

Abstract
Gene expression profiles of matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) were evaluated in peripheral blood leukocytes of children with nonalcoholic fatty liver disease (NAFLD). Gene expression patterns were correlated with their plasma protein counterparts, systemic parameters of liver injury, and selected markers of inflammation. The MMP-2, MMP-9, MMP-12, MMP-14, TIMP-1, TIMP-2, TGF-β, and IL-6 transcripts levels were tested by the real-time PCR. Plasma concentrations of MMP-9, TIMP-1, MMP-9/TIMP-1 ratio, MMP-2/TIMP-2 ratio, sCD14, leptin, resistin, IL-1 beta, and IL-6 and serum markers of liver injury were estimated by ELISA. The MMP-9, TIMP-2 expression levels, plasma amounts of MMP-9, TIMP-1, and the MMP-9/TIMP-1 ratio were increased in children with NAFLD. Concentrations of AST, ALT, GGT, and leptin were elevated in serum patients with NAFLD, while concentration of other inflammatory or liver injury markers was unchanged. The MMP-2 and MMP-9 levels correlated with serum liver injury parameters (ALT and GGT concentrations, respectively); there were no other correlations between MMP/TIMP gene expression profiles, their plasma counterparts, and serum inflammatory markers. Association of MMP-2 and MMP-9 expression with serum liver injury parameters (ALT, GGT) may suggest leukocyte engagement in the early stages of NAFLD development which possibly precedes subsequent systemic inflammatory responses.
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is a chronic inflammatory disorder, closely related to metabolic syndrome components, like obesity and type 2 diabetes mellitus. The disease is defined as liver fat accumulation (steatosis) that exceeds 5% of hepatocytes and is not secondary to genetic and metabolic disorders, infections, alcohol consumptions, or malnutrition [1]. NAFLD refers to the spectrum ranging from a simple steatosis to nonalcoholic steatohepatitis (NASH), fibrosis, and cirrhosis, with possible progression to hepatocellular carcinoma. Those histopathological changes not only occur within the hepatocytes [2] but primarily focus on active remodeling of the liver extracellular matrix (ECM) [3]. This process leads to excessive deposition of ECM compounds, such as type I, III, and IV collagens, and enhances expression of noncollagenous components including fibronectins, laminins, proteoglycans, and elastins [4]. Considering that all ECM compounds are degraded by endopeptidases called matrix metalloproteinases (MMPs), and whose activities are controlled by their endogenous tissue inhibitors (TIMPs), MMPs and equally TIMPs become the main players in rising fibrotic changes in the course of NAFLD progression [5, 6].
MMPs are involved in degradation and remodeling of ECM protein in both the physiological and pathological conditions. Natural, physiological MMP inhibitors (TIMPs) regulate the proteolytic MMP activity in tissues, forming with them stable noncovalent bonds. According to this, a healthy liver has a moderate ECM turnover that correlates with a small amount of metalloproteinases, as well as TIMPs constitutively expressed in normal livers [7], while any disruption of MMP activity and imbalance in the expression of MMP/TIMP system components [5] often leads to tissue damage and functional alternation triggering pathological conditions like liver fibrosis.
Liver MMP/TIMP components originate from stellate cells, sinusoidal endothelial cells (LECs), residual macrophages (Kupffer cells), and a variety of other cell types migrating to the liver from peripheral tissues including monocytes, neutrophils, and lymphocytes [8], as well as bone marrow-derived precursors [9]. It is worth noticing that leukocyte expression of the MMP/TIMP system plays an important role in the development of local inflammation, its resolution, and tissue repair.
Most types of leukocytes [10–13] have been shown to be engaged in liver injury and repair. Liver infiltration by inflammatory leukocytes is controlled by chemokines, which determine the composition and activation status of immune cells recruited to the injured liver [14, 15]. Therefore, we assumed that peripheral blood leukocytes in children with NAFLD will respond to liver injury through changes in their MMP/TIMP expression patterns, as well as IL-6 and TGF-β gene expression profiles, and that these changes will more or less correspond to plasma inflammatory markers related to liver injury.
Leukocyte expression of MMP-2, MMP-9 (gelatinases), and MMP-14 (a membrane-type MMP) enhances their abilities for liver infiltration during inflammatory responses (basement membrane degradation) where they participate in ECM remodeling and regulation of inflammatory responses by modifying chemokine and cytokine activities [16]. The MMP-2, MMP-9, and MMP-14 degrade denatured collagen (gelatin) and other collagen types, as well as noncollagenous ECM components such as elastin, fibronectin, and laminin [17]. Macrophage MMP-12 is characterized by especially strong antielastin activity and plays an important role during liver fibrosis and fibrosis resolution [18]. Additionally, TIMP-1 preferentially complexes with pro-MMP-9, while TIMP-2 interacts with pro-MMP-2 [19]. The purpose of this study was to assess whether the profile of the MMP/TIMP system expression in peripheral leukocytes and plasma samples of children with NAFLD may reflect the disease activity or serve as an early marker of an ongoing disease process that eventually precedes the appearance of clinical symptoms of NAFLD.
2. Materials and Methods
2.1. Study Subjects
This study has been conducted according to the Declaration of Helsinki and with the approval from the Children's Memorial Health Institute Ethics Committee. All patients and parents gave informed consent to participate in the study. A total of 35 patients with NAFLD (aged 14.2 ± 2.6 years; BMI 29.3 ± 4.7) and 37 healthy lean control volunteers (aged 14.7 ± 2.6; BMI 21.2 ± 3.7) were included in the study. The exclusion criteria were as follows: renal insufficiency, viral and autoimmune liver damage, storage disease, hepatic cirrhosis, type 1 and 2 diabetes, chronic diseases of the gastrointestinal tract, taking lipid-lowering or lipid metabolism-affecting drugs. Diagnostic criteria were (a) ultrasound findings of fatty liver disease [20] and (b) increased ALT activity. The upper limit of the normal ALT value established for healthy children was 25.8 U/L (boys) and 22.1 U/L (girls) [21]. In all patients and healthy adolescents, the general clinical examination was conducted including anthropometry with BMI calculation and waist and hip measurement with W/H ratio counting. Venous blood was collected from an antecubital vein after at least 12 h of fasting. This blood was used to measure serum levels of total cholesterol (TC), triglycerides (TG), low-density lipoprotein-cholesterol (LDL-C), high-density lipoprotein-cholesterol (HDL-C), γ-glutamyltransferase (GGT), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and high-sensitivity C-reactive protein (hs-CRP) levels by enzymatic commercial biochemical tests (Roche Diagnostics; Risch-Rotkreuz, Switzerland). The hexokinase method was used to measure glucose and immunoradiometric assays (Biosources; San Diego, CA, USA) for insulin concentration. Insulin resistance was calculated as follows: (fasting glucose × fasting insulin)/22.5, according to the homeostasis model assessment of insulin resistance (HOMA-IR) [22] (Table 1).
Table 1.
Anthropometric and clinical characteristics of the study (NAFLD) and control groups.
| NAFLD | Control | |
|---|---|---|
| No. | 35 | 37 |
| Age (years) | 14.2 ± 2.6 | 14.7 ± 2.6 |
| Height (cm) | 164.5 ± 15 | 171.7 ± 17 |
| Weight (kg) | 80.3 ± 20 | 63.0 ± 16.5 |
| BMI (kg/m2) | 29.3 ± 4.7 | 21.0 ± 3.0 |
| SDS-BMI | 2.2 ± 0.9 | 0.2 ± 0.8 |
| ALT (IU/L) | 66.7 ± 44.8 | 16.2 ± 7.2 |
| AST (IU/L) | 40.0 ± 20.9 | 19.6 ± 6.0 |
| GGT (IU/L) | 45 ± 32 | 21.6 ± 4.2 |
| TG (mg/dL) | 108 ± 52 | 84 ± 44 |
| TC (mg/dL) | 187 ± 53 | 163 ± 34 |
| HDL-C (mg/dL) | 43.7 ± 13 | 46.3 ± 14 |
| LDL-C (mg/dL) | 123 ± 46 | 101 ± 34 |
| hs-CRP (mg/dL) | 0.31 ± 0.2 | 0.17 ± 0.1 |
| Waist circumference | 97.3 ± 12.7 | 73.9 ± 10 |
| Hip circumference | 101 ± 12 | 92.4 ± 10 |
| WHR | 0.96 | 0.82 |
| Fasting glucose (mg/dL) | 82.5 ± 9.8 | 86.0 ± 6.8 |
| Fasting insulin (μIU/mL) | 15.5 ± 8.1 | 13.9 ± 5.9 |
| HOMA-IR | 3.2 ± 1.8 | 3.2 ± 1.5 |
ALT: alanine aminotransferase; AST: aspartate aminotransferase; GGT: gamma-glutamyltransferase; TG: triglycerides; TC: total cholesterol; HDL-C: high-density lipoprotein-cholesterol; LDL-C: low-density lipoprotein-cholesterol; hs-CRP: high-sensitivity C-reactive protein; WHR: waist to hip ratio; HOMA-IR: homeostasis model assessment of insulin resistance.
2.2. RNA Isolation and Real-Time PCR Technique
Details concerning the PCR technique used here were previously described [23]. Briefly, peripheral blood leukocytes were obtained by Histopaque (Sigma-Aldrich 1119; Saint Louis, MO, USA) gradient centrifugation. Total RNA was isolated with the Chomczyński method using TRIzol Reagent (Ambion; Carlsbad, CA, USA). By using absorbance at 260 nm and 280 nm, each sample RNA concentration was determined and purity/integrity was checked. One microgram of total RNA per sample was converted into cDNA via Reverse Transcription Polymerase Chain Reaction (RT-PCR) by using TaqMan Reverse Transcription Reagents. Quantitative RT-PCR (real-time PCR) was used for the following target genes: MMP-9, MMP-2, MMP-12, MMP-14, TIMP-1, TIMP-2, TGF-β, IL-6, and the endogenous control (reference gene) glyceraldehyde-3-phosphate dehydrogenase (G3PDH). The tests were performed using the ViiA 7 Real-Time System, according to the manufacturer's recommendation. For one reaction, 50 ng of cDNA was applied with SYBR Green PCR Master Mix and 10 nmol/L for each of the forward and reverse primers (Table 2). The sample was run twice, and the average was taken to analysis. The specificity of the amplification reaction was verified by analysis of the melting curve. Relative fold changes in target gene expression between the NAFLD patients and the control group were determined by normalization of expression of the reference gene G3PDH, by using Pfaffl's mathematical model [24]. All reagents, equipment, and other supplies used for the real-time PCR technique were provided by Applied Biosystems distributed by Thermo Fisher Scientific Inc. (Waltham, MA, USA).
Table 2.
Primer sequence of target genes and reference gene for SYBR Green real-time PCR.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| MMP-9 | CAA CAT CAC CTA TTG GAT CC | CGG GTG TAG AGT CTC TCG CT |
| MMP-2 | TGA TCT TGA CCA GAA TAC CAT CGA | GGC TTG CGA GGG AAG AAG TT |
| TIMP-1 | CTT CTG GCA TCC TGT TGT TG | AGA AGG CCG TCT GTG GGT |
| TIMP-2 | CGA CAT TTA TGG CAA CCC TAT CA | CAG GCC CTT TGA ACA TCT TTA TCT |
| MMP-12 | TTCCCCTGAACAGCTCTACAAGCCTGGAAA | GATCCAGGTCCAAAAGCATGGGCTAGGATT |
| MMP-14 | CGC TAC GCC ATC CAG GGT CTC AAA | CGC TAC GCC ATC CAG GGT CTC AAA |
| TGF-β | GGA AAC CCA CAA CGA AAT CTA TG | CGG GTT CAG GTA CCG CTT C |
| IL-6 | TGA AAG CAG CAA AGA GGC ACT | GGC AAG TCT CCT CAT TGA ATC C |
| G3PDH | GCG GGG CTC TCC AGA ACA TCA T | CCA GCC CCA GCG TCA AAG GTG |
MMP-2, 9, 12, and 14 indicate matrix metalloproteinase-2, 9, 12, or 14, respectively. TIMP-1 and 2 indicate tissue inhibitor of metalloproteinase-1 or 2. TGF-β: transforming growth factor beta; IL-6: interleukin-6; G3PDH: glyceraldehyde-3-phosphate dehydrogenase.
2.3. ELISA Method
Peripheral blood plasma was collected using EDTA as an anticoagulant. The DuoSet ELISA kits (R&D Systems; Minneapolis, MN, USA) were used to determine the total concentration of MMP-9 and TIMP-1, as well as MMP-9/TIMP-1 ratio, MMP-2/TIMP-2 ratio, and sCD14. Leptin and resistin levels were determined with the use of ELISA kits (DRG International (Springfield, NJ, USA) and Phoenix Pharmaceuticals (Burlingame, CA, USA), respectively), and plasma amounts of IL-1 beta and IL-6 were determined by the use of OptEIA Set ELISA kits (Becton Dickinson; Franklin Lake, NJ, USA) as previously described [25].
2.4. Statistical Analysis
The data were analyzed using the Statistica 13.1 software package. The data distribution pattern was tested by using the Shapiro-Wilk W test, and the appropriate statistical tests were chosen: parametric (Student's t-test) or nonparametric (Mann-Whitney U test). All data were not normally distributed. The differences between the two groups (patients vs. control) were estimated by using the nonparametric tests. The relation between the clinical, anthropometric, biochemical, molecular, and immunological parameters was assessed by Spearman's rank correlation analysis. The differences and correlation indexes were considered significant at p < 0.05.
3. Results and Discussion
We showed that the levels of leukocyte mRNA expression of MMP-9 and TIMP-2 but not TIMP-1 were significantly higher in patients with NAFLD than in their healthy counterparts (2.38 (p = 0.001) and 3.89 (p = 0.002) fold change, respectively) (Figure 1).
Figure 1.
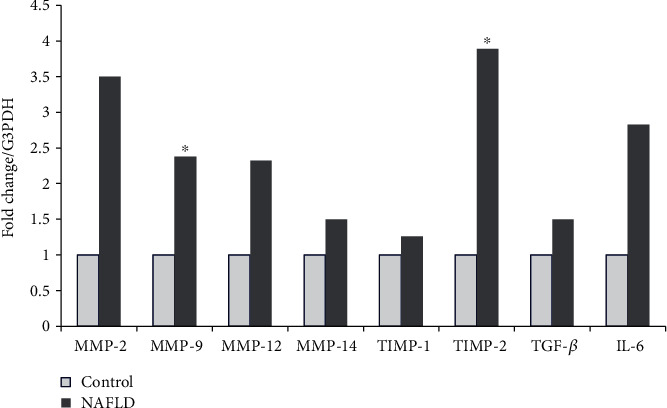
Gene expression in children with NAFLD. Transcript levels of MMP-9, TIMP-1, MMP-2, TIMP-2, MMP-12, MMP-14, TGF-β, and IL-6 in leukocytes from the control group (open bars) and children with NAFLD (solid bars). Data are expressed as fold changes from children with NAFLD versus the control (n = 35/37). ∗Statistically significant at <0.01 by the Mann-Whitney U test.
MMP-9 expression correlated inversely with TIMP-1 (r = −0.587, p < 0.05), TIMP-2 (r = −0.470, p < 0.05), and MMP-12 (r = −0.573, p < 0.05) and positively with GGT serum concentration (r = 0.432, p < 0.05). Leukocyte MMP-2 expression correlated positively with TIMP-1 (r = 0.404, p < 0.05), TIMP-2 (r = 0.375, p < 0.05), and serum ALT levels (r = 0.344, p < 0.05). Additionally, MMP-12 expression correlated positively with TIMP-1 (r = 0.598, p < 0.05), TIMP-2 (r = 0.811, p < 0.05), and IL-6 (r = 0.753, p < 0.05). Finally, TIMP-1 levels of expression correlated with TIMP-2 (r = 0.713, p < 0.05) (Figures 2(a)–2(k)).
Figure 2.
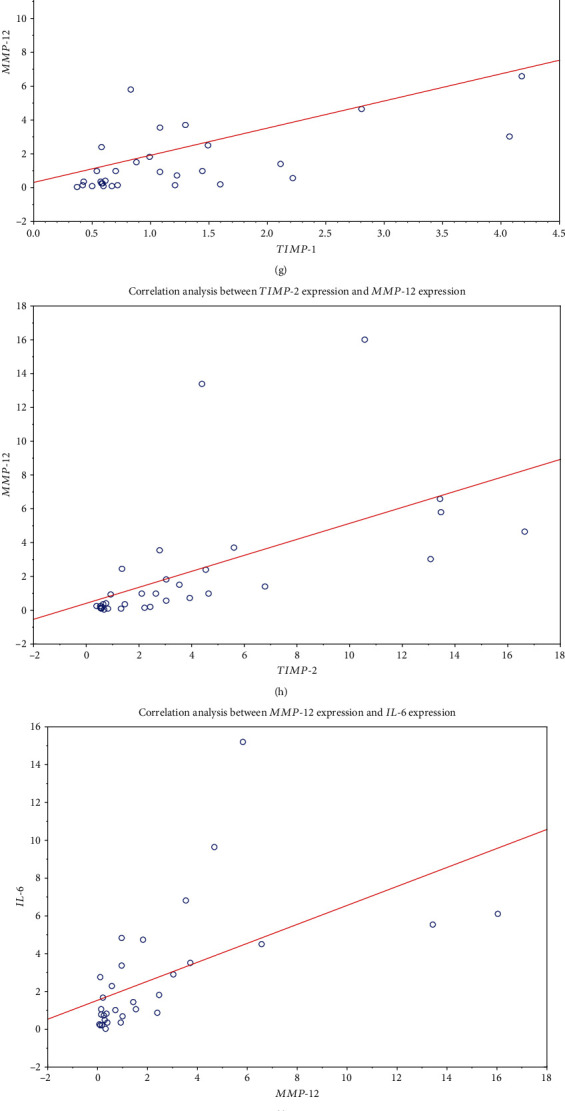
Spearman's correlation analysis between the expression of metalloproteinases, tissue inhibitors, their plasma protein counterparts, and liver injury markers: MMP-9 and TIMP-1 (a); MMP-9 and TIMP-2 (b); MMP-9 and MMP-12 (c); MMP-2 and TIMP-1 (d); MMP-2 and TIMP-2 (e); TIMP-1 and TIMP-2 (f); TIMP-1 and MMP-12 (g); TIMP-2 and MMP-12 (h); MMP-12 and IL-6 (i); MMP-9 and GGT (j); MMP-2 and ALT (k); MMP-9 and TIMP-1 (l); MMP-9 and MMP-9/TIMP-1 ratio (m); ALT and ASP (n); ALT and GGT (o).
MMP-9 and TIMP-1 plasma concentrations, as well as the plasma MMP-9/TIMP-1 ratio and leptin levels, were significantly increased in the patients with NAFLD as compared to the control group (Table 3).
Table 3.
Plasma levels of metalloproteinases and their tissue inhibitors as well as cytokines and adipokines in the children with NAFLD and control groups.
| Children group | sCD14 (ng/mL) | MMP-9 (ng/mL) | TIMP-1 (ng/mL) | MMP-2/TIMP-2 (ng/mL) | MMP-9/TIMP-1 (ng/mL) | IL-1 beta (pg/mL) | IL-6 (pg/mL) | Leptin (ng/mL) | Resistin (ng/mL) |
|---|---|---|---|---|---|---|---|---|---|
| NAFLD group size (n = 35) | 1316 ± 145 | 63 ± 30∗ | 194 ± 137∗ | 133 ± 79 | 9.6 ± 7.7∗ | 5.6 ± 5.3 | 7.7 ± 7.0 | 17.3 ± 14.4∗ | 6.2 ± 4.5 |
| Control group size (n = 37) | 1216 ± 340 | 25 ± 19 | 87 ± 44 | 136 ± 44 | 2.2 ± 1.7 | 5.8 ± 2.8 | 5.7 ± 4.7 | 2.0 ± 1.5 | 3.9 ± 1.2 |
∗Statistically significant at <0.01 by the Mann-Whitney U test.
The elevated MMP-9 plasma levels were positively correlated with TIMP-1 (r = 0.533, p < 0.05) and MMP-9/TIMP-1 ratio concentration (r = 0.848, p < 0.05). Moreover, ALT was correlated with concentrations of AST (r = 0.848, p < 0.05) and GGT (r = 0.494, p < 0.05) (Figures 2(l)–2(o)).
We found that the children with NAFLD were characterized by increased leukocyte MMP-9 and TIMP-2 expression, negative correlation of leukocyte MMP-9 with TIMP-1 and TIMP-2, and elevated plasma levels of MMP-9, TIMP-1, and plasma MMP-9/TIMP-1 ratio. The leukocyte MMP-2 expression correlated with ALT serum levels, and the leukocyte MMP-9 expression correlated with GGT serum concentrations. There were no other associations between leukocyte MMP/TIMP gene expression levels, their plasma protein counterparts, and serum markers of liver injury (ALT, AST, and GGT) or systemic inflammation. Leukocyte MMP-9 upregulation may precede further stages of NAFLD development and subsequent systemic inflammatory responses reflected here by elevated plasma levels of total MMP-9, TIMP-1, and MMP-9/TIMP-1 ratio. However, the engagement of other proinflammatory immune response components remains limited (as demonstrated here by the unchanged plasma levels of IL-1 beta and IL-6).
Increased leukocyte MMP-9 levels, unchanged TIMP-1 expression, and positive correlation of MMP-9 expression with plasma GGT and MMP-2 expression with serum ALT concentrations indicate the role of MMP-9 and MMP-2 gelatinases in NAFLD progression. MMP-9 is an inducible gelatinase expressed by all leukocyte types and other tissues including the native liver, albeit at very low levels [26]. In inflammatory conditions such as NAFLD, MMP-9 may play a role as an important mediator of leukocyte-induced liver damage [27, 28].
ECM degrading potential depends on the balance between TIMPs and MMPs [29]. Unchanged leukocyte TIMP-1 levels with high MMP-9 expression and elevated plasma MMP-9/TIMP-1 ratio may indicate increased MMP-9 activity (here high concentration of the MMP-9/TIMP-1 ratio) that should subsequently result in clearance of the fibrotic matrix. In contrast, TIMP-1 overexpression inhibits fibrotic matrix degradation and leads to extensive accumulation of interstitial ECM [30, 31]. Normal serum MMP-2/TIMP-2 ratio levels along with increased MMP-9/TIMP-1 concentrations may suggest rather low profibrotic MMP-2 activities at this stage of the disease. It has previously been found that leukocyte MMP-9 and MMP-9 plasma expression levels correlated with increased IL-6 and IL-1 beta production. In contrast, both MMP-2 and MMP-2 levels were associated with the production of anti-inflammatory cytokines (IL-4, IL-10) with profibrotic potential [32] indirectly confirming the role of MMP-2 in the development of liver fibrosis. These data may suggest that leukocyte MMP-9 and MMP-2 levels can mutually control each other to balance the inflammatory and anti-inflammatory activities necessary to regulate functions of the liver. Here, we found a positive correlation between leukocyte MMP-2 expression and plasma ALT levels, confirming the role of leukocyte MMP-2 in liver damage [33]. As previously reported, MMP-2 is strongly engaged in liver ECM remodeling and fibrosis [34]. We found that the levels of the plasma MMP-2/TIMP-2 ratio remained unchanged in patients with NAFLD in comparison to the control group. It may suggest that in the early stage of NAFLD (child model), the liver's profibrotic response to injury was rather low because the increase in MMP-2 plasma levels has previously been considered a sensitive marker of fibrosis in the early stage of NASH [35]. As described here, the positive correlations between leukocyte MMP-12 expression and leukocyte IL-6, TIMP-1, and TIMP-2 levels suggest that IL-6-induced MMP-12 expression may be counterbalanced by TIMPs. MMP-12 plays a key role in elastin degradation, so this observation may suggest that peripheral monocytes in children with NAFLD have decreased MMP-12 activities that result in diminution of their capacity for fibrosis resolution [36]. On the other hand, the expression of leukocyte TGF-β (strong fibrosis inducer) remained unchanged, suggesting rather low ability for its profibrotic action in the early stage of NAFLD [37].
4. Conclusions
Altogether, our data suggest the following: (a) changes in leukocyte MMP/TIMP expression profiles are mostly unrelated to their plasma levels but may represent early markers of leukocyte subset activation that eventually may precede the subsequent liver response to damage in the early stage of NAFLD, and (b) the subsequent increase in plasma levels of MMP-9 and TIMP-1 as well as the elevation of MMP-9/TIMP-1 ratios may reflect NAFLD's progress towards fibrosis, which is in agreement with the recent data on the role of TIMPs (especially TIMP-1) in the regulation of the matrix turnover [38].
Acknowledgments
The authors thank Hanna Gregorek from the Children's Memorial Health Institute for her insightful review and valuable comments. This study was supported by a grant from Polish National Science Centre (2011/01/B/NZ6/02661).
Data Availability
All data generated or analyzed during this study are included in this published article.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
The contributions of the authors involved in this study are as follows: conception and design of the study: JBT, JM, and PS; acquisition, analysis, and interpretation of the data: JBT, RG, JM, WJ, LG, IK, AH, AW, MS, and PS; drafting the article or revising it critically for important intellectual content: JM, JBT, RG, WJ, IK, AW, and PS; and final approval of the version to be published: JBT, JM, RG, WJ, AW, and PS.
References
- 1.Demir M., Lang S., Steffen H. M. Nonalcoholic fatty liver disease - current status and future directions. Journal of Digestive Diseases. 2015;16(10):541–557. doi: 10.1111/1751-2980.12291. [DOI] [PubMed] [Google Scholar]
- 2.Brown G. T., Kleiner D. E. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism. 2016;65(8):1080–1086. doi: 10.1016/j.metabol.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iredale J. P., Thompson A., Henderson N. C. Extracellular matrix degradation in liver fibrosis: biochemistry and regulation. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2013;1832(7):876–883. doi: 10.1016/j.bbadis.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Mas V. R., Fisher R. A., Archer K. J., Maluf D. G. Proteomics and liver fibrosis: identifying markers of fibrogenesis. Expert Review of Proteomics. 2014;6(4):421–431. doi: 10.1586/epr.09.59. [DOI] [PubMed] [Google Scholar]
- 5.Duarte S., Baber J., Fujii T., Coito A. J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biology. 2015;44-46:147–156. doi: 10.1016/j.matbio.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yilmaz Y., Eren F. Serum biomarkers of fibrosis and extracellular matrix remodeling in patients with nonalcoholic fatty liver disease. European Journal of Gastroenterology & Hepatology. 2019;31(1):43–46. doi: 10.1097/MEG.0000000000001240. [DOI] [PubMed] [Google Scholar]
- 7.Roderfeld M. Matrix metalloproteinase functions in hepatic injury and fibrosis. Matrix Biology. 2018;68-69:452–462. doi: 10.1016/j.matbio.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka M., Miyajima A. Liver regeneration and fibrosis after inflammation. Inflammation and Regeneration. 2016;36(1):1–19. doi: 10.1186/s41232-016-0025-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higashiyama R., Inagaki Y., Hong Y. Y., et al. Bone marrow-derived cells express matrix metalloproteinases and contribute to regression of liver fibrosis in mice. Hepatology. 2007;45(1):213–222. doi: 10.1002/hep.21477. [DOI] [PubMed] [Google Scholar]
- 10.Tacke F., Zimmermann H. W. Macrophage heterogeneity in liver injury and fibrosis. Journal of Hepatology. 2014;60(5):1090–1096. doi: 10.1016/j.jhep.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 11.Xu R., Huang H., Zhang Z., Wang F.-S. The role of neutrophils in the development of liver diseases. Cellular & Molecular Immunology. 2014;11(3):224–231. doi: 10.1038/cmi.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang W., Tao Y., Wu Y., et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nature Communications. 2019;10(1):p. 1076. doi: 10.1038/s41467-019-09046-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narayanan S., Surette F. A., Hahn Y. S. The immune landscape in nonalcoholic steatohepatitis. Immune Network. 2016;16(3):147–158. doi: 10.4110/in.2016.16.3.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ju C., Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cellular & Molecular Immunology. 2016;13(3):316–327. doi: 10.1038/cmi.2015.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma P.-F., Gao C.-C., Yi J., et al. Cytotherapy with M1-polarized macrophages ameliorates liver fibrosis by modulating immune microenvironment in mice. Journal of Hepatology. 2017;67(4):770–779. doi: 10.1016/j.jhep.2017.05.022. [DOI] [PubMed] [Google Scholar]
- 16.Fingleton B. Matrix metalloproteinases as regulators of inflammatory processes. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2017;1864(11):2036–2042. doi: 10.1016/j.bbamcr.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 17.Verspaget H. W., Kuyvenhoven J. P., Sier C. F. M., van Hoek B. Matrix metalloproteinases in chronic liver diseases and liver transplantation. In: Lendeckel U., Hooper N. M., editors. Proteases in Gastrointestinal Tissue. The Netherlands: Springer; 2006. pp. 209–234. [Google Scholar]
- 18.Shapiro S. D. Elastolytic metalloproteinases produced by human mononuclear phagocytes. Potential roles in destructive lung disease. American Journal of Respiratory and Critical Care Medicine. 1994;150, 6, Part 2:S160–S164. doi: 10.1164/ajrccm/150.6_pt_2.s160. [DOI] [PubMed] [Google Scholar]
- 19.Duffy M. J., McCarthy K. Matrix metalloproteinases in cancer: prognostic and targets for therapy. International Journal of Oncology. 1998;12:1343–1348. doi: 10.3892/ijo.12.6.1343. [DOI] [PubMed] [Google Scholar]
- 20.Saverymuttu S. H., Joseph A. F., Maxwell J. D. Ultrasound scanning in the detection of hepatic fibrosis and steatosis. BMJ. 1986;292(6512):13–15. doi: 10.1136/bmj.292.6512.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwimmer J. B., Behling C., Newbury R., et al. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology. 2005;42(3):641–649. doi: 10.1002/hep.20842. [DOI] [PubMed] [Google Scholar]
- 22.Wallace T. M., Levy J. C., Matthews D. R. Use and abuse of HOMA Modeling. Diabetes Care. 2004;27(6):1487–1495. doi: 10.2337/diacare.27.6.1487. [DOI] [PubMed] [Google Scholar]
- 23.Trojanek J. B., Cobos-Correa A., Diemer S., et al. Airway mucus obstruction triggers macrophage activation and matrix metalloproteinase 12-dependent emphysema. American Journal of Respiratory Cell and Molecular Biology. 2014;51(5):709–720. doi: 10.1165/rcmb.2013-0407OC. [DOI] [PubMed] [Google Scholar]
- 24.Pfaffl M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research. 2001;29(9):2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Litwin M., Michałkiewicz J., Trojanek J., Niemirska A., Wierzbicka A., Szalecki M. Altered Genes profile of Renin–Angiotensin system, immune system, and adipokines receptors in leukocytes of children with primary hypertension. Hypertension. 2013;61(2):431–436. doi: 10.1161/HYPERTENSIONAHA.111.00181. [DOI] [PubMed] [Google Scholar]
- 26.Niemirska A., Litwin M., Trojanek J., et al. Altered matrix metalloproteinase 9 and tissue inhibitor of metalloproteinases 1 levels in children with primary hypertension. Journal of Hypertension. 2016;34(9):1815–1822. doi: 10.1097/HJH.0000000000001024. [DOI] [PubMed] [Google Scholar]
- 27.Litwin M., Michałkiewicz J., Niemirska A., et al. Inflammatory activation in children with primary hypertension. Pediatric Nephrology. 2010;25(9):1711–1718. doi: 10.1007/s00467-010-1548-4. [DOI] [PubMed] [Google Scholar]
- 28.Coito A. J. Leukocyte transmigration across endothelial and extracellular matrix protein barriers in liver ischemia/reperfusion injury. Current Opinion in Organ Transplantation. 2011;16(1):34–40. doi: 10.1097/MOT.0b013e328342542e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamada T., Fondevila C., Busuttil R. W., Coito A. J. Metalloproteinase-9 deficiency protects against hepatic ischemia/reperfusion injury. Hepatology. 2008;47(1):186–198. doi: 10.1002/hep.21922. [DOI] [PubMed] [Google Scholar]
- 30.Iredale J. P. Tissue inhibitors of metalloproteinases in liver fibrosis. The International Journal of Biochemistry & Cell Biology. 1997;29(1):43–54. doi: 10.1016/S1357-2725(96)00118-5. [DOI] [PubMed] [Google Scholar]
- 31.Arpino V., Brock M., Gill S. E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biology. 2015;44-46:247–254. doi: 10.1016/j.matbio.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 32.Compana L., Iredale J. P. Regression of liver fibrosis. Seminars in Liver Disease. 2017;37(1):1–10. doi: 10.1055/s-0036-1597816. [DOI] [PubMed] [Google Scholar]
- 33.Toyoda H., Kumada T., Kiriyama S., et al. Higher hepatic gene expression and serum levels of matrix metalloproteinase-2 are associated with steatohepatitis in non-alcoholic liver disease. Biomarkers. 2012;18(1):82–87. doi: 10.3109/1354750x.2012.738249. [DOI] [PubMed] [Google Scholar]
- 34.Xue M., March L., Sambrook P. N., Jackson C. J. Differential regulation of matrix metalloproteinase 2 and matrix metalloproteinase 9 by activated protein C: relevance to inflammation in rheumatoid arthritis. Arthritis & Rheumatism. 2007;56(9):2864–2874. doi: 10.1002/art.22844. [DOI] [PubMed] [Google Scholar]
- 35.Naim A., Pan Q., Baig M. S. Matrix metalloproteinases (MMPs) in liver injury. Journal of Clinical and Experimental Hepatology. 2017;7:367–372. doi: 10.1016/j.jceh.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fingleton B. MMPs as therapeutic targets-still a viable option? Seminars in Cell & Developmental Biology. 2008;19(1):61–68. doi: 10.1016/j.semcdb.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chambers A. F., Matrisian L. M. Changing views of the role of matrix metalloproteinases in metastasis. JNCI Journal of the National Cancer Institute. 1997;89(17):1260–1270. doi: 10.1093/jnci/89.17.1260. [DOI] [PubMed] [Google Scholar]
- 38.Boeker K. H. W., Haberkorn C. I., Michels D., Flemming P., Manns M. P., Lichtinghagen R. Diagnostic potential of circulating TIMP-1 and MMP-2 as markers of liver fibrosis in patients with chronic hepatitis C. Clinica Chimica Acta. 2002;316(1-2):71–81. doi: 10.1016/S0009-8981(01)00730-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.