

Abstract
Three paramagnetic Co(II) macrocyclic complexes containing 2-hydroxypropyl pendant groups, 1,1’,1”,1”’-(1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetrayl)tetrakis(propan-2-ol) ([Co(L1)]2+, 1,1’-(4,11-dibenzyl-1,4,8,11-tetraazacyclotetradecane-1,8-diyl)bis(propan-2-ol) ([Co(L2)]2+(, and 1,1’-(4,11-dibenzyl-1,4,8,11-tetraazacyclotetradecane-1,8-diyl)bis(octadecan-2-ol) ([Co(L3)]2+( were synthesized to prepare transition metal liposomal chemical exchange saturation transfer (lipoCEST) agents. In solution, )[Co(L1)]2+( forms two isomers as shown by 1H NMR spectroscopy. X-ray crystallographic studies show one isomer with 1,8-pendants in cis-configuration and a second isomer with 1,4-pendants in trans-configuration. The [Co(L2)]2+ complex has 1,8-pendants in a cis-configuration. Remarkably, the paramagnetic induced shift of water 1H NMR resonances in the presence of the [Co(L1)]2+ complex is as large as that observed for one of the most effective Ln(III) water proton shift agents. Incorporation of [Co(L1)]2+ into the liposome aqueous core, followed by dialysis against a solution of 300 mOsm/L produces a CEST peak at 3.5 ppm. Incorporation of the amphiphilic [Co(L3)]2+ complex into the liposome bilayer produces a more highly shifted CEST peak at −13 ppm. Taken together, these data demonstrate the feasibility of preparing Co(II) lipoCEST agents.
Keywords: ParaCEST, Water Shift Agents, LipoCEST, amphiphilic shift reagent
Graphical Abstract

Paramagnetic Co(II) complexes produce water 1H shifts that rival those of lanthanide(III) complexes. Incorporation of these complexes into the interior and bilayer of shrunken liposomes gives highly shifted intraliposomal water protons for chemical exchange saturation transfer (CEST) experiments as the first examples of transition metal ion lipoCEST agents.
Introduction
Paramagnetic chemical exchange saturation transfer (CEST) agents or paraCEST agents are a class of compounds that are under development as MRI contrast agents.[1] CEST and paraCEST agents contain protons, generally as OH or NH groups, that exchange with bulk water protons. Saturation of these exchangeable protons with a selective radiofrequency pulse reduces the water proton signal to produce contrast.[1a] While most examples of paraCEST agents are paramagnetic lanthanide complexes, paramagnetic transition metal complexes have also shown promise as paraCEST agents.[2] Transition metal paraCEST agents are uniquely suited for development as responsive MRI probes that switch oxidation state or spin state according to biological environment.[3] However, the low sensitivity of the agents must be addressed in order to realize the full potential of paraCEST agents in MRI. Strategies to increase paraCEST agent signal have long been pursued.[4]
The incorporation of paramagnetic MRI probes into supramolecular complexes or nanoparticles can improve their sensitivity by several orders of magnitude.[4b, 5] Liposomal CEST approaches, or “lipoCEST” agents, have shown CEST signal at nanomolar concentrations of probe.[4a] LipoCEST agents have a pool of intraliposomal water molecules with proton resonances that are shifted by the paramagnetic complex. An increase in sensitivity is produced by loading high concentrations of the shift reagent into a small intraliposomal volume.[6] Typically, the paramagnetic complex contains a trivalent lanthanide ion (Ln(III)) with a rapidly exchanging bound water that produces a large shift of the proximal water proton resonances.[7] The shift of the intraliposomal water protons may be further increased by osmotically shrinking the liposome into an oblong shape and by incorporating additional Ln(III) complex into the lipid bilayer by attachment of a long alkyl chain.[13, 17]
High spin Co(II) and Fe(II) macrocyclic complexes are alternatives to Ln(III) complexes for the production of highly paramagnetically shifted water protons.[2b–g] Interestingly, while paramagnetic complexes with inner-sphere water ligands produce strongly shifted water protons, so do complexes that interact with water through second-sphere interactions.[2a–c] Here we report three new Co(II) complexes that produce highly shifted water proton resonances. The large shift is attributed to an unusual geometry at Co(II) and to the hydroxyalkyl groups. Two Co(II) complexes presented here shift water protons to nearly the same extent as the best lanthanide shift agents, and when incorporated into an osmotically-shrunken liposome, produce a lipoCEST effect.
Results and Discussion
Co(II) complexes with 2-hydroxypropyl pendants were prepared from cyclam (1,4,7,11-tetrazacyclododecane) and also from 1,8-bis(benzyl) cyclam and S-propylene oxide (Scheme 1). Solution studies were consistent with high spin Co(II) complexes as shown by magnetic moments of 4.98 ±0.07 and 4.84±0.12, for [Co(L1)]2+ and [Co(L2)]2+, respectively at 25 °C, characteristic of S = 3/2 Co(II) complexes. The 1H NMR resonances of the Co(II) complexes range from −100 to +340 ppm (Figure S1). [Co(L2)]2+ has 14 strongly paramagnetically shifted proton resonances assigned to macrocycle backbone and methylenes of benzyl and hydroxypropyl pendants, consistent with apparent C2 symmetry of the complex with both hydroxy pendants bound. In contrast, there are approximately 34 proton resonances for [Co(L1)]2+. The unequal intensity of the resonances suggests that there is more than one isomer in solution. Comparison with the 1H NMR spectrum of [Co(L2)]2+ suggests that one set of resonances is produced by a 1,8-isomer, as the major species, and the other set of resonances is produced by a smaller amount of a different isomer of lower symmetry. Heating of the [Co(L1)]2+ complex to 37 °C or 60 °C, followed by cooling does not change the percentage of the isomers as gauged by 1H NMR spectroscopy, suggesting equilibrium is attained. [Co(L1)]2+ is inert towards loss of Co(II) at pH 7.12, 37 °C for 18 hours in the presence of carbonate and phosphate, but dissociates appreciably in acid at pD 3.4 (Figures S8–S10, Table S3). The greater degree of dissociation between 1 hour and 18 hours is consistent with a kinetic effect. However 1H NMR studies and pH potentiometric studies show that Co(L1) reforms as the pH is returned to neutral, consistent with thermodynamic stability under these conditions (S10). A third Co(II) complex [Co(L3)]2+ which is amphiphilic was prepared and characterized by NMR spectroscopy. The 1H NMR spectrum of [Co(L3)]2+ resembled that of [Co(L2)]2+ , consistent with the formation of a complex with similar geometry (Figure S2). This hydrophobic complex was prepared for the purpose of making liposome type B which has [Co(L3)]2+ incorporated into the liposome bilayer with core loaded [Co(L1)]2+.
Scheme 1.

X-ray crystallographic analysis indicated two isomers of the [Co(L1)]2+ cation including a complex with 1,4-pendants separated by an ethylene group bound in trans-configuration and a complex containing coordinating 1,8-pendants in a cis-configuration (Figure 1 and Figure S26). The complex cation of the bis-benzyl substituted complex [Co(L2)]2+ has the hydroxypropyl pendants coordinated in a cis-configuration. The cis-complex of [Co(L1)]2+ and [Co(L2)]2+ have the Co(II) ion displaced by 0.705(6) Å and 0.751(18) Å, respectively, from the centroid of the plane formed by the cyclam nitrogens, whereas the trans-isomer has the Co(II) nearly in the plane of the cyclam macrocycle. The 1,4-[Co(L1)]2+ isomer has the macrocycle in the common trans-III form (S,S,R,R at nitrogen). This form is the most stable for divalent transition metal ion complexes.[8] Previous examples of Co(II) cyclam complexes with four pendant amides show the 1,8-isomer of [Co(L4)]2+ in the trans-III form but the 1,4-isomer in the trans-II form.[9] Both the [Co(L2)]2+ complex cation and the 1,8-[Co(L1)]2+ complex cation have the rare folded cis-I configuration with all nitrogen pendant groups oriented up.[10] This configuration has been observed for a Cd(II) complex of cyclam which has a bidentate carbonate,[8] but has not been observed for a divalent metal ion cyclam complex with coordinating pendant groups, to the best of our knowledge. Crystallographic data is given in Table S6. Bond lengths, bond angles and additional description of structures for all three complexes are given in Tables S7–S12 and Figures S26–S36.
Figure 1.
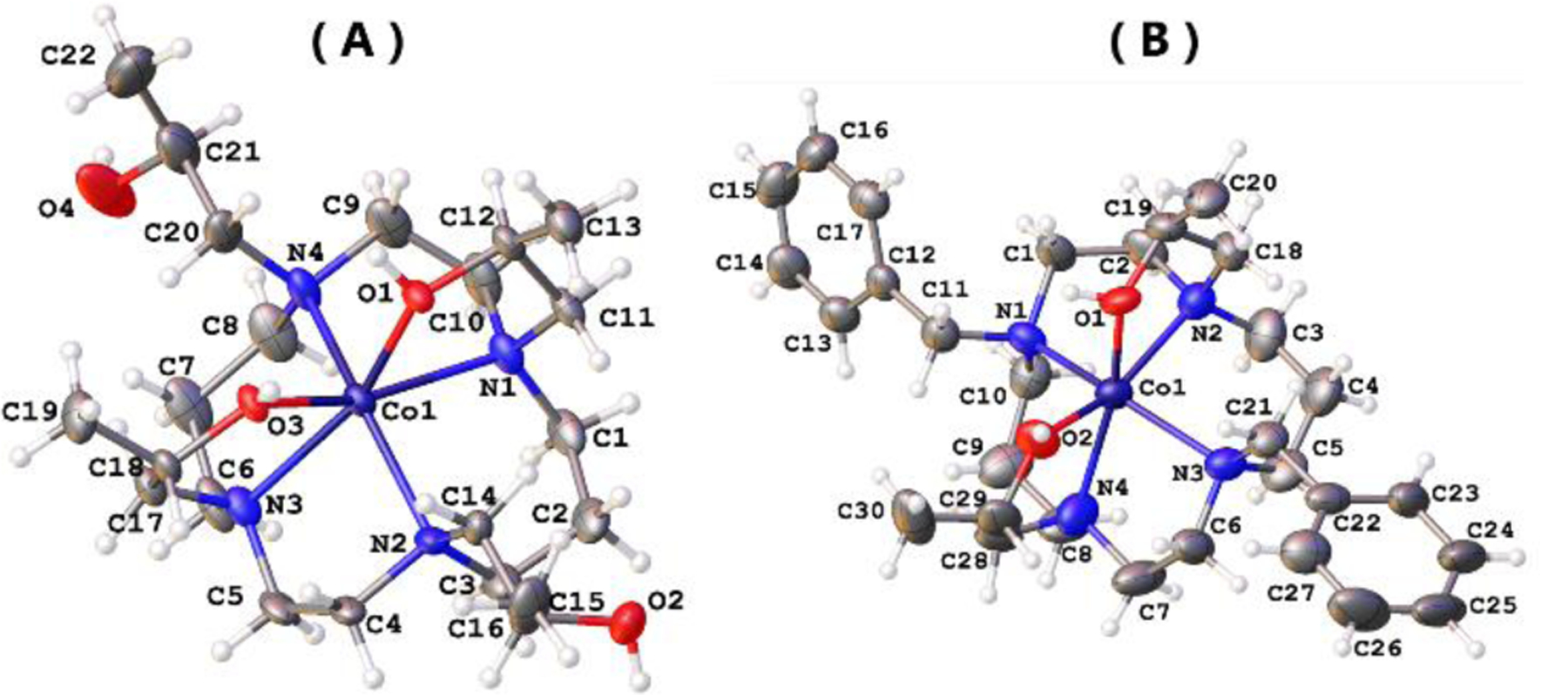
Molecular structures of cis-1,8-[Co(L1)]2+ (A, only one of two nearly identical cations shown) and [Co(L2)]2+ (B) cations with thermal ellipsoids drawn at 50% probability level.
The solution 1H NMR data for [Co(L1)]2+ and [Co(L2)]2+, as described above, in conjunction with the structural data support the predominance of the cis-I form in solution. The minor form of [Co(L1)]2+ is most likely the 1,4-isomer in trans-III form based on comparison of proton shifts to that of the [Co(L4)]2+ isomers.[9b] The z-spectra of the two complexes also support this analysis by showing weak CEST peaks characteristic of OH exchangeable protons bound to Co(II).[2a, 2b] [Co(L2)]2+ shows a single CEST peak at 33 ppm versus bulk water, consistent with a single isomer. [Co(L1)]2+ produces two CEST peaks consistent with the two isomers, one at 33 and one at 58 ppm. The pH dependence of the CEST effect increases with pH up to pH 7.2 with exchange rate constants ranging from 800 to 3900 s−1, characteristic of ligand donor groups that have exchanging OH protons.[2a, 2b] The low intensity of CEST peaks suggest that these complexes would not be useful as paraCEST agents. However Co(II) complexes with hydroxypropyl pendants are excellent water proton shift reagents (SR).[2b] Encapsulation of such SRs in the interior of a liposome magnifies the number of exchangeable protons and produces a more effective CEST agent.
Both [Co(L1)]2+ and [Co(L2)]2+ complexes are effective water 1H resonance SRs. Plots of the water 1H shift as a function of concentration of the complexes in solution show that the slopes are close to that of [Tm(DOTA)]− (Figure 2). In comparison, [Co(L4)]2+, which contains amide pendants, does not have such a large effect on the water proton shift. To elucidate the basis for these paramagnetic induced chemical shift changes, variable temperature 17O NMR spectroscopy was used to study the nature of the water interactions with the Co(II) complexes. As shown in Figure S11, a broad 17O resonance is observed in the presence of 50 mM [Co(L1)]2+ complex which shifts downfield on raising the temperature to 60 °C and then sharpens. In comparison, the [Co(L4)]2+ complex shows only a slight upfield shift of the 17O NMR resonance upon heating over the same temperature range. The behaviour of [Co(L1)]2+ and [Co(L2)]2+ is characteristic of complexes with an exchangeable inner-sphere water molecule, whereas that of [Co(L4)]2+ is characteristic of a complex with no innersphere waters. A Swift-Connick plot of the reduced shift (q = 1) and linewidth for [Co(L1)]2+ and [Co(L2)]2+ gives kex of 4.47 × 106 s−1 and 3.54 × 106 s−1, respectively, for an inner-sphere water, similar to rate constants reported previously for Co(II) macrocyclic complexes (Figure S12 and Table S3).[2a–2c]
Figure 2.
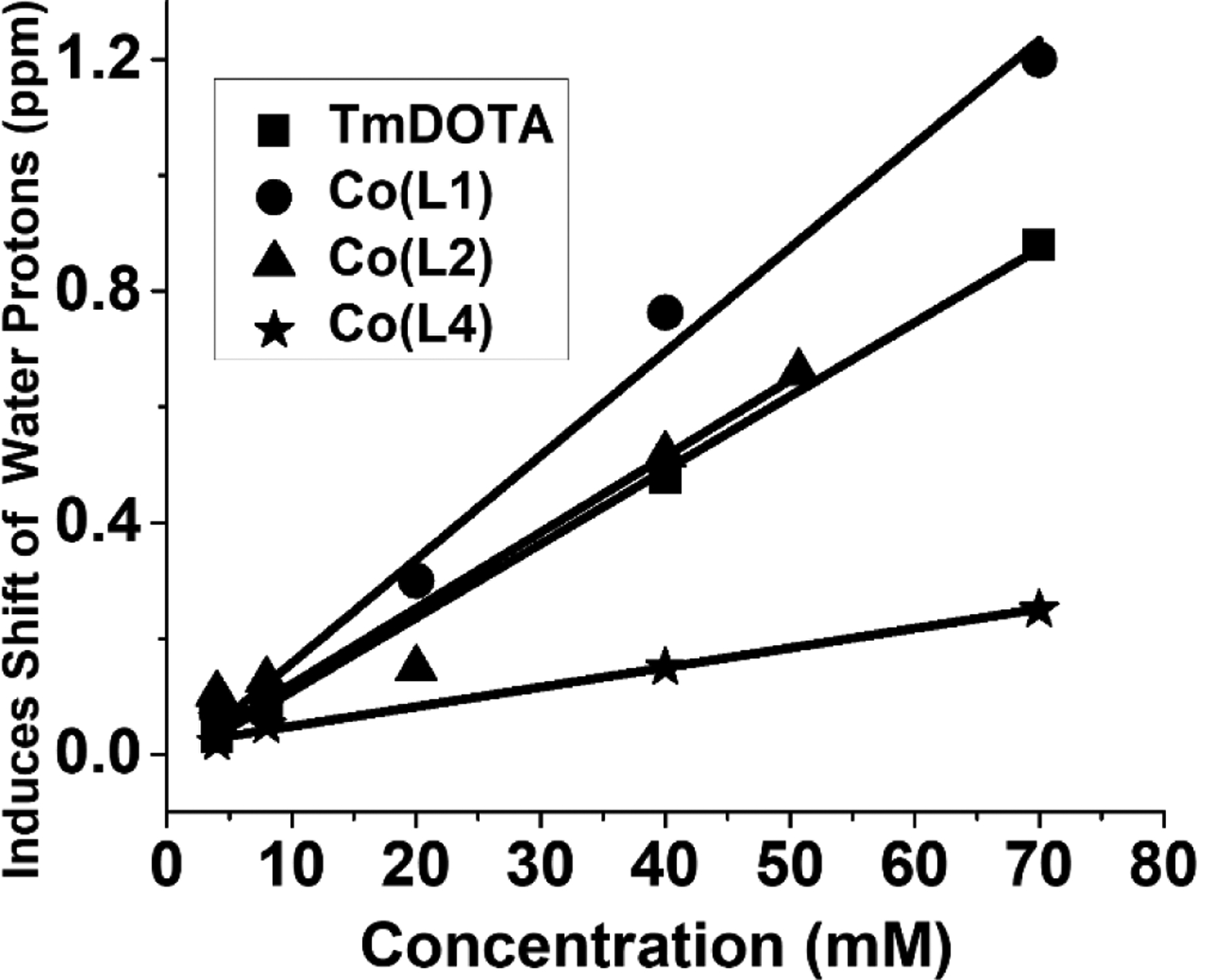
Plot of the 1H NMR water shift for Co(II) complexes (non-liposomal system) at 25 °C. Slopes of lines are 0.0128, 0.0179, 0.0131 and 0.003 for [Tm(DOTA)]−, [Co(L1)]2+, [Co(L2)]2+ and [Co(L4)]2+, respectively. The shift was corrected for bulk magnetic susceptibility effects.
Plots of the shift of the 17O resonance as a function of complex are shown in Figure S13 for [Co(L1)]2+, [Co(L2)]2+, [Co(L4)]2+ and Co(NO3)2, as a classic method to determine the number of bound water molecules. Two pH values, 7.0 and 7.5 were studied in order to probe the effect of deprotonation of the hydroxypropyl pendants at near neutral pH (7.6 for [Co(L1)]2+ and 7.4 for [Co(L2)]2+; Table S5, Figures S14, S15). The slope decreases for both complexes at pH 7.5 compared to 7.0, consistent with a decrease in q number at the higher pH. If we assume that Co(II)aq has six inner-sphere waters, then q is one for [Co(L1)]2+ at pH 7.5, a pH close to the first pKa, but is higher (q = 2) at pH 7, at 37 °C. Similarly, studies with [Co(L2)]2+ gave a q value of 1.8 at pH 7. However, estimation of inner-sphere waters by 17O NMR spectroscopy is complicated by second-sphere contributions and by the position of the water ligand with respect to the effective magnetic axes.[11] Nonetheless, both the variable temperature 17O NMR studies and the plots of the shift of the 17O resonance as a function of Co(II) complex concentration are most consistent with an inner-sphere water for [Co(L1)]2+ and [Co(L2)]2+ which may result from formation of a seven-coordinate Co(II) complex, or by displacement of a hydroxypropyl pendant. Thus, we attribute the large proton water shift produced by [Co(L1)]2+ and [Co(L2)]2+ to the unusual cis-I geometry and to the large second sphere effects associated with hydroxylalkyl pendants.[2a, 2b, 2m]
LipoA was formed with [Co(L1)]2+ loaded into the liposomal cavity using an ethanol injection method, followed by extrusion through polycarbonate membranes.[12] Osmotically-shrunken liposomes were then prepared by dialysing the liposome suspension against different concentrations of NaCl and HEPES buffer. Intraliposomal water chemical shift increases upon changing the shape of the liposome from spherical to oblong by increasing osmotic stress.[13] We chose small-sized liposomes to increase surface area to volume ratio which increases the exchange rate between the intraliposomal water and bulk water to produce an enhanced CEST effect.[14] Sizes less than 200 nm are also preferable for in vivo applications to increase the circulation time.[14–15] Dynamic light scattering measurements were consistent with a diameter of 125 nm for [Co(L1)]2+-based lipoA (Figure S16)[16] with a low polydispersity index, 0.064, indicating a narrow size distribution. LipoA contained 4 mM [Co(L1)]2+ as determined by ICP-MS and NMR spectroscopy (Evans method). In LipoA, the intraliposomal chemical shift is a sum of two contributions: the hyperfine interaction between the intraliposomal water protons and the paramagnetic center, and bulk magnetic susceptibility effects which are an important for aspherical liposomes (Eq.1).[13b]
| Eq.1 |
Z-spectra, plotted as the percent decrease in water proton magnetization as a function of presaturation pulse frequency, were taken to study the CEST effect (Fig. 3). Several different conditions with varying osmotic pressure were studied to produce shrunken liposomes. Shrinking the liposome increases the concentration of the SR inside the liposome and also increases the bulk magnetic susceptibility.[13b, 17] Moreover, the CEST peak position increases upon shrinking the liposome from 3.5 ppm at 300 mOsm/L to 10 ppm at 1200 mOsm/L and 18 ppm at 2000 mOsm/L. This CEST peak disappears upon disrupting the liposomes with addition of 1% TX100 detergent (Figure S21), confirming that the Co(II) complex is in the interior of the liposome. LipoA containing [Co(L2)]2+ (6 mM) as an alternative to [Co(L1)]2+ showed a similar asymmetry in its Z-spectrum (Figure S23).
Figure 3.

Z-spectra obtained from the liposome type A encapsulating [Co(L1)]2+ with an osmotic pressure of 300 mOsm/L to 2000 mOsm/L. Experiments were run at 25 °C, pH 6.50, 11 μT and 2 second irradiation time.
In order to increase the intraliposomal chemical shift without using an osmolality larger than that found in blood (300 mOsm/L), liposome type B (lipoB) with an amphphilic SR [Co(L3)]2+ and core loaded with [Co(L1)]2+ was prepared. Notably in lipoB, two factors contribute to the intraliposomal chemical shift; the hyperfine interaction of intraliposomal water protons with the paramagnetic encapsulated SR, and the hyperfine interaction of intraliposomal water protons with the incorporated SR pointing into the liposome cavity, (Eq.2). These two hyperfine contributions may produce proton shifts in different directions as shown for Ln(III) reagents.[17] The bulk magnetic suceptibility is directly proportional to the total concentration of Co(II) complex loaded inside the cavity or incorporated into the internal layer of the liposome and pointing inward. As a result, the incorporation of [Co(L3)]2+ complex in the liposome bilayer increases both the bulk magnetic suceptibility contribution (BMS) and the hyperfine shift contribution.
| Eq.2 |
The incorporation of paramagnetic amphiphilic complex in the lipid bilayer along with core loaded [Co(L1)]2+ (12 mM total Co) changes the intraliposomal chemical shift from 3.5 ppm observed for lipoA at 300 mOsm/L to −13 ppm at 300 mOsm/L (Figure 4). CEST experiments of LipoB were run at two different temperatures (25 °C and 37 °C) and little change in the CEST peak intensity was observed (Figure S25). The change in the sign of the CEST peak for lipoB suggests a change in orientation of the liposome with respect to the static magnetic field.[13] The alignment of shrunken paramagnetic liposomes in a magnetic field is influenced by the sign of the magnetic susceptibility anisotropy of the paramagnetic metal complex, which is modulated by the electronic configuration and geometry of the metal complex, and the mode of insertion of the complex into the lipid bilayer.[4a, 13, 17] For example, the magnetic field alignment of liposomes filled with Ln(III) complexes changes from parallel to perpendicular upon incorporation of an amphiphilic Ln(III) complex, to reverse the direction of the intraliposomal chemical shift.[13] Thus, the incorporation of [Co(L3)]2+ into the liposomal membrane not only increased the CEST peak shift, but also changed the sign which suggests that the major population of shrunken liposomes is oriented with the longer axis perpendicular to the external magnetic field.[4a, 17]
Figure 4.
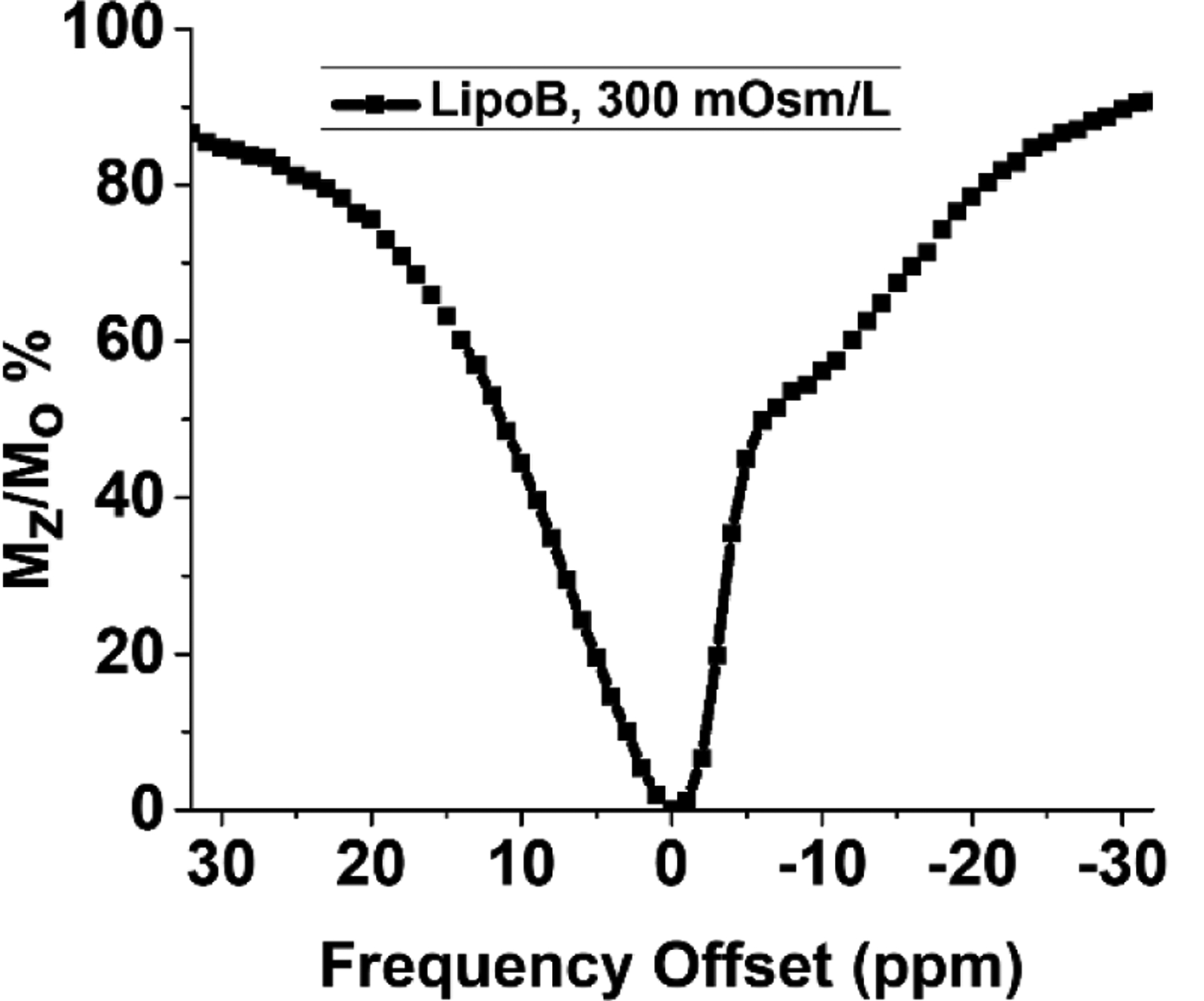
Z-spectra of liposome type B (lipoB containing surface conjugated [Co(L3)]2+ & core loaded with [Co(L1))]2+ and dialyzed against osmotic pressure of 300 mOsm/L, Experiment was run at 17 μT, pH 6.5 with 2 second irradiation.
The Co(II) lipoCEST agents described here compare favorably to those based on lanthanides. For comparison, shrunken liposomes with 40 mM encapsulated Tm(III) complex produce shifts of up to 10 ppm, and incorporation of amphiphilic Tm(III) complexes into the bilayer produces shifts as large as −31 ppm.[18] A DPPC liposome containing a Tm(III)[19] amphiphilic SR and core loaded 20 mM (Dy)-1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA)-Na produced a CEST peak at 4 to 6 ppm at 300 mOsm/L and a CEST peak at 15 ppm at 1200 mOsm/L.[19]
Conclusion
The Co(II) containing liposomes presented here are the first examples of transition metal-based lipoCEST agents, to the best of our knowledge. Shrunken liposomes with bilayer incorporated amphiphilic and core loaded complexes are especially promising as they demonstrate that Co(II) complexes, despite having a lower effective magnetic moment than Dy(III) or Tm(III) complexes, show a pronounced lipoCEST signal. Our studies suggest ways to improve Co(II)-based lipoCEST agents including development of Co(II) complexes of increased kinetic inertness by modifying the macrocycle framework.[2b, 2c, 9] The preparation of complexes with an overall neutral charge and with multiple Co(II) centers will further improve the liposome loading efficiency in order to produce increased CEST peak shifts.18 Moreover, new applications may be realized with transition metal ion liposome based agents, such as capitalizing on changes in oxidation and spin state for the development of biochemically responsive agents.[3, 20] In the long term, paramagnetic transition metal liposomes that generate a combination of T1, T2 or CEST MR contrast would be beneficial for in vivo applications.[21]
Supplementary Material
Acknowledgements
JRM acknowledges the NSF (CHE-1710224 and CHE-1310374) for support. This project used ICP-MS and FTMS instruments purchased with funding from NSF CHE-0959565 and National Institutes of Health (S10 RR029517). We thank Alejandra P. Lopez for assistance in fitting potentiometric data
References
- [1].a) Hancu I, Dixon WT, Woods M, Vinogradov E, Sherry AD, Lenkinski RE, Acta radiologica, 2010, 51, 910–923; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Longo D, Sun P, Consolino L, Michelotti F, Uggeri F, Aime S, J. Am. Chem. Soc, 2014, 136; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) M Ward K, Aletras A, Balaban R, J Magn Reson 2000, 143, 79–87; [DOI] [PubMed] [Google Scholar]; d) Sherry AD, Woods M, Annu Rev Biomed Eng, 2008, 10, 391–411; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Vinogradov E, Sherry AD, Lenkinski RE, J. Magn. Reson, 2013, 229, 155–172; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Woods M, Woessner DE, Sherry AD, Chem. Soc. Rev, 2006, 35, 500–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Abozeid SM, Snyder EM, Lopez AP, Steuerwald CM, Sylvester E, Ibrahim KM, Zaky RR, Abou-El-Nadar HM, Morrow JR, Eur. J. Inorg, 2018, 2018, 1902–1908; [Google Scholar]; b) Abozeid SM, Snyder EM, Tittiris TY, Steuerwald CM, Nazarenko AY, Morrow JR, Inorg. Chem, 2018, 57, 2085–2095; [DOI] [PubMed] [Google Scholar]; c) Bond CJ, Sokolow GE, Crawley MR, Burns PJ, Cox JM, Mayilmurugan R, Morrow JR, Inorg. Chem, 2019, 58, 8710–8719; [DOI] [PubMed] [Google Scholar]; d) Burns PJ, Cox JM, Morrow JR, Inorg. Chem, 2017, 56, 4545–4554; [DOI] [PubMed] [Google Scholar]; e) Olatunde AO, Bond CJ, Dorazio SJ, Cox JM, Benedict JB, Daddario MD, Spernyak JA, Morrow JR, Chem. Eur. J,2015, 21, 18290–18300; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Tsitovich PB, Gendron F, Nazarenko AY, Livesay BN, Lopez AP, Shores MP, Autschbach J, Morrow JR, Inorg. Chem, 2018, 57, 8364–8374; [DOI] [PubMed] [Google Scholar]; g) Tsitovich PB, Tittiris TY, Cox JM, Benedict JB, Morrow JR, Dalton Trans, 2018, 47, 916–924; [DOI] [PubMed] [Google Scholar]; h) Dorazio SJ, Olatunde AO, Tsitovich PB, Morrow JR, J. Biol. Inorg. Chem, 2014, 19, 191–205; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Du K, Waters EA, Harris TD, Chem. Sci, 2017, 8, 4424–4430; [DOI] [PMC free article] [PubMed] [Google Scholar]; j Du K, Harris TD, J. Am. Chem. Soc, 2016, 138, 7804–7807; [DOI] [PubMed] [Google Scholar]; k) Srivastava K, Ferrauto G, Young VG, Aime S, Pierre VC, Inorg. Chem, 2017, 56, 12206–12213; [DOI] [PubMed] [Google Scholar]; l) Thorarinsdottir AE, Harris TD, Chem. Commun, 2019, 55, 794–797. [DOI] [PubMed] [Google Scholar]; m) Botta M, Eur. J. Inorg. Chem, 2000, 2000, 399–407. [Google Scholar]
- [3].a) Jeon IR, Park JG, Haney CR, Harris TD, Chem. Sci, 2014, 5, 2461–2465; [Google Scholar]; b) Du K, Waters EA, Harris TD, Chem. Sci, 2017, 8, 4424–4430; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tsitovich PB, Spernyak JA, Morrow JR, Angew. Chem. Int. Ed, 2013, 52, 13997–14000. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Thorarinsdottir AE, Gaudette AI and Harris TD Chem. Sci, 2017, 8, 2448–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Ferrauto G, Delli Castelli D, Di Gregorio E, Terreno E, Aime S, Angew. Chem. Int. Ed, 2016, 8, 602–618; [DOI] [PubMed] [Google Scholar]; b) Wu Y, Zhou Y, Ouari O, Woods M, Zhao P, Soesbe TC, Kiefer GE, Sherry AD, J. Am. Chem. Soc, 2008, 130, 13854–13855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Aime S, Delli Castelli D, Terreno E, Angew. Chem. Int. Ed, 2003, 42, 4527–4529; [DOI] [PubMed] [Google Scholar]; b) Ferrauto G, Di Gregorio E, Ruzza M, Catanzaro V, Padovan S, Aime S, Angew. Chem. Int. Ed, 2017, 56, 12170–12173; [DOI] [PubMed] [Google Scholar]; c) Aime S, Delli Castelli D, Terreno E, Angew. Chem. Int. Ed, 2005, 44, 5513–5515. [DOI] [PubMed] [Google Scholar]
- [6].Terreno E, Delli Castelli D, Viale A, Aime S, Chem. Rev, 2010, 110, 3019–3042. [DOI] [PubMed] [Google Scholar]
- [7].a) Chahid B, Vander Elst L, Flament J, Boumezbeur F, Medina C, Port M, Muller RN, Lesieur S, Contrast Media Mol. Imaging, 2014, 9, 391–399; [DOI] [PubMed] [Google Scholar]; b) Zhang S, Merritt M, Woessner DE, Lenkinski RE, Sherry AD, Acc. Chem. Res, 2003, 36, 783–790. [DOI] [PubMed] [Google Scholar]
- [8].Liang X, Parkinson JA, Parsons S, Weishäupl M, Sadler PJ, Inorg. Chem, 2002, 41, 4539–4547. [DOI] [PubMed] [Google Scholar]
- [9].a) Freeman GM, Barefield EK, Van Derveer DG, Inorg. Chem, 1984, 23, 3092–3103; [Google Scholar]; b) Bond CJ, Cineus R, Nazarenko AY, Spernyak JA, Morrow JR, Dalton Trans, 2020, 49, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bosnich B, Poon CK, Tobe ML, Inorg. Chem, 1965, 4, 1102–1108. [Google Scholar]
- [11].a) Helm L, Morrow JR, Bond CJ, Carniato F, Botta M, Braun M, Baranyai Z, Pujales-Paradela R, Regueiro-Figueroa M, Esteban-Gómez D, Platas-Iglesias C, Scholl TJ, in Contrast Agents for MRI: Experimental Methods, The Royal Society of Chemistry, 2018, pp. 121–242; [Google Scholar]; b) Ferrauto G, Aime S, McMahon MT, Morrow JR, Snyder EM, Li A, Bartha R, in Contrast Agents for MRI: Experimental Methods, The Royal Society of Chemistry, 2018, pp. 243–317. [Google Scholar]
- [12].Opina ACL, Ghaghada KB, Zhao P, Kiefer G, Annapragada A, Sherry AD, PLOS ONE, 2011, 6, e27370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Terreno E, Cabella C, Carrera C, delli castelli D, Mazzon R, Rollet S, Stancanello J, Visigalli M, Aime S, Angew. Chem. Int. Ed, , 2007, 46, 966–968; [DOI] [PubMed] [Google Scholar]; b) Terreno E, Delli Castelli D, Violante E, Sanders HM, Sommerdijk NA, Aime S, Chem Eur. J, 2009, 15, 1440–1448. [DOI] [PubMed] [Google Scholar]
- [14].Zhao JM, Har-el Y.-e., McMahon MT, Zhou J, Sherry AD, Sgouros G, Bulte JWM, van Zijl PCM, J. Am. Chem. Soc, 2008, 130, 5178–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Litzinger DC, Buiting AMJ, van Rooijen N, Huang L, BBA-BIOMEMBRANES, 1994, 1190, 99–107. [DOI] [PubMed] [Google Scholar]
- [16].Mozafari M, Danaei M, Dehghankhold M, Ataei S, Hasanzadeh Davarani F, Javanmard R, Dokhani A, Khorasany S, Pharmaceutics, 2018, 10, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Aime S, Castelli DD, Terreno E, in Methods in enzymology, Academic Press, 2009, 464, 193–210. [DOI] [PubMed] [Google Scholar]
- [18].Terreno E, Barge A, Beltrami L, Cravotto G, Castelli DD, Fedeli F, Jebasingh B, Aime S, Chem. Commun, 2008, 600–602. [DOI] [PubMed] [Google Scholar]
- [19].Maruyama S, Ueda J, Kimura A, Murase K, Magn. Reson. Med. Sci, 2016, 15, 324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20.Wang H, Jordan VC, Ramsay IA, Sojoodi M, Fuchs BC, Tanabe KK, Caravan P, Gale EM.
- [21].Castelli DD, Dastru W, Terreno E, Cittadino E, Mainini F, Torres E, Spadaro M, Aime S, J. Control Release 2010, 114, 271–279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.