

Abstract
O-GlcNAcylation is a posttranslational modification involving the addition of the single monosaccharide N-acetylglucosamine (GlcNAc) onto serine and threonine residues of intracellular proteins. Though O-GlcNAc is found on ~1,000 proteins in mammals, its specific function on individual substrates remains largely a mystery. To overcome this shortcoming, work has been put toward developing metabolic chemical reporters (MCRs) to label O-GlcNAcylated proteins for subsequent biochemical analysis. Typically, these MCRs are GlcNAc or GalNAc analogs functionalized with azide or alkyne handles. These unnatural sugar moieties can be metabolically incorporated directly on to protein substrates. The protocols outlined in this article describe how to use MCRs as tools for visualizing and identifying potentially O-GlcNAc modified proteins via in-gel fluorescence, Western blotting, and mass spectrometry. Taken together, MCR labeling provides a powerful tool to discover where and when substrates are O-GlcNAc modified.
INTRODUCTION
This article describes a general procedure for metabolic labeling and analysis of O-GlcNAcylated proteins. This metabolic labeling approach involves the use of metabolic chemical reporters (MCRs) (Chuh and Pratt, 2015; Chuh et al., 2016). O-GlcNAc MCRs consist of a modified monosaccharide bearing azide or alkyne functionalized handles. Typically, these sugar analogues can be added to normal growth media where they passively diffuse through cell membranes. A successful MCR should be similar enough to GlcNAc that it can be biosynthesized into UDP-GlcNAc and directly transferred on to endogenous protein substrates. The bioorthogonal azide or alkyne handle can then be reacted with detection tags like fluorophores or affinity tags to analyze metabolic labeling and find specific O-GlcNAc modified substrates. Several different MCRs have been developed for the investigation of O-GlcNAcylated proteins (Figure 1) (Vocadlo et al., 2003; Zaro et al., 2011; 2017; Chuh et al., 2014; 2017; Li et al., 2016; Hang et al., 2003; Boyce et al., 2011; Hao et al., 2019). Basic Protocol 1 describes a basic procedure for incorporation of metabolic chemical reporters on to live cells, cell harvesting techniques, and CuAAC click chemistry. Basic Protocol 2 describes visualization of MCR-treated cell lysate through fluorophore tagging and in-gel fluorescence. Basic Protocol 3 describes a basic biotin enrichment protocol, followed by Western blotting. Basic Protocol 4 describes the biotin-mediated enrichment of MCR-labeled proteins coupled to on-bead trypsinolysis and identification of modified proteins using mass spectrometry. Together these methods provide a robust and user-friendly approach to the identification of potentially O-GlcNAc modified proteins (Figure 2).
Figure 1.
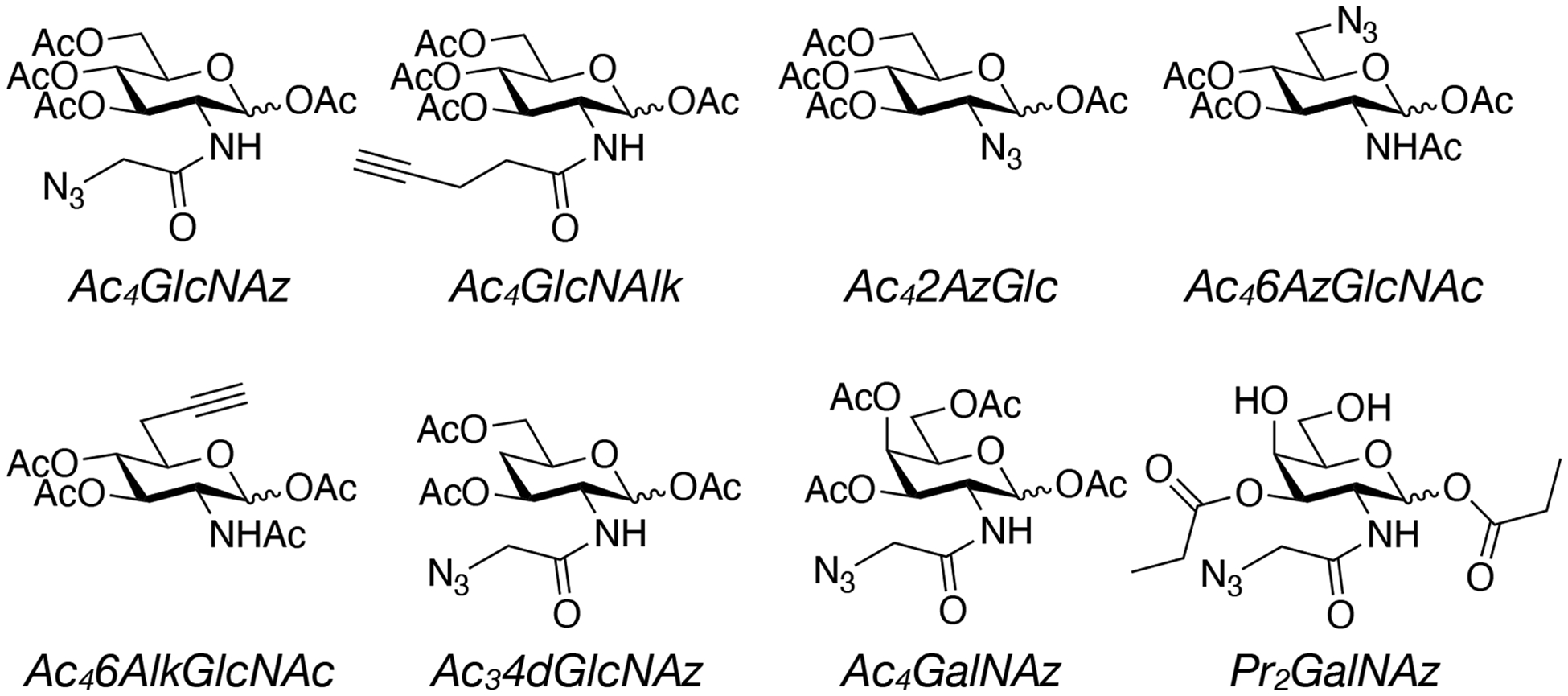
Chemical structures of different MCRs used to identify and characterize O-GlcNAc modified proteins.
Figure 2.
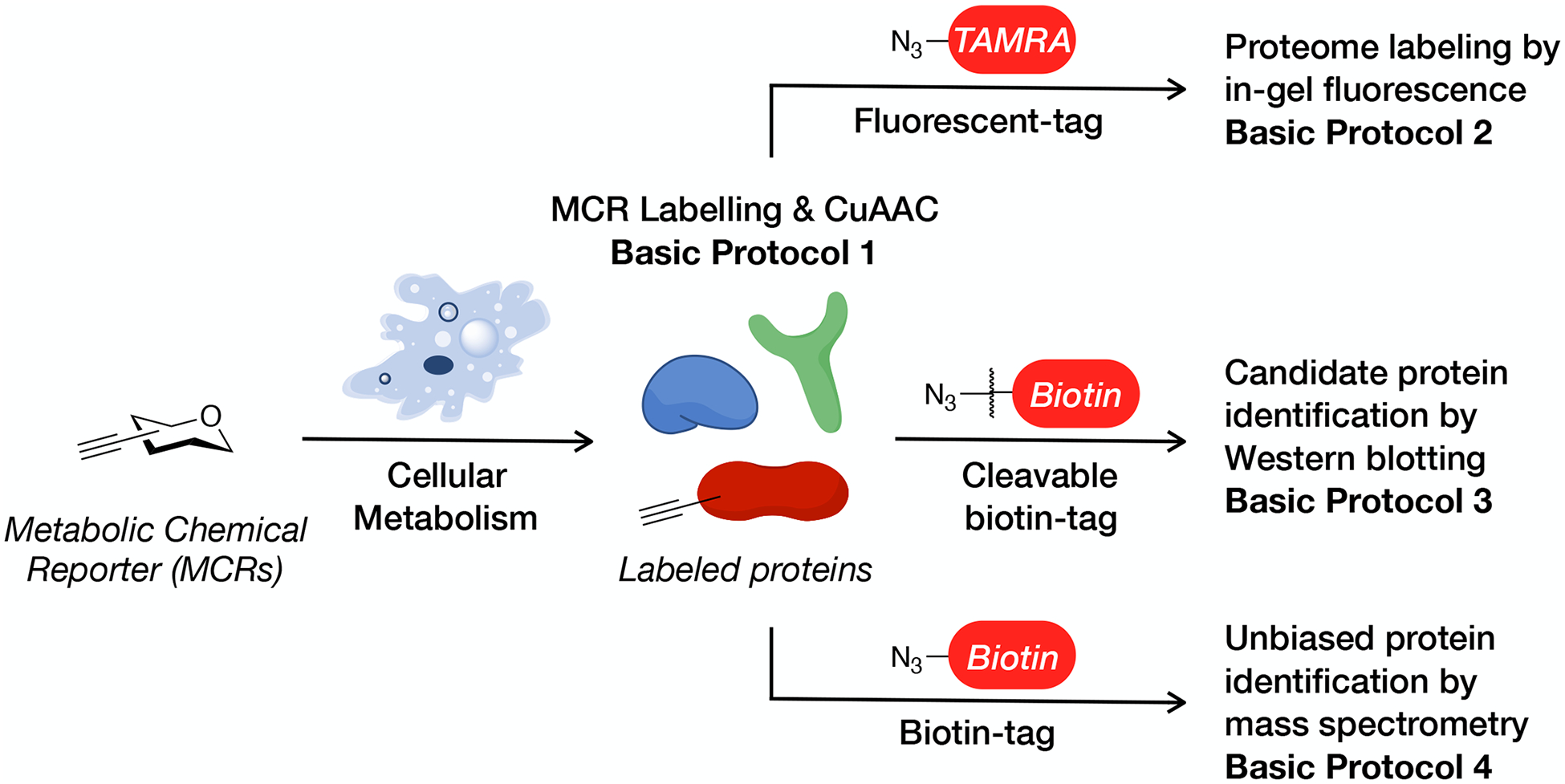
Overview of the MCR protocol. Basic protocol one describes the labeling of living cells by an MCR and the subsequent bioorthogonal labeling of modified proteins using a general CuAAC reaction. Basic protocol 2 describes the use of fluorescent tags to visualize the proteome-wide labeling by MCRs. Basic protocol 3 describes the application of cleavable biotin-tags to characterize the labeling of candidate proteins by Western blotting. Basic protocol 4 describes the use of a biotin tag and mass spectrometry to perform an unbiased identification of labeled proteins.
The exact choice of reporter is up to the end user, and each MCR has its own advantages and drawbacks. For example, alkyne-containing MCRs result in less background labeling during the CuAAC reaction and therefore display better signal to noise. This is largely due to a background reaction between alkyne-tags and cysteine residues in proteins(Speers and Cravatt, 2004). Another important factor is the inherent selectivity of a given MCR for different classes of glycans. Several, MCRs are incorporated into both O-GlcNAc and cell-surface glycans, including GlcNAz, GlcNAlk, and GalNAz, while the others are more selective for O-GlcNAc. Finally, only a subset of MCRs are commercially available, Click Chemistry Tools as a good selection, while the others require synthetic expertise to prepare. As a good starting point, we recommend either GlcNAz or GalNAz, as they are commercially available from multiple vendors and result in high levels of metabolic labeling in most cell-lines.
BASIC PROTOCOL 1
Treatment of cells and CuAAC
Basic Protocol 1 describes a representative procedure for the incorporation of metabolic chemical reporters and optimized conditions for the CuAAC click chemistry in cell lysate. Once an MCR has been designed and synthesized, the first step in characterizing its metabolic labeling efficiency is to treat, or “feed”, the reporter to cells. Typical treatment involves growing cells in normal growth media supplemented with the MCR. This is followed by an incubation period long enough for adequate metabolic incorporation. Experiments with new reporters require optimization of incubation time and reporter concentration to achieve optimal labeling. For O-GlcNAc reporters, labeling typically increases in the first 24 hours followed by a decrease as labeled proteins are turned over.
Following treatment, cells should be lysed in appropriate lysis buffer in preparation for the ensuing CuAAC reaction. Consideration of the buffer components is important for guaranteeing successful click chemistry. SDS buffers containing HEPES, for alkyne-MCRs, or TEA, for azide-MCRs, have been reported to give the highest signal to noise ratio for their respective orientations.
Once lysed, samples are ready to be submitted to CuAAC click chemistry. While the cargo bound to the alkyne or azido-probe will depend on subsequent analysis, the basic steps in this section remain the same. Carrying out of the reaction requires four main components, an alkyne or azide bound tag (e.g., TAMRA-Azide or -Alkyne), a reducing agent, a stabilizing ligand, and a source of copper. In biological applications, in situ reduction of Cu(II) to catalytically active Cu(I) is necessary. Reagents described in the following protocol are optimized for cell lysate labeling.
Materials
Solutions and buffers should be prepared with 18 MΩ H2O at at 25 °C. Reagents should be stored at room temperature unless otherwise noted. Dispose of hazardous waste appropriately.
1× solution of DPBS (Thermo Fisher Scientific cat. no. 21600010)
- HeLa Cells (ATCC cat. no. CCL-2)
- Cell line of interest may vary depending on nature of experiment. Optimized culturing conditions including growth media, supplements, and tissue culture flask, can be found using ATCC’s website (www.atcc.org) or another reliable resource.
SDS buffers (See Reagents and Solutions)
Ac4GlcNAz (Click Chemistry Tools cat. no. 1085–5) or alternative MCR (Click Chemistry Tools)
- If continuing on to Basic Protocol 2 use one of the following probes depending on orientation of MCR (i.e., TAMRA Azide paired with Alkyne MCR):
- TAMRA Azide (Click Chemistry Tools cat. no. AZ109–1)
- TAMRA Alkyne (Click Chemistry Tools cat. no. TA108–1)
- If continuing on to Basic Protocol 3 or 4 use one of the following probes depending on orientation of MCR (i.e., Azide-PEG4-biotin paired with Alkyne MCR):
- 5 mM Azide-PEG3-biotin (3.56 mg/mL in DMSO, Click Chemistry Tools cat. no. AZ104–5)
- 5 mM Alkyne-PEG4-biotin (3.98 mg/mL in DMSO (Click Chemistry Tools cat. no. TA105–5)
50 mM TCEP (14.34 mg/mL in H2O, Thermo Fisher Scientific cat. no. 20490)
10 mM TBTA (5.31 mg/mL in DMSO, Click Chemistry Tools cat. no. 1061–100)
50 mM CuSO4*5H2O (12.47 mg/mL in H2O, Sigma-Aldrich cat. no. 7758–99-8)
2× SDS-free loading buffer.
Trypsin EDTA (VWR cat. no. 45000–662)
cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail (Millipore Sigma cat. no. 11836170001)
0.5 M EDTA.
Tip sonicator (Q Sonica model no. Q125)
Pierce BCA Protein Assay Kit (Thermo Fisher Scientific cat. no. 23225)
Bench top centrifuge (e.g. VWR)
Preparation of cell lysates
-
1
Change media on cells grown to ~80% confluency with fresh media containing 200 μM of Ac4GlcNAz or DMSO vehicle.
Most cell lines require a minimum of one 150 mM dish to harvest enough protein for a 1 mg enrichment.
-
2
Incubate HeLa cells at 37 °C for 16 hours for efficient incorporation of Ac4GlcNAz.
Exact timing depends on reporter. Optimize reporter concentration and treatment time depending on your reporter and experiment.
-
3
Harvest cells.
For HeLa cells and other adherent cells, use trypsin EDTA (VWR) to detach cells from the dish. For a 150 mM dish, add 2 mL of trypsin, incubate at room temperature for 5 – 10 min or until cells are visibly detached, and wash with 8 mL of DPBS. Collect total volume into a 15 mL centrifuge tube and pellet cells at 2000 × g for 5 min at 4 °C.
For suspension cells, transfer growth media (e.g., DMEM with 10% fetal bovine serum) containing cells into a 15 mL centrifuge tube and pellet cells at 2000 × g for 5 min at 4 °C.
-
4
Aspirate media and resuspend cell pellets in 1 mL of ice-cold DPBS buffer. Transfer pellets to pre-cooled microcentrifuge tubes on ice.
-
5
Centrifuge suspension at 2000 × g for 5 min at 4 °C. Aspirate supernatant. Repeat steps 4 and 5 once more for a total of three washes.
Once washed, cell pellets can be stored at −20 °C. When resuming experiments, thaw pellets on ice.
-
6
Remove from ice and add 200 μL of 4% SDS buffer supplemented with 5 mg/mL cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail (Millipore Sigma).
-
7
Lyse cells via tip sonication. Three cycles of at 35% amplitude for 5 s on, 5 s off.
-
8
Pellet any cell debris to clear lysate by centrifugation, 10000 × g for 10 min at 15 °C. Transfer supernatant (soluble fraction) to fresh microcentrifuge tubes.
-
9
Normalize lysate protein concentrations using a Pierce BCA assay (Thermo Fisher Scientific).
Prepare master mix by mixing 50 parts (1 mL) of Reagent A with 1 part (20 μL) of Reagent B. Enough master mix should be made to account for 1 mL per sample plus six (five for standards plus one extra to account for pipetting error). Aliquot 1 mL into fresh microcentrifuge tubes per sample. Each assay requires an independently preformed standard curve using concentrations of albumin standard (provided with kit at a concentration of 2 mg/mL) ranging from 0 to 16 μg. Generate five 1 mL aliquots to account for this. Add 0, 1, 2, 5, and 8 μL of albumin standard to respective tubes. Add 1 μL of individual cell lysate samples to respective 1 mL master mix tubes. Vortex. Place each BCA sample on a heating block at 60 °C for 30 min. Following incubation, read each sample using a UV spectrophotometer at 562 nm. Use the 0 μg standard sample as the blank.
-
10
Based on BCA results, dilute samples to 4 mg/mL in 4% SDS lysis buffer.
Harvesting a confluent 150 mM dish of HeLa cells will typical afford 1 – 2 mg of total protein.
Remove 20 μL and add 20 μL 2× SDS-free loading buffer to generate inputs at a final concentration of 2 mg/mL. Scale up or down as needed.
CuAAC click chemistry reaction (1 mg scale)
Enriching labeled lysates from 1 mg of total protein is usually sufficient for all subsequent experiments described in this series of protocols. The exact amount of protein will vary depending on metabolic reporter, cell line, and incubation time.
-
11
Prepare working solutions of protein lysates at 4 mg/mL in 4% SDS buffer. In a 15 mL centrifuge tube, dilute 250 μL of each sample with 130 μL of 1.25% SDS buffer followed by 550 μL 0% SDS buffer for a final concentration of 1 mg/mL in 1.25% SDS buffer.
For azide-containing reporters use SDS buffers containing TEA and for alkyne-containing reporters use SDS buffers containing HEPES.
-
12
Add 70 μL CuAAC cocktail to each sample.
CuAAC cocktail is freshly prepared as a master mix for each experiment. Stock solutions are described in detail in the Basic Protocol 1 Materials section above. In order, add 20 μL per sample of alkyne tag stock, 20 μL TCEP, 10 μL TBTA, and 20 μL CuSO4*5H2O into a 15 mL falcon tube. Vortex between additions. Add 70 μL of this master mix to each CuAAC reaction.
Specific azide or alkyne tags for each type of experiment will be described in subsequent sections.
-
13
Incubate protein lysate treated with CuAAC master mix in the dark for 1 h at room temperature.
Optional: to quench the reaction, add 10 μL of 0.5 M EDTA to each sample.
-
14Precipitate proteins using MeOH/CHCl3/H2O by performing the following steps:
- Add 3000 μL MeOH, vortex.
- Add 750 μL CHCl3, vortex.
- Add 2000 μL H2O, vortex.
- Centrifuge samples at 6000 × g for 5 min at 15 °C.
- Carefully pipet top layer, discard.
- Add 2250 μL MeOH, vortex.
- Centrifuge samples at 10000 × g for 10 min at 15 °C.
-
Decant supernatant carefully.At this point, 1000 μL fresh MeOH can be added to pellets. Suspension can be stored at −20 °C. When continuing, centrifuge suspension at 6000 × g for 10 min at 15 °C, decant supernatant, and continue.
-
15
Air-dry pellets for 5 min.
Air-dried pellets cannot be stored.
BASIC PROTOCOL 2
In-gel fluorescence of labeled cell lysates (1 mg scale)
One of the simplest and most straightforward approaches to evaluate the efficiency a given MCR’s labeling is through in-gel fluorescence. Here, MCR-treated cell lysate submitted to the CuAAC reaction can be run on an SDS-PAGE gel. Samples are prepared by diluting cell lysate with a loading buffer containing bromophenol blue dye and glycerol. Diluted samples are boiled to disrupt secondary and tertiary structures before being loaded into gel wells.
SDS-PAGE separates proteins by size and charge. Lysate that have incorporated the tagged MCR will be distributed down the gel with the smallest proteins migrating the farthest allowing for a crude analysis. Depending on the efficiency of metabolic incorporation, adjustments in the total amount of protein loaded can be made. A good starting point is 40 μg per sample (20 μL of a 2 mg/mL solution). This can be scaled up or down as necessary. Metabolic incorporation is visualized by in-gel fluorescence scanning. A commonly used fluorophore, and the one discussed in the following section is TAMRA conjugated to an azide or alkyne handle.
Materials
Solutions and buffers should be prepared with18 MΩ H2O at at 25 °C. Reagents should be stored at room temperature unless otherwise noted. Dispose of hazardous waste appropriately.
SDS buffers (see Reagents and Solutions)
2× SDS-free loading buffer.
Criterion TGX 4–20% protein gel (Bio-Rad cat. no. 5671094)
Electrophoresis Cell (e.g. Bio-Rad)
Gel imaging system (e.g. GE Healthcare)
Coomassie Brilliant Blue R-250 Staining Solution (Bio-Rad cat. no. 161–0436)
Destaining buffer: 40% H2O, 50% MeOH, 10% acetic acid.
Preparation of gel samples for SDS-PAGE separation (1 mg scale)
-
Following CuAAC treatment described in Basic Protocol 1 (Steps 11–15), add 250 μL 4% SDS buffer to dried 1 mg precipitated protein pellets.
The use of either TEA or HEPES 4% buffer works at this step.
Completely resuspend protein pellet by incubating samples in a bath sonicator for 5 – 10 min.
Add 250 μL of 2× SDS-free loading buffer with BME. Boil for 5 min at 95 °C. Gel samples will end at a final protein concentration of 2 mg/mL.
-
Load 20 μL per sample into a Criterion TGX 4–20% protein gel (Bio-Rad) and run the gel for 200 V for 1 h at room temperature in the dark.
SDS-PAGE gel should be run in dark to avoid photobleaching of TAMRA dye.
-
Visualize the gel by scanning it on a gel imaging system such as a Typhoon Variable Mode Imager (GE Healthcare) or comparable product (example in Figure 3).
Use the following settings for TAMRA fluorescence: 534 nm excitation and 30 nm bandpass filter centered at 610 nm for detection.
- Stain gels in coomassie blue (Bio-Rad) to evaluate protein loading.
- Incubate gel in coomassie blue stain for 10 min on rocker
- Rinse with H2O
- Incubate gel in destaining buffer on rocker
- Rinse with MeOH and H2O
Figure 3.

Example of in-gel fluorescence result. Cells were treated with either Ac4GlcNAz (200 μM) or DMSO vehicle for 16 h before undergoing Basic Protocols 1 and 2.
BASIC PROTOCOL 3
Enrichment of labeled proteins, trypsinolysis, and collection of peptides for proteomics
Basic Protocol 3 describes a basic procedure biotin-enrichment of labeled proteins followed by either Western blot analysis or further processing via on-bead trypsinolysis. MCR-treated cell lysate is submitted to a CuAAC reaction between the reporter and a biotin-tag followed by incubation with streptavidin beads. This enables enrichment of labeled proteins due to the highly specific interaction between avidin and biotin. Post incubation, a series of washes removes all unbound proteins. Experiments with new reporters require optimization of incubation time and number of washes to achieve an optimal balance of signal-to-noise and pull-down efficiency.
To identify specific O-GlcNAcylated substrates, Western blot analysis can directly follow enrichment. Eluted samples are loaded on to an SDS-PAGE gel and separated by size and charge. However, instead of full lysate visualization, separated proteins are transferred to a PDVF membrane using an electrophoretic transfer cell. Transferred samples are then incubated with antibodies raised against proteins of interested. Enriched O-GlcNAc modified proteins are readily visualized using chemiluminescence reagents. Depending on both the efficiency of metabolic incorporation and target protein’s modification stoichiometry, adjustments in the total amount of protein loaded can be made. In order to assess successful enrichment, it is necessary to blot for both positive and negative control proteins. Standard positive control proteins are Nup62 and CREB, both of which are known to be highly O-GlcNAc modified. The standard negative control protein is β-actin, which is not modified.
Materials
Solutions and buffers should be prepared with 18 MΩ H2O at at 25 °C. Reagents should be stored at room temperature unless otherwise noted. Dispose of hazardous waste appropriately.
Resuspension buffer: 6 M urea, 2 M thiourea, 10 mM HEPES, pH 8.0.
1× solution of DPBS (Thermo Fisher Scientific cat. no. 21600010)
1% SDS buffer: 1% SDS (m/v) in DPBS.
Elution buffer: 1% SDS (m/v), 25 mM sodium dithionite in DPBS.
NeutrAvidin beads (Thermo Fisher Scientific cat. no. PI29204)
Dolphin-nosed tubes (VWR cat. no. 490010–618)
2× SDS-free loading buffer
4% SDS: 4% SDS (m/v), 150 mM NaCl, 50 mM TEA, pH 7.4.
Criterion TGX 4–20% protein gel (Bio-Rad cat. no. 5671094)
Electrophoresis Cell (e.g. Bio-Rad)
PVDF membrane (Bio-Rad cat. no. 162–0177)
Electrophoretic Transfer cell (e.g. Bio-Rad)
Blotting paper (VWR cat. no. 28297–990)
1× solution of TBST (Cell Signaling cat. no. 9997S)
Clarity Western ECL substrate (Bio-Rad cat. no. 170–5061)
Nup62 monoclonal antibody (Thermo Fisher Scientific cat. no. MA1–071)
Anti-Rabbit secondary HRP-conjugated antibody (VWR cat. no. 102649–670)
ChemiDoc XRS+ molecular imager (e.g. Bio-Rad)
Enrichment of labeled proteins (1 mg scale)
-
1
Following CuAAC treatment described in Basic Protocol 1 (Steps 11–15), add 800 μL of resuspension buffer to 1 mg precipitated protein pellet.
Ensure complete resuspension by bath sonicating pellets until pellet is no longer visible.
-
2Prepare streptavidin beads in 2 mL dolphin-nosed tubes. Between washes, carefully discard supernatant.
- Measure out 25 μL of a 50% slurry per sample.
- Wash 2 × with 1 mL of DPBS by vortexing.
- Centrifuge for at 2000 × g for 2 min at 15 °C.
- Wash 1 × with 1 mL of resuspension buffer by vortexing.
- Centrifuge for at 2000 × g for 2 min at 15 °C.
- Resuspend beads in 200 μL of resuspension buffer.
-
3
Add protein samples to dolphin nosed tube containing prepared beads.
-
4
Incubate samples on a rotator for 2 h at room temperature with full rotation.
Biotin-Cleavable Elution for Western Blot Analysis
-
5Following incubation, wash beads by vortexing. Centrifuge samples for at 2000 × g for 2 min at 15 °C. Between washes, carefully discard supernatant.
- 2 × with 1 mL of resuspension buffer
- 2 × with 1 mL of DPBS
- 2 × with 1 mL of 1% SDS in PBS buffer
-
6
Incubate beads in 25 μL of elution buffer on a rotator for 30 min at room temperature with half rotation to elute captured proteins. Centrifuge at 2000 × g for 2 min at 15 °C. Collect eluent in fresh microcentrifuge tube.
-
7
Repeat elution step (step 2) and pool corresponding eluent fractions.
-
8Precipitate proteins using MeOH/CHCl3/H2O by performing the following steps:
- Add 3000 μL MeOH, vortex.
- Add 750 μL CHCl3, vortex.
- Add 2000 μL H2O, vortex.
- Centrifuge samples 6000 × g for 5 min at 15 °C.
- Carefully pipet top layer, discard.
- Add 2250 μL MeOH, vortex.
- Decant supernatant carefully.
-
9
Air-dry pellets for 5 min.
-
10
Add 30 μL of 4% SDS buffer.
Ensure complete resuspension by bath sonicating pellets until pellet is no longer visible.
-
11
Add 30 μL of 2× SDS-free loading buffer with BME. Boil for 5 min at 95 °C. Gel samples will end at a final protein concentration of 2 mg/mL.
-
12
Load 15 μL of input and 30 μL of IP for each sample onto a protein gel for separation by SDS-PAGE at 200 V for 50 min.
-
13
Transfer SDS-PAGE gel onto PDVF membrane using a Trans-Blot SD Semi-Dry Electrophoretic Transfer cell (Bio-Rad) or comparable transfer cell at 20 V for 1 h.
Create a transfer sandwich using the following materials in order from the bottom up: blotting paper (Bio-Rad), PDVF membrane (Bio-Rad), SDS-PAGE gel, blotting paper (Bio-Rad). Between addition of layers, remove air bubbles that might form to ensure even transfer from gel to membrane.
-
14Perform standard Western blot. Western blot parameters described here are for imaging Nup62, a common positive control for GlcNAc MCRs. Optimization of the Western blotting procedure will depend on cell line, primary and secondary antibodies, and efficiency of pull down.
- Block in One-Block Western-CL buffer (Genesee Scientific) buffer for 1 h at room temperature on a rocker. Other commonly used blocking buffers are 5% BSA in TBST buffer or 5% milk in TBST buffer.
- Incubate with Nup62 primary antibody (Thermo Fisher Scientific) in blocking buffer at a dilution of 1:1000 for 12 – 16 h at 4 °C on a rocker.
- Wash 3 × 10 min in TBST buffer at room temperature on a rocker.
- Incubate with anti-rabbit secondary antibody (VWR) in blocking buffer at a dilution of 1:10000 for 1 h at room temperature on a rocker.
- Wash 3 × 10 min in TBST buffer at room temperature on a rocker.
- Incubate in Clarity Western ECL substrate (Bio-Rad) for 5 min at room temperature on a rocker.
- Image using ChemiDoc XRS+ molecular imager (Bio-Rad).
-
15Stain membrane in Coomassie blue (Bio-Rad) to evaluate protein loading. Alternatively, Ponsceau stain can be used.
- Incubate gel in coomassie blue stain for 10 min on rocker
- Rinse with H2O
- Incubate gel in destaining buffer for 10 min on rocker
- Rinse with MeOH, H2O
BASIC PROTOCOL 4
Basic Protocol 4 describes the general steps for preparation of peptides generated from reporter-modified proteins for mass spectrometry (MS)-based proteomic analysis. Reporter-modified proteins, which are biotinylated through CuAAC, are enriched with streptavidin beads and purified away from unmodified proteins. Bead-bound proteins are next digested with trypsin (Promega). Supernatant containing tryptic peptides is collected and desalted on a C18 column before being lyophilized to dryness. Following concentration, samples are resuspended in Buffer A (98/2/0.1% Water/MeCN/Formic Acid) and peptide concentration determined before MS acquisition. Once data has been acquired, the raw data is processed through analysis software, taking into account a few standard differential and static modifications that may have been made to the peptide during sample preparation or posttranslationally within the cell. It is important to note here that modified peptides are not retrieved in this process. They remain attached to the streptavidin beads, and therefore we do not search for the reporter modification in this analysis.
Materials
Solutions and buffers should be prepared with 18 MΩ H2O at at 25 °C. Reagents should be stored at room temperature unless otherwise noted. Dispose of hazardous waste appropriately.
0.2% SDS buffer: 0.2% SDS (m/v), 150 mM NaCl, 50 mM TEA, pH 7.4.
4 M urea in DPBS.
50 mM NH4HCO3.
50 mM NH4HCO3, 10 mM TCEP, pH 8.0.
200 mM CaCl2.
Sequencing grade trypsin (Promega cat. no. V5111)
Mini Bio-Spin columns (Bio-Rad cat. no. 7326207)
1% formic acid.
ACN buffers (see Reagents and Solutions)
Pierce™ C18 Spin Columns (Thermo Fisher Scientific cat. no. 89870)
Pierce Quantitative Fluorometric Peptide Assay (Thermo Fisher Scientific cat. no. 23290)
Screw top vials (VWR cat. no. 89093–854)
On-Bead Trypsinolysis for Proteomics Analysis (1 mg scale)
-
Following CuAAC treatment described in Basic Protocol 1 steps 11–15, add 100 μL of 4% SDS buffer (TEA) to 1 mg precipitated protein pellet.
Ensure complete resuspension by bath sonicating pellets until pellet is no longer visible.
Once resuspended, add 1.9 mL of 150 mM NaCl, 50 mM TEA, pH 7.4 to each sample. Total volume of suspension is now 2 mL.
- Prepare streptavidin beads in 2 mL dolphin-nosed tubes. Between washes, carefully discard supernatant.
- Measure out 50 μL of a 50% slurry per sample.
- Wash 3 × with 1 mL of 0.2% SDS buffer (TEA) by vortexing.
- Centrifuge for at 2000 × g for 2 min at 15 °C.
Add protein samples to dolphin nosed tube containing prepared beads and incubate samples on a rotator for 1.5 h at room temperature with full rotation.
- Transfer beads and supernatant to a fritted gravity-flow column (Bio-Rad) and drain supernatant by aspiration or gentle centrifugation (800 × g, 1 min, 25 ℃) before subjecting beads to the following washes:
- Wash 6 × with 2 mL 1% SDS buffer (TEA) in DPBS
- Wash 3 × with 2 mL 4 M urea in DPBS
- Wash 8 × with 2 mL 50 mM NH4HCO3
Resuspend beads in 1 mL of 50 mM NH4HCO3, 10 mM TCEP, pH 8.0 and incubate for 30 min with gentle shaking at room temperature. Following incubation, wash samples 3 × with 2 mL of 50 mM NH4HCO3 and resuspended in 100 μL of 50 mM NH4HCO3.
Elution of Tryptic Peptides
Transfer bead suspensions from previous section to MS screw top vials and add 2 μL of CaCl2 (200 mM stock in H2O) 2 μL of trypsin (Sequencing Grade, Promega, 0.1 mg/mL in H2O) to each sample. Incubate for 18 h at 37 °C.
Transfer resulting suspension of peptides and beads to Mini Bio-Spin columns (Bio-Rad) and collect the eluent by centrifugation at 1000 × g for 1 min at 15 °C.
- Elute remaining peptides by adding the below washes to spin columns followed by immediate centrifugation at 1000 × g for 1 min at 15 °C into corresponding capture tube for each sample.
- 100 μL of 1% formic acid in H2O
- 100 μL of 15% acetonitrile in H2O
- 100 μL of 1% formic acid H2O
- Prepare C18 spin columns (Pierce, Thermo Fisher Scientific) for sample desalting as follows:
- Wash 2 × with 200 μL of 50% ACN:H2O and centrifuging at 1500 × g for 1 min at 15 °C.
- Wash 2 × with 200 μL of 5% ACN, 0.5% TFA in H2O and centrifuging 1500 × g for 1 min at 15 °C.
Add eluent from Step 3 to prepared C18 spin columns and centrifuge at 1500 × g for 1 min at 15 °C. Wash column 3 × with 200 μL of 5% ACN, 0.5% TFA in H2O and centrifuging 1500 × g for 1 min at 15 °C. Discard flow through.
Transfer C18 spin column to fresh microcentrifuge tube and elute peptides from spin column by adding 20 μL of 70% ACN in H2O and centrifuging 1500 × g for 1 min at 15 °C. Repeat elution step two more times pooling elution fractions for corresponding samples.
Freeze-dry sample on a lyophilizer for subsequent analysis of tryptic peptides. The sample can be stored at −80 °C indefinitely.
Prior to MS analysis, resuspend sample in 100 μL of Buffer A. Briefly sonicate peptides into solution.
Determine the peptide concentration of each sample using the Pierce Quantitative Fluorometric Assay. To minimize use of the sample, scale down to 50 μL working solution and 5 μL of sample.
For reporter-enriched samples, normalize peptide concentration to desired injection volume according to your mass spectrometer’s best practices with Buffer A (typically 1 −2 μL. On average expect approximately ~5–10% recovery of total input following enrichment and cleanup. For example, from 1 150mm plate of confluent HeLa cells treated with 6AlkGlcNAc, a minimum of 1 mg of lysate is generated and approximately 5 μg of peptide sample isolated from enriched proteins. Sample injection amount can vary from 100 ng – 800 ng depending on quality of instrumentation. For control (No reporter) samples, scale amount injected, dilute as calculated for the reporter samples with Buffer A.
REAGENTS AND SOLUTIONS
Solutions and buffers should be prepared with 18 MΩ H2O at at 25 °C. Reagents should be stored at room temperature unless otherwise noted. Dispose of hazardous waste appropriately.
- Sample preparation and CuAAC buffers
- 1× solution of DPBS: 9.6 g DPBS powder to 1 L volume of H2O (Thermo Scientific cat. no. 21600010)
- SDS buffers
- For azide-containing reporters:
- 4% SDS: 4% SDS (m/v), 150 mM NaCl, 50 mM TEA, pH 7.4. Add 20 g SDS, 4.38 g NaCl, and 3.73 g TEA to 400 mL of H2O. Adjust pH to 7.4. Add additional H2O for a final volume of 500 mL. Readjust pH to 7.4 if needed.
- 1.25% SDS buffer: 1.25% SDS (m/v), 150 mM NaCl, 50 mM TEA pH 7.4. Add 6.25 g SDS, 4.38 g NaCl, and 3.73 g TEA to 400 mL of H2O. Adjust pH to 7.4. Add additional H2O for a final volume of 500 mL. Readjust pH to 7.4 if needed
-
0% SDS buffer: 150 mM NaCl, 50 mM TEA pH 7.4. Add 4.38 g NaCl, and 3.73 g TEA to 400 mL of H2O. Adjust pH to 7.4. Add additional H2O for a final volume of 500 mL. Readjust pH to 7.4 if neededFor alkyne-containing reporters:
- 4% SDS buffer: 4% SDS (m/v), 150 mM NaCl, 50 mM HEPES, pH 7.4. Add 20 g SDS, 4.38 g NaCl, and 5.96 g HEPES to 400 mL of H2O. Adjust pH to 7.4. Add additional H2O for a final volume of 500 mL. Readjust pH to 7.4 if needed.
- 1.25% SDS buffer: 1.25% SDS (m/v), 150 mM NaCl, 50 mM HEPES, pH 7.4. Add 20 g SDS, 4.38 g NaCl, and 5.96 g HEPES to 400 mL of H2O. Adjust pH to 7.4. Add additional H2O for a final volume of 500 mL. Readjust pH to 7.4 if needed.
- 0% SDS buffer: 150 mM NaCl, 50 mM HEPES, pH 7.4. Add 20 g SDS, 4.38 g NaCl, and 5.96 g HEPES to 400 mL of H2O. Adjust pH to 7.4. Add additional H2O for a final volume of 500 mL. Readjust pH to 7.4 if needed.
- 2× SDS-free loading buffer: 20% glycerol (v/v), 0.2% bromophenol blue (m/v), 100 mM TRIS, pH 6.8. Add 100 mL glycerol, 1 g bromophenol blue, 6.06 g TRIS to 300 mL of H2O. Adjust pH to 6.8. Add additional H2O for a final volume of 500 mL. Readjust pH to 7.4 if needed. Add 14μL of β-mercaptoethanol (BME) to a 1mL aliquot directly before use.
- 0.5 M EDTA: 73.06 g EDTA to 500 mL H2O
- Destaining buffer: 40% H2O, 50% MeOH, 10% acetic acid.
- Biotin enrichment and Western blotting buffers
- Resuspension buffer: 6 M urea, 2 M thiourea, 10 mM HEPES, pH 8.0. Add 180.18 g urea, 76.12 g thiourea, 1.1915 g HEPES to 400 mL H2O. Adjust pH to 8.0. Add additional H2O for a final volume of 500 mL. Readjust pH to 7.4 if needed.
- 1% SDS buffer: 1% SDS (m/v), pH 7.4. Add 10 g SDS and 9.6 g DPBS powder to 900 mL of H2O. Adjust pH to 7.4. Add additional H2O for a final volume of 1 L. Readjust pH to 7.4 if needed.
- Elution buffer: 1% SDS (m/v), 25 mM Na2S2O4. To already prepared 1% SDS buffer, add 4.35 mg Na2S2O4 directly before use. Stable for 2 h at room temperature. Discard solid Na2S2O4 after 3 months.
- 1× solution of TBST: 100 mL 10× TBST to 900 mL H2O (Cell Signaling cat. no. 9997S)
- Trypsinolysis buffers:
- 0.2% SDS buffer: 0.2% SDS (m/v), 150 mM NaCl, 50 mM TEA, pH 7.4. Add 1 g SDS, 4.38 g NaCl, and 3.73 g TEA to 400 mL of H2O. Adjust pH to 7.4. Add additional H2O for a final volume of 500 mL. Readjust pH to 7.4 if needed.
- 4 M urea: 120.12 g urea to 500 mL H2O
- 50 mM NH4HCO3: 1.98 g NH4HCO3 to 500 mL H2O
- 50 mM NH4HCO3, 10 mM TCEP buffer: 1.98 g NH4HCO3, 1.25 g TCEP to 500 mL H2O
- 200 mM CaCl2 buffer: 11.10 g CaCl2 to 500 mL H2O
- 1% FA: Add 1 mL FA to 99 mL H2O
- ACN buffers
- 15% ACN in H2O
- 50% ACN in H2O
- 5% ACN, 0.5% TFA, 94.5% in H2O
- 70% ACN in H2O
- Buffer A: 98/2/0.1% Water/MeCN/Formic Acid
BACKGROUND INFORMATION
O-GlcNAcylation is a posttranslational modification (PTM) resulting from the addition of the monosaccharide N-acetylglucosamine (GlcNAc) to serine and threonine residues of nuclear, cytosolic, and mitochondrial proteins (Bond and Hanover, 2015; Yang and Qian, 2017; Zachara, 2018). O-GlcNAcylation is dynamically modified through the action of two enzymes; O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA), which catalyze the addition and removal of GlcNAc respectively (Levine and Walker, 2016; Joiner et al., 2019; King et al., 2019). The reversible nature of O-GlcNAcylation allows it to play important roles in modulating gene expression, signal transduction, stress response, and protein stability. O-GlcNAcylation has been shown to be essential for normal growth and development of nearly all multicellular organisms. Genetic knockout of OGT results in embryonic lethality in Drosophila and mice. Tissue specific knockout of OGT in T-cells results in their apoptosis and loss of OGT in neurons is associated with neurodegeneration in mice (O’Donnell et al., 2004; Shafi et al., 2000; Sinclair et al., 2009; Wang et al., 2016). Even small shifts in global O-GlcNAcylation levels have been implicated in a variety of diseases. O-GlcNAc levels are consistently elevated in all types of cancer compared to healthy tissue (Ma and Vosseller, 2013; Ferrer et al., 2016; Vasconcelos-dos-Santos et al., 2018; Hanover et al., 2018) whereas the modification levels are lower in Alzheimer’s disease (Liu et al., 2004; 2009; Aguilar et al., 2017; Pinho et al., 2019). Taken together, these observations demonstrate the vital role of O-GlcNAc in modulating biochemical pathways. With all that is known about the importance of O-GlcNAcylation, understanding the effects of specific modifications on protein regulation and biological pathways remains a major challenge.
There are several advantages of using MCRs over other visualization techniques. First, splitting up the labeling moiety and probe allows for an almost traceless incorporation of the reporter with minimal perturbation on a cell’s endogenous machinery. The functionalized handle can then be used in a variety of ways by exploiting the same bioorthogonal reaction, expanding the utility of a single probe. Second, the reaction that occurs between the probe and tag are selective, leading to low background signal. Finally, because they are metabolically incorporated, MCRs offer unique insight into modification dynamics using pulse and pulse-chase experiments.
It is important to note, however, that MCRs can have certain drawbacks. One complication is that the UDP-GlcNAc product is distributed across almost all different forms of glycosylation. As such, it is important to design MCRs to be a biased substrate for OGT transfer over other glycosyltransferases. For example, we have demonstrated that subtle changes in the MCR structure can make them preferential substrates for OGT. This approach has led to the development of multiple more-selective O-GlcNAc MCRs including Ac4GlcNAlk, Ac36AzGlcNAc, Ac36AlkGlcNAc, and Ac42AzGlc (Zaro et al., 2011; Chuh et al., 2014; 2017; Zaro et al., 2017). Second, treatment of cells with MCRs may cause increased flux into the corresponding donor sugar, resulting in higher than typical amounts of protein modification. Finally, recent analyses of certain MCRs has shown that they can label proteins in a non-enzymatic fashion, leading to false-positives (Qin et al., 2018). Fortunately, this “background labeling” can be reduced through the use of specific placement of the acetyl protecting groups (Hao et al., 2019). Regardless, once a protein has been identified as being O-GlcNAcylated using an MCR, the endogenous modification status should always be confirmed using a complementary technique, including anti-O-GlcNAc antibodies or chemoenzymatic modification (Clark et al., 2008).
CRITICAL PARAMETERS
Optimal conditions for treating/harvesting cells
Success of a MCR labeling experiment relies on carefully selecting the cell line and harvesting conditions. The first step in characterizing any new MCR is testing whether or not it can be metabolically incorporated. Different cell lines have different incorporation efficiencies and so it is important to select one that has historically high incorporation rates. For subsequent experiments, selecting a cell line that expresses target proteins in order to investigate physiological relevance is necessary.
Similarly, optimal harvesting and lysis conditions depend on the goal of the experiment. Harsher detergents, such as SDS, can be used if one is simply checking whether or not incorporation of the MCR occurs at all. These will yield the largest number of proteins from all cellular compartments. More mild, non-denaturing conditions are useful if one is investigating protein-protein or protein-substrate interactions.
Optimal labeling time and concentration
The optimal time and concentration of a given MCR will depend on both cell line and system with which one is investigating. Choice of probe-tag orientation is important. Alkyne-probes clicked with azide-tags yield a lower signal-to-noise ratio. When possible, synthesizing probes in this orientation will generate cleaner data. It is recommended to treat cells with a range of concentrations for different amounts of time to determine the optimal treatment conditions. A normal starting point for O-GlcNAc probes is to treat at concentrations ranging from 1 to 200 μM in increments of 4 hours up to 24 hours. During these initial experiments, taking note of the solubility of the probe in aqueous growth media is important. Ensuring the probe is fully dissolved in media heated to 37 °C before adding the mixture to cells will ensure reproducible data and prevent errors.
Optimal CuAAC reaction conditions
CuAAC click chemistry reaction should be performed using freshly prepared solutions of TCEP and CuSO4 as both of these reagents can oxidized overtime. For reactions using fluorescent tags, it is important to keep samples in the dark as exposure to light can lead to photobleaching. For cellular experiments, Cu(I) is required. Depending on the orientation of the click reaction, choice in SDS buffer varies. For alkyne-probes, SDS buffer containing HEPES is required whereas for azide-probes SDS buffers containing TEA as these show the lowest signal-to-noise for their respective reporters.
Optimal enrichment and blotting conditions
An experiment that relies on selective enrichment and elution steps has the chance of generating background labeling that could convolute any actual results. Therefore, using appropriate negative controls is important for critically evaluating the experiment. For reporter experiments, including a sample that was treated with DMSO-vehicle as well as blotting for proteins that should not pull-down are crucial. Ensuring the sample buffer is compatible with biotin streptavidin binding is important for adequate enrichment. For samples lysed in SDS buffer, washing precipitated pellets twice with methanol and resuspending them in a diluted SDS buffer attenuates this step.
Not all antibodies bind the same to proteins transferred to PDVF membranes. Optimizing the concentration of primary and secondary antibodies, length of incubation, as well as using an appropriate blocking buffer is necessary for generating optimal blot images.
Optimal biotin enrichment conditions for mass spectrometry
For biotin enrichment conditions it is critical that all insoluble debris are removed prior to enrichment and on-bead digestion. We recommend that upon resuspension of the precipitated protein pellet following methanol-chloroform precipitations, the resuspended protein sample be centrifuged to pellet any remaining insoluble debris (maximum speed, 2 min, 4 °C). The supernatant can be then be transferred to a fresh tube prior to streptavidin bead incubation.
Suggested mass spectrometry analysis search parameters
Depending on the mass spectrometer and analysis software utilized, search parameters may change accordingly. However, there are certain modifications that we recommend including in your search parameters to improve detection. These include
Fixed modification: +57.02146 C - from cysteine alkylation by iodoacetamide.
Variable modifications: Acetyl +42.010565 N-term, pyro-Glu −17.026549 N-term Q, and pyro-Glu −18.010565 N-term E.
Precursor tolerance set at 30.0 ppm.
False discovery rate set to 0.01 with significance calculated using ANOVA.
Troubleshooting
| Protocol # | Problem | Cause | Solution |
|---|---|---|---|
| 1 | Insufficient number of cells | Treatment was toxic | Lower concentration, reduce treatment time Plate more cells |
| Cells lost during harvesting/lysing | Be more careful Switch techniques Plate more cells |
||
| CuAAC reaction failed | Reagents have gone bad | Prepare fresh stock concentrations Store all reagents based on manufacturers recommendations |
|
| MCR did not label cells | Optimize treatment time and concentration Ensure MCR is soluble in cell media Ensure cell line has enzymatic machinery to metabolically incorporate cells |
||
| 2 | No fluorescence signal | No MCR incorporation | See above |
| CuAAC reaction failed | See above Carry out the reaction in the dark |
||
| High background fluorescence signal | Excess dye in gel | Destain the gel before imaging Precipitate protein pellets using MeOH |
|
| Wrong choice of buffer | Remake buffers and ensure HEPES is used for alkyne probes and TEA is used for azide probes | ||
| Background CuAAC reactivity with cysteine residues | Cap cysteine residues before submission to CuAAC reaction | ||
| 3 | Positive controls did not pull-down | Inefficient biotinylation | Store biotin stocks at 4 °C, check expiration date |
| No MCR incorporation | See above | ||
| Negative controls pull down | Beads insufficiently washed | Increase number of washes post incubation Ensure beads are fully resuspended between washes |
|
| Weak/no Western blot signal | Inefficient transfer to membrane | Remake transfer buffer Ensure tight sandwhich making in transfer apparatus |
|
| Ineffective antibody | Add more protein to gel sample Optimize antibody dilution and incubation time |
||
| 4 | No proteins enriched by in positive control as detected by MS analysis | Positive controls did not pull-down | (see Protocol 3 Trouble shooting) |
| MS detection is poor | Run QC sample on instrument, perform leak and flow restriction tests. | ||
| Efficiency of reporter incorporation is poor | Pull down from 10 mg of starting material | ||
| Proteins enriched by negative control as detected by MS analysis | Negative controls pull-down | (see Protocol 3 Trouble shooting) | |
| MS is dirty | Run blank sample on instrument and analyze to see if proteins detected are on due to column or source contamination. |
ANTICIPATED RESULTS
Success of Basic Protocols 1 and 2 will yield in-gel fluorescence signal of MCR-treated cell lysate. If the signal is greater than DMSO-treated control lanes, the MCR is thought to have been efficiently incorporated onto protein substrates. In-gel fluorescence intensity should be modulated by changing the treatment time and MCR concentration. If no labeling is detected, using the positive control of 200 μM Ac4GalNAz, a robust GlcNAC MCR, can help distinguishing between failed MCR incorporation versus failed CuAAC reaction. Efficient and selective pull-down can be established by using positive control proteins, CREB and Nup62, and negative control protein β-actin. A successful Western blot will appear as a discrete and distinct band corresponding to the molecular weight of the protein of interest. Incubation time and temperature as well as total protein may need to be adjusted depending on target protein expression, MCR metabolism, and chosen cell line.
The number of proteins detected by mass spectrometry will vary based off of the promiscuity of the reporter selected and robustness of the instrument itself. For low-efficiency reporters we expect approximately 1000 proteins detected to be significantly enriched in the reporter sample compared to the control sample. For higher efficiency reporters we would expect over 3000 proteins when utilizing highly-efficiency dual-cycle MS instrumentation capable of utilizing ion mobility.
TIME CONSIDERATIONS
Once MCR treatment has is complete, Basic Protocol 1 can be completed in 6 h. Work can be stopped after harvesting as cell pellets, post-lysis as cell lysate, or after the CuAAC reaction is completed, stored overnight in MeOH. Completion of Basic Protocol 2 takes 1.5 h as it only requires setting up, running, and imaging an SDS-PAGE gel. Basic Protocols 1 and.2 can easily be completed in one day. Basic Protocol 3, starting from resuspending cell pellets after CuAAC, will add a second day. For Western blotting, once protein samples have been eluted from streptavidin beads they can be frozen and reboiled. Standard time for blocking, primary antibody, and secondary antibody incubation is 1 hr at rt however each of these steps can be extended overnight at 4 °C.
Biotin enrichment of reporter-modified proteins in Basic Protocol 4 should take approximately 5 hours to perform. Trypsin digestion is an overnight process (~16 h). Sample recovery and cleanup will take 2 h and lyophilization can be performed for an additional 4 h. Resuspension of the MS sample and quantification of peptide concentration can be performed within 1 h and MS acquisition varies according to your instrument’s best practices. In general, we perform 2 or 4 h gradients. Data analysis time will vary based off of computation power and software used. We recommend MaxQuant as a free option for MS analysis and Peaks or Byonic as purchase options.
ACKNOWLEDGEMENTS
Our research in this area is supported by the National Institutes of Health (R01GM125939 to M.R.P.) and the University of California San Francisco. N.J.P. is supported by NIGMS T32GM118289
ABBREVIATIONS
- Ac36AzGlcNAc
1,3,4-tri-O-acetyl-2-deoxy-2-N-acetyl-6-deoxy-azido-glucopyranose
- Ac42AzGlc
1,3,4,6-tetra-O-acetyl-N-azidoglucose
- Ac4GlcNAz
1,3,4,6-tetra-O-acetyl-N-azidoacetylglucosamine
- ACN
acetonitrile
- BCA
bicinchoninic acid assay
- BME
β-mercaptoethanol
- CaCl2
calcium chloride
- CHCl2
dichloromethane
- CREB
cAMP response element binding
- CuAAc
copper(I)-catalyzed azide-alkyne cycloaddition
- CuSO4*5H2O
copper sulfate pentahydrate
- DMSO
dimethyl sulfoxide
- DPBS
Dulbecco’s phosphate-buffered saline, no calcium, no magnesium
- EDTA
ethylenediaminetetraacetic acid
- FA
formic acid
- GlcNAc
N-acetylglucosamine
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- H2O
water
- MCR
metabolic chemical reporter
- MeOH
methanol
- MS
mass spectrometry
- NaCl
sodium chloride
- Na2S2O4
sodium dithionite
- NH4HCO3
ammonium bicarbonate
- Nup62
Nucleoporin 62
- OGA
O-GlcNAcase
- OGT
O-GlcNAc transferase
- PEG
polyethylene glycol
- PTM
posttranslational modification
- SDS
sodium dodecyl sulfate
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- TBST
tris buffered saline with tween
- TBTA
tris[(1-benzyl-1-H-1,2,3-triazol-4-yl)methyl]amine
- TCEP
tris(2-carboxyethyl)phosphine hydrochloride
- TEA
triethanolamine
- TFA
trifluoricacetic acid
- UDP
uridine diphosphate
REFERENCES
- Aguilar AL, Hou X, Wen L, Wang PG, and Wu P 2017. A Chemoenzymatic Histology Method for O-GlcNAc Detection. ChemBioChem 18:2416–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond MR, and Hanover JA 2015. A little sugar goes a long way: the cell biology of O-GlcNAc. The Journal of cell biology 208:869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce M, Carrico IS, Ganguli AS, Yu S-H, Hangauer MJ, Hubbard SC, Kohler JJ, and Bertozzi CR 2011. Metabolic cross-talk allows labeling of O-linked beta-N-acetylglucosamine-modified proteins via the N-acetylgalactosamine salvage pathway. Proceedings of the National Academy of Sciences of the United States of America 108:3141–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuh KN, and Pratt MR 2015. Chemical methods for the proteome-wide identification of posttranslationally modified proteins. Current opinion in chemical biology 24:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuh KN, Batt AR, and Pratt MR 2016. Chemical Methods for Encoding and Decoding of Posttranslational Modifications. Cell Chemical Biology 23:86–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuh KN, Batt AR, Zaro BW, Darabedian N, Marotta NP, Brennan CK, Amirhekmat A, and Pratt MR 2017. The New Chemical Reporter 6-Alkynyl-6-deoxy-GlcNAc Reveals O-GlcNAc Modification of the Apoptotic Caspases That Can Block the Cleavage/Activation of Caspase-8. Journal of the American Chemical Society 139:7872–7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuh KN, Zaro BW, Piller F, Piller V, and Pratt MR 2014. Changes in metabolic chemical reporter structure yield a selective probe of O-GlcNAc modification. Journal of the American Chemical Society 136:12283–12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark PM, Dweck JF, Mason DE, Hart CR, Buck SB, Peters EC, Agnew BJ, and Hsieh-Wilson LC 2008. Direct In-Gel Fluorescence Detection and Cellular Imaging of O-GlcNAc-Modified Proteins. Journal of the American Chemical Society 130:11576–11577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer CM, Sodi VL, and Reginato MJ 2016. O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. Journal of molecular biology 428:3282–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang HC, Yu C, Kato DL, and Bertozzi CR 2003. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proceedings of the National Academy of Sciences of the United States of America 100:14846–14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanover JA, Chen W, and Bond MR 2018. O-GlcNAc in cancer: An Oncometabolism-fueled vicious cycle. Journal of bioenergetics and biomembranes 50:155–173. [DOI] [PubMed] [Google Scholar]
- Hao Y, Fan X, Shi Y, Zhang C, Sun D-E, Qin K, Qin W, Zhou W, and Chen X 2019. Next-generation unnatural monosaccharides reveal that ESRRB O-GlcNAcylation regulates pluripotency of mouse embryonic stem cells. Nature Communications 10:4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner CM, Li H, Jiang J, and Walker S 2019. Structural characterization of the O-GlcNAc cycling enzymes: insights into substrate recognition and catalytic mechanisms. Curr. Opin. Struct. Biol 56:97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DT, Males A, Davies GJ, and Vocadlo DJ 2019. Molecular mechanisms regulating O-linked N-acetylglucosamine (O-GlcNAc)–processing enzymes. Current Opinion in Chemical Biology 53:131–144. [DOI] [PubMed] [Google Scholar]
- Levine ZG, and Walker S 2016. The Biochemistry of O-GlcNAc Transferase: Which Functions Make It Essential in Mammalian Cells? Annual Review of Biochemistry 85:631–657. [DOI] [PubMed] [Google Scholar]
- Li J, Wang J, Wen L, Zhu H, Li S, Huang K, Jiang K, Li X, Ma C, Qu J, et al. 2016. An OGA-Resistant Probe Allows Specific Visualization and Accurate Identification of O-GlcNAc-Modified Proteins in Cells. ACS chemical biology 11:3002–3006. [DOI] [PubMed] [Google Scholar]
- Liu F, Iqbal K, Grundke-Iqbal I, Hart G, and Gong C 2004. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America 101:10804–10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Shi J, Tanimukai H, Gu J, Grundke-Iqbal I, Iqbal K, and Gong CX 2009. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain 132:1820–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, and Vosseller K 2013. O-GlcNAc in cancer biology. Amino Acids 45:719–733. [DOI] [PubMed] [Google Scholar]
- O’Donnell N, Zachara NE, Hart GW, and Marth JD 2004. Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Molecular and cellular biology 24:1680–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho TS, Correia SC, Perry G, Ambrósio AF, and Moreira PI 2019. Diminished O-GlcNAcylation in Alzheimer’s disease is strongly correlated with mitochondrial anomalies. BBA - Molecular Basis of Disease 1865:2048–2059. [DOI] [PubMed] [Google Scholar]
- Qin W, Qin K, Fan X, Peng L, Hong W, Zhu Y, Lv P, Du Y, Huang R, Han M, et al. 2018. Artificial Cysteine S-Glycosylation Induced by Per-O-Acetylated Unnatural Monosaccharides during Metabolic Glycan Labeling. Angewandte Chemie (International ed in English) 57:1817–1820. [DOI] [PubMed] [Google Scholar]
- Shafi R, Iyer SP, Ellies LG, O’Donnell N, Marek KW, Chui D, Hart GW, and Marth JD 2000. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proceedings of the National Academy of Sciences of the United States of America 97:5735–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DAR, Syrzycka M, Macauley MS, Rastgardani T, Komljenovic I, Vocadlo DJ, Brock HW, and Honda BM 2009. Drosophila O-GlcNAc transferase (OGT) is encoded by the Polycomb group (PcG) gene, super sex combs (sxc). Proceedings of the National Academy of Sciences of the United States of America 106:13427–13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speers A, and Cravatt B 2004. Profiling enzyme activities in vivo using click chemistry methods. Chemistry & Biology 11:535–546. [DOI] [PubMed] [Google Scholar]
- Vasconcelos-dos-Santos A, Queiroz RM, Rodrigues BC, Todeschini AR, and Dias WB 2018. Hyperglycemia and aberrant O-GlcNAcylation: contributions to tumor progression. 1–13. [DOI] [PubMed]
- Vocadlo D, Hang H, Kim E, Hanover J, and Bertozzi C 2003. A chemical approach for identifying O-GlcNAc-modified proteins in cells. Proceedings of the National Academy of Sciences of the United States of America 100:9116–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AC, Jensen EH, Rexach JE, Vinters HV, and Hsieh-Wilson LC 2016. Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America 113:15120–15125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, and Qian K 2017. Protein O-GlcNAcylation: emerging mechanisms and functions. Nature Reviews Molecular Cell Biology 18:452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachara NE 2018. Critical observations that shaped our understanding of the function(s) of intracellular glycosylation (O‐Glc NAc). FEBS letters 592:3950–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaro BW, Batt AR, Chuh KN, Navarro MX, and Pratt MR 2017. The Small Molecule 2-Azido-2-deoxy-glucose Is a Metabolic Chemical Reporter of O-GlcNAc Modifications in Mammalian Cells, Revealing an Unexpected Promiscuity of O-GlcNAc Transferase. ACS chemical biology 12:787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaro BW, Yang Y-Y, Hang HC, and Pratt MR 2011. Chemical reporters for fluorescent detection and identification of O-GlcNAc-modified proteins reveal glycosylation of the ubiquitin ligase NEDD4–1. Proceedings of the National Academy of Sciences of the United States of America 108:8146–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]