

Abstract
Recruitment of RNA polymerase II to hypoxia-inducible factor (HIF) target genes under normoxia is a prerequisite for HIF-mediated transactivation. However, the underlying mechanism of this recruitment remains unknown. Here we report that chromodomain helicase DNA-binding protein 4 (CHD4) physically interacts with αand β subunits of HIF-1 and HIF-2 and enhances HIF-driven transcriptional programs to promote breast cancer progression. Loss of HIF-1/2α abolished CHD4-mediated breast tumor growth in mice. In breast cancer cells under normoxia, CHD4 enrichment at HIF target gene promoters increased RNA polymerase II loading through p300. Hypoxia further promoted CHD4 binding to the chromatin via HIF-1/2α, where CHD4 in turn enhanced recruitment of HIF-1α, leading to HIF target gene transcription. CHD4 was upregulated and correlated with HIF target gene expression in human breast tumors; upregulation of CHD4 and other known HIF coactivators in human breast tumors was mutually exclusive. Furthermore, CHD4 was associated with poor overall survival of breast cancer patients. Collectively, these findings reveal a new fundamental mechanism of HIF regulation in breast cancer, which has clinical relevance.
Keywords: Hypoxia, HIF, Co-activator, RNA polymerase II, Breast cancer
Introduction
Hypoxia, a hallmark of the microenvironment of breast tumors, contributes to the pathogenesis of human breast cancer and has been associated with increased risk of mortality in breast cancer patients (1). The transcription factor hypoxia-inducible factor (HIF), consisting of HIF-α and HIF-β subunits (2), is a master regulator of response to hypoxia in breast tumors and activates its target genes to promote angiogenesis, cell proliferation/survival, metabolic reprogramming, epigenetic reprogramming, pH homeostasis, immune evasion, tumor invasion and metastasis (3). Three HIF family members have been identified in mammals, and HIF-1 and HIF-2 are highly expressed in breast tumors (4–6). The transcriptional activity of HIF-1 and HIF-2 is regulated indirectly by the ubiquitin-dependent protein degradation of α subunit (7). Several ubiquitin E3 ligases including cullin 2, CHIP, HAF, and Parkin have been shown to regulate the protein stability of HIF-1α and/or HIF-2α (8–11). Accumulating studies uncover an essential role of epigenetic regulators in transactivation of HIF-1 and HIF-2 in cancer cells (12). Multiple histone acetyltransferases/deacetylases, histone/DNA methyltransferases/ demethylases, and histone readers, including p300, CBP, ZMYND8, BRD4, JMJD2C, CDK8, Tip60, JMJD1A, G9a, HDAC7, and TET1, are known to function as HIF coregulators (13–19). We and others recently found that RNA polymerase II (RNA Pol II) is pre-loaded and paused at the promoter of HIF target genes under normoxia and then released upon hypoxia to mediate transcriptional elongation of HIF target genes in breast cancer cells (14,20). HIF coactivator ZMYND8 controls RNA Pol II-mediated elongation of HIF target genes in breast cancer cells under hypoxia by recruiting BRD4 (14). However, the fundamental mechanism underlying RNA Pol II recruitment to HIF target genes for its pausing under normoxia remains unknown.
Chromodomain helicase DNA-binding protein 4 (CHD4) is a key catalytic subunit of the ATP-dependent chromatin remodeler nucleosome remodeling and deacetylase (NuRD) complex, which plays a critical role in gene regulation by modulating chromatin structure and assembly (21). It has been well documented that CHD4 is involved in gene repression in normal and cancer cells. CHD4 together with the NuRD complex controls the accessibility of the chromatin and mediates downregulation of many genes, whose products are involved in DNA damage response, tumor malignancy, cardiac development, and stem cell fate (22–25). However, the role of CHD4 in gene activation is less studied.
In the present study, we found that CHD4 physically interacts with HIF-1α and HIF-2α and enhances HIF downstream target gene expression by increasing recruitment of HIF and RNA Pol II in breast cancer cells. CHD4 increases breast tumor growth in vitro and in mice through coactivating HIF. Together, these studies uncover a new fundamental mechanism of HIF activation in breast cancer.
Materials and Methods
Plasmid constructs
Full-length CHD4 cDNA was generated by PCR using mouse CHD4 cDNA (Harvard plasmid, MmCD00320214) as a template and cloned into HpaI/NheI-linearized pcFUGW-3XFLAG vector. FLAG-CHD4 (K750R) was generated by site-directed PCR mutagenesis. Human MBD3 and MTA2 cDNAs were amplified from MDA-MB-321 cell cDNA by PCR and cloned into pcDNA3.1-V5 vector. cDNAs encoding human truncated HIF-2α domains were cloned to pGEX-6P-1 vector. DNA oligonucleotides of the short hairpin RNAs (shRNAs) targeting CHD4 (Table S1) were annealed and ligated into AgeI/EcoRI-linearized pLKO.1 vector (Addgene, #8435) or Teton-pLKO (Addgene, #21915). Other constructs have been described previously (14,15,26). The DNA sequences of all recombinant plasmids were confirmed by nucleotide sequence analysis.
Cell culture, transfection, and virus production
MDA-MB-231 (from Rolf Brekken at UT Southwestern in 2015), MCF-7 (from ATCC in 2014), T47D, HeLa, HEK293T (from Gregg L. Semenza at Johns Hopkins in 2014), HEK293FT (from ThermoFisher in 2014) cells were cultured in DMEM or RPMI1640 supplemented with 10% heat-inactivated fetal bovine serum at 37°C in a 5% CO2/95% air incubator. Hypoxic cells were placed in a modular incubator chamber (Billups-Rothenberg) flushed with a gas mixture of 1% O2, 5% CO2 and balanced N2. Cells within six passages were used for experiments. Cells were transfected using PolyJet (SignaGen) or FuGENE6 (Promega). All cell lines are mycoplasma-free and have been authenticated by STR DNA profiling analysis in 2016–2017. Lentiviruses encoding scrambled control shRNA (shSC), shCHD4#1, or shCHD4#2 were generated by transfecting HEK293FT cells with the transducing vector and packaging vectors pMD2.G and psPAX2. 48 hours later, virus particles in the medium were harvested, filtered, and transduced into cancer cells. 48 hours after infection, cells were selected for 4 days using 2.5 μg/ml puromycin. CHD4 protein levels were verified by immunoblot assay.
Quantitative reverse transcription-polymerase chain reaction (RT-qPCR) assay
Total RNA was extracted using Trizol reagent (ThermoFisher), treated with DNase I (ThermoFisher), and reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad). Real-time PCR was performed by a CFX-96 real-time system (Bio-Rad) using iTaq universal SYBR green supermix (Bio-Rad) with primers listed in Table S2. The fold change in target mRNA expression was calculated based on the threshold cycle (Ct) as 2−Δ(ΔCt), where ΔCt = Cttarget – Ct18S rRNA and Δ(ΔCt) = ΔCt1% O2 − ΔCt20% O2.
RNA-sequencing (RNA-seq) assay
MDA-MB-231-shSC and -shCHD4 cells were exposed to 20% or 1% O2 for 24 hours. Total RNA was isolated using a RNeasy Mini kit and treated with DNase (Qiagen). The quality of total RNA was confirmed with a RIN score 8.5 or higher by the Agilent Tapestation 4200. 200ng of DNA-free total RNA was used for library preparation with a Kapa mRNA HyperPrep kit (Roche). Briefly, mRNA with poly(A) was purified and fragmented before strand specific cDNA synthesis. cDNAs were end repaired, A-tailed, and ligated with the indexed sequencing adapters. After cDNAs were amplified by PCR and purified with Kapa pure beads, they were sequenced on the Illumina NextSeq500 with the read configuration as 75 bp, single end. Each sample had approximately 25 million to 35 million reads. The Fastq files were subjected to quality check using fastqc (version 0.11.2, http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and fastq_screen (version 0.4.4, http://www.bioinformatics.babraham.ac.uk/projects/fastq_screen), and trimmed using fastq-mcf (ea-utils/version 1.1.2–806, https://github.com/ExpressionAnalysis/ eautils). Trimmed fastq files were mapped to hg19 (UCSC version from igenomes) using Tophat. Duplicates were marked using picard-tools (version 1.127, https://broadinstitute.github.io/picard/). Read counts were generated using featureCounts and the differential expression analysis was performed using edgeR with a false discovery rate (FDR) < 0.05 and mRNA fold change > 1.5 as a cutoff.
Chromatin immunoprecipitation (ChIP)-qPCR assay
Cells were exposed to 20% or 1% O2 for 24 hours, crosslinked with 1% formaldehyde for 10 minutes at room temperature, and quenched in 0.125 M glycine. Cells were lyzed in lysis buffer (50 mM Tris-HCl, 10 mM EDTA, 1% SDS, protease inhibitor cocktail), sonicated, and subjected to immunoprecipitation overnight in the presence of salmon sperm DNA/protein A beads with antibodies against CHD4 (Proteintech, 14173–1-AP), HIF-1α (homemade), RNA polymerase II (Abcam, ab817), or IgG at 4 °C. Precipitated chromatin DNA was extensively washed, eluted with freshly prepared elution buffer (0.1 M NaHCO3, 1% SDS), decrosslinked at 65°C for 4 hours followed by treatment with proteinase K at 45°C for 45 minutes, purified with phenol/chloroform/isoamyl alcohol (25:24:1, v/v), and quantified by real-time qPCR assay. The primers used for ChIP-qPCR are listed in Table S2. Fold enrichment was calculated based on Ct as 2−Δ(ΔCt), where ΔCt = CtIP − CtInput and Δ(ΔCt) = ΔCtantibody − ΔCtIgG.
Co-immunoprecipitation (IP) and immunoblot assays
Cells were lysed in NETN lysis buffer (150 mM NaCl, 1 mM EDTA, 10 mM Tris-HCl, pH 8.0, 0.5% NP-40 and protease inhibitor cocktail) and subjected to IP with indicated antibodies at 4 °C for 2 hours, and then protein G magnetic beads (Bio-Rad) were added for further incubation overnight. Next day, proteins bound on the beads were washed four times with NETN lysis buffer, boiled in 1x Laemmli buffer for 6 minutes, and fractionated by SDS-PAGE, followed by immunoblot assay using indicated antibodies.
Glutathione-S-transferase (GST) pull-down assay
GST-HIF-1α and GST-HIF-2α domain proteins were expressed in Escherichia coli BL21-Gold (DE3) and purified by binding to glutathione Sepharose beads (GE Healthcare). The purified GST-HIF-1α and GST-HIF-2α domains and GST proteins were incubated overnight at 4 °C in the presence of glutathione Sepharose beads with lysates from HEK293T cells transfected with FLAG-CHD4 vector. Proteins bound on the beads were washed, boiled in 1x Laemmli buffer for 6 minutes, and subsequently subjected to SDS-PAGE and immunoblot assay.
Luciferase reporter assay
Cells were plated on 48-well plates and transiently transfected with HIF luciferase reporter plasmid p2.1 containing a HRE (5’-ACGTG-3’) from human ENO1 gene (27); control reporter plasmid pSV-Renilla; shSC, shCHD4#1, shCHD4#2, pcFUGW-3xFLAG-CHD4 [wild-type (WT) or K750R], pcDNA3.1-MBD3, pcDNA3.1-MTA2, or empty vector (EV). 24 hours later, cells were exposed to 20% or 1% O2 for 24 hours. The firefly and Renilla luciferase activities were measured using the Dual Luciferase Reporter Assay System (Promega).
Clonogenic assay
100 cells were seeded on a 6-well plate and cultured under 20% or 1% O2 for 14 days. Subsequently, the colonies were stained with 0.1% crystal violet (Sigma). Only positive colonies (diameter > 50 μm) were counted.
Boyden chamber invasion assay
5 × 104 of cells were re-suspended in serum-free medium and seeded in a Matrigel-coated transwell insert. Cell culture medium with 10% FBS will be placed at the bottom chamber. Cells were exposed to 20% or 1% O2 for 24 hours. Cells that invade to the lower side of the transwell insert will be fixed with methanol, stained with 0.1% crystal violet, and counted.
Animal studies
Animal experiments were approved by the Animal Care and Use Committee at UT Southwestern Medical Center. 2 × 106 of cells in 100 μL PBS/Matrigel (1:1, Corning) were injected into the second left mammary fat pad of female NSG or NOD/SCID mice (6–8 weeks old). Mice that were implanted with MCF-7 cells were administrated subcutaneously with a slow-release 17β-estradiol pellet (0.72 mg/60-day release/pellet) one day before cell implantation. Tumor volume was measured with a caliper every three days starting day 14–16 after cell implantation and calculated according to the formula: V = 0.52 × L × H × W (V: volume, L: length, H: height, W: width). Primary tumors were harvested, photographed, and weighed when their volume reached to about 1500 mm3.
Statistical analysis
Statistical analysis was performed by two-tailed Student’s t test between two groups, and one-way or two-way ANOVA with multiple testing correction within multiple groups. Kaplan-Meier survival curve was analyzed by log-rank test. RNA-seq was repeated twice, and other experiments were repeated at least three times. Data were expressed as mean ± SEM. p < 0.05 is considered significant.
Accession number
The RNA-seq data were deposited at the GEO database with the accession number GSE129293.
Results
CHD4 physically interacts with HIF-1 and HIF-2 in human breast cancer cells
To determine the role of CHD4 in HIF transactivation in human breast cancer, we investigated whether CHD4 physically interacts with HIF-1α and HIF-2α in human breast cancer cells. MDA-MB-231 cells were exposed to 1% O2 for 6 hours and the whole cell lysates were subjected to co-IP assay with anti-HIF-1α antibody or control antibody IgG, followed by immunoblot analysis. HIF-1α immunoprecipitation pulled down endogenous CHD4 (Fig. 1A), indicating the physical interaction of HIF-1α and CHD4. Likewise, co-IP assay showed that endogenous HIF-2α interacted with endogenous CHD4 in hypoxic MDA-MB-231 cells (Fig. 1B). We further detected the physical interaction between endogenous HIF-1β and CHD4 in hypoxic MDA-MB-231 cells (Fig. 1C). Similar results were also observed in another breast cancer cell line T47D (Supplemental Fig. S1A–C). We next performed a reciprocal co-IP assay in MDA-MB-231 cells exposed to 1% O2 for 6 hours. Anti-CHD4 antibody, but not control IgG, coprecipitated endogenous CHD4, HIF-1α, and HIF-2α in hypoxic MDA-MB-231 cells (Fig. 1D). These data indicate that CHD4 physically interacts with both HIF-1 and HIF-2 in human breast cancer cells under hypoxia.
Figure 1.
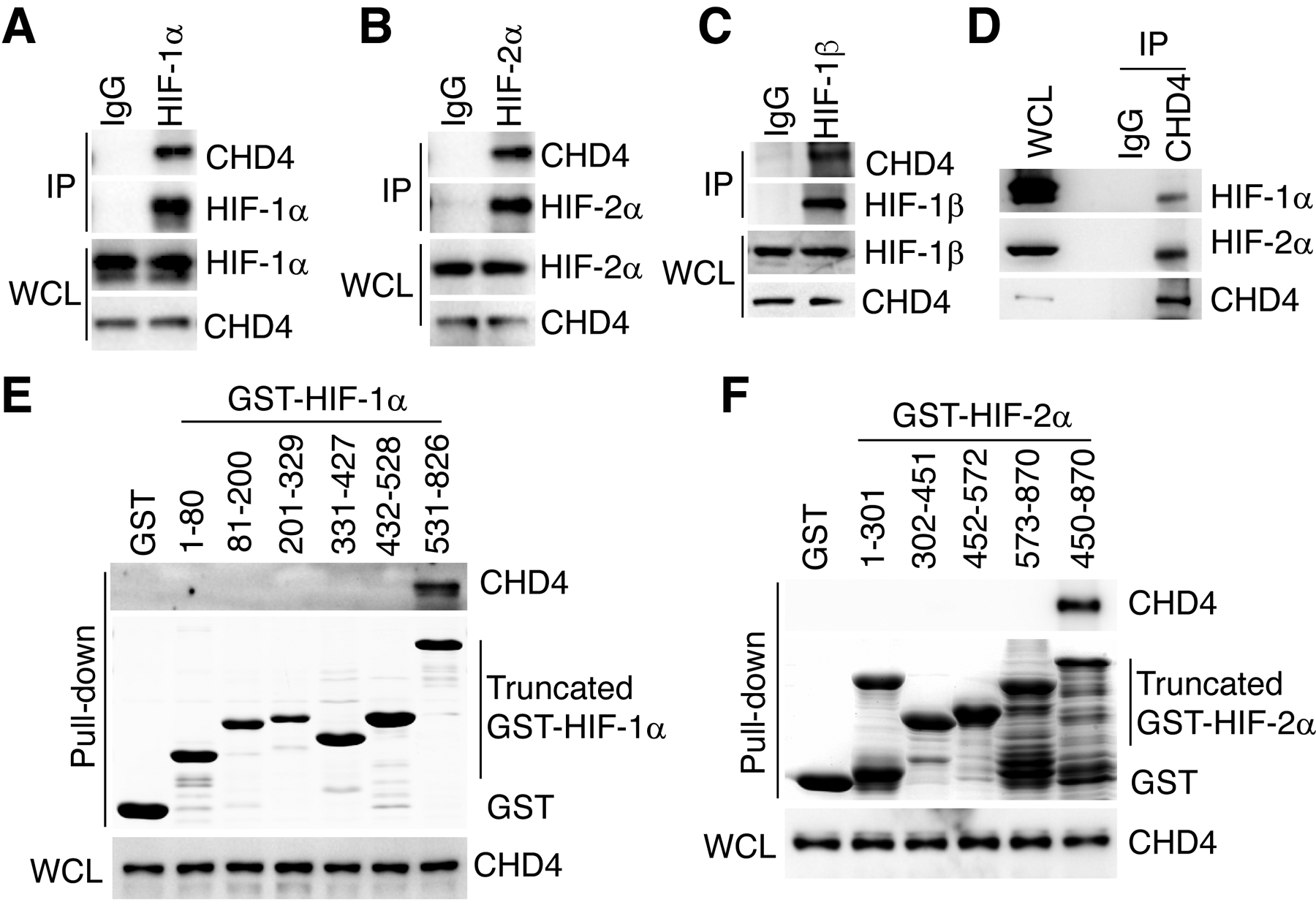
CHD4 interacts with HIF-1 and HIF-2. (A-D) Co-IP with IgG, anti-HIF-1α (A), anti-HIF-2α (B), anti-HIF-1β (C), or anti-CHD4 (D) antibody in MDA-MB-231 cells exposed to 1% O2 for 6 hrs. WCL, whole cell lysate. (E and F) Purified GST, GST-HIF-1α domains (E), or GST-HIF-2α domains (F) were incubated in the presence of glutathione Sepharose beads with cell lysates from HEK293T cells transfected with FLAG-CHD4, followed by immunoblot assay with anti-CHD4 antibody or Ponceau S staining.
Next, we mapped the HIF-1α and HIF-2α domains binding to CHD4. HEK293T cells were transfected with FLAG-CHD4 vector and the whole cell lysates were incubated with a series of truncated GST-HIF-1α or GST-HIF-2α proteins that were expressed and purified from bacteria, followed by immunoblot assay with anti-CHD4 antibody or Ponceau S staining. CHD4 had a robust interaction with the transactivation domain of HIF-1α (531–826 amino acids) or HIF-2α (450–870 amino acids), but not other domains of HIF-1α or HIF-2α and GST (Fig. 1E and F). These data indicate that CHD4 solely binds to the C-terminal transactivation domain of HIF-1α and HIF-2α.
CHD4 enhances HIF transactivation independent of its helicase activity
To determine whether CHD4 increases HIF transcriptional activity, we first performed HIF luciferase reporter assay. HeLa cells were selected for the assay because of their high transfection efficiency. Cells were co-transfected with a HIF luciferase reporter (p2.1), pSV-Renilla, and EV or FLAG-CHD4 expression vector, and exposed to 20% or 1% O2 for 24 hours. Ectopic expression of WT FLAG-CHD4 significantly increased HIF luciferase reporter activity in hypoxic HeLa cells, compared to EV (Fig. 2A). HIF-1/2α double knockout (DKO) completely abolished CHD4’s effect on HIF transcriptional activity in hypoxic HeLa cells (Fig. 2B), indicating specific coactivation of HIF-1/2 by CHD4. To determine whether the NuRD complex is involved in CHD4-mediated HIF activation, we studied the effect of helicase-dead CHD4 mutant (K750R) and other subunits of the NuRD complex on HIF transcriptional activity. CHD4 (K750R) significantly increased HIF transcriptional activity, similar to WT CHD4 (Fig. 2A). In line with this, CHD4 (K750R) interacted with FLAG-HIF-1α as efficiently as WT CHD4 in hypoxic HEK293T cells (Fig. 2C). The other two subunits of the NuRD complex MBD3 and MTA2 both failed to increase HIF transcriptional activity in HeLa cells under hypoxia (Fig. 2D), further suggesting that the NuRD complex does not control HIF transcriptional activity. To complement gain-of-function studies, we performed loss-of-function studies using two independent CHD4 shRNAs. Knockdown (KD) of CHD4 by either of the shRNAs significantly decreased HIF transcriptional activity in hypoxic HeLa cells, compared to shSC (Fig. 2E). Note that the incomplete suppression of HIF transcriptional activity in CHD4 KD cells is possibly due to the knockdown efficiency of CHD4 by transient transfection of CHD4 shRNAs as well as the redundancy of other HIF coactivators. CHD4 KD had no effect on protein levels of HIF-1α or HIF-2α in MDA-MB-231 cells (Fig. 2F), indicating that HIF activation by CHD4 is not caused by HIF-1/2α protein stability. Together, these data indicate that CHD4 enhances HIF transcriptional activity independent of its helicase activity and the NuRD complex.
Figure 2.
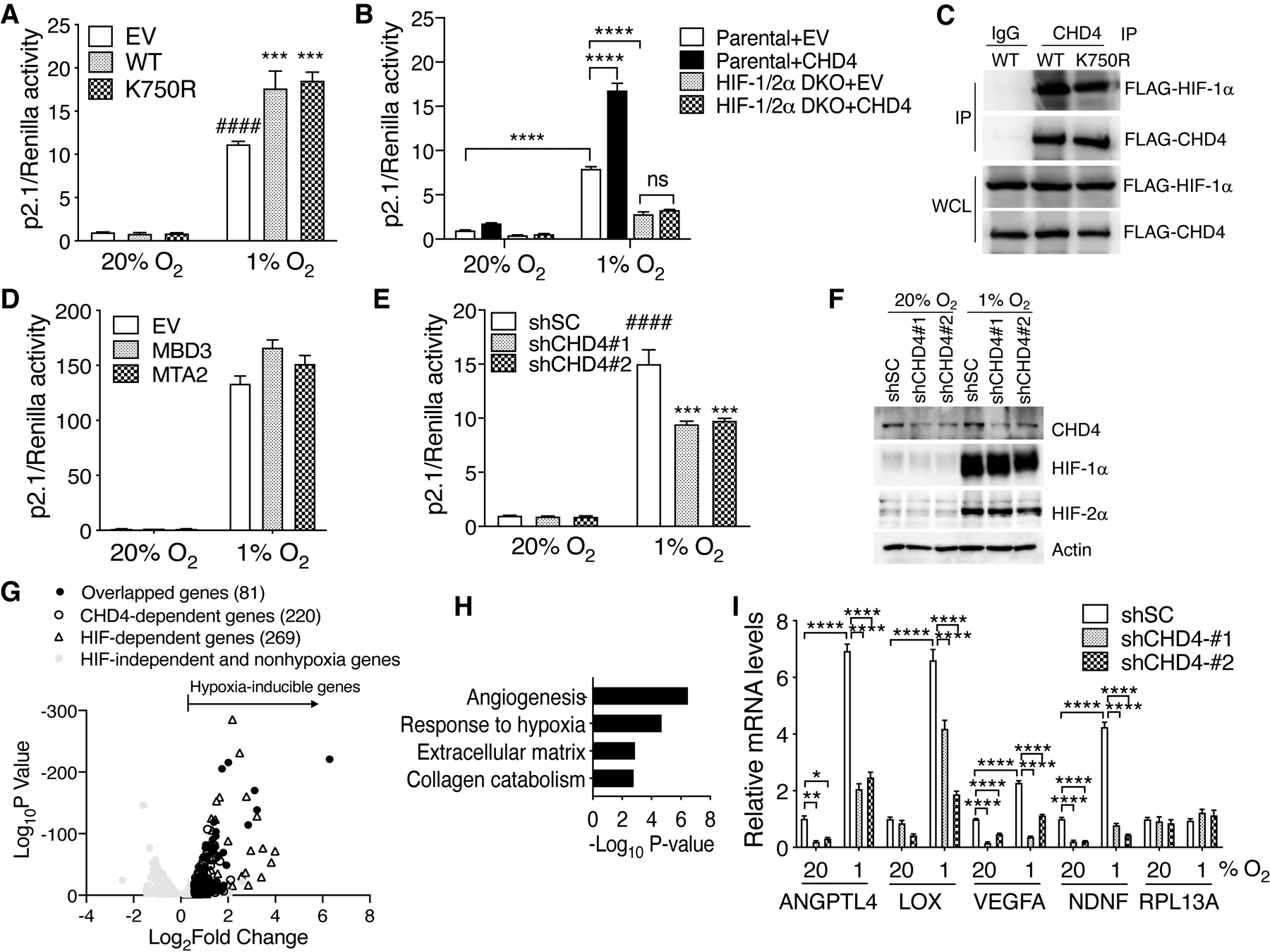
CHD4 enhances HIF transcriptional activity independent of its helicase activity. (A) HeLa cells were co-transfected with a HIF-1 luciferase reporter (p2.1), pSV-Renilla, and vector encoding WT or K750R FLAG-CHD4 or EV. Cells were exposed to 20% or 1% O2 for 24 hrs and subjected to dual-luciferase reporter assays (n = 3, mean ± SEM). ***p < 0.001 vs. EV at 1% O2, ####p < 0.0001 vs. EV at 20% O2, by 2-way ANOVA with Turkey’s test. (B) Parental and HIF-1/2α DKO HeLa cells were co-transfected with p2.1, pSV-Renilla, and vector encoding FLAG-CHD4 or EV. Cells were exposed to 20% or 1% O2 for 24 hrs and subjected to dual-luciferase reporter assays (n = 3, mean ± SEM). ****p < 0.0001, by 2-way ANOVA with Turkey’s test. ns, not significant. (C) Co-IP with IgG and anti-CHD4 antibody in transfected HEK293T cells exposed to 1% O2 for 6 hrs. (D) HeLa cells were co-transfected with p2.1, pSV-Renilla, and vector encoding MBD3-V5, MTA2-V5, or EV. Cells were exposed to 20% or 1% O2 for 24 hrs and subjected to dual-luciferase reporter assays (n = 3, mean ± SEM). (E) HeLa cells were co-transfected with p2.1, pSV-Renilla, and vector encoding shSC, shCHD4#1, or shCHD4#2. Cells were exposed to 20% or 1% O2 for 24 hrs and subjected to dual-luciferase reporter assays (n = 3, mean ± SEM). ***p < 0.001 vs. shSC at 1% O2, ####p < 0.0001 vs. shSC at 20% O2, by 2-way ANOVA with Turkey’s test. (F) Immunoblot analysis of indicated proteins in shSC or CHD4 KD MDA-MB-231 cells exposed to 20% or 1% O2 for 24 hrs. (G) Volcano plot of the overlapped HIF and CHD4 target genes in MDA-MB-231 cells under hypoxia. (H) Gene ontology analysis of CHD4-induced HIF target genes. The partial list of the enriched biological function is shown. See Table S3 for the full list. (I) RT-qPCR analysis of indicated mRNAs in shSC or CHD4 KD MDA-MB-231 cells exposed to 20% or 1% O2 for 24 hrs (n = 3, mean ± SEM). *p < 0.05; **p < 0.01; ****p < 0.0001, by 2-way ANOVA with Turkey’s test.
Next, we analyzed the effect of CHD4 on HIF downstream target gene expression in breast cancer cells. To this end, we generated two independent CHD4 KD MDA-MB-231 cell lines by transducing lentivirus carrying CHD4 shRNA#1 or #2. Either of the CHD4 shRNAs robustly reduced CHD4 protein levels in MDA-MB-231 cells under 20% or 1% O2 (Fig. 2F). RNA-seq was performed in shSC and CHD4 KD#1 MDA-MB-231 cells exposed to 20% or 1% O2 for 24 hours. We found that 220 genes were downregulated in CHD4 KD cells, compared to shSC cells under hypoxia (mRNA fold change > 1.5, FDR < 0.05) (Fig. 2G). Likewise, we identified 269 HIF-1/2-inducible genes in MDA-MB-231 cells (Fig. 2G). Intriguingly, 81 genes were upregulated by both CHD4 and HIF-1/2, which represented 30% of HIF-inducible genes (Fig. 2G). Gene ontology analysis showed that these overlapped genes participated in several HIF-dependent biological processes, including angiogenesis, response to hypoxia, extracellular matrix organization, collagen catabolic process, apoptosis, and cell proliferation (Fig. 2H; Table S3). RT-qPCR analysis validated that the transcription of HIF target genes, VEGFA, ANGPTL4, LOX, and NDNF, was significantly decreased by CHD4 KD#1 or #2 in hypoxic MDA-MB-231 cells (Fig. 2I). However, mRNA transcription of the non-HIF target gene RPL13A was not altered by CHD4 KD#1 or #2 in MDA-MB-231 cells (Fig. 2I), indicating specificity of activation of HIF target genes by CHD4 under hypoxia. These data indicate that CHD4 enhances expression of a subset of HIF downstream target genes in breast cancer cells under hypoxia.
Mutual recruitment of CHD4 and HIF-1α to HIF target genes in breast cancer cells under hypoxia
To study the role of CHD4 in HIF-1 binding to the HRE, we first examined whether CHD4 localizes on the chromatin near the HRE by ChIP-qPCR assay in MDA-MB-231 cells exposed to 20% or 1% O2 for 24 hours. CHD4 was associated with the chromatin regions near the HRE of VEGFA, LOX, and ANGPTL4, as compared with control IgG in nonhypoxic MDA-MB-231 cells, and hypoxia significantly increased CHD4 occupancy on these HIF target genes by 2–3.7 folds, but not on RPL13A (Fig. 3A–D). To determine if HIF-1/2α are required for hypoxia-induced CHD4 binding to HIF target genes, we compared CHD4 occupancy in parental and HIF-1/2α DKO MDA-MB-231 cells exposed to 20% or 1% O2 for 24 hours (Fig. 3E). HIF-1/2α DKO abolished hypoxia-induced CHD4 occupancy on VEGFA, LOX, and ANGPTL4 but not RPL13A (Fig. 3A–D). These findings indicate that HIF-1/2α control CHD4 binding to HIF target genes in breast cancer cells under hypoxia.
Figure 3.
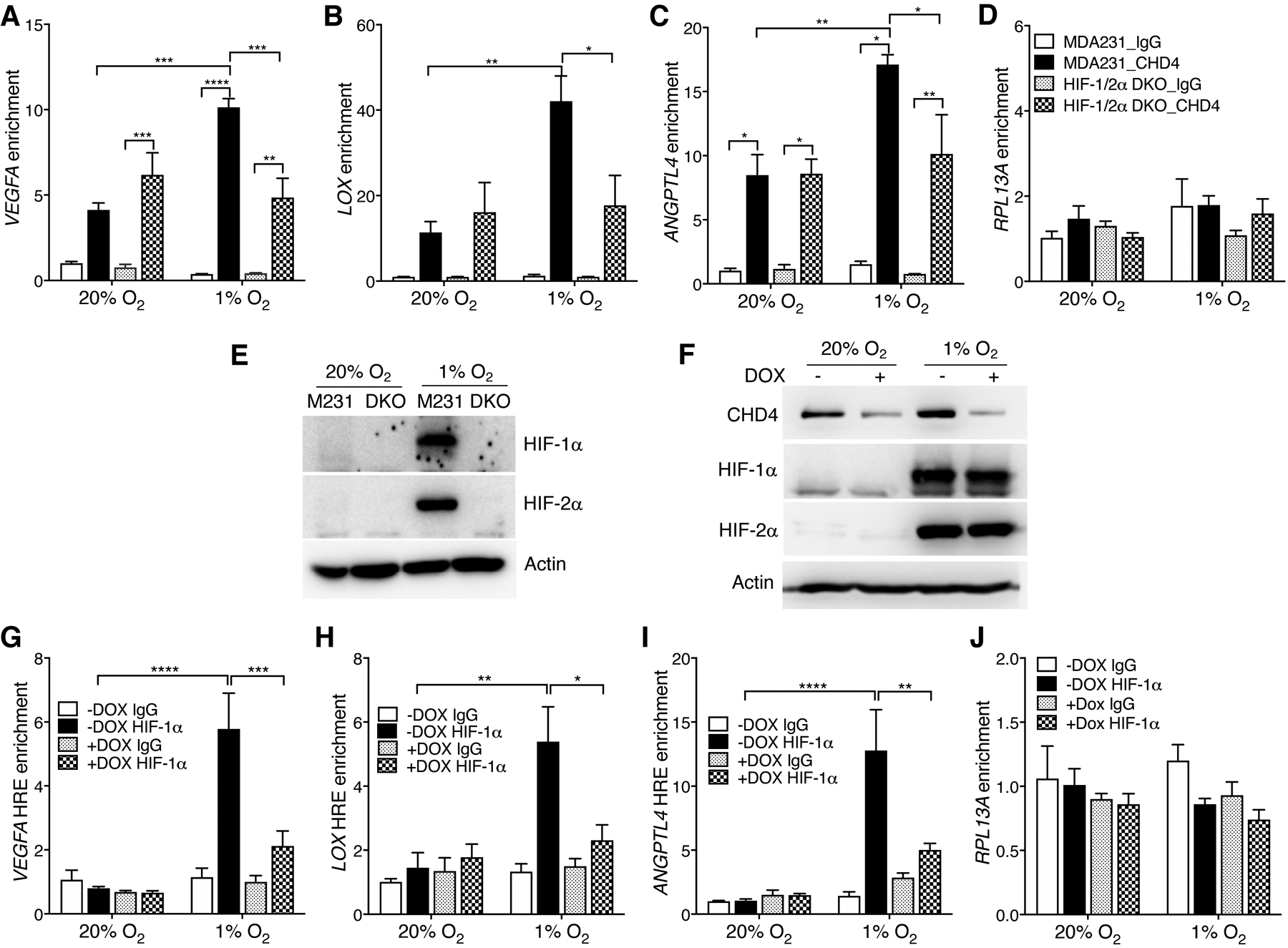
Mutual recruitment of CHD4 and HIF-1 to HIF target genes under hypoxia. (A-D) CHD4 ChIP-qPCR assay in parental and HIF-1/2α DKO MDA-MB-231 (MDA231) cells exposed to 20% or 1% O2 for 24 hrs (n = 3, mean ± SEM). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, by 2-way ANOVA with Turkey’s test. (E) Immunoblot analysis of indicated proteins in parental and HIF-1/2α DKO MDA-MB-231 (M231) cells exposed to 20% or 1% O2 for 24 hrs. (F) Immunoblot analysis of indicated proteins in MDA-MB-231-Teton CHD4 KD cells exposed to 20% or 1% O2 for 24 hrs in the presence or absence of DOX (1 μg/ml). (G-J) HIF-1α ChIP-qPCR assay in MDA-MB-231-Teton CHD4 KD cells exposed to 20% or 1% O2 for 24 hrs in the presence or absence of DOX (1 μg/ml) (n = 3, mean ± SEM). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, by 2-way ANOVA with Turkey’s test.
We next generated doxycycline (DOX)-inducible CHD4 KD MDA-MB-231 cells (Fig. 3F) and studied whether CHD4 KD attenuates HIF-1 binding to the HRE. ChIP-qPCR results showed that HIF-1α strongly bound to the HRE of VEGFA, LOX, and ANGPTL4 genes in hypoxic MDA-MB-231 cells, as expected (Fig. 3G–I). CHD4 KD almost completely abolished HIF-1α occupancy at these HREs under hypoxia (Fig. 3G–I). Neither hypoxia nor CHD4 KD affected HIF-1α enrichment on the non-HIF target gene RPL13A (Fig. 3J), indicating the specific effect of CHD4 on HIF-1 recruitment to the HRE. These findings indicate that CHD4 promotes HIF-1 binding to the HRE of its target genes in hypoxic breast cancer cells.
CHD4 increases recruitment of RNA Pol II to HIF target genes through p300 in breast cancer cells
To investigate whether CHD4 regulates RNA Pol II recruitment to HIF target genes, we first studied the physical interaction between CHD4 and RNA Pol II by co-IP assay. Anti-CHD4 antibody, but not IgG, precipitated RNA Pol II in MDA-MB-231 cells under 20% O2, and hypoxia had no effect on CHD4-RNA Pol II interaction (Fig. 4A). We further found by ChIP-qPCR assay that CHD4 KD#1 or #2 significantly decreased RNA Pol II enrichment at the promoter of the VEGFA and LOX genes in MDA-MB-231 cells under 20% O2 (Fig. 4B and C). Although reduced RNA Pol II occupancy at VEGFA and LOX was also found in CDH4 KD MDA-MB-231 cells under 1% O2, it was less robust as compared to that under normoxic conditions (Fig. 4B and C). These data indicate that CHD4 increases recruitment of RNA Pol II to HIF target genes to enhance their transcription in breast cancer cells.
Figure 4.
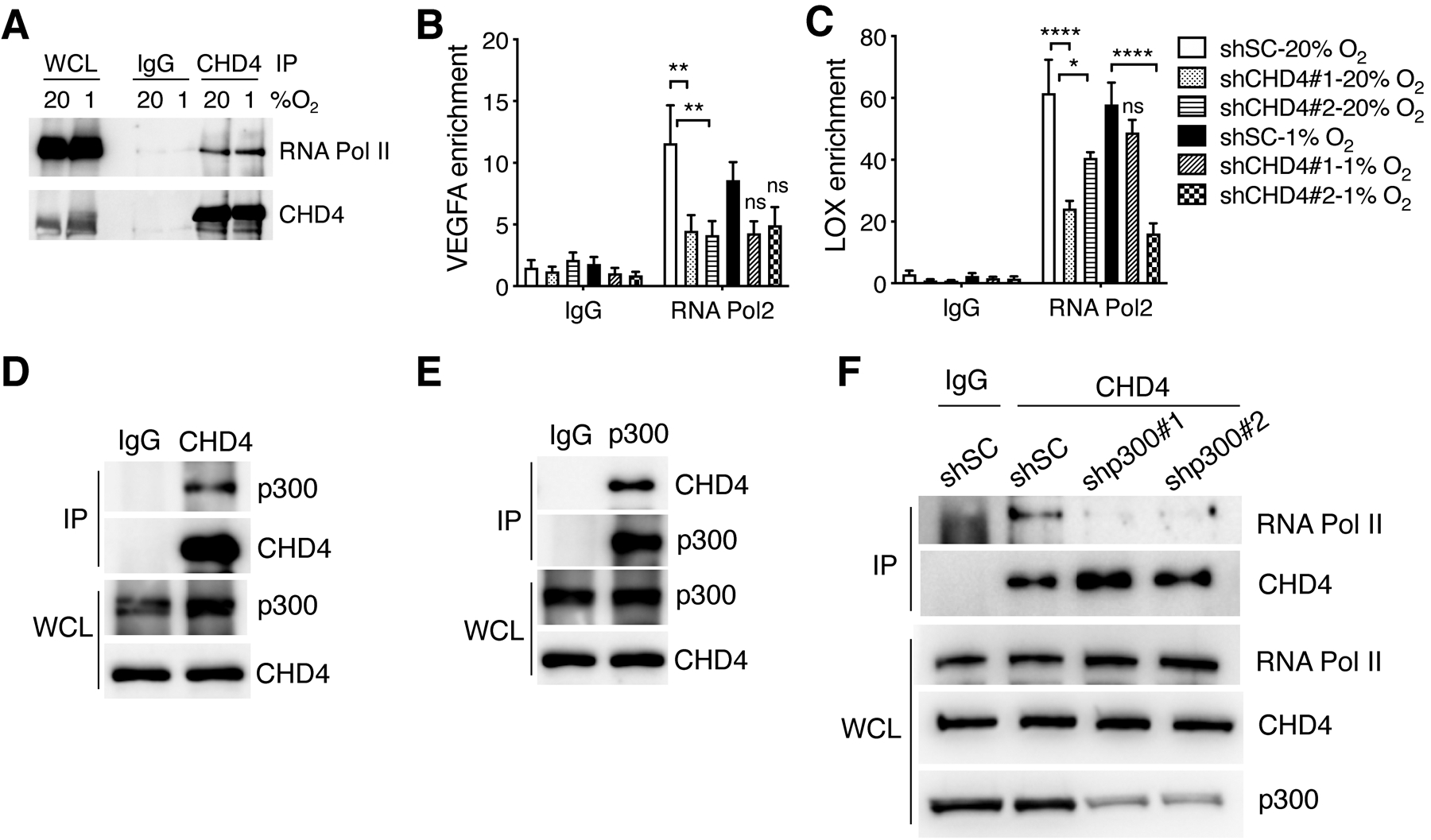
CHD4 increases RNA Pol II loading to HIF target genes through p300. (A) Co-IP with IgG or anti-CHD4 antibody in MDA-MB-231 cells exposed to 20% or 1% O2 for 24 hrs. (B and C) RNA Pol II ChIP-qPCR assay in shSC and CHD4 knockdown MDA-MB-231 cells exposed to 20% or 1% O2 for 24 hrs (n = 3, mean ± SEM). *p < 0.05; **p < 0.01; ****p < 0.0001, by 2-way ANOVA with Turkey’s test. ns, not significant. (D and E) Co-IP with IgG, anti-CHD4 (D), or anti-p300 (E) antibody in MCF-7 cells. (F) Co-IP with IgG or anti-CHD4 antibody in shSC and p300 KD MCF-7 cells.
RNA Pol II recruitment to HIF target genes under normoxia is a prerequisite for HIF-mediated transactivation. Thus, we investigated the molecular mechanism of CHD4-mediated RNA Pol II recruitment under normoxia. It was recently reported that the homolog of p300/CREB-binding protein regulates RNA Pol II pausing and release in Drosophila (28). Given that p300 is a well-known HIF coactivator (13), we hypothesized that p300 modulates CHD4-RNA Pol II interaction for RNA Pol II recruitment under normoxia. To address this, we studied whether CHD4 interacts with p300 in breast cancer cells under normoxia. Co-IP assay showed that anti-CHD4 antibody pulled down endogenous p300 in MCF-7 cells (Fig. 4D). Conversely, anti-p300 antibody also precipitated endogenous CHD4 in MCF-7 cells (Fig. 4E). KD of p300 completely abolished CHD4-RNA Pol II interaction in MCF-7 cells (Fig. 4F). These data indicate that p300 is required for CHD4 binding to RNA Pol II in breast cancer cells.
CHD4 increases colony formation and invasion of breast cancer cells in vitro
To determine the oncogenic role of CHD4 in breast cancer cells in vitro, we first performed clonogenic assay. Single cell suspension of MDA-MB-231-shSC, -CHD4 KD#1 and -KD#2 cells were cultured under 20% or 1% O2 for 14 days. Hypoxia markedly increased colony formation of MDA-MB-231-shSC cells (Supplemental Fig. S2A and B). CHD4 KD#1 or #2 almost eliminated colony growth under 20% and 1% O2 (Supplemental Fig. S2A and B). To determine specificity of CHD4 KD on colony growth, we generated CHD4-rescued MDA-MB-231 cells (Fig. 5A). Re-expression of FLAG-CHD4 in MDA-MB-231-CHD4 KD#1 cells by lentiviral transduction completely rescued cell growth under 20% and 1% O2 (Fig. 5B and C). These findings demonstrate that CHD4 increases breast cancer cell colony formation in vitro.
Figure 5.
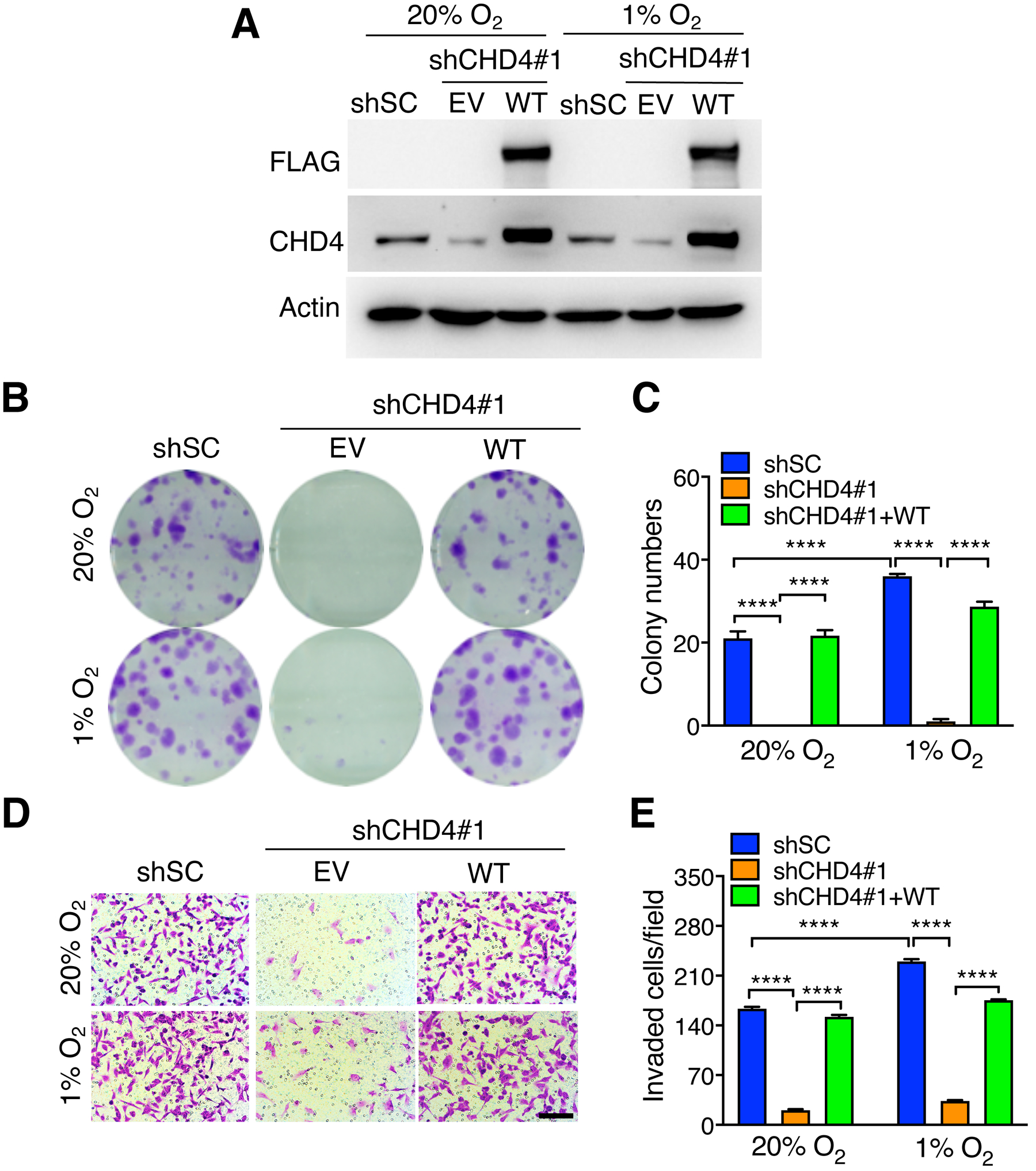
CHD4 promotes colony formation, migration, and invasion of breast cancer cells. (A) Immunoblot analysis of indicated proteins in shSC, CHD4 KD and rescued MDA-MB-231 cells exposed to 20% or 1% O2 for 24 hrs. (B and C) Clonogenic assay in shSC, CHD4 KD and rescued MDA-MB-231 cells exposed to 20% or 1% O2 for 14 days. Representative images from three independent experiments are shown in B. The data are quantified in C (n = 3, mean ± SEM). ****p < 0.0001, by 2-way ANOVA with Turkey’s test. (D and E) Invasion assay in shSC, CHD4 KD and rescued MDA-MB-231 cells exposed to 20% or 1% O2 for 24 hrs. Representative images from three independent experiments are shown in D. The data are quantified in E (n = 3, mean ± SEM). ****p < 0.0001, by 2-way ANOVA with Turkey’s test. Scale bar, 100 μm.
Next, we performed Boyden chamber invasion assay to investigate the effect of CHD4 on breast cancer cell invasion in vitro. Hypoxia significantly increased invasion of MDA-MB-231-shSC cells (Fig. 5D and E; Supplemental Fig. S2C and D). CHD4 KD#1 or #2 significantly decreased cell invasion under 20% and 1% O2 (Supplemental Fig. S2C and D). Importantly, re-introduction of FLAG-CHD4 to MDA-MB-231-CHD4 KD#1 cells rescued cell invasion under 20% and 1% O2 (Fig. 5D and E). These findings indicate that CHD4 increases breast cancer cell motility in vitro.
CHD4 augments breast tumor growth in mice through coactivating HIF
We next investigated the role of CHD4 in tumor growth in an orthotopic breast cancer xenograft mouse model. CHD4 KD in MDA-MB-231 cells by either of the CHD4 shRNAs significantly attenuated breast tumor growth in NSG mice (Supplemental Fig. S3A–C). Immunoblot assay confirmed the CHD4 KD efficiency in CHD4 KD breast tumors as compared with shSC tumors (Supplemental Fig. S3D). Re-expression of FLAG-CHD4 reversed tumor inhibition conferred by CHD4 KD#1 (Fig. 6A–C). Consistently, CHD4 KD#1 in MCF-7 cells also significantly suppressed tumor growth in NSG mice implanted with slow-released 17β-estradiol pellets (Supplemental Fig. S3E–H). Expression of HIF target genes ANGPTL4, NDNF, and LOX, but not the non-HIF target gene RPL13A, seemed to be reduced in CHD4 KD MCF-7 tumors (Fig. 6D). To study whether HIF is required for CHD4-mediated breast tumor progression, we generated parental and HIF-1/2α DKO MDA-MB-231 cells overexpressing FLAG-CHD4 or EV (Fig. 6E) and these cells were implanted into the mammary fat pad of female NOD/SCID mice. Overexpression of FLAG-CHD4 in MDA-MB-231 cells significantly increased breast tumor growth in mice, which was abolished by HIF-1/2α DKO (Fig. 6F–H). Therefore, these data indicate that CHD4 promotes breast tumor growth in mice through coactivating HIF.
Figure 6.
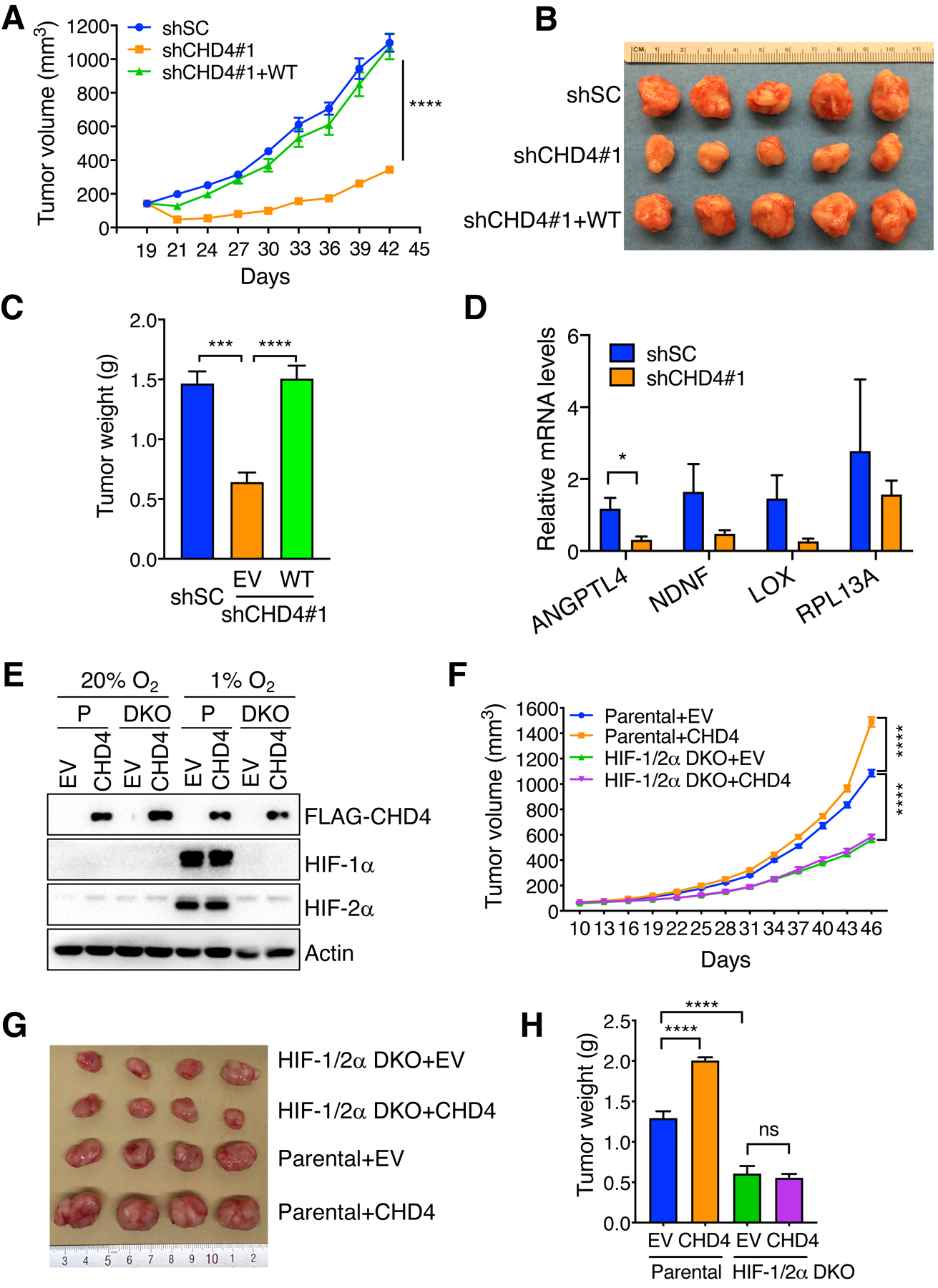
CHD4 promotes breast tumor growth in mice through coactivating HIF. (A–C) shSC, CHD4 KD and rescued MDA-MB-231 cells were orthotopically implanted into the mammary fat pad of female NSG mice, respectively. Tumor growth curve (A), image (B), and weight (C) are shown (n = 5, mean ± SEM). ***p < 0.001, ****p < 0.0001, by 2-way ANOVA with Turkey’s test. (D) RT-qPCR analysis of indicated mRNAs in shSC and CHD4 KD#1 MCF-7 tumors (n = 5, mean ± SEM). *p < 0.05, by Student’s t test. (E) Immunoblot analysis of indicated proteins in parental (P) and HIF-1/2α DKO MDA-MB-231 cells expressing EV or CHD4 exposed to 20% or 1% O2 for 24 hrs. (F-H) Parental and HIF-1/2α DKO MDA-MB-231 cells expressing EV or CHD4 were orthotopically implanted into the mammary fat pad of female NOD/SCID mice, respectively. Tumor growth curve (F), image (G), and weight (H) are shown (n = 5, mean ± SEM). ****p < 0.0001, by 2-way ANOVA with Turkey’s test.
CHD4 is highly expressed in breast cancer and positively correlated with HIF target genes and poor clinical outcomes in patients with breast cancer
To determine clinical relevance of CHD4 in human breast cancer, we retrieved the CHD4 expression data from The Cancer Genome Atlas (TCGA) breast cancer dataset and found that CHD4 was amplified along with the significantly high mRNA expression in a subset of human breast tumors (Fig. 7A). Consistently, CHD4 mRNA levels were significantly elevated in both primary and metastatic breast tumors as compared to adjacent normal breast tissues (Fig. 7B). CHD4 mRNA upregulation was observed in all breast cancer subtypes including luminal A, luminal B, HER2+ and basal-like, and stages 1–4 of human breast tumors (Fig. 7C and D). Remarkably, CHD4 upregulation was mutually exclusive to other known HIF coactivators ZMYND8, KDM4C (JMJD2C), CDK8, CREBBP (CBP), and KAT5 whose mRNAs were also upregulated in human breast tumors, but not with EP300 (p300) that was not upregulated in breast tumors (Fig. 7E), suggesting heterogeneity of epigenetic regulation of HIF in breast cancer. Furthermore, we found that CHD4 expression was positively correlated with expression of HIF target genes LOX and VEGFA in human breast tumors (Fig. 7F and G), which supports our findings about upregulation of LOX and VEGFA by CHD4 in hypoxic breast cancer cells (Fig. 2I). Lastly, Kaplan-Meier analysis of the TCGA dataset showed that high CHD4 mRNA levels were positively correlated with poor overall survival of patients with breast cancer (Fig. 7H). Collectively, these data indicate that CHD4 is upregulated and positively associated with HIF target genes in human breast cancer, and that CHD4 is an independent risk factor for women with breast cancer.
Figure 7.
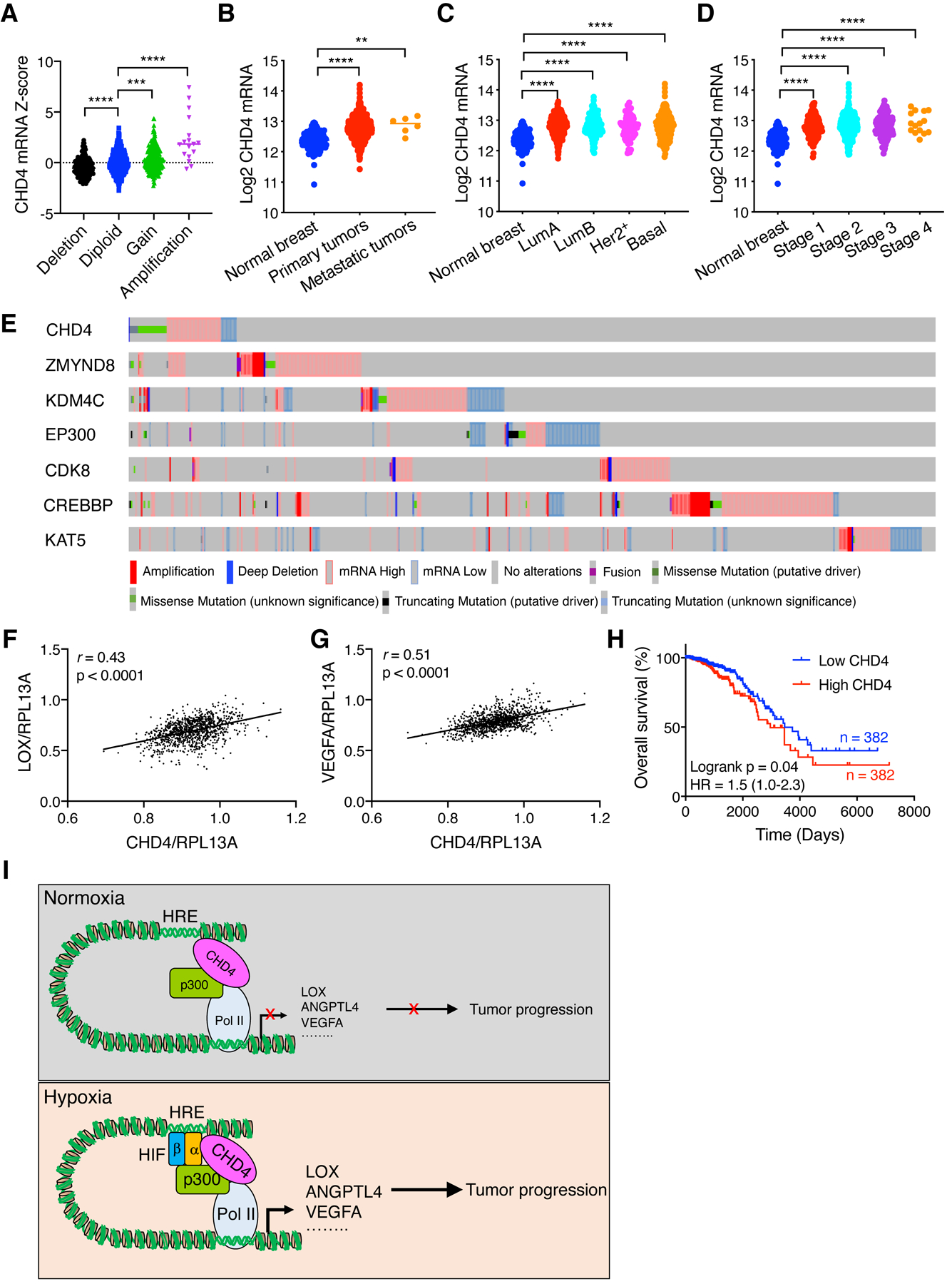
CHD4 is correlated with HIF target genes and a potential prognostic factor in breast cancer. (A) Association of CHD4 copy number alteration with its mRNA expression in the TCGA breast cancer cohort. ***p < 0.001, ****p < 0.0001, by 1-way ANOVA with Dunnett’s test. (B-D) Analysis of CHD4 mRNA levels in normal breast tissues and breast tumors. The data were retrieved from the TCGA. **p < 0.01, ****p < 0.0001, by 1-way ANOVA with Turkey’s or Dunnett’s test. (E) Upregulation and genetic alterations of HIF coactivators in human breast tumors. The data were retrieved from the TCGA in the cBioPortal. (F and G) Pearson correlation analysis of CHD4 mRNA with LOX (F) and VEGFA (G) mRNAs in human breast tumors from the TCGA dataset. Their gene expression was normalized to non-HIF target gene RPL13A. (H) Kaplan-Meier survival analysis for patients with breast cancer. Patients were divided by median expression levels of CHD4. The data were retrieved from the TCGA. HR, hazard ratio. (I) A proposed model of CHD4-mediated HIF activation in breast cancer. CHD4 interacts with p300 and mediates recruitment of RNA Pol II to HIF target genes for pausing under normoxia. Upon hypoxia, CHD4 interacts with HIF-1/2α and increases their recruitment to the HRE, thereby enhancing transcription of a subset of HIF downstream target genes that promote breast cancer progression.
Discussion
In the present study, we identified CHD4 as a novel HIF coactivator in breast cancer cells. CHD4 is enriched at the chromatin regions near the HRE and induces a subset of HIF target genes in breast cancer cells under hypoxia and in xenograft tumors. These genes are involved in angiogenesis, collagen remodeling, apoptosis, and cell proliferation, which are the classical HIF-mediated biological functions involved in breast tumor progression. CHD4 is also positively correlated with HIF target genes LOX and VEGFA in human breast tumors, strongly supporting HIF transactivation by CHD4 in vivo. CHD4 is known to mediate gene repression through the NuRD complex. A recent study showed that CHD4 represses hypoxia-induced expression of Ripk3 in murine embryonic endothelial cells (29). Although we did not observe the same regulation for Ripk3 as Ripk3 expression is almost undetectable in human breast cancer cells, we found by RNA-seq that several CHD4-repressed genes can be induced by hypoxia. Neither hypoxia nor HIF is involved in CHD4-mediated gene repression, indicating specificity of CHD4 co-activator function for HIF under hypoxia. Our present studies showed that the coactivator function of CHD4 is independent of its helicase activity and the NuRD complex in hypoxic cells. These findings support an idea that CHD4 is a peripheral component of the NuRD complex and can function independently (21). A recent study also reported coactivation of PAX3-FOXO1 target genes by CHD4 in alveolar rhabdomyosarcoma, although it remains unclear about the role of the NuRD complex in this context (30). Importantly, we found that CHD4 physically interacts with p300, suggesting that free CHD4 outside the NuRD complex is associated with the transcriptional coactivator complex to exert its coactivator function in breast cancer cells.
Our mechanistic studies uncover two layers of HIF regulation by CHD4 in breast cancer cells: (1) increased HIF binding to the HRE and (2) increased recruitment of RNA Pol II. We showed that CHD4 occupies at the promoter of HIF target genes under normoxia. Previous studies reported that the plant zinc finger homeodomain mediates CHD4 binding to histone H3 and their interaction is enhanced by acetylation of lysine 9 of histone H3 (H3K9ac) (31). We have previously shown H3K9ac at the promoter of HIF target genes (26), suggesting that H3K9ac may be responsible for the recruitment of CHD4 to HIF target genes under normoxia. Interestingly, CHD4 and HIF-1/2α are mutually recruited to HIF target genes in breast cancer cells under hypoxia. Our biochemical studies showed that CHD4 binds to the transactivation domain of HIF-1/2α. This physical interaction may mediate HIF recruitment to the HRE, thereby enhancing transcription of HIF downstream target genes. Identification of the amino acid residues of HIF-1/2α and CHD4 involved in their protein-protein interaction would help address this hypothesis.
RNA Pol II is pre-loaded and paused on HIF target genes under normoxia (14,20), which offers a fine and rapid regulatory mechanism of HIF target gene transcription in response to hypoxia. Our recent studies showed that ZMYND8 triggers RNA Pol II-mediated transcriptional elongation of HIF target genes to enhance their expression in breast cancer cells (14). Unlike ZMYND8, CHD4 is required for RNA Pol II loading to HIF target genes in breast cancer cells and this effect is more robust under normoxia as compared to that under hypoxic conditions, indicating its critical role in RNA Pol II recruitment for pausing under normoxia.
Previous studies have discovered many transcriptional coactivators for HIF in breast cancer cells (14,16,17,32). These coactivators are often upregulated in breast tumors and few gene mutations have been identified. Moreover, upregulation of CHD4 and other known HIF coactivators including ZMYND8, KDM4C (JMJD2C), CDK8, CREBBP (CBP), and KAT5 occurs in the mutually exclusive subset of breast cancer patients according to the TCGA breast cancer dataset, suggesting that the individual HIF coactivator may have a dominant function for HIF transcriptional activity in the particular population of breast cancer patients. Although some of HIF coactivators (e.g., CHD4 and ZMYND8) have the redundant function for induction of HIF target genes (e.g. LOX and VEGFA), the heterogenous pattern highlights a great importance of comprehensive identification of HIF coactivators including CHD4 towards understanding of regulation of hypoxia responses in breast cancer cells.
Our in vitro functional studies here showed that CHD4 KD decreases colony growth and cell invasion under both normoxia and hypoxia. CHD4’s oncogenic functions under normoxia may also require HIF as MDA-MB-231 cells express the low but detectable levels of HIF-1/2α under normoxia, but we cannot exclude a possible involvement of the gene repression and DNA damage functions of CHD4 in colony growth and cell invasion under normoxia (33–35). It is worth pointing out that our in vivo xenograft studies showed that HIF is required for CHD4-mediated breast tumor growth in mice. As breast tumor is hypoxic (36), the CHD4-HIF axis appears to play a key role in tumor progression in vivo.
In conclusion, our biochemical, cancer biology, and bioinformatics studies in human breast cancer cells, breast cancer mouse models, and human breast cancer patients suggest a working model (Fig. 7I): CHD4 interacts with p300 and mediates the recruitment of RNA Pol II to HIF target genes for pausing under normoxia. Upon hypoxia, CHD4 interacts with HIF-1/2α and increases their recruitment to the HRE, thereby enhancing transcription of a subset of HIF downstream target genes that promote breast cancer progression. CHD4 is highly expressed in breast tumors and a potential prognostic factor of survival of breast cancer patients. Importantly, CHD4 is positively correlated with HIF target genes LOX and VEGFA in human breast tumors. Therefore, the CHD4-HIF pathway has clinical relevance and disrupting this signaling pathway may be an invaluable approach for the treatment of breast cancer.
Supplementary Material
Significance:
This study identifies CHD4 as a HIF coactivator and elucidates the fundamental mechanism underlying CHD4-mediated HIF transactivation in breast tumors.
Acknowledgments
This work was supported by grants from the Susan G. Komen® (CCR16376227), the NIH (R01CA222393 and R00CA168746), the CPRIT (RR140036 and RP190358), the Mary Kay Foundation (08-19), and the Welch Foundation (I-1903) to W.L., and the NIH (R00NS078049, R35GM124693, and R01AG066166), the CPRIT (RP170671), and the Welch Foundation (I-1939) to Y.W.. W.L. is a CPRIT Scholar in Cancer Research.
Financial support: YJ.W. (CPRIT); Y.C. (Susan G Komen); L.B. (NIH, Welch); B.Z. (CPRIT); J.E.W. (NIH); A. K. (UTSW); C. X. (UTSW); Y.W. (NIH, CPRIT, Welch, UTSW); W.L. (NIH, CPRIT, Welch, Susan G Komen, Mary Kay, UTSW)
Footnotes
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Vaupel P, Mayer A, Höckel M. Tumor hypoxia and malignant progression. Methods Enzymol 2004;381:335–54 [DOI] [PubMed] [Google Scholar]
- 2.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 1995;92:5510–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 2012;148:399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 1992;12:5447–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev 1997;11:72–82 [DOI] [PubMed] [Google Scholar]
- 6.Gu YZ, Moran SM, Hogenesch JB, Wartman L, Bradfield CA. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr 1998;7:205–13 [PMC free article] [PubMed] [Google Scholar]
- 7.Kaelin WG Jr., Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 2008;30:393–402 [DOI] [PubMed] [Google Scholar]
- 8.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001;292:464–8 [DOI] [PubMed] [Google Scholar]
- 9.Koh MY, Darnay BG, Powis G. Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1a, leading to its oxygen-independent degradation. Mol Cell Biol 2008;28:7081–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo W, Zhong J, Chang R, Hu H, Pandey A, Semenza GL. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1a but not HIF-2a. J Biol Chem 2010;285:3651–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Zhang C, Zhao Y, Yue X, Wu H, Huang S, et al. Parkin targets HIF-1alpha for ubiquitination and degradation to inhibit breast tumor progression. Nat Commun 2017;8:1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo W, Wang Y. Epigenetic regulators: multifunctional proteins modulating hypoxia-inducible factor-alpha protein stability and activity. Cell Mol Life Sci 2018;75:1043–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, et al. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci U S A 1996;93:12969–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Zhang B, Bao L, Jin L, Yang M, Peng Y, et al. ZMYND8 acetylation mediates HIF-dependent breast cancer progression and metastasis. J Clin Invest 2018;128:1937–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bao L, Chen Y, Lai HT, Wu SY, Wang JE, Hatanpaa KJ, et al. Methylation of hypoxia-inducible factor (HIF)-1alpha by G9a/GLP inhibits HIF-1 transcriptional activity and cell migration. Nucleic Acids Res 2018;46:6576–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci U S A 2012;109:E3367–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai YP, Chen HF, Chen SY, Cheng WC, Wang HW, Shen ZJ, et al. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol 2014;15:513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galbraith MD, Allen MA, Bensard CL, Wang X, Schwinn MK, Qin B, et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 2013;153:1327–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kato H, Tamamizu-Kato S, Shibasaki F. Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J Biol Chem 2004;279:41966–74 [DOI] [PubMed] [Google Scholar]
- 20.Choudhry H, Schodel J, Oikonomopoulos S, Camps C, Grampp S, Harris AL, et al. Extensive regulation of the non-coding transcriptome by hypoxia: role of HIF in releasing paused RNApol2. EMBO Rep 2014;15:70–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Low JK, Webb SR, Silva AP, Saathoff H, Ryan DP, Torrado M, et al. CHD4 Is a Peripheral Component of the Nucleosome Remodeling and Deacetylase Complex. J Biol Chem 2016;291:15853–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilczewski CM, Hepperla AJ, Shimbo T, Wasson L, Robbe ZL, Davis IJ, et al. CHD4 and the NuRD complex directly control cardiac sarcomere formation. Proc Natl Acad Sci U S A 2018;115:6727–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao H, Han Z, Liu X, Gu J, Tang F, Wei G, et al. The chromatin remodeler Chd4 maintains embryonic stem cell identity by controlling pluripotency- and differentiation-associated genes. J Biol Chem 2017;292:8507–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xia L, Huang W, Bellani M, Seidman MM, Wu K, Fan D, et al. CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell 2017;31:653–68 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKenzie LD, LeClair JW, Miller KN, Strong AD, Chan HL, Oates EL, et al. CHD4 regulates the DNA damage response and RAD51 expression in glioblastoma. Sci Rep 2019;9:4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011;145:732–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem 1996;271:32529–37 [DOI] [PubMed] [Google Scholar]
- 28.Boija A, Mahat DB, Zare A, Holmqvist PH, Philip P, Meyers DJ, et al. CBP Regulates Recruitment and Release of Promoter-Proximal RNA Polymerase II. Mol Cell 2017;68:491–503 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colijn S, Gao S, Ingram KG, Menendez M, Muthukumar V, Silasi-Mansat R, et al. The NuRD chromatin-remodeling complex enzyme CHD4 prevents hypoxia-induced endothelial Ripk3 transcription and murine embryonic vascular rupture. Cell Death Differ 2020;27:618–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bohm M, Wachtel M, Marques JG, Streiff N, Laubscher D, Nanni P, et al. Helicase CHD4 is an epigenetic coregulator of PAX3-FOXO1 in alveolar rhabdomyosarcoma. J Clin Invest 2016;126:4237–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Musselman CA, Mansfield RE, Garske AL, Davrazou F, Kwan AH, Oliver SS, et al. Binding of the CHD4 PHD2 finger to histone H3 is modulated by covalent modifications. Biochem J 2009;423:179–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JS, Kim Y, Bhin J, Shin HJ, Nam HJ, Lee SH, et al. Hypoxia-induced methylation of a pontin chromatin remodeling factor. Proc Natl Acad Sci U S A 2011;108:13510–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arends T, Dege C, Bortnick A, Danhorn T, Knapp JR, Jia H, et al. CHD4 is essential for transcriptional repression and lineage progression in B lymphopoiesis. Proc Natl Acad Sci U S A 2019;116:10927–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo CW, Wu CC, Chang SJ, Chang TM, Chen TY, Chai CY, et al. CHD4-mediated loss of E-cadherin determines metastatic ability in triple-negative breast cancer cells. Exp Cell Res 2018;363:65–72 [DOI] [PubMed] [Google Scholar]
- 35.Qi W, Chen H, Xiao T, Wang R, Li T, Han L, et al. Acetyltransferase p300 collaborates with chromodomain helicase DNA-binding protein 4 (CHD4) to facilitate DNA double-strand break repair. Mutagenesis 2016;31:193–203 [DOI] [PubMed] [Google Scholar]
- 36.Luo W, Wang Y. Hypoxia Mediates Tumor Malignancy and Therapy Resistance. Adv Exp Med Biol 2019;1136:1–18 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.