

Abstract
Inflammasomes are a class of cytosolic protein complexes. They act as cytosolic innate immune signal receptors to sense pathogens and initiate inflammatory responses under physiological and pathological conditions. The NLR-family pyrin domain-containing protein 3 (NLRP3) inflammasome is the most characteristic multimeric protein complex. Its activation triggers the cleavage of pro-interleukin (IL)-1β and pro-IL-18, which are mediated by caspase-1, and secretes mature forms of these mediators from cells to promote the further inflammatory process and oxidative stress. Simultaneously, cells undergo pro-inflammatory programmed cell death, termed pyroptosis. The danger signals for activating NLRP3 inflammasome are very extensive, especially reactive oxygen species (ROS), which act as an intermediate trigger to activate NLRP3 inflammasome, exacerbating subsequent inflammatory cascades and cell damage. Vascular endothelium at the site of inflammation is actively involved in the regulation of inflammation progression with important implications for cardiovascular homeostasis as a dynamically adaptable interface. Endothelial dysfunction is a hallmark and predictor for cardiovascular ailments or adverse cardiovascular events, such as coronary artery disease, diabetes mellitus, hypertension, and hypercholesterolemia. The loss of proper endothelial function may lead to tissue swelling, chronic inflammation, and the formation of thrombi. As such, elimination of endothelial cell inflammation or activation is of clinical relevance. In this review, we provided a comprehensive perspective on the pivotal role of NLRP3 inflammasome activation in aggravating oxidative stress and endothelial dysfunction and the possible underlying mechanisms. Furthermore, we highlighted the contribution of noncoding RNAs to NLRP3 inflammasome activation-associated endothelial dysfunction, and outlined potential clinical drugs targeting NLRP3 inflammasome involved in endothelial dysfunction. Collectively, this summary provides recent developments and perspectives on how NLRP3 inflammasome interferes with endothelial dysfunction and the potential research value of NLRP3 inflammasome as a potential mediator of endothelial dysfunction.
Subject terms: Cell biology, Cardiovascular diseases
Facts
NLRP3 inflammasome is involved in a wide range of pathological conditions and diseases as intracellular innate immune sensors.
NLRP3 inflammasome-mediated inflammation and pyroptosis play a pivotal role in endothelial dysfunction.
Noncoding RNA, as an emerging disease biomarker, may regulate endothelial function by mediating the NLRP3 inflammasome signaling pathway.
Certain drugs, such as statins, hypoglycemic agents, and other anti-inflammatory or antioxidant drugs, can improve vascular dysfunction by inhibiting the NLRP3 inflammasome signaling pathway.
Open questions
What is the molecular mechanism underlying NLRP3 inflammasome-related endothelial dysfunction?
How does NLRP3 inflammation perceive information about different mediators?
Whether different agonists affect endothelial function through a single cascade of events or through distinct pathways to activate NLRP3 inflammation?
Do other inflammasomes such as NLRP1, NLRC4, NLRP6, AIM2, noncanonical inflammasomes, and upstream pathways affect endothelial function?
Introduction
The innate immune system is the primary mechanism by which most organisms respond immediately to infections or injury. Pattern-recognition receptors (PRRs) in the host are activated, which recognize molecules released by pathogens or damaged cells. These molecular signals are known as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs)1. The PRR family has many members, including Toll-like receptors (TLRs), NOD-like receptors (NLRs), C-type lectin receptors, retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), as well as several intracellular DNA sensors2. Innate immune cells play critical roles in PRR-initiated innate inflammatory response, but the effect of nonimmune cells, such as endothelial cells (ECs), is also a force to be reckoned with3. The transcriptional upregulation of pro-inflammatory genes by PRRs triggers a cascade of inflammatory responses. Although inflammation has the beneficial effect of limiting cellular and organ damage, disruption in its regulation may result in a sustained inflammatory response and ultimately, local or systemic inflammation4.
The inflammasome, a type of PRR, constitutes an essential component of the innate immunity5. Abnormal activation of inflammasomes is the pathogenesis of various inflammatory diseases6. The inflammasome is a high-molecular-weight protein complex that acts as a cytosolic innate immune signaling receptor that senses PAMPs or DAMPs and mediates a highly inflammatory state. The first inflammasome was discovered in 20025 following which, various inflammasomes have been identified, including NLR-family pyrin domain-containing protein (NLRP) 1, NLRP3, NLRP6, NLR-family caspase recruitment domain (CARD)-containing protein 4 (NLRC4), absent in melanoma 2 (AIM2), and pyrin inflammasomes7. Among them, the NLRP3 inflammasome is the most well-characterized, largest multimeric protein complex to date.
Microvascular ECs at a site of inflammation are both active participants and regulators of inflammatory processes. During the transition from acute inflammation to chronic inflammation or from innate immunity to adaptive immunity, the characteristics of ECs change through EC activation, rapid recruitment of neutrophils, and increased vascular leakage of plasma proteins. These changes eventually lead to endothelial dysfunction8. Interleukin (IL)-1β is an important pro-inflammatory cytokine released during the endothelial inflammatory response9. Interestingly, activation of NLRP3 inflammasome can produce high numbers of IL-1β.
Additionally, high-quality research has shown that NLRP3 inflammasome activation is correlated with multiple chronic inflammatory diseases and metabolic disorders, including obesity, hypertension, diabetes, atherosclerosis, neuroinflammation, retinopathy, stroke, and cancer10–16. These diseases also have a close connection with the dysregulation of the endothelium17. These studies indicate that the activation of NLRP3 inflammasome in ECs under pathophysiological conditions may aggravate endothelial dysfunction, leading to various diseases. In recent years, endothelial function research has increasingly focused on the regulatory role of NLRP3 inflammasome. In this review, we attempt to summarize the latest progress and development trends in the NLRP3 inflammasome research, highlighting its role in redox regulation and endothelial dysfunction. Also, we emphasize the current knowledge of the factors that mediate and influence NLRP3-related endothelial dysfunction and potential therapeutic targets, which may be helpful for the treatment of endothelial dysfunction-related diseases.
Inflammasome family
The inflammasome is a molecular platform that drives effector caspase-1 activation, which is assembled by NLRs, AIM2-like receptors (ALRs), or pyrin that can directly or indirectly (via the adaptor apoptosis-associated speck-like protein containing a CARD (ASC)), activate caspase-1 (Fig. 1)18. Structurally, the N-terminal domain of these sensors includes a CARD or a pyrin domain (PYD). The adaptor ASC is composed of a PYD and a CARD, while caspase-1 contains a CARD19. Recently, studies have elucidated that NLRP1, NLRP3, NLRP6, AIM2, and pyrin carry a PYD in their N-terminal region, whereas NLRP1b and NLRC4 contain a CARD. The sensor protein, containing a PYD, binds to the PYD of ASC, allowing the ASC to activate caspase-1 by interacting with the CARD of pro-caspase-1. In contrast, a CARD-containing sensor protein may activate caspase-1 by directly binding to the CARD of pro-caspase-1 without ASC20. However, the presence of ASC can enhance the assembly of the sensor protein containing a CARD and the activation of caspase-1 (Fig. 2)21.
Fig. 1. Domain structure of representative inflammasome.

Inflammasome is a protein complex formed by the aggregation of inflammasome sensor, adaptor protein ASC, and effector protein caspase-1. PYD pyrin domain, NBD nucleotide-binding domain, LRR leucine-rich-repeat domain, FIIND function-to-find domain, CARD caspase activation and recruitment domain, C–C coiled-coil domain, B B-box domain, BIR baculovirus inhibitor of apoptosis repeat, ASC apoptosis-associated speck-like protein containing a CARD, CASP1 caspase-1.
Fig. 2. The canonical inflammasome activation pathway occurs by sensing diverse pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs).

a NLRP3, NLRP6, AIM2, and pyrin, containing a PYD, binds to the PYD of ASC, allowing the ASC to activate caspase-1 by interacting with the CARD of pro-CASP1. Activated caspase-1 triggers the cleavage of pro-interleukin (IL)-1β, pro-IL-18, and gasdermin D (GSDMD), and then releases the N-terminal domain of GSDMD to induce pyroptosis, followed by the release of IL-1β and IL-18. b NLRP1b and NLRC4, containing a CARD, activate caspase-1 by directly binding the CARD of pro-caspase-1 without ASC or binding the paired ASC scaffold. The presence of ASC can enhance the assembly of the sensor protein containing a CARD and the activation of caspase-1. The molecular process after caspase-1 activation is the same as part A.
Furthermore, different sensor proteins respond to different activators (Fig. 2). NLRP1 inflammasome is activated by specific pathogens, such as anthrax lethal toxin, muramyl dipeptide (MDP), Toxoplasma gondii, Shigella flexneri, and Listeria monocytogenes, which cause proteolysis of the NLRP1 N terminal21,22. NLRC4 inflammasome can be activated by bacterial ligands, including flagellin and components of the type III secretion system (T3SS), which are sensed and bound by NOD-like receptor family apoptosis-inhibitory protein (NAIP) to induce NLRP4 oligomerization23. NLRP6, one of the newest and less-researched members of the NLR family, is overexpressed explicitly in human and mouse intestinal epithelial cells. Microbial metabolites secreted by intestinal commensal bacteria can modulate NLRP6 activity and IL-18 secretion24. Moreover, certain pathogens, such as Listeria monocytogenes, Salmonella typhimurium, and Staphylococcus aureus, have been shown to activate the NLRP6 inflammasome as part of the host defense25. AIM2 acts as a cytoplasmic sensor that can detect and directly bind to double-stranded (ds) DNA of any sequence produced by pathogens or cellular perturbations, inducing the capability of self-oligomerization and then recruiting ASC for activation of inflammasome26. In 2004, pyrin was first shown to self-assemble into an inflammasome by recognizing the inactivation of RhoA GTPase, a molecular switch that controls the dynamics of the cytoskeleton27. Recently, studies found that bacterial toxins can modify the Switch-I region of RhoA and trigger pyrin activation, such as TcdA and TcdB secreted from Clostridium difficile27. Additionally, a most recent study demonstrated that bile acid analogs of microbial origin, such as BAA473 and BAA485, can also activate the pyrin inflammasome28. NLRP3 inflammasome is triggered by numerous stimuli, including endogenous molecules, crystalline substances, pathogenic microbes, and ATP. NLRP3 inflammasome functions as a signal integrator to sense several cellular signals, including ion fluxes such as potassium (K+) efflux and calcium (Ca2+) influx, lysosomal leakage, mitochondrial dysfunction, and ROS production29.
Overall, different inflammasomes play unique roles in the defense against specific pathogens. Overactivation of inflammasomes can lead to significant inflammatory responses and pathological changes that are closely related to autoimmunity and autoinflammation. Therefore, inflammasomes are involved in a wide range of pathological conditions and diseases as intracellular innate immune sensors for conditions like infection or aseptic inflammation, metabolic syndrome, age-related chronic inflammatory disease, and autoimmune disease7,10.
NLRP3 inflammasome activation
NLRP3 inflammasome contains innate immune sensor NLRP3, adaptor molecule ASC, and effector protease pro-caspase-130. The activation of NLRP3 inflammasome is essentially the autocatalytic activation of caspase-1. Once activated, NLRP3 acts as a sensor molecule that occurs in self-oligomerization and recruits ASC via homotypic PYD–PYD interaction, which induces the assembly of ASC into large speck-like structures. Subsequently, aggregated ASC recruits pro-caspase-1 via CARD–CARD contact, leading to autocatalytic activation of caspase-1. The function of activated caspase-1 heterotetramers is proteolytic activation of the pro-inflammatory cytokines IL-1β and IL-18, and of a soluble cytosolic protein gasdermin D (GSDMD). Upon proteolysis, the oligomerized gasdermin N can bind membrane lipids and form membrane pores to mediate the nonconventional secretion of IL-1β and IL-18. In parallel, cells undergo a pro-inflammatory type of cell death known as pyroptosis17,31,32.
Multiple cellular signals are thought to account for NLRP3 inflammasome activation, including ion fluxes such as K+ efflux, Cl− efflux, Ca2+ influx, and Na+ influx, lysosomal leakage, mitochondrial dysfunction, and ROS production33. These various upstream signaling pathways may be interrelated or independent (Fig. 3). Additionally, noncanonical NLRP3 inflammasome activation and alternative NLRP3 inflammasome activation are two other forms of activation34. Noncanonical NLRP3 inflammasome activation is induced by cytosolic LPS that is sensed by noncanonical caspases 4/5 in humans and caspase-11 in mice. Subsequently, caspases 4/5/11 are activated to cleave GSDMD, resulting in K+ efflux and pyroptosis35. Alternative NLRP3 inflammasome activation is species-specific, as it only occurs in human and porcine monocytes. This activation is induced by TLR4–TIR-domain-containing adaptor-inducing interferon-β (TRIF)–receptor-interacting serine/threonine-protein kinase 1 (RIPK1)–Fas-associated protein with death domain (FADD)–CASP8 signaling pathway. Of note, this pathway does not depend on K+ efflux and does not induce ASC speck formation and pyroptosis36.
Fig. 3. Reactive oxygen species (ROS) contribute to the NLRP3 inflammasome activation in endothelial cells.

A wide range of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) trigger NLRP3 inflammasome activation by inducing potassium (K+) efflux, calcium (Ca2+) influx, lysosomal leakage, mitochondrial dysfunction, and ROS production. ROS mainly derived from endoplasmic reticulum (ER) stress, damaged mitochondria, and NADPH oxidase. ER stress could activate the NF-κB, TXNIP, and SREBP signaling pathways, Ca2+ release, and ROS production. Moreover, athero-prone flow can also mediate SREBP signaling pathways. The release of mtROS and mtDNA in damaged mitochondria might activate NLRP3 inflammasome. Furthermore, mitochondrial antiviral signaling protein (MAVS) is capable of molulating the recruitment and localization of NLRP3. However, NF-E2-related factor 2 (Nrf2) activated under ROS-induced stress conditions can inhibit NLRP3 inflammasome activation. Taken together, ROS is an intermediate factor involved in multiple signaling pathways and can trigger the activation of NLRP3 inflammasome.
Among the aforementioned multiple activation mechanisms, ROS bridges the interaction between NLRP3 inflammasome and endothelial dysfunction. Specifically, ROS are one of the first intermediates produced by many known NLRP3 inflammasome activators, and are involved in the mechanisms that trigger the formation and activation of the NLRP3 inflammasome, thereby promoting tissue inflammation and activating immune response37. Thioredoxin-interacting protein (TXNIP), nuclear factor-kappaB (NF-κB), and the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) are proteins involved in the response to oxidative stress, which links ROS to NLRP3 activation (Fig. 3)38. Moreover, aberrant production of ROS may increase nitric oxide (NO) catabolism, leading to a decrease in NO bioavailability. This imbalance in NO–ROS leads to the expression of inflammation-related genes and upregulation of inflammatory proteins, which in turn destroys endothelial function39.
ROS production in the mitochondria is the primary source of cellular ROS. Indeed, mitochondrial ROS (mtROS) are involved in NLRP3 inflammasome activation (Fig. 3). The endothelial mitochondria act as critical signaling organelles that play a crucial role in endothelial function, including subcellular location, dynamics, biogenesis, mitophagy, autophagy, ROS production, calcium homeostasis, regulation of EC death, and heme synthesis40. Intriguingly, the accumulation of damaged mitochondria has also been proposed to be essential for NLRP3 activation. In addition to increased mtROS, the exposure of mtDAMPs, such as mtDNA and cardiolipin to the cytosol, alterations in metabolite levels, and mitochondrial antiviral signaling protein, can also contribute to NLRP3 activation38.
ROS also bridge the interaction between endoplasmic reticulum (ER) stress and NLRP3 inflammasome activation (Fig. 3). ER stress has been proven to activate NLRP3 inflammasome and promote the development of endothelial dysfunction. Specifically, ER stress-induced protein misfolding, imbalance of sterol synthesis and distribution, and release of Ca2+ and ROS can trigger NLRP3 inflammasome activation41. Meanwhile, multiple molecular pathways during severe ER stress are also involved in the activation of the NLRP3 inflammasome, including p38 mitogen-activated protein kinase (MAPK) pathway, Jun-N-terminal kinase (JNK) signaling, X-box-binding protein-1 (XBP1), CCAAT/enhancer-binding protein–homologous protein (CHOP), NF-κB, and TXNIP signaling pathways, which are terminal signals in the unfolded protein response (UPR). The function of UPR is to detect protein misfolding to restore ER homeostasis under cellular perturbations41. Additionally, several studies have reported that ER, as an intracellular metabolic regulator, plays a pivotal role in endothelial dysfunction42–44. ER stress also acts as an active inflammatory trigger, accelerating the progression of endothelial dysfunction via inflammation and oxidative stress45. Besides, ER Ca2+ released from stressed cells increases the mtROS production and amplifies the activation of the NLRP3 inflammasome46. Accordingly, the mutual promotion between mitochondrial dysfunction and ER stress contributes to the activation of NLRP3 inflammasome and endothelial dysfunction, both of which are involved in oxidative stress and the production of ROS.
Implications of NLRP3 inflammasome activation in endothelial dysfunction
NLRP3 inflammasome activation in endothelial inflammation
Generally, microbes, exogenous and endogenous crystals, and metabolic dysbiosis could trigger NLRP3 inflammasome activation, which initiates the secretion of mature forms of IL-1β and IL-18 from cells to promote further inflammatory processes and oxidative stress in the endothelium (Fig. 4)47. This intense inflammasome activation increases the release of new DAMPs, forming a negative-feedback loop48. The cytokine output of inflammasomes, specifically IL-1β, is a key inflammatory mediator in response to microbial invasion and tissue damage. EC is a target cell of IL-1β, and it also produces IL-1β during inflammation49. The activation of IL-1β can trigger the activation of secondary inflammatory mediators, such as IL-6 and C-reactive protein50. Moreover, IL-1β can also promote the secretion of adhesion molecules and chemokines in ECs, inducing a potent pro-inflammatory response50. Endothelial inflammation may initiate the occurrence and progression of endothelial dysfunction and promote one another in subsequent processes.
Fig. 4. The role of NLRP3 inflammasome activation in endothelial dysfunction.

a The secretion of mature forms of IL-1β, IL-18, and HMGB1 is the result of activation of NLRP3 inflammasome. These mediators possess properties of pro-inflammatory activation. Therefore, NLRP3 inflammasome can induce a potent inflammatory response, oxidative stress, and pro-inflammatory cell death called pyroptosis. b Inflammation and oxidative stress can induce endothelium DNA damage, activate NF-κB signaling pathway, increase P53/P21/P16 transcription, and inhibit autophagy, which may promote the process of endothelial cell senescence. Senescent cells can secrete senescence-associated secretory phenotype (SASP), including pro-inflammatory mediators, which may promote endothelial dysfunction and eventually lead to vascular sclerosis. c IL-1β binding to its cell surface receptor IL-1 receptor 1 (IL-1R1) recruits IL-1 receptor accessory protein (IL-1RacP) to activate intracellular signaling molecules, including myeloid differentiation factor 88 (MyD88), IL-1 receptor-associated kinase 1/4 (IRAK1/4), and TNF receptor-associated factor (TRAF), which then causes NF-κB activation. IL-18 binding to its cell surface receptor IL-18 receptor α chain (IL-18Rα) recruits IL-18 receptor β chain (IL-18Rβ) to activate similar intracellular signaling molecules. HMGB1 can also activate the NF-κB signaling pathway, which is downstream of toll-like receptor (TLR)2/4 activation. The activation of NF-κB signaling pathway increases the secretion of pro-inflammatory mediators such as cytokines and chemokines to mediate the adhesion of leukocyte and promote leukocyte extravasation. Additionally, the binding of HMGB1 to the RAGE receptor leads to the activation of downstream p38 MAP kinase, resulting in phosphorylation of the actin-binding protein Hsp27 and caldesmon, which causes actin stress fibers to form, cytoskeletal remodeling, and endothelial contraction. All of these reactions increase endothelial permeability by altering cell contractility and disrupting intercellular connections.
A growing number of agonists have been demonstrated to promote vascular inflammation by activating the NLRP3 inflammasome, including the endogenous medium. For example, visfatin has been demonstrated to promote endothelial inflammation as a pro-inflammatory adipokine51. Xia et al. found that NLRP3 inflammasome activation is an underlying cause of visfatin-induced endothelial inflammation52. This inflammatory response can cause endothelial dysfunction, initiating atherosclerosis during obesity. Moreover, visfatin has also been shown to increase neointimal formation both in vitro and in vivo, which is partly attributed to the formation and activation of NLRP3 inflammasome and IL-1β secretion52.
NLRP3 inflammasome activation is also involved in exogenous substance-mediated endothelial inflammation. Xia et al. demonstrated that NLRP3 inflammasome activation is involved in tetrachlorobenzoquinone (TCBQ)-induced endothelial inflammation53. TCBQ not only promotes the post-translational activation of NLRP3 and IL-1β secretion in HUVECs, but also downregulates the ubiquitination of NLRP3, which facilitates its activation, as an active metabolite of pentachlorophenol (PCP). This study also reported that K+ efflux, mtROS production, and mtDNA damage might contribute to TCBQ-induced NLRP3 inflammasome activation53. Further research revealed that GSDMD and mixed-lineage kinase domain-like (MLKL) play crucial roles in TCBQ-induced atypical inflammatory pathway activation54. The results showed that TCBQ aggravated NLRP3 inflammasome activation via the disruption of intracellular and extracellular ion homeostasis, which was caused by the leakage of GSDMD and MLKL and the release of cell contents54. Many agonists that target the NLRP3 inflammasome signaling pathway and facilitate endothelial dysfunction are listed in Table 1.
Table 1.
Summary of agonists targeting NLRP3 inflammasome signaling pathway and facilitating endothelial dysfunction.
| Agonist | Category | Effects and potential mechanisms | Disease | References |
|---|---|---|---|---|
| TMAO | Choline metabolite |
•↑ Endothelial inflammation •↑ ROS–TXNIP–NLRP3 pathway •↓ eNOS expression •↓ NO roduction |
AS | 124 |
| Visfatin | Adipokine |
•↑ Endothelial inflammation •↑ Neointimal formation •↑ NLRP3 formation and activation |
Obesity | 52 |
| Acetate, propionate | SCFAs |
•↑ Endothelial inflammation •↑ Neointimal formation •↑ NLRP3 formation and activation |
AS | 125 |
| TCBQ | Toxicant |
•↑ Endothelial inflammation •↑ NLRP3 activation, IL-1β secretion •↑ NLRP3 deubiquitination •↑ GSDMD and MLKL leakage |
/ | 53,54 |
| LCWE | L. casei cell wall extract |
•↑ Endothelial inflammation •↑ Lysosome membrane permeability, cathepsin B release •↑ NLRP3 formation and activation |
Coronary arteritis | 126 |
| Heme | Iron porphyrins |
•↑ Endothelial inflammation •↑ NLRP3 activation, IL-1β secretion |
Hemolytic diseases | 127 |
| Endothelial GPR124 | Orphan receptor |
•↑ Atherosclerosis progression •↑ Nitrosative stress •↑ NLRP3 activation |
AS | 128 |
| AGEs | Glycation products |
•↑ Endothelial inflammation •↑ ROS/TXNIP/NLRP3 pathway •↓ EPC numbers, NO level, and eNOS expression |
Chronic kidney disease | 129 |
| SREBP2 | Transcription factor |
•↑ Endothelial inflammation •↑ The production of toxic lipids, cholesterol crystals, and necrotic cells •↑ NLRP3 activation |
AS | 130 |
| Adora2a | Adenosine receptor |
•↑ Cerebral endothelial inflammation •↑ NLRP3 activation |
Ischemic stroke | 131 |
| Prdx6-aiPLA2 activity | Enzyme |
•↑ Endothelial inflammation •↑ Endothelial NOX2 activation •↑ ROS/NF-κB/NLRP3 pathway |
ALI | 132 |
| PGE2 | Prostaglandin |
•↑ Endothelial inflammation, apoptosis •↑ NLRP3 activation •↑ Inflammatory chemokines |
Diabetic retinopathy | 133 |
| Acrolein | Unsaturated aldehyde |
•↑ Pyroptosis •↓ Cell migration •↓ ROS-dependent autophagy •↑ NLRP3 activation |
AS | 57 |
| Cadmium | Metal toxicant |
•↑ Endothelial inflammation, pyroptosis •↑ mtROS-mediated NLRP3 activation |
CVD | 58 |
| Nicotine | Alkaloid |
•↑ Endothelial inflammation, pyroptosis •↑ ROS production, NLRP3 activation |
AS | 134 |
|
•↑ Endothelial barrier dysfunction •↑ Cathepsin B-dependent NLRP3 activation •↑ HMGB1 release |
69 |
NLRP3 inflammasome activation in endothelial cell death
Typically, cell death mainly includes apoptosis and necrosis; however, an emerging type of cell-intrinsic death has recently been found, termed pyroptosis55. Pyroptosis, an inflammatory programmed cell death process, is another functional outcome of NLRP3 inflammasome activation, which is mediated by the membrane pore-forming activity of the GSDMD N-terminal domain released by caspase-1 cleavage56. Pyroptosis is manifested by cell membrane rupture, cellular lysis, and release of pro-inflammatory contents, including IL-1β, IL-18, as well as high-mobility group box 1 (HMGB1), signifying the inflammatory nature of pyroptosis, and distinguishing pyroptosis from other forms of cell death55. NLRP3 inflammasome-mediated pyroptosis has been identified as a potential cause of EC death (Fig. 4).
Our previous study reported that NLRP3-mediated pyroptosis is involved in EC death caused by certain stimuli. Acrolein, an exogenous toxin, is produced in response to environmental pollution. Jiang et al. reported that acrolein-induced EC death is attributed to a decrease in ROS-dependent autophagy, which promotes NLRP3 inflammasome activation and pyroptosis in HUVECs. It is generally known that autophagy is a negative regulator of NLRP3 inflammasome activation. Consequently, NLRP3 inflammasome activation plays a crucial role in this process57. Chen et al. illustrated a new molecular mechanism of EC death and inflammation caused by cadmium (Cd), an independent risk factor for various cardiovascular diseases. Cd exposure induces EC death and inflammatory response via mitochondrial ROS-mediated NLRP3 activation and pyroptosis in HUVECs58.
A recent study reported that genetic copy number variations (CNVs) might be linked to the activation of NLRP3 inflammasome and endothelial pyroptosis. Chen et al. found that the human neutrophil peptide (HNP)-encoding gene (DEFA1/DEFA3) CNVs play specific roles in sepsis59. Interestingly, they reported that transgenic mice carrying more copies of the DEFA1/DEFA3 suffer from more severe sepsis and mortality than those with low copy numbers of DEFA1/DEFA3 and wild-type mice, which is caused by broader endothelial barrier dysfunction and EC pyroptosis. Also, in a mouse lung microvascular endothelial cell line model, it was reported that HNP-1 induces EC pyroptosis and endothelial barrier dysfunction by directly targeting the purinergic receptor P2X ligand-gated ion channel 7 (P2X7) and subsequently activating the canonical NLRP3/caspase-1 pathway. Functional blockade of HNP1–3 alleviates endothelial pyroptosis and protects the mice from sepsis59. This study provided a novel molecular mechanism for NLRP3 activation to mediate endothelial pyroptosis.
NLRP3 inflammasome activation in endothelial barrier dysfunction
The vascular ECs cover the intima of blood vessels, forming a semipermeable barrier between circulating blood and extravascular matrix. This barrier maintains the transport of solutes, fluids, and cells by coordinating the opening and closing of cell junctions60. The endothelial barrier dysfunction is characterized by the loss of local contact between ECs, and extravasation of plasma proteins, cells, or solutes61. Sustained inflammatory activation of the endothelium initiates the secretion of various inflammatory mediators, including IL-1, tumor necrosis factor (TNF), vascular endothelial growth factor, histamine, and bradykinin, leading to the internalization of VE cadherin and disruption of inter-endothelial junctions62. Therefore, endothelial barrier dysfunction is considered to be an important mechanism underlying many diseases associated with inflammation, where inflammatory mediators activate diverse signaling pathways in the endothelium, resulting in the increased endothelial permeability and inappropriate loss of solutes and cells62.
Moreover, IL-1β, IL-18, and HMGB1 released by NLRP3 activation may activate NF-κB pathway, which in turn promotes the transcriptional activation of chemokines and adhesion molecules, increases leukocyte adhesion, and changes in cell permeability, thereby playing a crucial role in endothelial barrier dysfunction. IL-1β and IL-18 are products of NLRP3 inflammasome activation. As inflammatory mediators, they can enhance the expression of adhesion molecules and chemokines in the endothelium via binding to their cell surface receptors respectively, and activating the NF-κB pathway (Fig. 4)63. Endothelial NF-κB activation initiates transcriptional activation of numerous genes, including chemokines and adhesion molecules64. These reactions can increase the adhesion and rolling of leukocyte and promote leukocyte extravasation. Additionally, activated leukocytes can secrete cytokines that mediate persistent inflammatory responses, such as IL-1 or TNF, which can increase vascular endothelial permeability by altering cell contractility and disrupting intercellular connections62.
HMGB1 is closely related to NLRP3 inflammasome activation and endothelial barrier dysfunction (Fig. 4). NLRP3 inflammasome activation can also release DAMPs such as HMGB1 to the extracellular environment65. Extracellular release of HMGB1 may be caused by noncanonical mechanisms along with IL-1β and IL-18, or it may be a passive consequence of cell lysis during pyroptosis66. Extracellular HMGB1 can bind to its receptors, including toll-like receptor 2 (TLR2), TLR4, and RAGE, thereby inducing inflammation and repair responses66. Moreover, HMGB1 can induce endothelial barrier disruption by increasing the contractile activity and endothelial permeability. These effects are mediated by binding of HMGB1 to the RAGE receptor, leading to downstream p38 MAPK activation as well as actin-binding protein Hsp27 phosphorylation67. Interestingly, HMGB1 was also demonstrated to increase the secretion of pro-inflammatory mediators such as cytokines and chemokines via mediating NF-κB signaling pathway, at the downstream of TLR2 and TLR4 activation. This may indirectly cause endothelial barrier dysfunction. However, this needs to be thoroughly investigated in the future67.
Previous studies have unraveled that endothelial barrier dysfunction induced by certain risk factors is linked to NLRP3 inflammasome activation, which is achieved via the release of HMGB1. Chen et al. reported that the activation of NLRP3 inflammasome and subsequent release of HMGB1 are the underlying causes of intercellular junction fracture in mouse vascular ECs (MVECs) treated with high glucose (HG)68. In contrast, NLRP3 deficiency has a protective effect on intercellular junction interruption in the coronary arterial ECs of diabetic mice68. Zhang et al. revealed that NLRP3 inflammasome is involved in triggering nicotine-induced endothelial barrier dysfunction69. This study suggested that nicotine enhances NLRP3 inflammasome activation and causes the release of HMGB1, leading to the damage of inter-endothelial junction and hyperpermeability69.
NLRP3 inflammasome activation in endothelial cells’ senescence
Tissue aging is often accompanied by chronic inflammation, and the same goes for endothelial senescence70. By inducing vascular structural and functional changes, EC senescence actively regulates aging-associated vascular dysfunction (Fig. 4)71. Specifically, the disruption of the cell cycle increases ROS and oxidative stress, vascular inflammation, causes impaired Ca2+ signaling, high serum uric acid levels, and activates renin–angiotensin–aldosterone system, which is closely related to the premature senescence of ECs71. Recently, NLRP3 inflammasome has been demonstrated to correlate mild systemic inflammation with age-related functional decline72. NLRP3 inflammasome can sense the accumulation of DAMPs during aging and mediate the pro-inflammatory cascade both inside and outside the brain, which is independent of the noncanonical caspase-11 pathway72. Inhibiting abnormal NLRP3 inflammasome activity during aging reduces age-associated innate immune activation, attenuates multiple age-related chronic diseases, and prolongs the health span, consistent with the previous results72. Furthermore, Youm et al. showed that NLRP3 inflammasome activation also causes age-related thymic involution and immunosenescence73.
A most recent study demonstrated that NLRP3 inflammasome-mediated IL-1β has a pathogenic role in multiple distinct ocular aging diseases74. Nevertheless, little is known about whether NLRP3 inflammasome contributes to EC senescence and the underlying molecular mechanisms. Yin et al. explored the roles and potential mechanisms of NLRP3 inflammasome in EC senescence75. It was found that NLRP3 inflammasome activation promotes bleomycin-induced EC senescence by increasing the IL-1β secretion. Additionally, secreted IL-1β also significantly upregulates the expression of the senescence-related marker p53/p21 protein. During these processes, ROS plays a key role in inducing TXNIP–NLRP3 interaction75. The molecular mechanism and role of NLRP3 inflammasome in endothelial senescence remain to be further studied.
Recently, some pieces of literature have reported that some beneficial substances can reduce the senescence of ECs, at least in part, by inhibiting NLRP3 activation. Sun et al. demonstrated that purple sweet potato color (PSPC), one type of flavonoid derived from purple sweet potato, ameliorates endothelium senescence by restraining ROS production and NLRP3 inflammasome activation, and then reduces atherosclerotic lesions in insulin-resistant mice76. Additionally, they further investigated the correlation between NLRP3 inflammasome and autophagy and the underlying mechanisms of EC senescence. It was found that PSPC inhibits endothelial senescence via enhancing autophagy and keeping inflammasome activation in check77. With the clarification of the molecular mechanism and role of NLRP3 inflammasome in EC aging in the future, we believe that more drugs will target the NLRP3 inflammasome signaling pathway to treat endothelial aging-related diseases.
Noncoding RNAs (ncRNAs) targeting the NLRP3 inflammasome signaling pathway in endothelial dysfunction
ncRNAs are a broad spectrum of RNA molecules that have high transcriptional activity, and regulatory and structural functions, but do not encode proteins. ncRNAs, such as microRNAs (miRNAs), long noncoding RNAs (lncRNAs), and circular RNAs (circRNAs), are novel regulators of cardiovascular risk factors and cell functions78–81. Additionally, ncRNAs have been demonstrated to precisely control gene expression and have gradually become indispensable regulators of inflammation and immunity82,83. Several studies have provided evidence that ncRNAs may regulate endothelial function by mediating the NLRP3 inflammasome signaling pathway; these studies were summarized in this section (see Table 2).
Table 2.
Summary of ncRNAs targeting NLRP3 inflammasome signaling pathway in endothelial dysfunction.
| ncRNA | Target gene | Effect on NLRP3 activation | Functions | Disease/research context | References |
|---|---|---|---|---|---|
| miR-22 | NLRP3 | Down |
•↓ EC apoptosis •↓ Pro-inflammatory cytokines •↑ Tube formation |
Coronary atherosclerosis | 135 |
| miR-495 | NLRP3 | Down |
•↓ EC inflammation •↓ EC apoptosis •↑ EC proliferation |
Myocardial ischemia/reperfusion injury | 136 |
| miR-15a | FOXO1 | Down | •↓ EC inflammation | Diabetic retinopathy | 137 |
| miR-126 | HMGB1 | Down | •↓ EC inflammation | Diabetic retinopathy | 138 |
| miR-590-3p | NLRP1, NOX4 | Down | •↓ EC inflammation, pyroptosis | Diabetic retinopathy | 139 |
| miR-20a | TXNIP, TLR4 | Down |
•↓ EC inflammation •↓ ROS generation |
Atherosclerosis | 140 |
| miR-30c-5p | FOXO3 | Down | •↓ EC inflammation, pyroptosis | Atherosclerosis | 141 |
| miR-383-3p | IL1R2 | Down |
•↓ EC inflammation •↓ EC apoptosis •↑ Tube formation |
Coronary atherosclerosis | 142 |
| miR-20b | TXNIP | Down | •↓ EC senescence | H2O2-induced EC senescence | 143 |
| miR-101-3p | Bim | Down | •↓ EC apoptosis | Serum deprivation-induced EC apoptosis | 144 |
| miR-200a-3p | Keap1, NLRP3 | Up | •↑ EC inflammation | Sepsis-induced brain injury | 145 |
| miR-92a | SIRT1, KLF2, and KLF4 | Up |
•↑ Endothelial innate immunity •↓ NO bioavailability |
Atherosclerosis | 146 |
| miR-125a-5p | TET2 | Up | •↑ EC inflammation, pyroptosis | Atherosclerosis | 147 |
| LncRNA MALAT1 | miR-22 | Up | •↑ EC inflammation, pyroptosis | Atherosclerosis | 148 |
| LncRNA MEG3 | miR-223 | Up | •↑ EC pyroptosis | Atherosclerosis | 149 |
| LncRNA NEXN-AS1 | / | Down | •↓ EC pyroptosis | Atherosclerosis | 94 |
| Hsa_circ_0068087 | miR-197 | Up |
•↑ EC inflammation •↑ Tube formation |
T2DM | 150 |
Some exploratory research has demonstrated the potential of ncRNAs as clinical biomarkers or drug targets in various diseases78,84–86. Additionally, the distinctive roles of ncRNAs in the regulation of endothelial function have been widely explored87–92. Nevertheless, research on ncRNAs regulating endothelial function via the NLRP3 inflammasome signaling pathway is still limited (Fig. 5). ncRNAs tested in clinical samples have more potential as disease biomarkers in clinical applications. A great deal of clinical validation and mechanistic research is needed to elucidate the roles of ncRNAs regulating NLRP3 inflammasome signaling pathway in endothelial function.
Fig. 5. The involvement of noncoding RNAs (ncRNAs) in regulating NLRP3 inflammasome-mediated endothelial dysfunction and diseases.
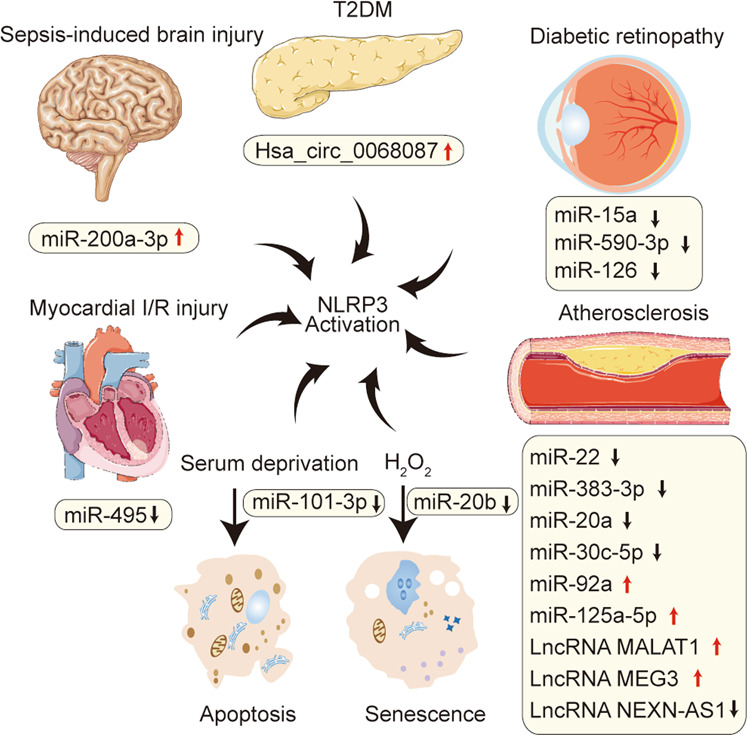
Schematic representation of ncRNAs upregulated or downregulated in multiple endothelial dysfunction-related disease models. These ncRNAs mediate endothelial dysfunction by directly or indirectly activating the NLRP3 inflammasome signaling pathway. The reported diseases mainly include myocardial ischemia/reperfusion (I/R) injury, sepsis-induced brain injury, type 2 diabetes mellitus (T2MD), diabetic retinopathy, and atherosclerosis. Moreover, miR-20b can be downregulated in H2O2-treated endothelial cells (ECs) and induces EC senescence by activating NLRP3 inflammasome. In addition, miR-101-3p is downregulated in serum deprivation-induced EC apoptosis, which is partially induced by activating the NLRP3 inflammasome signaling pathway. In this figure, the red upward arrow indicates upward adjustment, and the black downward arrow indicates downward adjustment.
Clinical drugs and NLRP3 inflammasome and endothelial dysfunction-related diseases
Statins
Statins are widely applied in the clinics because of their lipid-level-lowering properties. The functions of statins have been extended to include anticoagulant, anti-inflammatory, and immunomodulatory, suggesting that statins may have other benefits besides cholesterol reduction, as numerous clinical studies showed93. Wu et al. found that atorvastatin inhibited EC pyroptosis via increasing the expression of lncRNA NEXN-antisense RNA 1 (AS1) and its cognate gene NEXN, and inhibiting the expression of NLRP3 and pyroptosis-related molecules such as caspase-1 and IL-1β94. Simvastatin has been demonstrated to ameliorate endothelial dysfunction by inhibiting the activation of NLRP3 inflammasome in HG conditions. Mechanistically, simvastatin treatment remarkably enhanced the expression of zonula occluden-1 (ZO-1) and VE cadherin by inhibiting the NLRP3 inflammasome-dependent HMGB1 release, leading to the alleviation of vascular endothelial hyperpermeability95. Additionally, simvastatin can also exert an atheroprotective action via modulating the endothelial kruppel-like factor 2 (Klf2)-forkhead box P transcription factor 1 (Foxp1)–NLRP3 inflammasome network to suppress NLRP3 inflammasome activation96. These studies shed light on new mechanisms of statins to improve atherosclerosis and diabetic angiopathy by inhibiting NLRP3 inflammasome activation.
Hypoglycemic agents
In recent years, studies have found that antidiabetic drugs can reduce endothelial disorders via blocking NLRP3 inflammasome activation, providing the potential for treating diabetic vasculopathy. The classification of these drugs based on pharmacological mechanisms is summarized as follows.
Dipeptidyl peptidase-4 (DPP-4) inhibitors are a class of effective hypoglycemic agents for the treatment of type 2 diabetes mellitus (T2DM). DPP-4 inhibitors have proven to be effective in improving endothelial function, reducing oxidative and pro-inflammatory states, suggesting beneficial effects on cardiovascular function. Qi et al. found that DPP-4 inhibitor vildagliptin ameliorates endothelial dysfunction, induced by high free fatty acid (FFA) levels through inhibiting the AMPK–NLRP3–HMGB1 pathway97. Moreover, vildagliptin also reduces the cellular lactate dehydrogenase (LDH) release and ROS levels, as well as protecting mitochondrial function and recovering eNOS levels, indicating its protective roles on endothelial dysfunction97. Recently, anagliptin, a novel DPP-4 inhibitor licensed for treating T2DM, was found to restore HG-induced endothelial dysfunction via SIRT1-dependent inhibition of NLRP3 inflammasome activation and suppression of NOX4–ROS–TXNIP–NLRP3 signaling, which suggests that anagliptin may have broad pharmacological effects in cardiovascular diseases and other metabolic disorders98.
Dulaglutide, a kind of glucagon-like peptide-1 receptor agonist (GLP-1 RA), has been approved for use in the treatment of T2DM. Luo et al. found that dulaglutide inhibits HG-induced endothelial dysfunction via SIRT1-dependent inhibition of NLRP3 inflammasome activation and expression of NOX4 and TXNIP99.
Acarbose, a well-known α-glucosidase inhibitor, is a postprandial acting antidiabetic drug. It has been reported that acarbose has protective effects against vascular endothelial dysfunction by inhibiting the production of NOX4 oxidase-dependent O2•−, which contributes to blocking NLRP3 inflammasome activation100. Furthermore, the reduced expression of NLRP3 inflammasome is also involved in the amelioration of vascular hyperpermeability by acarbose, which is attributed to the enhanced expression of junction protein ZO-1 and VE cadherin100.
Fenofibrate is a selective agonist of peroxisome proliferator-activated receptor α (PPARα) that prevents the progression of microvascular complications in T2DM. Deng et al. found that fenofibrate promotes wound healing via alleviating EPC dysfunction and stimulating angiogenesis in diabetic mice induced by streptozotocin (STZ), the effects of which are attributed to the inhibition of NLRP3 inflammasome pathway101.
Additionally, cilostazol, a phosphodiesterase-3 inhibitor, also alleviates the adverse vascular effects of living with diabetes. Cilostazol significantly reduces NLRP3 inflammasome activation and the activity of NOX4, TXNIP, HMGB1, IL-1β, and IL-18 in HAECs induced with FFA. Cilostazol also protected the function of SIRT1, which serves to limit the activity of NLRP3 inflammasome102.
Glibenclamide is a sulfonylurea drug widely prescribed to treat T2DM. Studies have discovered that glibenclamide attenuates blood–brain barrier (BBB) disruption in experimental intracerebral hemorrhage model by inhibiting NLRP3 inflammasome activation in microvessel ECs, thereby maintaining the integrity of the BBB103.
Other anti-inflammatory or antioxidant drugs
Other anti-inflammatory or antioxidant drugs have also been investigated in different disease contexts. Aspirin is one of the most commonly used drugs for the secondary prevention of cardiovascular disease104. Zhou et al. found that aspirin protects the expression of ZO-1 and ZO-2 by inhibiting NLRP3 inflammasome formation and activation in LPS-induced MVECs and coronary arterial endothelium, thereby alleviating endothelial gap junction dysfunction105. This study unraveled a novel mechanism of aspirin in ameliorating endothelial dysfunction by blocking redox signaling and NLRP3 inflammasome activation, providing a new viewpoint on the clinical potential of aspirin in the early prevention of cardiovascular diseases105. The mitochondrion-targeting antioxidant mitoquinone (MitoQ) was found to inhibit endothelial inflammation and barrier injury in cigarette smoke extract (CSE)-treated HUVECs via restoring mitochondrial damage106. Mechanistically, the protective effects of MitoQ depend on suppressing the internalization of VE cadherin and cytoskeleton remodeling, and inhibiting the activation of NF-κB/NLRP3 inflammasome pathway, as well as restraining the production of mtROS and autophagy106.
All these drugs ameliorate endothelial dysfunction by inhibiting NLRP3 inflammasome activation. Accordingly, we summarized the drugs targeting NLRP3 inflammasome signaling pathway and their targets in endothelial dysfunction-related diseases (Fig. 6). However, it is necessary to further study the molecular mechanism of these drugs inhibiting NLRP3 inflammasome pathway. Together, these studies provide new perspectives on the pharmacological mechanisms of drugs and therapeutic strategies for endothelial dysfunction-related diseases.
Fig. 6. Drugs targeting NLRP3 inflammasome signaling pathway and their targets in endothelial dysfunction-related diseases.
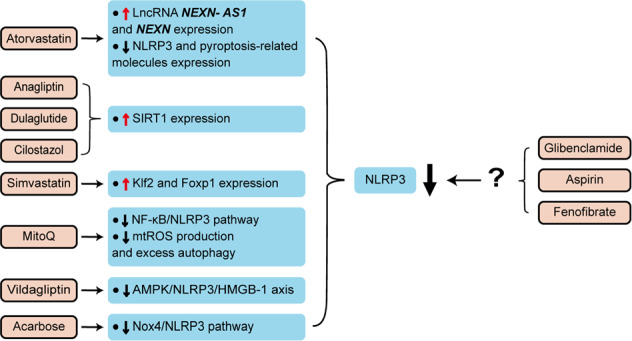
These drugs, including statins, hypoglycemic agents, and other anti-inflammatory or antioxidant drugs, ameliorate endothelial dysfunction by inhibiting NLRP3 inflammasome signaling pathway. The drugs on the left of the figure have been proved to inhibit NLRP3 inflammasome activation by mediating different targets, while the targets of the drug on the right of the figure still need further study to verify.
NLRP3 inflammasome-specific pharmacological inhibitors for the treatment of endothelial dysfunction
NLRP3 inflammasome is involved in the progress of a variety of disorders, which promotes the discovery of NLRP3 inflammasome inhibitors. A large number of inhibitors have been demonstrated to improve endothelial dysfunction by inhibiting NLRP3 inflammasome signaling pathway, including endogenous and exogenous inhibitors, but with low specificity107–109. Here, we show the potential of specific NLRP3 inhibitors to ameliorate endothelial dysfunction (Table 3), which are briefly discussed below.
Table 3.
Potential specific NLRP3 inflammasome inhibitors for the treatment of endothelial dysfunction.
| Agents | Targets | Host | Functions | Disease | References |
|---|---|---|---|---|---|
| MCC950 | ASC oligomerization | HRECs |
•↑ Endothelial viability •↓ Endothelial apoptosis |
Diabetic retinopathy | 112 |
| BMVECs |
•↓ BBB permeability •↑ Vascular integrity |
Diabetes mellitus | 114 | ||
| Diabetic mice |
•↓ Vascular dysfunction •↑ ACh vasodilation |
Diabetes mellitus | 113 | ||
| CLP rat | •↑ Endothelial permeability | Sepsis | 115 | ||
| HUVECs/CLP mice |
•↑ Aortic vasodilation •↑ p-eNOS expression |
Sepsis | 116 | ||
| Oridonin | Cysteine 279 of NLRP3 | HUVECs | •↓ Vascular inflammation | Vascular inflammation | 119 |
| HUVECs | •↓ Angiogenesis | Breast cancer | 120 | ||
| Tranilast | NLRP3 oligomerization | ApoE−/− mice |
•↓ Vascular inflammation •↓ Atherosclerosis |
Atherosclerosis | 118 |
MCC950 is one of the NLRP3 inflammasome-specific inhibitors by blocking ASC oligomerization and NLRP3 ATP hydrolysis110,111. A previous study reported that MCC950 treatment alleviated HG-induced human retinal endothelial cell (HREC) dysfunction probably in part by suppressing the NEK7–NLRP3 interaction112. Treatment of MCC950 also attenuated diabetes-related vascular dysfunction in diabetic mice model113. Moreover, treatment of diabetic rats after stroke with MCC950 ameliorated cognitive function, vascular permeability, and neurovascular remodeling114. More recently, it has been found that sepsis-related endothelial dysfunction was also improved with MCC950 intervention in in vivo and in vitro models115,116. Additionally, CY-09, OLT1177, 3,4-methylenedioxy-β-nitrostyrene (MNS), N-[3′,4′-dimethoxycinnamoyl]-anthranilic acid (Tranilast), and oridonin are also specific inhibitors of NLRP3 inflammasome33,117. Remarkably, a recent study found that tranilast exhibited antivascular inflammation and anti-atherosclerosis properties via increasing NLRP3 ubiquitination and impeding NLRP3 inflammasome activation118. Besides, oridonin was found to blunt endothelial inflammation through restraining the activation of MAPK and NF-κB signaling pathways119. It was also reported that oridonin diminished cell migration and angiogenesis in VEGF-treated HUVECs120.
Hence, NLRP3 inflammasome-specific pharmacological inhibitors may be the optimal choice for the treatment of endothelial dysfunction, providing a new strategy to treat endothelial dysfunction-related disease. In the future, the effects of these inhibitors are needed to be verified at the animal level and in clinical trials.
Conclusions
In recent years, a group of evidence has accumulated linking NLRP3 inflammasome activation to the control of endothelial dysfunction. The findings reviewed here highlight the tremendous potential of direct or indirect modulation of NLRP3 inflammasome activation in combating various endothelial dysfunctions. When ECs are stimulated by exogenous substances or endogenous mediators, in addition to triggering oxidative stress, ER stress, mitochondrial dysfunction, and immune activation, these effects may also trigger a common signaling pathway of NLRP3 inflammasome activation, thereby exacerbating endothelial dysfunction. Of note, emerging literature indicates that certain well-known drugs, such as statins, hypoglycemic agents, and other anti-inflammatory or antioxidant drugs, can improve vascular dysfunction by inhibiting NLRP3 inflammasome signaling pathway. We have also summarized ncRNAs that involved in the regulation of endothelial function by directly or indirectly regulating NLRP3 inflammasome activation; this helps to understand the molecular mechanisms underlying NLRP3 inflammasome-related endothelial dysfunction, and may provide novel targets for the development of future therapeutics.
With the advancement of the research, our understanding of potential mechanisms that affect endothelial function through the NLRP3 inflammasome activation pathway is expanding. Meanwhile, the interactions between endothelial dysfunction and the NLRP3 inflammasome-regulated pathways may open up a new avenue for the treatment of cardiovascular diseases. While our understanding of the influence of NLRP3 inflammasome activation on endothelial dysfunction is continuously growing, our ideas on how NLRP3 inflammation perceives information about different mediators are still limited. Furthermore, it is unclear whether different agonists affect endothelial function through a single cascade of events or through distinct pathways to activate NLRP3 inflammation. In parallel, the effects of other inflammasomes, such as NLRP1, NLRC4, NLRP6, and AIM2, noncanonical inflammasomes, and upstream pathways on endothelial function, have not been thoroughly investigated so far.
Of the ncRNAs that regulate NLRP3 inflammasome pathway, miRNAs are currently the most studied. Accumulating research has indicated that miRNAs have tremendous potential as an indicative molecular biomarker or drug target in the diagnosis and treatment of diseases related to NLRP3 dysfunction. Nevertheless, plenty of thorny issues remain to be resolved to achieve this goal, including poor target specificity, poor miRNA stability, and the side effects involving liver damage121. Also, their effects may be restricted and removed by the kidney and liver due to the absence of protection around the carrier molecule122. Besides, the innate immune system may recognize and eliminate miRNA drugs123. However, it is encouraging that miRNA drug efficacy, delivery, and safety issues are currently being addressed. Shortly, miRNAs as therapeutic targets will be the focus of most clinical research. In the long run, as our understanding of other ncRNA mechanisms deepens, new ncRNAs may emerge as therapeutic targets. Additionally, the possible interaction between ncRNAs and NLRP3 inflammasome is only a preliminary study, and further research is needed in the future.
Excitingly, the expanding research on the role of NLRP3 inflammasome in endothelial dysfunction will hopefully enrich the understanding of the vital role of NLRP3 inflammasome in endothelial dysfunction-related inflammatory diseases. Furthermore, elucidating the molecular mechanisms of such interactions will be favorable for developing novel prevention approaches and more effective therapeutic strategies for diseases related to endothelial dysfunction, development of which is an exciting challenge and goal for future research.
Acknowledgements
This work was supported by The National Natural Science Foundation of China (grant nos. 81870331, 31701208, and 91849209), The Natural Science Foundation of Shandong Province (grant no. ZR2017MC067), The Project of Shandong Province Higher Educational Science and Technology Program (no. J18KA285), and The Qingdao municipal science and technology bureau project (grant no. 18-2-2-65-jch).
Author contributions
B.B., Y.Y., and Q.W. collected materials and wrote the paper. T.Y., X.C., and Y.Y. provided the idea. B.B., M.L., and Y.L. are responsible for the schematic diagram within this paper. T.Y., C.T., L.H.H.A., and P.L. helped with the final revision of the paper. All authors reviewed and approved the final paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by Y. Shi
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Baochen Bai, Yanyan Yang
Contributor Information
Tao Yu, Email: yutao0112@qdu.edu.cn.
Xian-ming Chu, Email: 18661801698@163.com.
References
- 1.Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020;20:95–112. doi: 10.1038/s41577-019-0215-7. [DOI] [PubMed] [Google Scholar]
- 2.Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2016;16:35–50. doi: 10.1038/nri.2015.8. [DOI] [PubMed] [Google Scholar]
- 3.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 4.Han J, Ulevitch RJ. Limiting inflammatory responses during activation of innate immunity. Nat. Immunol. 2005;6:1198–1205. doi: 10.1038/ni1274. [DOI] [PubMed] [Google Scholar]
- 5.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 6.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009;10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prochnicki T, Latz E. Inflammasomes on the crossroads of innate immune recognition and metabolic control. Cell Metab. 2017;26:71–93. doi: 10.1016/j.cmet.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 8.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007;7:803–815. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 9.Godo S, Shimokawa H. Endothelial functions. Arterioscler Thromb. Vasc. Biol. 2017;37:e108–e114. doi: 10.1161/ATVBAHA.117.309813. [DOI] [PubMed] [Google Scholar]
- 10.Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 11.Rheinheimer J, de Souza BM, Cardoso NS, Bauer AC, Crispim D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: a systematic review. Metabolism. 2017;74:1–9. doi: 10.1016/j.metabol.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 12.Sun HJ, et al. NLRP3 inflammasome activation contributes to VSMC phenotypic transformation and proliferation in hypertension. Cell Death Dis. 2017;8:e3074. doi: 10.1038/cddis.2017.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Z, et al. A novel role of NLRP3-generated IL-1beta in the acute-chronic transition of peripheral lipopolysaccharide-elicited neuroinflammation: implications for sepsis-associated neurodegeneration. J. Neuroinflammation. 2020;17:64. doi: 10.1186/s12974-020-1728-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loukovaara S, et al. NLRP3 inflammasome activation is associated with proliferative diabetic retinopathy. Acta Ophthalmol. 2017;95:803–808. doi: 10.1111/aos.13427. [DOI] [PubMed] [Google Scholar]
- 15.Fann DY, et al. Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res. Rev. 2013;12:941–966. doi: 10.1016/j.arr.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer. 2018;17:158. doi: 10.1186/s12943-018-0900-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32:373–379. doi: 10.1016/j.it.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 19.Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. 2019;40:1035–1052. doi: 10.1016/j.it.2019.09.005. [DOI] [PubMed] [Google Scholar]
- 20.Hayward, J.A., Mathur, A., Ngo, C. & Man, S.M. Cytosolic recognition of microbes and pathogens: inflammasomes in action. Microbiol. Mol. Biol. Rev. 82, e00015-18 (2018). [DOI] [PMC free article] [PubMed]
- 21.Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe. 2010;8:471–483. doi: 10.1016/j.chom.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chavarría-Smith J, Vance RE. The NLRP1 inflammasomes. Immunol. Rev. 2015;265:22–34. doi: 10.1111/imr.12283. [DOI] [PubMed] [Google Scholar]
- 23.Duncan JA, Canna SW. The NLRC4 inflammasome. Immunol. Rev. 2018;281:115–123. doi: 10.1111/imr.12607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levy M, Shapiro H, Thaiss CA, Elinav E. NLRP6: a multifaceted innate immune sensor. Trends Immunol. 2017;38:248–260. doi: 10.1016/j.it.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 25.Ghimire, L., Paudel, S., Jin, L. & Jeyaseelan, S. The NLRP6 inflammasome in health and disease. Mucosal. Immunol.133,388–398 (2020). [DOI] [PMC free article] [PubMed]
- 26.Lugrin J, Martinon F. The AIM2 inflammasome: sensor of pathogens and cellular perturbations. Immunol. Rev. 2018;281:99–114. doi: 10.1111/imr.12618. [DOI] [PubMed] [Google Scholar]
- 27.Xu H, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the pyrin inflammasome. Nature. 2014;513:237–241. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 28.Alimov I, et al. Bile acid analogues are activators of pyrin inflammasome. J. Biol. Chem. 2019;294:3359–3366. doi: 10.1074/jbc.RA118.005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He Y, Hara H, Nunez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016;41:1012–1021. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mangan MSJ, et al. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018;17:588–606. doi: 10.1038/nrd.2018.97. [DOI] [PubMed] [Google Scholar]
- 31.Ding J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–116. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 32.He W-t, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25:1285–1298. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019;10:128. doi: 10.1038/s41419-019-1413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019;19:477–489. doi: 10.1038/s41577-019-0165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kayagaki N, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 36.Gaidt MM, et al. Human monocytes engage an alternative inflammasome pathway. Immunity. 2016;44:833–846. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 37.Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015;22:1111–1129. doi: 10.1089/ars.2014.5994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dan Dunn J, Alvarez LA, Zhang X, Soldati T. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. 2015;6:472–485. doi: 10.1016/j.redox.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shihata WA, Michell DL, Andrews KL, Chin-Dusting JP. Caveolae: a role in endothelial inflammation and mechanotransduction? Front. Physiol. 2016;7:628. doi: 10.3389/fphys.2016.00628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ. Res. 2013;112:1171–1188. doi: 10.1161/CIRCRESAHA.111.300233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X, et al. ER stress activates the NLRP3 inflammasome: a novel mechanism of atherosclerosis. Oxid. Med. Cell Longev. 2019;2019:3462530. doi: 10.1155/2019/3462530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cunard R. Endoplasmic reticulum stress, a driver or an innocent bystander in endothelial dysfunction associated with hypertension? Curr. Hypertens. Rep. 2017;19:64. doi: 10.1007/s11906-017-0762-x. [DOI] [PubMed] [Google Scholar]
- 43.Maamoun, H., Abdelsalam, S. S., Zeidan, A., Korashy, H. M. & Agouni, A. Endoplasmic reticulum stress: a critical molecular driver of endothelial dysfunction and cardiovascular disturbances associated with diabetes. Int. J. Mol. Sci. 20, 1658 (2019). [DOI] [PMC free article] [PubMed]
- 44.Luchetti F, et al. Endothelial cells, endoplasmic reticulum stress and oxysterols. Redox Biol. 2017;13:581–587. doi: 10.1016/j.redox.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Battson ML, Lee DM, Gentile CL. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2017;312:H355–H367. doi: 10.1152/ajpheart.00437.2016. [DOI] [PubMed] [Google Scholar]
- 46.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 47.Chen Z, Martin M, Li Z, Shyy JY. Endothelial dysfunction: the role of sterol regulatory element-binding protein-induced NOD-like receptor family pyrin domain-containing protein 3 inflammasome in atherosclerosis. Curr. Opin. Lipido. 2014;25:339–349. doi: 10.1097/MOL.0000000000000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bortolotti P, Faure E, Kipnis E. Inflammasomes in tissue damages and immune disorders after trauma. Front. Immunol. 2018;9:1900. doi: 10.3389/fimmu.2018.01900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang JG, et al. Monocytic microparticles activate endothelial cells in an IL-1beta-dependent manner. Blood. 2011;118:2366–2374. doi: 10.1182/blood-2011-01-330878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ. Res. 2018;122:1722–1740. doi: 10.1161/CIRCRESAHA.118.311362. [DOI] [PubMed] [Google Scholar]
- 51.Lee WJ, et al. Visfatin-induced expression of inflammatory mediators in human endothelial cells through the NF-kappaB pathway. Int. J. Obes. 2009;33:465–472. doi: 10.1038/ijo.2009.24. [DOI] [PubMed] [Google Scholar]
- 52.Xia M, et al. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am. J. Pathol. 2014;184:1617–1628. doi: 10.1016/j.ajpath.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia X, et al. Tetrachlorobenzoquinone stimulates NLRP3 inflammasome-mediated post-translational activation and secretion of IL-1beta in the HUVEC endothelial cell line. Chem. Res. Toxicol. 2016;29:421–429. doi: 10.1021/acs.chemrestox.6b00021. [DOI] [PubMed] [Google Scholar]
- 54.Xia X, et al. Atypical gasdermin D and mixed lineage kinase domain-like protein leakage aggravates tetrachlorobenzoquinone-induced nod-like receptor protein 3 inflammasome activation. Chem. Res. Toxicol. 2018;31:1418–1425. doi: 10.1021/acs.chemrestox.8b00306. [DOI] [PubMed] [Google Scholar]
- 55.Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 2017;42:245–254. doi: 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 56.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 2009;7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang C, et al. Acrolein induces NLRP3 inflammasome-mediated pyroptosis and suppresses migration via ROS-dependent autophagy in vascular endothelial cells. Toxicology. 2018;410:26–40. doi: 10.1016/j.tox.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 58.Chen H, et al. Cadmium induces NLRP3 inflammasome-dependent pyroptosis in vascular endothelial cells. Toxicol. Lett. 2016;246:7–16. doi: 10.1016/j.toxlet.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 59.Chen Q, et al. Increased gene copy number of DEFA1/DEFA3 worsens sepsis by inducing endothelial pyroptosis. Proc. Natl Acad. Sci. USA. 2019;116:3161–3170. doi: 10.1073/pnas.1812947116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Komarova YA, Kruse K, Mehta D, Malik AB. Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circulation Res. 2017;120:179–206. doi: 10.1161/CIRCRESAHA.116.306534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Radeva, M. Y. & Waschke, J. Mind the gap: mechanisms regulating the endothelial barrier. Acta Physiol.222, e12860 (2018). [DOI] [PubMed]
- 62.Wettschureck N, Strilic B, Offermanns S. Passing the vascular barrier: endothelial signaling processes controlling extravasation. Physiol. Rev. 2019;99:1467–1525. doi: 10.1152/physrev.00037.2018. [DOI] [PubMed] [Google Scholar]
- 63.Barker BR, Taxman DJ, Ting JP. Cross-regulation between the IL-1beta/IL-18 processing inflammasome and other inflammatory cytokines. Curr. Opin. Immunol. 2011;23:591–597. doi: 10.1016/j.coi.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mussbacher M, et al. Cell type-specific roles of NF-kappaB linking inflammation and thrombosis. Front. Immunol. 2019;10:85. doi: 10.3389/fimmu.2019.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Keyel PA. How is inflammation initiated? Individual influences of IL-1, IL-18 and HMGB1. Cytokine. 2014;69:136–145. doi: 10.1016/j.cyto.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 66.Vande Walle L, Kanneganti TD, Lamkanfi M. HMGB1 release by inflammasomes. Virulence. 2011;2:162–165. doi: 10.4161/viru.2.2.15480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wolfson RK, Chiang ET, Garcia JG. HMGB1 induces human lung endothelial cell cytoskeletal rearrangement and barrier disruption. Microvasc. Res. 2011;81:189–197. doi: 10.1016/j.mvr.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Y, et al. Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. J. Mol. Med. 2016;94:1335–1347. doi: 10.1007/s00109-016-1481-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y, Chen Y, Zhang Y, Li PL, Li X. Contribution of cathepsin B-dependent Nlrp3 inflammasome activation to nicotine-induced endothelial barrier dysfunction. Eur. J. Pharm. 2019;865:172795. doi: 10.1016/j.ejphar.2019.172795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014;69:S4–9. doi: 10.1093/gerona/glu057. [DOI] [PubMed] [Google Scholar]
- 71.Jia G, Aroor AR, Jia C, Sowers JR. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2019;1865:1802–1809. doi: 10.1016/j.bbadis.2018.08.008. [DOI] [PubMed] [Google Scholar]
- 72.Youm YH, et al. Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab. 2013;18:519–532. doi: 10.1016/j.cmet.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Youm YH, et al. The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell Rep. 2012;1:56–68. doi: 10.1016/j.celrep.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marneros AG. Increased VEGF-A promotes multiple distinct aging diseases of the eye through shared pathomechanisms. EMBO Mol. Med. 2016;8:208–231. doi: 10.15252/emmm.201505613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yin Y, et al. Vascular endothelial cells senescence is associated with NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation via reactive oxygen species (ROS)/thioredoxin-interacting protein (TXNIP) pathway. Int J. Biochem. Cell Biol. 2017;84:22–34. doi: 10.1016/j.biocel.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 76.Sun C, et al. Purple sweet potato color inhibits endothelial premature senescence by blocking the NLRP3 inflammasome. J. Nutr. Biochem. 2015;26:1029–1040. doi: 10.1016/j.jnutbio.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 77.Sun C, et al. Purple sweet potato color attenuated NLRP3 inflammasome by inducing autophagy to delay endothelial senescence. J. Cell Physiol. 2019;234:5926–5939. doi: 10.1002/jcp.28003. [DOI] [PubMed] [Google Scholar]
- 78.Poller W, et al. Non-coding RNAs in cardiovascular diseases: diagnostic and therapeutic perspectives. Eur. Heart J. 2018;39:2704–2716. doi: 10.1093/eurheartj/ehx165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang Q, et al. Long noncoding RNA XXYLT1-AS2 regulates proliferation and adhesion by targeting the RNA binding protein FUS in HUVEC. Atherosclerosis. 2020;298:58–69. doi: 10.1016/j.atherosclerosis.2020.02.018. [DOI] [PubMed] [Google Scholar]
- 80.Liu Y, et al. Insights into the regulatory role of circRNA in angiogenesis and clinical implications. Atherosclerosis. 2020;298:14–26. doi: 10.1016/j.atherosclerosis.2020.02.017. [DOI] [PubMed] [Google Scholar]
- 81.Liu S, et al. Understanding the role of non-coding RNA (ncRNA) in stent restenosis. Atherosclerosis. 2018;272:153–161. doi: 10.1016/j.atherosclerosis.2018.03.036. [DOI] [PubMed] [Google Scholar]
- 82.Chew CL, Conos SA, Unal B, Tergaonkar V. Noncoding RNAs: master regulators of inflammatory signaling. Trends Mol. Med. 2018;24:66–84. doi: 10.1016/j.molmed.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 83.Yu T, et al. The kinase inhibitor BX795 suppresses the inflammatory response via multiple kinases. Biochem. Pharm. 2020;174:113797. doi: 10.1016/j.bcp.2020.113797. [DOI] [PubMed] [Google Scholar]
- 84.Slack FJ, Chinnaiyan AM. The role of non-coding RNAs in oncology. Cell. 2019;179:1033–1055. doi: 10.1016/j.cell.2019.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Anfossi S, Babayan A, Pantel K, Calin GA. Clinical utility of circulating non-coding RNAs—an update. Nat. Rev. Clin. Oncol. 2018;15:541–563. doi: 10.1038/s41571-018-0035-x. [DOI] [PubMed] [Google Scholar]
- 86.Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017;16:167–179. doi: 10.1038/nrd.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lopez-Ramirez MA, Reijerkerk A, de Vries HE, Romero IA. Regulation of brain endothelial barrier function by microRNAs in health and neuroinflammation. FASEB J. 2016;30:2662–2672. doi: 10.1096/fj.201600435RR. [DOI] [PubMed] [Google Scholar]
- 88.Njock MS, Fish JE. Endothelial miRNAs as cellular messengers in cardiometabolic diseases. Trends Endocrinol. Metab. 2017;28:237–246. doi: 10.1016/j.tem.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 89.Hulshoff MS, Xu X, Krenning G, Zeisberg EM. Epigenetic regulation of endothelial-to-mesenchymal transition in chronic heart disease. Arterioscler Thromb. Vasc. Biol. 2018;38:1986–1996. doi: 10.1161/ATVBAHA.118.311276. [DOI] [PubMed] [Google Scholar]
- 90.Pantsulaia I, Ciszewski WM, Niewiarowska J. Senescent endothelial cells: potential modulators of immunosenescence and ageing. Ageing Res Rev. 2016;29:13–25. doi: 10.1016/j.arr.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 91.Uchida S, Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ. Res. 2015;116:737–750. doi: 10.1161/CIRCRESAHA.116.302521. [DOI] [PubMed] [Google Scholar]
- 92.Jae N, Heumuller AW, Fouani Y, Dimmeler S. Long non-coding RNAs in vascular biology and disease. Vasc. Pharm. 2019;114:13–22. doi: 10.1016/j.vph.2018.03.003. [DOI] [PubMed] [Google Scholar]
- 93.Parihar SP, Guler R, Brombacher F. Statins: a viable candidate for host-directed therapy against infectious diseases. Nat. Rev. Immunol. 2019;19:104–117. doi: 10.1038/s41577-018-0094-3. [DOI] [PubMed] [Google Scholar]
- 94.Wu LM, et al. Atorvastatin inhibits pyroptosis through the lncRNA NEXN-AS1/NEXN pathway in human vascular endothelial cells. Atherosclerosis. 2020;293:26–34. doi: 10.1016/j.atherosclerosis.2019.11.033. [DOI] [PubMed] [Google Scholar]
- 95.Lv Z-H, Phuong TA, Jin S-J, Li X-X, Xu M. Protection by simvastatin on hyperglycemia-induced endothelial dysfunction through inhibiting NLRP3 inflammasomes. Oncotarget. 2017;8:91291–91305. doi: 10.18632/oncotarget.20443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhuang T, et al. Endothelial Foxp1 suppresses atherosclerosis via modulation of Nlrp3 inflammasome activation. Circ. Res. 2019;125:590–605. doi: 10.1161/CIRCRESAHA.118.314402. [DOI] [PubMed] [Google Scholar]
- 97.Qi Y, Du X, Yao X, Zhao Y. Vildagliptin inhibits high free fatty acid (FFA)-induced NLRP3 inflammasome activation in endothelial cells. Artif. Cells Nanomed. Biotechnol. 2019;47:1067–1074. doi: 10.1080/21691401.2019.1578783. [DOI] [PubMed] [Google Scholar]
- 98.Jiang T, Jiang D, Zhang L, Ding M, Zhou H. Anagliptin ameliorates high glucose-induced endothelial dysfunction via suppression of NLRP3 inflammasome activation mediated by SIRT1. Mol. Immunol. 2019;107:54–60. doi: 10.1016/j.molimm.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 99.Luo X, et al. Dulaglutide inhibits high glucose- induced endothelial dysfunction and NLRP3 inflammasome activation. Arch. Biochem. Biophys. 2019;671:203–209. doi: 10.1016/j.abb.2019.07.008. [DOI] [PubMed] [Google Scholar]
- 100.Li XX, et al. Protective effects of acarbose against vascular endothelial dysfunction through inhibiting Nox4/NLRP3 inflammasome pathway in diabetic rats. Free Radic. Biol. Med. 2019;145:175–186. doi: 10.1016/j.freeradbiomed.2019.09.015. [DOI] [PubMed] [Google Scholar]
- 101.Deng Y, et al. PPARalpha agonist stimulated angiogenesis by improving endothelial precursor cell function Via a NLRP3 inflammasome pathway. Cell Physiol. Biochem. 2017;42:2255–2266. doi: 10.1159/000479999. [DOI] [PubMed] [Google Scholar]
- 102.Wang X, Huang H, Su C, Zhong Q, Wu G. Cilostazol ameliorates high free fatty acid (FFA)-induced activation of NLRP3 inflammasome in human vascular endothelial cells. Artif. Cells Nanomed. Biotechnol. 2019;47:3704–3710. doi: 10.1080/21691401.2019.1665058. [DOI] [PubMed] [Google Scholar]
- 103.Xu F, Shen G, Su Z, He Z, Yuan L. Glibenclamide ameliorates the disrupted blood-brain barrier in experimental intracerebral hemorrhage by inhibiting the activation of NLRP3 inflammasome. Brain Behav. 2019;9:e01254. doi: 10.1002/brb3.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Raber I, et al. The rise and fall of aspirin in the primary prevention of cardiovascular disease. Lancet. 2019;393:2155–2167. doi: 10.1016/S0140-6736(19)30541-0. [DOI] [PubMed] [Google Scholar]
- 105.Zhou X, et al. Aspirin alleviates endothelial gap junction dysfunction through inhibition of NLRP3 inflammasome activation in LPS-induced vascular injury. Acta Pharm. Sin. B. 2019;9:711–723. doi: 10.1016/j.apsb.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen S, et al. The antioxidant MitoQ protects against CSE-induced endothelial barrier injury and inflammation by inhibiting ROS and autophagy in human umbilical vein endothelial cells. Int. J. Biol. Sci. 2019;15:1440–1451. doi: 10.7150/ijbs.30193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang C, et al. The protective effects of orexin a against high glucose-induced activation of NLRP3 inflammasome in human vascular endothelial cells. Arch. Biochem. Biophys. 2019;672:108052. doi: 10.1016/j.abb.2019.07.017. [DOI] [PubMed] [Google Scholar]
- 108.Zhang L, Yuan M, Zhang L, Wu B, Sun X. Adiponectin alleviates NLRP3-inflammasome-mediated pyroptosis of aortic endothelial cells by inhibiting FoxO4 in arteriosclerosis. Biochem. Biophys. Res. Commun. 2019;514:266–272. doi: 10.1016/j.bbrc.2019.04.143. [DOI] [PubMed] [Google Scholar]
- 109.Lian D, et al. Puerarin inhibits hyperglycemia-induced inter-endothelial junction through suppressing endothelial Nlrp3 inflammasome activation via ROS-dependent oxidative pathway. Phytomedicine. 2019;55:310–319. doi: 10.1016/j.phymed.2018.10.013. [DOI] [PubMed] [Google Scholar]
- 110.Coll RC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015;21:248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Coll RC, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019;15:556–559. doi: 10.1038/s41589-019-0277-7. [DOI] [PubMed] [Google Scholar]
- 112.Zhang Y, et al. Protection of Mcc950 against high-glucose-induced human retinal endothelial cell dysfunction. Cell Death Dis. 2017;8:e2941. doi: 10.1038/cddis.2017.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ferreira, N. S. et al. NLRP3 inflammasome and mineralocorticoid receptors are associated with vascular dysfunction in type 2 diabetes mellitus. Cells8, 1595 (2019). [DOI] [PMC free article] [PubMed]
- 114.Ward R, et al. NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharm. Res. 2019;142:237–250. doi: 10.1016/j.phrs.2019.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cornelius DC, et al. NLRP3 inflammasome inhibition attenuates sepsis-induced platelet activation and prevents multi-organ injury in cecal-ligation puncture. PLoS ONE. 2020;15:e0234039. doi: 10.1371/journal.pone.0234039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Luo M, et al. Role of the nucleotide-binding domain-like receptor protein 3 inflammasome in the endothelial dysfunction of early sepsis. Inflammation. 2020;43:1561–1571. doi: 10.1007/s10753-020-01232-x. [DOI] [PubMed] [Google Scholar]
- 117.Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological inhibitors of the NLRP3 inflammasome. Front. Immunol. 2019;10:2538. doi: 10.3389/fimmu.2019.02538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen S, et al. Novel role for tranilast in regulating NLRP3 ubiquitination, vascular inflammation, and atherosclerosis. J. Am. Heart Assoc. 2020;9:e015513. doi: 10.1161/JAHA.119.015513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huang W, Huang M, Ouyang H, Peng J, Liang J. Oridonin inhibits vascular inflammation by blocking NF-kappaB and MAPK activation. Eur. J. Pharm. 2018;826:133–139. doi: 10.1016/j.ejphar.2018.02.044. [DOI] [PubMed] [Google Scholar]
- 120.Li J, et al. Oridonin synergistically enhances the anti-tumor efficacy of doxorubicin against aggressive breast cancer via pro-apoptotic and anti-angiogenic effects. Pharm. Res. 2019;146:104313. doi: 10.1016/j.phrs.2019.104313. [DOI] [PubMed] [Google Scholar]
- 121.Chakraborty C, Sharma AR, Sharma G, Doss CGP, Lee SS. Therapeutic miRNA and siRNA: moving from bench to clinic as next generation medicine. Mol. Ther. Nucleic Acids. 2017;8:132–143. doi: 10.1016/j.omtn.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev. Pharm. Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 123.Diebold, S. S., Kaisho, T., Hemmi, H., Akira, S. & e Sousa, C. R. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531 (2004). [DOI] [PubMed]
- 124.Sun X, et al. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016;481:63–70. doi: 10.1016/j.bbrc.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 125.Yuan X, Wang L, Bhat OM, Lohner H, Li PL. Differential effects of short chain fatty acids on endothelial Nlrp3 inflammasome activation and neointima formation: antioxidant action of butyrate. Redox Biol. 2018;16:21–31. doi: 10.1016/j.redox.2018.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen Y, et al. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim. Biophys. Acta. 2015;1853:396–408. doi: 10.1016/j.bbamcr.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Erdei J, et al. Induction of NLRP3 inflammasome activation by heme in human endothelial cells. Oxid. Med. Cell Longev. 2018;2018:4310816. doi: 10.1155/2018/4310816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gong DM, et al. Endothelial GPR124 exaggerates the pathogenesis of atherosclerosis by activating inflammation. Cell Physiol. Biochem. 2018;45:547–557. doi: 10.1159/000487032. [DOI] [PubMed] [Google Scholar]
- 129.Yeh WJ, Yang HY, Pai MH, Wu CH, Chen JR. Long-term administration of advanced glycation end-product stimulates the activation of NLRP3 inflammasome and sparking the development of renal injury. J. Nutr. Biochem. 2017;39:68–76. doi: 10.1016/j.jnutbio.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 130.Li Y, Xu S, Jiang B, Cohen RA, Zang M. Activation of sterol regulatory element binding protein and NLRP3 inflammasome in atherosclerotic lesion development in diabetic pigs. PLoS ONE. 2013;8:e67532. doi: 10.1371/journal.pone.0067532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhou Y, et al. Inactivation of endothelial adenosine A2A receptors protects mice from cerebral ischaemia-induced brain injury. Br. J. Pharm. 2019;176:2250–2263. doi: 10.1111/bph.14673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vazquez-Medina JP, et al. Genetic inactivation of the phospholipase A2 activity of peroxiredoxin 6 in mice protects against LPS-induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2019;316:L656–l668. doi: 10.1152/ajplung.00344.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang Y, Tao J, Yao Y. Prostaglandin E2 activates NLRP3 inflammasome in endothelial cells to promote diabetic retinopathy. Horm. Metab. Res. 2018;50:704–710. doi: 10.1055/a-0664-0699. [DOI] [PubMed] [Google Scholar]
- 134.Wu X, et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018;9:171. doi: 10.1038/s41419-017-0257-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Huang WQ, Wei P, Lin RQ, Huang F. Protective effects of microrna-22 against endothelial cell injury by targeting NLRP3 through suppression of the inflammasome signaling pathway in a rat model of coronary heart disease. Cell Physiol. Biochem. 2017;43:1346–1358. doi: 10.1159/000481846. [DOI] [PubMed] [Google Scholar]
- 136.Zhou T, et al. MicroRNA-495 ameliorates cardiac microvascular endothelial cell injury and inflammatory reaction by suppressing the NLRP3 inflammasome signaling pathway. Cell Physiol. Biochem. 2018;49:798–815. doi: 10.1159/000493042. [DOI] [PubMed] [Google Scholar]
- 137.Curtiss E, Liu L, Steinle JJ. miR15a regulates NLRP3 inflammasome proteins in the retinal vasculature. Exp. Eye Res. 2018;176:98–102. doi: 10.1016/j.exer.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang W, Wang Y, Kong Y. Exosomes derived from mesenchymal stem cells modulate miR-126 to ameliorate hyperglycemia-induced retinal inflammation via targeting HMGB1. Investig. Ophthalmol. Vis. Sci. 2019;60:294–303. doi: 10.1167/iovs.18-25617. [DOI] [PubMed] [Google Scholar]
- 139.Gu C, et al. miR-590-3p inhibits pyroptosis in diabetic retinopathy by targeting NLRP1 and inactivating the NOX4 signaling pathway. Investig. Ophthalmol. Vis. Sci. 2019;60:4215–4223. doi: 10.1167/iovs.19-27825. [DOI] [PubMed] [Google Scholar]
- 140.Chen M, Li W, Zhang Y, Yang J. MicroRNA-20a protects human aortic endothelial cells from Ox-LDL-induced inflammation through targeting TLR4 and TXNIP signaling. Biomed. Pharmacother. 2018;103:191–197. doi: 10.1016/j.biopha.2018.03.129. [DOI] [PubMed] [Google Scholar]
- 141.Li P, et al. MicroRNA-30c-5p inhibits NLRP3 inflammasome-mediated endothelial cell pyroptosis through FOXO3 down-regulation in atherosclerosis. Biochem. Biophys. Res. Commun. 2018;503:2833–2840. doi: 10.1016/j.bbrc.2018.08.049. [DOI] [PubMed] [Google Scholar]
- 142.Lian Z, Lv FF, Yu J, Wang JW. The anti-inflammatory effect of microRNA-383-3p interacting with IL1R2 against homocysteine-induced endothelial injury in rat coronary arteries. J. Cell Biochem. 2018;119:6684–6694. [Google Scholar]
- 143.Dong F, et al. miR‑20b inhibits the senescence of human umbilical vein endothelial cells through regulating the Wnt/β‑catenin pathway via the TXNIP/NLRP3 axis. Int. J. Mol. Med. 2020;45:847–857. doi: 10.3892/ijmm.2020.4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kim J-H, et al. A miRNA-101-3p/Bim axis as a determinant of serum deprivation-induced endothelial cell apoptosis. Cell Death Dis. 2017;8:e2808–e2808. doi: 10.1038/cddis.2017.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yu J, Chen J, Yang H, Chen S, Wang Z. Overexpression of miR200a3p promoted inflammation in sepsisinduced brain injury through ROSinduced NLRP3. Int. J. Mol. Med. 2019;44:1811–1823. doi: 10.3892/ijmm.2019.4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen Z, et al. Oxidative stress activates endothelial innate immunity via sterol regulatory element binding protein 2 (SREBP2) transactivation of microRNA-92a. Circulation. 2015;131:805–814. doi: 10.1161/CIRCULATIONAHA.114.013675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhaolin Z, et al. OxLDL induces vascular endothelial cell pyroptosis through miR-125a-5p/TET2 pathway. J. Cell Physiol. 2019;234:7475–7491. doi: 10.1002/jcp.27509. [DOI] [PubMed] [Google Scholar]
- 148.Song Y, et al. Long noncoding RNA MALAT1 promotes high glucose-induced human endothelial cells pyroptosis by affecting NLRP3 expression through competitively binding miR-22. Biochem. Biophys. Res. Commun. 2019;509:359–366. doi: 10.1016/j.bbrc.2018.12.139. [DOI] [PubMed] [Google Scholar]
- 149.Zhang, Y. et al. Melatonin prevents endothelial cell pyroptosis via regulation of long noncoding RNA MEG3/miR-223/NLRP3 axis. J. Pineal. Res.64, e12449 (2018). [DOI] [PubMed]
- 150.Cheng J, et al. Downregulation of hsa_circ_0068087 ameliorates TLR4/NF-kappaB/NLRP3 inflammasome-mediated inflammation and endothelial cell dysfunction in high glucose conditioned by sponging miR-197. Gene. 2019;709:1–7. doi: 10.1016/j.gene.2019.05.012. [DOI] [PubMed] [Google Scholar]