

Abstract
The mitochondrion performs critical roles in eukaryotic cells including ATP production, cell growth, survival, apoptosis, and differentiation. Many human diseases can be traced to dysfunction within the mitochondria, but selective delivery of therapeutics into the mitochondria has been challenging. This chapter describes the detailed protocols for the synthesis of a new family of mitochondrion-targeting, cell-penetrating molecules (CPMs) and their application for the delivery of small-molecule and peptidyl cargos into the mitochondrial matrix. Live-cell confocal microscopic imaging of HeLa cells treated with a variety of CPM-cargo conjugates revealed that the CPMs efficiently and specifically deliver membrane-impermeable linear and cyclic peptidyl cargos into the mitochondrial matrix, as long as the cargo carries no more than two negative charges.
Keywords: Cell-penetrating molecule, cyclic peptide, drug delivery, mitochondrion targeting, peptide
1. Introduction
Mitochondria play critical roles in many biochemical pathways including ATP production, heat production, apoptosis, calcium storage and signaling, cell cycle and cell differentiation (Fulda et al., 2010). Dysfunction of the mitochondria is associated with some 300 different human diseases including several rare genetic diseases, obesity, cancers, diabetes, Alzheimer’s and Parkinson’s diseases (Arruda et al., 2014; Castellani et al., 2002; Martin & McGee, 2014; Sarparanta et al., 2017; Vyas et al., 2016; Yan et al., 2013; Yang et al., 2016). The mitochondrial matrix is surrounded by two lipid bilayers, an outer membrane which is readily permeable to small molecules (due to the presence of membrane pores) and an inner membrane impermeable to most molecules (Mejia & Hatch, 2016). This renders the development of mitochondrion-specific drugs exceedingly difficult, because a drug molecule must traverse at least two lipid bilayers (the plasma membrane and the inner mitochondrial membrane) in order to gain access to a target inside the mitochondrial matrix. Several mitochondrion-targeting strategies have been reported in the literature. For membrane-permeable small molecules, covalent conjugation with lipophilic cations, such as triphenylphosphonium (TPP) ion, results in the accumulation of the conjugates inside the mitochondrial matrix, due to the presence of a negative membrane potential across the mitochondrial membrane (Murphy, 2008). However, this method is not applicable to cargo molecules that are inherently membrane impermeable, such as peptides and proteins. The latter must be genetically modified with an N-terminal mitochondrion-targeting sequence (MTS) and expressed inside eukaryotic cells, making it unsuitable for therapeutic applications. Previous researchers have reported short mitochondrion-penetrating peptides (MPPs) which, when covalently attached to small-molecule or peptide cargoes, can deliver the latter into the mitochondrial matrix with modest specificity and efficiency (Chuah et al., 2015; Horton et al., 2008; Jean et al., 2016). More recently, we discovered a family of non-peptidic cell-penetrating molecules (e.g., CPM1-3; Figure 1) which are capable of delivering both membrane-permeable small molecules and membrane-impermeable peptidyl cargoes into the mitochondrial matrix with high specificity and efficiency (Appiah Kubi et al., 2018). Finally, nanoparticle-based (Pathak et al., 2015) and liposomal-based mitochondrial delivery systems have been reported (Yamada & Harashima, 2012). In this chapter, we provide a detailed protocol for the preparation and application of CPM-cargo conjugates and evaluate the cargo capacity of the CPMs by using a set of d-peptides of different sequences.
Figure 1.
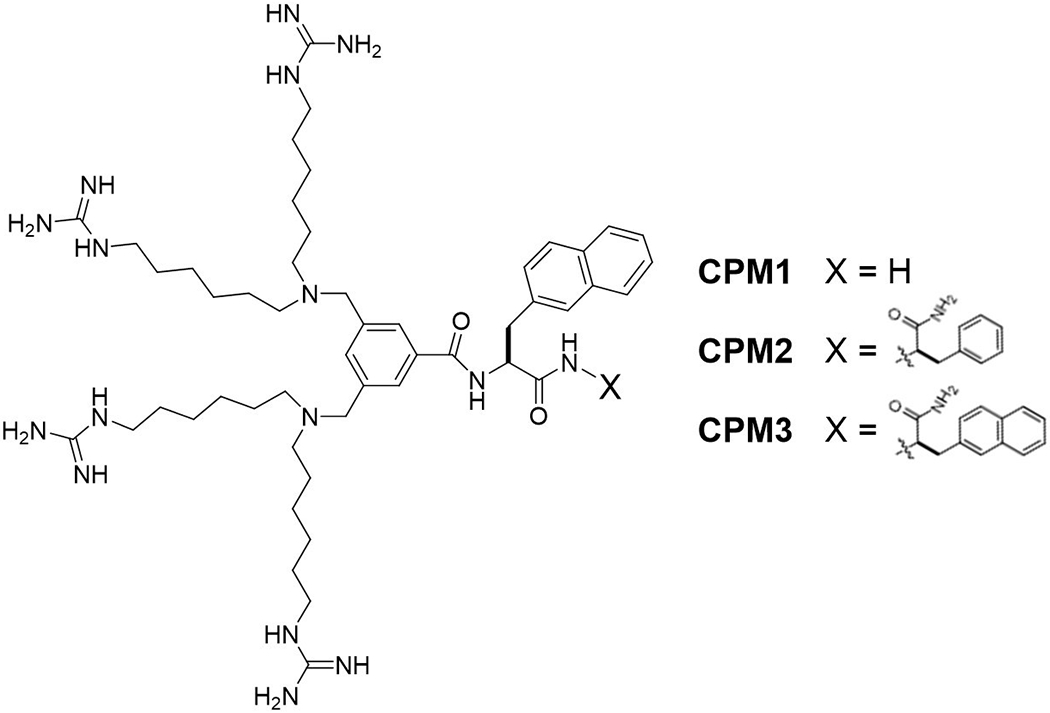
Structures of CPM1-3.
2. Synthesis of CPM-Cargo Conjugates
CPMs and CPM-cargo conjugates are most conveniently synthesized on solid phase. To minimize any mutual interference between the CPM and a cargo molecule, we recommend the use of one or more 8-amino-3,6-dioxaoctanoyl-lysine (miniPEG-K) moieties as a flexible linker between the CPM and the cargo (Figure 2). Figure 2 illustrates the synthesis of CPM3-miniPEG-K, starting with the coupling of Fmoc-miniPEG-Lys onto the solid support (e.g., Rink amide resin). This is followed by the addition of d-2-naphthylalanine (nal) and l-2-naphthylalanine (Nal), by using the standard Fmoc-based peptide chemistry. Next, 3,5-bis(bromomethyl)benzoic acid is coupled to the N-terminal amine by using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) as the coupling agent. Finally, the two resin-bound benzyl bromides are reacted with excess 1,1’-(azanediylbis(hexane-6,l-diyl))diguanidine (via SN2 reactions) to install the four guanidinium groups. Subsequent treatment with trifluoroacetic acid (TFA) removes the Boc group from the Fys side chain and releases CPM3-miniPEG-K from the resin.
Figure 2.
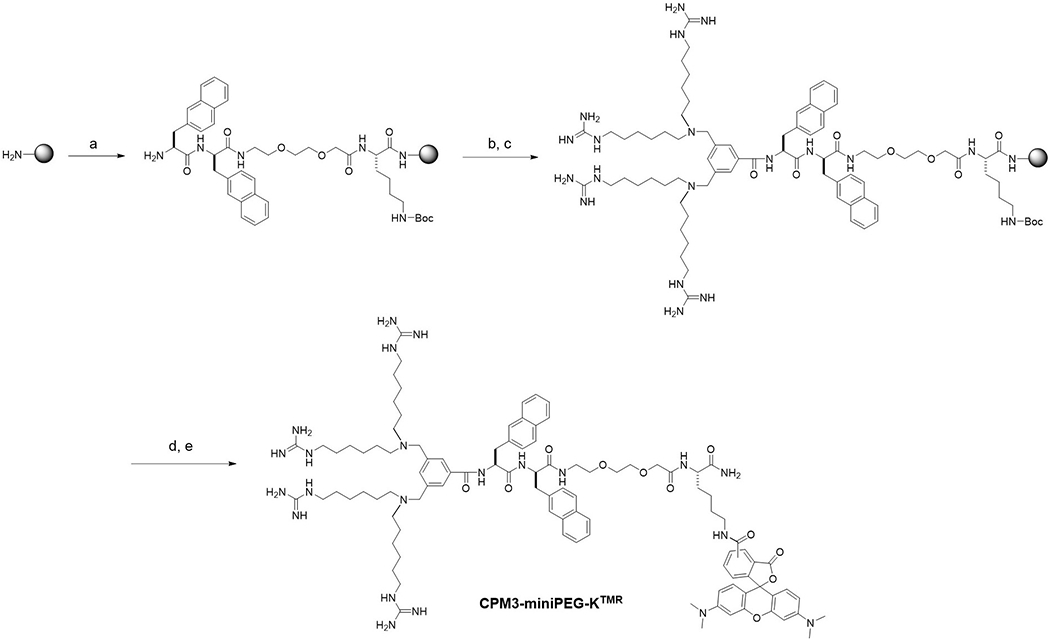
Solid-phase synthesis of CPM3-miniPEG-KTMR. Reagents and conditions: a) Solid-phase peptide synthesis by standard Fmoc/HATU chemistry; b) 3,5-bis(bromomethyl)benzoic acid, HATU; c) 1,1’-(azanediylbis(hexane-6,1-diyl))diguanidine in 2:1 (v/v) 0.5 M NaHCO3 (pH 9)/acetonitrile; d)TFA; and e)TMR-NHS.
CPM-cargo conjugates are similarly synthesized. When the cargo is a fluorescent dye molecule, it may be attached to the lysine side chain by simply treating CPM-miniPEG-K with an activated ester of the dye, e.g., 5(6)-carboxytetramethylrhodamine-N-hydroxysuccinimide (TMR-NHS). Small-molecule drugs may be conjugated to the lysine side chain in a similar manner. Alternatively, the lysine residue may be replaced with a cysteine, which is then conjugated to a cargo molecule via a disulfide or thioether bond (e.g., by reacting with a maleimide) (Appiah Kubi et al., 2018). When the cargo is a peptide (or cyclic peptide), the entire CPM-cargo conjugate is synthesized on solid phase. Typically, the peptidyl cargo is synthesized first and CPM-miniPEG-K is next added to the N-terminus of the peptidyl cargo (or a side chain of the cyclic peptide). Figure 3 shows the preparation of TMR-labeled CPM1-miniPEG-LB76 as an example. LB76 is a cyclopentapeptidyl inhibitor of heat shock protein 90 (HSP90) (Rahimi et al., 2019). The synthesis begins with coupling the sidechain carboxyl group of Fmoc-Asp-O-All (where -All is allyl) to Rink amide resin. After removal of the N-Fmoc group, Fmoc-Ser(tBu), Fmoc-Tyr(tBu), methyltrityl (Mtt)-protected Fys [Fmoc-Fys(Mtt)-OH], and Boc-protected lysine [Fmoc-Fys(Boc)-OH] are sequentially coupled to form the pentapeptide. The C-terminal allyl group is removed by treatment with Pd(PPh3)4 and the N-terminal Fmoc group is removed by treatment with piperidine. The pentapeptide is cyclized on resin by treatment with benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate (PyBOP). Next, the Mtt group is selectively removed from the first Fys side chain with 2% (v/v) TFA in DCM and the synthesis is continued onto the Fys side chain to construct the CPM1-miniPEG moiety, as described above. Treatment of the resin with TFA simultaneously releases the peptide off the solid support and removes the sidechain protecting groups. After purification by reversed-phase HPFC, the compound is labeled with TMR on the side chain of the second lysine residue by reacting with TMR-NHS. The purity of the peptides is usually checked by analytical HPFC on a C18 column (typically ≥95%) and the authenticity of the peptides is confirmed by high-resolution mass spectrometry (e.g., MAFDI-ICR).
Figure 3.
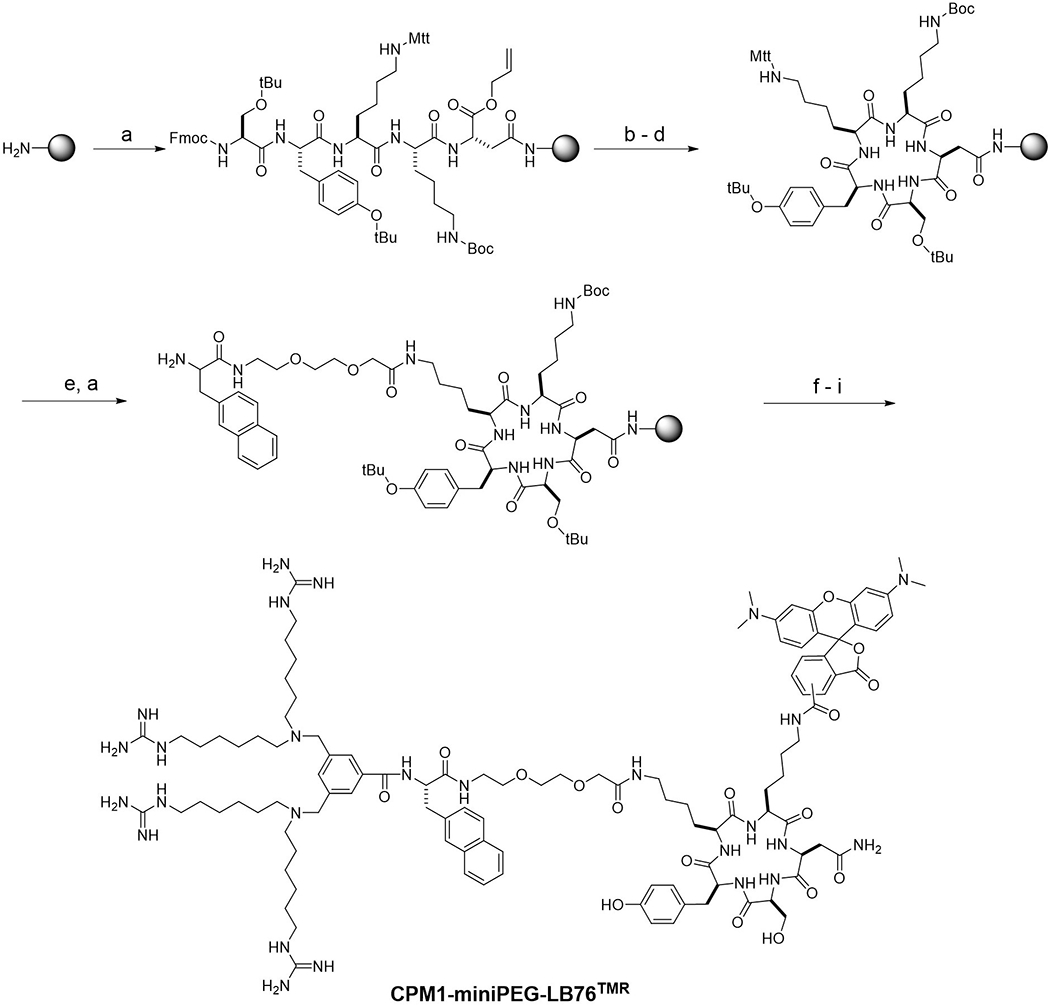
Synthesis of CPM1-miniPEG-LB76TMR. Reagents and conditions: a) Solid-phase peptide synthesis using Fmoc/HATU chemistry; b) Pd(PPh3)4; c) piperidine; d) PyBOP; e) 2% (v/v) TFA in DCM; f) 3,5-bis(bromomethyl)benzoic acid, HATU; g) 1,1’-(azanediylbis(hexane-6,1-diyl))diguanidine in 2:1 (v/v) 0.5 M NaHCO3 (pH 9)/acetonitrile; h) TFA; and i) TMR-NHS.
3. Mitochondrion-Specific Cargo Delivery by CPMs
The mechanism by which the CPMs enter mammalian cells has not yet been extensively investigated. Preliminary studies indicate that they enter the cells by endocytosis, followed by highly efficient escape from the endosome into the cytosol (Appiah Kubi et al., 2018). Five-cell confocal microscopic imaging of HeFa cells treated with 2 μM CPM3TMR showed that CPM3TMR was almost exclusively localized inside the mitochondria, with only a small fraction apparently remaining inside the endosomes/lysosomes (Figure 4). High-resolution confocal imaging further demonstrated that a CPM3-peptide conjugate was localized within the mitochondrial matrix (Appiah Kubi et al., 2018). Their apparent absence from the cytosol suggests that the CPMs specifically target the mitochondria and that, once escaping the endosome, they are rapidly internalized by the mitochondria.
Figure 4.
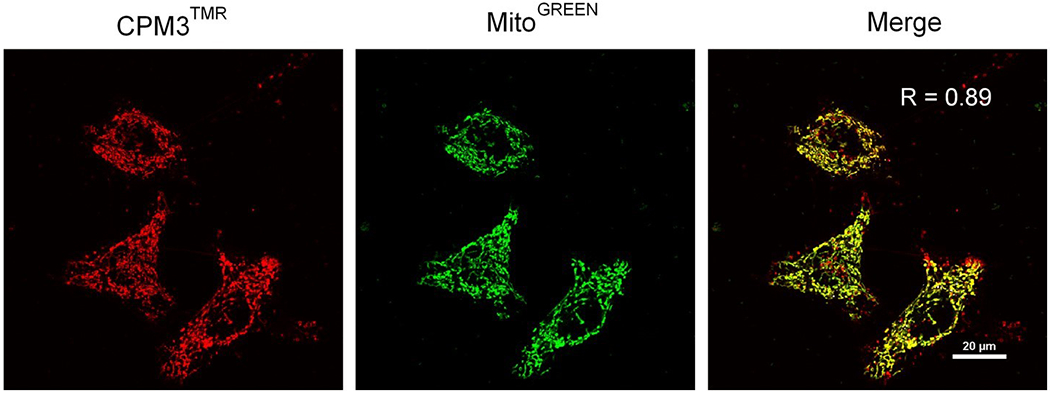
Live-cell confocal microscopic images of HeLa cells after treatment with 2 μM CPM3TMR for 2 h (red) and incubation with MitoTracker Green for 15 min (green). A merged image is shown on the right with the R value representing Pearson’s correlation coefficient for co-localization. This figure was reproduced from Appiah Kubi et al. (2018) with permission.
The CPMs have been evaluated for their capacity to deliver small-molecule and peptidyl cargoes. Conjugation of geldanamycin, an HSP90 inhibitor, to CPM3 selectively delivered the small-molecule drug into the mitochondrial matrix and greatly enhanced its anticancer activity (Appiah Kubi et al., 2018). To test whether the CPMs can deliver membrane-impermeable cargoes into the mitochondria, Appiah Kubi et al. (2018) conjugated CPM3 to the N-terminus of a d-peptide, ala-asn-ala-asn-ala-asn-miniPEG-Lys (ananan), which is more stable against proteolytic degradation than the corresponding L-peptide. High-resolution confocal imaging of He La cells treated with the peptide (CPM3-anananTMR) showed that the peptide cargo was specifically delivered into the mitochondrial matrix. Labeling of the same peptide with a pH-sensitive dye, naphthofluorescein (NF), allowed its cytosolic/mitochondrial entry efficiency to be quantitated by flow cytometry. It was found that CPM3-anananNF entered the cytosol/mitochondria of HeLa cells with remarkable efficiency, being 170- to 1300-fold more efficient than some of the most widely used mitochondria-targeting vehicles, such as (Fxr)3 (where Fx is cyclohexylalanine and r is d-arginine) and TPP.
In this work, we further evaluated the cargo capacity of CPM3 by conjugating it to a panel of d-peptides including dnnnnnK, dqdqdqK, dddddqK, and rararaK (where d is d-aspartic acid, n is d-asparagine, q is d-glutamine, r is d-arginine and a is d-alanine). The peptides were labeled with 5(6)-carboxyfluorescein-N-hydroxysuccinimide or TMR-NHS to give CPM3-dnnnnnKTMR, CPM3-dqdqdqKF1, CPM3-dddddqKF1 and CPM3-rararaKF1, which contain −1, −3, −5 and +3 charged cargoes, respectively, at physiological pH. HeLa cells were treated with the peptides and MitoTracker Green (or MitoTracker Red) and imaged by live-cell confocal microscopy. CPM3-dnnnnnKTMR produced intracellular fluorescence that overlapped well with MitoTracker Green with a Pearson’s correlation coefficient (R) of 0.83 (Figure 5a). Similarly, CPM3-rararaKF1, which carries a positively charged cargo (+3), effectively entered the mitochondria, although it exhibited significant cytotoxicity at concentrations above 3 μM (Figure 5b). We previously observed that CPM3 labeled with Alexa488, which carries a −2 charge at physiological pH, was also specifically localized inside the mitochondria of HeLa cells (Appiah Kubi et al., 2018). On the other hand, He La cells treated with CPM3-dqdqdqKF1 and CPM3-dddddqKF1 produced weak, punctate fluorescence signals which poorly overlapped with those of MitoTracker Red (Pearson’s correlation coefficient R = 0.19 and 0.16, respectively) (Figure 5c and 5d).
Figure 5.
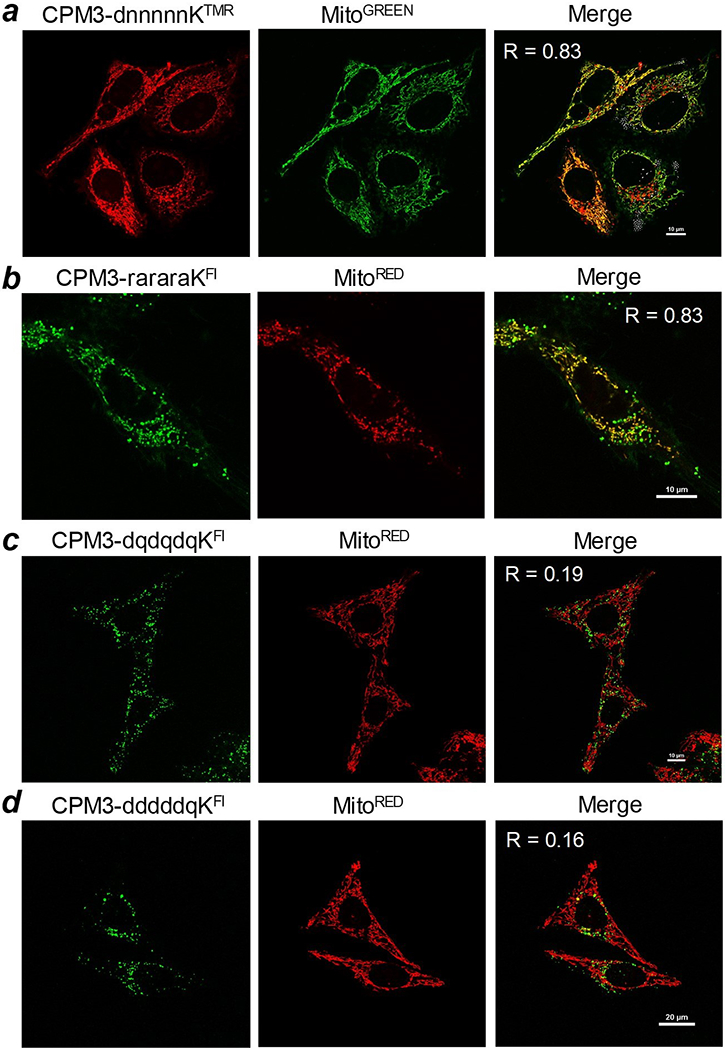
Live-cell confocal microscopic images of HeLa cells after treatment with various CPM3-linear peptide conjugates (2 μM) for 2 h and MitoTracker for 15 min. Merge, overlap of peptide and MitoTracker signals; R, Pearson’s correlation coefficient.
Finally, we tested whether CPMs can deliver cyclic peptides into the mitochondria. To this end, we conjugated CPM1 to cyclic pentapeptide LB76, an Hsp90 inhibitor (Rahimi et al., 2019) and labeled LB76 with TMR (Figure 3). Live-cell confocal microscopic imaging of HeLa cells treated with CPM1-miniPEG-LB76TMR showed efficient uptake and excellent co-localization with MitoTracker Green (Pearson’s correlation coefficient R = 0.83) (Figure 6a). To further examine the effect of negative charges on CPM delivery efficiency, we conjugated CPM3 to two fluorescein-labeled cyclic peptides, cyclo(Lys-asn-Asp-asp-Asp-asn-Gln)-LysFL and cyclo(Lys-asp-Asp-asp-Asp-asp-Gln)-LysFL (where asn is d-asparagine and asp is d-aspartic acid), through their lysine side chains. The resulting conjugates [CPM3-c(KnDdDnQ)-KF1 and CPM3-c(KdDdDdQ)-KF1] carry −3 and −5 charges, respectively, in their cargo moieties. HeLa cells treated with CPM3-c(KnDdDnQ)-KF1 exhibited weak, punctate fluorescence, which had little overlap with the MitoTracker Red signals (R = 0.09) (Figure 6b). Cells treated with CPM3-c(KdDdDdQ)-KF1 showed little intracellular fluorescence (Figure 6c). Taken together, these data demonstrate that the CPMs are capable of efficiently delivering both small-molecule and peptidyl (linear or cyclic) cargoes into the mitochondrial matrix. However, for most effective delivery, the cargo should carry no more than two negative charges. Highly negatively charged cargoes (with ≥3 negative charges) presumably interact with the positively charged CPMs (either intra- or intermolecularly) and inhibit their interaction with the plasma and/or endosomal membranes, reducing the cellular uptake and/or endosomal escape efficiencies. Consequently, the small amount of conjugates that enter the cell remain entrapped inside the endosomal/lysosomal pathway, producing punctate but non-overlapping signals with the mitochondria.
Figure 6.
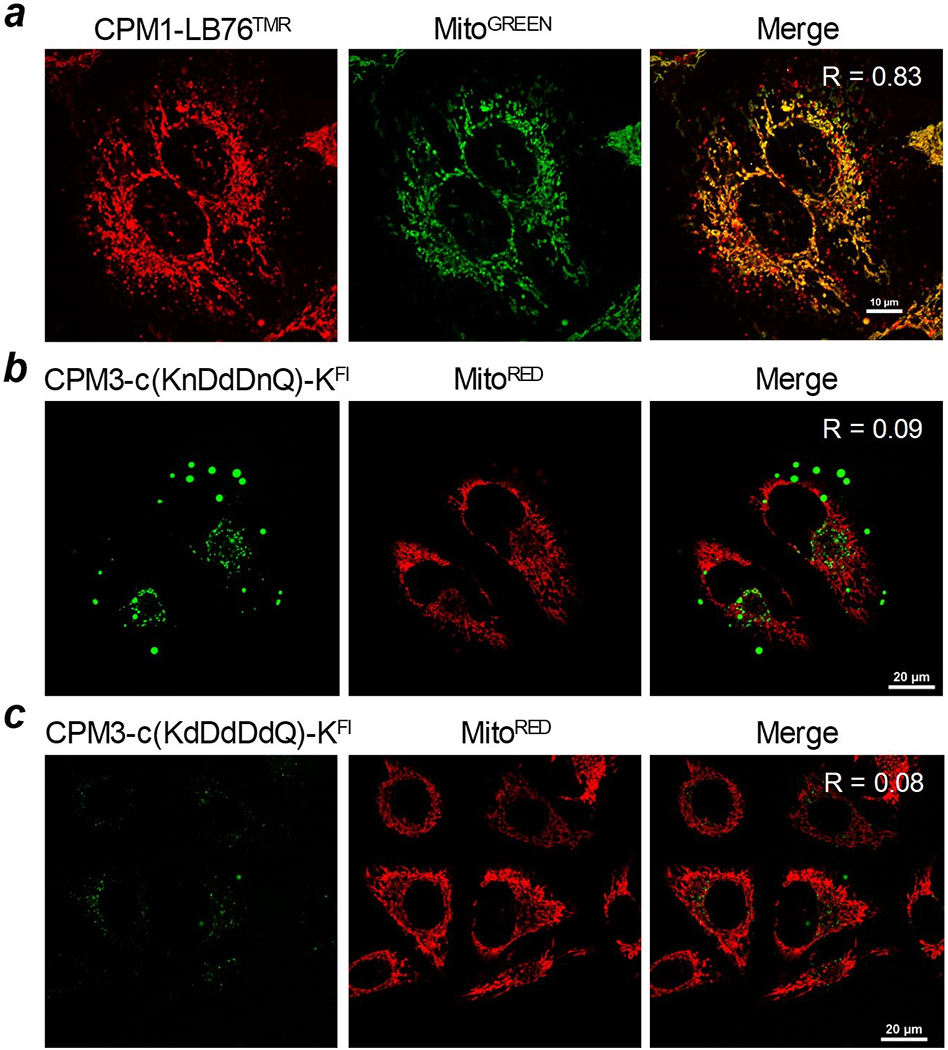
Live-cell confocal microscopic images of HeLa cells after treatment with 2 μM CPM-cyclic peptide conjugates for 2 h and MitoTracker for 15 min. Merge, overlap of peptide and MitoTracker signals; R, Pearson’s correlation coefficient.
4. Protocol
Synthesis of 1,1’-(azanediylbis(hexane-6,1-diyl))diguanidine and 3,5-bis(bromomethyl)benzoic acid has been described elsewhere (Appiah Kubi et al., 2018) and will not be detailed in this chapter. Below we outline the detailed protocols for the synthesis of CPM3 and CPM1-miniPEG-LB76. All experiments are performed under ambient temperature unless otherwise stated. This protocol may be used to synthesize other CPMs (e.g., CPM1 and CPM2) and their peptide conjugates. The protocol assumes that the user is knowledgeable about standard organic chemistry techniques and has access to basic organic laboratory equipment.
4.1. Materials and Reagents
TentaGel S NH2 resin (0.26 mmol/g)
Coupling reagents: 4 eq. 1 -[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU), 4 eq. N-hydroxybenzotriazole (HOBt), 8 eq. N,N-diisopropylethylamine (DIPEA) as solids or as prepared as stock solutions in N,N-dimethylformamide (DMF)
Fmoc deprotection reagent: 20% (v/v) piperidine in DMF
Solvents for synthesis: Dichloromethane (DCM) and DMF
Amino acids: 4 eq. of the desired Fmoc-protected amino acids dissolved in DMF (2 mF), to be used on the same day.
Cyclization reagent: 5 eq. (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP), 5 eq. HOBt and 10 eq. DIPEA, dissolved in 2 mF of DMF
Cleavage/deprotection cocktail: 2.5:2.5:2.5:92.5 (v/v) doubly distilled water (H2O)/triisopropyl silane (TIS)/1,3-dimethoxybenzene (DMB)/trifluoroacetic acid (TFA)
Methyltrityl (Mtt) deprotection reagent: 2:2:96 (v/v) TFA/TIS/DCM
Fluorescent dyes: 5(6)-carboxytetramethylrhodamine N-succinimidyl ester (TMR-NHS), 5(6)-carboxyfluorescein N-succinimidyl ester (F1-NHS), MitoTracker Red, MitoTracker Green
HPLC solvents: 0.05% (v/v) TFA in doubly distilled water and 0.05% (v/v) TFA in acetonitrile
4.2. Solid-phase synthesis of CPM3
-
Swell 100 mg of TentaGel S amine resin in 3 mL of DMF for 20 min.
Tip: TentaGel resin is recommended, but other hydrophilic resins (e.g., aminomethyl ChemMatrix resin) may also be used.
Drain the DMF solution and wash the resin twice with DCM and DMF (3 mL each).
-
Add 4 eq. of Rink amide linker, 4 eq. of HATU and 8 eq. of DIPEA and allow the contents to mix for 1 h.
Tip: First dissolve the Rink amide linker and HATU in 2 mL of DMF, add DIPEA to the solution, vortex and then add the mixture to the swollen resin. The uronium-based coupling reagents (e.g., HATU, HBTU, and HCTU) provide cost- and time-effective synthesis with minimal racemization. However, phosphonium- (e.g., PyBOP), or carbenium- (e.g., COMU) reagents are also effective.
Drain the mixture and wash the resin exhaustively with DMF, DCM and DMF (3 x 3 mL each).
-
Add 20% (v/v) piperidine in DMF (3 mL) and incubate for 10 min. Repeat this step once.
Tip: If Fmoc deprotection with 20% piperidine in DMF is incomplete (as revealed by MS), a mixture of 2:2:96 (v/v) 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)/piperidine/DMF is recommended. A 3 mL solution of 20% piperazine in DMF is also an acceptable alternative.
-
Drain the mixture and wash the resin exhaustively with DMF, DCM and DMF (3 × 3 mL each).
Tip: Make sure that any residual piperidine on the vessel cap is completely removed by inverting the capped vessel several times. Even trace amounts of piperidine may complicate the next coupling reaction.
-
Add 4 eq. of Fmoc-Lys(Boc)-OH, 4 eq. HATU and 8 eq. of DIPEA and allow the contents to mix for 1 h.
Tip: Refer to Tip under step 3.
Repeat steps 4 through 7 with different amino acids to obtain resin-bound peptide sequence Fmoc-Nal-nal-miniPEG-Lys.
Remove the N-terminal Fmoc group by following steps 5 and 6.
Add 4 eq. of 3,5-bis(bromomethyl) benzoic acid, 4 eq. of HATU, and 8 eq. of DIPEA and allow the contents to mix for 30 min.
Drain and wash the resin exhaustively with DMF, DCM and DMF (3 x 3 mL each).
Incubate the resin in 3 mL of 1:1 (v/v) water/DMF for 20 min.
Drain the water/DMF mixture from the resin and resuspend it in 3 mL of water. Incubate for 20 min.
Drain the water and add 10 eq. of 1,1’-(azanediylbis(hexane-6,1-diyl))diguanidine in 5 mL of 2:1 (v/v) mixture of 0.5 M NaHCCh (pH 9) and acetonitrile. Incubate the contents at 50 °C for 14 h with mixing.
Drain the solution and wash the resin exhaustively with water, DMF and DCM (5 x 3 mL each). Make sure that the final washing step is carried out with DCM.
Add 10 mL of 2.5:2.5:2.5:92.5 (V/v) H2O/TIS/DMB/TFA and incubate for 2 h to deprotect the lysine side chain and release CPM3 from the solid support.
-
Drain the released compound solution into a 15-mL Falcon tube and evaporate the solvents by gently blowing an inert gas (nitrogen or argon) over the liquid until a semi-solid is obtained.
Tip: Safety precautions should be taken to prevent TFA burns. This step must be performed inside a well-ventilated fume hood.
Gently add 10 mL of chilled diethyl ether to precipitate the product.
Centrifuge at 7,000 rpm for 5 min. Carefully remove the diethyl ether with a glass pipette, while leaving the precipitated crude product behind.
Repeat steps 18 and 19 twice.
Dissolve the crude product (3-5 mg) in a minimal volume of DMF (30 μL). Dilute the solution with 400 μL of 3:1 (v/v) water/acetonitrile and purify the crude product by reversed-phase HPLC by using a semi-preparative C-18 column.
Lyophilize the purified CPM3-miniPEG-Lys solution to obtain a white powder.
4.3. Fluorescent labeling of CPM3
Weigh ~2 mg (1.4 μmol) of HPLC purified CPM3-miniPEG-Lys in a microcentrifuge tube.
Add 20 μL of DMF to dissolve the compound. Adjust the pH to ~8 by adding ~15 μL of 0.5 M NaHCCO3 buffer.
Weigh 1.2 eq. of TMR-NHS into another microcentrifuge tube and dissolve it in 20 μL of DMF.
-
Mix the two solutions, wrap the tube with aluminum foil, and allow the reaction to proceed with mixing for 2 h.
Tip: If precipitate forms, add a small amount of DMF to solubilize the precipitate.
Purify the fluorescently labeled CPM3-miniPEG-KTMR by reversed-phase HPLC using a semi-preparative C18 column.
Lyophilize the purified CPM3-miniPEG-KTMR to obtain a red solid.
4.4. Solid-phase synthesis of CPM3-peptide cargo conjugates
The protocol described below was employed for the preparation of CPM1-miniPEG-LB76. Other CPM3-peptide cargo conjugates were similarly synthesized.
Swell 100 mg of TentaGel S amine resin in 3 mL of DMF for 20 min.
Drain the solvent and wash the resin with DCM and DMF (2 x 3 mL each).
Dissolve 4 eq. of Rink amide linker and 4 eq. of HATU in 2 mL of DMF. Add 8 eq. of DIPEA and vortex to dissolve all the contents. Add the reagent solution to the swollen resin and incubate for 1 h with constant mixing.
Drain the mixture and wash the resin exhaustively with DMF, DCM and DMF (3 x 3 mL each).
Add 20% (v/v) piperidine in DMF (3 mL) and incubate for 10 min. Repeat this step once.
Drain the mixture and wash the resin exhaustively with DMF, DCM and DMF (3 x 3 mL each).
Dissolve 4 eq. of Fmoc-Asp-O-All and 4 eq. of HATU in 2 mL of DMF. Add 8 eq. of DIPEA and vortex to dissolve all the contents. Add the solution to the resin and incubate for 1 h with constant mixing.
Repeat steps 4 through 7 for the other four amino acids to obtain the resin-bound peptide sequence Fmoc-Ser(tBu)-Tyr(tBu)-Lys(Mtt)-Lys(Boc)-Asp-O-All.
Remove the allyl protecting group on the C-terminal Asp residue by treating the resin with 0.3 eq. of tetrakis(triphenylphosphine)palladium and 10 eq. of phenylsilane in 2 mL of dry DCM for 15 min. Wash the resin with 3 mL of dry DCM after each incubation. Repeat this step twice.
Drain and wash resin with DMF, DCM, and DMF (3 x 3 mL each).
Incubate the resin in 10% (wt/v) sodium dimethyldithiocarbamate in 3 mL of DMF for 10 min. Repeat this step once.
Drain and wash resin with DMF, DCM and DMF (3 x 3 mL each).
Remove the N-terminal Fmoc group by treating with 3 mL of 20% piperidine in DMF for 10 min. Repeat this step once.
Drain and wash resin with DMF, DCM and DMF (3 x 3 mL each).
Add 3 mL of 1 M HOBt in DMF to the resin and incubate for 10 min. Repeat this step once.
Drain the HOBt solution. Add 5 eq. of PyBOP, 5 eq. of HOBt, and 10 eq. of DIPEA in 2 mL of DMF to the resin and incubate for 2 h with mixing. Repeat this step once.
Drain and wash the resin exhaustively with DMF and DCM (4 x 3 mL each)
Remove the Mtt group by treating the resin with 3 mL of 2:2:96 (v/v) TFA/TIPS/DCM for 5 min. Wash the resin with dry DCM (3 mL). Repeat this step five times.
Incubate the resin in 3 mL of 5% (v/v) DIPEA in DMF for 10 min.
Dissolve 4 eq. of Fmoc-miniPEG-OH and 4 eq. of HATU in 2 mL of DMF. Add 8 eq. of DIPEA and vortex to dissolve all the contents. Add the solution to the resin and incubate for 1 h with mixing.
Repeat steps 4 through 6.
Dissolve 4 eq. of Fmoc-L-2-Nal-OH and 4 eq. of HATU in 2 mL of DMF. Add 8 eq. of DIPEA and vortex to dissolve all the contents. Add the solution to the resin and incubate for 1 h with mixing.
Repeat steps 4 through 6.
Dissolve 4 eq. of 3,5-bis(bromomethyl)benzoic acid and 4 eq. HATU in 2 mL of DMF. Add 8 eq. of DIPEA and vortex to dissolve the contents. Add the solution to the resin and incubate for 30 min with mixing.
Drain and wash the resin exhaustively with DMF, DCM and DMF (3 x 3 mL each).
Incubate the resin in 3 mL of 1:1 (v/v) water/DMF mixture for 20 min.
Drain the water/DMF mixture and incubate the resin in water (3 mL) for 20 min.
Drain the water and add 10 eq. of 1,1’-(azanediylbis(hexane-6,1-diyl))diguanidine dissolved in 5 mL of 2:1 (v/v) 0.5 M NaHCO3 (pH 9)/acetonitrile to the resin. Incubate at 50 °C for 14 h with mixing.
Drain and wash the resin exhaustively with H2O, DMF and DCM (5 x 3 mL each). Make sure that the final washing step is carried out with DCM.
Add 10 mL of 2.5:2.5:2.5:92.5 (v/v) H2O/TIS/DMB/TFA and incubate for 2 h with mixing.
Carefully drain the resin and collect the solution into a clean 15-mL Falcon tube. Evaporate the solvents by gently blowing an inert gas (nitrogen or argon) over the liquid until a semi-solid is obtained.
Gently add 10 mL of chilled diethyl ether to precipitate the product.
Centrifuge at 7,000 rpm for 5 min. Carefully remove the diethyl ether with a glass pipette, while leaving the precipitated crude product behind.
Repeat steps 31 and 32 twice.
Dissolve the crude product (3-5 mg) in a minimal volume of DMF (~30 μL). Dilute the solution with ~400 μL of 3:1 (v/v) water/acetonitrile and purify the crude product by reversed-phase HPLC by using a semi-preparative C-18 column on a linear gradient of 10 - 50% acetonitrile over 40 min.
Lyophilize the purified CPM1-miniPEG-LB76 to obtain a white solid.
4.5. Confocal microscopic imaging
Culture HeLa cells by following the supplier’s instructions. Other cell lines may be used.
Seed 5 x 104 cells/mL in a 35-mm glass-bottomed microwell (MatTek; 500 μL) and culture for 24 h.
Gently wash the cells twice with Dulbecco’s phosphate buffer saline (DPBS).
-
Add 500 μL of 2 μM CPM3-miniPEG-KTMR in Dulbecco’s modified eagle medium (DMEM) to the cells and incubate at 37 °C for 1 h 45 min in the presence of 5% CO2.
Tip: Use DMEM (phenol red free, equipped with 1% fetal bovine serum (FBS) and 1% streptomycin/penicillin) to prepare 500 μL of 2 μM CPM3-miniPEG-KTMR and add to the cells.
Prepare a 200 nM MitoTracker green solution in DMEM (phenol red free, equipped with 1% FBS and 1% streptomycin/penicillin) and add 500 μL to the cells to give a final volume of 1000 μL.
Incubate the cells at 37 °C for 15 min in the presence of 5 % CO2.
Gently wash the cells with DPBS twice. Add 500 μL of fresh DMEM (equipped with 1% FBS and 1% penicillin/streptomycin).
Image the cells using live-cell confocal microscope equipped with 100X oil objective.
5. Summary
To the best of our knowledge, the CPMs described in this chapter represent the most powerful mitochondrion-targeting vehicles that have been reported to date. They are effective for delivering small-molecule and peptidyl cargoes that are positively charged, neutral, or if negatively charged, contain no more than two negative charges. For these cargoes, delivery is both highly efficient and mitochondrion-specific. Synthesis of the CPMs and CPM-cargo conjugates is straightforward and most conveniently performed on the solid phase, by following the detailed protocols provided in this chapter. Their capacity for delivering other types of cargoes, e.g., proteins and nucleic acids, as well as their mechanism of cellular/mitochondrial entry remain to be determined.
Acknowledgment
This work was supported by a grant from the NIH (GM122459).
References
- Appiah Kubi G, Qian Z, Amiar S, Sahni A, Stahelin RV, & Pei D (2018). Non-Peptidic Cell-Penetrating Motifs for Mitochondrion-Specific Cargo Delivery. Angew Chem Int Ed Engl 57(52), 17183–17188. doi: 10.1002/anie.201811940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, & Hotamisligil GS (2014). Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nature Medicine, 20, 1427. doi: 10.1038/nm.3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani R, Hirai K, Aliev G, Drew KL, Nunomura A, Takeda A, … M. A. Smith (2002). Role of mitochondrial dysfunction in Alzheimer’s disease. Journal of Neuroscience Research, 70(3), 357–360. doi: 10.1002/jnr.10389 [DOI] [PubMed] [Google Scholar]
- Chuah JA, Yoshizumi T, Kodama Y, & Numata K (2015). Gene introduction into the mitochondria of Arabidopsis thaliana via peptide-based carriers. Sci Rep, 5, 7751. doi: 10.1038/srep07751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S, Galluzzi L, & Kroemer G (2010). Targeting mitochondria for cancer therapy. Nat Rev Drug Discov, 9(6), 447–464. doi: 10.1038/nrd3137 [DOI] [PubMed] [Google Scholar]
- Horton KL, Stewart KM, Fonseca SB, Guo Q, & Kelley SO (2008). Mitochondria-penetrating peptides. Chem Biol, 75(4), 375–382. doi: 10.1016/j.chembiol.2008.03.015 [DOI] [PubMed] [Google Scholar]
- Jean SR, Ahmed M, Fei EK, Wisnovsky SP, & Kelley SO (2016). Peptide-Mediated Delivery of Chemical Probes and Therapeutics to Mitochondria. Acc Chem Res, 49(9), 1893–1902. doi: 10.1021/acs.accounts.6b00277 [DOI] [PubMed] [Google Scholar]
- Martin SD, & McGee SF (2014). The role of mitochondria in the aetiology of insulin resistance and type 2 diabetes. Biochimica et Biophvsica Acta (BBA) - General Subjects, 1840(4), 1303–1312. doi: 10.1016/j.bbagen.2013.09.019 [DOI] [PubMed] [Google Scholar]
- Mejia EM, & Hatch GM (2016). Mitochondrial phospholipids: role in mitochondrial function. Journal of Bioenergetics and Biomembranes, 48(2), 99–112. doi: 10.1007/s10863-015-9601-4 [DOI] [PubMed] [Google Scholar]
- Murphy MP (2008). Targeting lipophilic cations to mitochondria. Biochimica et Biophvsica Acta (BBA) - Bioenergetics, 1777(1), 1028–1031. doi: 10.1016/j.bbabio.2008.03.029 [DOI] [PubMed] [Google Scholar]
- Pathak RK, Kolishetti N, & Dhar S (2015). Targeted nanoparticles in mitochondrial medicine. Wiley Interdiscip Rev Nanomed Nanobiotechnol, 7(3), 315–329. doi: 10.1002/wnan.1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi MN, Foster HG, Farazi SN, Chapman R, & McAlpine SR (2019). Polymer mediated transport of the Hsp90 inhibitor FB76, a polar cyclic peptide, produces an Hsp90 cellular phenotype. Chem Commun (Camb), 55(31), 4515–4518. doi: 10.1039/c9cc00890j [DOI] [PubMed] [Google Scholar]
- Sarparanta J, Garcia-Macia M, & Singh R (2017). Autophagy and Mitochondria in Obesity and Type 2 Diabetes. Current Diabetes Reviews, 13(4), 352–369. doi: 10.2174/1573399812666160217122530 [DOI] [PubMed] [Google Scholar]
- Vyas S, Zaganjor E, & Haigis MC (2016). Mitochondria and Cancer. Cell, 166(3), 555–566. doi: 10.1016/j.cell.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, & Harashima H (2012). Delivery of bioactive molecules to the mitochondrial genome using a membrane-fusing, liposome-based carrier, DF-MITO-Porter. Biomaterials, 33(5), 1589–1595. doi: 10.1016/j.biomaterials.2011.10.082 [DOI] [PubMed] [Google Scholar]
- Yan MH, Wang X, & Zhu X (2013). Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radical Biology and Medicine, 62, 90–101. doi: 10.1016/i.freeradbiomed.2012.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Karakhanova S, Hartwig W, D’Haese JG, Philippov PP, Werner J, & Bazhin AV (2016). Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. Journal of Cellular Physiology, 231(12), 2570–2581. doi: 10.1002/jcp.25349 [DOI] [PubMed] [Google Scholar]