

Summary
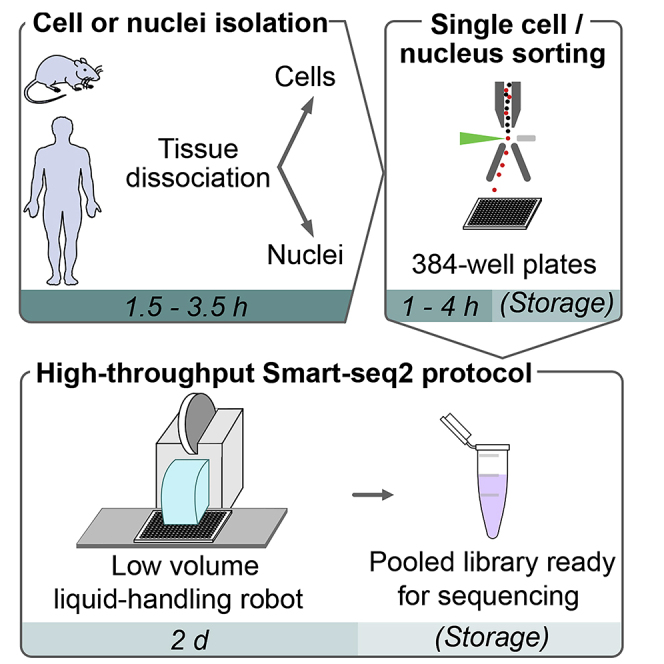
This protocol presents a plate-based workflow to perform RNA sequencing analysis of single cells/nuclei using Smart-seq2. We describe (1) the dissociation procedures for cell/nucleus isolation from the mouse brain and human organoids, (2) the flow sorting of single cells/nuclei into 384-well plates, and (3) the preparation of libraries following miniaturization of the Smart-seq2 protocol using a liquid-handling robot. This pipeline allows for the reliable, high-throughput, and cost-effective preparation of mouse and human samples for full-length deep single-cell/nucleus RNA sequencing.
For complete details on the use and execution of this protocol, please refer to Bowers et al. (2020).
Graphical Abstract
Highlights
-
•
Isolation of cells or nuclei from the mouse brain and human forebrain organoids
-
•
Flow sorting of single cells or nuclei into 384-well plates
-
•
Miniaturization of the Smart-seq2 protocol using a liquid-handling robot
-
•
Protocol enables high-throughput full-length single-cell/nucleus RNA sequencing
This protocol presents a plate-based workflow to perform RNA sequencing analysis of single cells/nuclei using Smart-seq2. We describe (1) the dissociation procedures for cell/nucleus isolation from the mouse brain and human organoids, (2) the flow sorting of single cells/nuclei into 384-well plates, and (3) the preparation of libraries following miniaturization of the Smart-seq2 protocol using a liquid-handling robot. This pipeline allows for the reliable, high-throughput, and cost-effective preparation of mouse and human samples for full-length deep single-cell/nucleus RNA sequencing.
Before You Begin
This protocol describes:
-
•
Tissue dissociation protocols for cell and nuclei isolation
-
•
Single-cell/nucleus sorting into 384-well plates
-
•
Miniaturized Smart-seq2 protocol
Note: Tissue dissociation and single-cell/nucleus sorting should be performed on the same day and as fast as possible to preserve the mRNA quality. After flow sorting, 384-well plates containing the samples can be stored at −80°C for up to 3 months before performing the Smart-seq2 protocol. At the end of Part III, pooled libraries can be kept at −20°C for up to 6 months before sequencing.
Choose the Appropriate Dissociation Protocol
We provide here three different methods for the isolation of nuclei and cells from the mouse brain or human forebrain organoids. Both nuclei and whole cells can be isolated from the embryonic and early postnatal brain (until postnatal day 12, P12). Nucleus isolation should be used when additional antibody stainings against nuclear antigens are needed for the identification of a specific population. In contrast, whole-cell preparation allows the isolation of fluorescently labeled cells in which the fluorescent signal is not retained inside the nucleus and would therefore be lost during nucleus isolation. In the adult mouse brain, whole-cell isolation results in low yield and can elicit non-specific neuronal activation reflected by the up-regulation of RNA and protein expression of Immediate Early Genes (Lacar et al., 2016). Nucleus isolation represents a fast and reliable isolation method to investigate transcription consistent with the behavioral experience of the mouse. To study human brain development and human-specific diseases, we provide a method for the dissociation of human forebrain-regionalized organoids into single cells.
Once you have chosen the appropriate dissociation protocol (mouse nuclei isolation, mouse cell isolation, or human forebrain organoid cell isolation) follow the corresponding steps below before you begin the protocol.
For Mouse Nuclei Isolation: Prepare the Working Space
Timing: 10 min
-
1.
Clean the bench, dissection tools, and the glass Dounce homogenizers with 70% ethanol and a cleaning agent to remove RNases (e.g., RNaseZap™).
-
2.
For each sample prepare and number a Petri dish, a glass Dounce homogenizer and a 1.5 mL Eppendorf tube.
-
3.
Prechill all tubes and tools on ice.
-
4.
Plunge the Pestles in a FACS tube to avoid contamination from ice.
-
5.
Pre-cool the table top centrifuge to 4°C.
For Mouse Nuclei Isolation: Prepare Solutions
-
6.
Prepare 50 mL of Nuclei Isolation Media (NIM) and 50 mL of Nuclei Storage Buffer (NSB) stock solutions.
-
7.
Filter the stock solutions with sterile 0.22 μm syringe filters.
-
8.
On experiment day prepare NIM-DP (2 mL/sample) and NSB-DP (2 mL/sample + extra 2 mL/sample if antibody staining is performed) working solutions for the appropriate number of samples.
-
9.
Filter the working solutions with sterile 0.22 μm syringe filters before use.
-
10.
Fill each glass Dounce homogenizer with 1 mL of NIM-DP working solution + 10 μL of Triton X-100 (0.1% final concentration) + 0.2 μL DNA staining dye (e.g., Hoechst, Propidium Iodide). (Continue to step 24).
CRITICAL: The pH of the stock and working solutions should be 7.4.
Note: Stock solutions can be stored at 4°C for no more than 3 months.
Note: All steps must be carried out on ice.
Note: Use RNase-free filter tips during the entire protocol.
For Mouse Cell Isolation: Prepare Fire-Polished Pasteur Pipettes
-
11.
Using a Bunsen burner, fire-polish the tip of glass Pasteur pipettes to an opening size of approximately 600, 300, and 150 μm. Check the size under a stereoscope using a fine-grade ruler. You need a set of three sizes per sample. Store in a plastic box previously cleaned with 70% ethanol and a cleaning agent to remove RNases (e.g., RNaseZap™). A video tutorial can be found at: https://www.youtube.com/watch?v=aMUs1UEJUdY
For Mouse Cell Isolation: Prepare the Working Space
-
12.
Clean the bench and dissection tools with 70% ethanol and a cleaning agent to remove RNases (e.g., RNaseZap™).
-
13.
Prepare an ice bucket, Petri dishes and as many Eppendorf tubes as there are different samples.
-
14.
Pre-cool the table top centrifuge to 4°C.
For Mouse Cell Isolation: Prepare Solutions
-
15.
Prepare 100 mL of 1× Artificial Cerebrospinal Fluid Solution (ACSF) as follows:
-
a.
Dilute 10 mL of 10× ACSF Stock Solution in 90 mL of Milli-Q water.
-
b.
Add 0.4 g of Glucose and stir well.
-
c.
Transfer the solution to a clean 100 mL bottle. Remove the lid of the bottle and insert a clean tubing connected to a gas bottle containing 95% O2/5% CO2. Open the valve at ∼1 bar pressure and bubble the solution thoroughly at room temperature for 20 min.
-
d.
While bubbling, add 0.1 mL of 1 M MgCl2 solution and 0.1 mL of 2 M CaCl2 solution. Bubble for 10 more minutes.
-
e.
Filter with a syringe connected to a 0.22 μm filter. Cool down on ice.
Optional: You can use any ultrapure water that has the same quality standard as the Milli-Q water.
Note: Be sure that glassware is clean; flush three times with Milli-Q water to remove any detergent residue.
Note: Use a volumetric flask to be more precise.
-
16.
Prepare Washing Solution (2 mL per sample) and store on ice.
-
17.
Prepare Pronase Solution (1 mL per sample) and store at room temperature.
-
18.
Prepare Quenching Solution (1 mL per sample) and store on ice.
-
19.
Prepare DNase Solution (2 mL per sample) and store on ice (continue to step 24).
Note: All solutions must be prepared right before use.
For Human Organoid Cell Isolation: Prepare the Working Space
-
20.
Clean the hood and pipets with 70% ethanol and a cleaning agent to remove RNases (e.g., RNaseZap™).
-
21.
Prepare an ice bucket containing labeled 5 mL FACS tubes with 35 μm Cell Strainer Snap Cap for each sample.
For Human Organoid Cell Isolation: Prepare Solutions
-
22.
Prepare 2 mL Accutase containing DNase per sample:
Add 20 μL of DNase to 2 mL of Accutase
-
23.
Prepare 500 μL DPBS containing EDTA per sample:
Add 1 μL of 0.5 M EDTA to 500 μL DPBS (Continue to step 24).
Note: All solutions must be prepared right before use.
Prepare for the Miniaturized Smart-seq2 Protocol
-
24.
Dissolve all the primers in TE buffer to a 100 μM concentration. Aliquot them and store them at −20°C for up to 6 months. TSO primers should be stored at −80°C, avoiding freeze-thaw cycles.
-
25.
Resuspend the control RNA (Human Liver Total RNA) in nuclease-free water to a 50 ng/μL concentration. Aliquot and store at −80°C for up to 6 months, avoiding freeze-thaw cycles.
-
26.
Prepare the barcoding plate containing 384 different Nextera XT-compatible barcoding primer combinations. The primer concentration should be 5 μM each. The barcoding plate can be stored at −20°C and reused for up to 6 months. The specific sequence for the barcodes in each well will be used to assign the reads to the different cells upon de-multiplexing.
-
27.
Program the thermocyclers with the different protocols in advance. For all the steps included in the protocol, heat the lid to 105°C to avoid excessive evaporation.
-
28.
Calibrate the STP Labtech Mosquito HV liquid-handling robot according to the manufacturer’s indications and change the spool containing the tips early enough to avoid running out of tips in the middle of the protocol.
Note: Use RNase-free filter tips during the entire protocol.
-
29.
Prepare the lysis buffer. Adapt the volumes to the number of plates that you want to prepare.
-
30.
For every 384-well sample-collection plate to be prepared, distribute 24 μL of lysis buffer to each well of a column of an empty 384-well plate.
-
31.
Use the Mosquito HV liquid-handling robot to distribute 0.8 μL from each column into an entire new 384-well sample-collection plate. Repeat this step with the rest of the columns of the master plate until you prepared the desired number of sample-collection plates.
-
32.
Cover the plates with the adhesive PCR plate seals, spin them down and store them at −20°C for up to 3 months.
Prepare for Single-Cell/Nucleus Flow Sorting
-
33.
Bring your frozen sample-collection plate in a Styrofoam box with dry ice.
-
34.
Carefully clean with 70% ethanol and a cleaning agent to remove RNases (e.g., RNaseZap™) all elements of the FACSAria III sorter that are located downstream of the sort block. In particular, the Automated Cell Deposition Unit (ACDU) adaptor, the ACDU, the sort collection chamber and the filter lid.
-
35.
Turn on the cooling water bath to bring the surface of the ACDU to 4°C.
Optional: The following steps describe how to practice your sorting into 384-well plates
A colorimetric assay, adapted from (Rodrigues and Monard, 2016), can be performed to assess the accuracy of the sorting in 384-well plates. Use the FACSAria III setup described in the step-by-step method and follow the indications below:
-
36.
Resuspend the Pierce Horseradish Peroxidase (HRP) in PBS to a 6.25 mg/mL concentration (100× stock). Aliquot and store at −20°C for up to 6 months, avoiding freeze-thaw cycles.
-
37.
Use the Mosquito HV liquid-handling robot to prepare a 384-well plate containing 0.8 μL of TMB solution per well.
-
38.
Right before sorting, add the HRP to your cells diluting it 1:100.
-
39.
Calibrate the sorter and sort the cells in the 384-well plate containing TMB.
-
40.
Evaluate the sorting efficiency by counting the number of wells that turned blue.
-
41.
Practice you sorting efficiency until it gets close to 100%.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ambion RNase inhibitor (40 U/μL) | Thermo Fischer | AM2682 |
| Accutase | Sigma-Aldrich | A6964 |
| AMPure XP beads | Beckman Coulter | A63881 |
| Betaine solution | Sigma-Aldrich | B0300 |
| Buffer EB | QIAGEN | 19086 |
| Calcium chloride dihydrate (CaCl2) | Sigma-Aldrich | C8106 |
| cOmplete Protease inhibitor Cocktail | Roche | 11697498001 |
| D-(+)-Glucose | Sigma-Aldrich | G5767 |
| DNase I recombinant, RNase-free | Roche | 4716728001 |
| dNTP Mix | Promega | U1515 |
| DTT (HSCH2CH(OH)CH(OH)CH2SH) | Sigma-Aldrich | D9779 |
| Dulbecco's phosphate-buffered saline (DPBS) |
Thermo Fisher | 14190-094 |
| Ethanol | Thommen-Furler AG | 02860 |
| Ethylenediaminetetraacetic acid (EDTA) | Sigma-Aldrich | EDS-500G |
| Fetal Bovine Serum (FBS) | Gibco | 10270106 |
| Hoechst 3342 | Thermo Fischer | H3570 |
| Human Liver Total RNA | Thermo Fischer | AM7960 |
| MgCl2 (1 M) | Thermo Fischer | AM9530G |
| Pierce™ Horseradish Peroxidase (HRP) | Thermo Fischer | 31491 |
| Potassium chloride (KCl) | Sigma-Aldrich | P9541 |
| Pronase from Streptomyces griseus | Roche | 10165921001 |
| Propidium Iodide | Thermo Fischer | R37108 |
| Protector RNase Inhibitor | Roche | 03335402001 |
| RNase-Free DNase Set | QIAGEN | 79254 |
| RNase ZAP™, cleaning agent to remove RNases | Sigma-Aldrich | R2020 |
| RNasin Plus | Promega | N2615 |
| Sodium bicarbonate (NaHCO3) | Sigma-Aldrich | S8875 |
| Sodium chloride (NaCl) | Sigma-Aldrich | 71380 |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich | S0751 |
| Sucrose | Sigma-Aldrich | S0389 |
| Tris hydrochloride (NH2-C(CH2-OH)3 · HCl) | Sigma-Aldrich | T5941 |
| Triton X-100 | Sigma-Aldrich | 93443 |
| UltraPure DNase/RNase-Free Distilled Water | Thermo Fischer | 10977035 |
| Zombie dye (Live/dead marker) with a different fluorescence then your cells of interest | Biolegend | 423113 |
| 3,3′,5,5′-tetramethyl-benzidine (TMB) | ThermoFischer | N301 |
| Critical Commercial Assays | ||
| Agilent High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 |
| D1000 Reagents | Agilent Technologies | 5067-5583 |
| D1000 ScreenTape | Agilent Technologies | 5067-5582 |
| KAPA HiFi HotStart ReadyMix | Roche | KK2601 |
| Nextera XT DNA Library Preparation Kit | Illumina | FC-131-1096 |
| SuperScript II Reverse Transcriptase | Thermo Fischer | 18064014 |
| Oligonucleotides | ||
| Oligo-dT30VN: Biot–AAGCAGTGGTATCAACGCAGA GTACT30VN |
Picelli et al., 2014 | N/A |
| TSO: Biot-AAGCAGTGGTATCAACGCA GAGTACATrGrG+G (rG=riboguanosine; +G=LNA-modified guanosine) |
Picelli et al., 2014 | N/A |
| ISPCR oligo: Biot-AAGCAGTGGTATCAACGCAGAGT |
Picelli et al., 2014 | N/A |
| IDT Illumina Nextera XT-compatible 384 UDIs (10 bp) primer set (see Table S1) | This paper (Integrated DNA Technologies) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mus musculus | N/A | N/A |
| Human ESC line H9 | Wicell | WA09-PCBC |
| Other | ||
| Adhesive PCR Plate Foils | Thermo Fischer | AB0626 |
| Adhesive PCR Plate Seals | Thermo Fischer | AB0558 |
| Bunsen burner | Falc Instruments™ | 140205010 |
| CoolRack XT PCR384 | Biocision | BCS-538 |
| C1000 Touch™ Thermal Cycler with 384-Well Reaction Module | BioRad | 1851138 |
| DNA LoBind Tubes (1.5 mL) | Eppendorf | 0030108051 |
| DNA LoBind Tubes (2 mL) | Eppendorf | 0030108078 |
| DynaMag-2 Magnet | Thermo Fischer | 12321D |
| Epifluorescence stereoscope | Leica | MZ FL III |
| Eppendorf twin.tec PCR Plate 384 | Eppendorf | 0030128508 |
| Falcon® 5mL Test Tube with Cell Strainer Snap Cap | Corning | 352235 |
| FACSAria III | BD Biosciences | N/A |
| Fine-grade ruler | N/A | N/A |
| Magnetic bead separation block | SPT Labtech | N/A |
| Millex-GP, 0.22 μm filters | Millipore | SLGP033RS |
| Mosquito HV liquid-handling robot | SPT Labtech | N/A |
| Osmometer | Advanced Instruments | Model 3250 |
| Pasteur pipettes with cotton plugs | Hilgenberg | 3177102 |
| pH-Meter | Accumet | AB150 |
| Spool of Mosquito HV pipette tips at 4.5 mm pitch (18,500 per spool) | SPT Labtech | N/A |
| Stereoscope | Zeiss | Stemi 2000-C |
| Sterilin™ Standard 90mm Petri Dishes | Thermo Fisher | 101R20 |
| Syringe Omnifix® 50 mL, Luer lock attachment | B. Braun | 4617509F |
| Table top centrifuge for 1.5-2.0 mL tubes | Eppendort | 5424 R |
| Water bath at 37°C | Thermo Fischer | TSGP02 |
| 2100 Bioanalyzer Instrument | Agilent Technologies | G2939BA |
| 384-well Low Volume Serial Dilution (LVSD) plates |
SPT Labtech | N/A |
| 4200 TapeStation System | Agilent Technologies | G2991AA |
Materials and Equipment
NIM Stock Solution
| Reagent | Final Concentration | Add to 50 mL |
|---|---|---|
| Sucrose (1.5 M) | 250 mM | 8.33 mL |
| KCl (1 M) | 25 mM | 1.25 mL |
| MgCl2 (1 M) | 5 mM | 250 μL |
| TrisCl pH 7.4 (1 M) | 10 mM | 500 μL |
| Milli-Q water | N/A | up to 50 mL |
NSB Stock Solution
| Reagent | Final Concentration | Add to 50 mL |
|---|---|---|
| Sucrose (dry) | 166.5 mM | 2.85 g |
| MgCl2 (1 M) | 5 mM | 250 μL |
| TrisCl pH 7.4 (1 M) | 10 mM | 500 μL |
| Milli-Q water | N/A | up to 50 mL |
NIM-DP Working Solution (2 mL/sample)
| Reagent | Final Concentration |
|---|---|
| NIM Stock solution | 1× |
| DTT | 1 μM |
| Protease Inhibitor | 1× |
| RNase Inhibitor | 1:1,000 |
NSB-DP Working Solution (2 mL/Sample + Extra 2 mL/Sample if Antibody Staining Is Performed)
| Reagent | Final Concentration |
|---|---|
| NSB Stock solution | 1× |
| DTT | 1 μM |
| Protease Inhibitor | 1× |
| RNase Inhibitor | 1:1,000 |
10× ACSF Stock Solution
| Reagent | Final Concentration | Add to 1 L |
|---|---|---|
| NaCl | 1,250 mM | 73.05 g |
| KCl | 25 mM | 1.86 g |
| NaH2PO4 | 12.92 mM | 1.55 g |
| NaHCO3 | 256 mM | 21.5 g |
| Milli-Q water | N/A | up to 1 L |
Note: 10× ACSF Stock Solution can be stored at 4°C for no more than 2 months.
Note: For each new batch of 10× ACSF Stock Solution, check whether the final 1× ACSF has the right pH (between 7.35 and 7.45) and osmolarity (between 305 and 315 mOsm). To do this, prepare 100 mL of 1× ACSF Solution and measure it with a pH-meter and an osmometer within 2 min after having bubbled with 95% O2/5% CO2.
1× ACSF Solution
| Reagent | Final Concentration | Add to 100 mL |
|---|---|---|
| 10× ACSF Stock Solution | 1× | 10 mL |
| Glucose | 22 mM | 0.4 g |
| MgCl2 1 M | 1 mM | 0.1 mL |
| CaCl2 2 M | 2 mM | 0.1 mL |
| Milli-Q water | N/A | up to 100 mL |
Note: Prepare 1× ACSF Solution following the instructions from the “Cell dissociation from the neonatal mouse brain” section.
Washing Solution
| Reagent | Final Dilution |
|---|---|
| 1× ASCF | 1× |
| RNase inhibitor (RNAsin Plus) | 1:1,000 |
Pronase Solution
| Reagent | Final Dilution |
|---|---|
| 1× ASCF | 1× |
| RNase inhibitor (RNAsin Plus) | 1:1,000 |
| Pronase | 1 mg/mL |
Note: Vortex thoroughly to dissolve Pronase
Quenching Solution
| Reagent | Final Dilution |
|---|---|
| 1× ASCF | 1× |
| RNase inhibitor (RNAsin Plus) | 1:1,000 |
| Fetal Bovine Serum (FBS) | 1% |
DNase Solution
| Reagent | Final Dilution |
|---|---|
| 1× ASCF | 1× |
| RNase inhibitor (RNAsin Plus) | 1:1,000 |
| DNase | 1:100 |
Lysis Buffer
| Reagent | Final Concentration | Volume per Well (μL) | Volume for a 384-Well Plate |
|---|---|---|---|
| Triton X-100 (10%) | 0.1% | 0.008 | 4 μL |
| Nuclease-free water | N/A | 0.552 | 277 μL |
| RNasin Plus (40 U/μL) | 1 U/μL | 0.02 | 10 μL |
| Biotinylated Oligo-dT (100 μM) | 2.5 μM | 0.02 | 10 μL |
| dNTP Mix (10 mM each) | 2.5 mM | 0.2 | 100 μL |
| Total | N/A | 0.8 μL | 401 μL |
RT Mix
| Reagent | Final Concentration | Volume per Well (μL) | Volume per 384-Well Plate (μL) |
|---|---|---|---|
| SuperScript II reverse transcriptase (200 U/μL) | 20 U | 0.1 | 50 |
| RNasin Plus (40 U/μL) | 2 U | 0.05 | 25 |
| Superscript II first-strand buffer (5×) | 1× | 0.4 | 200 |
| DTT (100 mM) | 5 mM | 0.1 | 50 |
| Betaine (5 M) | 1 M | 0.4 | 200 |
| MgCl2 (1 M) | 6 mM | 0.012 | 6 |
| TSO (100 μM) | 1 μM | 0.02 | 10 |
| Nuclease-free water | N/A | 0.058 | 29 |
| Total | N/A | 1.14 | 570 |
PCR Mix
| Reagent | Final Concentration | Volume per Well (μL) | Volume per 384-Well Plate (μL) |
|---|---|---|---|
| KAPA HiFi HotStart ReadyMix (2×) | 1× | 2.5 | 1,250 |
| IS PCR primers (100 μM) | 0.1 μM | 0.005 | 2.5 |
| Nuclease-free water | N/A | 0.495 | 250 |
| Total | N/A | 3 | 1,502.5 |
Tagmentation Mix
| Reagent | Final Concentration | Volume per Well (μL) | Volume per 384-Well Plate (μL) |
|---|---|---|---|
| Tagment DNA Buffer (TD, 2×) | 1× | 2 | 1,000 |
| Amplicon Tagment Mix (ATM) | N/A | 1 | 500 |
| Total | N/A | 3 | 1,500 |
Indexing PCR mix
| Reagent | Final Concentration | Volume per Well (μL) | Volume per 384-Well Plate (μL) |
|---|---|---|---|
| Nextera PCR Master Mix (NPM) | N/A | 3 | 1,500 |
| Nuclease-free water | N/A | 1 | 500 |
| Total | N/A | 4 | 2,000 |
-
•
Pronase (Roche 10165921001) causes serious eye and skin irritation and may cause respiratory irritation and breathing difficulties if inhaled. Handle it using appropriate safety equipment.
-
•
DNase (QIAGEN 79254) may cause an allergic skin reaction and breathing difficulties if inhaled. Handle it using appropriate safety equipment.
-
•
Triton X-100 (Sigma-Aldrich, T9284) can cause skin irritation and serious eye damage if spilled. Handle it using appropriate safety equipment.
-
•
SuperScript II Reverse Transcriptase kit (Thermo Fischer, 18064014DTT) contains DTT. Avoid inhaling fumes or contact with the skin. Handle it using appropriate safety equipment.
-
•
Ethanol (Thommen-Furler AG02860-1L) is flammable. Handle it inside a hood or in a well-ventilated room using appropriate safety equipment.
-
•
Keep hands clear of the instrument while the Mosquito HV liquid-handling robot is pipetting.
Alternatives:
-
•
The standard reagents in this protocol (e.g., dNTPs, RNase inhibitor, ethanol, salt solutions) can be replaced by equivalent products from other manufacturers.
-
•
This protocol is based on the full volume version of Smart-seq2 (Picelli et al., 2014). It uses SuperScript II Reverse Transcriptase (Thermo Fischer, 18064014) but it can be replaced by SuperScript IV Reverse Transcriptase (Thermo Fischer, 18090050) with minor adjustments.
-
•
In this protocol, Bioanalyzer is used to assess the abundance and quality of cDNA and the 4200 TapeStation System is used to determine library quality. Any other solution for accurate nucleic acid analysis, such as Fragment Analyzer Systems (Agilent technologies), could also be used.
Step-By-Step Method Details
Nucleus Dissociation from the Mouse Brain
This protocol allows the isolation of nuclei from the embryonic or adult mouse brain (Figure 1). At the end of the dissociation procedure, optional antibody staining against nuclear antigens can be performed to allow the isolation of specific populations.
Note: For complete details on the use and execution of this protocol, please refer to (Jaeger et al., 2018).
-
1.
Sacrifice the embryo or adult mouse through decapitation and dissect the brain into a cold Petri dish previously placed on ice. Quickly dissect the region of interest.
-
2.
Transfer the dissected tissue into a glass Dounce homogenizer containing NIM-DP, Triton and the DNA-binding dye.
-
3.
Dissociate the tissue with the loose pestle (pestle A) 5–7 strokes without introducing bubbles.
-
4.
Apply 8–10 strokes gently with the tight pestle (pestle B). (See troubleshooting section for additional information).
-
5.
Transfer the contents to pre-chilled 1.5 mL Eppendorf tubes and spin for 7 min at 90 × g and 4°C.
-
6.
Aspirate the supernatant and gently resuspend in 1 mL of NIM-DP.
-
7.
Repeat spin for 7 min at 90 × g and 4°C to remove more Triton.
-
8.
Aspirate the supernatant and resuspend in 500 μL of NSB-DP.
-
9.
Strain through pre-chilled 5 mL FACS tubes with 35 μm Cell Strainer Snap Cap on ice.
-
10.
Count the nuclei and adjust the concentration to 10 × 106 nuclei/mL in NSB-DP.
-
11.
Keep tubes on ice, protected from light, until sorting (continue to step 57) or proceed to antibody staining (continue to step 12 below).
Note: The volume of NSB-DP must be adjusted to prevent excessive clogging during the filtering step.
Note: The slow centrifugation speed is critical to avoid damaging the nuclei.
Note: Nuclei preparation generates numerous cellular debris, the size of which is in the similar range as the nuclei. To facilitate counting, nuclei can be visualized via the DNA staining dye used in step 10.
Figure 1.

Schematic Representation of the Nucleus Dissociation Protocol for the Mouse Brain
Optional Antibody Staining
Note: If directly labeled antibodies against your antigen of interest are available, perform direct staining (skip steps 22–29). If there are no directly labeled antibodies available, indirect staining must be performed. Nuclei will first be stained with a primary antibody against your antigen of interest and subsequently with a fluorescently labeled secondary antibody which recognizes the primary antibody.
-
12.
Aliquot 100 μL of your nucleus suspension (1 × 106 nuclei /well) into as many wells of a 96 well V-bottom plate as required.
Note: If you want to sort a high number of nuclei or if the estimated abundance of your population of interest within your sample is low, prepare multiple wells that will be stained with the same antibody panel. At the end of the staining procedure, wells with the same condition will be pooled into a single FACS tube for sorting.
Note: Prepare additional wells for staining controls needed to properly interpret your flow cytometry data. This includes unstained nuclei, single stained nuclei and Fluorescence Minus One (FMO) controls.
-
13.
Spin down the plate for 5 min at 200 × g and 4°C.
-
14.
While your plate is in the centrifuge, prepare your primary antibody mixes in NSB-DP (100 μL of antibody mix/well) at the manufacturer-recommended dilutions.
Note: Final antibody concentrations usually range between 0.1–10 μg/mL but optimum concentrations should be determined by antibody titration.
-
15.
Remove the supernatant by “flicking” the plate into the sink.
Note: “Flicking” the plate is a very fast and efficient technique to remove the supernatant that works best with 96-well V-bottom plates. At the end of the centrifugation, firmly hold the plate upright in the palm of your hand. In a single motion, move the plate upwards, turn it over and bring it straight down, stopping abruptly above the sink.
-
16.
Resuspend the nuclei in 100 μL of the primary antibody mix/well.
-
17.
Incubate for 30 min at 4°C in the dark.
-
18.
Spin down the plate for 5 min at 200 × g and 4°C.
-
19.
Remove the supernatant by “flicking” the plate into the sink.
-
20.
Resuspend in 200 μL NSB-DP/well.
-
21.
Spin down the plate for 5 min at 200 × g and 4°C.
-
22.
While your plate is in the centrifuge, prepare your fluorochrome-labeled secondary antibody mixes in NSB-DP (100 μL of antibody mix/well) at the manufacturer-recommended dilutions.
-
23.
Remove the supernatant by “flicking” the plate into the sink.
-
24.
Resuspend the nuclei in 100 μL of the secondary antibody mix/well.
-
25.
Incubate for 20–30 min at 4°C in the dark.
-
26.
Spin down the plate for 5 min at 200 × g and 4°C.
-
27.
Remove the supernatant by “flicking” the plate into the sink.
-
28.
Resuspend in 200 μL NSB-DP/well.
-
29.
Spin down the plate for 5 min at 200 × g and 4°C.
-
30.
Resuspend the nuclei with 100 μL NSB-DP/well.
-
31.
Transfer the samples to FACS tubes and pool wells stained with the same conditions.
-
32.
Dilute samples with NSB-DP to 5–30 × 106 nuclei/tube and keep on ice and protected from the light until sorting (continue to step 57).
Cell Dissociation from the Neonatal Mouse Brain
This protocol allows the isolation of fluorescently labeled neurons from the neonatal mouse brain (P6-P12) (Figure 2).
Note: This is a faster adaptation from a previous protocol (Hempel et al., 2007).
Note: Do not process more than 6 brains per time.
Note: All solutions but Pronase Solution must be ice-cold.
Note: All steps but incubation in Pronase Solution must be carried out on ice.
Note: Use RNase-free filter tips in all the steps.
-
33.
Sacrifice the pup through decapitation and dissect the brain into a cold Petri dish previously placed on ice. Quickly dissect the region of interest.
-
34.
Check the quality of the dissection under the fluorescent stereoscope. Take a picture to keep track of the procedure, but try to be as fast as possible.
-
35.
Transfer the dissected tissue into a 1.5 mL Eppendorf tube.
Note: If you process more than one brain, clean carefully the dissection tools between each sample; use ethanol 70% and a cleaning agent to remove RNases. Leave the samples on ice until you have dissected all of them.
-
36.
Add 1 mL per brain of Pronase Solution and incubate at room temperature. Short incubation with Pronase digests the extracellular matrix. Incubation time depends on the age: 21 min at P6, 24 min at P9 and 27 min at P12.
-
37.
Remove Pronase Solution and rinse once with 1 mL of Washing Solution. (See troubleshooting section for additional information).
-
38.
Remove Washing Solution and add 1 mL of Quenching Solution. FBS contained in the Quenching solution inactivates the residual Pronase. Incubate for 5 min.
-
39.
Remove Quenching Solution and rinse once with 1 mL of Washing Solution.
-
40.
Remove Washing Solution and add 0.5 mL of DNase Solution.
-
41.
Gently triturate the sample pipetting up and down 10 times with the fire-polished 600 μm Pasteur pipettes. Repeat 10 times with the 300 μm pipettes and 10 times with 150 μm pipettes. Avoid making bubbles. Do not touch the bottom of the tube to avoid a tighter closing of the pipette tip.
Note: During this step a lot of cellular debris are generated and many cells die. Damaged cells release DNA which tends to create floccules. The DNase contained in the solution will degrade any released DNA, preventing the clogging of glass Pasteur pipettes.
-
42.
Strain through pre-chilled 5 mL FACS tubes with 35 μm Cell Strainer Snap Cap on ice.
-
43.
Count the cells and resuspend at 1–10 million cells per tube in DNase Solution.
-
44.
Add a live/dead discrimination marker (e.g., Zombie dyes) at a 1:100 to 1:1,000 dilution range and incubate for 15 min at room temperature, protected from light.
-
45.
Wash and resuspend in DNase Solution at–5 to 30 × 106 cells/tube and keep on ice and protected from the light until sorting (continue to step 57).
Figure 2.

Schematic Representation of the Cell Dissociation Protocol for the Neonatal Mouse Brain
Cell Dissociation from Human Forebrain Organoids
This method allows the isolation of single cells from human forebrain-regionalized organoids (Figure 3).
Note: The protocol is partially adapted from the dissociation described in (Camp et al., 2015).
Note: The protocol is optimized for the dissociation of approximately 10 organoids. However, it can be scaled up accordingly.
Note: Use RNase-free filter tips in all the steps.
-
46.
Collect organoids in a 2 mL Eppendorf tube.
-
47.
Wash organoids 3 times with DPBS.
Note: Due to the density of the organoids they tend to sink to the bottom of the Eppendorf otherwise briefly spin the tube in a mini-centrifuge.
-
48.
Transfer organoids into 2 mL Accutase containing DNase.
-
49.
Incubate in a water bath for 45 min.
-
50.
After the first 10 min of incubation treat them every 5 min by alternating gentle shaking (vortex at slow speed) and pipetting up and down using a P200 pipet (Figure 3).
Note: Damaged cells release DNA which tends to create floccules; the DNase contained in the solution will degrade any released DNA, preventing the clogging of the pipette tip.
-
51.
Collect cells by centrifugation at 100 × g for 4 min.
-
52.
Wash cells twice using DPBS.
-
53.
Resuspend cell pellet in 500 μm DPBS containing EDTA.
-
54.
Strain through pre-chilled 5 mL FACS tubes with 35 μm Cell Strainer Snap Cap on ice.
-
55.
Add a live/dead discrimination marker (e.g., Zombie dyes) at a 1:100 to 1:1,000 dilution range and incubate for 15 min at room temperature, protected from light.
-
56.
Wash and resuspend in DPBS containing EDTA at 5–30 × 106 cells/tube and keep on ice and protected from the light until sorting (continue to step 57).
Figure 3.

Schematic Representation of the Cell Dissociation Protocol for Human Forebrain Organoids
Single-Cell/Nuclei Flow Sorting
In this section, we describe the procedure to sort single cells/nuclei into 384-well plates using a FACS Aria III, BD Biosciences (Figure 4).
-
57.
Calibrate the FACSARIA III sorter using the 85 μm nozzle and a sheath pressure of 45psi.
-
58.
Following stream set up and drop delay adjustments, use the far-left side stream to distribute the sample into the 384-well plate. The far-left stream should be targeted to the inner left 4-way position using the voltage sliders (Figure 4A).
-
59.
Calibrate the “Home” position of the plate (well A1). To do so, use the same type of 384-well plate as the one you will be sorting your sample in (e.g., Eppendorf twin.tec PCR Plate 384) and cover it with a transparent adhesive plate-sealing film. Perform a very brief test sort with the aspirator drawer open to aim the side stream to the 384-well plate. Observe the location of the drop on the transparent plate-sealing film at the A1 well position. Wipe-off the drop with a cotton swab. Adjust the position of the ACDU. Repeat until the drop is positioned as shown in Figure 4B.
Note: Although using bigger nozzles is gentler on the cells, in our hands, it decreases the accuracy of the stream and leads to more wells with poor RNA content. Drops that do not immediately reach the lysis buffer during sorting are likely to evaporate on the sides of the well which affects the RNA quality.
-
60.
The FACSAria III is ready for sorting. You can define your experiment and gating strategy. Representative FACS plots showing nuclei obtained from the adult mouse brain and cells isolated from the neonatal mouse brain or human forebrain organoids are shown in Figure 5.
Figure 4.

Schematic Representation of the Key Settings Recommended for Single-Cell/Nucleus Sorting into 384-Well Plates Using the FACS Aria III (BD Biosciences)
(A) Schematic representation of the stream and the nozzle to be used in the protocol.
(B) Drop location in home position.
(C) Recommended plate layout.
Figure 5.

Gating Strategies to Identify Single Cells/Nuclei Prior to Sorting
Representative FACS plots showing examples of gating strategies for the isolation of NeuN+ nuclei from the adult hippocampus (A), tdTom-labeled neurons (whole cells) from the postnatal mouse brain at P9 (B), and tdTom+ cells isolated from human forebrain-regionalized organoids after 36 days in culture (C).
-
61.
Thaw the sample-collection plate for 1 min at room temperature and spin it down for 2 min at 1,500–2,000 × g and 4°C.
-
62.
Remove the plate-sealing film and carefully place the 384-well plate on the ACDU.
-
63.
Verify your gating strategy (e.g., singlets, live cells) and hierarchy.
-
64.
Select the population to be sorted and define your sort layout leaving wells A1 and P24 empty for RNA spike-in controls.
Note: To limit the evaporation of the low volume of lysis buffer (0.8 μL/well) in the sample-collection plate, the ACDU cooling system is essential and the sorting time should be reduced as much as possible (maximum ∼1h per plate). Long sorting time might affect the quality of the samples and result in plate biases (please see the Critical point below for additional explanation).
-
65.
Set the “precision mode” on “single”.
-
66.
Activate the “index sorting” mode to allow the retrospective identification of each single cell/ nuclei’s high-dimensional phenotype in your sample-collection plate.
-
67.
Start acquisition and sort. (See troubleshooting section for additional information).
-
68.
At the end of your sort, cover your plate with an adhesive PCR plate foil.
-
69.
Annotate the side of your plate with the date/experimental information and spin it down for 5 min at 1,500–2,000 × g and 4°C.
-
70.
Put your plate back in the Styrofoam box containing dry ice. Store at −80°C as soon as possible.
Pause Point: The sorted plates can be stored at −80°C for up to 3 months before proceeding to the miniaturized Smartseq-2 protocol.
Note: When using a stringent sorting mask (e.g., precision mode on “single cell”), the drop-out rate during sorting can be especially high. For this reason, one will need to start with a number of cells/nuclei that is significantly larger than the desired amount of final sorted single cells/nuclei.
Miniaturized Smart-seq2
Here, we describe a miniaturized and semi-automated version of the Smart-seq2 protocol (Picelli et al., 2013, Picelli et al., 2014) using the low-volume liquid-handling robot Mosquito HV, SPT Lab Tech (Figure 6).
Figure 6.

Schematic Representation of the Main Steps of the Miniaturized Smart-seq2 Protocol
Miniaturized Smart-seq2: Reverse Transcription
Perform reverse transcription to obtain cDNA from single cells/nuclei previously sorted into lysis buffer in 384-well sample-collection plates.
-
71.
If the collection plate containing the cells previously sorted in lysis buffer is frozen at −80°C, spray it with RNase decontamination solution and thaw it slowly using a 384-well cooling rack on ice. If you are working with a freshly sorted plate, proceed to the next step.
-
72.
Thaw and dilute the control RNA in nuclease-free water to 25 pg/μL (1:2,000).
-
73.
Add 0.5 μL of the diluted control RNA to wells A1 and P24 (these wells should have been left empty during the cell sorting and only contain lysis buffer).
Note: If more wells are needed to evaluate the performance of the method, add 0.5 μL of the control RNA to the selected wells.
-
74.
Cover the 384-well plate with an adhesive seal and incubate it in a thermocycler for 3 min at 72°C. Transfer the plate back to ice immediately afterwards.
-
75.
Thaw the required reagents and prepare the RT mix.
-
76.
Distribute 35 μL of RT mix to each well of a column in a 384-well plate (master plate).
-
77.
Use the Mosquito HV liquid-handling robot to distribute 1.14 μL from the column in the master plate into all the columns of the 384-well sample-collection plate. The final volume per well is 2 μL.
-
78.
Cover the plate with an adhesive PCR plate seal, vortex briefly, and spin down.
-
79.
Run the RT reaction following the program below:
| Reverse Transcription Program | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| RT and template-switching | 42°C | 90 min | 1 |
| Unfolding of RNA secondary structures | 50°C | 2 min | 10 |
| RT and template-switching | 42°C | 2 min | |
| Enzyme inactivation | 70°C | 15 min | 1 |
| Hold | 4°C | Forever | |
Miniaturized Smart-Seq2: cDNA Amplification
Perform a PCR reaction to amplify the cDNA generated in the reverse transcription.
-
80.
Thaw the required reagents and prepare the PCR mix.
-
81.
Because the volume for an entire plate does not fit into a single column, distribute 30 μL of PCR mix to each of the wells of three columns of a 384-well master plate.
-
82.
Use the Mosquito HV liquid-handling robot to distribute 3 μL from each column of the master plate into 8 columns of the 384-well sample-collection plate containing the cDNA. Repeat the process until the PCR mix has been added to the whole plate. The final volume per well is 5 μL.
-
83.
Cover the plate with an adhesive PCR plate seal, vortex briefly, and spin down.
-
84.
Run the PCR reaction following the program below:
| cDNA Amplification Program | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial denaturation | 98°C | 3 min | 1 |
| Denaturation | 98°C | 20 s | 20–24 |
| Annealing | 67°C | 15 s | |
| Extension | 72°C | 6 min | |
| Final extension | 72°C | 5 min | 1 |
| Hold | 4°C | Forever | |
Miniaturized Smart-Seq2: cDNA Purification (Bead Clean-Up)
Purify the cDNA using Ampure XP beads.
-
85.
Before starting, equilibrate the Ampure XP beads at room temperature for 30 min. Vortex thoroughly to ensure that the beads are properly mixed with the buffer.
-
86.
Prepare an 80% (vol/vol) ethanol solution in nuclease-free water.
-
87.
Distribute 40 μL of Ampure XP bead suspension to each well of three columns of an LVSD 384-well plate (master plate).
-
88.
Use the Mosquito HV liquid-handling robot to distribute 4 μL of beads from each column of the master plate into 8 columns of the 384-well sample-collection plate containing the amplified cDNA. Repeat the process until the beads have been added to the whole plate. The final volume per well is 9 μL (1:0.8 cDNA to beads ratio).
-
89.
Cover the plate with an adhesive PCR plate seal, vortex, and incubate 5 min at room temperature.
-
90.
Spin down the plate and incubate in a 384-well magnetic rack using the magnetic bead separation block from SPT Labtech for 5 min.
-
91.
Using the Mosquito HV liquid-handling robot, wash the magnetic beads as follows:
-
a.
To avoid evaporation problems, the beads will be consecutively washed in 3 sets of 8 columns (1–8, 9–16, 17–24).
-
b.Prepare a clean LVSD 384-well master plate containing the following reagents and install it on the robot:
-
-6 columns containing 55 μL of 80% ethanol solution (columns 1–6 in the master plate).
-
-3 columns containing 50 μL of EB buffer (columns 7–9 in the master plate).
-
-
-
c.
Install the 384-well magnetic rack with the cDNA plate on the robot.
-
d.
Remove 9 μL of supernatant of wells 1–8. The supernatants of the different washing steps can be transferred to an empty column of the same LDSD 384-well plate.
-
e.
Add 5 μL of 80% ethanol from column 1 to the master plate to columns 1–8 in the cDNA plate.
-
f.
Repeat steps d-e for columns 9–16 and 17–24, pipetting the ethanol from columns 2 and 3 respectively.
-
g.
Remove the ethanol (5 μL) from well 1–8 in the cDNA plate.
-
h.
Add 5 μL of 80% ethanol from column 4 in the master plate to columns 1–8 in the cDNA plate.
-
i.
Repeat steps g and h for columns 9–16 and 17–24, pipetting the ethanol from columns 5 and 6 respectively.
-
j.
Remove the ethanol (5 μL) from wells 1–8 in the cDNA plate.
-
k.
Add 5 μL of EB buffer from columns 7 in the master plate to columns 1–8 in the cDNA plate.
-
l.
Repeat steps j-k for columns 9–16 and 17–24, pipetting the EB buffer from columns 8 and 9 respectively.
-
92.
Cover the plate with an adhesive PCR plate seal, vortex well and incubate 5 min at room temperature (out of the magnetic rack).
-
93.
Install the plate back on the 384-well magnetic rack on the robot.
-
94.
Transfer 5 μL of the supernatant containing the clean cDNA into a new 384-well plate.
Note: The volume of EB containing clean cDNA might be less than 5 μL.
Miniaturized Smart-Seq2: cDNA Quality Control and Quantification
Evaluate the quality and concentration of the cDNA.
Note: Assessing the quality of a whole plate is expensive and time consuming. Consequently, adapt the number of samples to analyze to the throughput and price of the QC instrument you are using. When using an Agilent High Sensitivity DNA Kit in an Agilent 2100 Bioanalyzer Instrument, the chip can measure 11 samples per run. Try to cover the maximum number of columns diagonally to assess the general quality of the plate. If using only one chip, for instance, analyze wells A1 (RNA control), B2, C3, D4, E5, F6, L20, M21, N22, O23 and P24 (RNA control).
-
95.
Follow manufacturer’s instruction to check the size distribution of the cDNA from selected wells using an Agilent High Sensitivity DNA Kit in an Agilent 2100 Bioanalyzer Instrument. A good cDNA preparation should be almost free of small fragments (<300 bp) and should peak at ∼1.5–2 kb (Figure 7).
Figure 7.

Examples of cDNA Profiles Obtained on the Bioanalyzer Instrument
Miniaturized Smart-Seq2: cDNA Tagmentation
Fragment the cDNA and introduce the adaptor sequences for the final library preparation.
-
96.
Thaw the required reagents and prepare the tagmentation mix.
-
97.
Distribute 30 μL of tagmentation mix to each well of three columns of a 384-well master plate.
-
98.
Use the Mosquito HV liquid-handling robot to distribute 3 μL from each column of the master plate into 8 columns of a new empty 384-well plate. Repeat the process until the tagmentation mix has been added to the whole plate.
-
99.
Use the Mosquito HV liquid-handling robot to distribute 1 μL from each well of the 384-well sample-collection plate containing the clean cDNA, to the plate containing the tagmentation mix. The final volume per well is 4 μL.
-
100.
Cover the plate with an adhesive PCR plate seal, vortex briefly, and spin down.
-
101.
Run the tagmentation reaction following the program below:
| cDNA Tagmentation Program | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Tagmentation | 55°C | 5 min | 1 |
| Hold | 4°C | Forever | |
-
102.
Once the tagmentation is done, use the Mosquito HV liquid-handling robot to distribute 1 μL of Neutralize Tagment Buffer (NT) into each well of the tagmentation plate. The final volume per well is 5 μL.
Note: This is the most expensive part of the protocol. The above described reactions are performed using a whole Nextera XT DNA Library Preparation Kit (96 rxn) for a 384-well plate. If both the quality and abundance of cDNA are high, the tagmentation can be performed with half the amount of cDNA and half the volumes of reagents indicated in this section, which significantly reduces the cost of the whole protocol.
Miniaturized Smart-Seq2: Index Addition and Amplification of Libraries
Barcode and amplify the libraries from the individual wells.
-
103.
Thaw the required reagents and prepare the Indexing PCR.
-
104.
Distribute 40 μL of the indexing PCR mix to each well of three columns of a master 384-well plate.
-
105.
Use the Mosquito HV liquid-handling robot to distribute 4 μL from each column of the master plate into 8 columns of the 384-well plate containing the tagmented cDNA. Repeat the process until the indexing PCR mix has been added to the whole plate.
-
106.
Use the Mosquito HV liquid-handling robot to distribute 1 μL from each well in the barcoding plate to the corresponding well in the 384-well plate containing the tagmented cDNA together with the indexing PCR mix. The final volume per well is 10 μL.
-
107.
Cover the plate with an adhesive PCR plate seal, vortex briefly, and spin down.
-
108.
Run the indexing PCR reaction following the program included in the following table.
| Indexing PCR program | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial extension | 72°C | 3 min | 1 |
| Initial denaturation | 95 | 30 s | 1 |
| Denaturation | 95°C | 10 s | 10 |
| Annealing | 55°C | 30 s | |
| Extension | 72°C | 30 s | |
| Final extension | 72°C | 5 min | 1 |
| Hold | 4°C | Forever | |
-
109.
Once the indexing PCR is done, use the Mosquito HV liquid-handling robot to pool the whole 384-well plate into three columns of an empty LVSD plate.
-
110.
Manually pool the content of all the wells in the three columns into two 2 mL tubes (the expected volume is around 3.5 mL).
Note: The number of cycles in the indexing PCR can be adjusted from 8 to 12 depending on the amount of cDNA used as an input for the tagmentation. The whole plate is pooled prior to bead clean-up and library QC. However, for the implementation of the protocol, the libraries in the different wells can be individually purified and QCed if needed.
Miniaturized Smart-Seq2: Final Library Purification (Bead Clean-Up)
Double-sided size selection of final libraries.
-
111.
Before starting, equilibrate the Ampure XP beads at room temperature for 30 min. Vortex thoroughly to ensure that the beads are properly mixed with the buffer.
-
112.
Prepare an 80% (vol/vol) ethanol solution in nuclease-free water.
-
113.
Distribute the final libraries into four 2 mL tubes (the expected volume is around 900 μL in each of them). Process all tubes in parallel following the subsequent steps in the protocol.
-
114.
Add 450 μL of beads to each tube (1:0.5 library to beads ratio).
-
115.
Vortex briefly and incubate them 5 min at room temperature.
-
116.
Put the tubes in a compatible magnetic rack and incubate them until the solution clears.
-
117.
Transfer the supernatants to new 2 mL tubes.
-
118.
Add 270 μL of beads to each tube (1:0.3 library to beads ratio).
-
119.
Vortex briefly and incubate them 5 min at room temperature.
-
120.
Put the tubes in a compatible magnetic rack and incubate them until the solution becomes clear.
-
121.
Add 1 mL of 80% ethanol to each of the tubes and incubate for 30 s.
-
122.
Remove the ethanol.
-
123.
Repeat steps 62 and 63 for a total of two washes.
-
124.
Remove the tubes from the magnetic rack and add 50 μL of EB buffer.
-
125.
Vortex briefly and incubate them for 5 min at room temperature.
-
126.
Put the tubes back into a compatible magnetic rack and incubate them until the solution clears.
-
127.
Collect the supernatant of all tubes and transfer them into a new 1.5 mL tube. The libraries are clean and ready for QC and sequencing.
Miniaturized Smart-Seq2: Final Library QC
Evaluate the quality and concentration of the libraries prior to sequencing.
-
128.
Follow the manufacturer’s instruction to check the size distribution of the library pool using D1000 kit in an Agilent 4200 TapeStation System. A good library pool should show a broad distribution with a wide peak at around 300–800 bp (Figure 8).
Figure 8.

Examples of Library Profiles Obtained on the TapeStation Instrument
Note: The libraries can be sequenced single-end or paired-end, although single-end (75–125 bp) is sufficient for standard single-cell transcriptomics analysis. Depending on the analytical needs, the sequencing depth can be adjusted from 250K to 1 Million reads per cell. Illumina HiSeq2500/4000, NextSeq500/550 or NovaSeq6000 are suitable instruments to achieve the recommended depth.
Expected Outcomes
At the end of this protocol your single-cell/nucleus libraries are ready for sequencing. After sequencing and demultiplexing, each cell/nucleus will have its own raw sequencing FASTQ file based on the unique index combination used for each well of your 384-well plate. Downstream bioinformatic analysis—using for instance the Seurat pipeline (Stuart et al., 2019)—will allow you to uncover the transcriptional content of your individual cells/nuclei. If you activated the “index” mode during flow sorting, you will be able to correlate the protein expression levels measure by FACS with the transcriptional information for each cell/nucleus.
Limitations
If important differences in processing time between the first well and the last well of the plate occur during sample dissociation, flow sorting or Smart-seq2 preparation, intra-plate biases might occur. For this reason, the existence of potential plate biases must be tested during the bioinformatic analysis to avoid interpretation errors. We recommend to evaluate the number of genes detected per cell/nucleus with regard to the position of each cell/nucleus on the plate. This QC will help you visualize potential areas, lanes or rows on the plate that could have been improperly processed and should be excluded for downstream analysis. When analyzing cells, the percentage of mitochondrial genes detected in each cell across the plate can also be used as an indicator of sample quality. Finally, plate artifacts should also be verified during cluster identification. Depending on your sorting strategy, clusters formed by cells/nuclei coming exclusively from one unique location on the plate should be carefully investigated. In projects with multiple 384-well plates, potential inter-plate biases should also be assessed. If possible, sort samples from your different conditions on the same plates (e.g., Both plate I and plate II contain: condition A (192 cells/nuclei) + condition B (192 cells/nuclei), instead of plate I: condition A (384 cells/nuclei) and plate II: condition B (384 cells/nuclei)). For projects with numerous plates, having one sample consistently present in each plate could facilitate subsequent downstream bioinformatic analysis.
Troubleshooting
Problem
During nuclei dissociation, chunks of tissue might remain after the Dounce homogenization procedure.
Potential Solution
Perform more strokes with the tight pestle but be aware that additional strokes will also affect the integrity of the nuclei. You can observe the DNA staining dye fluorescence under a microscope to verify the quality of the nuclei preparation after the final filtering step.
Problem
During cell isolation from the neonatal mouse brain, the tissue becomes very sticky after incubation with Pronase and gets easily stuck inside the pipette tips when changing solutions.
Potential Solution
Before changing solution, spin the sample at max 100 × g in a table top refrigerated centrifuge at 4°C for 1 min.
Problem
Depending on the size and rigidity of the organoids not all parts are completely dissolved.
Potential Solution
The incubation step in Accutase can be prolonged or the pipetting during the incubation can be enhanced. There is a balance between dissociating as many cells as possible and losing cells due to cell death.
Problem
Nozzle clogs during sorting.
Potential Solution
Nucleus and cell suspensions from the mouse brain tend to contain numerous debris. To prevent clogging, run at a low flow rate (1,000–3,000 events/s). If clogged, sonicate the nozzle and dilute the sample.
Problem
The sample-collection plates are empty or the volume between wells is inconsistent.
Potential Solution
Calibrate the Mosquito HV liquid-handling robot so that the tips get in contact with the plate while dispensing.
Problem
The volume of the different wells is inconsistent after PCR.
Potential Solution
Make sure that the adhesive foil is tightly attached to the plate, paying special attention to the wells that are close to the edges of the plate.
Problem
Beads are lost during the clean-up using the robot. The cDNA yield is inconsistent between wells.
Potential Solution
Calibrate the robot so that it pipettes slightly above the beads (without touching the bottom of the wells) during bead clean-up.
Problem
The quality of the cDNA is poor.
Potential Solution
If the positive controls (RNA spike-ins) included in the plate look as expected, try to improve your sample preparation and sorting protocols. If the positive controls look degraded, prepare new sample-collection plates, and verify the performance of some columns of a plate of the new batch.
Problem
Libraries are under/over-tagmented.
Potential Solution
Adjust the amount of cDNA used for tagmentation. Make sure the Neutralize Tagment Buffer (NT) is added right after tagmentation is finished.
Problem
A clear bias between columns/rows is observed in the initial QC of the sequencing data.
Potential Solution
If a clear bias between rows is observed, reduce the time of sample preparation and sorting, and review the sorting parameters. If a bias between columns is observed, double-check the different pipetting steps of the miniaturized Smart-seq2 protocol.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sebastian Jessberger (jessberger@hifo.uzh.ch).
Data and Code Availability
All data are available from the authors upon request.
Materials Availability
This study did not generate new unique reagents.
Acknowledgments
This work was supported by the European Research Council (STEMBAR to S.J. and ERC_679175 to T.K.), the Swiss National Science Foundation (BSCGI0_157859 to S.J. and SNSF 31003A_17003 to T.K.), the Zurich Neuroscience Center, and the University of Zurich (UZH) Forschungskredit fellowship (B.N.J.). We thank the Functional Genomics Center Zurich (UZH and ETHZ), the Cytometry Facility of UZH, Stefanie Chie for the human forebrain organoid preparation, and Tong Liang for mouse brain microdissections.
Author Contributions
Conceptualization, B.N.J., E.Y., L.G., A.D.-L., and S.J.; Investigation, B.N.J., E.Y., L.G., A.D.-L., and M.K.; Writing B.N.J., E.Y., L.G., A.D.-L., and S.J.; Funding Acquisition, T.K. and S.J.; Supervision, B.N.J., E.Y., T.K., and S.J.
Declaration of Interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xpro.2020.100081.
Contributor Information
Baptiste N. Jaeger, Email: jaeger@hifo.uzh.ch.
Sebastian Jessberger, Email: jessberger@hifo.uzh.ch.
Supplemental Information
References
- Bowers M., Liang T., Gonzalez-Bohorquez D., Zocher S., Jaeger B.N., Kovacs W.J., Rohrl C., Cramb K.M.L., Winterer J., Kruse M., Dimitrieva S., Overall R.W. FASN-dependent lipid metabolism links neurogenic stem/progenitor cell activity to learning and memory deficits. Cell Stem Cell. 2020;27:98–109.e11. doi: 10.1016/j.stem.2020.04.002. [DOI] [PubMed] [Google Scholar]
- Camp J.G., Badsha F., Florio M., Kanton S., Gerber T., Wilsch-Brauninger M., Lewitus E., Sykes A., Hevers W., Lancaster M., Knoblich J.A., Lachmann R. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. U S A. 2015;112:15672–15677. doi: 10.1073/pnas.1520760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempel C.M., Sugino K., Nelson S.B. A manual method for the purification of fluorescently labeled neurons from the mammalian brain. Nat. Protoc. 2007;2:2924–2929. doi: 10.1038/nprot.2007.416. [DOI] [PubMed] [Google Scholar]
- Jaeger B.N., Linker S.B., Parylak S.L., Barron J.J., Gallina I.S., Saavedra C.D., Fitzpatrick C., Lim C.K., Schafer S.T., Lacar B., Jessberger S., Gage F.H. A novel environment-evoked transcriptional signature predicts reactivity in single dentate granule neurons. Nat. Commun. 2018;9:3084. doi: 10.1038/s41467-018-05418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacar B., Linker S.B., Jaeger B.N., Krishnaswami S., Barron J., Kelder M., Parylak S., Paquola A., Venepally P., Novotny M., O'connor C., Fitzpatrick C.H. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun. 2016;7:11022. doi: 10.1038/ncomms11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S., Bjorklund A.K., Faridani O.R., Sagasser S., Winberg G., Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- Picelli S., Faridani O.R., Bjorklund A.K., Winberg G., Sagasser S., Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Rodrigues O.R., Monard S. A rapid method to verify single-cell deposition setup for cell sorters. Cytometry A. 2016;89:594–600. doi: 10.1002/cyto.a.22865. [DOI] [PubMed] [Google Scholar]
- Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the authors upon request.