 696–704, doi:10.1016/j.chom.2016.04.013 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Sirohi D et al. The 3.8 A resolution cryo-EM structure of Zika virus. Science 352, 467–470, doi:10.1126/science.aaf5316 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fibriansah G et al. DENGUE VIRUS. Cryo-EM structure of an antibody that neutralizes dengue virus type 2 by locking E protein dimers. Science 349, 88–91, doi:10.1126/science.aaa8651 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Therkelsen MD et al. Flaviviruses have imperfect icosahedral symmetry. Proc Natl Acad Sci U S A 115, 11608–11612, doi:10.1073/pnas.1809304115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.