

Abstract
A next generation total synthesis of vancomycin aglycon is detailed that was achieved in 17 steps (longest linear sequence, LLS) from the constituent amino acid subunits with kinetically-controlled diastereoselective introduction of all three elements of atropisomerism. In addition to new syntheses of three of the seven amino acid subunits, highlights of the approach include a ligand-controlled atroposelective one-pot Miyaura borylation-Suzuki coupling sequence for introduction of the AB biaryl axis of chirality (>20:1 dr), an essentially instantaneous and scalable macrolactamization of the AB ring system nearly free of competitive epimerization (>30:1 dr), and two room temperature atroposelective intramolecular SNAr cyclizations for sequential CD (8:1 dr) and DE ring closures (14:1 dr) that benefit from both preorganization by the preformed AB ring system and subtle substituent effects. Combined with a protecting group free two-step enzymatic glycosylation of vancomycin aglycon, this provides a 19-step total synthesis of vancomycin. The approach paves the way for large scale synthetic preparation of pocket modified vancomycin analogues that directly address the underlying mechanism of resistance to vancomycin.
Graphical Abstract
INTRODUCTION
Vancomycin (1), first disclosed1 and introduced into the clinic over 60 years ago, remains one of the clinically most effective and important antibiotics for treatment of life-threatening Gram-positive bacterial infections,2 including methicillin-resistant S. aureus (MRSA). Its complex structure,3 the three strained macrocyclic ring systems interwoven into the highly functionalized and rigid tricyclic heptapeptide core, the unusual centers of axial or planar chirality (atropisomers), and its glycosylation present formidable synthetic challenges (Figure 1). Three total syntheses of vancomycin aglycon4–6 and two total syntheses of vancomycin7,8 have been disclosed to date. Related efforts have detailed total syntheses of orienticin C,9 teicoplanin10,11 and ristocetin A12 aglycons and a near endless number of methodology studies have been conducted that address synthetic challenges posed by their structures. The comprehensive reviews of Perkins,13 Williams,14,15 Nicolaou,16 Courvalin,17,18 Walsh,19,20 Kahne,2 ourselves,21,22 and others23–25 provide summaries of the rich history on the isolation, structure elucidation, biosynthesis, semisynthetic and synthetic studies, mechanism of action, and mechanisms of resistance of the glycopeptide antibiotics.
Figure 1.
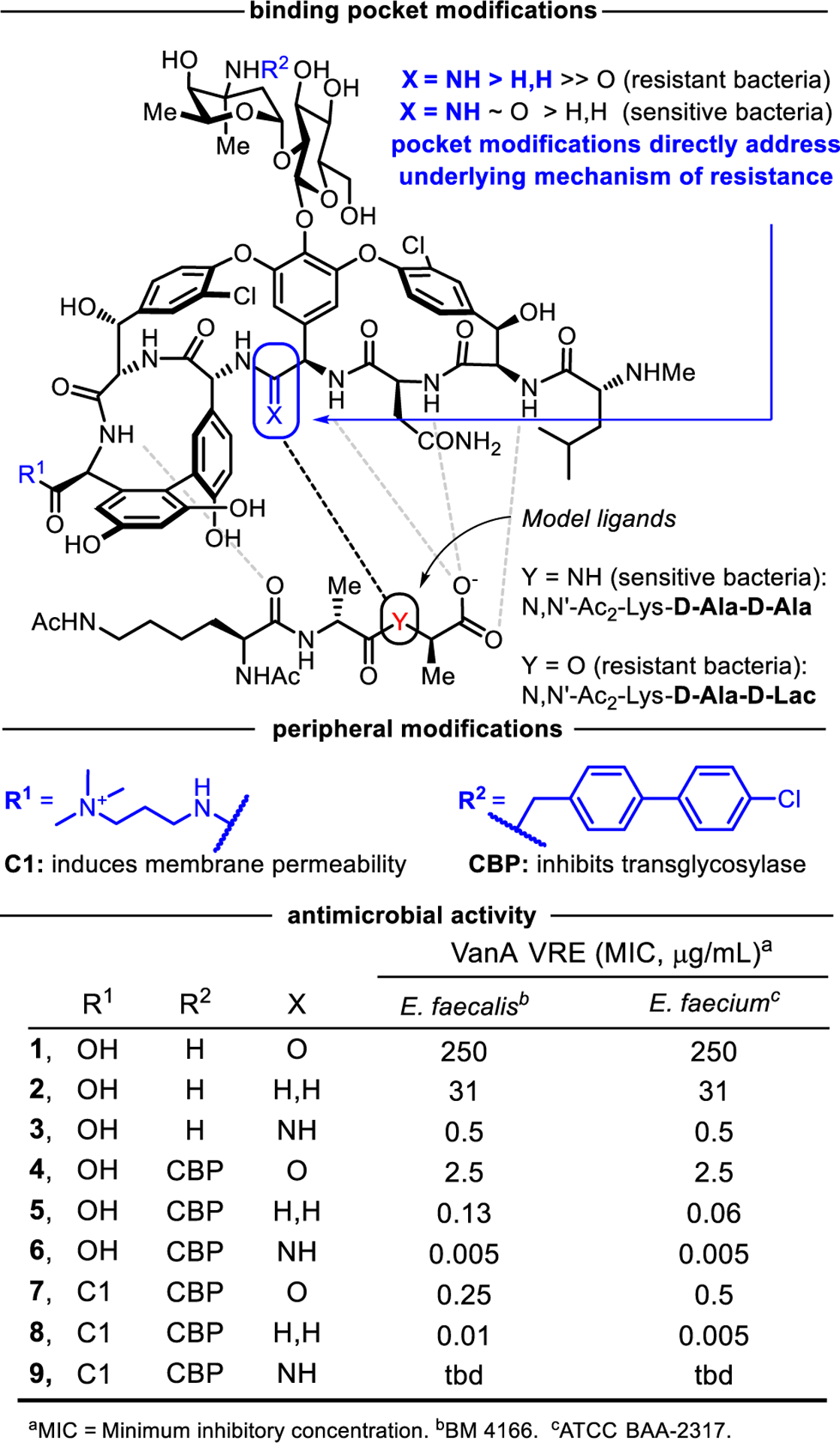
Top: Structure of vancomycin and key analogues with H-bonding interactions between vancomycin and model ligands indicated by dashed lines. Middle: Added peripheral modifications that synergistically improve activity by inducing membrane permeabilization (C1) or directly inhibiting transglycosylase (CBP), both independent of D-Ala-D-Ala/D-Lac binding. Bottom: VanA VRE antimicrobial activity.
The only clinically significant resistance to vancomycin even after >60 years of use first emerged in enterococci (VanA and VanB VRE, 1987)26 and more recently in S. aureus (VRSA, 2002).18 It was co-opted from nonpathogenic source organisms that use an intricate mechanism to protect themselves while producing vancomycin.27 It is induced upon detection28–31 of a glycopeptide challenge that initiates an orchestrated response, resulting in late-stage remodeling of the N-terminus of peptidoglycan precursors from D-Ala-D-Ala to D-Ala-D-Lac.32 This single atom exchange in the cell wall precursors reduces vancomycin binding (1000-fold) and its derived antimicrobial activity (1000-fold).33
An extension of our efforts on the total synthesis of the glycopeptide antibiotics has targeted vancomycin analogues, containing deep-seated compensatory single atom exchanges in the binding pocket,34 that exhibit dual D-AlaD-Ala/D-Ala-D-Lac binding and antimicrobial activity against both vancomycin sensitive and resistant organisms (235 and 336). These efforts, along with peripheral modifications that introduce additional independent mechanisms of action,37,38 have provided extraordinarily potent analogues that display especially durable antimicrobial activity (Figure 1). We have shown that incorporation of two simple peripheral modifications (CBP and C1) independently (e.g., 4-6) or simultaneously (8) into the more accessible of our pocket modified analogues at the time, [Ψ[CH2NH]Tpg4]vancomycin (2), provided potent antimicrobial agents with up to three synergistic mechanisms of action, each of which is individually effective against both vancomycin-resistant and vancomycin-sensitive bacteria and two of which are independent of DAla-D-Ala/D-Lac binding.37 The exceptional potency (MIC 0.01–0.005 μg/mL, VRE) and remarkable durability of the prototypical members, CBP-[Ψ[C(=NH)NH]Tpg4]vancomycin (6) and C1,CBP-[Ψ[CH2NH]Tpg4]vancomycin (8), has inspired our continued examination of new analogues, including 9, and their expanded preclinical examination.
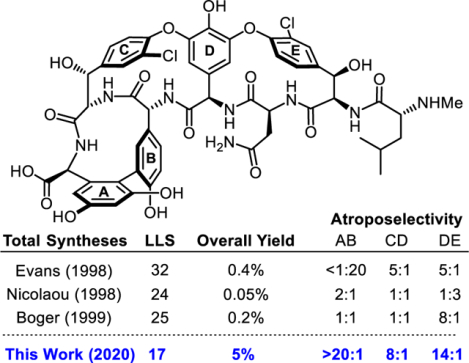
In order to facilitate these studies, we have now developed a scalable total synthesis that could provide sufficient amounts of each analogue within the confines of an academic lab for initial preclinical assessment of in vivo efficacy and safety. In contrast to what many might think would be the most challenging feature, we earlier reported a scalable enzymatic conversion of the aglycon to the fully decorated natural product8 (2 steps) or its pocket-modified analogues39 that is conducted without protecting groups. However, the total synthesis of vancomycin aglycon (10) and its residue 4 modified analogues required redesign to overcome practical issues, including a long step count (25), low overall yield (0.2% without atropisomer recycling), lengthy syntheses of several of the unnatural amino acid subunits, and lack of kinetic atroposelectivity in the construction of the AB and CD ring systems. While the latter issue was originally addressed in our efforts by a defined order of macrocyclizations that permitted thermal equilibration40 and recycling of the unnatural atropisomers, it required two atropisomer separations that limit material throughput. Therefore, we embarked on and herein detail a streamlined 17-step total synthesis of vancomycin aglycon (10) that achieves high kinetic diastereoselectivity for formation of each macrocyclic ring and improves the overall yield of 10 more than 20-fold.
RESULTS AND DISCUSSION
Improved synthesis of the A, C and D ring amino acid subunits.
Our synthesis of vancomycin aglycon begins with seven unnatural amino acid subunits (11-17), four of which are either commercially available or readily prepared. The remaining three were each prepared in modest overall yield (ca. 20%) in our previous efforts (Figure 2). The A-ring6 (14) and D-ring41 (12) subunits were accessed in 5 and 9 total steps, respectively, with the requisite absolute stereochemistry introduced by Sharpless asymmetric aminohydroxylations,42,43 while the C-ring subunit12 (11) was prepared by a modestly diastereoselective (5:1 syn:anti) Schöllkopf aldol reaction12,44 with full control of the α-amino acid stereochemistry. Substantially improved syntheses of these three subunits were developed during the course of this work. With these improvements, five of the subunits are now derived from inexpensive chiral pool starting materials and only two require asymmetric synthesis. All but one of the subunits are now available in >50% overall yield.
Figure 2.
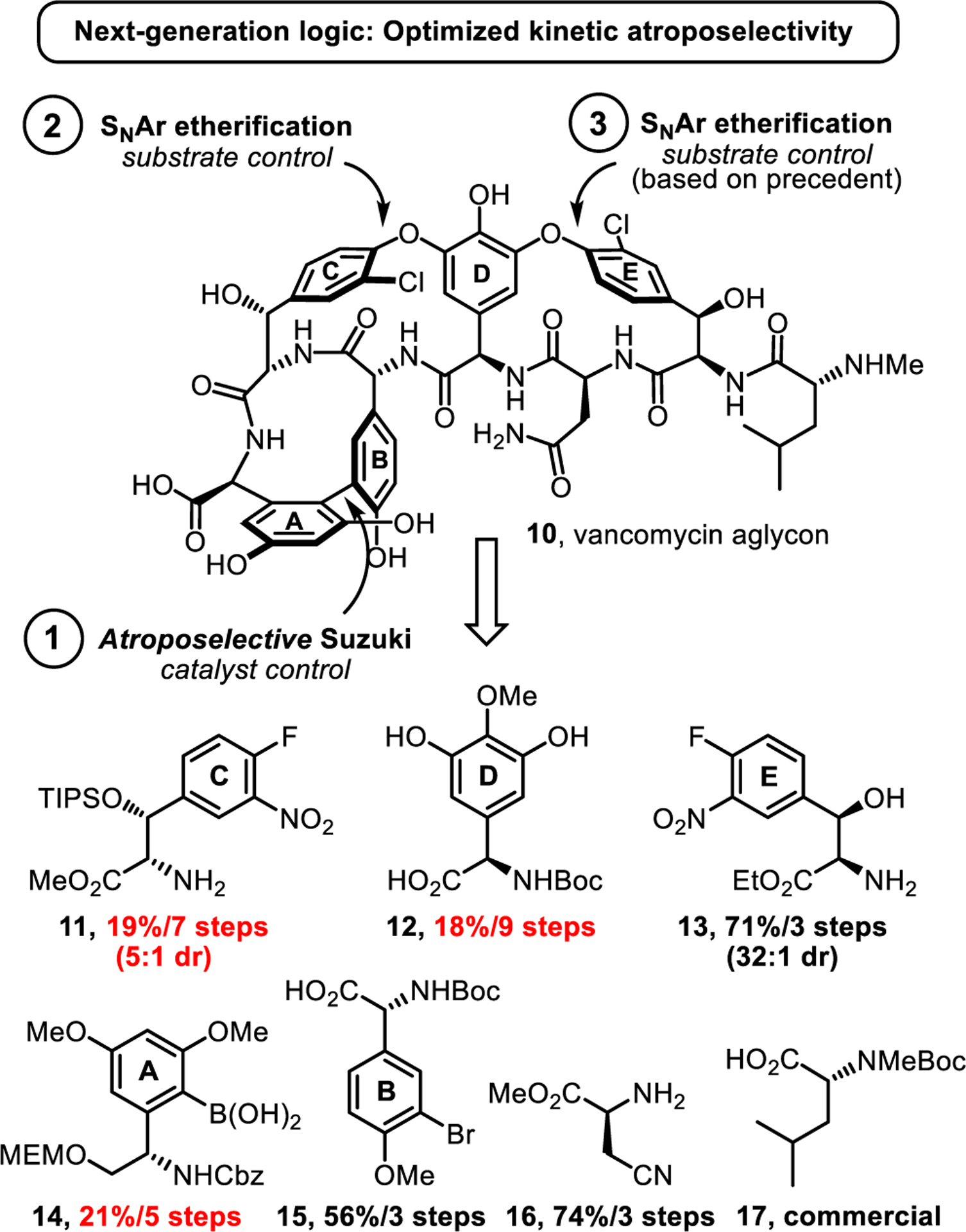
Key retrosynthetic disconnections and initial starting subunits.
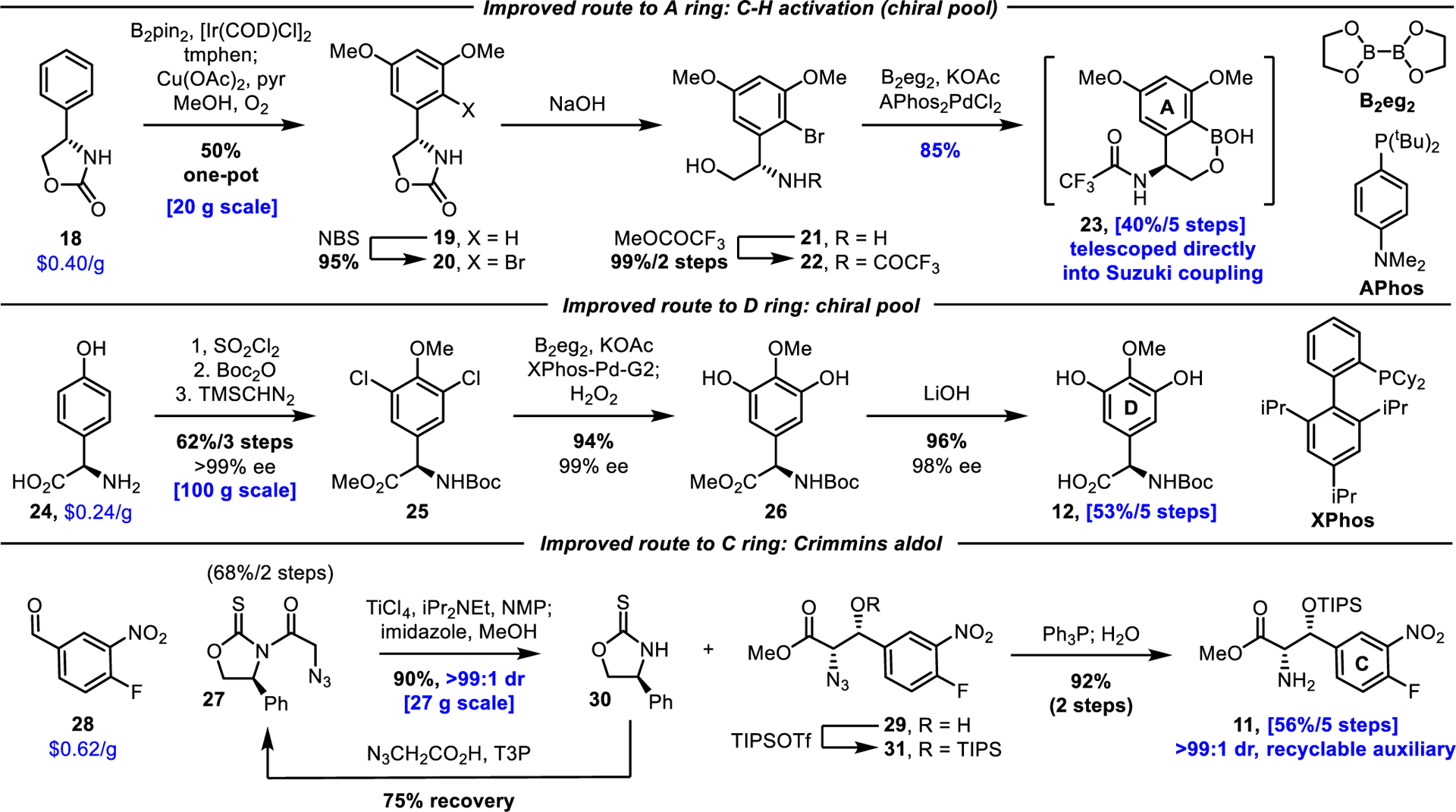
An asymmetric aminohydroxylation (AA) reaction reported by Sharpless42 was used to prepare the A-ring amino acid in our prior efforts6 and proceeded in 68% yield (90% ee) on 2 mmol (328 mg) scale and provided regioisomeric products (3:1 rr). However, the yield of this reaction diminished on larger scales (>10 g, 33–41%) in our hands and the product was invariably contaminated with an additional inseparable ring-chlorinated byproduct. An alternative, chiral pool route with complete control of the absolute stereochemistry was developed based on C-H activation and meta-functionalization of the commercially available inexpensive ($0.40/g) phenylglycine-derived Evans auxiliary 18 (Scheme 1). A one-pot sequential Ir-mediated bis C-H borylation45/Chan-Lam methoxylation46 furnished 19 (50% overall) in a reaction that was easily scaled to >20 g. Bromination of 19 followed by recrystallization provided 20 (95%). To date, this three-step sequence has provided more than 300 g of intermediate 20. Subsequent oxazolidinone hydrolysis and N-trifluoroacetamide protection of 21 provided 22 quantitatively. The lithium-halogen exchange/B(OMe)3-trapping procedure previously used6 to prepare the boronic acid 14 (59% yield) was superseded by a modified Miyaura borylation, providing high yields (85%) of cyclic boronate 23. Additionally and importantly, the crude reaction mixture containing boroxane 23 could be telescoped directly into a subsequent atroposelective Suzuki coupling (see below). Hence, the new route to the A-ring subunit provides the cyclic boronate 23 in 40% yield (5 steps), a 2-fold improvement over the asymmetric aminohydroxylation route (21%) with complete (chiral pool) control of the absolute stereochemistry. Further, its implementation provides direct access to 23, which is functionalized (trifluoroacetamide vs NHCbz) for more effective use in the final streamlined total synthesis detailed herein. The successful implementation of this route hinged on the use of 3,4,7,8-tetramethyl-1,10-phenanthroline (tmphen) as the ligand in the C-H borylation,47,48 the inclusion of pyridine in the Chan-Lam methoxylation (Supporting Information Figure S1),49 and the development of an efficient Miyaura borylation of the hindered aryl bromide 22 with bis(ethyleneglycolato)diboron (B2eg2), whereas typical Miyaura borylation conditions [B2pin2/PdCl2(dppf)]50 delivered only trace product.
Scheme 1.
Improved synthesis of starting subunits
The D-ring phenylglycine 12 was previously the most cumbersome subunit to prepare on scale, requiring 9 steps and relying on an asymmetric aminohydroxylation42,43 (AA) to install the needed chiral center (69%, 96% ee, 7:1 regioselectivity).41 We developed a new chiral pool 5-step route to the D-ring subunit starting from inexpensive ($0.24/g) D-4-hydroxyphenylglycine (24), which was first converted to dichlorinated methyl ester 25 in 62% yield (59% for 3 steps on 100 g scale, Scheme 1) by modification of a reported procedure.51 Although 25 proved sensitive to racemization (Et3N, 23 °C), net hydroxylation of both chlorides in 25 could be accomplished in a one-pot Miyaura borylation/oxidation sequence (94%) with negligible loss of optical purity (99% ee for 26). Saponification (LiOH, 96%, 98% ee) provided the required 3,5-dihydroxy-4-methoxyphenylglycine 12 now available in a much shorter (5 vs 9 steps) and easily scalable sequence with a 3-fold improvement in yield (56% yield for 5 steps) over our previous route (18% for 9 steps) and with complete control (chiral pool) of the absolute stereochemistry. Notably, the use of dichloride 25 as a substrate for the key borylation-oxidation reaction was essential, whereas use of the corresponding dibromide or diiodide substrates resulted in considerable ring halogenation upon exposure to H2O2.
Preparation of the C-ring subunit 11 previously was achieved by zirconium-mediated addition of a Schöllkopf reagent44 (commercially available for >$100/g, or in 4 steps52 from D-valine), proceeding in 5:1 dr (syn:anti) and 50% yield (syn isomer). In order to avoid the needed separation of the alcohol diastereomers and lengthy preparation or costly reagent purchsae, we turned to a Ti-mediated aldol reaction of the known oxazolidinethione 27,53 which proceeds with near perfect diastereoselection and enlists a more readily prepared (2 steps, 68%) and recyclable chiral auxiliary (27) (Scheme 1). Addition of 27 to 4-fluoro-3-nitrobenzaldehyde (28) under conditions designed and disclosed by Crimmins54 to provide the syn aldol product, followed by in situ methanolysis, cleanly provided methyl ester 29 (90%, >99:1 dr, 27 g scale). In addition, the recovered auxiliary 30 was recycled in a single step to regenerate 27, without prior separation of 29 from 30. Subsequent TIPS protection of alcohol 29 and Staudinger reduction of 31 provided 11 in high yield (92% for 2 steps, >99% ee, 56% overall for 5 steps) virtually free of anti-diastereomer (>99% de). To date, >320 g of 11 has been prepared by this route.
Formal total synthesis of vancomycin aglycon: kinetically-controlled diastereoselective introduction of all three elements of atropisomerism.
Concurrent with the above efforts, we undertook studies on the atroposelective construction of the AB biaryl axis of chirality as well as diastereoselective formation of the CD and DE macrocyclic diaryl ethers. Efficient elements of the synthesis of the vancomycin core structure developed in our prior efforts were maintained, enlisting a macrolactamization6 for closure of the 12-membered biaryl AB ring system and two aromatic nucleophilic substitution reactions for macrocyclization of the 16-membered diaryl ethers in the CD42/DE6 ring systems but conducted in an altered order. The availability now of robust methods for atroposelective Suzuki biaryl coupling suggested that we set the AB biaryl stereochemistry first. Then, following macrolactamization and using the preformed AB macrocycle as an element of preorganization, we hoped to achieve high kinetic atroposelectivity in the closure of the CD ring system under substrate control, although precedent suggested this might be improbable.9,55 If successful and based on our previous work,6 we could anticipate that the subsequent DE ring closure would proceed with excellent substrate-controlled atroposelectivity (see Figure 2).
Although not incorporated into their total synthesis of vancomycin, Nicolaou and coworkers later reported56 an atroposelective Suzuki biaryl coupling conducted on model substrates similar to our own that was mediated by (R)BINAP and Pd(OAc)2 (3:1 ligand:Pd). We suspected that the active catalyst was actually the palladium-ligated bisphosphine mono-oxide of (R)-BINAP [(R)-BINAP(O)-Pd0] based on the frequently underappreciated role of such ligand complexes57,58 that are most often unknowingly generated in situ. Consistent with this proposal, the coupling of 32 with 14 that employed a 2:1 combination of (R)-BINAP(O):Pd2dba359 (1:1 ligand:Pd) afforded biaryl 33 with consistently high yield and atroposelectivity (>20:1 dr, up to 89% yield on 8 g scale) (Figure 3). In contrast, the combination of (R)-BINAP and Pd2dba3 was ineffective, confirming that mono-oxidation of the BINAP ligand was essential to catalytic activity. While the (R)-BINAP(O):Pd2dba3 system provided more than sufficient material for initial studies, the use of a noncommercial ligand as well as the variable quality of commercial Pd2dba3 (Pd-black contamination)60 introduced practical challenges that we hoped to circumvent by direct in situ generation of the catalyst [(R)-BINAP(O)-Pd0] from 1:1 (R)-BINAP/Pd(OAc)2. Our initial screen of available chiral ligands for the coupling of 32 with 14 revealed that the (R)-BINAP/Pd(OAc)2 combination was among the most effective of those examined (Supporting Information Figure S2). However, these initially favorable results from the PdII/BINAP system proved difficult to maintain as we scaled the reaction, which we found was due to precipitation of the active (R)-BINAP(O)-Pd0 catalyst as a cherry-red solid prior to substrate addition. Fortunately, highly reproducible results were obtained by first heating a mixture of Pd(OAc)2, (R)-BINAP and aryl bromide in the presence of aqueous NaHCO3, thereby trapping the active catalyst as the stable and soluble oxidative addition complex, followed by slow addition of the boronic acid. The superiority of this latter procedure was confirmed in subsequent studies of the coupling of 34 with 23 that consistently provided high yields of 35 (82–93%) on scales up to 25 g. Finally, the oxidative addition complex 36 was isolated in excellent yield (91%) from the reaction of (R)-BINAP, Pd(OAc)2, and 32 in the absence of boronic acid, and 36 was demonstrated to be catalytically competent in the coupling of 32 with 14 (TON = 60, Supporting Information Figure S4). In comparison, the oxidative addition to 32 employing Pd2dba3 and (R)-BINAP(O) furnished 36 in a more modest yield (24%), although it is worth highlighting the simplicity and effectiveness of the “dump-and-stir” Pd2dba3/BINAP(O) method, which may be preferable for small-scale experiments.
Figure 3.
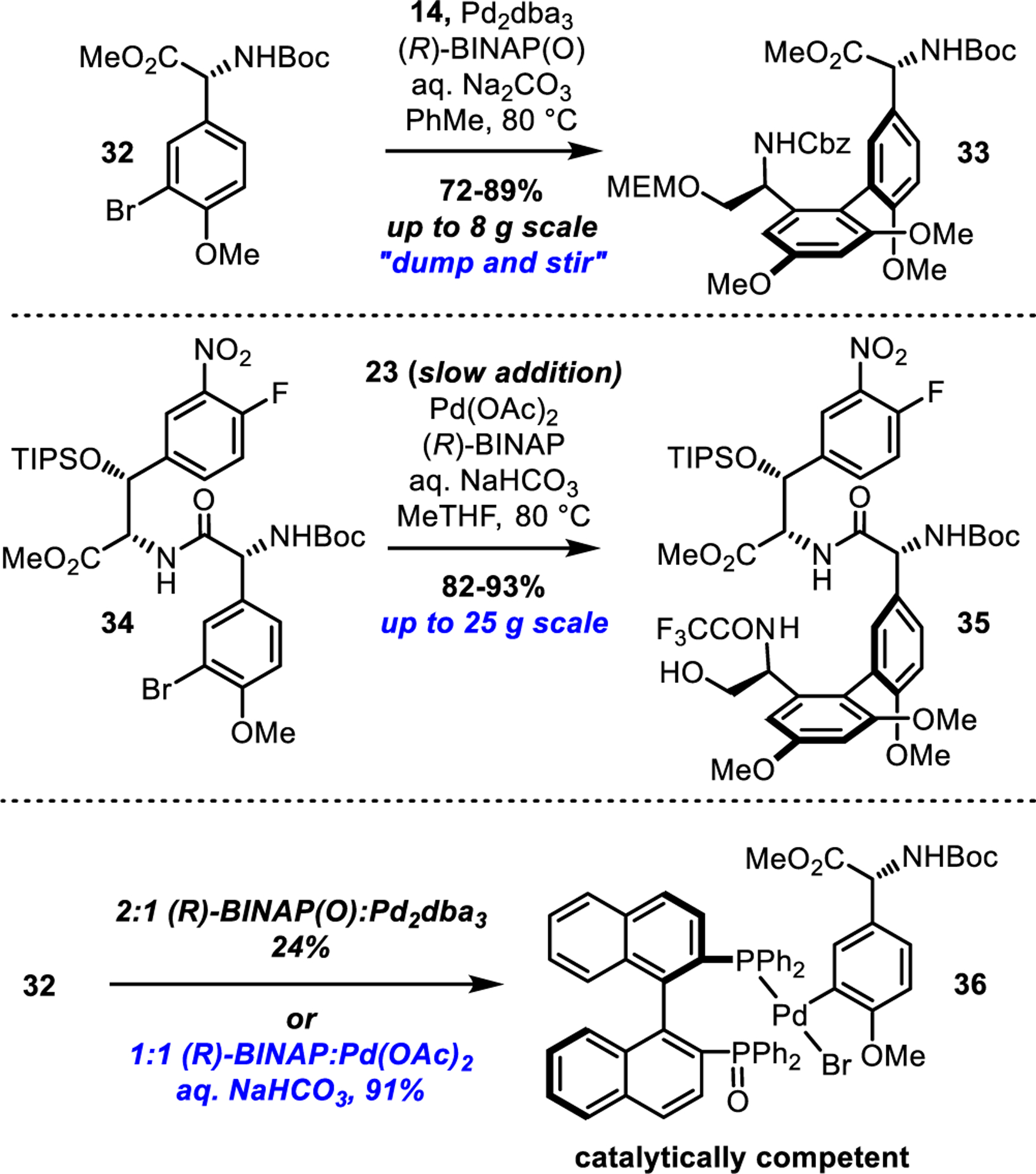
Key observations in the development of a catalyst-controlled diastereoselective synthesis of AB axis of chirality.
Biaryl 33 was converted to the macrolactamization precursor 42 as shown in Figure 4. The A-ring Cbz protecting group was exchanged for Alloc (H2, Pd/C; AllocCl, 83% for 2 steps) to allow eventual amine deprotection under conditions that avoid reduction of the C-ring nitro group. Subsequent saponification (LiOH, 99%), coupling of the carboxylic acid with 11 (DMTMM, 91%), Alloc deprotection (PhSiH3, Pd(PPh3)4, 91%), and methyl ester saponification (LiOH, 99%) provided 41. Macrolactamization of 41 under previously optimized conditions36 [3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT),61 N-methylmorpholine (NMM), 1 mM in THF, 3:1 dr (inseparable), 48% combined yield] proved disappointing. Prior iterations on this route with smaller C-ring alcohol silyl ether protecting groups (TBS, TBDPS) did not suffer from low yields or competitive epimerization (60%, no epimerization and 60%, 13:1 dr, respectively), demonstrating that the increased steric bulk of the TIPS protecting group impeded productive cyclization. However, TIPS protection proved essential to the success of a subsequent diastereoselective CD ring closure, requiring an improved method for the macrolactamization of 41 that avoids epimerization of the C ring α-stereocenter.
Figure 4.
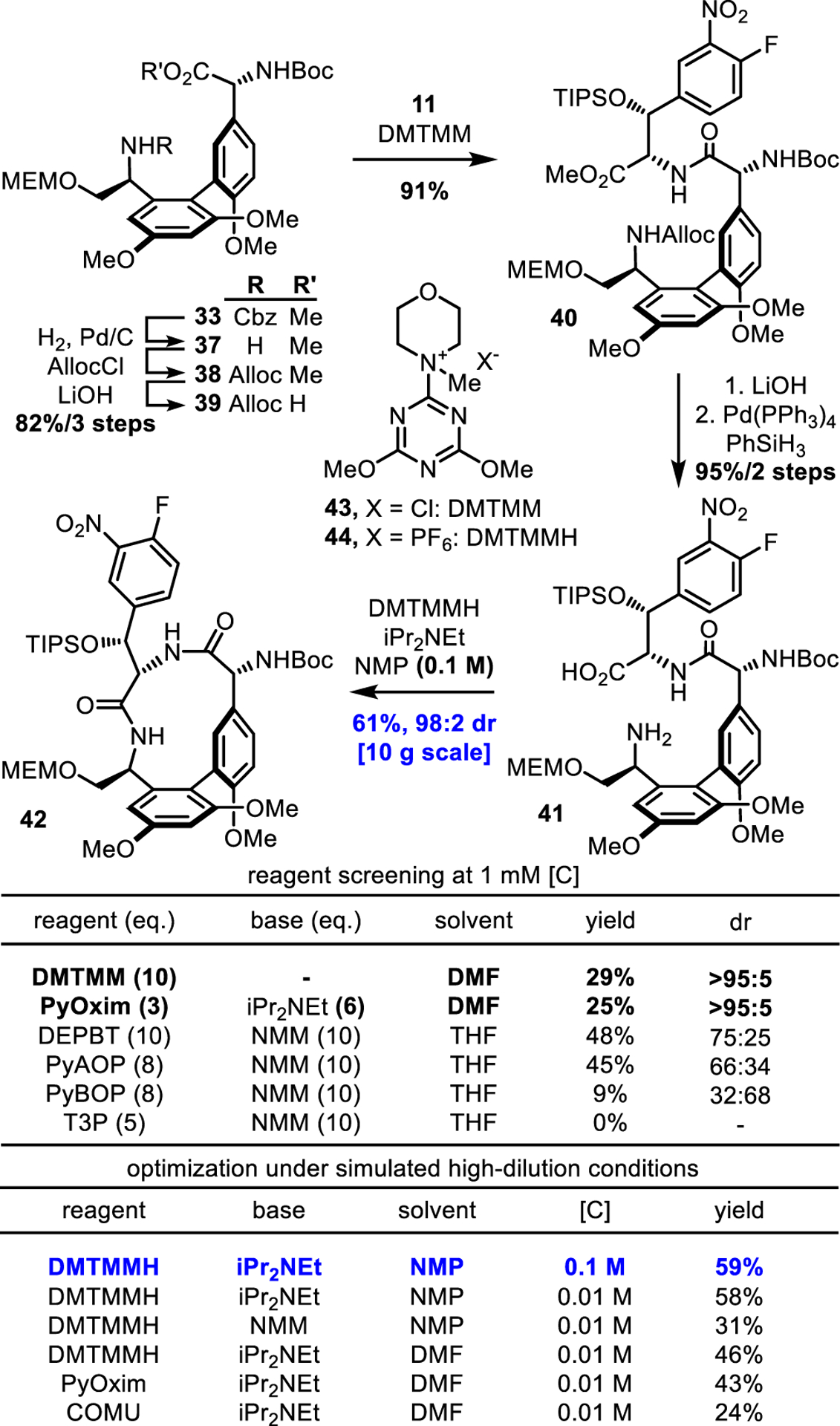
Top: Synthesis of the AB ring system. Bottom: Optimization of the macrolactamization reaction
Extensive reagent screening identified DMTMM62 (43) and its PF6− salt63 (DMTMMH) (44), PyOxim,64 and COMU65 as the only reagents capable of suppressing epimerization (>95:5 dr) in the macrolactamization of 41, consistent with their capabilities for promoting the couplings of hindered carboxylic acids (Figure 4). Further optimization of solvent, base, and reagent under simulated high dilution conditions (i.e., slow addition of substrate to reagents) revealed that DMTMMH provided the highest yields, particularly under conditions that promote fast cyclization (base: iPr2NEt > NMM, and solvent: NMP (N-methylpyrrolidone) > DMF > MeCN). These studies culminated in a rapid macrolactamization reaction that is complete on addition, conducted at a practical concentration (0.1 M), and capable of furnishing 42 in 61% yield on 10 g scale with negligible (≤2%) epimerization. Subsequent Boc deprotection (HCO2H) of 42 and coupling of the free amine with the D-ring subunit carboxylic acid 12 (DEPBT, 72% for 2 steps) set the stage for studies on the key CD ring closure.
The proposal to use the AB macrocycle as an element of preorganization for the CD ring closure was first validated by an improvement in the CD cyclization dr from 1:142 in the absence of the AB macrocycle to 3:1 in its presence with preferential formation of natural atropisomer 46b when the C-ring alcohol was protected as a TBS ether. The macrocyclization of substrate 45b was also unusually facile, proceeding readily at 23 °C in DMF (Scheme 2). Unfortunately, thorough screening of solvent, base, temperature, and additives did not improve the reaction diastereoselectivity beyond 3:1. Moreover, the TBS ether proved sensitive to desilylation as noted in our previous work. Conducting the cyclization on the unprotected alcohol 45a reduced the diastereoselectivity to 2:1, suggesting that the sterically bulky TBS ether improved the SNAr atroposelectivity. Indeed, a progressive increase in the size of the silyl ether protecting group improved the diastereoselectivity (TBS: 3:1 dr, TBDPS: 5:1 dr, TIPS: 7:1 dr). As an added benefit, TIPS-protected 45d was considerably less prone to desilylation than either 45b or 45c, although strict anhydrous conditions were still required for optimal results. The challenge of installation and removal of even larger silyl ether protecting groups (e.g., BIBS66 and supersilyl), as well as their potential to further decelerate the macrolactamization, led us to select the TIPS ether as the preferred alcohol protecting group.
Scheme 2.
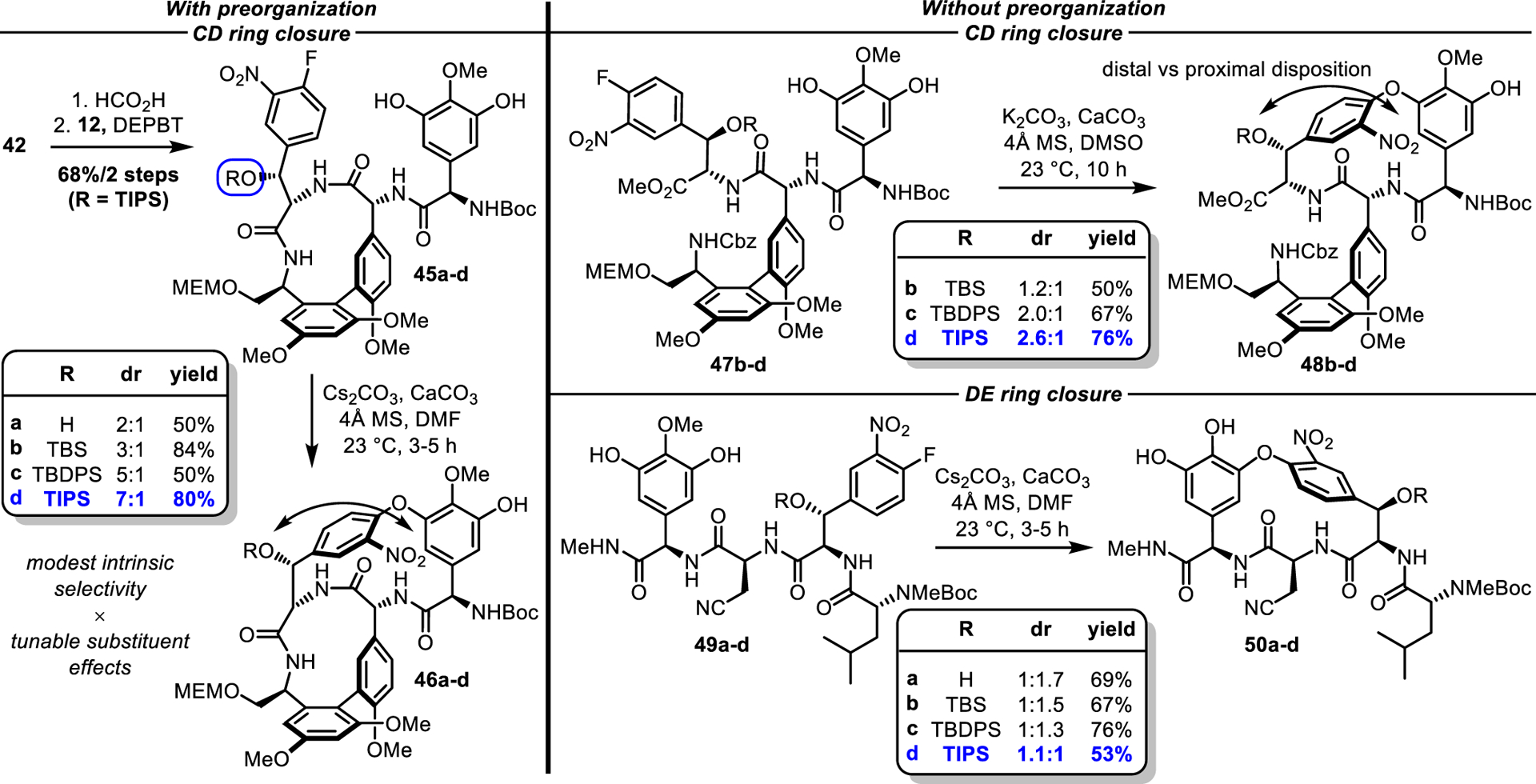
CD and DE ring macrocyclization studies
A similar study of the CD macrocyclization of substrates 47b-d prior to macrolactamization of the AB ring system displayed the same trends, where increasing the size of the silyl ether protecting group improved the selectivity, although diastereoselections were lower than those obtained for substrates 45a-d that contain the intact AB ring system (Scheme 2). Additionally, the cyclizations of 47b-d proceeded 2–3 times more slowly than 45b-d bearing the intact AB ring system under identical conditions (Cs2CO3¸ DMF, 23 °C, 10 h vs 3–5 h, respectively). Collectively, these studies illustrate that the rate and diastereoselectivity of the CD ring closures of 45a-d benefit from a combined preorganization provided by the AB ring system, which preferentially adopts an inherent cis amide conformation6 poised for cyclization, and a subtle tunable substituent effect that sterically further directs the nitro group distal to the large silyl ether. An analogous study of the SNAr cyclization of the isolated DE ring system (substrates 49a-d) demonstrated that an increase in the size of the silyl ether protecting group on the E ring hydroxyl also progressively favored the natural DE atropisomers 50a-d. However, the diastereoselectivities achieved were modest and only the TIPS-protected substrate 49d favored formation of the natural atropisomer (1.1:1 dr), whereas all other substrates examined slightly favored formation of the unnatural DE atropisomer (1:1.7–1:1.3 dr). Nonetheless, the consistent correlation between the size of the silyl ether protecting group and reaction diastereoselectivity was ultimately key to realizing the highly diastereoselective construction of the CD and DE biaryl ethers.
Completion of the formal synthesis required introduction of the DE ring system (Scheme 3). The high diastereoselection (8:1) obtained for the room temperature DE ring closure in our previous work6 with substrates bearing the intact ABCD ring system was observed with the E-ring free alcohol substrate (7:1) herein and was further improved to 11:1 by the subtle impact of TIPS protection of the E-ring hydroxyl group, mirroring the results obtained above. Simultaneous C/E ring TIPS deprotection (Bu4NF/HOAc, 98%), nitro group reduction (Zn/NH4Cl) and double Sandmeyer chlorination (tBuONO, HBF4; CuCl/CuCl2, 54% for 2 steps) provided known intermediate 56, thereby completing a formal total synthesis of vancomycin aglycon 10 in 26 projected steps6 from the individual amino acid subunits and 0.7% overall yield (major diastereomer only) without thermal atropisomer equilibrations. This result compares favorably with our previous synthesis,6 which proceeded in 25 steps but with a more modest 0.2% overall yield when material recovered by thermal atropisomer equilibrations is excluded.
Scheme 3.
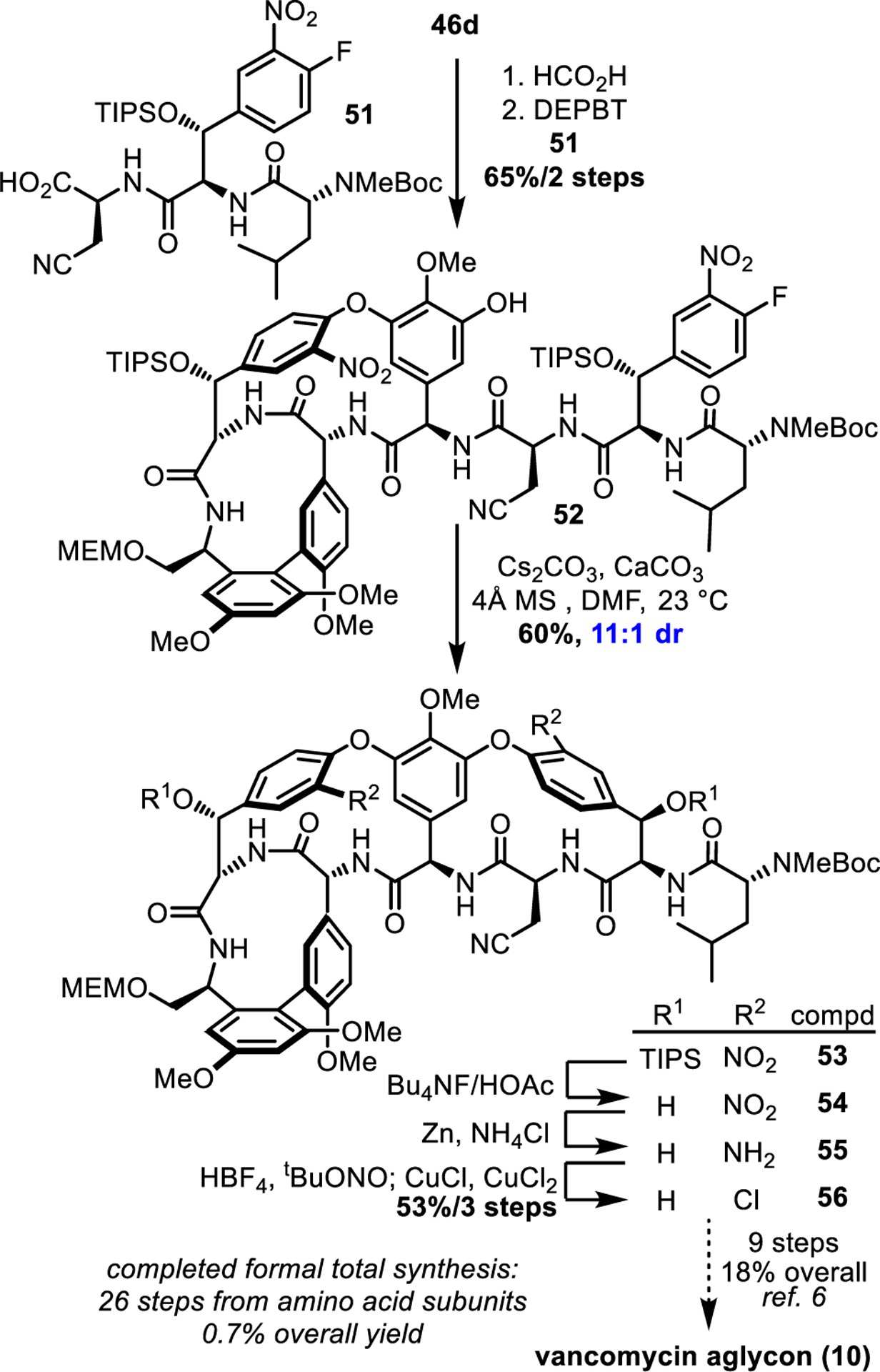
Completions of formal total synthesis
Concurrent with the above efforts, we undertook further optimization of the Sandmeyer chlorination to minimize protodediazoniation, which in practice often leads to copolar deschloro reaction byproducts. In previous efforts, we mitigated this issue by employing a large excess of CuCl/CuCl2 in a mixed CH3CN/H2O solvent system at low temperature.39,42 In these prior studies, we also observed that Sandmeyer chlorination of the C-ring substituent was especially challenging, whereas the less-hindered D-ring substitution was more straightforward. In this work, Sandmeyer substitution of model substrate 57 for the challenging C-ring substitution was problematic at 0 °C (3:1 58:59, 65%) (Figure 5). Further lowering the reaction temperature (−35 °C) suppressed protodediazoniation to acceptable levels (18:1), although the more difficult model substrate 60 bearing the fully functionalized ABCD ring system yielded a less satisfactory but good 9:1 ratio of 61:62 under identical conditions. Disappointingly, the inclusion of CCl4 as an additional chlorine atom donor increased the amount of reduction byproduct by >2-fold, contrary to earlier claims by Zhu.67 Fortunately, the product ratio improved >3-fold (to >32:1) in CD3CN at −35 °C, indicating that reduction primarily occurs by solvent acetonitrile C-H abstraction. A full study of factors influencing the reaction is summarized in Supporting Information Figure S5. These optimal conditions (CD3CN, −35 °C) translated smoothly to the simultaneous double Sandmeyer substitution of substrate 72 bearing the fully functionalized ABCDE ring system in the further streamlined total synthesis below, providing the dichlorinated product 73 in 63% yield. Importantly, CD3CN is inexpensive and available in bulk quantities, such that its use in the Sandmeyer chlorination of 1 g of a late-stage intermediate such as 72 would cost only $20.
Figure 5.
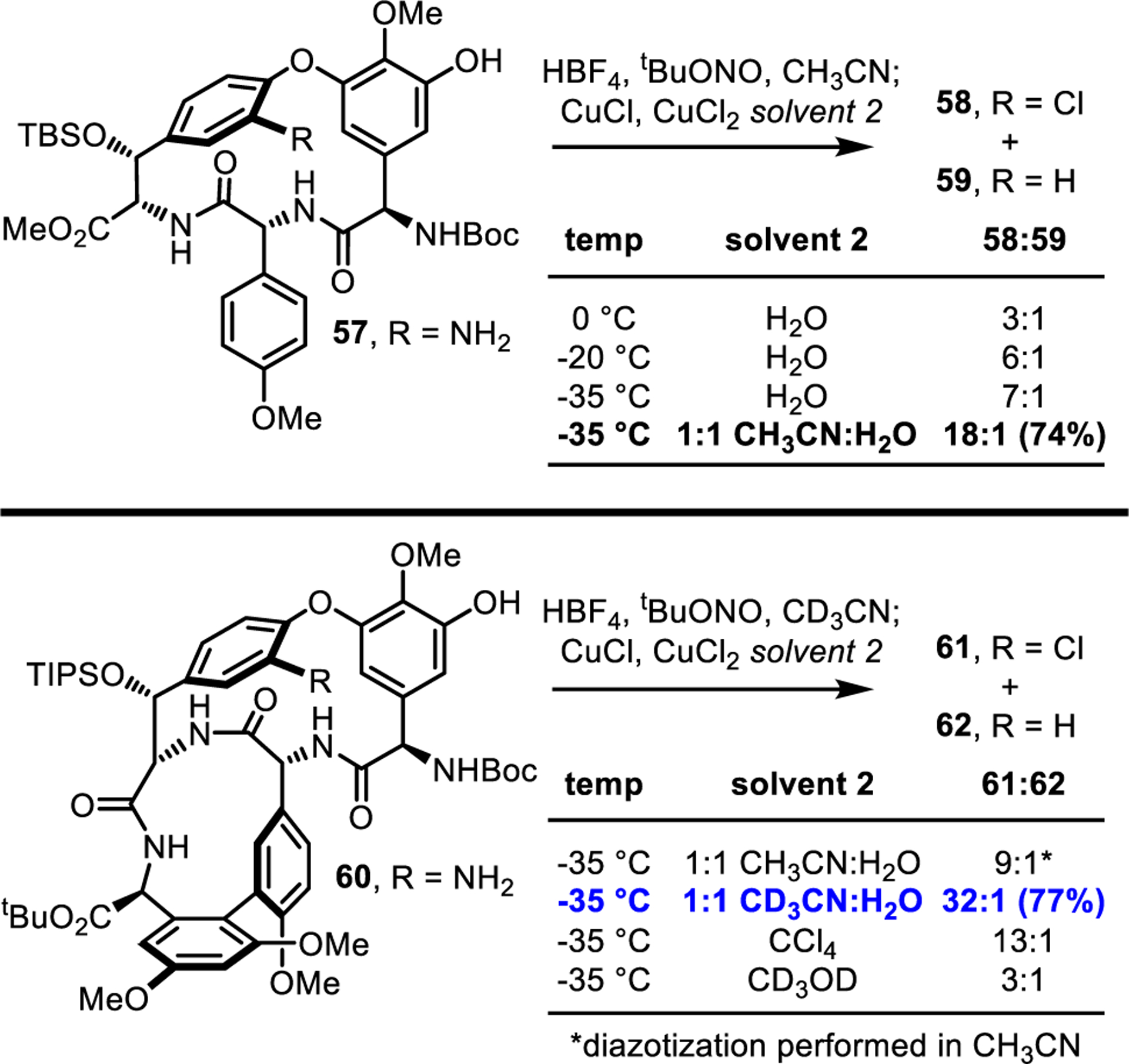
Model studies of the Sandmeyer chlorination reaction. General conditions: HBF4 (1.3 equiv), tBuONO (1.3 equiv), solvent 1, 0 °C, 30 min; cooled to specified temperature, then treated with CuCl (50 equiv) and CuCl2 (60 equiv) in solvent 2, and warmed to room temperature over 1 h. Product ratios were determined by LC/MS analysis of the crude reaction mixture. Isolated yields are given in parentheses.
Next generation total synthesis of vancomycin.
With high kinetic diastereoselectivity achieved for all three atropisomer elements as well as access to large quantities of the amino acid subunits, we proceeded to streamline the approach in anticipation of preparing significant quantities of pocket modified analogues by total synthesis. Foremost among our concerns with the redesigned formal synthesis above were the step count (26 steps LLS), low overall yield (<1%), unnecessary A-ring amine protecting group exchange, and late-stage conversion of the C-terminus MEM ether to the carboxylic acid. Although the latter feature was used to preclude C-terminus epimerization of a precursor methyl ester in prior studies, it introduced an inefficient deprotection-oxidation sequence (53%, 4 steps)6 conducted toward the end of the synthesis that is even more challenging for substrates containing the oxidation prone residue 4 thioamide36 used to access pocket modified analogues.
We incorporated the A-ring subunit as the unprotected alcohol 23 also now bearing an orthogonal trifluoroacetamide protecting group (Scheme 4). The use of 22 in a direct, one-pot borylation/atroposelective Suzuki coupling with BC dipeptide 34, prepared by coupling 11 with 15 (DMTMM, 88%), provided 35 in superb yield and outstanding diastereoselectivity (93%, unnatural atropisomer not detected), the development of which is detailed in Figure 3. To date, this reaction has been scaled to 25 g without loss of efficiency and avoids isolation and handling of the sensitive boronic acid 23. Subsequent primary alcohol oxidation and esterification (TEMPO, PhI(OAc)2;68 tert-butyl trichloroacetimidate,69 82% for 2 steps, 26 g scale) secured the C-terminus in the required oxidation state in high yield and at an early stage prior to introduction of the D-ring subunit (residue 4) that will bear a key thioamide in future syntheses of pocket-modified analogues. Alternative efforts to incorporate the C-terminus tert-butyl ester into the A-ring subunit prior to the biaryl coupling resulted in racemization in studies conducted to date. Through judicious choice of the amine protecting group, simultaneous single-step deprotection of the C-ring methyl ester and A-ring trifluoroacetamide of 64 (LiOH, 82%) provided 65 without competitive tert-butyl ester hydrolysis and set the stage for closure of the AB ring system. Gratifyingly, the macrolactamization conditions developed herein (see Figure 4) proved most effective for 65 bearing the C-terminal tert-butyl ester, despite the more electron-deficient nature of the reacting amine. Not only did the macrocyclization of 65 (DMTMMH, iPr2NEt, NMP) proceed essentially instantaneously to provide 66 in higher yield (up to 85% yield, on scales of up to 16.4 g) than the C-terminal MEM ether (61% for 42), but alternative substrates that bear a C-terminal free alcohol or TBS-protected alcohol cyclized in inferior yields (33% and 12%, respectively), revealing that the substrate containing the least nucleophilic amine (65) cyclized most effectively (Figure 6). Although this trend may appear counterintuitive, adducts of the less effective starting amines with the coupling reagent DMTMMH were invariably observed, indicating that an amine triazinylation side reaction was competitive with macrocyclizations of substrates bearing more nucleophilic amines. Finally, the structure, relative stereochemistry, and the cis amide linking residues 5 and 6 (vancomycin numbering) were confirmed in a single crystal X-ray structure determination of 66.70 In addition, the suggested distal disposition of the large TIPS ether and aryl nitro substituent poised for diastereoselective CD ring closure was observed in the X-ray crystal structure of 66. In total, the AB macrocycle was prepared in 53% yield (5 steps), representing a significant improvement over the formal synthesis above (37%/8 steps) with the added advantage of early stage oxidation of the C-terminus that avoids 4 additional late-stage steps in the overall synthesis.
Scheme 4.
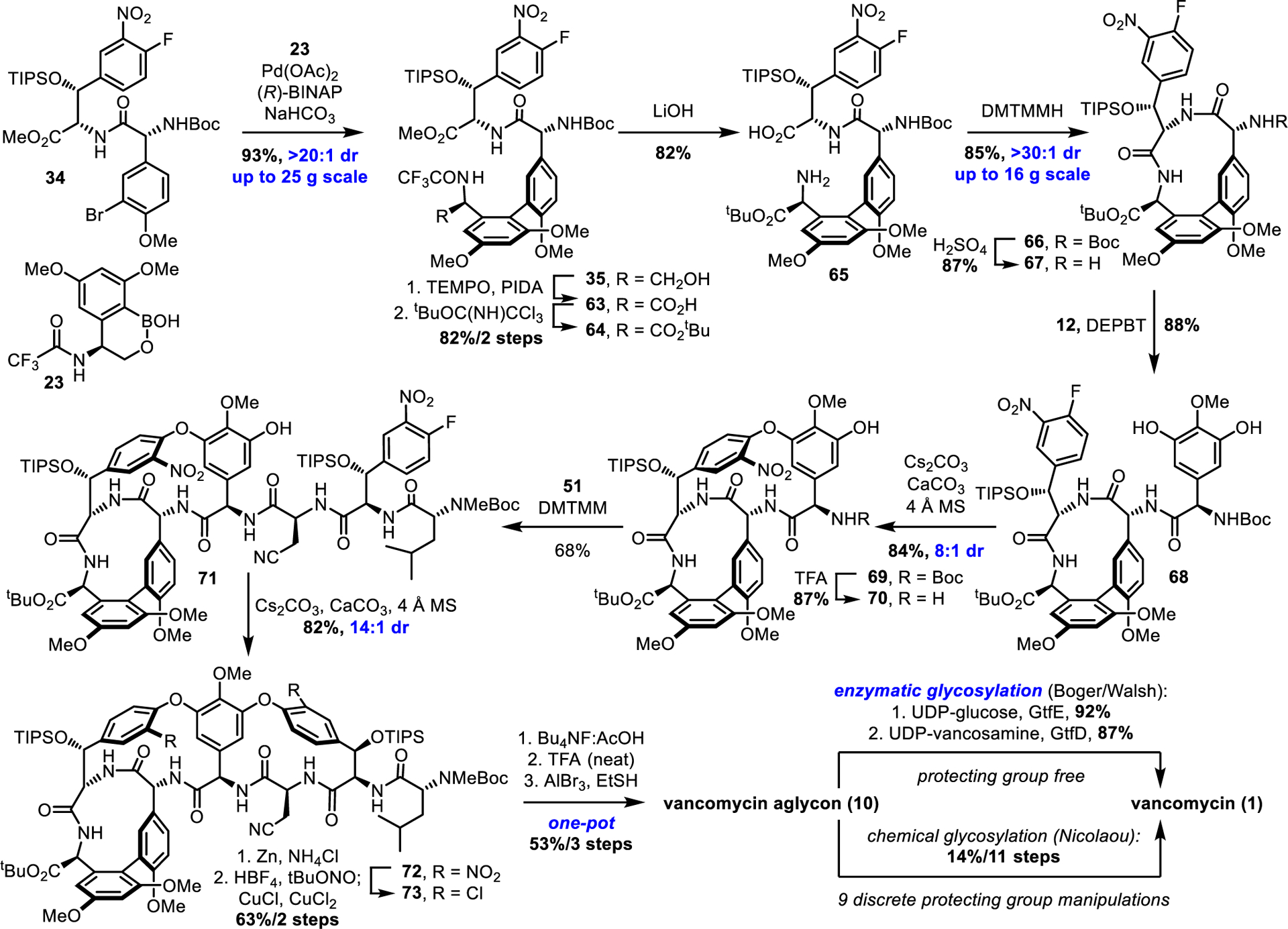
Next generation vancomycin total synthesis
Figure 6.
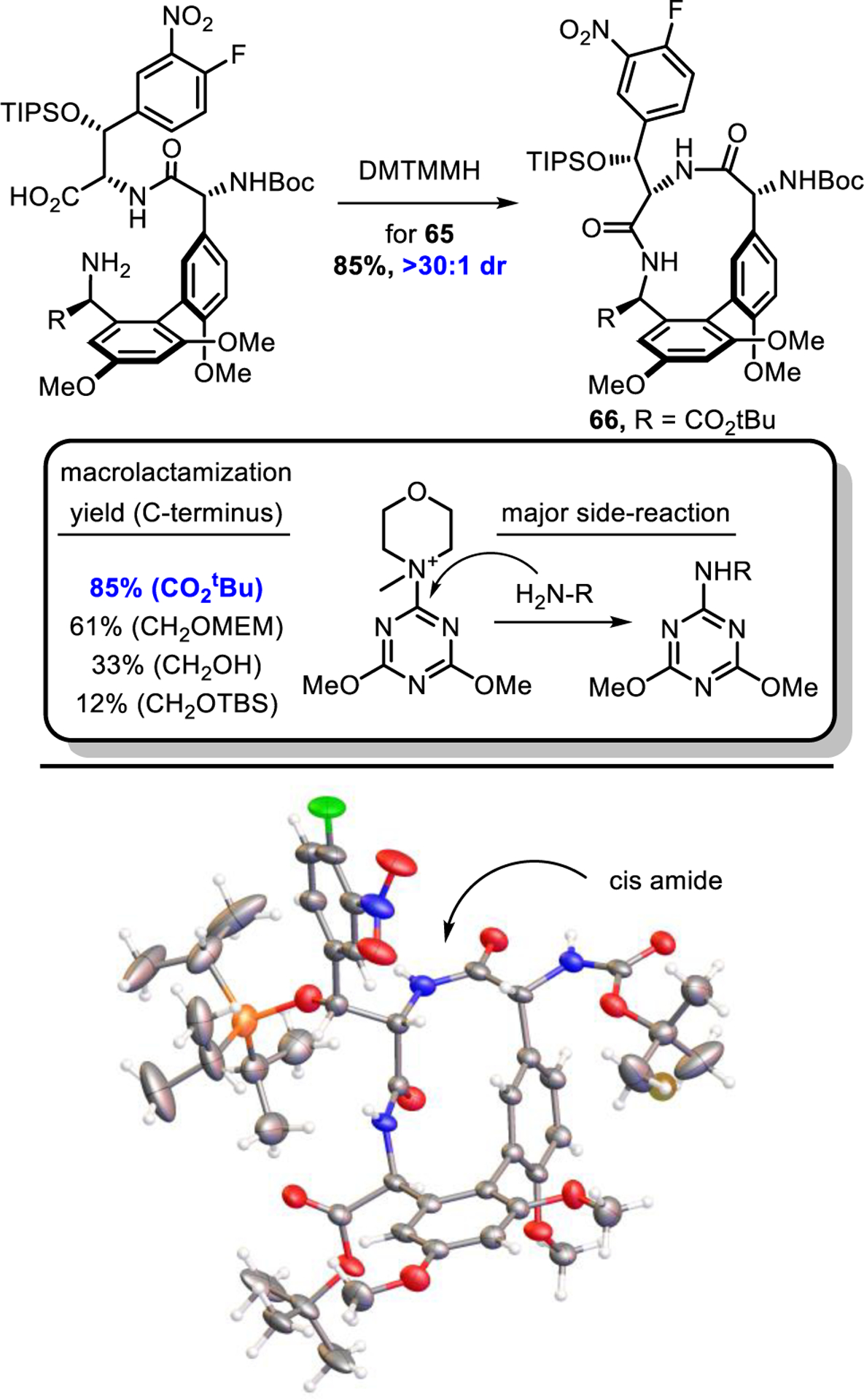
Top: Substituent impact on macrolactamization of AB ring system. Bottom: X-ray crystal structure of 66.
The completion of our next-generation total synthesis of vancomycin aglycon 10 is outlined in Scheme 4. Selective N-Boc deprotection of 66 under conditions that may permit reversible tert-butyl ester deprotection/reprotection69 (H2SO4, tBuOAc, 86% yield, 98% brsm, 6.9 g scale) followed by coupling of amine 67 with the D-ring subunit 12 provided 68 (88%) poised for CD ring closure. Further refinement of conditions for the room temperature macrocyclization of this substrate provided the ABCD ring system 69 in superb yield and excellent diastereoselectivity (84%, 8:1 dr) without detectable epimerization of the C-terminal tert-butyl ester. Attentive reaction monitoring permitted N-Boc deprotection of 69 without significant tert-butyl ester deprotection (5% TFA-CH2Cl2, 87%). Coupling of the amine 70 with the E-ring tripeptide 51 promoted by DMTMM provided 71 in excellent yield with minimal epimerization of the β-cyanoalanine residue (6:1 dr, 68% major diastereomer) that is ordinarily problematic for this segment coupling.36,10 Room temperature macrocyclization of the DE ring system proceeded with near exclusive formation of the desired DE atropisomer (14:1 dr) in high yield (82%), where the rate and substrate-controlled diastereoselectivity benefit from both the preorganization provided by the ABCD ring system observed in prior studies6 as well as the now added influence of the E ring TIPS alcohol protecting group discovered in this work. Dual nitro group reduction followed by Sandmeyer chlorination under the conditions developed herein (tBuONO/HBF4; CuCl/CuCl2, CD3CN:H2O, −35 °C) yielded dichloride 73 (63% for 2 steps), and this proved more effective than Sandmeyer substitution following removal of the two TIPS protecting groups. Without optimization, final conversion to vancomycin aglycon 10 was conducted in a now simplified 3-step, one-pot sequence, consisting of desilylation (Bu4NF/HOAc, THF, 23 °C), nitrile hydration with concomitant Boc and tert-butyl ester deprotections (neat TFA, 23 °C), and global O-demethylation (AlBr3, EtSH, 23 °C; 53–58% for 3 steps with removal of 9 protecting groups). This next generation total synthesis of vancomycin aglycon was completed in 17 steps and in 5% overall yield from the amino acid subunits with excellent kinetic diastereoselectivity achieved for introduction of each atropisomer element.
Just as importantly, we previously implemented a practical solution to what many may think would be the most challenging feature of developing synthetic glycopeptide antibiotics, the late-stage steps used for scalable conversion of vancomycin aglycon to the fully decorated natural product or its analogues. This entailed enzymatic glycosylation of the aglycon(s) with the biosynthetic enzymes (glycosyltransferases GtfE and GtfD)71–73 and commercial (UDP-glucose)74 or readily accessible (UDP-vancosamine)75 glycosyl donors for introduction of the disaccharide.8 The two enzymes have been stably overexpressed in E. coli by Walsh, making them readily available as reagents for use on any scale, like the UDP-glycosyl donors.71–73 Moreover, this utilizes the fully functionalized aglycon without introduction or removal of protecting groups and was found to work effectively on the vancomycin aglycon residue 4 thioamide that serves as our immediate precursor to binding pocket analogues.8,38 Combined, this provides a 19-step total synthesis of vancomycin 1 that proceeds in 3.7% overall yield, substantially improving on prior approaches (Figure 7).
Figure 7.
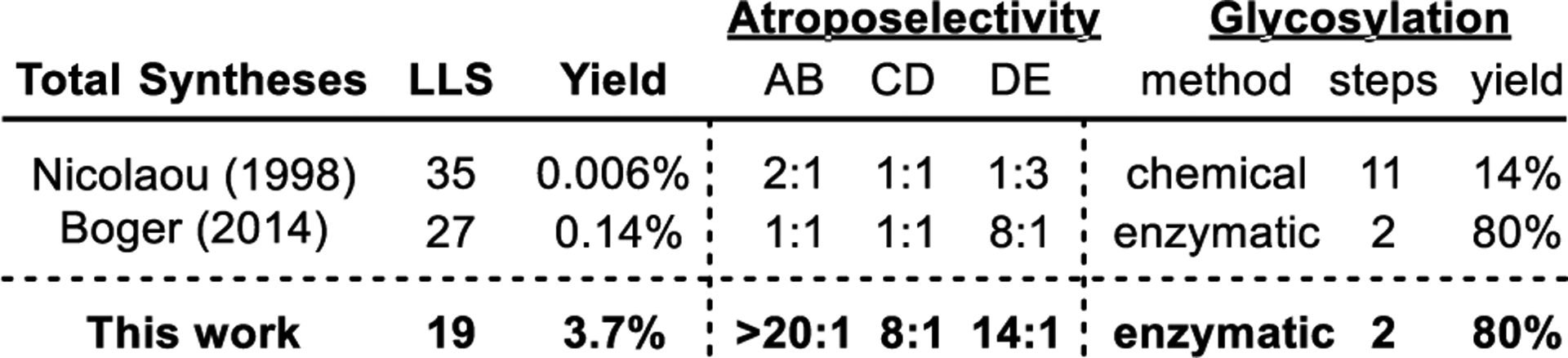
Comparison of total syntheses of vancomycin (1), highlighting the late-stage enzymatic glycosylation. Yield and LLS start from individual amino acid subunits.
CONCLUSION
Despite the exceptional antimicrobial potency and durability of the pocket modified vancomycin analogues discovered by our group, their preclinical evaluation has been slowed by the challenges presented by their total synthesis. Herein, we address this issue through the development of a scalable atroposelective total synthesis of vancomycin that considerably reduced the step count and increased the overall yield. With this new approach in hand, our efforts are focused on the preparation of new durable vancomycin analogues (e.g., 9) and preclinical evaluation of the most promising candidates, including those that express three synergistic mechanisms of action (MOAs), two of which are independent of D-Ala-D-Ala/D-Lac binding and each of which is independently effective against vancomycin-resistant and vancomycin-sensitive bacteria. It is possible such durable analogues that act by up to three MOAs might provide extraordinarily potent antibiotics, which display clinical lifetimes measured not in decades, or even the halfcentury of vancomycin, but perhaps in centuries. The total synthesis of vancomycin detailed herein provides the opportunity to prepare and assess such analogues.76,77
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge financial support from the NIH (CA041101, D.L.B.), postdoctoral support from Nippon Chemiphar (Y.M.), and graduate fellowship support from the Kellogg family (M.J.M.). We especially wish to acknowledge and thank Dr. Shouliang Yang for contributions to the synthetic studies early in development; Dr. Jason Chen, Brittany Sanchez, and Emily Sturgell for assistance with HRMS, LC, and chiral SFC analysis; Dr. Thomas Razler (Bristol Myers Squibb) for helpful discussion and for generously providing ligands; Dr. Dee Huang and Dr. Laura Pasternack for NMR assistance; and Professor Arnold L. Reingold (UCSD) for the X-ray structure determination of 66.
Footnotes
Supporting Information
Full experimental details and copies of 1H, 13C, and 2D NMR spectra (pdf).
The Supporting Information is available free of charge on the ACS Publications website at DOI: XX.
The authors declare no competing financial interest.
REFERENCES
- 1.McCormick MH; McGuire JM; Pittenger GE; Pittenger RC; Stark WM, Vancomycin, a new antibiotic. I. Chemical and biologic properties. Antibiot. Ann 1955, 3, 606–611. [PubMed] [Google Scholar]
- 2.Kahne D; Leimkuhler C; Lu W; Walsh C, Glycopeptide and lipoglycopeptide antibiotics. Chem. Rev 2005, 105, 425–448. [DOI] [PubMed] [Google Scholar]
- 3.Harris CM; Kopecka H; Harris TM, Vancomycin: Structure and transformation to CDP-I. J. Am. Chem. Soc 1983, 105, 6915–6922. [Google Scholar]
- 4.Evans DA; Wood MR; Trotter BW; Richardson TI; Barrow JC; Katz JL, Total syntheses of vancomycin and eremomycin aglycons. Angew. Chem. Int. Ed 1998, 37, 2700–2704. [DOI] [PubMed] [Google Scholar]
- 5.Nicolaou KC; Takayanagi M; Jain NF; Natarajan S; Koumbis AE; Bando T; Ramanjulu JM, Total synthesis of vancomycin aglycon—part 3: Final stages. Angew. Chem. Int. Ed 1998, 37, 2717–2719. [DOI] [PubMed] [Google Scholar]
- 6.Boger DL; Miyazaki S; Kim SH; Wu JH; Castle SL; Loiseleur O; Jin Q, Total synthesis of the vancomycin aglycon. J. Am. Chem. Soc 1999, 121, 10004–10011. [Google Scholar]
- 7.Nicolaou KC; Mitchell HJ; Jain NF; Winssinger N; Hughes R; Bando T, Total synthesis of vancomycin. Angew. Chem. Int. Ed 1999, 38, 240–244. [Google Scholar]
- 8.Nakayama A; Okano A; Feng Y; Collins JC; Collins KC; Walsh CT; Boger DL, Enzymatic glycosylation of vancomycin aglycon: Completion of a total synthesis of vancomycin and N-and C-terminus substituent effects of the aglycon substrate. Org. Lett 2014, 16, 3572–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans DA; Barrow JC; Watson PS; Ratz AM; Dinsmore CJ; Evrard DA; DeVries KM; Ellman JA; Rychnovsky SD; Lacour J, Approaches to the synthesis of the vancomycin antibiotics. Synthesis of orienticin C (bis-dechlorovancomycin) aglycon. J. Am. Chem. Soc 1997, 119, 3419–3420. [Google Scholar]
- 10.Boger DL; Kim SH; Mori Y; Weng J-H; Rogel O; Castle SL; McAtee JJ, First and second generation total synthesis of the teicoplanin aglycon. J. Am. Chem. Soc 2001, 123, 1862–1871. [DOI] [PubMed] [Google Scholar]
- 11.Evans DA; Katz JL; Peterson GS; Hintermann T, Total synthesis of teicoplanin aglycon. J. Am. Chem. Soc 2001, 123, 12411–12413. [DOI] [PubMed] [Google Scholar]
- 12.Crowley BM; Mori Y; McComas CC; Tang D; Boger DL, Total synthesis of the ristocetin aglycon. J. Am. Chem. Soc 2004, 126, 4310–4317. [DOI] [PubMed] [Google Scholar]
- 13.Perkins H, Vancomycin and related antibiotics. Pharmacol. Ther 1982, 16, 181–197. [DOI] [PubMed] [Google Scholar]
- 14.Williams DH, The glycopeptide story–how to kill the deadly ‘ superbugs’. Nat. Prod. Rep 1996, 13, 469–477. [DOI] [PubMed] [Google Scholar]
- 15.Williams DH; Bardsley B, The vancomycin group of antibiotics and the fight against resistant bacteria. Angew. Chem. Int. Ed 1999, 38, 1172–1193. [DOI] [PubMed] [Google Scholar]
- 16.Nicolaou K; Boddy CN; Bräse S; Winssinger N, Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew. Chem. Int. Ed 1999, 38, 2096–2152. [DOI] [PubMed] [Google Scholar]
- 17.Courvalin P, Resistance of enterococci to glycopeptides. Antimicrob. Agents Chemother 1990, 34, 2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Courvalin P, Vancomycin resistance in gram-positive cocci. Clin. Infect. Dis 2006, 42, S25–S34. [DOI] [PubMed] [Google Scholar]; (b) Weigel LM; Clewell DB; Gill SR; Clark NC; McDougal LK; Flannagan SE; Kolonay JF; Shetty J; Killgore GE; Tenover FC Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 2003, 302, 1569–1571. [DOI] [PubMed] [Google Scholar]
- 19.Walsh CT, Vancomycin resistance: Decoding the molecular logic. Science 1993, 261, 308–310. [DOI] [PubMed] [Google Scholar]
- 20.Walsh C, Deconstructing vancomycin. Science 1999, 284, 442–443. [DOI] [PubMed] [Google Scholar]
- 21.James RC; Pierce JG; Okano A; Xie J; Boger DL, Redesign of glycopeptide antibiotics: Back to the future. ACS Chem. Biol 2012, 7, 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okano A; Isley NA; Boger DL, Total syntheses of vancomycin-related glycopeptide antibiotics and key analogues. Chem. Rev 2017, 117, 11952–11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Binda E; Marinelli F; Marcone GL, Old and new glycopeptide antibiotics: Action and resistance. Antibiot. 2014, 3, 572–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butler MS; Hansford KA; Blaskovich MA; Halai R; Cooper MA, Glycopeptide antibiotics: Back to the future. J. Antibiot 2014, 67, 631–644. [DOI] [PubMed] [Google Scholar]
- 25.Boneca IG; Chiosis G, Vancomycin resistance: occurrence, mechanisms and strategies to combat it. Expert Opin. Ther 2003, 7, 311–328. [DOI] [PubMed] [Google Scholar]
- 26.Leclercq R; Derlot E; Duval J; Courvalin P, Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N. Engl. J. Med 1988, 319, 157–161. [DOI] [PubMed] [Google Scholar]
- 27.Marshall C; Broadhead G; Leskiw B; Wright G, D-Ala-D-Ala ligases from glycopeptide antibiotic-producing organisms are highly homologous to the enterococcal vancomycin-resistance ligases VanA and VanB. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 6480–6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ikeda S; Hanaki H; Yanagisawa C; Ikeda-Dantsuji Y; Matsui H; Iwatsuki M; Shiomi K; Nakae T; Sunakawa K; Ōmura S, Identification of the active component that induces vancomycin resistance in MRSA. J. Antibiot 2010, 63, 533–538. [DOI] [PubMed] [Google Scholar]
- 29.Hong H-J; Hutchings MI; Buttner MJ, Vancomycin resistance VanS/VanR two-component systems In Bacterial Signal Transduction: Networks and Drug Targets, Springer: 2008; pp 200–213. [Google Scholar]
- 30.Koteva K; Hong H-J; Wang XD; Nazi I; Hughes D; Naldrett MJ; Buttner MJ; Wright GD, A vancomycin photoprobe identifies the histidine kinase VanSsc as a vancomycin receptor. Nat. Chem. Biol 2010, 6, 327. [DOI] [PubMed] [Google Scholar]
- 31.Kwun MJ; Novotna G; Hesketh AR; Hill L; Hong H-J, In vivo studies suggest that induction of VanSdependent vancomycin resistance requires binding of the drug to D-Ala-D-Ala termini in the peptidoglycan cell wall. Antimicrob. Agents Chemother 2013, 57, 4470–4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bugg TDH; Wright GD; Dutka-Malen S; Arthur M; Courvalin P; Walsh CT, Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry 1991, 30, 10408–10415. [DOI] [PubMed] [Google Scholar]
- 33.McComas CC; Crowley BM; Boger DL, Partitioning the loss in vancomycin binding affinity for D-Ala-D-Lac into lost H-bond and repulsive lone pair contributions. J. Am. Chem. Soc 2003, 125, 9314–9315. [DOI] [PubMed] [Google Scholar]
- 34.Boger DL, The difference a single atom can make: synthesis and design at the chemistry–biology interface. J. Org. Chem 2017, 82, 11961–11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crowley BM; Boger DL, Total synthesis and evaluation of [Ψ[CH2NH]Tpg4]vancomycin aglycon: Reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J. Am. Chem. Soc 2006, 128, 2885–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie J; Okano A; Pierce JG; James RC; Stamm S; Crane CM; Boger DL, Total synthesis of [Ψ[C(═S)NH]Tpg4]vancomycin aglycon, [Ψ[C(═NH)NH]Tpg4]vancomycin aglycon, and related key compounds: Reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J. Am. Chem. Soc 2012, 134, 1284–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okano A; Isley NA; Boger DL, Peripheral modifications of [Ψ[CH2NH]Tpg4]vancomycin with added synergistic mechanisms of action provide durable and potent antibiotics. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E5052E5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okano A; Nakayama A; Schammel AW; Boger DL, Total synthesis of [Ψ[C(=NH)NH]Tpg4] vancomycin and its (4-chlorobiphenyl)methyl derivative: Impact of peripheral modifications on vancomycin analogues redesigned for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J. Am. Chem. Soc 2014, 136, 13522–13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okano A; Nakayama A; Wu K; Lindsey EA; Schammel AW; Feng Y; Collins KC; Boger DL, Total syntheses and initial evaluation of [Ψ[C(=S)NH]Tpg4] vancomycin, [Ψ[C(=NH)NH]Tpg4]vancomycin, [Ψ[CH2NH]Tpg4] vancomycin, and their (4-chlorobiphenyl) methyl derivatives: synergistic binding pocket and peripheral modifications for the glycopeptide antibiotics. J. Am. Chem. Soc 2015, 137, 3693–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boger DL; Miyazaki S; Kim SH; Wu JH; Loiseleur O; Castle SL, Diastereoselective total synthesis of the vancomycin aglycon with ordered atropisomer equilibrations. J. Am. Chem. Soc 1999, 121, 3226–3227. [Google Scholar]
- 41.Boger DL; Borzilleri RM; Nukui S; Beresis RT, Synthesis of the vancomycin CD and DE ring systems. J. Org. Chem 1997, 62, 4721–4736. [Google Scholar]
- 42.Reddy KL; Sharpless KB, From styrenes to enantiopure α-arylglycines in two steps. J. Am. Chem. Soc 1998, 120, 1207–1217. [Google Scholar]
- 43.Li GG; Chang HT; Sharpless KB Catalytic asymmetric aminohydroxylation (AA) of olefins. Angew. Chem. Int. Ed. Engl 1996, 35, 451–454. [Google Scholar]
- 44.Schöllkopf U; Nozulak J; Grauert M, Asymmetric synthesis via heterocyclic intermediates; XXIV1. Asymmetric synthesis of enantiomerically and diastereomerically pure D-threonine by the bis-lactim ether method. Synthesis 1985, 1985, 55–56. [Google Scholar]
- 45.Ishiyama T; Takagi J; Hartwig JF; Miyaura N, A stoichiometric aromatic C-H borylation catalyzed by iridium(I)/2,2′-bipyridine complexes at room temperature. Angew. Chem. Int. Ed 2002, 41, 3056–3058. [DOI] [PubMed] [Google Scholar]
- 46.Feng Y; Holte D; Zoller J; Umemiya S; Simke LR; Baran PS, Total synthesis of verruculogen and fumitremorgin A enabled by ligand-controlled C–H borylation. J. Am. Chem. Soc 2015, 137, 10160–10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Preshlock SM; Ghaffari B; Maligres PE; Krska SW; Maleczka RE Jr; Smith III MR, High-throughput optimization of Ir-catalyzed C–H borylation: A tutorial for practical applications. J. Am. Chem. Soc 2013, 135, 7572–7582. [DOI] [PubMed] [Google Scholar]
- 48.Oeschger RJ; Larsen MA; Bismuto A; Hartwig JF, Origin of the difference in reactivity between Ir catalysts for the borylation of C–H bonds. J. Am. Chem. Soc 2019, 141, 16479–16485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chan DMT; Monaco KL; Wang R-P; Winters MP, New N- and O-arylations with phenylboronic acids and cupric acetate. Tetrahedron Lett. 1998, 39, 2933–2936. [Google Scholar]
- 50.Ishiyama T; Murata M; Miyaura N, Palladium(0)-catalyzed cross-coupling reaction of alkoxydiboron with haloarenes: A direct procedure for arylboronic esters. J. Org. Chem 1995, 60, 7508–7510. [Google Scholar]
- 51.Yamada Y; Akiba A; Arima S; Okada C; Yoshida K; Itou F; Kai T; Satou T; Takeda K; Harigaya Y, Synthesis of linear tripeptides for right-hand segments of complestatin. Chem. Pharm. Bull 2005, 53, 1277–1290. [DOI] [PubMed] [Google Scholar]
- 52.Chen J; Corbin SP; Holman NJ, An improved large scale synthesis of the Schöllkopf chiral auxiliaries: (2R)- and (2S)-2,5-Dihydro-3,6-dimethoxy-2-isopropylpyrazine. Org. Process Res. Dev 2005, 9, 185–187. [Google Scholar]
- 53.Patel J; Clavé G; Renard P-Y; Franck X, Straightforward access to protected syn α-amino-β-hydroxy acid derivatives. Angew. Chem. Int. Ed 2008, 47, 4224–4227. [DOI] [PubMed] [Google Scholar]
- 54.Crimmins MT; She J, An improved procedure for asymmetric aldol additions with N-acyl oxazolidinones, oxazolidinethiones and thiazolidinethiones. Synlett 2004, 2004, 1371–1374. [Google Scholar]
- 55.Evans DA; Dinsmore CJ; Watson PS; Wood MR; Richardson TI; Trotter BW; Katz JL, Nonconventional stereochemical issues in the design of the synthesis of the vancomycin antibiotics: challenges imposed by axial and nonplanar chiral elements in the heptapeptide aglycons. Angew. Chem. Int. Ed 1998, 37, 2704–2708. [DOI] [PubMed] [Google Scholar]
- 56.Nicolaou KC; Li H; Boddy CNC; Ramanjulu JM; Yue T-Y; Natarajan S; Chu X-J; Bräse S; Rübsam F, Total synthesis of vancomycin—part 1: Design and development of methodology. Chem. Eur. J 1999, 5, 2584–2601. [Google Scholar]
- 57.Ji Y; Plata RE; Regens CS; Hay M; Schmidt M; Razler T; Qiu Y; Geng P; Hsiao Y; Rosner T; Eastgate MD; Blackmond DG, Mono-oxidation of bidentate bis-phosphines in catalyst activation: kinetic and mechanistic studies of a Pd/Xantphos-catalyzed C–H functionalization. J. Am. Chem. Soc 2015, 137, 13272–13281. [DOI] [PubMed] [Google Scholar]
- 58.Beutner G; Carrasquillo R; Geng P; Hsiao Y; Huang EC; Janey J; Katipally K; Kolotuchin S; La Porte T; Lee A; Lobben P; Lora-Gonzalez F; Mack B; Mudryk B; Qiu Y; Qian X; Ramirez A; Razler TM; Rosner T; Shi Z; Simmons E; Stevens J; Wang J; Wei C; Wisniewski SR; Zhu Y, Adventures in atropisomerism: total synthesis of a complex active pharmaceutical ingredient with two chirality axes. Org. Lett 2018, 20, 3736–3740. [DOI] [PubMed] [Google Scholar]
- 59.Grushin VV, Synthesis of hemilabile phosphine–phosphine oxide ligands via the highly selective Pd-catalyzed mono-oxidation of bidentate phosphines: Scope, limitations, and mechanism. Organometallics 2001, 20, 3950–3961. [Google Scholar]
- 60.Zalesskiy SS; Ananikov VP, Pd2(dba)3 as a precursor of soluble metal complexes and nanoparticles: Determination of palladium active species for catalysis and synthesis. Organometallics 2012, 31, 2302–2309. [Google Scholar]
- 61.Li H; Jiang X; Ye Y.-h.; Fan C; Romoff T; Goodman M, 3-(Diethoxyphosphoryloxy)-1, 2, 3-benzotriazin-4 (3 H)-one (DEPBT): A new coupling reagent with remarkable resistance to racemization. Org. Lett 1999, 1, 91–94. [DOI] [PubMed] [Google Scholar]
- 62.Kunishima M; Kawachi C; Iwasaki F; Terao K; Tani S, Synthesis and characterization of 4-(4,6-dimethoxy1,3,5-triazin-2-yl)-4-methylmorpholinium chloride. Tetrahedron Lett. 1999, 40, 5327–5330. [Google Scholar]
- 63.Raw SA, An improved process for the synthesis of DMTMM-based coupling reagents. Tetrahedron Lett. 2009, 50, 946–948. [Google Scholar]
- 64.Subirós-Funosas R; El-Faham A; Albericio F, PyOxP and PyOxB: The Oxyma-based novel family of phosphonium salts. Org. Biomol. Chem 2010, 8, 3665–3673. [DOI] [PubMed] [Google Scholar]
- 65.Albericio F; El-Faham A, Choosing the right coupling reagent for peptides: A twenty-five-year journey. Org. Process Res. Dev 2018, 22, 760–772. [Google Scholar]
- 66.Liang H; Hu L; Corey EJ, Di-tert-butylisobutylsilyl, another useful protecting group. Org. Lett 2011, 13, 4120–4123. [DOI] [PubMed] [Google Scholar]
- 67.Vergne C; Bois-Choussy M; Zhu J, An efficient chloro-deamination procedure, application to the synthesis of advanced vancomycin CODOE ring. Synlett 1998, 1998, 1159–1161. [Google Scholar]
- 68.De Mico A; Margarita R; Parlanti L; Vescovi A; Piancatelli G, A versatile and highly selective hypervalent iodine (III)/2,2,6,6-tetramethyl-1-piperidinyloxyl-mediated oxidation of alcohols to carbonyl compounds. J. Org. Chem 1997, 62, 6974–6977. [Google Scholar]
- 69.(a) Armstrong A; Brackenridge I; Jackson RF; Kirk JM, A new method for the preparation of tertiary butyl ethers and esters. Tetrahedron Lett. 1988, 29, 2483–2486. [Google Scholar]; (b) Lin LS; Lanza T; de Laszlo SE; Truong Q; Kamenecka T; Hagmann WK, Deprotection of N-tert-butoxycarbonyl (Boc) groups in the presence of tert-butyl esters. Tetrahedron Lett. 2000, 41, 7013–7016. [Google Scholar]
- 70. The structure, relative stereochemistry, and the cis amide conformation of 66 were confirmed with a singlecrystal X-ray structure determination conducted on crystals grown from methanol (colorless plates). The structure has been deposited with the Cambridge Crystallographic Data Center (CCDC 2014740).
- 71.Losey HC; Peczuh MW; Chen Z; Eggert US; Dong SD; Pelczer I; Kahne D; Walsh CT, Tandem action of glycosyltransferases in the maturation of vancomycin and teicoplanin aglycones: Novel glycopeptides. Biochemistry 2001, 40, 4745–4755. [DOI] [PubMed] [Google Scholar]
- 72.Losey HC; Jiang J; Biggins JB; Oberthür M; Ye X-Y; Dong SD; Kahne D; Thorson JS; Walsh CT, Incorporation of glucose analogs by GtfE and GtfD from the vancomycin biosynthetic pathway to generate variant glycopeptides. Chem. Biol 2002, 9, 1305–1314. [DOI] [PubMed] [Google Scholar]
- 73.Oberthür M; Leimkuhler C; Kruger RG; Lu W; Walsh CT; Kahne D, A systematic investigation of the synthetic utility of glycopeptide glycosyltransferases. J. Am. Chem. Soc 2005, 127, 10747–10752. [DOI] [PubMed] [Google Scholar]
- 74. Commercially available from several vendors. As of April 2020, the price of UDP-glucose is $36.23/100 mg (Medchemexpress LLC).
- 75.Oberthür M; Leimkuhler C; Kahne D, A practical method for the stereoselective generation of β−2-deoxy glycosyl phosphates. Org. Lett 2004, 6, 2873–2876. [DOI] [PubMed] [Google Scholar]
- 76.Wright PM; Seiple IB; Myers AG, The evolving role of chemical synthesis in antibacterial drug discovery. Angew. Chem. Int. Ed 2014, 53, 8840–8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Atropisomer assignments are not discussed in the text but are summarized in the Supporting Information (SI Figure S6). The data is summarized in the individual compound experimentals and copies of key 1H-1H ROESY NMR spectra displaying the data are presented.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.