

Abstract
Omic technologies have enabled the complete readout of the molecular state of a cell at different biological scales. In principle, the combination of multiple omic data types can provide an integrated view of the entire biological system. This integration requires appropriate models in a systems biology approach. Here, we focus on genome-scale models (GEMs) as one computational systems biology approach for interpreting and integrating multi-omic data. GEMs convert the reactions (related to metabolism, transcription and translation) that occur in an organism to a mathematical formulation that can be modeled using optimization principles. We review a variety of genome-scale modeling methods used to interpret multiple omic data types, including genomics, transcriptomics, proteomics, metabolomics, and meta-omics. The ability to interpret omics in the context of biological systems has yielded important findings for human health, environmental biotechnology, bioenergy, and metabolic engineering. We find that concurrent with advancements in omic technologies, genome-scale modeling methods are also expanding to enable better interpretation of omic data. Therefore, we expect continued synthesis of valuable knowledge through the integration of omic data with GEMs.
Keywords: systems biology, genome-scale model, computational model, machine learning, genomics
1. Introduction
Omic technologies aim to measure the molecular composition of a cell in its entirety. These measurements profile the functional potential (genomics) and activity (transcriptomics, proteomics, metabolomics) of an organism at the systems scale. These entities (genome, transcriptome, proteome, metabolome) are interrelated through expression, metabolism, signaling and regulation. Understanding and interpreting each of these omic data types individually and combined could help unravel the mechanistic intricacies of biological systems. However, the interconnectedness among these different levels of function within a biological system poses significant challenges for studying the underlying mechanisms and relationships.
Each individual omic data type only describes part of the larger system. Therefore, integrative omic platforms are being developed. For instance, proteogenomics (proteomics with genomics/transcriptomics) can address genetic polymorphisms[1], improve the detection of novel genes or identify misannotated ORFs[2] and address the “missing protein problem,” which refers to predicted proteins that are not detected in proteomic data[3]. Likewise, metabolomics has been combined with other omic platforms to demonstrate the environmental effects on post-translational modification (PTM) rates[4], to understand the regulation of metabolite levels[5] and to elucidate complex interactions between the host, commensal bacteria, and pathogens[6]. These diverse datasets can yield a comprehensive understanding of biological mechanisms when they are contextualized and unified into a systems view of biology.
Systems biology is an interdisciplinary field that aims to predict the behavior of biological systems (i.e., phenotype) by considering interactions among biological parts in the context of the whole system. One approach to predicting system behavior is computational modeling such as genome-scale modeling. Genome-scale models (GEMs) have been used to analyze individual and multi-omic data sets[7]. GEMs can be analyzed using various methods including COnstraint-Based Reconstruction and Analysis (COBRA) methods (Figure 1) [8].
Figure 1. Building genome-scale models (GEMs) and integrating them with various omic data types as constraints.

Genome-scale models are systems representations of interactions occurring between different molecular components (e.g., metabolites, proteins). These models are built using the annotated genomes of respective organisms. Other omic data can also be used to refine GEMs (A). GEMs need to be constrained to obtain biologically relevant information (B). General/environmental constraints such as mass balance constraints and and flux bounds can be added to GEMs. For ME-models, additional constraints such as coupling constraints and biomass dilution constraints need to be applied[9](B-I). Transcriptional regulatory networks (TRNs) combined with transcriptome data can also be used to constrain a GEM to create integrated regulatory genome-scale model. It should be noted that gene expression thresholds are applied in this case as well (B-II). Likewise, multiple omic data (transcriptomic, proteomic and metabolomic) can be utilized to constrain the model using various approaches. The integration of data leads to new optimization problems (e.g., minimization of inconsistency between fluxes and expression states, maximization of total sum of fluxes through core reactions, etc.) subjected to their own sets of constraints including mass balance and flux constraints (B-III). The resulting models (C) can be simulated to investigate the genotype-phenotype-environment relationship in the biological system being studied.
In general, for COBRA analysis, first the molecular composition of an organism can be represented as a network of interactions in which nodes represent specific entity (e.g., metabolites) and edges represent the interaction between these entities (such as substrate-product conversion). To implement modeling using COBRA framework, these networks are converted to stoichiometric matrix (S-matrix) in which rows represent molecular entities and columns represent their interactions. Then, the S-matrix can be analyzed using mathematical optimization as formulated in the COBRA framework[8]. In this approach, the steady state of an organism can be solved by optimizing an objective function (Z). Without adding constraints to this optimization problem, we can get infinite number of solutions (fluxes) that can satisfy the steady-state assumption. Therefore, the optimization problem is subjected to certain constraints which are: Sv = 0 (mass balance constraints), and l ≤ vi ≤ u (flux bounds). Here, vi is the flux vector and l, u are the lower and upper bounds of the flux of the ith reaction. Hence, by optimizing Z, one can approximate the flux state of an organism, and identify molecular interactions that lead to such state.
GEMs were traditionally used to model the metabolic state of an organism (metabolic or M-model). In recent years, however, GEMs have also been utilized to compute the metabolic and proteomic state of an organism (metabolism and macromolecular expression or ME-model). Since ME-models deal with metabolism and proteome allocation, additional constraints including coupling constraints and biomass dilution constraints are added (check [9] for more information). In this article, we review how modeling in systems biology has yielded new insights from omic data, and how GEMs can be used to interpret large-scale data. Modeling platforms in systems biology can integrate multiple omics and synthesize knowledge. Such modeling platforms include kinetic modeling (stochastic or deterministic), Boolean formalisms, Bayesian approaches, and COBRA[8b]. We focus on the use of COnstraint-Based Reconstruction and Analysis (COBRA) methods in which steady state of a biological system are modeled by optimizing an objective function subjected to constraints including thermodynamic, stoichiometric, and enzymatic ones. We highlight recent advances in COBRA that were made to integrate multi-omic data types. We organize our review by the omic data types analyzed and COBRA methods used (Figure 2).
Figure 2.
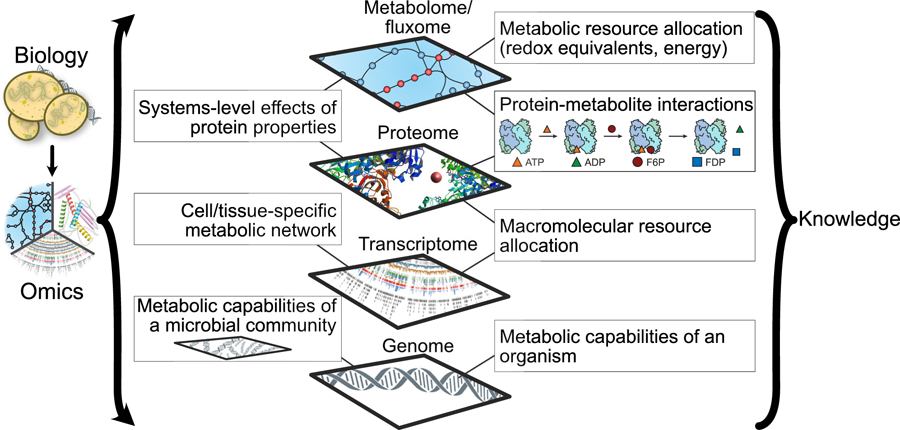
Graphical overview of synthesizing knowledge using omic data and genome-scale models
2. From annotated genome sequences to genome-scale models of cell metabolism
The genome encodes the functional capabilities of an organism. Genomics is the study of the whole genome of an organism. Since the first genome sequence of human mitochondria in 1981, there has been a steady increase in the publications that contribute to this field[10] (Figure 3). With the explosion of sequenced genomes, tools in comparative genomics have been developed to annotate sequences of previously uncharacterized genomes to unveil their functional potential[11]. With the advent of next generation sequencing technologies, sequencing genomes has become relatively quick, easy and cheap[12]. However, even though we can sequence an organism, we still do not understand the full functional potential of organisms[13].
Figure 3.
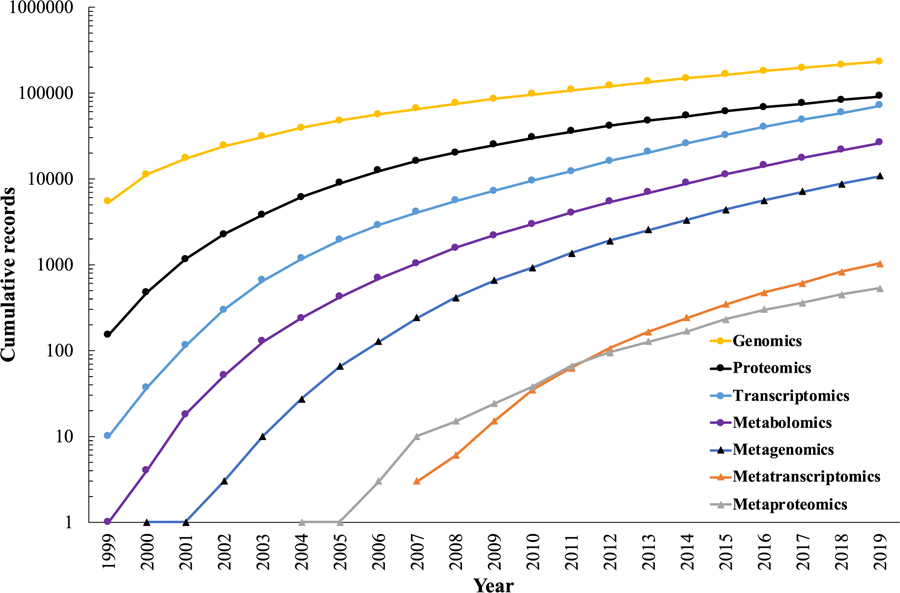
Web of Science publications for various omic technologies.
A genome-scale model (GEM) is a modeling approach for mathematically describing all possible functions that are encoded by the genome, and their interactions, within the context of the full interaction network. For reconstruction of the network of an organism, genomic data and proper annotation are crucial in order to represent the correct interaction between various molecular entities. Following the reconstruction, COBRA methods can be utilized to analyze the state of the network to identify and predict important features of the organism such as genotype-phenotype-environment relationships, including growth rate, metabolite exchange rates, and gene essentiality[14]. GEMs have also been useful in predicting and analyzing the end result of adaptive evolution[15]. At present, GEMs have been manually reconstructed for at least 183 organisms [16], and methods are being developed to model microbial communities[17].
3. Using omic data to refine the genome-scale models
Three broad approaches exist to improve genome-scale models by utilizing omic data. First and foremost, omic data can be directly compared with the flux distribution derived by simulating GEMs to identify any discrepancies between the predicted and experimental data. For instance, one can compare exometabolome with modeling result to determine how accurately the model can predict the secretion profile of an organism under given media condition.
Next, omic data can be used as additional flux constraints on the GEMs to create context- and tissue-specific models[7a, 7b, 18] (Figure 1). For such purpose, numerous methods have been developed which can be divided into three subcategories— 1) use omic data to either indicate presence or absence of enzymes or put relative constraints on enzyme activities (GIMME[19], GIM3E[20], REMI[21]), 2) use expression datasets to create context-specific models without prior knowledge of objective function (e.g. iMAT[22], INIT[23]) and 3) prune non-functional reactions (as extracted from the expression data) to create tissue-specific models (e.g. MBA[24], mCADRE[25], CORDA[26]). One inherent issue with these approaches is that such models can only describe the regulation of metabolism for which the data is integrated into the model.
For predictive models, integrated regulatory metabolic models need to be built. Methods such as Probabilistic Regulation Of Metabolism (PROM) [27] exist that can integrate expression datasets (normally transcriptomes) with a transcriptional regulatory network (TRN) and superimpose the information onto a GEM to create an integrated model. In such method, the maximum allowable fluxes of reactions catalyzed by particular enzymes are constrained by the probability of the expression of respective genes given the expression state of a controlling transcription factor (TF). Such TF expression is calculated from the expression datasets across multiple conditions. The rules of TF-target enzyme interaction are governed by the structure of TRN. Since PROM requires a pre-constructed TRN, in recent times, Integrated Deduced REgulation AND Metabolism (IDREAM) has been developed. IDREAM can create a TRN followed by integration of TRN with GEM and high-throughput expression data (using PROM framework) to create integrated models [28].
In the next few sections, we will discuss individual omic data types and novel methods that are being introduced to integrate and improve the predictive capabilities of GEMs.
4. Using transcriptomics to build context-specific models
Transcriptomics is the quantitative study of all expressed RNA in an organism. Transcriptomic technologies have advanced from the use of microarrays (i.e., methods involving use of probes to detect specific markers) to exploiting next generation sequencing in the form of RNA-sequencing (RNA-seq). RNA-seq has become more popular because of the ease and availability of advanced sequencing machines[29]. Multiple RNA-Seq algorithms are available with varying degree of accuracy and precision[30].
Transcriptomics can be used to refine GEMs by integrating the data into model constraints. In this approach, transcriptomics is analyzed by specialized algorithms to create context-specific models by determining the subset of genes that are expressed in a specific cell type, cell line, or tissues[31]. These context-specific models accurately capture tissue specific genotype-phenotype relationships, including gene essentiality[31]. Multiple algorithms are available for building context-specific models, each making different assumptions. Opdam et al.[31] compared six algorithms and showed that the choice of algorithm (and assumptions) had the greatest impact on the model’s accuracy of gene essentiality predictions[31]. Once constructed appropriately, context-specific models have yielded important insights into the metabolic mechanisms underlying human diseases.
Gatto et al.[32] constructed context-specific models for 917 primary tumor samples across 13 cancer types, using RNA-Seq and the tINIT algorithm[33]. The models indicated that although cancers can differ in gene expression, their metabolic capabilities are largely similar. Furthermore, cancer metabolic networks overlapped largely with matched normal tissues, suggesting that the metabolic reprogramming—a hallmark of cancer—may reflect cancer cell plasticity to varying conditions. The study also identified a smaller set of 18 metabolic reactions that are present in all the cancers included in the study but without housekeeping functions (such as growth, energy generation and metabolism) present in normal tissues[32].
5. Integrating metabolomics with genome-scale models
Metabolomic technologies quantify the small molecules (molecular mass < 1500 Da) involved in energy metabolism (“metabolites”), representing the most direct way to profile a cell’s biochemical activity[34]. Metabolites are involved in the regulation of expression, metabolism and function of DNA, RNA, and proteins[35]. Research using metabolomic approaches has increased over the past decade (Figure 3) as studies involving identification of disease biomarkers[36] and other important applications[34a, 35] have risen. To identify and quantify metabolites, methods such as Nuclear Magnetic Resonance (NMR) and mass spectrometry (MS) are used. In MS, targeted (hypothesis-driven), untargeted (discovery-based), and recently introduced pseudo-targeted approaches are available[34b, 37]. Identification of a metabolite through its spectral signature is crucial to understanding its biological role. However, this approach is limited by the number of available spectra in the available databases[38]. Therefore, multiple methods have been developed for predicting metabolites of which machine learning methods appear promising [39]. Furthermore, a metabolite’s function depends on its context-specific interactions with other biological entities. Various computational methods including pathway mapping and network modeling[40], and GEMs are addressing this need.
Multiple recent studies have used GEMs to integrate metabolomic data. In a recent study, the authors analyzed metabolomic data using GEMs of hepatocytes and identified dopa decarboxylase (DDC) as one of the major cancer-causing enzymes. Following this discovery, authors used the Library of Integrated Network-Based Cellular Signatures (LINCS) program to identify possible drugs that could inhibit expression of DDC[41]. In another study, time-course metabolomic data from human red blood cells (RBCs) stored at different temperatures was analyzed using an RBC genome-scale model. The analysis revealed temperature-dependent metabolic states of RBCs in storage conditions[42]. Recently, a new COBRA method called unsteady-state flux balance analysis (uFBA) has been developed to integrate time-course metabolomic data with GEMs to study the metabolism of RBCs stored in blood bags. The uFBA method predicted that stored RBCs metabolize citric acid cycle intermediates to regenerate key cofactors. These predictions were experimentally confirmed using 13C-metabolic flux analysis[43].
6. Measuring and predicting proteome allocation using ME-models
The proteome represents the functional state of a cell. Proteomics is the quantitative study of all expressed proteins in an organism. Out of several methods used in proteomics, mass spectrometry (MS) is one of the most common platforms. MS can be used in tandem (MS/MS) to provide additional information about a given peptide[44], and it can be coupled with chromatographic methods to reduce sample complexity and to improve quantification accuracy[45]. Furthermore, it allows multiple properties of the proteome—such as expression, interactions, and modification—to be studied[46].
Depending on the goal of a study, either targeted or untargeted proteomic approaches can be applied, and sometimes combined for improved analysis[47]. In untargeted proteomics, all possible proteins expressed from a sample are detected and quantified without a priori knowledge. On the other hand, a targeted platform is used to detect specific proteins especially when the desired proteins are known to be present in low abundance a priori[48]. Therefore, targeted approaches are more precise but have lower coverage than untargeted methods[47]. For data acquisition using tandem mass spectrometry (MS/MS), two modes exist – data-dependent acquisition (DDA) mode and data-independent acquisition (DIA) mode. In DDA, a subset of the most abundant precursor ions that exceed a predefined intensity threshold are selected from the first MS scan to the next MS scan. For targeted proteomics, alternative approaches called multiple reaction monitoring (MRM) and parallel reaction monitoring (PRM) which select precursor ions for a small set of predetermined peptides for subsequent MS scan are used[49]. DIA, on the other hand, relies on successive isolation and subsequent fragmentation of peptides within a defined mass-to-charge (m/z) window throughout the entire m/z range[50]. SWATH-MS is a DIA method combined with targeted proteomic analysis, and provides good coverage with comparable accuracy and reproducibility [51]. Both DDA and DIA approaches have their advantages and disadvantages related to sensitivity, dynamic range, accuracy, flexibility and ease of use[49, 51–52]. Finally, for quantification of proteins in a given sample, either relative or absolute quantification methods can be used (please refer to Calderón-Celis et al.[53] for review).
Genome-scale models of metabolism and macromolecular expression (ME-models)[7c, 9b, 54] directly predict protein expression and proteome allocation (i.e., the relative mass or mole fractions of expressed proteins in a cell). These predictions are validated directly using proteomics, or indirectly using transcriptomics. ME-models predict fluxes for reactions spanning metabolism, transcription, translation, protein modifications, translocation[55], and protein folding[56]. ME models compute up to 85% protein mass in E. coli[57]. ME models are now available for three organisms: Thermotoga maritima[58], E. coli[54a, 54b], and Clostridium ljungdahlii[59]. Proteomic data has been used to calibrate a ME-model of E. coli, decreasing prediction errors of growth rate and metabolic fluxes by 69% and 14%[54d], and to validate proteomes predicted by a ME-model updated with machine learning-based enzyme turnover rates[54c].
Recently, ME-models were extended to predict cellular response to three stresses: thermal (FoldME)[56], oxidative (OxidizeME)[57], and acid (AcidifyME)[60]. By mechanistically reconstructing key molecular responses to each stress, the models successfully predicted phenotypic response (change in growth rate) and differential expression in various growth conditions (i.e., media, supplements, etc.) and stress intensities. These models have been used to explain biological mechanisms by interpreting omics.
A ME-model accounting for the proteostasis network, FoldME[56], was used to study the global effects caused by the protein stability of dihydrofolate reductase (DHFR). The experimental (transcriptomic data) and predicted data were quantitatively correlated for the major clusters of orthologous groups (COG). Further analysis using the ME-model suggested that protein destabilizing mutations can lead to chaperone-mediated strategy of systems-level proteome reallocation including downregulation of coenzyme biosynthetic pathways[56].
In another study, a ME-model accounting for the effect of ROS on metalloproteins, OxidizeME[57], was used to explain why the growth rate of E. coli was limited when using naphthoquinone (NQ) instead of ubiquinone (UQ) in the electron transport system (ETS)[15b]. NQ autoxidizes more readily than ubiquinone (UQ), generating superoxide in the periplasm. OxidizeME showed that the metabolic and protein expression cost of detoxifying periplasmic superoxide strongly decreased growth rate. The reduced electron transport system efficiency due to electron leakage from NQ toward superoxide generation decreased growth rate further; however, the cost of detoxification was demonstrated to be the primary reason for reduced growth rate.
A modeling approach called metabolism and macromolecular mechanisms (MM) was developed recently for human RBCs[61]. Unlike ME-models, the reactions related to transcription and translation are not present in RBC-MM. In RBC-MM, proteomic data were used to constrain enzyme abundances, which constrained the reaction fluxes. This model simulates metabolism, hemoglobin binding, and the formation and detoxification of reactive oxygen species (ROS)[61].
7. Integrating multi-omic data with genome-scale models
Studies are now combining multi-omic platforms with GEMs to study complex interactions that occur at the molecular level within organisms. In a recent study, metabolomics combined with proteomics was integrated in genome-scale model of E. coli to identify pathway engineering strategies to improve biofuel production[62]. A ME-model was recently used to analyze multi-omic (genomic, transcriptomic, ribosomal profiling, proteomic, and fluxomic) data to discover two biological regularities associated with enzyme turnover rates and translation in E. coli [7c]. Likewise, a laboratory rat genome-scale model was integrated with transcriptomic, metabolomic and fluxomic data to identify plasma metabolites that are associated with acetaminophen-induced liver injury[63].
8. Using meta-omic data to build and refine microbial community models
Meta-omic technologies (metagenomics, metatranscriptomics and metaproteomics) measure the molecular makeup of an entire sample, which can include unculturable organisms. This area has grown steadily since the mid-2000s (Figure 3). Metagenomics provides tools to analyze genomic DNA to determine the abundance of all detectable organisms present in a sample[64]. In metatranscriptomics, RNA is sequenced and analyzed to reveal the functionally active members in a microbial community[65]. Metaproteomics provides platform for analysis of proteins expressed by the organisms in a given sample[66]. Meta-omic approaches have been applied to environmental (including marine and soil communities)[67], waste management[68] and clinical samples[69]. These platforms are crucial for generating and analyzing data to understand the dynamics within a community and to study biological systems in nature.
Multiple recent studies have integrated meta-omic data with GEMs to study microbial communities in finer detail. Computational tools have been developed to automatically reconstruct microbial community models using meta-omics. For example, human gut microbiome models can be efficiently reconstructed using metagenomics through the Microbiome Modeling Toolbox[17b]. Another method, MICOM (MIcrobial COMmunity), was developed to build personalized metabolic models for the human gut microbiomes of 186 people using their individual metagenomic samples. The models revealed that changes in microbiome composition and diet have highly personalized effects[70]. Meta-omics in combination with GEMs have also been applied to environmental samples. For example, meta-genomics and meta-proteomics were used to build GEMs of two microbial communities in polyaromatic hydrocarbon contaminated soil[71].
9. Models provide a systems context for protein structures
Structural genomics aims to determine all 3D structures of proteins expressed from an organism’s genome, and this field has yielded over 150,000 structures in the Protein Data Bank (PDB)[72]. Recent studies have shown that this increasingly abundant data types can be integrated into GEMs. This integration has expanded the scope of mechanisms and biological questions addressable by computational systems biology. In particular, all three of the recent ME-models that account for stress functions in E. coli use 3D structures to perform key computations[56–57, 60].
In the FoldME[56] model that predicts E. coli’s thermal stress response, a key feature is to predict protein thermostability. This task required fitting thermodynamic contributions from each type of amino acid using 3D structures of E. coli proteins available from PDB. The OxidizeME[57] model that predicts E. coli’s response to oxidative stress required a method to predict metal cofactor damage for approximately 43 metalloproteins. Since experimental measurements for every metalloprotein were not available, the probability of metal cofactor damage was computed using protein 3D structural properties. A key feature of the AcidifyME[60] model is to compute (periplasmic) protein stability as a function of pH. This task required applying the multi-conformation continuum electrostatics method to 3D protein structures. In all of the studies above, the availability of high-quality 3D structures was necessary to predict systems-level response to macromolecule properties that change in response to physical and chemical stimuli.
10. Using machine learning to improve structure-function predictions and to enhance the predictive accuracy of GEMs
One gap between structural proteome and cell phenotype is that functional alterations due to the variations in protein structure are still expensive or difficult to predict. More efficient structure-function prediction models, which predict functions of a protein based on its structure, would enable routine computation of mutation effects on function and properties (e.g., solubility, stability, activity, etc.) of proteins in the whole-cell context.
Machine learning (ML) has been successfully used in the computer vision and the natural language processing field. Recently, there has been a significant interest in applying machine ML in the research of protein structure-function prediction[73]. Motivated by the expensive and time-consuming experimental protein functions annotations and aiming to improve the traditional computational approaches, a variety of machine learning methods have been developed to predict protein functions[73e]. The traditional approaches (relying on sequence similarity) might not produce accurate predictions because some proteins might have similar function even with low sequence similarity[73e]. ML methods have improved the prediction performance of such in silico methods that make prediction solely based on the amino acid sequence similarity between proteins by focusing on protein structure itself[74]. Other ML methods focus on predicting the properties of proteins based on more comprehensive features, like protein 3D structure and biological process information[73a]. Current state-of-the-art ML methods for protein structure-function prediction formulate the problem as a supervised classification task. The use of additional information such as the hierarchical structure of gene ontology and protein-protein interactions have been proved helpful to improve prediction capability[73a, 73c–e]. However, developing these ML methods is challenging because real biological data tend to be incomplete, noisy, biased and multi-modal[73e]. Nonetheless, continued development of ML techniques to address these data limitations and, more directly, increase the availability of data for ML analysis will make ML a promising approach for predicting protein function from structures[73a, 73c].
ML has already been used to improve genome-scale model predictions, by predicting catalytic turnover rates in E. coli from a diverse set of features[75]. These features included network context, protein structure, biochemistry, and assay conditions. The study identified important features for turnover rate prediction: structural (active site depth, active site solvent accessibility, active site exposure), network context (predicted reaction fluxes, reflecting evolutionary selection pressure on turnover rate), and the number of reactions an enzyme promiscuously catalyzes. Using these ML-predicted turnover rates improved the accuracy of genome-scale model predictions: by 20–34% [75].
11. Using GEMs to delineate the network-level effects of post-translational modifications
Proteoforms are proteins expressed from one gene but altered through post-translational modifications (PTMs) and that may possess different functions from each other[76]. There are more than 200 types of PTMs recorded in various databases[77]. For proteoform detection, top-down MS-based proteomic approaches which require intact protein separation through methods including serial size exclusion chromatography[78] and capillary zone electrophoresis[79] have been considered. To have a comprehensive understanding of a biological system, knowledge of global effects of PTMs is essential.
There have been some modeling efforts that have examined the network-level effect of PTMs. Brunk et al.[80] identified important branch point enzymes in the metabolic network using a genome-scale model. The authors then integrated the genome-scale model predictions, multiplex automated genome editing (MAGE), and molecular dynamic simulations to elucidate the mechanisms by which PTMs can affect the protein activity and overall cellular fitness. The authors demonstrated that PTMs can modulate protein interactions (in serine hydroxymethyltransferase), impact substrate binding (transaldolase) and regulate catalytic residues (enolase). These mechanistic insights elucidated how specific PTMs regulate cellular function at multiple biological scales, from individual enzymes to pathway usage and, ultimately, cellular phenotypes.
12. Conclusions and future perspectives
Advancements in omic technologies continue to extend our ability to read out the complete molecular makeup of a cell under various conditions of relevance to health, engineering, and knowledge expansion. Each omic technology measures a specific molecular category (RNA, protein, metabolite, etc.) as the cell is “taken apart” and analyzed. Computational systems biology provides a platform to “put together” these disparate data sets and to synthesize knowledge. Literature indicates that this pipeline of measure–model–synthesize is yielding knowledge with consistency and improving accuracy. However, we are also gaining more appreciation of the complexities associated with integrating multi-omics. Specifically, as the types of omic data types increase, so do the number of interactions we must consider across the different biological layers. Systems biology models, including the genome-scale models that we focused on here, help to navigate complexity by consolidating existing knowledge to provide context for data. Not all omic types can be interpreted with equal fidelity and resolution, however. Hence, mechanism-elucidating models are used routinely to study metabolic processes using multi-omics, while gene regulation, epigenetics, and signalling require more data-driven or statistical modeling approaches to study system-level phenomena. Furthermore, while structural proteomics has become invaluable for genome-scale modeling in recent years, we require more efficient algorithms to compute the functional effects of genetic and structural perturbations. Recent advances in machine learning in this area show promise. With better predictions and availability of more data, the predictive power of genome-scale models will continue to rise making GEMs incredibly powerful tools in decoding biological systems.
Acknowledgements
This research was supported by Queen’s University (S.D., H.X., and L.Y.), the Institute for Systems Biology’s Translational Research Fellows Program (J.T.Y.), and the National Institute of General Medical Sciences of the National Institutes of Health Grant R01GM057089 (B.O.P.).
Footnotes
Conflict of interest
The authors have declared no conflict of interest.
References
- [1].Low TY, Mohtar MA, Ang MY, Jamal R, Proteomics 2019, 19, e1800235. [DOI] [PubMed] [Google Scholar]
- [2].a) Omasits U, Varadarajan AR, Schmid M, Goetze S, Melidis D, Bourqui M, Nikolayeva O, Québatte M, Patrignani A, Dehio C, Genome research 2017, 27, 2083; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mao Y, Yang X, Liu Y, Yan Y, Du Z, Han Y, Song Y, Zhou L, Cui Y, Yang R, The American journal of tropical medicine and hygiene 2016, 95, 562; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McAfee A, Harpur BA, Michaud S, Beavis RC, Kent CF, Zayed A, Foster LJ, Journal of proteome research 2016, 15, 411; [DOI] [PubMed] [Google Scholar]; d) Chapman B, Bellgard M, Proteomics 2017, 17, 1700197. [DOI] [PubMed] [Google Scholar]
- [3].a) Manda SS, Nirujogi RS, Pinto SM, Kim M-S, Datta KK, Sirdeshmukh R, Prasad TK, Thongboonkerd V, Pandey A, Gowda H, Journal of proteome research 2014, 13, 3166; [DOI] [PubMed] [Google Scholar]; b) Pinto SM, Manda SS, Kim M-S, Taylor K, Selvan LDN, Balakrishnan L, Subbannayya T, Yan F, Prasad TK, Gowda H, Journal of proteome research 2014, 13, 2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kori Y, Sidoli S, Yuan Z-F, Lund PJ, Zhao X, Garcia BA, Scientific reports 2017, 7, 10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, Cell metabolism 2013, 17, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kaiser BLD, Li J, Sanford JA, Kim Y-M, Kronewitter SR, Jones MB, Peterson CT, Peterson SN, Frank BC, Purvine SO, PloS one 2013, 8, e67155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Vivek-Ananth RP, Samal A, Biosystems 2016, 147, 1; [DOI] [PubMed] [Google Scholar]; b) Cho J, Gu C, Han T, Ryu J, Lee S, Current Opinion in Systems Biology 2019, 15, 1; [Google Scholar]; c) Ebrahim A, Brunk E, Tan J, O’Brien EJ, Kim D, Szubin R, Lerman JA, Lechner A, Sastry A, Bordbar A, Feist AM, Palsson BO, Nat Commun 2016, 7, 13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Ebrahim A, Lerman JA, Palsson BO, Hyduke DR, BMC systems biology 2013, 7, 74; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bordbar A, Monk JM, King ZA, Palsson BO, Nature Reviews Genetics 2014, 15, 107; [DOI] [PubMed] [Google Scholar]; c) Palsson B, Systems biology, Cambridge university press, 2015; [Google Scholar]; d) Heirendt L, Arreckx S, Pfau T, Mendoza SN, Richelle A, Heinken A, Haraldsdottir HS, Wachowiak J, Keating SM, Vlasov V, Magnusdottir S, Ng CY, Preciat G, Zagare A, Chan SHJ, Aurich MK, Clancy CM, Modamio J, Sauls JT, Noronha A, Bordbar A, Cousins B, El Assal DC, Valcarcel LV, Apaolaza I, Ghaderi S, Ahookhosh M, Ben Guebila M, Kostromins A, Sompairac N, Le HM, Ma D, Sun Y, Wang L, Yurkovich JT, Oliveira MAP, Vuong PT, El Assal LP, Kuperstein I, Zinovyev A, Hinton HS, Bryant WA, Aragon Artacho FJ, Planes FJ, Stalidzans E, Maass A, Vempala S, Hucka M, Saunders MA, Maranas CD, Lewis NE, Sauter T, Palsson BO, Thiele I, Fleming RMT, Nat Protoc 2019, 14, 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Lloyd CJ, Ebrahim A, Yang L, King ZA, Catoiu E, O’Brien EJ, Liu JK, Palsson BO, PLoS Comput Biol 2018, 14, e1006302; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang L, Yurkovich JT, King ZA, Palsson BO, Current Opinion in Microbiology 2018, 45, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Nature 1981, 290, 457. [DOI] [PubMed] [Google Scholar]
- [11].a) Parkhill J, Wren B, Mungall K, Ketley J, Churcher C, Basham D, Chillingworth T, Davies R, Feltwell T, Holroyd S, Nature 2000, 403, 665; [DOI] [PubMed] [Google Scholar]; b) Patrick S, Parkhill J, McCoy LJ, Lennard N, Larkin MJ, Collins M, Sczaniecka M, Blakely G, Microbiology 2003, 149, 915. [DOI] [PubMed] [Google Scholar]
- [12].a) Metzker ML, Nature reviews genetics 2010, 11, 31; [DOI] [PubMed] [Google Scholar]; b) Loman NJ, Pallen MJ, Nature Reviews Microbiology 2015, 13, 787; [DOI] [PubMed] [Google Scholar]; c) Besser J, Carleton HA, Gerner-Smidt P, Lindsey RL, Trees E, Clinical microbiology and infection 2018, 24, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ghatak S, King ZA, Sastry A, Palsson BO, Nucleic acids research 2019, 47, 2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Price ND, Reed JL, Palsson BO, Nat Rev Microbiol 2004, 2, 886. [DOI] [PubMed] [Google Scholar]
- [15].a) Harcombe WR, Delaney NF, Leiby N, Klitgord N, Marx CJ, PLoS Comput Biol 2013, 9, e1003091; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Anand A, Chen K, Yang L, Sastry AV, Olson CA, Poudel S, Seif Y, Hefner Y, Phaneuf PV, Xu S, Szubin R, Feist AM, Palsson BO, Proc Natl Acad Sci U S A 2019, 116, 25287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gu C, Kim GB, Kim WJ, Kim HU, Lee SY, Genome biology 2019, 20, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Machado D, Andrejev S, Tramontano M, Patil KR, Nucleic Acids Res 2018, 46, 7542; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Baldini F, Heinken A, Heirendt L, Magnusdottir S, Fleming RMT, Thiele I, Bioinformatics 2019, 35, 2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Opdam S, Richelle A, Kellman B, Li S, Zielinski DC, Lewis NE, Cell Syst 2017, 4, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Becker SA, Palsson BO, PLoS Comput Biol 2008, 4, e1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schmidt BJ, Ebrahim A, Metz TO, Adkins JN, Palsson BO, Hyduke DR, Bioinformatics 2013, 29, 2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pandey V, Hadadi N, Hatzimanikatis V, PLoS computational biology 2019, 15, e1007036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zur H, Ruppin E, Shlomi T, Bioinformatics 2010, 26, 3140. [DOI] [PubMed] [Google Scholar]
- [23].Agren R, Bordel S, Mardinoglu A, Pornputtapong N, Nookaew I, Nielsen J, PLoS Comput Biol 2012, 8, e1002518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jerby L, Shlomi T, Ruppin E, Mol Syst Biol 2010, 6, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang Y, Eddy JA, Price ND, BMC systems biology 2012, 6, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schultz A, Qutub AA, PLoS Comput Biol 2016, 12, e1004808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chandrasekaran S, Price ND, Proc Natl Acad Sci U S A 2010, 107, 17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang Z, Danziger SA, Heavner BD, Ma S, Smith JJ, Li S, Herricks T, Simeonidis E, Baliga NS, Aitchison JD, Price ND, PLoS Comput Biol 2017, 13, e1005489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Byrne A, Cole C, Volden R, Vollmers C, Philos Trans R Soc Lond B Biol Sci 2019, 374, 20190097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].a) Baruzzo G, Hayer KE, Kim EJ, Di Camillo B, FitzGerald GA, Grant GR, Nature methods 2017, 14, 135; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Van Den Berge K, Hembach KM, Soneson C, Tiberi S, Clement L, Love MI, Patro R, Robinson MD, 2019. [Google Scholar]
- [31].S. R. Opdam Anne; Kellman Benjamin; Li Shanzhong; Zielinski Daniel; Lewis Nathan E, Cell Systems 2017, 4, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gatto F, Ferreira R, Nielsen J, Metab Eng 2020, 57, 51. [DOI] [PubMed] [Google Scholar]
- [33].Agren R, Mardinoglu A, Asplund A, Kampf C, Uhlen M, Nielsen J, Molecular systems biology 2014, 10. [DOI] [PMC free article] [PubMed]
- [34].a) Wishart DS, Physiol Rev 2019, 99, 1819; [DOI] [PubMed] [Google Scholar]; b) Yan M, Xu G, Anal Chim Acta 2018, 1037, 41. [DOI] [PubMed] [Google Scholar]
- [35].a) Rinschen MM, Ivanisevic J, Giera M, Siuzdak G, Nat Rev Mol Cell Biol 2019, 20, 353; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Simithy J, Sidoli S, Garcia BA, Proteomics 2018, 18, e1700309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].a) López-López Á, López-Gonzálvez Á, Barker-Tejeda TC, Barbas C, Expert Review of Molecular Diagnostics 2018, 18, 557; [DOI] [PubMed] [Google Scholar]; b) Rangel-Huerta OD, Pastor-Villaescusa B, Gil A, Metabolomics 2019, 15, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fenaille F, Barbier Saint-Hilaire P, Rousseau K, Junot C, J Chromatogr A 2017, 1526, 1. [DOI] [PubMed] [Google Scholar]
- [38].a) Allen F, Greiner R, Wishart D, Metabolomics 2015, 11, 98; [Google Scholar]; b) da Silva RR, Dorrestein PC, Quinn RA, Proceedings of the National Academy of Sciences 2015, 112, 12549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].a) Dührkop K, Shen H, Meusel M, Rousu J, Böcker S, Proceedings of the National Academy of Sciences 2015, 112, 12580; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ji H, Xu Y, Lu H, Zhang Z, Analytical chemistry 2019, 91, 5629; [DOI] [PubMed] [Google Scholar]; c) Nguyen DH, Nguyen CH, Mamitsuka H, Briefings in Bioinformatics 2018, 20, 2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Johnson CH, Ivanisevic J, Siuzdak G, Nature reviews Molecular cell biology 2016, 17, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wu H-Q, Cheng M-L, Lai J-M, Wu H-H, Chen M-C, Liu W-H, Wu W-H, Chang PM-H, Huang C-YF, Tsou A-P, PLoS computational biology 2017, 13, e1005618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yurkovich JT, Zielinski DC, Yang L, Paglia G, Rolfsson O, Sigurjónsson ÓE, Broddrick JT, Bordbar A, Wichuk K, Brynjólfsson S, Palsson S, Gudmundsson S, Palsson BO, Journal of Biological Chemistry 2017, 292, 19556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bordbar A, Yurkovich JT, Paglia G, Rolfsson O, Sigurjónsson ÓE, Palsson BO, Scientific reports 2017, 7, 46249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].El-Aneed A, Cohen A, Banoub J, Applied Spectroscopy Reviews 2009, 44, 210. [Google Scholar]
- [45].Bantscheff M, Lemeer S, Savitski MM, Kuster B, Analytical and bioanalytical chemistry 2012, 404, 939. [DOI] [PubMed] [Google Scholar]
- [46].Han X, Aslanian A, Yates JR III, Current opinion in chemical biology 2008, 12, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sobsey CA, Ibrahim S, Richard VR, Gaspar V, Mitsa G, Lacasse V, Zahedi RP, Batist G, Borchers CH, Proteomics 2019, 10.1002/pmic.201900029e1900029. [DOI] [PubMed]
- [48].a) Borras E, Sabido E, Proteomics 2017, 17; [DOI] [PubMed]; b) Saleh S, Staes A, Deborggraeve S, Gevaert K, Proteomics 2019, 19, e1800435. [DOI] [PubMed] [Google Scholar]
- [49].Hu A, Noble WS, Wolf-Yadlin A, F1000Res 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Venable JD, Dong M-Q, Wohlschlegel J, Dillin A, Yates JR III, Nature methods 2004, 1, 39. [DOI] [PubMed] [Google Scholar]
- [51].Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, Aebersold R, Molecular & Cellular Proteomics 2012, 11, O111. 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kawashima Y, Watanabe E, Umeyama T, Nakajima D, Hattori M, Honda K, Ohara O, International journal of molecular sciences 2019, 20, 5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].a) Calderón-Celis F, Encinar JR, Sanz-Medel A, Mass spectrometry reviews 2018, 37, 715; [DOI] [PubMed] [Google Scholar]; b) Ankney JA, Muneer A, Chen X, Annual Review of Analytical Chemistry 2018, 11, 49. [DOI] [PubMed] [Google Scholar]
- [54].a) Thiele I, Fleming RMT, Que R, Bordbar A, Diep D, Palsson BO, PLOS One 2012, 7, e45635; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) O’Brien EJ, Lerman JA, Chang RL, Hyduke DR, Palsson BØ, Mol Syst Biol 2013, 9, 693; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Heckmann DL, Colton J; Mih Nathan; Ha Yuanchi; Zielinski Daniel C; Haiman Zachary B; Desouki Abdelmoneim Amer; Lercher Martin J; Palsson Bernhard O, Nature Communications 2018, 9, 5252; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yang L, Yurkovich JT, Lloyd CJ, Ebrahim A, Saunders MA, Palsson BO, Sci Rep 2016, 6, 36734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Liu JK, O’Brien EJ, Lerman JA, Zengler K, Palsson BO, Feist AM, BMC Syst Biol 2014, 8, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chen K, Gao Y, Mih N, O’Brien EJ, Yang L, Palsson BO, Proc Natl Acad Sci U S A 2017, 114, 11548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yang L, Mih N, Anand A, Park JH, Tan J, Yurkovich JT, Monk JM, Lloyd CJ, Sandberg TE, Seo SW, Kim D, Sastry AV, Phaneuf P, Gao Y, Broddrick JT, Chen K, Heckmann D, Szubin R, Hefner Y, Feist AM, Palsson BO, Proc Natl Acad Sci USA 2019, 116, 14368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lerman JA, Hyduke DR, Latif H, Portnoy VA, Lewis NE, Orth JD, Schrimpe-Rutledge AC, Smith RD, Adkins JN, Zengler K, Palsson BO, Nature Communications 2012, 3, 929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Liu JK, Lloyd C, Al-Bassam MM, Ebrahim A, Kim JN, Olson C, Aksenov A, Dorrestein P, Zengler K, PLoS Comput Biol 2019, 15, e1006848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Du B, Yang L, Lloyd CJ, Fang X, Palsson BO, PLoS Comput Biol 2019, 15, e1007525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Yurkovich JT, Yang L, Palsson BO, bioRxiv 2019, 797258.
- [62].Brunk E, George KW, Alonso-Gutierrez J, Thompson M, Baidoo E, Wang G, Petzold CJ, McCloskey D, Monk J, Yang L, Cell systems 2016, 2, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Pannala VR, Wall ML, Estes SK, Trenary I, O’Brien TP, Printz RL, Vinnakota KC, Reifman J, Shiota M, Young JD, Wallqvist A, Sci Rep 2018, 8, 11678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Simon HY, Siddle KJ, Park DJ, Sabeti PC, Cell 2019, 178, 779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Shakya M, Lo C-C, Chain PS, Frontiers in genetics 2019, 10, 904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].a) Wilmes P, Heintz-Buschart A, Bond PL, Proteomics 2015, 15, 3409; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Isaac NI, Philippe D, Nicholas A, Raoult D, Eric C, Clinical Mass Spectrometry 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].a) Eloe-Fadrosh EA, Ivanova NN, Woyke T, Kyrpides NC, Nature Microbiology 2016, 1, 15032; [DOI] [PubMed] [Google Scholar]; b) Miller IJ, Weyna TR, Fong SS, Lim-Fong GE, Kwan JC, Scientific reports 2016, 6, 34362; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Holmes DE, Shrestha PM, Walker DJ, Dang Y, Nevin KP, Woodard TL, Lovley DR, Appl. Environ. Microbiol 2017, 83, e00223; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jones DS, Flood BE, Bailey JV, The ISME journal 2016, 10, 1015; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kleindienst S, Herbst F-A, Stagars M, Von Netzer F, Von Bergen M, Seifert J, Peplies J, Amann R, Musat F, Lueders T, The ISME journal 2014, 8, 2029; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, Kassabgy M, Huang S, Mann AJ, Waldmann J, Science 2012, 336, 608. [DOI] [PubMed] [Google Scholar]
- [68].a) Bouhajja E, Agathos SN, George IF, Biotechnology advances 2016, 34, 1413; [DOI] [PubMed] [Google Scholar]; b) Delforno TP, Macedo TZ, Midoux C, Lacerda GV Jr, Rué O, Mariadassou M, Loux V, Varesche MB, Bouchez T, Bize A, Science of The Total Environment 2019, 649, 482; [DOI] [PubMed] [Google Scholar]; c) Wilmes P, Wexler M, Bond PL, PLoS One 2008, 3, e1778; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hagen LH, Frank JA, Zamanzadeh M, Eijsink VG, Pope PB, Horn SJ, Arntzen MØ, Appl. Environ. Microbiol 2017, 83, e01955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].a) Pascal V, Pozuelo M, Borruel N, Casellas F, Campos D, Santiago A, Martinez X, Varela E, Sarrabayrouse G, Machiels K, Gut 2017, 66, 813; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schirmer M, Franzosa EA, Lloyd-Price J, McIver LJ, Schwager R, Poon TW, Ananthakrishnan AN, Andrews E, Barron G, Lake K, Nature microbiology 2018, 3, 337; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lai LA, Tong Z, Chen R, Pan S, in Functional Proteomics, Springer; 2019, p. 123; [Google Scholar]; d) Pinto E, Anselmo M, Calha M, Bottrill A, Duarte I, Andrew PW, Faleiro ML, Microbiology 2017, 163, 161. [DOI] [PubMed] [Google Scholar]
- [70].Diener C, Gibbons SM, Resendis-Antonio O, mSystems 2020, 5. [DOI] [PMC free article] [PubMed]
- [71].Tobalina L, Bargiela R, Pey J, Herbst F-A, Lores I, Rojo D, Barbas C, Peláez AI, Sánchez J, von Bergen M, Bioinformatics 2015, 31, 1771. [DOI] [PubMed] [Google Scholar]
- [72].Mih N, Palsson BO, Mol Syst Biol 2019, 15, e8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].a) Townshend R, Bedi R, Suriana P, Dror R, presented at Advances in Neural Information Processing Systems 2019;; b) Fout A, Byrd J, Shariat B, Ben-Hur A, presented at Advances in neural information processing systems 2017;; c) Gligorijević V, Barot M, Bonneau R, Bioinformatics 2018, 34, 3873; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kovács IA, Luck K, Spirohn K, Wang Y, Pollis C, Schlabach S, Bian W, Kim D-K, Kishore N, Hao T, Calderwood MA, Vidal M, Barabási A-L, Nature Communications 2019, 10, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhang F, Song H, Zeng M, Li Y, Kurgan L, Li M, Proteomics 2019, 19, 1900019; [DOI] [PubMed] [Google Scholar]; f) Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Žídek A, Nelson AW, Bridgland A, Nature 2020, 1. [DOI] [PubMed] [Google Scholar]
- [74].Cozzetto D, Minneci F, Currant H, Jones DT, Scientific reports 2016, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Heckmann D, Lloyd CJ, Mih N, Ha Y, Zielinski DC, Haiman ZB, Desouki AA, Lercher MJ, Palsson BO, Nature Communications 2018, 9, 5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kumar D, Bansal G, Narang A, Basak T, Abbas T, Dash D, Proteomics 2016, 16, 2533. [DOI] [PubMed] [Google Scholar]
- [77].Kim MS, Zhong J, Pandey A, Proteomics 2016, 16, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Chen X, Ge Y, Proteomics 2013, 13, 2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].a) Shen X, Yang Z, McCool EN, Lubeckyj RA, Chen D, Sun L, TrAC Trends in Analytical Chemistry 2019, 115644; [DOI] [PMC free article] [PubMed]; b) McCool EN, Lubeckyj RA, Shen X, Chen D, Kou Q, Liu X, Sun L, Analytical chemistry 2018, 90, 5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Brunk E, Chang RL, Xia J, Hefzi H, Yurkovich JT, Kim D, Buckmiller E, Wang HH, Cho B-K, Yang C, Palsson BO, Church GM, Lewis NE, Proc Natl Acad Sci U S A 2018, 43, 11096. [DOI] [PMC free article] [PubMed] [Google Scholar]