

Abstract
Diabetes is characterized by hyperglycemia, loss of functional islet beta-cell mass, deficiency of glucose-lowering insulin and persistent alpha-cell secretion of gluconeogenic glucagon. Still, no therapies that target these underlying processes are available. We therefore performed high-throughput screening of 300,000 compounds and extensive medicinal chemistry optimization and here report the discovery of SRI-37330, an orally bioavailable, non-toxic small molecule, which effectively rescued mice from streptozotocin- and obesity-induced (db/db mice) diabetes. Interestingly, in rat cells and in mouse and human islets, SRI-37330 inhibited expression and signaling of thioredoxin-interacting protein, which we have previously found to be elevated in diabetes and to have detrimental effects on islet function. In addition, SRI-37330 treatment inhibited glucagon secretion and function, reduced hepatic glucose production and reversed hepatic steatosis. Thus, these studies describe a newly designed chemical compound that, compared to currently available therapies, may provide a distinct and effective approach to treating diabetes.
eTOC
Here, Thielen et al. show that a newly designed orally available small molecule inhibited pancreatic islet TXNIP expression, glucagon secretion, hepatic glucagon action, glucose production and steatosis and exhibited strong anti-diabetic effects in mouse models of type 1 and type 2 diabetes, promising a distinct and innovative diabetes treatment approach.
Graphical Abstract
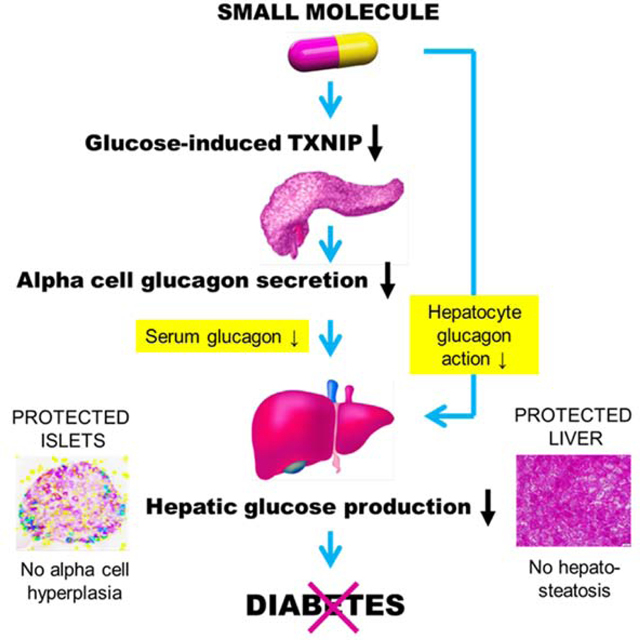
Type 2 diabetes (T2D) has become a worldwide epidemic and poses major therapeutic challenges. Over the last several decades it has been recognized as a bihormonal disease distinguished by the combination of hypoinsulinemia due to loss of functional pancreatic beta cell mass and hyperglucagonemia due to persistent alpha cell-mediated glucagon secretion (Unger and Orci, 1975). Insufficient production of the glucose-lowering hormone insulin and high levels of the gluconeogenic hormone glucagon combine to exacerbate the hyperglycemia that is the hallmark of T2D, leading to a vicious cycle of glucose toxicity that furthers beta cell dysfunction (Poitout and Robertson, 2002). However, while T2D management has improved significantly over the years, therapies that target these underlying processes are still lacking.
We previously identified thioredoxin-interacting protein (TXNIP) as the top glucose-induced gene in a human islet gene expression microarray study (Shalev et al., 2002) and as a critical link between glucose toxicity and beta cell loss (Chen et al., 2008b), suggesting that this arrestin protein may play an important role in T2D. Indeed, TXNIP expression is not only upregulated by glucose (Minn et al., 2005; Shalev et al., 2002), it is also increased in different mouse models of T2D and in human islets from individuals with diabetes (Chen et al., 2008a; Chen et al., 2008b; Minn et al., 2005; Xu et al., 2013). Moreover, genetic TXNIP deletion has been shown to protect mice from diabetes and have beneficial effects on islet biology (Chen et al., 2008a; Lerner et al., 2012; Xu et al., 2013). Together this suggested that TXNIP inhibition may represent an attractive novel approach to T2D treatment, but no specific TXNIP inhibitors were available. The goal of the present studies was therefore to identify an orally bioavailable, small molecule TXNIP inhibitor and test its effects in human islets and different mouse models of diabetes.
RESULTS AND DISCUSSION
High-throughput screening of 300,000 small molecules and several medicinal chemistry iterations, modifications and optimizations led to the identification of SRI-37330 (Fig. 1a), a lead compound, based on its efficacy of inhibiting TXNIP expression, its pharmacokinetic properties, favorable safety profile and lack of off-target liabilities at least in the currently available panel (Supplemental Table S1a). Transient transfection experiments showed that SRI-37330 inhibited the activity of the human TXNIP promoter in INS-1 cells by ~70% (Fig. 1b), consistent with inhibition at the transcriptional level. SRI-37330 also inhibited endogenous TXNIP mRNA expression as shown by 7-point dose-response qRT-PCR assays (Fig. 1c), revealing an IC50 of 0.64 μM and no cytotoxicity (Supplemental Table S1a). Similarly, SRI-37330 also inhibited TXNIP protein levels in a dose-dependent manner (Fig. 1d). In order to mimic diabetic conditions, we also conducted experiments in the context of high (25 mM) glucose and found that SRI-37330 was able to significantly inhibit TXNIP expression in rat INS-1 cells (Fig. 1e), primary mouse islets (Fig. 1f) and isolated human islets (Fig. 1g).
Figure 1. Substituted quinazoline sulfonamide (SRI-37330) controls islet TXNIP expression and associated signaling pathway genes.

(a) Chemical structure of the substituted quinazoline sulfonamide, SRI-37330 (the HCl salt is shown).
(b) Effect of SRI-37330 on the activity of the full-length human TXNIP promoter as assessed by dual luciferase and compared to no SRI-37330 control (t2 = 6.71, *P = 0.0215).
(c) Inhibition of TXNIP mRNA expression in INS-1 cells by SRI-37330 as assessed by qPCR 7-point dose response curve.
(d) Dose-dependent inhibition of TXNIP protein expression in INS-1 cells by SRI-37330 as assessed by immunoblotting (F3,8 = 9.65, *P = 0.0049); a representative immunoblot (n = 3 total blots analyzed) is shown.
(e) SRI-37330 effects on TXNIP expression in the context of high (25 mM) glucose in rat INS-1 cells (t10 = 3.63, *P = 0.0046) as assessed by qPCR; mean ± s.e.m., n = 6 independent experiments.
(f) SRI-37330 effects on TXNIP expression in the context of high glucose in primary mouse islets (t4 = 3.67, *P = 0.0213), n = 3 independent experiments.
(g) SRI-37330 effects on TXNIP expression in the context of high glucose in human islets (t5 = 9.11, *P = 0.0003), n = 6 different human islet donors.
(h) RNA sequencing was performed on human islets from three different donors (A-C) treated with or without SRI-37330 in the context of high (25 mM) glucose. Volcano plot contains all genes with a baseMean expression of > 5. Genes with a FDR <0.05 are shown in color.
(i) Heat map and pathway showing TXNIP-associated genes that are significantly (adjusted DESeq2 P< 0.05) changed after treatment with SRI-37330.
(j) Confirmation by qPCR of changes in the expression of TXNIP-associated genes (NLRP1 *P = 0.0374, MLXIPL *P = 0.0309, CTGF *P = 0.0031, MAFA *P = 0.0075, IGF1R *P = 0.0061, BCL2L1 *P = 0.0004). The arrestin proteins ARRB1 and AADC3 as well as thioredoxin were run as negative controls (N.S. not significant), n = 6 human islet donors.
To further assess the overall effects of SRI-37330 on human islet gene expression, we performed RNA sequencing experiments using islets from three different donors. As shown in a volcano plot (Fig. 1h), a similar number of genes were up- as well as down-regulated, which is consistent with the notion that SRI-37330 does not inhibit general transcription. Genes that were significantly up- or down-regulated by SRI-37330 (adjusted P-value < 0.05) are listed in Supplemental Table S1b. Interestingly, gene ontology analysis further revealed a number of genes previously shown to be involved in TXNIP regulation and signaling pathways, including TXNIP itself (Supplemental Table S1c, Fig. 1i). We confirmed these findings in additional human islets by qRT-PCR, and demonstrated that unlike TXNIP, other arrestin genes, such as ARRB1 and ARRDC3 as well as thioredoxin were not affected by SRI-37330, supporting the specificity of the effects (Fig. 1j). MLX-interacting protein-like (MLXIPL), which has previously been shown to be part of a complex that promotes TXNIP transcription via a specific E-box motif in the proximal promoter of TXNIP (Cha-Molstad et al., 2009; Minn et al., 2005), was downregulated, whereas the insulin-like growth factor-1 receptor (IGF-1R), which recently was found to inhibit TXNIP expression (Nagaraj et al., 2018), was upregulated. In addition, V-MAF avian musculoaponeurotic fibrosarcoma oncogene homolog A (MAFA), an insulin transcription factor previously shown to be downregulated by TXNIP (Xu et al., 2013), was upregulated by our TXNIP inhibitor. Similarly, Bcl-2-like 1 (BCL2L1), which is involved in the control of beta cell apoptosis and increased in beta cell-specific TXNIP knockout mice (Chen et al., 2008a), and connective tissue growth factor (CTGF), which is involved in developmental beta cell proliferation (Riley et al., 2015) and upregulated by TXNIP inhibition (Wu et al., 2018) were both upregulated in response to SRI-37330. Finally, we also observed lower expression of the inflammasome, NACHT, LRR and PYD domain-containing protein 1 (NLRP1), consistent with the fact that TXNIP has been shown to promote inflammasome activation (Zhou et al., 2010).
We have previously shown that TXNIP expression, especially in the context of high glucose and diabetes is regulated via a distinct E-box motif in the TXNIP promoter that is well conserved across species (Cha-Molstad et al., 2009; Minn et al., 2005). We therefore next investigated whether SRI-37330 confers its effects through this element. Indeed, while human TXNIP promoter deletions of over 1000 bp did not diminish the inhibitory effects of SRI-37330 as assessed by luciferase activity, mutation of the E-box motif completely blunted these effects (Fig. 2a). Moreover, this E-box motif was able to confer SRI-37330 responsiveness to a heterologous SV40 promoter (Fig. 2b). Furthermore, chromatin immunoprecipitation revealed that SRI-37330 effectively inhibited polymerase II (Pol II) binding to the E-box motif region of the TXNIP promoter, but not to an upstream TXNIP promoter region or to an established Pol II binding site in the insulin receptor substrate 2 (IRS2) (Fig. 2c).
Figure 2. SRI-37330 acts via a distinct E-box motif in the TXNIP promoter.

(a) Effects of SRI-37330 on the activity of the full-length (FL-1518) human TXNIP promoter, the D-420 and D-400 deletions and the mutD-400 E-box mutation as assessed by dual luciferase assays and compared to no SRI-37330 control, FL-1518 (*P = 0.0275); D-420 (*P = 0.0010); D-400 (*P = 0.0157); mutD-400 (N.S. not significant); mean ± s.e.m, n = 3 independent experiments.
(b) Effects of SRI-37330 on relative luciferase activity driven by a SV40 control promoter (N.S.) or the SV40 promoter coupled with the TXNIP-E-box motif (F3, 20 =17.82,*P< 0.0001), n = 6 independent experiments.
(c) Chromatin immunoprecipitation (ChIP) analysis of polymerase II (Pol II) binding to the TXNIP promoter in the E-box motif region (TXNIP E-box t4 = 2.88, *P = 0.0451), an upstream promoter region (TXNIP Upstream, N.S.) and an established Pol II binding site in the insulin receptor substrate 2 (IRS2, N.S.) in response to SRI-37330, n = 3 independent experiments.
Consistent with the lack of cytotoxicity observed in vitro, SRI-37330 also showed an excellent safety profile in vivo (Supplemental Table S1a). In addition, 3 weeks of oral administration at 100 mg/kg/day in the drinking water to male C57BL/6J mice was well tolerated, caused no change in body weight (Fig. 3a), and no gross abnormalities in any organs were noticed at sacrifice. Interestingly, a small but significant decrease in blood glucose levels was observed in mice receiving SRI-37330 within 3 days of starting the treatment, which persisted for the 3 weeks of the experiment (Fig. 3b). Intraperitoneal glucose tolerance tests (GTTs) revealed a consistently and significantly improved glucose tolerance in response to SRI-37330 administration (Fig. 3c), whereas insulin tolerance tests (ITTs) remained unaltered (Fig. 3d). Fasting blood glucose was slightly, but significantly lower in SRI-37330-treated mice (Fig. 3e). Notably, while fasting serum insulin levels were unchanged in SRI-37330-treated mice as compared to controls (Fig. 3f), fasting serum glucagon levels were lower (Fig. 3g), suggesting that SRI-37330 may also affect glucagon secretion from pancreatic alpha cells. Of note, in TXNIP-deficient mice, SRI-37330 treatment was no longer able to further improve glucose homeostasis or lower serum glucagon levels (Supplemental Figure S1a-c), underscoring the role of TXNIP and of the associated reduction in glucagon levels in the mechanism of action of this small molecule.
Figure 3. Oral administration of SRI-37330 lowers serum glucagon levels, inhibits hepatic glucose production and improves glucose homeostasis in mice.

(a) Effects of oral SRI-37330 on body weight as compared to no treatment (N.S. not significant) in wild-type C57BL/6J mice, mean ± s.e.m, n = 16 mice per group.
(b) Non-fasting blood glucose (F1,124 = 17.14, *P < 0.0001), n = 16 mice per group.
(c) I.p glucose tolerance tests (GTT) (F1,155 = 11.25, *P = 0.0010), n = 16 mice per group.
(d) I.p. insulin tolerance tests (ITT) (N.S.), n = 16 mice per group.
(e) Fasting blood glucose (t22 = 2.20, *P = 0.0389), n =15–16 mice per group.
(f) Fasting serum insulin (N.S.), n =15–16 mice per group.
(g) Fasting serum glucagon (t29 = 2.91, *P = 0.0069), n =15–16 mice per group.
(h) Basal hepatic glucose production in SRI-37330 treated versus untreated control mice (t9 = 2.35, *P = 0.0432) as assessed by hyperinsulinemic euglycemic clamp studies, n = 5–6 mice per group.
(i) Insulin-stimulated glucose uptake in skeletal muscle (SM; gastrocnemius) (N.S.), n = 5–6 mice per group.
(j) Insulin-stimulated glucose uptake in white adipose tissue (WAT; epididymal) (N.S.), n = 5–6 mice per group.
(k) Glucose infusion rates (N.S.), n = 5–6 mice per group.
(l) Hepatic insulin action (N.S.), n = 5–6 mice per group.
Based on these findings and to further investigate the potential mechanisms by which SRI-37330 may lower blood glucose levels, we performed hyperinsulinemic-euglycemic clamp studies in awake mice treated with SRI-37330 to assess insulin sensitivity and glucose metabolism in individual organs. Importantly, we found that basal hepatic glucose production was significantly downregulated in mice receiving SRI-37330 as compared to controls (Fig. 3h), while no alteration in insulin sensitivity was observed and insulin-stimulated glucose uptake in skeletal muscle and white adipose tissue as well as glucose infusion rates and hepatic insulin action were comparable between groups (Fig. 3i-l).Consistent with these findings of downregulated hepatic glucose production, we also observed lower expression of the key enzymes of hepatic gluconeogenesis, phosphoenolpyruvate carboxykinase 1 (Pck1) and glucose-6-phosphatase (G6pc) as well as higher glycogen content in livers of SRI-37330 treated mice as compared to controls (Supplemental Figure S2a-d). In contrast, SRI-37330 led to lower liver and serum triglyceride levels and did not cause any elevation in liver transaminase levels (Supplemental Figure S2e-g). Together, these results suggest that SRI-37330 lowers blood glucose levels, at least in part, by decreasing serum glucagon levels and hepatic glucose production without having detrimental effects on the liver.
Next we studied how SRI-37330 may downregulate glucagon levels. Interestingly, incubation of the mouse alpha cell line alpha TC1-6 cells with SRI-37330 did not lower proglucgon gene expression or glucagon content (Fig. 4a-b), but resulted in significantly lower glucagon secretion (Fig. 4c). Of note, TXNIP knockdown was able to mimic the inhibitory effects of SRI-37330 on alpha cell glucagon secretion, while TXNIP overexpression promoted glucagon secretion (Fig. 4d-e). In contrast, when glucagon secretion was maximally stimulated by the potent glucagon secretagogues, arginine and norepinephrine (Hauge-Evans et al., 2010), SRI-37330 was no longer able to inhibit glucagon secretion (Fig. 4f). Similarly, SRI-37330 had no effect on glucagon secretion in the context of low glucose (Fig. 4g). Together, these results suggest that SRI-37330 inhibits alpha cell glucagon secretion via TXNIP inhibition and that this effect does not occur under low glucose conditions or in the context of glucagon secretion stimulated by counterregulatory hormones such as norepinephrine. These features may help limit the hypoglycemic liability of SRI-37330 and explain why we did not notice any problems with blood glucose levels dropping too low under any circumstances. In fact, even in the context of insulin-induced hypoglycemia, SRI-37330 treated mice were able to maintain their blood glucose levels equally well to untreated controls (Fig. 4h).
Figure 4. SRI-37330 controls alpha cell glucagon secretion.

(a) Proglucagon mRNA expression (NS, not significant) from αTC1-6 alpha cells incubated with or without SRI-37330, mean ± s.e.m. of 3 independent experiments.
(b) Glucagon content (N.S.) in αTC1-6 alpha cells incubated with or without SRI-37330, n = 3 independent experiments.
(c) Glucagon secretion from αTC1-6 alpha cells incubated with or without SRI-37330 (t4 =3.84, *P = 0.0185), n = 3 independent experiments.
(d) Glucagon secretion from αTC1-6 alpha cells with or without TXNIP knockdown (KD) (t4 = 3.50, *P = 0.0248), n = 3 independent experiments.
(e) Glucagon secretion from αTC1-6 alpha cells with or without TXNIP overexpression (OE) (t4 = 3.35, *P = 0.0285), n = 3 independent experiments.
(f) Effects of SRI-37330 on glucagon secretion stimulated by arginine (20 mM) and norepinephrine (10μM) (N.S.).
(g) Effects of SRI-37330 on glucagon secretion stimulated by low glucose (1.67 mM) (N.S.).
(h) Comparison of hypoglycemia as assessed by blood glucose 15min after a bolus of insulin (1U/kg) in fasting SRI-37330 treated and untreated mice (N.S.). n=4 mice per group.
To further investigate whether SRI-37330 may also have any direct hepatic effects, we used isolated primary mouse hepatocytes to assess glucose output under basal conditions (no substrate) as a measure of glycogenolysis as well as under stimulated conditions (addition of lactate as a substrate for gluconeogenesis). Of note, SRI-37330 did not affect basal or stimulated glucose output from primary hepatocytes in the absence of glucagon (Fig. 5a-b). However, in the presence of glucagon in the culture media, SRI-37330 led to a dose-dependent decrease in glucose output suggesting that it inhibited glucagon function and glucagon-induced glucose output (Fig. 5a-b). In fact, glucagon-induced production of cAMP, the major second messenger conferring glucagon function, was also significantly downregulated in a dose-dependent manner by SRI-37330 in primary hepatocytes (Fig. 5c). In addition, glucagon-induced expression of the gluconeogenic genes Pck1 and G6pc was inhibited by SRI-37330 in primary hepatocytes of wild-type C57BL/6J mice (Fig. 5d-e) consistent with the findings in liver. However, this SRI-37330 effect was completely blunted in primary hepatocytes of liver-specific glucagon receptor knockout mice (GcgR−/−L) (Fig. 5f-g). Moreover, in the absence of GcgR in the liver, SRI-37330 was no longer able to inhibit hepatocyte glucose output or cAMP production (Fig. 5h-j). In contrast, in primaryhepatocytes of TXNIP-deficient mice, SRI-37330 maintained its ability to block glucose output and glucagon-induced cAMP production (Fig. 5k-m), suggesting that these hepatic SRI-37330 effects were not TXNIP-dependent. Indeed, TXNIP signaling genes found to be regulated by SRI-37330 in primary islets were not affected in primary hepatocytes or livers of SRI-37330 treated mice (Supplemental Figure S2h-i). These results suggest that while the SRI-37330-mediated inhibition of alpha cell glucagon secretion is TXNIP-dependent, the decrease in hepatic glucagon sensitivity is mediated by the glucagon receptor.
Figure 5. SRI-37330 inhibits glucagon-induced glucose output from primary hepatocytes.

(a) Primary mouse hepatocytes were incubated for 24h with SRI-37330 at the indicated doses and glucose output was assessed with no substrate and in the absence of glucagon (N.S., not significant) as well as in the presence of glucagon (100 nM) (F2,6 = 14.11, *P = 0.0054), mean ± s.e.m, n = 3–4 mice per group.
(b) SRI-37330 effects on glucose output in the presence of substrate (10 nM lactate) and in the absence of glucagon (N.S.) as well as in the presence of glucagon (100 nM) (F2,6 = 16.41, *P = 0.0037), n = 3–4 mice per group.
(c) cAMP production in primary hepatocytes incubated for 24h in the presence of glucagon with or without SRI-37330 at the indicated doses (F2,6 = 9.06, *P = 0.0154), n = 3 independent experiments.
(d) Expression of phosphoenolpyruvate carboxykinase (Pck1) in hepatocytes of wild-type mice incubated in the presence of glucagon with or without SRI-37330 (5 μM) for 24h (t4 = 4.49, *P = 0.0109), n = 3.
(e) Expression of glucose-6-phosphatase (G6pc) inhepatocytes of wild-type mice in the presence of glucagon with or without SRI-37330 (t4 = 7.38, *P = 0.0018), n = 3.
(f) Pck1 expression inhepatocytes of liver-specific glucagon receptor knockout mice (GcgR−/−L) incubated in the presence of glucagon with or without SRI-37330 (N.S.), n = 3.
(g) G6pc expression inGcgR−/−L hepatocytes incubated in the presence of glucagon with or without SRI-37330 (N.S.), n = 3.
(h) Glucose output from GcgR−/−L hepatocytes incubated in the presence of glucagon with or without SRI-37330 and no substrate (N.S.), n = 3.
(i) Glucose output from GcgR−/−L hepatocytes incubated in the presence of glucagon with or without SRI-37330 and with lactate (N.S.), n = 3.
(j) cAMP production from GcgR−/−L hepatocytes incubated in the presence of glucagon with or without SRI-37330 (N.S.), n = 3.
(k) Glucose output from primary mouse hepatocytes of Txnip-deficient mice (Txnip−/−) incubated in the presence of glucagon, with or without SRI-37330 and with no substrate (t4 = 4.27, *P = 0.0130), n = 3.
(l) Glucose output from Txnip−/− hepatocytes incubated in the presence of glucagon, with or without SRI-37330 and with lactate (t2.1 = 28.63, *P = 0.0010), n = 3.
(m) cAMP production from Txnip−/− hepatocytes incubated in the presence of glucagon, with or without SRI-37330 (t4 = 8.71, *P = 0.0010), n = 3.
Next, to evaluate the potential effects of SRI-37330 in the context of T2D, we used obese, insulin-resistant and diabetic, leptin receptor-deficient db/db mice. Similar to our observations in lean mice, SRI-37330 treatment for 4 weeks did not affect body weights and both groups of mice continued to gain weight (Fig. 6a). Notably, blood glucose levels decreased significantly within 3 days of starting SRI-37330 treatment and returned to euglycemic levels for the remaining 4 weeks of the experiment (Fig. 6b). Fasting blood glucose levels were also significantly downregulated in SRI-37330 treated mice (Fig. 6c), while fasting serum insulin levels remained unchanged (Fig. 6d). In contrast, a significant downregulate in fasting serum glucagon levels was again observed with SRI-37330 treatment (Fig. 6e). While compared to non-diabetic lean animals, the average glucagon levels were higher in the db/db mice consistent with the elevation of glucagon associated with diabetes (Unger and Orci, 1975), the observed downregulation in response to SRI-37330 was similar to the findings in lean mice suggesting that it may also contribute to the improved glucose homeostasis found in the context of diabetes. Indeed, hyperinsulinemic-euglycemic clamp studies in diabetic db/db mice revealed again significantly lower basal hepatic glucose production in mice receiving SRI-37330 as compared to controls (Fig. 6f), whereas insulin-stimulated glucose uptake in skeletal muscle and white adipose tissue remained unchanged (Fig. 6g-h), consistent with the findings in lean mice. Also, the glucose infusion rate and hepatic insulin action during the clamp were not significantly different and very low in both groups in alignment with the remaining severe insulin resistance of these db/db mice (Fig. 6i-j). These results demonstrate that SRI-37330 downregulates serum glucagon levels and hepatic glucose production in lean as well as obese, diabetic mice suggesting that these effects may contribute to the improvement in glucose homeostasis observed.
Figure 6. SRI-37330 rescues mice from obesity-induced diabetes.

(a) Body weight of obese, insulin resistant and diabetic male 8-week old db/db mice treated with or without oral SRI-37330 (N.S. not significant), mean ± s.e.m, n = 7–12 mice per group.
(b) Non-fasting blood glucose in db/db mice treated with or without oral SRI-37330 (F1,107 = 23.63, *P < 0.0001), n = 7–12 mice per group.
(c)Fasting blood glucose in db/db mice treated with or without oral SRI-37330 (t15 = 5.61, P < 0.0001), n = 7–10 mice per group.
(d) Fasting serum insulin in db/db mice treated with or without oral SRI-37330,(N.S.), n = 4–7 mice per group.
(e) Fasting serum glucagon in SRI-37330 treated and untreated db/db mice, (t8.2 = 2.35, *P = 0.0459), n = 4–8 mice per group.
(f) Basal hepatic glucose production in SRI-37330 treated and untreated db/db mice as assessed by hyperinsulinemic euglycemic clamp studies, (t9 = 2.31, *P = 0.0460), n = 5–6 mice per group.
(g) Insulin-stimulated glucose uptake in skeletal muscle (SM; gastrocnemius) in SRI-37330 treated and untreated db/db mice (N.S.), n = 5–6 mice per group.
(h) Insulin-stimulated glucose uptake in white adipose tissue (WAT; epididymal) in SRI-37330 treated and untreated db/db mice (N.S.), n = 5–6 mice per group.
(i) Glucose infusion rates in SRI-37330 treated and untreated db/db mice (N.S.), n = 5–6 mice per group.
(j) Hepatic insulin action in SRI-37330 treated and untreated db/db mice (N.S.), n = 5–6 mice per group.
(k) Representative histological liver sections (n = 12 total sections analyzed) of SRI-37330 treated and untreated db/db mice, scale bar = 20 μm.
The potential benefits of disrupting glucagon signaling for glucose homeostasis have been well recognized, but glucagon receptor antagonism has in some cases also been shown to cause alpha cell hyperplasia and hepatic steatosis limiting the applicability of these approaches (Scheen et al., 2017). To determine whether SRI-37330 may have similar problems, we also assessed islet cell morphometry in treated and untreated db/db mice. However, the results revealed that rather than causing additional alpha cell hyperplasia, SRI-37330 treatment led to a small, but significant decline in alpha cell mass and a clear trend towards lower alpha cell number, while beta cell mass and number remained unchanged (Supplemental Figure S3a-e). Moreover, analysis of liver histology revealed that SRI-37330 not only did not lead to any fatty liver in lean mice (Supplemental Figure S2d), but dramatically improved the severe steatosis observed in db/db mice (Fig. 6j). This now raises the intriguing possibility that SRI-37330 might also be beneficial in the context of non-alcoholic fatty liver disease, a complication frequently associated with diabetes and/or obesity.
To investigate whether SRI-37330 might also have therapeutic effects in the context of a non-genetic, non-obese model of diabetes, we used C57BL/6J mice rendered diabetic by multiple low-dose streptozotocin (STZ)-induced beta cell loss. Indeed, animals receiving treatment with SRI-37330 starting after the 5-day STZ regimen maintained normal and significantly lower blood glucose levels as compared to untreated mice (Fig. 7a). These results are consistent with our previous findings demonstrating that genetic deletion of TXNIP protected mice from STZ-induced diabetes (Chen et al., 2008a).In addition, to compare the effects of SRI-37330 with other available oral anti-diabetic drugs, we also tested the effects of metformin, currently the first-line, and most widely used, oral anti-diabetic and of empagliflozin, a sodium-glucose cotransporter-2 (SGLT2) inhibitor and as such a member of the newest class of oral anti-diabetic drugs. Consistent with previous reports, both metformin and empagliflozin resulted in improved glucose homeostasis (Daems et al., 2019; Han et al., 2017). However, SRI-37330 achieved even slightly better blood glucose control than metformin or empagliflozin (Fig. 7a). Also, urine glucose was more than 10-fold lower in SRI-37330 treated mice than in untreated or empagliflozin-treated animals, confirming that unlike SGLT2 inhibitors, SRI-37330 does not lower blood glucose by increasing urinary glucose excretion (Fig. 7b). Moreover, SRI-37330 was the only treatment that resulted in significantly lower serum triglycerides (Fig. 7c). Furthermore, as compared to the untreated group, mice receiving SRI-37330 were able to maintain higher body weight and serum insulin levels and lower fasting blood glucose (Fig. 7d-f). Most strikingly though, fasting serum glucagon levels were 3.6-fold lower in SRI-37330 treated than untreated STZ-diabetic mice (Fig. 7g), again consistent with our findings in db/db diabetic (Fig. 6e) as well as non-diabetic mice (Fig. 3g).
Figure 7. SRI-37330 protects mice from STZ-induced diabetes.

(a) C57BL/6J mice were rendered diabetic by multiple low-dose STZ injections and randomized to receive oral SRI-37330, metformin, empagliflozin or no additional treatment and their non-fasting blood glucose levels at the designated time points are shown (F3,17 = 19.24, P < 0.0001, STZ+37330 vs STZ control *P = 0.0003 (day 12), *P = 0.0224 (day 15), *P < 0.0001 (day 17 and 18); vs STZ+metformin #P = 0.0112 (day 12), #P = 0.0003 (day 17); vs STZ+empagliflozin &P = 0.0017 (day 17), mean ± s.e.m, n = 5–6 mice per group.
(b) Urine glucose (F3,12 = 186.5, P < 0.0001), STZ+37330 vs STZ control and vs STZ+empagliflozin *&P < 0.0001), n = 5–6 mice per group.
(c) Serum triglycerides in the different treatment groups (F3,16 = 8.28, P = 0.0015, STZ+37330 vs STZ control *P = 0.0008, vs STZ+empagliflozin &P = 0.0020, vs STZ+metformin #P = 0.0079), n = 5–6 mice per group.
(d) Body weight ofSRI-37330 treated and untreated mice at the end of the experiment, (t9 = 4.34, *P = 0.0010), n = 5–6 mice per group.
(e) Fasting blood glucose ofSRI-37330 treated and untreated mice (t9 = 3.33, *P = 0.0088), n = 5–6 mice per group.
(f) Fasting serum insulin ofSRI-37330 treated and untreated mice (t8 = 3.86, *P = 0.0048), n = 5–6 mice per group.
(g) Fasting serum glucagon ofSRI-37330 treated and untreated mice (t5,9 = 4.88, *P = 0.0029), n = 5–6 mice per group.
(h) Blood glucose levels in C57BL/6J mice rendered diabetic by multiple low-dose STZ injections and treated with or without oral SRI-37330 starting after development of overt diabetes (F7,50 = 4.70, *P = 0.0004) n = 4–5 mice per group.
This combination of dramatically lower glucagon levels and in this case also clearly higher insulin levels in response to SRI-37330 helps explain the improved glucose homeostasis observed even in the context of this beta cell loss-induced diabetes model. Importantly, we found that SRI-37330 was also effective in improving glucose homeostasis in mice with STZ-induced diabetes even when treatment was started after two weeks when the animals had already developed overt diabetes (Fig. 7h). Of note, we also performed oral gavage experiments with SRI-37330 administered twice a day and the results confirmed its effectiveness in obesity- as well as STZ-induced diabetes (Supplemental Figure S4a-b). These findings are promising in terms of potential feasibility of SRI-37330 dosing and also demonstrated the dose-dependence of the glucose-lowering effects of SRI-37330 (Supplemental Figure S4b). Together, these results show that the anti-diabetic effects of SRI-37330 are distinct, robust and consistent across different models of diabetes and suggest that they might offer attractive additional benefits as compared to other currently available oral anti-diabetic drugs.
In summary, our studies have identified a novel substituted quinazoline sulfonamide, SRI-37330 that is orally bioavailable, has a favorably safety profile and inhibits TXNIP expression and signaling in mouse and human islets, inhibits glucagon secretion and function, lowers hepatic glucose production and hepatic steatosis and exhibits strong anti-diabetic effects in mouse models of type 1 diabetes (T1D) (STZ) and T2D (db/db).
These findings are consistent with our previous observations that genetic deletion or non-specific pharmacological inhibition of TXNIP expression by blockage of L-type calcium channels with verapamil protected and rescued mice from STZ- and obesity-induced diabetes (Chen et al., 2008a; Xu et al., 2012). Interestingly, verapamil has also recently been shown to have beneficial effects in a randomized controlled trial in human subjects with T1D (Ovalle et al., 2018), to be associated with reduced incidence of newly diagnosed T2D (Cooper-Dehoff et al., 2006; Yin et al., 2017) and to be associated with lower blood glucose levels in subjects with diabetes (Khodneva et al., 2016).However, verapamil is a potent blood pressure drug and its TXNIP-lowering capacity is intricately linked to its function as an L-type calcium channel blocker (Xu et al., 2012), which in turn limits its use in certain patient populations, especially those with hypotension or ventricular dysfunction. Of note, in contrast to verapamil, SRI-37330 is not a calcium channel blocker and is therefore not expected to have these limitations. While the safety and efficacy of SRI-37330 in humans still remains to be determined, it is highly effective in human islets, is orally bioavailable and is well tolerated in mice. Also, in alignment with our present data, TXNIP deficiency has previously been reported to decrease hepatic glucose production in mice (Chutkow et al., 2008) and to have beneficial effects in the cardiovascular system (Chen et al., 2016; Xiang et al., 2005; Yoshioka et al., 2012) and in the context of diabetic nephropathy (Shah et al., 2015) and retinopathy (Perrone et al., 2010), suggesting that systemic inhibition rather than a cell-specific approach may actually be desirable. Still, while in general translating therapeutic findings from rodent studies to humans can be problematic, in this case the beneficial effects of targeting TXNIP signaling in the context of diabetes have already been shown to be translatable to human subjects (Ovalle et al., 2018).
Decreased blood glucose levels can be the result of increased glucose disposal and/or decreased glucose production. Interestingly, SRI-37330 did not affect insulin sensitivity or glucose uptake into skeletal muscle or white adipose tissue as demonstrated by hyperinsulinemic-euglycemic clamp studies in non-diabetic as well as obese, diabetic mice. Instead, SRI-37330 selectively downregulated basal hepatic glucose production, hepatic glucagon signaling and serum glucagon levels. These results suggest that these effects may at least in part be responsible for the observed improvement in glucose homeostasis. Of note, such results are consistent with the notion that reducing hepatic glucose production improves glucose homeostasis (Sharabi et al., 2017) and that inhibition of glucagon action ameliorates glucose control in diabetes (Scheen et al., 2017). Interestingly, in contrast to some approaches of glucagon receptor antagonism (Scheen et al., 2017) (Liang et al., 2004), treatment with SRI-37330 did not lead to any liver enzyme abnormalities, alpha cell hyperplasia or hyperglucagonemia, but rather to a lowering of serum glucagon levels, which may explain the more pronounced effects on glucose homeostasis observed. In addition, this strong glucose lowering effect via inhibition of alpha cell glucagon secretion and hepatic glucose production may have rendered any increase in insulin secretion or production unnecessary to achieve normal glucose homeostasis. Obviously though, these effects may not be sufficient in the context of severely compromised functional beta cell mass and indeed, in the STZ studies we also observed a significant almost two-fold elevation in serum insulin levels, consistent with previously reported beneficial effects of TXNIP inhibition on insulin production and secretion (Jo et al., 2018; Xu et al., 2013).
Notably, in SRI-37330-treated TXNIP-deficient mice, the lack of beneficial effects on glucose homeostasis was associated with a lack of decline in serum glucagon levels, supporting the notion that lowering serum glucagon is critical and that both effects are TXNIP-dependent. The fact that serum glucagon levels were even elevated in this context is consistent with the previously reported hyperglucagonemia in response to glucagon receptor antagonism (Scheen et al., 2017) and our observation that SRI-37330 led to a glucagon receptor-dependent (but TXNIP-independent) inhibition of hepatic glucagon sensitivity and action. Thus, our findings not only reveal a previously unappreciated link between TXNIP signaling and glucagon secretion, but also raise the conceptually novel idea that combined inhibition of both alpha cell glucagon secretion and hepatic glucagon action can have pronounced beneficial effects on glucose homeostasis and diabetes without some of the problems associated with previous attempts of glucagon receptor antagonism. In this regard it is further noteworthy that no issues with hypoglycemia were noticed at any point with SRI-37330 treatment and mice were able to maintain blood glucose levels that were indistinguishable from untreated mice even in the context of insulin-induced hypoglycemia. This apparent low hypoglycemic risk may be explained by the fact that SRI-37330 did not seem to impair glucagon secretion under low glucose conditions or in response to counterregulatory hormones, such as norepinephrine.
Importantly, SRI-37330 treatment also led to a marked improvement in diabetes- and obesity-associated hepatic steatosis and therefore may have additional therapeutic potential in the context of non-alcoholic fatty liver disease, another rapidly growing public health problem.
Over the years, several classes of oral anti-diabetic drugs have been developed for the treatment of T2D. Some of them are not dependent on beta cell function, such as metformin and SGLT2 inhibitors, and therefore have a limited ability to lower blood glucose even in the context of beta cell loss and T1D (Daems et al., 2019; Han et al., 2017) consistent with our present findings. However, their adjunctive use in human T1D has proven problematic (Meng et al., 2018; Taylor et al., 2019), leaving insulin still as the only approved treatment of T1D. Our findings that SRI-37330 was able to protect against STZ-induced diabetes and even resulted in better control of glucose homeostasis as compared to these oral anti-diabetic drugs is therefore particularly promising. Together with the fact that it was also effective after the onset of overt diabetes as well as when just dosed twice a day by oral gavage, raises the possibility that SRI-37330 may ultimately lead to a much needed oral drug that could also be used for T1D.
In conclusion, our studies describe a newly designed chemical compound that controls TXNIP, glucagon signaling, hepatic glucose production and hepatic steatosis and has strong antidiabetic properties and suggest that SRI-37330 may provide an attractive approach that is very different from any currently available therapy to ameliorate human diabetes.
LIMITATIONS OF STUDY
At this point it is unclear why a drug that was identified in beta cells as an inhibitor of TXNIP has an effect on glucagon action in hepatocytes independently of TXNIP. It is possible that the same molecular target of the drug acts in beta cells to affect E-box occupancy and thus TXNIP expression, but in hepatocytes affects glucagon action in a different way that is not dependent on TXNIP expression in that cell type. But that means the molecular target of the compound still needs to be identified. Another limitation is that the drug has not been tested in high fat fed mice as another relevant model of human T2D or in a spontaneous, immunological model of T1D such as the non-obese diabetic mice.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact Anath Shalev (shalev@uab.edu).
Materials Availability
There are restrictions to the availability of SRI-37330 due to patent/licensing and production limitations. Any inquiries may be mailed to Ms. T. Grimes, 1825 University Blvd. Birmingham, AL 35294.
Data and Code Availability
The RNA sequencing datasets generated during this study are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151588.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Studies
All mouse studies were approved by the University of Alabama at Birmingham (UAB) Animal Care and Use Committee and conformed to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice were housed under 12 h light and 12 h dark cycles under temperature and humidity controlled conditions and fed a regular chow diet ad libitum. They were in general good health.
Wild-type, 8-week-old, male C57BL/6J mice (The Jackson Laboratory) were used in healthy mouse experiments or were rendered diabetic by multiple low-dose STZ injections (40 mg/kg/day for 5 days; freshly prepared in 0.1mmol/L sodium citrate at pH 4.5) (Chen et al., 2008a).
Obese, insulin-resistant and diabetic leptin receptor-deficient male B6.BKS(D) – Leprdb/J (db/db) mice (The Jackson Laboratory) were used as a model of T2D.
The whole body TXNIP-deficient mice have been described previously (Chen et al., 2008a).
The liver-specific glucagon receptor knockout mouse has been previously described (Kim et al., 2018) and was a generous gift of Dr. Kirk Habegger.
Mice were randomly assigned to their treatment or control group. The dose was chosen in analogy to our previous verapamil studies using the same mouse models and SRI-37330 was administered in the drinking water, resulting in a dose of 100 mg/kg/day (Xu et al., 2012) and an average plasma concentration of 5 μmol/L. Metformin (Sigma) and empagliflozin (Sigma) were also administered in the drinking water at the previously established effective doses of 250 mg/kg/day and 10 mg/kg/day, respectively (Daems et al., 2019; Han et al., 2017). Control mice were housed under identical conditions, but without SRI-37330 or any additional treatment. For gavage experiments, mice were dosed twice a day, 12 h apart. A Freestyle Lite glucometer was used to determine blood glucose levels.
Tissue Culture
All cells were grown at 37°C in a 5% CO2 humid atmosphere. Rat INS-1 cells (Asfari et al., 1992) were validated for retention of their beta cell-like character by their continued ability to produce insulin. They were grown in RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, 1 mmol/L sodium pyruvate, 2 mmol/L L-glutamine, 10 mmol/L HEPES, 0.05 mmol/L 2-mercaptoethanol and containing 11.1 mM glucose as previously described (Minn et al., 2005) and were treated with the indicated doses of SRI-37330 for 24h.
Mouse pancreatic islets were isolated from C57BL/6J mice as described previously (Chen et al., 2008b). In brief, pancreata were inflated with 5 mL collagenase solution (0.45mg/mL type XI collagenase, Sigma, St. Louis, MO) in Hanks’ balanced salt solution (HBSS) (Corning, Tewksbury, MA) with 0.02% radioimmunoassay-grade bovine serum albumin (BSA) (Sigma), placed in 25mL of the same solution in a 37°C water bath shaker for 5 min and starting at 6min vigorously shaken for 20 seconds every 2 min for an additional 10min. After a quick spin at 1500 rpm, the tissue pellet was washed twice with 10 mL cold HBSS, passed through a 925-micron Spectra mesh filter (Spectrum Labs, Cincinnati, OH) to remove large debris, and resuspended in 5mL of 25% Ficoll (type 400-DL; Sigma) prepared with HBSS in a 50-mL conical tube, 2.5 mL each of 23, 20.5, and 11% Ficoll were layered carefully on top of each other and the gradient was centrifuged for 5 min at 1800 rpm. Layers above the 25% Ficoll containing the isolated islets were collected, washed with HBSS, and the islets pelleted by a 1 min centrifugation at 1500 rpm. To further exclude contamination by exocrine tissue, islets were handpicked under stereomicroscopic observation and used prior to all experiments.
Human islets were obtained from the Integrated Islet Distribution Program (IIDP). Islets from at least three different donors were used per experiment and always islets of the same donor were used as controls. For experiments conducted in the context of high (25 mM) glucose, INS-1 cells, isolated mouse islets and primary human islets were maintained overnight in 5 mM glucose RPMI 1640 medium and then incubated for 24 h in 25 mM glucose RPMI 1640 medium with or without 1 μM of SRI-37330.
Mouse αTC1-6 alpha cells (ATCC CRL-2934, sex unknown) were grown in Dulbecco’s Modified Eagle Medium (Thermo Fisher) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, 16.7 mM glucose, 1mM sodium pyruvate and 15 mM HEPES. Primary hepatocytes were isolated from anesthetized lean chow-fed male C57BL/6J mice or from liver-specific glucagon receptor knockout or Txnip-deficient mice as previously described (Kim et al., 2018). Primary hepatocytes were then cultured in collagen-coated 12-well plates at a density of 3 × 105 cells/well with serum-free DMEM medium. After cells were allowed to attach for 4 h, the medium was changed and they were incubated with different concentrations of SRI-37330 for 24h.
METHOD DETAILS
High-throughput screening
To develop a screening assay, the human TXNIP promoter region including −1518bp upstream of the ATG start codon was subcloned into the pGL4.17 [luc2/Neo] vector (Promega) and confirmed by sequencing to generate the luciferase reporter plasmid. INS-1 rat beta-like cells were then transfected with this plasmid (0.4μg/well) using DharmaFECT Duo transfection reagent (1μL/well, Dharmacon) and 48h after transfection, geneticin (100 μg/mL, Thermo Fisher) was added to select stable transfected cells. After all geneticin-sensitive (untransfected) cells had died, single clones were picked and the dose of geneticin was reduced to the maintenance dose of 50 ug/ml, which was used thereafter for expansion and culture of this stably transfected cell line. The luciferase assay using this monoclonal cell line was then miniaturized and optimized for robotics going from a 12-well to a 384-well plate.
For the primary high-throughput screening assay, these INS-1 cells stably transfected with the human TXNIP promoter luciferase reporter system were maintained overnight in 5 mM glucose media and then incubated for 24 h in 25 mM glucose media with or without the small molecule compounds (10 μM) to be tested. Over 300,000 small molecules from an in-house Southern Research Institute (SRI) library were screened and compounds decreasing TXNIP promoter activity by ≥ 50% were classified as hits (~1%). They were then filtered to select those inactive in another luciferase-based general transcription counter screen and those with a CC50/72h ≥ 5 μM in a cellular cytotoxicity assay (Cell Titer-Glo Luminescent Cell Viability Assay kit, Promega). Hits were further validated by assessing effects on endogenous TXNIP expression using 10 point qRT-PCR dose response experiments. To this end RNA was extracted from INS-1 cells cultured in 96-well plates using RNeasy 96 Kit (Qiagen) following the manufacturer’s protocol using spin technology. QuantiFast SYBR Green RT-PCR Kit (Qiagen) was used as a one-step qRT-PCR amplification system using primers listed in Supplemental Table S2. Gene expression results were corrected for the 18S ribosomal subunit run as an internal standard. From this narrowed down list of compounds, quinazolines, including the initial hit compound (Supplemental Figure S5a) emerged as a core chemical structure for further analysis. Multiple derivatives of this structure were produced (Supplemental Figure S5a) and medicinal chemistry efforts and analysis of structure-activity-relationship were focused on optimizing potency, minimizing cytotoxicity, targeting a solubility of ≥ 10 μM and a Log D of 2–4 and increasing metabolic stability. To this end composite site lability values were generated in silico using the StarDrop software and the information was used to guide and prioritize the design of compounds by estimating their rate of metabolism prior to being synthesized and tested in mouse and human liver microsomal stability assays and ultimately in vivo for oral bioavailability. In addition, promising derivatives were validated for their TXNIP lowering capacity by manual transient transfection luciferase assays, qRT-PCR and immunoblotting. This lengthy procedure ultimately yielded the lead compound SRI-37330.
Compound synthesis
A schematic of the chemical synthesis pathway of SRI-37330.HCl is shown in Supplemental Figure S5b and a brief description is below.
Preparation of tert-butyl 3-(methylsulfonamidomethyl)piperidine-1-carboxylate: 2.
To a stirred solution of tert-butyl 3-(aminomethyl)piperidine-1-carboxylate 1 (25.0 g, 116.65 mmol) in CH2Cl2 (250 mL) were added Et3N (49.09 mL, 349.97 mmol) and methane sulphonyl chloride (15 mL, 174.98 mmol) at room temperature (RT). The reaction mixture was stirred at RT for 3 h. Upon complete consumption of starting material, the reaction mixture was poured into water (250 mL), extracted with CH2Cl2 (2 × 250 mL).The organic extracts were washed with saturated NaHCO3 (200 mL), water (200 mL), brine (200 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford 2 (36.0 g, crude) as thick colorless liquid. MS (MM): m/z = 293 [M+H]+.
Preparation of 1-(tert-butoxycarbonyl)-3-(methylsulfonamidomethyl)piperidin-1-ium chloride: 3
To a stirred solution of 2 (36.0 g, 123.28 mmol) in 1, 4-dioxane HCl (4M, 72 mL) was stirred at RT for 16 h. Upon complete consumption of starting material, the reaction mixture was concentrated under reduced pressure. The residue was co-distilled with toluene (3 × 250 mL) to afford 3 (30.0 g, crude) as thick colorless liquid. Hydrogen-1 nuclear magnetic resonance (1HNMR) (300 MHz, DMSO-d6): δ 9.18–8.98 (m, 1H), 7.23 (t, J = 6.0 Hz, 1H), 3.23–3.15 (m, 2H), 3.23–315 (m, 2H), 2.89–2.73 (m, 1H), 2.70 (s, 3H), 2.68–2.56 (m, 3H), 1.91–1.62 (m, 4 H), 1.22–1.16 (m, 1H); MS (MM): m/z = 193 [M+H]+. The crude product directly used in the next step.
Preparation of 6-(trifluoromethyl)quinazolin-4-ol: 5.
To a solution of 2-amino-5-(trifluoromethyl)benzoic acid 4 (25.0 g, 121.95 mmol) in Ethoxy Ethanol (250 mL) was added Farmamidine acetate (25.39 mL, 243.90 mmol)) and stirred at 130°C for 30 h, the reaction mixture was poured into water (500 mL), The solid was precipitated, filtered and dried to afforded 5 (15.0 g, 60%) as an off-white solid. 1H-NMR (400 MHz, DMSO-d6) δ: 12.62 (s, 1H), 8.36 (d, J = 0.8 Hz, 1H), 8.26 (s, 1H),8.12 (dd, J = 2 Hz, 8.4 Hz), 7.87 (d, J = 8.8 Hz, 1H); MS (MM): m/z = 215 [M + H]+.
Preparation of 4-chloro-6-(trifluoromethyl)quinazoline: 6.
To a stirred solution of 5 (10.0 g, 46.72 mmol) in Thionyl chloride(100 mL) was added DMF (1.0 mL, Catalytic) at 0°C and stirred for 2 h at 90°C.Upon complete consumption of starting material, the reaction mixture was concentrated under reduced pressure. Co-distilled with toluene (3 × 50 mL) to afford 6 (10 g, crude) as an off-white solid. The crude product directly used in the next step.
Preparation of 3-(methylsulfonamidomethyl)-1-(6-(trifluoromethyl)quinazolin-4-yl)piperidin-1-ium Chloride: SRI-37330.HCl.
To a stirred solution of 6 (10 g, 43.10 mmol)in NMP (100 mL) were added 3 (9.8 g, 43.10 mmol) and DIPEA (22.54 mL, 129.31 mmol) at RT for 3 h in inert gas atmosphere. Upon complete consumption of the starting material, the reaction mixture was poured into water (200 mL), extracted with EtOAc (2 × 200 mL). The organic extracts were washed with saturated NaHCO3 (100 mL), water (100 mL), brine (100 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by combi-flash column chromatography using 120 g column (elution of 100% EtOAc) to give N-((1-(6-(trifluoromethyl)quinazolin-4-yl)piperidin-3-yl)methyl)methanesulfonamide(5.4 g, 32 %) as off-white solid. 1H NMR (400 MHz, DMSO-d6): δ 8.67 (s, 1H), 8.2 (s, 1H), 8.05 (dd, J = 1.6 Hz, 8.8 Hz), 7.95 (d, J = 8.8 Hz, 1H), 7.10 (t, J = 6.4 Hz, 1H), 4.39 (d, J = 12.0 Hz, 1H), 4.22 (d, J = 12.8 Hz, 1H), 3.29 (s, 1H), 3.06–2.88 (m, 5H), 1.89–1.80 (m, 3H), 1.7–1.61 (m, 1H), 1.37–1.29 (m, 1H); FABMS (M + H) calculated for C16H19F3N4O2S.H was 389.1254 found 389.1243; HPLC: 96.0% (% of ACU). To as a stirred solution of the free base N-((1-(6-(trifluoromethyl)quinazolin-4-yl)piperidin-3-yl)methyl)methanesulfonamide (19.0 g, 48.96 mmol) in 1, 4-dioxane (100 mL) were added 1, 4-dioxane HCl (4M, 38 mL) was stirred at RT for 2 h. Upon complete consumption of starting material, the reaction mixture was concentrated under reduced pressure. The residue was co-distilled with toluene(50 mL), DCM(50 mL) and MTBE (50 mL), solid formed, filtered, dried to afford SRI-37330.HCl(21 g, 91%) as an off-white solid. 1H-NMR (400 MHz, DMSO-d6): δ 8.87 (s, 1H), 8.37 (s, 1H), 8.28 (dd, J = 1.2 Hz, 8.8 Hz, 1H), 8.02 (d, J = 8.8 Hz 1H), 7.17 (t, J = 6.4 Hz 1H), 4.69 (d, J = 11.2 Hz, 1H), 4.5 (d, J = 11.2 Hz, 1H), 3.64 (t, J = 12.0 Hz 1H), 3.38 (t, J = 10.4 Hz 1H), 2.98–2.92 (m, 1H), 2.85 (s, 3H), 1.90 (m, 3H) 1.74–1.65 (m, 1H) 1.46–1.39 (m, 1H); MS (MM): m/z = 388.1 [M + H]+; FABMS (M + H) calculated for C16H19F3N4O2S.H was 389.1254 found 389.1253; HPLC = 97.1 (% of ACU); Elemental analysis (% calculated, % found for C16H19F3N4O2S*1.1 HCl): C (44.85, 44.45), H (4.73, 4.81), Cl (9.10, 9.33), N (13.07, 12.70).
Alpha Cell Glucagon Expression and Secretion
For assessment of proglucagon expression αTC1-6 alpha cells were incubated with 5 μM SRI-37330 or vehicle for 24 h. For glucagon content and secretion assays, cells were incubated with 5μM SRI-37330 or vehicle for 1h. For TXNIP knockdown, cells were transfected with specific siRNA oligos for mouse Txnip (L-040441-01-0005, SMARTpool: ON-TARGETplus Txnip siRNA, Dharmacon, Lafayette, CO) or scrambled oligo (D-001810-01-20, Dharmacon) using DharmaFECT 1 Transfection Reagent (Dharmacon/Thermo Scientific, Chicago, IL). For TXNIP overexpression, cells were transfected with the TXNIP expression plasmid or control plasmid described previously (Minn et al., 2005) using DharmaFECT Duo Transfection Reagent (T-2010-03, Dharmacon, Lafayette, CO). For maximal stimulation of glucagon secretion, the established secretagogues arginine (20mM) and norepinephrine (10μM) were added as described previously (Hauge-Evans et al., 2010). To assess glucagon secretion under low glucose conditions, experiments were performed at 1.67mM glucose. Glucagon secretion was measured using Mouse Glucagon ELISA Kit (Crystal Chem) following the manufacturer’s instructions and protein levels were determined by Pierce BCA Protein Assay (Thermo Fisher).
Hepatocyte Glucose Output
Glucose release from primary mouse hepatocytes into the culture media was assessed during the last 6h after cells were washed twice with phosphate buffer saline and incubated in serum-, glucose- and phenol red-free DMEM in the presence or absence of lactate (10 mM) and/or glucagon (100 nM) at the different SRI-37330 concentrations. Glucose concentrations in the media were measured with Glucose Colorimetric Detection Kit (Thermo Fisher) and normalized to total protein content.
Hepatocyte cAMP Production
For assessment of cAMP production, primary hepatocytes were washed with phosphate buffer saline twice and harvested with 0.1M hydrogen chloride. Intracelluar cAMP levels were measured using cAMP Direct Immunoassay Kit (Abcam, Cambridge, MA) by following the manufacturer’s instructions.
Luciferase Assays and Quantitative Real-time RT-PCR
INS-1 cells were grown in 12-well plates and transiently transfected with the pGL3 human TXNIP promoter firefly luciferase plasmid (0.4 μg/mL) described previously (Minn et al., 2005) and the pRL-TK renilla luciferase control plasmid (10 ng/mL) using DharmaFECT Duo transfection reagent (1μL/mL; Dharmacon). 4h after transfection, SRI-37330 (1 μM) was added to the medium and after another 24h, cells were harvested and luciferase activity was determined by Dual-Luciferase Reporter Assay System (Promega). RNA isolation and quantitative real-time RT-PCR (qRT-PCR) were performed as previously described (Chen et al., 2008a; Xu et al., 2013) using the primers listed in Supplemental Table S2. Gene expression results were corrected for the 18S ribosomal subunit run as an internal standard.
Immunoblotting
Whole cell protein extracts from INS-1 cells were prepared as described previously (Chen et al., 2008b). Protein concentrations were determined by Pierce BCA Protein Assay (Thermo Fisher) and equal amounts of protein were loaded per lane. The following antibodies were used: mouse anti-TXNIP IgG (1:1000, K0205-3; MBL International), mouse anti-actin IgG (1:3000, ab3280; Abcam), and goat anti-mouse IgG-HRP (1:5000, sc-2005; Santa Cruz Biotechnology). Bands were visualized by ECL Plus (GE Healthcare) and quantified by ImageQuant TL software (GE Healthcare).
Promoter Analysis
Reporter plasmids containing the full length and deletion constructs of the TXNIP promoter as well as the SV40-E-box and SV40-Control plasmids (Promega) have been previously described (Minn et al., 2005). To determine the TXNIP promoter region responsible for conferring the effects of SRI-37330, INS-1 cells were grown in 12-well plates and transfected using DharmaFECT Duo transfection reagent (1 μL/mL; GE Dharmacon) with various TXNIP promoter deletion reporter plasmids (0.4 μg/mL) together with pRL-TK control plasmid (10 ng/mL) and incubated for 4h. To determine whether the identified E-box region could confer responsiveness to SRI-37330 to a heterologous promoter, INS-1 cells were transfected with the SV40-E-box or SV40-Control plasmid (0.4 μg/mL) together with pRL-TK control plasmid (10 ng/mL). After 18 h, the media was changed to 5 mM glucose for 6h and then to 25 mM glucose for the remainder of the experiment. All cells were treated with or without 5 μM SRI-37330 for 24 h, harvested and luciferase activity was determined by Dual-Luciferase Reporter Assay System (Promega).
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed as previously described (Cha-Molstad et al., 2009). In brief, INS-1 cells were cross-linked by adding 10 mL 1% formaldehyde (Sigma) in Phosphate-Buffered Saline (PBS, Thermo Fisher) to the cells for 15 min. 0.525 mL 2.5 M Glycine (Promega) was added to terminate the cross-linking reaction. Plates were placed on ice and washed 2 times with 10mL ice-cold PBS. Cells were scraped in 3mL ice-cold PBS and centrifuged at 2000 x g for 5 min at 4 °C. Cell pellets were resuspended in 1mL Lysis Buffer (0.1% SDS, 0.5% Triton X-100, 20mM Tris-HCl, 150mM NaCl, pH8.1) with 1X Proteinase inhibitors (PI, Roche) and incubated on ice for 30min. Sonication was performed with a Branson Sonifier 250 (Branson) (30 s pulse, 30 s rest) for 10 times at 17.5% output. Supernatant was harvested by centrifugation at 12000 rpm for 10min at 4 °C. Protein concentrations were determined by Pierce BCA Protein Assay (Thermo Fisher), and 500μg of precleared lysates were incubated with 4 μg of anti-RNA polymerase II antibody (ab5131, Abcam) or normal control IgG overnight at 4 °C. 50μg of lysates (1/10) were taken as inputs and stored in −80°C. Immune complexes were captured with 50 μL of a 50% Protein A-Sepharose slurry (Sigma) for 3 h at 4 °C. Protein A-Sepharose was spun down (1000rpm, 1 min, 4 °C) and washed with the following buffers: (1) 1mL Lysis Buffer, 5 min; (2) 1mL Washing Buffer (0.1% SDS, 0.5% Triton X-100, 2 mM EDTA, 20mM Tris-HCl, 150 mM NaCl, pH 8.1), 5 min; (3) 1 mL Lithium Chloride (LiCl) Buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl, pH 8.1), 5 min; (4) 1mL TE Buffer (1 mM EDTA, 10 mM Tris-HC, pH 8.1), 30 min; (5) 1 mL TE Buffer, 5 min. After the final washing, 100 μl Elution Buffer (1% SDS, 0.1 M NaHCO3, pH 8.0) was added and incubated at room temperature for 15 min. Supernatant was centrifuged (13000 rpm, 1 min) and transferred to a new tube. This was repeated one more time with another 100 μl Elution Buffer to yield 200 μl of eluate. To reverse the cross-links 10 μL of 5 M NaCl and 2 μL of Proteinase K (Thermo Fisher) were added to the eluates and incubated overnight in a heating block at 65°C and 850 rpm. DNA fragments were purified using a PCR purification kit (Qiagen) and quantified by real-time PCR with the ChIP primers listed in Supplemental Table S2.
RNA Sequencing
After overnight incubation at 5 mM glucose, isolated human islets were incubated at 25 mM glucose in the presence or absence of 1 μM SRI-37330 for 24 h prior to RNA extraction by using a miRNeasy Mini Kit (Qiagen). RNA sequencing was performed by Exiqon/Qiagen and included preparation of libraries using TruSeq stranded mRNA sample preparation kit (Illumina Inc.) and single-end sequencing was performed with an average of ~43 million reads obtained per sample. RNA-sequencing reads were aligned to the H. sapiens reference genome (GRCh38.p7) using STAR (v2.4.2a) with an average ~90% reads uniquely mapped. Alignments were quantified using Salmon (v0.8.2) and differential expression analysis was performed using DESeq2. The DESeq2 model accounted for the experimental design of paired treated and untreated samples from each donor.
Glucose and Insulin Tolerance Tests
Glucose tolerance tests (GTTs) and insulin tolerance tests (ITTs) were performed following a 4 h fast using 3g /kg glucose, i.p. and 1 U/kg insulin, i.p., respectively (Chen et al., 2008a).
Serum Insulin, Glucagon and Triglyceride Tests
Serum insulin and glucagon levels were determined by the Mouse Ultrasensitive Insulin ELISA kit (ALPCO) and Mouse Glucagon ELISA Kit (Crystal Chem), respectively. In addition, the Serum Triglyceride Assay Kit (Abcam) and the ALT Activity Assay (Sigma) were used. Urine glucose levels were assessed using a Mouse Glucose Assay Kit (Crystal Chem).
Liver Glycogen Assessment
To assess mouse liver glycogen levels, tissue samples were washed with cold phosphate buffer saline, resuspended in 200 μl of ddH2O and homogenized. Homogenized samples were boiled for 10 minutes to inactivate enzyme and centrifuged at 18,000 × g for 10 min at 4°C to collect glycogen. Glycogen levels were measured using Glycogen Colorimetric Assay Kit (Abcam, Cambridge, MA) by following the manufacturer’s instructions. Liver paraffin sections were also assessed by Periodic Acid Schiff (PAS) staining. Rehydrated slides were immersed in 0.5% periodic acid solution for 5 minutes, rinsed in distilled water and placed in Schiff reagent for 15 minutes, and washed in lukewarm tap water for 5 minutes. Then slides were counterstained with hematoxylin for 1 minute, dehydrated and mounted.
Hyperinsulinemic-Euglycemic Clamp Studies
Whole body fat and lean mass were non-invasively measured using 1H-MRS (Echo Medical Systems, Houston, TX). 5–6 days before the clamp experiments, survival surgery was performed in anesthetized lean C57BL/6J and obese, diabetic db/db mice to establish an indwelling catheter in the jugular vein. Prior to the clamp experiment, mice were treated with SRI-37330 and were fasted overnight. A 2 h hyperinsulinemic-euglycemic clamp was conducted in awake mice with a primed and continuous infusion of human insulin (150 mU/kg body weight priming followed by 2.5 mU/kg/min; Humulin, Eli Lilly, IN) (Kim, 2009). To maintain euglycemia, 20% glucose was infused at variable rates during clamps. Whole body glucose turnover was assessed with a continuous infusion of [3-3H]glucose (PerkinElmer, Waltham, MA), and 2-deoxy-D-[1-14C]glucose (2-[ C]DG) (PerkinElmer, Waltham, MA) was administered as a bolus (10 μCi) at 75 min after the start of the clamp to measure insulin-stimulated glucose uptake in individual organs. At the end of the clamps, mice were anesthetized, and tissues were taken for biochemical analysis. Glucose concentrations during clamps were analyzed using 10 μl plasma by a glucose oxidase method on Analox GM9 Analyser (Analox Instruments Ltd., London, UK). Plasma concentrations of [3-3H]glucose, 2-[14C]DG, and 3H2O were determined following deproteinization of plasma samples as previously described(Kim, 2009). For the determination of tissue 2-[14C]DG-6-Phosphate content, tissue samples were homogenized, and the supernatants were subjected to an ion-exchange column to separate 2-[14C]DG-6-P from 2-[14C]DG. Rates of basal hepatic glucose production and insulin-stimulated whole body glucose turnover as well as glucose infusion rates were determined as previously described (Kim, 2009). Insulin-stimulated glucose uptake in individual tissues was assessed by determining the tissue (e.g., skeletal muscle, white adipose tissue) content of 2-[14C]DG-6-phosphate and plasma 2-[14C]DG profile.
Alpha and Beta Cell Morphometry
Immunofluorescence for insulin and glucagon was performed as previously described with minor modifications (Saxena et al., 2010). Briefly, insulin was visualized by insulin staining using rabbit anti-insulin antibody (1:100, 4590S, Cell Signaling Technology, Danvers, MA) and Alexa Fluor 488-conjugated anti-rabbit IgG (1:100, 711-545-152, Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Glucagon was visualized by glucagon staining using mouse anti-glucagon antibody (1:100, G2654, MilliporeSigma, Burlington MA) and Alexa Fluor 594-conjugated anti-mouse IgG (1:100, 715-585-150, Jackson ImmunoResearch Laboratories Inc., West Grove, PA). The Mouse on mouse Detection Kit (BMK-2202, Vector Laboratories Inc., Burlingame, CA) was used according to the manufacturer’s protocol with minor modifications to reduce background and non-specific fluorescent signals. Slides were mounted using DAPI Fluoromount-G (0100-20, SouthernBiotech, Homewood, AL) to visualize the nuclei. To assess alpha- and beta-cell mass, immunostained pancreata were imaged using an Olympus IX81 inverted fluorescent microscope (Olympus Corporation, Tokyo, Japan) and analyzed using the cellSens Dimension imaging software (v.1.18, build 16686, Olympus Corporation, Tokyo, Japan). Images were stitched to produce a fluorescent image of the entire pancreas section. The signals corresponding to insulin, glucagon, and nuclei were then separated and quantified through thresholding. All thresholded images were manually inspected to remove background signal. Alpha cell and beta cell mass was calculated as (alpha cell area/total pancreas area)*pancreas weight and (beta cell area/total pancreas area)*pancreas weight, respectively. Relative alpha cell and beta cell numbers were determined by counting the glucagon and insulin positive cells per islet using at least two different sectioning positions for each mouse pancreas.
QUANTIFICATION AND STATISTICAL ANALYSIS
To calculate the significance of a difference between two groups, two-tailed Student t-tests or paired t-tests were used for independent or paired samples, respectively. For data sets of more than two groups, we performed analysis of variance (ANOVA) followed by post-hoc tests. A two-way repeated measures ANOVA was used for all repeated measures. The normality assumption was examined before the analysis. Sample sizes were determined based on analysis of our and other’s previous experience using similar models and settings. We did not exclude any available numbers from analysis and all statistical analyses were performed with the use of SAS 9.4 (Cary, NC). A P-value of < 0.05 was considered significant. For histological analyses experimenters were blinded to the treatment of the mice.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-TXNIP | MBL International | K0205-3 |
| Mouse anti-actin | Abcam | ab3280 |
| Goat anti-mouse IgG-HRP | Santa Cruz Biotechnology | sc-2005 |
| Rabbit anti-RNA polymerase II | Abcam | ab5131 |
| Rabbit normal IgG | Santa Cruz Biotechnology | sc-2027 |
| Rabbit anti-insulin | Cell Signaling Technology | 4590S |
| Alexa Fluor 488-conjugated anti-rabbit IgG | Jackson ImmunoResearch Laboratories Inc | 711-545-152 |
| Mouse anti-glucagon | MilliporeSigma | G2654 |
| Alexa Fluor 594-conjugated anti-mouse IgG | Jackson ImmunoResearch Laboratories Inc | 715-585-150 |
| Chemicals | ||
| Type XI collagenase | Sigma Aldrich | C7657 |
| Hanks’ balanced salt solution (HBSS) | Corning | 21-023-CM |
| 0.02% radioimmunoassay-grade BSA | Sigma Aldrich | A7888 |
| 925-micron Spectra mesh filter | Spectrum Labs | 146410 |
| Ficoll 400 | Sigma Aldrich | 9378 |
| Secretagogues arginine | Sigma Aldrich | A6969-100G |
| Norepinephrine | Sigma Aldrich | A0937-5G |
| Lactate | Sigma Aldrich | L7022 |
| Glucagon | Sigma Aldrich | G2044 |
| Hydrogen chloride | Thermo Fisher Scientific | A144-500 |
| Formaldehyde | Sigma Aldrich | F8775-500ML |
| Glycine | Promega | H5071 |
| 10% SDS | Caliber | GR155-1 |
| Triton X-100 | Promega | H5141 |
| 1 M Tris-HCl (PH8.1) | Thermo Fisher Scientific | BP1758-500 |
| 5 M NaCl | Promega | V4221 |
| Proteinase inhibitor cocktail tablet | Roche | 4693132001 |
| Protein A-Sepharose | Sigma Aldrich | P3391 |
| 0.5 M EDTA | Thermo Fisher Scientific | BP2482-500 |
| Lithium Chloride | Thermo Fisher Scientific | L121-100 |
| NP-40 | US Biological | N3500 |
| Deoxycholate | Sigma Aldrich | 264101-100GM |
| NaHCO3 | Boston BioProducts | BB-2612 |
| Proteinase K | Thermo Fisher Scientific | AM2546 |
| Streptozocin | Sigma Aldrich | S0130 |
| Metformin | Sigma Aldrich | D150959-5G |
| Empagliflozin | Sigma Aldrich | A112500-5G |
| Human insulin | Eli Lilly | N/A |
| Periodic acid | Sigma Aldrich | P7875 |
| Schiff Reagent | Sigma Aldrich | 3952016 |
| Mayer’s Hematoxylin Solution | Sigma Aldrich | MHS 16 |
| [3-3H]glucose | PerkinElmer | NET331C005MC |
| 2-deoxy-D-[1-14C]glucose (2-[14C]DG) | PerkinElmer | NEC495001MC |
| SRI-37330 | Southern Research/this paper | N/A |
| Critical Commercial Assays | ||
| Dual-Luciferase Reporter Assay System | Promega | E1960 |
| Cell Titer-Glo Luminescent Cell Viability Assay Kit | Promega | G7571 |
| RNeasy 96 Kit | Qiagen | 74181 |
| QuantiFast SYBR Green RT-PCR Kit | Qiagen | 204154 |
| Mouse Glucagon ELISA Kit | Crystal Chem | 81518 |
| Pierce BCA Protein Assay | Thermo Fisher Scientific | 23227 |
| Glucose Colorimetric Detection Kit | Thermo Fisher Scientific | EIAGLUC |
| cAMP Direct Immunoassay Kit | Abcam | ab65355 |
| miRNeasy Mini Kit | Qiagen | 217004 |
| First strand cDNA synthesis Kit | Roche | 04896866001 |
| PCR product purification kit | Qiagen | 28106 |
| Mouse Ultrasensitive Insulin ELISA Kit | ALPCO | 80-INSMSU-E01 |
| Serum Triglyceride Assay Kit | Abcam | ab65336 |
| ALT Activity Assay | Sigma Aldrich | MAK052 |
| Mouse Glucose Assay Kit | Crystal Chem | 81692 |
| Glycogen Colorimetric Assay Kit | Abcam | ab65620 |
| Mouse on Mouse Detection Kit | Vector Laboratories Inc | BMK-2202 |
| Deposited Data | ||
| RNA sequencing data | GEO | GSE151588 |
| Experimental Models: Cell Lines | ||
| Rat INS-1 beta-like cells | Asfari et al., 1992 | N/A |
| Mouse αTC1-6 alpha cells | ATCC | ATCC CRL-2934 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | The Jackson Laboratory | 000664 |
| GcgR-/-L mice | Kim et al., 2018 | N/A |
| Whole body Txnip-/- mice | Chen et al., 2008a | N/A |
| B6.BKS(D) – Leprdb/J (db/db) mice | The Jackson Laboratory | 000697 |
| Human islets | Integrated Islet Distribution Program (IIDP) | N/A |
| Oligonucleotides | ||
| Mouse TXNIP siRNA | Dharmacon, Horizon Discovery | L-040441-01-0005 |
| Scramble control siRNA | Dharmacon, Horizon Discovery | D-001810-01-20 |
| Primers | Sigma Aldrich/this paper | Supplemental Table S2 |
| Plasmids | ||
| pGL4.17 [luc2/Neo] vector | Promega | E6721 |
| TXNIP expression plasmid | Minn et al. 2005 | N/A |
| human TXNIP promoter plasmid (full length & deletions) | Minn et al. 2005 | N/A |
| SV40 E-box plasmid | Minn et al. 2005 | N/A |
| pRL-TK control plasmid | Promega | E2231 |
| Software and Algorithms | ||
| StarDrop | Optibrium | https://www.optibrium.com/ |
| ImageQuant TL | GE Healthcare | N/A |
| STAR (v2.4.2a) | N/A | N/A |
| Salmon (v0.8.2) | N/A | N/A |
| DESeq2 | Bioconductor | https://www.bioconductor.org/ |
| cellSens Dimension Imaging | Olympus Corporation | N/A |
| SAS 9.4 | SAS | https://www.sas.com/ |
| Other | ||
| DharmaFECT Duo transfection reagent | Dharmacon, Horizon Discovery | T-2010-03 |
| DharmaFECT 1 transfection reagent | Dharmacon, Horizon Discovery | T-2001-03 |
| ECL Plus Western Blotting Reagents | GE Healthcare | 45-002-401 |
| RPMI 1640 with glucose | Thermo Fisher Scientific | 11875119 |
| RPMI 1640 without glucose | Thermo Fisher Scientific | 11879020 |
| Dulbecco’s Modified Eagle Medium | Thermo Fisher Scientific | 11966025 |
| DMEM, no glucose, no glutamine, no phenol red | Thermo Fisher Scientific | A1443001 |
| Geneticin | Thermo Fisher Scientific | 10131027 |
| Fetal Bovine Serum | Thermo Fisher Scientific | 16140071 |
| Pen/Strep | Thermo Fisher Scientific | 15140122 |
| HEPES solution | Thermo Fisher Scientific | 15630130 |
| Sodium Pyruvate | Thermo Fisher Scientific | 11360070 |
| L-glutamine | Sigma Aldrich | G3126 |
| 2-Mercaptoethanol | Thermo Fisher Scientific | 19470583 |
| D(+)-Glucose | Thermo Fisher Scientific | AC410955000 |
| Trypsin-EDTA (0.25%) | Thermo Fisher Scientific | 25200056 |
| Phosphate-Buffered Saline | Thermo Fisher Scientific | 10010023 |
| Novex 10–20% Tris-Glycine Mini Gels | Thermo Fisher Scientific | XP10202BOX |
CONTEXT AND SIGNIFICANCE
The global health problem of diabetes is getting worse, but therapies that target the underlying causes are still lacking. By screening 300,000 small molecules, researchers at Birmingham, Alabama identified a compound that downregulates the detrimental, pro-diabetic protein, TXNIP in mouse and human pancreatic islets. When given orally to mice this small molecule also lowered serum levels of glucagon, a counter-regulatory hormone to insulin that is elevated in diabetes and increases blood glucose levels. Furthermore, the compound prevented fatty liver, inhibited unnecessary glucose production by the liver and reversed diabetes in different mouse models of type 1 and type 2 diabetes. Compared to currently available drugs, this compound may therefore, provide an effective and highly promising new approach to diabetes therapy.
HIGHLIGHTS
The small molecule SRI-37330 inhibits TXNIP expression in mouse and human islets
SRI-37330 decreases glucagon secretion and action and blocks hepatic glucose output
Oral SRI-37330 reverses obesity- and STZ-induced diabetes and hepatic steatosis in mice
Its antidiabetic effects and safety profile make SRI-37330 an attractive drug candidate
ACKNOWLEDGEMENTS
The work was supported by NIH grants U01DK120379 and R01DK078752 to A.S. and the Alabama Drug Discovery Alliance (ADDA). The UAB Center for Clinical and Translational Science (CCTS) is supported by UL1TR001417 and the National Mouse Metabolic Phenotyping Center (MMPC) at UMass is supported by NIH grant U2CDK093000 to J.K.K. The UAB Small Animal Phenotyping and Glycemic Clamp Cores are supported by the NIH Nutrition Obesity Research Center (P30DK056336).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
The following patent is related to this work: WO 2019/089693 A1
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Asfari M, Janjic D, Meda P, Li G, Halban PA, and Wollheim CB (1992). Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 130, 167–178. [DOI] [PubMed] [Google Scholar]
- Cha-Molstad H, Saxena G, Chen J, and Shalev A. (2009). Glucose-stimulated expression of Txnip is mediated by carbohydrate response element-binding protein, p300, and histone H4 acetylation in pancreatic beta cells. J Biol Chem 284, 16898–16905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Hui ST, Couto FM, Mungrue IN, Davis DB, Attie AD, Lusis AJ, Davis RA, and Shalev A. (2008a). Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB J 22, 3581–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Saxena G, Mungrue IN, Lusis AJ, and Shalev A. (2008b). Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes 57, 938–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Young ME, Chatham JC, Crossman DK, Dell’Italia LJ, and Shalev A. (2016). TXNIP regulates myocardial fatty acid oxidation via miR-33a signaling. Am J Physiol Heart Circ Physiol 311, H64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chutkow WA, Patwari P, Yoshioka J, and Lee RT (2008). Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J Biol Chem 283, 2397–2406. [DOI] [PubMed] [Google Scholar]
- Cooper-Dehoff R, Cohen JD, Bakris GL, Messerli FH, Erdine S, Hewkin AC, Kupfer S, Pepine CJ, and Investigators I. (2006). Predictors of development of diabetes mellitus in patients with coronary artery disease taking antihypertensive medications (findings from the INternational VErapamil SR-Trandolapril STudy [INVEST]). Am J Cardiol 98, 890–894. [DOI] [PubMed] [Google Scholar]
- Daems C, Welsch S, Boughaleb H, Vanderroost J, Robert A, Sokal E, and Lysy PA (2019). Early Treatment with Empagliflozin and GABA Improves beta-Cell Mass and Glucose Tolerance in Streptozotocin-Treated Mice. Journal of diabetes research 2019, 2813489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Tao YL, Deng YP, Yu JW, Cai J, Ren GF, Sun YN, and Jiang GJ (2017). Metformin ameliorates insulitis in STZ-induced diabetic mice. PeerJ 5, e3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauge-Evans AC, King AJ, Fairhall K, Persaud SJ, and Jones PM (2010). A role for islet somatostatin in mediating sympathetic regulation of glucagon secretion. Islets 2, 341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Chen J, Xu G, Grayson TB, Thielen LA, and Shalev A. (2018). miR-204 Controls Glucagon-Like Peptide 1 Receptor Expression and Agonist Function. Diabetes 67, 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodneva Y, Shalev A, Frank SJ, Carson AP, and Safford MM (2016). Calcium channel blocker use is associated with lower fasting serum glucose among adults with diabetes from the REGARDS study. Diabetes Res Clin Pract 115, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK (2009). Hyperinsulinemic-euglycemic clamp to assess insulin sensitivity in vivo. Methods Mol Biol 560, 221–238. [DOI] [PubMed] [Google Scholar]
- Kim T, Holleman CL, Nason S, Arble DM, Ottaway N, Chabenne J, Loyd C, Kim JA, Sandoval D, Drucker DJ, et al. (2018). Hepatic Glucagon Receptor Signaling Enhances Insulin-Stimulated Glucose Disposal in Rodents. Diabetes 67, 2157–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, et al. (2012). IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab 16, 250–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Osborne MC, Monia BP, Bhanot S, Gaarde WA, Reed C, She P, Jetton TL, and Demarest KT (2004). Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes 53, 410–417. [DOI] [PubMed] [Google Scholar]
- Meng H, Zhang A, Liang Y, Hao J, Zhang X, and Lu J. (2018). Effect of metformin on glycaemic control in patients with type 1 diabetes: A meta-analysis of randomized controlled trials. Diabetes Metab Res Rev 34, e2983. [DOI] [PubMed] [Google Scholar]
- Minn AH, Hafele C, and Shalev A. (2005). Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology 146, 2397–2405. [DOI] [PubMed] [Google Scholar]
- Nagaraj K, Lapkina-Gendler L, Sarfstein R, Gurwitz D, Pasmanik-Chor M, Laron Z, Yakar S, and Werner H. (2018). Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc Natl Acad Sci U S A 115, 1045–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovalle F, Grimes T, Xu G, Patel AJ, Grayson TB, Thielen LA, Li P, and Shalev A. (2018). Verapamil and beta cell function in adults with recent-onset type 1 diabetes. Nat Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone L, Devi TS, Hosoya KI, Terasaki T, and Singh LP (2010). Inhibition of TXNIP expression in vivo blocks early pathologies of diabetic retinopathy. Cell Death Dis 1, e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poitout V, and Robertson RP (2002). Minireview: Secondary beta-cell failure in type 2 diabetes--a convergence of glucotoxicity and lipotoxicity. Endocrinology 143, 339–342. [DOI] [PubMed] [Google Scholar]
- Riley KG, Pasek RC, Maulis MF, Peek J, Thorel F, Brigstock DR, Herrera PL, and Gannon M. (2015). Connective tissue growth factor modulates adult beta-cell maturity and proliferation to promote beta-cell regeneration in mice. Diabetes 64, 1284–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena G, Chen J, and Shalev A. (2010). Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J Biol Chem 285, 3997–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheen AJ, Paquot N, and Lefebvre PJ (2017). Investigational glucagon receptor antagonists in Phase I and II clinical trials for diabetes. Expert Opin Investig Drugs 26, 1373–1389. [DOI] [PubMed] [Google Scholar]
- Shah A, Xia L, Masson EA, Gui C, Momen A, Shikatani EA, Husain M, Quaggin S, John R, and Fantus IG (2015). Thioredoxin-Interacting Protein Deficiency Protects against Diabetic Nephropathy. J Am Soc Nephrol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalev A, Pise-Masison CA, Radonovich M, Hoffmann SC, Hirshberg B, Brady JN, and Harlan DM (2002). Oligonucleotide microarray analysis of intact human pancreatic islets: identification of glucose-responsive genes and a highly regulated TGFbeta signaling pathway. Endocrinology 143, 3695–3698. [DOI] [PubMed] [Google Scholar]
- Sharabi K, Lin H, Tavares CDJ, Dominy JE, Camporez JP, Perry RJ, Schilling R, Rines AK, Lee J, Hickey M, et al. (2017). Selective Chemical Inhibition of PGC-1alpha Gluconeogenic Activity Ameliorates Type 2 Diabetes. Cell 169, 148–160 e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SI, Blau JE, Rother KI, and Beitelshees AL (2019). SGLT2 inhibitors as adjunctive therapy for type 1 diabetes: balancing benefits and risks. The lancet. Diabetes & endocrinology 7, 949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger RH, and Orci L. (1975). The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet 1, 14–16. [DOI] [PubMed] [Google Scholar]
- Wu M, Li R, Hou Y, Song S, Han W, Chen N, Du Y, Ren Y, and Shi Y. (2018). Thioredoxin-interacting protein deficiency ameliorates kidney inflammation and fibrosis in mice with unilateral ureteral obstruction. Lab Invest. [DOI] [PubMed] [Google Scholar]
- Xiang G, Seki T, Schuster MD, Witkowski P, Boyle AJ, See F, Martens TP, Kocher A, Sondermeijer H, Krum H, et al. (2005). Catalytic degradation of vitamin D up-regulated protein 1 mRNA enhances cardiomyocyte survival and prevents left ventricular remodeling after myocardial ischemia. J Biol Chem 280, 39394–39402. [DOI] [PubMed] [Google Scholar]
- Xu G, Chen J, Jing G, and Shalev A. (2012). Preventing beta-cell loss and diabetes with calcium channel blockers. Diabetes 61, 848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Chen J, Jing G, and Shalev A. (2013). Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat Med 19, 1141–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Associated With Reduction of Newly Diagnosed Diabetes Mellitus. J Clin Endocrinol Metab Associated With Reduction of Newly Diagnosed Diabetes Mellitus. J Clin Endocrinol Metab 102, 2604–2610. [DOI] [PubMed] [Google Scholar]
- Yoshioka J, Chutkow WA, Lee S, Kim JB, Yan J, Tian R, Lindsey ML, Feener EP, Seidman CE, Seidman JG, et al. (2012). Deletion of thioredoxin-interacting protein in mice impairs mitochondrial function but protects the myocardium from ischemia-reperfusion injury. J Clin Invest 122, 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Tardivel A, Thorens B, Choi I, and Tschopp J. (2010). Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11, 136–140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing datasets generated during this study are available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE151588.