

Abstract
Cancer therapy utilizing adoptive transfer of chimeric antigen receptor (CAR) T cells has demonstrated remarkable clinical outcomes in hematologic malignancies. However, CAR T cell application to solid tumors has had limited success, partly due to the lack of tumor-specific antigens and an immune-suppressive tumor microenvironment. From the tumor tissues of gastric cancer patients, we found that intercellular adhesion molecule 1 (ICAM-1) expression is significantly associated with advanced stage and shorter survival. In this study, we report a proof-of-concept study using ICAM-1-targeting CAR T cells against gastric cancer. The efficacy of ICAM-1 CAR T cells showed a significant correlation with the level of ICAM-1 expression in target cells in vitro. In animal models of human gastric cancer, ICAM-1-targeting CAR T cells potently eliminated tumors that developed in the lungs, while their efficacy was more limited against the tumors in the peritoneum. To augment CAR T cell activity against intraperitoneal tumors, combinations with paclitaxel or CAR activation-dependent interleukin (IL)-12 release were explored and found to significantly increase anti-tumor activity and survival benefit. Collectively, ICAM-1-targeting CAR T cells alone or in combination with chemotherapy represent a promising strategy to treat patients with ICAM-1+ advanced gastric cancer.
Keywords: Chimeric antigen receptor, Gastric cancer, T cell imaging, inducible cytokines
Graphical Abstract
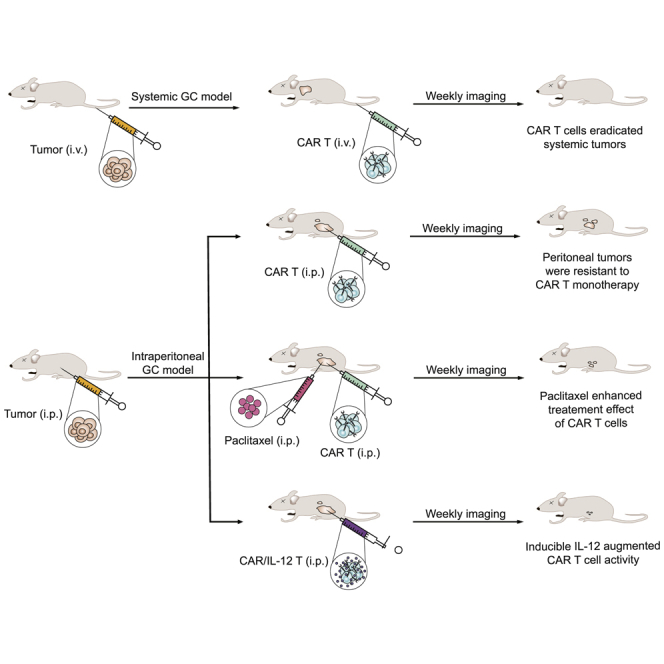
In this study, we demonstrated the therapeutic potential of CAR T cells targeting ICAM-1 in preclinical models of systemic and intraperitoneal metastases of gastric cancer. A combination with paclitaxel or armoring CAR T cells with inducible IL-12 was found to significantly enhance the treatment effect of CAR T cells.
Introduction
Gastric cancer (GC) is the fifth most common cancer and the third leading cause of death in the world.1 Surgical resection combined with adjuvant chemotherapy is the only method for treating localized GC.2,3 Moreover, more than 50% of GC patients progress to an advanced stage with dismal prognosis.4,5 The treatment of patients with locally advanced and metastatic disease is currently based on systemic therapy or palliative supportive care with a median overall survival of 1 year.6 The targeted agent for human epidermal growth factor receptor 2 (HER2), trastuzumab, has been used with modest efficacy for advanced disease in HER2+ patients, which accounts for less than 20% of patients with gastric tumors.7 In addition, patients with peritoneal carcinomatosis are largely excluded from trastuzumab therapy due to very low levels of HER2 expression, and show a median survival of 6 months due to a lack of any other effective therapeutic modalities.8 Therapies targeting vascular endothelial factor (VEGF) are also being explored with evidence of improved overall survival by 2 months.9 Yet, there is still a crucial need to develop new treatment modalities for GC.
Cancer immunotherapy with antibodies that block the interaction between programmed death-1 (PD-1) and its ligand (PD-L1) has produced durable responses in some types of solid tumors, particularly melanoma and lung cancer.10,11 In advanced GC, however, the efficacy of immune checkpoint inhibitors has been marginal, with objective response rates of 7%–26% across varying GC populations.12 CAR T cell therapy is another type of cancer immunotherapy that has substantially improved outcomes in patients with hematologic malignancies.13 In the setting of GC, a number of preclinical and clinical studies have examined the efficacy of CAR T cells that target antigens such as HER2, carcinoembryonic antigen (CEA), epithelial cell adhesion molecule (EpCAM), mesothelin (MSLN), and claudin 18.2 (CLDN18.2).14, 15, 16, 17, 18 However, the application of CAR T cell therapy for solid tumors in clinical studies is associated with limited success, partly due to the lack of tumor-specific antigens as well as inhibitory factors in the tumor microenvironments.19,20
Intercellular adhesion molecule 1 (ICAM-1) is a cell surface glycoprotein receptor involved in cell-cell and cell-matrix adhesive interactions.21 ICAM-1 is overexpressed in several cancers, including GC, and the survival rate of GC patients with high ICAM-1 expression is worse than that of those with low expression.22 Recently, we reported the robust results of CAR T cell therapy targeting ICAM-1 in thyroid cancer models. A single administration of ICAM-1 CAR T cells showed tumor killing that resulted in long-term remission and significantly improved survival in anaplastic thyroid cancer (ATC) xenograft and ATC patient-derived xenograft (PDX) models.23,24
In this study, we first examined the correlation between ICAM-1 expression and the prognostic outcomes of GC patients, which justifies ICAM-1 as an immunotherapeutic target for CAR T cell therapy in GC. The efficacy of ICAM-1 CAR T cells was then evaluated in systemic and peritoneal human GC xenografts. We further explored the combination of CAR T cell therapy with paclitaxel and interleukin (IL)-12 release to improve CAR T cell activity against difficult-to-treat peritoneal GC tumors. In summary, our study has demonstrated promising activity of ICAM-1 CAR T cells against GC solid tumors, and potential to improve CAR T cell efficacy by leveraging strategies to augment T cell functions.
Results
Patient Characteristics and ICAM-1 Expression in Tumor Tissues
The clinicopathologic parameters of GC patients enrolled in the CLASSIC trial who underwent radical gastrectomy with or without adjuvant chemotherapy were analyzed (n = 134; Table 1). The median age was 55 years (range, 20–76 years), with 93 patients (70.1%) being men. Fifty-four cases (40.3%) were stage II disease and 80 cases (59.7%) were stage III disease. Seventy patients (52%) underwent gastrectomy only, and 64 patients (48%) received gastrectomy and adjuvant chemotherapy. Positive ICAM-1 expression (histoscore [H score] ≥ 10; example images of ICAM-1 immunohistochemistry [IHC] are shown in Figure 1A) was found in 48 patients (35.8%) and was associated with a more advanced stage (20.4% in stage II versus 46.3% in stage III, p = 0.002) and nodal metastasis (26.7% in N stage 0, 27.8% in N stage 1, and 55% in N stage 2, p = 0.012). Ten patients (7.4%) were HER2 positive, and only two of them were ICAM-1 positive. According to the intensity of ICAM-1 staining, 12 of 54 cases (22.2%) of stage II and 39 of 80 cases (48.8%) of stage III were intensity 1 or higher (Table S1).
Table 1.
Baseline Characteristics of Patients According to ICAM-1 Expression
| Characteristics | ICAM-1 Negative (n = 86, 64%) | ICAM-1 Positive (n = 48, 36%) | p Value | ||
|---|---|---|---|---|---|
| Age | 0.371 | ||||
| median | 52 | 51 | |||
| range | 20–73 | 40–76 | |||
| Sex | 0.149 | ||||
| male | 56 (60.2) | 37 (39.8) | |||
| female | 30 (73.2) | 11 (26.8) | |||
| Lauren’s classification | 0.157 | ||||
| intestinal | 32 (69.6) | 14 (30.4) | |||
| diffuse | 43 (57.3) | 32 (42.7) | |||
| mixed and unclassifiable | 11 (84.6) | 2 (15.4) | |||
| HER2 status | 0.331 | ||||
| negative | 78 (62.9) | 46 (37.1) | |||
| positive | 8 (80) | 2 (20) | |||
| Surgery types | 0.262 | ||||
| subtotal gastrectomy | 58 (68.2) | 27 (31.8) | |||
| total gastrectomy | 28 (57.1) | 21 (42.9) | |||
| Stage | 0.002 | ||||
| II | 43 (79.6) | 11 (20.4) | |||
| III | 43 (53.8) | 37 (46.3) | |||
| T stage | 0.102 | ||||
| 1 | 2 (66.7) | 1 (33.3) | |||
| 2 | 36 (75) | 12 (25) | |||
| 3 | 47 (57.3) | 35 (42.7) | |||
| 4 | 1 (100) | 0 | |||
| N stage | 0.012 | ||||
| 0 | 11 (73.3) | 4 (26.7) | |||
| 1 | 57 (72.2) | 22 (27.8) | |||
| 2 | 18 (45) | 22 (55) | |||
| Adjuvant chemotherapy | 0.979 | ||||
| no | 45 (64.3) | 25 (35.7) | |||
| yes | 41 (64.1) | 23 (35.9) | |||
Figure 1.

ICAM-1 Expression Is Associated with Poor Prognosis in Gastric Cancer Patients
(A) ICAM-1 expression was graded as negative (0) (i), weak (1, light brown) (ii), moderate (2, brown) (iii) or strong (3, dark brown) (iv). Scale bars, 20 μm. (B and C) Kaplan-Meier plots indicating a significant difference in disease-free survival (B) and overall survival (C) between ICAM-1− (blue lines) and ICAM-1+ (green lines) gastric cancer patients (n = 134).
The Prognostic Value of ICAM-1 Expression in GC Patients
We evaluated the association between ICAM-1 expression and survival. Patients with positive ICAM-1 expression showed significantly shorter disease-free survival (DFS) than did those who were ICAM-1 negative (5-year DFS rate, 36.1% versus 75.7%, p < 0.001) (Figure 1B). In addition, ICAM-1 expression was significantly associated with worse overall survival (OS) (5-year OS rate, 49.7% versus 84.5%, p < 0.001) (Figure 1C).
ICAM-1 Expression of GC Cell Lines
We selected eight GC cell lines for evaluation of ICAM-1 expression and as target cells for validation of CAR T cell activity. According to the molecular subtypes by The Cancer Genome Atlas (TCGA),25,26 SNU-719 and NCC-24 were assigned to the Epstein-Barr virus (EBV) subtype, SNU-638 and SNU-1 to microsatellite instability (MSI), MKN-28 and SNU-5 to chromosomal instability (CIN), and Hs746t and SNU-601 to genomically stable (GS) (Table S2). By Lauren’s classification,27 SNU-1, SNU-5, SNU-601, NCC-24, and Hs746t were diffuse type, while SNU-638, SNU-719, and MKN-28 were intestinal type. Most GC cell lines were found to have an intermediate to high level of ICAM-1 expression, except for SNU-1 and SNU-5, which were negative for ICAM-1 (Figure 2A). Among the six ICAM-1+ GC cell lines, Hs746t showed the highest expression of ICAM-1, comparable to or slightly higher than ICAM-1 expression in a control cell line HeLa (∼106 molecules per cell24). The level of ICAM-1 expression in SNU-601, SNU719, and NCC-24 was moderate when compared to other GC cell lines with high ICAM-1 expression. However, there were no associations between the level of ICAM-1 expression and Lauren’s classification (intestinal type versus diffuse type) or TCGA genotypes (EBV, MSI, CIN, and GS).
Figure 2.

Correlation of ICAM-1 Expression with CAR T Cell Cytotoxicity in GC Cell Lines
(A) Flow cytometry histograms of ICAM-1 expression in ICAM-1− 293T, ICAM-1+ HeLa, and GC cell lines. (B) Schematic diagram of the ICAM-1 CAR lentiviral vector. Human SSTR2 was introduced after CAR and a ribosome skipping porcine teschovirus-1 2A (P2A) sequence for PET imaging of CAR T cell biodistribution. LTR, long terminal repeat; SS, signal sequence; TM, transmembrane; Cyt, cytoplasmid domain; EF1α, elongation factor 1α. (C) Flow cytometry plots showing CAR expression determined by anti-c-Myc and anti-SSTR2 antibodies. CD4/CD8 T cell subsets were determined with an anti-human CD3/CD4/CD8 antibody cocktail. (D) Effector-to-target assay measuring lysis of target cells by ICAM-1 CAR T cells from two different donors (CP1 and CP5) at an effector-to-target ratio of 2.5:1. The percentage of live cells was measured by bioluminescence intensity normalized to the level of target cells co-cultured with non-transduced T cells. Data represent mean ± SD of triplicate wells. (E) Comparison of target cell killing at 24 h based on Lauren’s classification, TCGA genotypes, or ICAM-1 expression (with inclusion of 293T and HeLa controls). Results were pooled from experiments with CP1 and CP5 as described in (D). Each value represents mean of triplicate wells. Statistical significance was determined by an unpaired, two-tailed t test or one-way ANOVA. ∗∗∗p < 0.001. ns, not significant.
ICAM-1 CAR T Cell Activity against GC Cell Lines In Vitro
Primary T cells from healthy donors were transduced (a schematic diagram of CAR vector is shown in Figure 2B) and expanded using an automated CliniMACS Prodigy system.28 Final CAR T cell products displayed comparable transduction efficiencies (51% ± 6%) and varying frequencies of CD4 and CD8 populations (Figure 2C; Table S3). We used CP1 and CP5 to test the ability of ICAM-1 CAR T cells to eliminate GC cells in vitro. For the effector-to-target (E:T) testing, we established target cell lines with stable expression of GFP and firefly luciferase (FLuc) via transduction with a FLuc-GFP lentivirus and cytometry-based sorting for GFP+ cells. E:T assays normalized to non-transduced (NT) cells demonstrated that the lytic activity of ICAM-1 CAR T cells was dependent on the level of ICAM-1 expression in target cells, regardless of Lauren’s classification or TCGA genotypes (Figures 2D and 2E). Overall, target cells with high ICAM-1 expression were eliminated more rapidly than cells with moderate levels of ICAM-1 expression, whereas minimal killing was observed for cells that were ICAM-1 negative (Figure 2E). In particular, SNU-638 was the most susceptible to CAR T cells, with approximately 50% of SNU-638 cells being lysed after 9 h of incubation with CP1 and CP5. Despite having the highest ICAM-1 expression, Hs746t displayed a delayed killing by CP5, while its lysis by CP1 was comparable to that of other ICAM-1-high target cells (e.g., MKN-28) or slightly slower than that of others (e.g., SNU-638, NCC-24). SNU-1 and SNU-5 with no ICAM-1 expression were largely insensitive to CAR T cell cytotoxicity, similar to the ICAM-1− 293T control cell line. With the exception of Hs746t, cytotoxic activities of CP1 versus CP5 were similar against all other GC cell lines (Figure 2D).
ICAM-1 CAR T Cell Activity against Systemic GC Xenografts in Mice
Prior to testing CAR T cell activity against GC xenografts in mice, we first examined the rate of tumor growth and engraftment patterns after intravenous (i.v.) versus intraperitoneal (i.p.) injections of GC cell lines. All eight cell lines exhibited robust engraftment in the peritoneal cavity after i.p. injection, confirmed by the growth of distinct masses detected by bioluminescence. In comparison, i.v. injection of cell lines led to two distinct growth patterns, one defined by engraftment in the lungs followed by growth in the peritoneum (SNU-601, MKN-28, SNU-719, and Hs746t) and the other by a growth in the peritoneal cavity only (SNU-638, SNU-5, NCC-24, and SNU-1) (Table S2). Hs746t was chosen for a systemic xenograft model due to its consistent engraftment in the thoracic cavity and high ICAM-1 expression (Figure 2A). Mice were xenografted by tail-vein injection of Hs746t cells (1.5 × 106 cells) and 8 days later were treated with either NT T cells (donor-matched NT, 10 × 106 cells, n = 4), ICAM-1 CAR T cells (CP5, 10 × 106 cells, n = 6), or left untreated (no treatment [no T], n = 4). In untreated mice or mice treated with NT cells, tumors continued to grow in the lungs and subsequently in the peritoneal cavity, causing death within 90 days after xenograft (Figures 3A and 3D). In comparison, mice treated with CAR T cells exhibited complete remission within 2 weeks of treatment, which lasted for variable times. However, tumor relapse in the CAR T cell cohort eventually occurred in two of the six animals. There was no obvious indication of toxicity in CAR T cell-treated mice, as assessed by no significant change in body weight (Figure 3C) and no sign of distress. However, two of the four tumor-free mice began to show signs of graft-versus-host disease (GvHD) starting 7–8 weeks after treatment, evidenced by weight loss, ruffled fur, and alopecia, and eventually expired without evidence of tumor relapse. Overall, the CAR T cell cohort displayed a significantly longer survival than did the no T or NT treatment cohorts (Figure 3D, p < 0.05). Positron-emission tomography (PET)/computed tomography (CT) imaging using a SSTR2 tracer, 18F-1,4,7-triazacyclononane-1,4,7-triacetic acid-octreotide (NOTAOCT),24 revealed the expansion of ICAM-1 CAR T cells in the lungs where tumor growth was observed by bioluminescence imaging (Figure 3E). Importantly, CAR T cells gradually contracted after eliminating tumors, as evidenced by decreasing PET signals in the lungs and lymph nodes over time. In contrast, untreated mice or mice treated with NT cells without SSTR2 expression showed no tracer retention in the lungs. A background signal of 18F-NOTAOCT was seen in the kidneys and bladder due to renal clearance, and less intensely in the gallbladder and intestinal track caused by hepatobiliary clearance.29
Figure 3.

Efficacy of ICAM-1 CAR T Cells in a Systemic Xenograft Model
(A) Whole-body bioluminescence images of Hs746t-engrafted NSG mice without treatment (no T), or treated with either NT or ICAM-1 CAR T cells (CP5). (B) Quantitation of total body bioluminescence (n = 4–6 mice per cohort). Thick lines indicate mean bioluminescence intensity, while thin lines represent data for each individual mouse. (C) Summary of body weight changes over time. Data represent mean ± SD (n = 4–6). (D) Kaplan-Meier survival curves (log-rank [Mantel-Cox] test; ∗p < 0.05, ∗∗p < 0.01). (E) Representative PET/CT (coronal view of 20-mm-thick plane, maximum intensity projection) images showing 18F-NOTAOCT uptake in mice left untreated or treated with either NT or ICAM-1 CAR T cells.
ICAM-1 CAR T Cell Activity against i.p. GC Xenografts in Mice
After validation of potent GC tumor elimination following i.v. injection of CAR T cells, we then examined CAR T cell efficacy against tumors developed in the peritoneal cavity. Peritoneal dissemination is the most common pattern of recurrence or metastasis in GC and is associated with poor prognosis.30 For an i.p. xenograft model, we chose SNU-638 for its aggressive growth in the peritoneal cavity and sensitivity to CAR T cells in vitro (Figure 2D). Mice were xenografted i.p. with SNU-638 cells (3 × 106 cells) and treated 5 days post-xenograft with either NT cells, CAR T cells (CP1), or left untreated (no T) (Figure 4A). To examine differences in CAR T cell efficacy by the route of injection, T cells were injected into mice 5 days after tumor injection via either the tail vein or i.p. cavity at two different doses (low dose, 1 × 106 CAR T cells; high dose, 10 × 106 CAR T cells). All untreated mice and the cohorts of i.v. and i.p. NT cell treatments showed continued tumor expansion and expired within 45 days of xenograft (Figures 4B and 4D). In comparison, CAR T cell-treated mice exhibited lower tumor burden and survived longer than did the no T or NT cell cohorts. i.p. delivery of CAR T cells resulted in a more pronounced treatment effect and survival benefit over i.v. delivery (Figures 4C and 4D). In the cohort treated with high-dose i.p. CAR T cells, the tumors seemed to be almost completely eradicated at 9 days post-xenograft; however, tumor relapse occurred as early as 2 weeks after complete response in most mice. The loss of body weight overall was caused by an increase in tumor burden (Figure 4E).
Figure 4.

Efficacy of ICAM-1 CAR T Cells in an Intraperitoneal Xenograft Model
(A) Whole-body bioluminescence image of SNU-638-engrafted NSG mice without treatment (no T), or treated with non-transduced T (NT) or low or high doses (LD or HD) of ICAM-1 CAR T cells. Mice were treated with T cells 5 days after tumor xenograft either by intravenous or intraperitoneal injection. LD, 1 × 106 CAR T cells; HD, 10 × 106 CAR T cells. (B) Quantitation of total body bioluminescence intensity. Data represent mean ± SD (n = 2–3). (C) Bioluminescence intensities on day 33 following xenograft. LD and HD cohorts were pooled for analysis. An unpaired, two-tailed t test was used for statistical comparisons. ∗p < 0.05, ∗∗p < 0.01. ns, not significant. (D) Kaplan-Meier survival curves. (E) Summary of body weight changes over time. Data represent mean ± SD (n = 2–3). (F) GFP images of tumors and gastrointestinal tracts acquired on day 85 post-xenograft. (G) Histologic images of H&E staining, GFP IHC, and CD3 IHC of tumor or spleen from mice treated with ICAM-1 CAR T cells.
Ex Vivo Validation of CAR T Cell Tumor Infiltration
Ex vivo images of the gastrointestinal organs further validated the treatment effect of CAR T cells against SNU-638 peritoneal tumors. In untreated mice, tumors appeared to form multiple lesions along the intestinal tract, identifiable by GFP imaging (Figure 4F). In comparison, tumor lesions in the intestinal tract of CAR T cell-treated mice were less frequent and smaller. IHC analysis of tumor nodules from the mice treated with ICAM-1 CAR revealed CD3+ T cells infiltrating GFP+ tumors (Figure 4G). Close inspection of the image revealed the snapshot of CAR T cell activity against tumors: the area with high CD3 density appeared to be largely devoid of tumor cells, while the area with sparsely distributed CD3 cells still contained a high density of tumor cells. In the spleen of the same mice, CD3+ human T cells were observed in high abundance even at 80 days after T cell infusion.
Combined Treatment of CAR T Cells with Paclitaxel in an i.p. Xenograft Model
Although ICAM-1 CAR T cells administered i.p. at a high dose appeared to have a survival benefit, the efficacy was modest and short-lived, and most treated animals eventually succumbed to tumor relapse and death. To examine whether the lower tumor burden at the time of CAR T cell treatment would lead to a better outcome, we first developed peritoneal tumors at different doses of SNU-638 cells (0.1 × 106, 0.5 × 106, or 3 × 106 cells per mouse) and analyzed the survival rate of each cohort. As expected, non-obese diabetic (NOD) severe combined immunodeficiency (SCID) gamma (NSG) mice survived proportionally longer when the xenograft dose was reduced (Figure 5A). In addition to reducing the xenograft dose to 0.1 × 106 SNU-638 cells, we explored the combination of CAR T cell therapy with chemotherapy, which has been shown to sensitize tumor cells to immunotherapy in preclinical studies.31,32 Among chemotherapeutic agents, we used paclitaxel, which is the most commonly used agent in advanced GC as a second-line therapy.33 In murine models, paclitaxel has been previously used, ranging up to 75 mg/kg for i.p. injection.34 In a pilot study, a weekly i.p. injection of 20 mg/kg paclitaxel for 3 weeks led to frequent severe toxicities or death (four out of six mice), while a dose of 10 mg/kg of paclitaxel was found to be well tolerated. For a combination study, a dose of 10 mg/kg for paclitaxel was chosen for i.p. weekly injection for 3 weeks, commencing on 2 days prior to CAR T cell infusion, and 5 and 12 days after infusion of CAR T cells. The cohorts of either T cells alone or T cells in combination with paclitaxel were infused with NT versus CAR T (CP1, 10 × 106) cells i.p. 7 days after tumor xenograft (n = 5 for each group; Figure 5B). Compared to the cohorts of no T or NT cells, paclitaxel monotherapy or paclitaxel with NT cells suppressed tumor growth; however, tumors reappeared once the treatment with paclitaxel ended. CAR T cell therapy alone produced complete to partial responses, which lasted for variable times before tumors began to grow back in most mice. In comparison, the combination therapy with CAR T cells and paclitaxel led to the most potent suppression of tumor growth, attaining complete response in two out of five mice. However, mice with tumor suppression in this cohort eventually succumbed to the regrowth of tumors (Figure 5B). Overall, the combination cohort achieved the longest survival, while CAR T cells alone, paclitaxel monotherapy, and paclitaxel with NT cohorts produced survival benefit only slightly better than the no T or NT cohort (Figure 5D). PET/CT imaging with 18F-NOTAOCT taken at 8 weeks after T cell injection confirmed the expansion and localization of CAR T cells in the lungs and even in peritoneal cavity, judging from significantly higher tracer uptake in the lower body compared to the levels seen in control mice (peritoneal areas other than kidneys and bladder in Figure 5E versus no T or NT mice in Figure 3B). Despite potential cytotoxic effects of paclitaxel on CAR T cells (Figure S1), our imaging supports the ability of CAR T cells to recover and persist in mice even after repeated administration of paclitaxel.
Figure 5.

Superior Efficacy of Combination Treatment of ICAM-1 CAR T Cells with Paclitaxel in an Intraperitoneal Xenograft Model
(A) Kaplan-Meier survival curves of untreated NSG mice intraperitoneally xenografted with different doses of SNU-638 cells (log-rank [Mantel-Cox] test; ∗∗p < 0.01; ns, not significant). (B) Whole-body bioluminescence images of SNU-638-engrafted NSG mice received either no treatment, or monotherapy with either T cells or paclitaxel, or combinatorial treatment. (C) Quantitation of total body bioluminescence intensity. Data represent mean ± SD (n = 5). (D) Kaplan-Meier survival curves (log-rank [Mantel-Cox] test; ∗p < 0.05, ∗∗p < 0.01; ns, not significant). (E) Representative PET/CT images of mice treated with ICAM-1 CAR T cells on 9 weeks post-xenograft (8 weeks after T cell injection). Coronal view of PET/CT images pseudo-colored with PET maximum intensity projection over a 20-mm-thick plane.
Inducible IL-12 Boosts CAR T Cell Activity against Peritoneal GC Tumors
As an alternative to combining CAR T cells with chemotherapy, we explored the incorporation of inducible cytokines into the CAR vector to promote CAR T cell proliferation and effector functions, and to improve CAR T cell activity against peritoneal tumors. We designed a lentiviral vector that incorporates the expression cassettes for CAR and CAR activation-dependent IL-12 to support the cytotoxic activity and survival of T cells and the T helper (Th)1 T cell response (Figure 6A).35,36 In order to locally deliver IL-12 upon CAR activation in the targeted tumor lesions while avoiding systemic toxicity that has been observed with constitutive secretion or systemic administration of IL-12,37, 38, 39 an inducible IL-12 gene was placed downstream of the synthetic promoter containing both a nuclear factor κB (NF-κB)-responsive promoter and IL-2 minimal promoter with six repeated binding sites for nuclear factor of activated T cells (NFAT) (designated as the NF-κB-NFAT promoter). An inducible GFP construct was also designed to validate the specificity of the NF-κB-NFAT promoter upon target cell stimulation. In Jurkat T cells transduced with ICAM-1 CAR/GFP, GFP expression was seen after co-incubation for 24 h at 37°C with human ICAM-1-transduced 293T cells (Figure 6B). Similarly, ICAM-1 CAR/IL-12 T cells secreted elevated levels of IL-12 after co-incubation with ICAM-1+ 293T cells or the addition of phorbol myristate acetate (PMA)/ionomycin to media, but not with ICAM-1− 293T cells, indicating that IL-12 secretion was specific to CAR signaling (Figure 6C). From the E:T assay measured at 24 h after incubation of target cells with T cells at a 1:1 ratio, CAR/IL-12 T cells mediated significantly higher killing of ICAM-1+ HeLa and SNU-638 cells compared to CAR T cells without IL-12 release (Figure 6D). Cytotoxicity of IL-12-armored CAR T cells was ICAM-1-dependent with minimal killing of ICAM-1− 293T cells. To compare the treatment efficacy in vivo, donor-matched CAR T (CP9, 15 × 106 total cells) and CAR/IL-12 T cells (15 × 106 total cells) were administered to mice i.p. 1 week after an i.p. xenograft with 0.5 × 106 of SNU-638 cells. Whole-body bioluminescence imaging revealed that CAR/IL-12 T cells reduced the tumor burden significantly more compared to untreated control or CAR T cells (Figures 6E and 6F). We collected peripheral blood at 2, 3, and 6 weeks after tumor xenograft to determine cytokine levels and the abundance of circulating T cells. As expected, IL-12 was detected only in the blood of mice treated with CAR/IL-12 T cells, which gradually increased to ∼1 ng/mL (Figure 6G). The improved tumor-lytic capacity of CAR/IL-12 in vivo was also accompanied with increased levels of serum interferon (IFN)-γ and tumor necrosis factor (TNF)-α (Figure 6G). Peripheral blood collected at 5–8 weeks after tumor xenograft revealed significant expansion of CD3+ T cells after treatment with CAR/IL-12 T cells compared to treatment with CAR T (CP9) cells (79.4% versus 19%) (Table S4). However, greater expansion of CD3+ T cells due to IL-12 secretion was not limited to CAR-transduced T cells (8% CAR positive in pre-infusion [Table S3] and 13% CAR positive in post-infusion [Table S4]), alluding to the systemic effect of IL-12, causing an expansion of all T cells. Collectively, our findings highlight the superior anti-tumor activity of CAR/IL-12 T cells against difficult-to-treat peritoneal tumors.
Figure 6.

Inducible IL-12 Release Augments Antitumor Activity of CAR T Cells in a GC Peritoneal Tumor Model
(A) Schematic diagram of the lentiviral vector encoding inducible IL-12. The expression of ICAM-1 CAR is driven by the EF1α promoter, while IL-12 expression is under the transcriptional control of a synthetic NF-κB-NFAT promoter. (B) GFP expression in Jurkat CAR/GFP T cells after coculture for 24 h with 293T cells with or without human ICAM-1 (hICAM-1) expression. CAR expression was estimated by staining with an anti-SSTR2 antibody. (C) IL-12 release by Jurkat CAR/IL-12 T cells after coculture with 293T cells with or without human ICAM-1 expression, or with the addition of PMA and ionomycin to media, measured in duplicate (mean ± SD). (D) Comparison of target cell lysis by CAR T (CP9) and CAR/IL-12 T cells during 24 h at an E:T ratio of 1:1. Data represent mean ± SD (n = 4; unpaired, two-tailed t test; ∗∗∗∗p < 0.0001; ns, not significant). (E) Whole-body bioluminescence images of NSG mice xenografted with 0.5 × 106 SNU-638 (two independent experiments, n = 6–7 per cohort). (F) Quantitation of bioluminescence intensity over time (n = 6–7 per cohort). Each line represents data for each individual mouse. (G) Cytokine levels in the plasma were measured from the blood collected at 2, 3, and 6 weeks post-xenograft. Data represent mean ± SD (n = 1–2).
Discussion
Currently, the treatment options for GC patients with locally advanced and metastatic disease are very limited, with a median survival of less than 1 year. Moreover, systemic and targeted therapies for peritoneal disease have very poor efficacy.8 In this study, we examined the therapeutic potential of CAR T cells targeting ICAM-1 in preclinical models of systemic and i.p. metastases of GC. Overall, a single infusion of ICAM-1-targeting CAR T cells was found to potently eliminate tumors generated in mice via i.v. injection that initiated tumor growth in the lungs. This is similar to the efficacy of ICAM-1-targeting CAR T cells observed against systemic ATC in our previous studies,23 providing a further proof of concept for this treatment modality against systemic tumors with high levels of ICAM-1 expression. In comparison, the same CAR T cells, delivered either i.v. or i.p., were less effective against tumors injected directly into the peritoneal cavity that developed into lesions along the intestinal tract and peritoneal membrane. The lower activity of CAR T cells against peritoneal tumor lesions is likely due to limitations hindering CAR T cell therapy against solid tumors, which include challenges to tumor penetration, an immune-suppressive milieu, and maintaining T cell activity in hypoxic and nutrient-depleted surroundings.40, 41, 42
We have chosen ICAM-1 as a target for our CAR T cells against GC based on the finding that ICAM-1 expression was seen in approximately 40% of the GC patients enrolled in a prospective trial,43 and it was directly associated with lymph node metastasis and higher stage disease. GC patients with positive ICAM-1 expression also showed significantly shorter OS and DFS. However, ICAM-1 expression has also been reported by others to be inversely correlated with the incidence of lymph node or distant metastases in colorectal, gastric, and breast cancer patients, and to be associated with a better prognosis.44, 45, 46 In one scenario, ICAM-1 expression in tumors may stimulate interaction with the immune cells via ICAM-1 interaction with integrins, including LFA-1,47 and boost immune surveillance. In contrast, ICAM-1 expression has also been observed in malignant, dedifferentiated tumors, including thyroid and breast cancers, as well as cancer stem cells.23,48, 49, 50 The reason for the discrepancy in the role of ICAM-1 on tumor growth and metastasis is unclear. It is possible that ICAM-1 expression at an early stage of tumor development may suppress tumor growth by elevating immune surveillance, while at a later stage ICAM-1 is further induced with tumor malignancy and loss of differentiation.
Aiming at achieving optimal response while avoiding toxicity toward normal cells with basal levels of ICAM-1 expression, in this study, we used affinity-tuned ICAM-1 CAR T cells (equilibrium dissociation constant [KD] of ∼20 μM).24 The use of low-affinity CAR against target antigen resulted in the lytic activity of CAR T cells to be in strong correlation with the level of ICAM-1 expression in GC cell lines in vitro. CAR T cells targeting other antigens, such as HER2, CEA, EpCAM, MSLN, and CLDN18.2, are being tested in gastric and colorectal cancer patients. Early clinical study of HER2 CAR T cells against metastatic colon cancer showed fatal on-target off-tumor toxicities mediated by recognition of HER2 in non-malignant pulmonary tissue.51 However, the newer generation of HER2 CAR T cells with lower affinity was well tolerated with no dose-limiting toxicity, although the treatment benefits were not obvious in HER2+ malignancies, including sarcoma and glioblastoma.14,52 CEA-targeting CAR T cells mediated tumor remission and inhibited tumor growth after re-challenges in preclinical colorectal peritoneal carcinomatosis models.15 This study has shown that a regional i.p. infusion resulted in superior anti-tumor response than did systemic delivery of CAR T cells. i.p. infused CEA CAR T cells are currently being studied in patients with CEA-expressing peritoneal metastases (ClinicalTrials.gov: NCT03682744).
Eight different GC cell lines developed tumors intrathoracically, i.p., or both when injected into mice i.v. via the tail vein. In comparison, all i.p. injections of cell lines led to tumor growth in the peritoneal space. The efficacy of ICAM-1-targeting CAR T cells was then evaluated in two different xenograft models, that is, an intrathoracic tumor growth model using the Hs746t cell line and i.p. tumor growth using SNU-638. In the intrathoracic xenograft model, the systemic administration of CP5 CAR T cells achieved a durable response with a 60% cure rate. Although CP5 cytotoxic activity against Hs746t seemed to be modest with a delay in the onset of killing in vitro, its activity in vivo was rapid, as demonstrated by complete tumor elimination within 2 weeks of treatment.
In the i.p. metastasis model, we compared the efficacy of CAR T cells delivered via the tail vein versus direct injection into the i.p. cavity. Similar to the previous report that intrapleural or i.p. injection of CAR T cells showed higher efficacy than did systemic administration against various tumor xenografts,15,53,54 we observed a significantly higher tumor response to CAR T cells delivered i.p. This is likely due to the ability of T cells to traffic more effectively and accumulate in the peritoneal space in higher numbers. However, despite the higher efficacy and longer survival after i.p. CAR T cell delivery, the overall response of peritoneal tumors was more modest compared to the response of tumors growing in the lungs. Incomplete response of CAR T cells against i.p. tumors was not due to a lack of CAR T cell persistence, as evidenced by histologic confirmation and PET/CT detection of CAR T cells in the body, including peritoneal space, lung, spleen, and lymph nodes. As shown from immunostaining of tumor tissues, the presence of sparsely distributed, single T cells interspersed among tumor cells may allude to the suppression of CAR T cell activity and proliferation by the tumor environment surrounding T cells.55
Preclinical studies have demonstrated that chemotherapy can sensitize tumor cells to immunotherapy, likely by improving tumor antigen recognition and inhibiting suppressive immune cells.31,32,56 In addition, CAR T cell therapy after conditioning chemotherapy has shown a better response rate and greater CAR T cell expansion.57,58 Therefore, we tested the combined treatment of paclitaxel and ICAM-1 CAR in an SNU-638 i.p. tumor model. Paclitaxel has been used as a standard second-line chemotherapy in GC worldwide,33 and it has a highly favorable concentration ratio of i.p. to plasma and remarkable drug penetration into cells.59 Monotherapy with paclitaxel or CAR T cells suppressed tumor growth at the early stage, yet tumor relapse eventually occurred in most animals. For the combination therapy, paclitaxel was given three times, 2 days prior to, as well as 5 and 12 days after CAR T cell infusion. The cytotoxic effect of paclitaxel would therefore not only be on tumor cells but also on CAR T cells, potentially limiting the full benefit of combination therapy. Paclitaxel was indeed confirmed to significantly reduce T cell proliferation in vitro at a dose (10 mg/kg, which is approximately 15–20 μM assuming 60% body weight being paclitaxel-accessible fluid) that would mimic the concentration of paclitaxel that T cells are exposed to (Figure S1). Despite potential cytotoxic effects by paclitaxel, CAR T cells appeared to have recovered and expanded, evidenced by PET/CT imaging of CAR T cell presence in the body. Therefore, incomplete elimination of tumor by combination therapy is not due to a lack of CAR T cell persistence, but more likely due to immune-suppressive effects caused by tumor growth. Our future studies will further explore optimization of the dose and frequency of CAR T cells and paclitaxel administration in order to further maximize the efficacy of combination therapy.
To improve the efficacy of CAR T cell treatment in solid tumors, combination therapy with other immunotherapeutic modalities, such as immune checkpoint inhibitors,60,61 oncolytic virus,62,63 and vaccines,64,65 has also been shown to improve the efficacy of CAR T cells. Some of these approaches are being used in clinical trials and may overcome the current problems encountered in the setting of solid tumors. One alternative approach is arming CAR T cells with T cell-activating cytokines, including IL-12, IL-7/CCL19, IL-15, IL-18, and IL-23.66, 67, 68, 69, 70 These cytokines were found to differentially affect T cell phenotype, persistence, and proliferation, and to improve CAR T cell treatment. IL-12-producing CAR T cells in particular resulted in longer survival and increased tumor cytotoxicity in preclinical models.66,71,72 Besides promotion of Th1 phenotype and expansion of T cells, IL-12 can augment CAR T cell activity by suppressing PD-1 upregulation, which is induced by T cell activation.73 Compared to prior studies of arming CAR T cells with constitutive expression of IL-12, we have designed our CAR T cells to release IL-12 in a manner dependent on T cell activation. The sequence or the number of repeats of NF-κB and/or NFAT binding motifs may be optimized to reduce the level of inducible cytokine secretion to further enhance safety. However, the mouse model used in this study, i.e., the use of human tumor and T cells in an immune-compromised NSG strain, is inadequate to fully evaluate potential toxicity arising from IL-12. This is partly due to the fact that human IL-12 and IFN-γ do not cross-react with murine receptors.74 Furthermore, xenogeneic transplants of human T cells into immunodeficient NSG mice invariably cause GvHD in time due to the recognition of mouse peptide/major histocompatibility complex (MHC) by human T cell receptor (TCR).75 We speculate that a huge spike in IL-12 release and T cell expansion of our CAR/IL-12 T cells may have been caused by xenogeneic TCR activation and subsequent activation of NF-κB-NFAT promoter and IL-12 release. Additional evidence for this hypothesis was seen in a subcutaneous thyroid cancer model (ATC23): low IL-12 levels (<250 pg/mL) were measured until tumor was completely eliminated, followed by a sudden spike of IL-12 (∼2 ng/mL) coincident with the onset of GvHD symptoms such as weight loss and ruffled fur (Figure S2). The use of a mouse strain that is less prone to GvHD (e.g., an MHC class I- and MHC class 2-deficient strain76) or mouse CAR T cells with inducible IL-12 in an immune-competent strain77 will be beneficial to further optimize cytokine delivery in conjunction with CAR, prior to ultimately testing the risks and benefits of arming CAR T cells with cytokine secretion to treat human patients.
The characteristics of pre-infusion CAR T cells can significantly impact CAR T cell efficacy in vivo. For instance, a comprehensive analysis of intrinsic CD19 CAR T cell characteristics has revealed that pre-infusion CAR T cell fitness can be a predictive marker of clinical response.78 This study has shown that a higher CAR T cell potency in mice, T cell memory-related gene signature, and specific T cell subsets (CD27+PD-1−CD8+) were found to correlate with clinical response. In our study, we used three batches of CAR T cells (CP1, CP9, and CAR/IL-12 T cells) produced from the blood of two different donors to evaluate CAR T cell activity against an i.p. xenograft, and one batch of CAR T (CP5) cells against a systemic xenograft. Given the significant difference in the CD4-to-CD8 ratios among these batches (Table S3) and other characteristics that have not been examined in this study, it is possible that the tumor response to CAR T cells may vary significantly from batch to batch. Despite some notable differences in T cell characteristics, however, our previous study has demonstrated fairly homogeneous activity of six different CAR T cell batches against advanced thyroid cancer cells in vitro and in vivo.28 Clearly, such a homogeneous response in a preclinical model is greatly ascribed to the use of healthy donor blood for CAR T cell manufacturing, and the use of a mouse model with tumor xenograft.
In conclusion, we have demonstrated a robust response to ICAM-1 CAR T cell monotherapy against systemic metastases using a GC mouse model. In addition, combination with paclitaxel was found to significantly enhance the treatment effect of CAR T cells in the more refractory peritoneal GC metastasis model. When our ICAM-1 CAR T cells were armored with inducible IL-12, the treatment effect was also more pronounced against peritoneal metastatic gastric tumors, accompanied with higher levels of IFN-γ and TNF-α detected in plasma. Our study warrants the development of ICAM-1 CAR T cells as a new treatment option for ICAM-1-high advanced GC patients.
Materials and Methods
Patients and Tissues
To investigate ICAM-1 expression in tumor tissues of GC patients, we evaluated the tumor tissues of patients who were enrolled in the CLASSIC trial in Yonsei Cancer Center, Seoul, Korea.3 All patients had undergone radical gastrectomy with D2 lymph node dissection for primary gastric tumors and were randomized to treatments with adjuvant chemotherapy or surgery alone, according to the CLASSIC protocol. The current study included 134 patients with stage II–III malignancies, based on the Sixth Edition of the American Joint Committee on Cancer/Union for International Cancer Control guidelines.79 Informed consents for the CLASSIC trial were obtained from all patients. Since this study was performed more than 5 years after the CLASSIC trial was finished, additional informed consents were waived by the Institutional Review Board (IRB) of Severance Hospital (IRB no. 4-2017-1111).
IHC Analysis
ICAM-1 expression in human tissue was evaluated by IHC using an anti-ICAM-1 antibody (G-5, Santa Cruz Biotechnology). Scoring was performed by an independent pathologist blinded to the patients’ clinical information. Protein expression was interpreted by the weighted histoscore method (H score).80 The intensity of protein expression was scored as 0 (negative), 1 (light brown), 2 (brown), or 3 (dark brown). The final score was calculated as follows: (0 × % of negative cells) + (1 × % of light brown cells) + (2 × % of brown cells) + (3 × % of dark brown cells). Tumors with an H score of equal to or more than 10 were defined as positive ICAM-1 expression, while tumors with a score of less than 10 were defined as negative ICAM-1 expression. HER2 expression was analyzed by IHC using a HercepTest kit (Dako, Denmark), fluorescence in situ hybridization (FISH) using a Vysis HER2/CEP17 FISH probe kit (Abbott, USA) and a Dako detection kit (Dako, Denmark), and silver-enhanced in situ hybridization (SISH) using the Ventana Discovery XT system (Ventana/Roche, USA) according to the manufacturers’ instructions. FISH and SISH scores were assessed by detecting the fluorescence signal in 50 malignant cell nuclei. HER2 positivity was defined as either HER2 IHC 3+ or IHC 2+ and FISH/SISH positive (HER2/CEP17 [centromere enumerator probe 17] ≥ 2).
Cell Culture
We evaluated eight GC cell lines for ICAM-1 expression. HeLa and HEK293T cell lines were used as positive and negative controls for ICAM-1 CAR, respectively. SNU series and MKN-28 were obtained from the Korean Cell Line Bank (Seoul National University, Seoul, Korea), while Hs746t, HeLa, and HEK293T cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). All target cells were transduced with lentivirus encoding FLuc-F2A-GFP (Biosettia), and GFP-expressing cells were isolated by fluorescence-activated cell sorting (FACS). All cells were maintained in RPMI 1640 medium (Lonza, Walkersville, MD, USA) supplemented with 10% fetal bovine serum (Omega Scientific, Tarzana, CA, USA) and cultured in a humidified incubator at 37°C with 5% CO2.
CAR T Cell Manufacturing
ICAM-1 CAR T cells were manufactured using the automated CliniMACS Prodigy (Miltenyi Biotec) as described previously.28 ICAM-1 CAR T cells manufactured from three different donors were designated as CP1, CP5, and CP9, respectively. ICAM-1 CAR T cells with inducible IL-12 were produced from the same donor as CP9 and manufactured using a 50-mL bioreactor tube (TubeSpin, TPP) on a bottle roller (Thermo Scientific) housed in a humidified incubator. Briefly, primary human T cells were transduced with lentivirus twice at 24 and 48 h after activation with anti-CD3/CD28 Dynabeads (Gibco). T cells were expanded for 10 days in TexMACS GMP (good manufacturing process) medium (Miltenyi Biotec) supplemented with 5% human AB serum (Sigma-Aldrich), 12.5 ng/mL IL-7 (Miltenyi Biotec), and 12.5 ng/mL IL-15 (Miltenyi Biotec). Cell products were cryopreserved on day 10 in a 1:2 mixture of T cell complete growth medium and CS10 (STEMCELL) for in vitro and in vivo experiments.
Flow Cytometry Analysis
Live cells were gated by calcein blue (Thermo Fisher Scientific) and stained with fluorophore-conjugated antibodies. ICAM-1 expression in GC cell lines was determined by staining with 5 μg/mL of allophycocyanin (APC)-conjugated ICAM-1 antibody (clone HA58, BioLegend). CAR expression was detected by fluorescein isothiocyanate (FITC)-conjugated anti-c-Myc antibody (clone SH1-26E7.1.3, Miltenyi Biotec). SSTR2 expression was detected by APC-conjugated anti-human SSTR2 antibody (clone 402038, R&D Systems). T cell products were also analyzed with a cocktail of anti-human phycoerythrin (PE)/Cy5-CD3, PE-CD4, and FITC-CD8 antibodies (clone UCHT1, RPA-T4, RPA-T8, BioLegend).
In Vitro Measurements of Inducible GFP and IL-12 Expression
To monitor inducible GFP and IL-12 expression upon CAR stimulation, Jurkat CAR T cells (5 × 104 Jurkat T cells per well) were co-incubated with 293T cells with or without human ICAM-1 (hICAM-1) expression at an E:T ratio of 1:1, or activated with 10 ng/mL PMA and 500 ng/mL ionomycin. GFP expression was analyzed by flow cytometry, and IL-12 secretion in the culture supernatant was detected by Bio-Plex MAGPIX (Bio-Rad) following the manufacturer’s instructions.
E:T Assay
Target cells (5 × 103) with FLuc expression were co-cultured with NT T cells or CAR T cells at the indicated E:T ratios in T cell media containing 150 μg/mL d-luciferin (Gold Biotechnology) with no cytokine supplement. Luminescence values were measured using a plate reader (Tecan Infinite M1000PRO), and the values of luminescence were normalized to the NT-treated target cells.
Animal Study
Animal studies were approved by Weill Cornell Medicine’s Institutional Animal Care and Use Committee. For the i.v. xenograft model, Hs746t cells were injected into 4- to 6-week-old male NSG mice (Jackson Laboratory) via the tail vein. Treatment cohorts consisted of no T, NT, or ICAM-1 CAR. NT and ICAM-1 CAR were injected via the tail vein 8 days after tumor cell injection. SNU-638 cells were used for the i.p. xenograft model. For combination treatment with paclitaxel in the i.p. xenograft model with SNU-638, paclitaxel was administered i.p. once a week at 10 mg/kg for 3 weeks. In the inducible IL-12 ICAM-1 CAR T cell study, NSG mice bearing peritoneal SNU-638 tumors were left untreated (no T) or treated i.p. with CAR T cells with and without IL-12 release on day 7 following tumor xenograft. Luminescence imaging was performed weekly using a whole-body optical imager (In-Vivo Xtreme 4MP, Bruker; or in vivo imaging system (IVIS) Spectrum, PerkinElmer) 15 min after subcutaneous injection of 200 μL of 15 mg/mL d-luciferin (GoldBio) in live xenograft mice under anesthesia using 2% isoflurane in 2 L/min O2. PET/CT was performed as described previously24 for the detection of T cell distribution using a micro-PET/CT scanner (Inveon, Siemens) at 2 h after 18F-NOTAOCT81 injection. Tracer uptake was visualized in the unit of % injection dose (ID)/cm3, determined by the counts in a reference tube.82 Mouse peripheral blood samples were collected, and serum cytokine levels were measured by Bio-Plex MAGPIX (Bio-Rad) following the manufacturer’s instruction. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Paque density gradient centrifugation and analyzed with a cocktail of anti-human PE/Cy5-CD3, PE-CD4, and FITC-CD8 antibodies (clone UCHT1, RPA-T4, RPA-T8, BioLegend). For survival studies, mice were euthanized when they had a weight loss of greater than 30% and exhibited signs of severe illness, including lethargy, hunched posture, and inactivity. Ex vivo images were obtained using a whole-body optical imager (In-Vivo F Pro, Bruker). Tumors were resected and fixed in 10% buffered formalin before being transferred to 70% ethanol. IHC analysis for CD3 (ab135372, Abcam) and GFP (A6455, Invitrogen) was performed in xenograft tumor tissues (Molecular Cytology Core Facility, Memorial Sloan Kettering).
Statistical Analysis
Differences between patients who were positive or negative with regard to ICAM-1 expression were compared using the chi-square or Fisher’s exact test. DFS was defined as the time from the date of the operation to the date of recurrence, death, or last follow-up. OS was defined as the time of operation to the date of death (from any cause), or last follow-up. DFS and OS were analyzed using the Kaplan-Meier method and the log-rank test. For in vitro experiments and animal studies, the values were analyzed by an unpaired, two-tailed Student’s t test (between two groups) or one-way ANOVA (between three or more groups). Mouse survival curves were generated using the method of Kaplan-Meier, and the significance was analyzed with the log-rank (Mantel-Cox) test. The statistical tests used in each experiment are indicated in the figure legends. Statistical analyses were performed using SPSS software (v.23.0, SPSS, Chicago, IL, USA) and Prism 8 software (GraphPad, San Diego, CA, USA). Differences with a p value of <0.05 were considered statistically significant.
Author Contributions
M.J., Y.Y., R.Z., and M.M.J. conceived and supervised the project. M.J., Y.Y, J.E.M., M.Z., Y.V., I.M.M., and K.D.G. performed experiments and analyzed data. M.J., X.Z., D.S., Y.Y.C., J.-H.C., S.H.N., S.Y.R., H.C.C., and M.M.J. managed patients and provided animals and facilities. M.J., Y.Y., J.E.M., and M.M.J wrote the manuscript. All authors reviewed the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by NIH grant R01CA217059, Emerson Collective Cancer Research Fund (ECCRF 191824-01), a sponsored research grant (AffyImmune), and by an institutional grant (MI3, Weill Cornell Medicine). We thank Dr. Eric von Hofe for critical reading of this manuscript.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.08.009.
Supplemental Information
References
- 1.Rawla P., Barsouk A. Epidemiology of gastric cancer: global trends, risk factors and prevention. Prz. Gastroenterol. 2019;14:26–38. doi: 10.5114/pg.2018.80001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakuramoto S., Sasako M., Yamaguchi T., Kinoshita T., Fujii M., Nashimoto A., Furukawa H., Nakajima T., Ohashi Y., Imamura H., ACTS-GC Group Adjuvant chemotherapy for gastric cancer with S-1, an oral fluoropyrimidine. N. Engl. J. Med. 2007;357:1810–1820. doi: 10.1056/NEJMoa072252. [DOI] [PubMed] [Google Scholar]
- 3.Bang Y.J., Kim Y.W., Yang H.K., Chung H.C., Park Y.K., Lee K.H., Lee K.W., Kim Y.H., Noh S.I., Cho J.Y., CLASSIC trial investigators Adjuvant capecitabine and oxaliplatin for gastric cancer after D2 gastrectomy (CLASSIC): a phase 3 open-label, randomised controlled trial. Lancet. 2012;379:315–321. doi: 10.1016/S0140-6736(11)61873-4. [DOI] [PubMed] [Google Scholar]
- 4.Digklia A., Wagner A.D. Advanced gastric cancer: current treatment landscape and future perspectives. World J. Gastroenterol. 2016;22:2403–2414. doi: 10.3748/wjg.v22.i8.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim H.S., Ryu M.H., Zang D.Y., Park S.R., Han B., Kang W.K., Rha S.Y., Jung M., Kim J.S., Kang B.W. Phase II study of oxaliplatin, irinotecan and S-1 therapy in patients with advanced gastric cancer: the Korean Cancer Study Group ST14-11. Gastric Cancer. 2018;21:802–810. doi: 10.1007/s10120-018-0794-1. [DOI] [PubMed] [Google Scholar]
- 6.Lordick F., Allum W., Carneiro F., Mitry E., Tabernero J., Tan P., Van Cutsem E., van de Velde C., Cervantes A. Unmet needs and challenges in gastric cancer: the way forward. Cancer Treat. Rev. 2014;40:692–700. doi: 10.1016/j.ctrv.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Bang Y.J., Van Cutsem E., Feyereislova A., Chung H.C., Shen L., Sawaki A., Lordick F., Ohtsu A., Omuro Y., Satoh T., ToGA Trial Investigators Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 8.Montori G., Coccolini F., Ceresoli M., Catena F., Colaianni N., Poletti E., Ansaloni L. The treatment of peritoneal carcinomatosis in advanced gastric cancer: state of the art. Int. J. Surg. Oncol. 2014;2014:912418. doi: 10.1155/2014/912418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilke H., Muro K., Van Cutsem E., Oh S.C., Bodoky G., Shimada Y., Hironaka S., Sugimoto N., Lipatov O., Kim T.Y., RAINBOW Study Group Ramucirumab plus paclitaxel versus placebo plus paclitaxel in patients with previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (RAINBOW): a double-blind, randomised phase 3 trial. Lancet Oncol. 2014;15:1224–1235. doi: 10.1016/S1470-2045(14)70420-6. [DOI] [PubMed] [Google Scholar]
- 10.Hamid O., Robert C., Daud A., Hodi F.S., Hwu W.J., Kefford R., Wolchok J.D., Hersey P., Joseph R., Weber J.S. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019;30:582–588. doi: 10.1093/annonc/mdz011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gandhi L., Rodríguez-Abreu D., Gadgeel S., Esteban E., Felip E., De Angelis F., Domine M., Clingan P., Hochmair M.J., Powell S.F., KEYNOTE-189 Investigators Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 2018;378:2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 12.Taieb J., Moehler M., Boku N., Ajani J.A., Yañez Ruiz E., Ryu M.-H., Guenther S., Chand V., Bang Y.-J. Evolution of checkpoint inhibitors for the treatment of metastatic gastric cancers: current status and future perspectives. Cancer Treat. Rev. 2018;66:104–113. doi: 10.1016/j.ctrv.2018.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y. Axicabtagene ciloleucel CAR T-Cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed N., Brawley V.S., Hegde M., Robertson C., Ghazi A., Gerken C., Liu E., Dakhova O., Ashoori A., Corder A. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol. 2015;33:1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katz S.C., Point G.R., Cunetta M., Thorn M., Guha P., Espat N.J., Boutros C., Hanna N., Junghans R.P. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016;23:142–148. doi: 10.1038/cgt.2016.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ang W.X., Li Z., Chi Z., Du S.H., Chen C., Tay J.C., Toh H.C., Connolly J.E., Xu X.H., Wang S. Intraperitoneal immunotherapy with T cells stably and transiently expressing anti-EpCAM CAR in xenograft models of peritoneal carcinomatosis. Oncotarget. 2017;8:13545–13559. doi: 10.18632/oncotarget.14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lv J., Zhao R., Wu D., Zheng D., Wu Z., Shi J., Wei X., Wu Q., Long Y., Lin S. Mesothelin is a target of chimeric antigen receptor T cells for treating gastric cancer. J. Hematol. Oncol. 2019;12:18. doi: 10.1186/s13045-019-0704-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang H., Shi Z., Wang P., Wang C., Yang L., Du G., Zhang H., Shi B., Jia J., Li Q. Claudin18.2-specific chimeric antigen receptor engineered T cells for the treatment of gastric cancer. J. Natl. Cancer Inst. 2019;111:409–418. doi: 10.1093/jnci/djy134. [DOI] [PubMed] [Google Scholar]
- 19.Newick K., O’Brien S., Moon E., Albelda S.M. CAR T cell therapy for solid tumors. Annu. Rev. Med. 2017;68:139–152. doi: 10.1146/annurev-med-062315-120245. [DOI] [PubMed] [Google Scholar]
- 20.Scarfò I., Maus M.V. Current approaches to increase CAR T cell potency in solid tumors: targeting the tumor microenvironment. J. Immunother. Cancer. 2017;5:28. doi: 10.1186/s40425-017-0230-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ziprin P., Ridgway P.F., Pfistermüller K.L., Peck D.H., Darzi A.W. ICAM-1 mediated tumor-mesothelial cell adhesion is modulated by IL-6 and TNF-alpha: a potential mechanism by which surgical trauma increases peritoneal metastases. Cell Commun. Adhes. 2003;10:141–154. [PubMed] [Google Scholar]
- 22.Jung W.C., Jang Y.J., Kim J.H., Park S.S., Park S.H., Kim S.J., Mok Y.J., Kim C.S. Expression of intercellular adhesion molecule-1 and E-selectin in gastric cancer and their clinical significance. J. Gastric Cancer. 2012;12:140–148. doi: 10.5230/jgc.2012.12.3.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Min I.M., Shevlin E., Vedvyas Y., Zaman M., Wyrwas B., Scognamiglio T., Moore M.D., Wang W., Park S., Park S. CAR T therapy targeting ICAM-1 eliminates advanced human thyroid tumors. Clin. Cancer Res. 2017;23:7569–7583. doi: 10.1158/1078-0432.CCR-17-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park S., Shevlin E., Vedvyas Y., Zaman M., Park S., Hsu Y.S., Min I.M., Jin M.M. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci. Rep. 2017;7:14366. doi: 10.1038/s41598-017-14749-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bass A.J., Thorsson V., Shmulevich I., Reynolds S.M., Miller M., Bernard B., Hinoue T., Laird P.W., Curtis C., Shen H., Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sohn B.H., Hwang J.-E., Jang H.-J., Lee H.-S., Oh S.C., Shim J.-J., Lee K.-W., Kim E.H., Yim S.Y., Lee S.H. Clinical significance of four molecular subtypes of gastric cancer identified by The Cancer Genome Atlas project. Clin. Cancer Res. 2017;23:4441–4449. doi: 10.1158/1078-0432.CCR-16-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma: an attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965;64:31–49. doi: 10.1111/apm.1965.64.1.31. [DOI] [PubMed] [Google Scholar]
- 28.Vedvyas Y., McCloskey J.E., Yang Y., Min I.M., Fahey T.J., Zarnegar R., Hsu Y.S., Hsu J.-M., Van Besien K., Gaudet I. Manufacturing and preclinical validation of CAR T cells targeting ICAM-1 for advanced thyroid cancer therapy. Sci. Rep. 2019;9:10634. doi: 10.1038/s41598-019-46938-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laverman P., D’Souza C.A., Eek A., McBride W.J., Sharkey R.M., Oyen W.J., Goldenberg D.M., Boerman O.C. Optimized labeling of NOTA-conjugated octreotide with F-18. Tumour Biol. 2012;33:427–434. doi: 10.1007/s13277-011-0250-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wei J., Wu N.D., Liu B.R. Regional but fatal: intraperitoneal metastasis in gastric cancer. World J. Gastroenterol. 2016;22:7478–7485. doi: 10.3748/wjg.v22.i33.7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michaud M., Martins I., Sukkurwala A.Q., Adjemian S., Ma Y., Pellegatti P., Shen S., Kepp O., Scoazec M., Mignot G. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–1577. doi: 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- 32.Alizadeh D., Trad M., Hanke N.T., Larmonier C.B., Janikashvili N., Bonnotte B., Katsanis E., Larmonier N. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014;74:104–118. doi: 10.1158/0008-5472.CAN-13-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hironaka S., Ueda S., Yasui H., Nishina T., Tsuda M., Tsumura T., Sugimoto N., Shimodaira H., Tokunaga S., Moriwaki T. Randomized, open-label, phase III study comparing irinotecan with paclitaxel in patients with advanced gastric cancer without severe peritoneal metastasis after failure of prior combination chemotherapy using fluoropyrimidine plus platinum: WJOG 4007 trial. J. Clin. Oncol. 2013;31:4438–4444. doi: 10.1200/JCO.2012.48.5805. [DOI] [PubMed] [Google Scholar]
- 34.Cividalli A., Cruciani G., Livdi E., Cordelli E., Eletti B., Tirindelli Danesi D. Greater antitumor efficacy of paclitaxel administered before epirubicin in a mouse mammary carcinoma. J. Cancer Res. Clin. Oncol. 1998;124:236–244. doi: 10.1007/s004320050160. [DOI] [PubMed] [Google Scholar]
- 35.Hsieh C.S., Macatonia S.E., Tripp C.S., Wolf S.F., O’Garra A., Murphy K.M. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 36.Trinchieri G. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu. Rev. Immunol. 1995;13:251–276. doi: 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- 37.Cohen J. IL-12 deaths: explanation and a puzzle. Science. 1995;270:908. doi: 10.1126/science.270.5238.908a. [DOI] [PubMed] [Google Scholar]
- 38.Leonard J.P., Sherman M.L., Fisher G.L., Buchanan L.J., Larsen G., Atkins M.B., Sosman J.A., Dutcher J.P., Vogelzang N.J., Ryan J.L. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90:2541–2548. [PubMed] [Google Scholar]
- 39.Kerkar S.P., Muranski P., Kaiser A., Boni A., Sanchez-Perez L., Yu Z., Palmer D.C., Reger R.N., Borman Z.A., Zhang L. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer Res. 2010;70:6725–6734. doi: 10.1158/0008-5472.CAN-10-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah N.N., Fry T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019;16:372–385. doi: 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rafiq S., Hackett C.S., Brentjens R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020;17:147–167. doi: 10.1038/s41571-019-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.D’Aloia M.M., Zizzari I.G., Sacchetti B., Pierelli L., Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Dis. 2018;9:282. doi: 10.1038/s41419-018-0278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maruo Y., Gochi A., Kaihara A., Shimamura H., Yamada T., Tanaka N., Orita K. ICAM-1 expression and the soluble ICAM-1 level for evaluating the metastatic potential of gastric cancer. Int. J. Cancer. 2002;100:486–490. doi: 10.1002/ijc.10514. [DOI] [PubMed] [Google Scholar]
- 44.Maeda K., Kang S.M., Sawada T., Nishiguchi Y., Yashiro M., Ogawa Y., Ohira M., Ishikawa T., Hirakawa-YS Chung K. Expression of intercellular adhesion molecule-1 and prognosis in colorectal cancer. Oncol. Rep. 2002;9:511–514. [PubMed] [Google Scholar]
- 45.Fujihara T., Yashiro M., Inoue T., Sawada T., Kato Y., Ohira M., Nishiguchi Y., Ishikawa T., Sowa M., Chung K.H. Decrease in ICAM-1 expression on gastric cancer cells is correlated with lymph node metastasis. Gastric Cancer. 1999;2:221–225. doi: 10.1007/s101200050067. [DOI] [PubMed] [Google Scholar]
- 46.Ogawa Y., Hirakawa K., Nakata B., Fujihara T., Sawada T., Kato Y., Yoshikawa K., Sowa M. Expression of intercellular adhesion molecule-1 in invasive breast cancer reflects low growth potential, negative lymph node involvement, and good prognosis. Clin. Cancer Res. 1998;4:31–36. [PubMed] [Google Scholar]
- 47.Reina M., Espel E. Role of LFA-1 and ICAM-1 in cancer. Cancers (Basel) 2017;9:153. doi: 10.3390/cancers9110153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buitrago D., Keutgen X.M., Crowley M., Filicori F., Aldailami H., Hoda R., Liu Y.F., Hoda R.S., Scognamiglio T., Jin M. Intercellular adhesion molecule-1 (ICAM-1) is upregulated in aggressive papillary thyroid carcinoma. Ann. Surg. Oncol. 2012;19:973–980. doi: 10.1245/s10434-011-2029-0. [DOI] [PubMed] [Google Scholar]
- 49.Figenschau S.L., Knutsen E., Urbarova I., Fenton C., Elston B., Perander M., Mortensen E.S., Fenton K.A. ICAM1 expression is induced by proinflammatory cytokines and associated with TLS formation in aggressive breast cancer subtypes. Sci. Rep. 2018;8:11720. doi: 10.1038/s41598-018-29604-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo P., Huang J., Wang L., Jia D., Yang J., Dillon D.A., Zurakowski D., Mao H., Moses M.A., Auguste D.T. ICAM-1 as a molecular target for triple negative breast cancer. Proc. Natl. Acad. Sci. USA. 2014;111:14710–14715. doi: 10.1073/pnas.1408556111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahmed N., Brawley V., Hegde M., Bielamowicz K., Kalra M., Landi D., Robertson C., Gray T.L., Diouf O., Wakefield A. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3:1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adusumilli P.S., Cherkassky L., Villena-Vargas J., Colovos C., Servais E., Plotkin J., Jones D.R., Sadelain M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 2014;6:261ra151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mayor M., Zeltsman M., McGee E., Adusumilli P.S. A regional approach for CAR T-cell therapy for mesothelioma: from mouse models to clinical trial. Immunotherapy. 2016;8:491–494. doi: 10.2217/imt-2016-0008. [DOI] [PubMed] [Google Scholar]
- 55.Martinez M., Moon E.K. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019;10:128. doi: 10.3389/fimmu.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma Y., Adjemian S., Mattarollo S.R., Yamazaki T., Aymeric L., Yang H., Portela Catani J.P., Hannani D., Duret H., Steegh K. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38:729–741. doi: 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 57.Turtle C.J., Hanafi L.A., Berger C., Hudecek M., Pender B., Robinson E., Hawkins R., Chaney C., Cherian S., Chen X. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 2016;8:355ra116. doi: 10.1126/scitranslmed.aaf8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sugarbaker P.H., Mora J.T., Carmignani P., Stuart O.A., Yoo D. Update on chemotherapeutic agents utilized for perioperative intraperitoneal chemotherapy. Oncologist. 2005;10:112–122. doi: 10.1634/theoncologist.10-2-112. [DOI] [PubMed] [Google Scholar]
- 60.Rafiq S., Yeku O.O., Jackson H.J., Purdon T.J., van Leeuwen D.G., Drakes D.J., Song M., Miele M.M., Li Z., Wang P. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018;36:847–856. doi: 10.1038/nbt.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chong E.A., Melenhorst J.J., Lacey S.F., Ambrose D.E., Gonzalez V., Levine B.L., June C.H., Schuster S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. 2017;129:1039–1041. doi: 10.1182/blood-2016-09-738245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wing A., Fajardo C.A., Posey A.D., Jr., Shaw C., Da T., Young R.M., Alemany R., June C.H., Guedan S. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol. Res. 2018;6:605–616. doi: 10.1158/2326-6066.CIR-17-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Watanabe K., Luo Y., Da T., Guedan S., Ruella M., Scholler J., Keith B., Young R.M., Engels B., Sorsa S. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight. 2018;3:e99573. doi: 10.1172/jci.insight.99573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ma L., Dichwalkar T., Chang J.Y.H., Cossette B., Garafola D., Zhang A.Q., Fichter M., Wang C., Liang S., Silva M. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science. 2019;365:162–168. doi: 10.1126/science.aav8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reinhard K., Rengstl B., Oehm P., Michel K., Billmeier A., Hayduk N., Klein O., Kuna K., Ouchan Y., Wöll S. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science. 2020;367:446–453. doi: 10.1126/science.aay5967. [DOI] [PubMed] [Google Scholar]
- 66.Chinnasamy D., Yu Z., Kerkar S.P., Zhang L., Morgan R.A., Restifo N.P., Rosenberg S.A. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin. Cancer Res. 2012;18:1672–1683. doi: 10.1158/1078-0432.CCR-11-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adachi K., Kano Y., Nagai T., Okuyama N., Sakoda Y., Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018;36:346–351. doi: 10.1038/nbt.4086. [DOI] [PubMed] [Google Scholar]
- 68.Hurton L.V., Singh H., Najjar A.M., Switzer K.C., Mi T., Maiti S., Olivares S., Rabinovich B., Huls H., Forget M.A. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA. 2016;113:E7788–E7797. doi: 10.1073/pnas.1610544113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hu B., Ren J., Luo Y., Keith B., Young R.M., Scholler J., Zhao Y., June C.H. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. 2017;20:3025–3033. doi: 10.1016/j.celrep.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma X., Shou P., Smith C., Chen Y., Du H., Sun C., Porterfield Kren N., Michaud D., Ahn S., Vincent B. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020;38:448–459. doi: 10.1038/s41587-019-0398-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koneru M., Purdon T.J., Spriggs D., Koneru S., Brentjens R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. OncoImmunology. 2015;4:e994446. doi: 10.4161/2162402X.2014.994446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chmielewski M., Kopecky C., Hombach A.A., Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697–5706. doi: 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- 73.Gerner M.Y., Heltemes-Harris L.M., Fife B.T., Mescher M.F. Cutting edge: IL-12 and type I IFN differentially program CD8 T cells for programmed death 1 re-expression levels and tumor control. J. Immunol. 2013;191:1011–1015. doi: 10.4049/jimmunol.1300652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zou J.J., Schoenhaut D.S., Carvajal D.M., Warrier R.R., Presky D.H., Gately M.K., Gubler U. Structure-function analysis of the p35 subunit of mouse interleukin 12. J. Biol. Chem. 1995;270:5864–5871. doi: 10.1074/jbc.270.11.5864. [DOI] [PubMed] [Google Scholar]
- 75.Poirot L., Philip B., Schiffer-Mannioui C., Le Clerre D., Chion-Sotinel I., Derniame S., Potrel P., Bas C., Lemaire L., Galetto R. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015;75:3853–3864. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 76.Brehm M.A., Kenney L.L., Wiles M.V., Low B.E., Tisch R.M., Burzenski L., Mueller C., Greiner D.L., Shultz L.D. Lack of acute xenogeneic graft- versus-host disease, but retention of T-cell function following engraftment of human peripheral blood mononuclear cells in NSG mice deficient in MHC class I and II expression. FASEB J. 2019;33:3137–3151. doi: 10.1096/fj.201800636R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yeku O.O., Purdon T.J., Koneru M., Spriggs D., Brentjens R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017;7:10541. doi: 10.1038/s41598-017-10940-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fraietta J.A., Lacey S.F., Orlando E.J., Pruteanu-Malinici I., Gohil M., Lundh S., Boesteanu A.C., Wang Y., O’Connor R.S., Hwang W.T. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018;24:563–571. doi: 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.American Cancer Society . 2002. AJCC Cancer Staging Manual, Sixth Edition (Springer) p. xiv. [Google Scholar]
- 80.Reiner A., Spona J., Reiner G., Schemper M., Kolb R., Kwasny W., Függer R., Jakesz R., Holzner J.H. Estrogen receptor analysis on biopsies and fine-needle aspirates from human breast carcinoma. Correlation of biochemical and immunohistochemical methods using monoclonal antireceptor antibodies. Am. J. Pathol. 1986;125:443–449. [PMC free article] [PubMed] [Google Scholar]
- 81.Laverman P., McBride W.J., Sharkey R.M., Eek A., Joosten L., Oyen W.J., Goldenberg D.M., Boerman O.C. A novel facile method of labeling octreotide with 18F-fluorine. J. Nucl. Med. 2010;51:454–461. doi: 10.2967/jnumed.109.066902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vedvyas Y., Shevlin E., Zaman M., Min I.M., Amor-Coarasa A., Park S., Park S., Kwon K.W., Smith T., Luo Y. Longitudinal PET imaging demonstrates biphasic CAR T cell responses in survivors. JCI Insight. 2016;1:e90064. doi: 10.1172/jci.insight.90064. e90064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.