

Summary
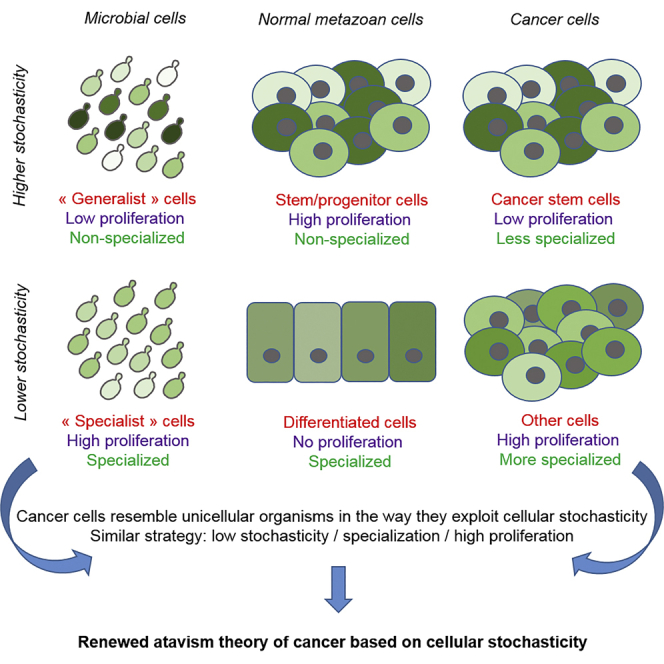
Similarities between microbial and cancer cells were noticed in recent years and serve as a basis for an atavism theory of cancer. Cancer cells would rely on the reactivation of an ancestral “genetic program” that would have been repressed in metazoan cells. Here we argue that cancer cells resemble unicellular organisms mainly in their similar way to exploit cellular stochasticity to produce cell specialization and maximize proliferation. Indeed, the relationship between low stochasticity, specialization, and quiescence found in normal differentiated metazoan cells is lost in cancer. On the contrary, low stochasticity and specialization are associated with high proliferation among cancer cells, as it is observed for the “specialist” cells in microbial populations that fully exploit nutritional resources to maximize proliferation. Thus, we propose a model where the appearance of cancer phenotypes can be solely due to an adaptation and a speciation process based on initial increase in cellular stochasticity.
Subject Areas: Ecology, Evolutionary Biology, Cancer
Graphical Abstract
Ecology; Evolutionary Biology; Cancer
Introduction
Parallels between cancer cells and microbial organisms have been noticed in past years, based either on various ecological and evolutionary considerations (Sprouffske et al., 2012; Thomas et al., 2017; Vincent, 2012) or on gene expression data (Trigos et al., 2017, 2018). Especially, some gene categories associated with multicellularity (extracellular matrix and adhesion) are downregulated in cancer, whereas those associated with unicellularity are upregulated (Trigos et al., 2017). These works favor the atavism hypothesis suggesting that cancer cells adopt a “selfish” unicellular mode of life through an active and directed process driven by selection (Thomas et al., 2017). This hypothesis proposes that cancer cells reactivate an “ancestral” gene expression program allowing survival in the highly stressful conditions such as low oxygen and pH found in cancer and resembling the pre-Cambrian environment, in which only cancer cells could flourish. In that sense, cells would return to a “pseudo-unicellular state” corresponding to the acquisition of phenotypes required in these primitive conditions. Coincidentally, these adaptations to cope with environmental stressors would provide an evolutionary explanation for the current difficulties of treating and curing cancer using aggressive treatments like radiation and chemotherapeutic drugs (Vincent, 2016).
Besides the acquisition of this ability to survive highly toxic environments, cancer cells are characterized by de-differentiation (loss of cellular characteristics mediating specialized somatic function) and de-specialization (loss of specialized somatic functionality by a tissue). Coupled with unlimited proliferation, these observations lead to the “reversion” concept where cancer cells deconstruct the metazoan phenotype and replace it by a behavior that recapitulates early life forms (Vincent, 2012). Cancer cells would be sufficiently autonomous to be considered unicellular. Nevertheless, cancer cells, although certainly losing their initial normal specialization, acquire during cancerization other specialized features that create intra-tumoral heterogeneity and enable various degrees of cooperation in a tumor between specialized subpopulations (Egeblad et al., 2010) (besides the general picture during cancer progression, where progression from benign to advanced stages of the disease is invariably linked with increased loss of differentiation). It appears that cancer cells remain able to specialize despite an initial and partial dedifferentiation. Only a particular subpopulation called cancer stem cells (CSCs) remain “de-specialized” and harbors high plasticity and stochasticity (Capp, 2019; Lee et al., 2016). Thus, although the malignant phenotype initiation might have links with loss-of-function ailments, tumorigenesis itself relies on cancer cells that acquire novel states that can be defined as abnormal cell types due to reshaping of the epigenetic landscape, making cancer a good example of disease with new combinations of cellular functions (Huang et al., 2009). This has been presented by proponents of the field of complex dynamical systems theory in their “cellular attractor” concept in the cancer context (Huang et al., 2009; Li et al., 2016).
In this article, we aim at giving a fresh look at the atavism hypothesis by reconsidering the way cancer cells lose their initial characteristics and acquire new ones allowing optimal growth in the tumoral microenvironment. To renew the parallelism between microbial and cancer cells, we propose to focus on cellular stochasticity (especially gene expression variability) and the way such stochasticity is exploited in microbial populations to produce specialized subpopulations with optimal growth capacity and better adaptation in a given environment. Indeed, cancer cells seem to revert the relationship between stochasticity, specialization, and proliferation found for normal metazoan cells. In metazoans, specialization and phenotypic stability are acquired in normal differentiating cells from unspecialized stem cells characterized by high proliferation ability and high intracellular stochasticity (see below). The process ends with proliferation arrest in mature tissues. In cancer cell populations, the same process of specialization associated with decrease cellular stochasticity occurs from similarly less specialized cells (the CSCs). However, instead of leading to proliferation arrest, it generates subpopulations with optimized capacity to proliferate and to benefit from both available nutritional resources and surrounding abnormal cooperating cells. This new relationship between specialization and proliferation in cancer cells is exactly what is observed in microbial populations that contain both less variable subpopulations specialized in the exploitation of a given environment but penalized if the environment changes and more variable subpopulations less adapted in this given environment but able to resume growth more rapidly after environmental changes (as the CSCs in a tumor). As specialization is, on the contrary, associated with proliferation arrest in normal metazoan organisms, cancer cells seem closer to microbial cells in the exploitation of cellular stochasticity to produce cell specialization and high proliferation.
After detailing the different situations briefly mentioned in this introduction, this article will finally discuss the potential origin of this new situation during oncogenesis, with an emphasis on the disruption of the normal micro-environment in cancer that is no more able to reciprocally interact with cellular stochasticity to ensure the specialization process of differentiating cells. In this tissue disruption-induced cell stochasticity (TiDiS) theory, being released from these environmental constraints that normally limit and canalize cellular stochasticity, pre-cancer cells would be free to explore all the possible combination of phenotypes thanks to their high plasticity. If heritable (see, for instance, Flavahan et al., 2017), the non-genetic heterogeneity produced would yield a suitable substrate fueling the somatic evolution of tumors (Brock et al., 2009), and thus produce fully malignant cells. This return of cancer cells to a behavior based on a heritable cellular stochasticity resembling the one of unicellular organisms is radically different from an explanation based on an existing “ancestral” genetic program that would need to be reactivated. Indeed, no activation of pre-existing “program” is required here. Finally, this hypothesis based on tissue disruption suggests new therapeutic perspectives: the restoration of relationships with the microenvironment resembling the ones that occur in healthy tissues should be able to revert the abnormal phenotypes of cancer cells.
Different Levels of Specialization Corresponds to Different Levels of Gene Expression Stochasticity across Kingdoms
Microbial Cells: Generalists (High Stochasticity) versus Specialists (Low Stochasticity)
Genetically identical microbial cells display heterogeneity in their morphology, the composition of their cellular components, and their growth dynamics (Ackermann, 2015). Previous works have demonstrated that growth rate heterogeneity can serve as a bet-hedging mechanism, providing a benefit to the population across changing environments, especially in yeast. Clonal Saccharomyces cerevisiae populations display broad distributions of growth rates with slow growth being predictive of resistance to heat killing in a probabilistic manner (Levy et al., 2012). A large component of growth rate heterogeneity is metastable and epigenetic in nature (Cerulus et al., 2016; Levy et al., 2012). Markers of slow versus fast growth are stochastically switched in yeast (Levy et al., 2012).
On the one hand, the fast-growing cells, that can be called “specialists,” express a restrained part of their genome with a small number of genes expressed at high level (van Dijk et al., 2015), allowing them to fully exploit the nutritional environment in which they grow thanks to the adequate strong expression of few genes (Siegal, 2015). Therefore they maximize proliferation in this environment through a specialized behavior, but they are heavily penalized when there is an environmental (especially nutritional) change. They do not resume growth because they do not harbor the adequate enzymatic and metabolic tools, even at low levels, to start growing in the new environment. This does not exclude the possibility that these specialist cells can collaborate with other microbial species in consortia for the reciprocal benefit of each species. However, they are still specialized in the present environment without bet-hedging anticipating potential uncertain futures.
On the other hand, the slow-growing subpopulation in S. cerevisiae expresses more genes in general, suggesting a more permissive chromatin and a more stochastic and plastic gene expression that may, in turn, allow them to explore a larger phenotypic space (van Dijk et al., 2015). This is detrimental for single cells in terms of growth rate in constant environments but advantageous when the cells need to shift to alternative carbon sources (faster transcriptional reprogramming and shorter lag phases) (Venturelli et al., 2015). In this so-called generalist strategy, more genes involved in alternative carbon and nitrogen source metabolism (less stringent catabolite repression) are expressed, and these cells are capable of growing on more heterogeneous environments (van Dijk et al., 2015). Only cells expressing “by chance” these genes allowing metabolization of the second nutritional resource continue growing, what is contrary to the classical models of progressive and general adaptation during these shifts (Siegal, 2015). Gene expression variability of catabolically active enzymes gives phenotypic heterogeneity in S. cerevisiae (New et al., 2014; Venturelli et al., 2015; Wang et al., 2015), Escherichia coli (Kotte et al., 2014), or Lactobacillus lactis (Solopova et al., 2014), which is an advantage when nutritional sources change compared with more homogeneous populations. This bet-hedging strategy of some cells allows better anticipation of environment changes. Finally, in microbial populations, the combinations high stochasticity/generalist strategy/low proliferation for the slow-growing cells and low stochasticity/specialist strategy/high proliferation for fast-growing cells are observed.
Normal Metazoan Cells: Stem Cells (High Stochasticity) versus Differentiated Cells (Low Stochasticity)
The same dichotomy is observed in metazoan organisms with stem cells and differentiated cells harboring high and low gene expression variability, respectively (for a review see Capp and Laforge, 2020). When examining embryonic stem cells (ESCs), an unusual nuclear structure where DNA is arranged in a less compacted chromatin structure and that allows rapid turnover of chromatin proteins is observed compared with differentiated cells (Meshorer et al., 2006). ESCs are enriched in epigenetic marks associated with elevated gene expression and possess less marks that compact chromatin enough to prevent any gene expression (Spivakov and Fisher, 2007). This is associated with the presence of high levels of proteins involved in chromatin remodeling and gene transcription (Efroni et al., 2008) and allows widespread, generalized, pervasive, and highly variable (stochastic) gene expression (Efroni et al., 2008; Fisher and Fisher, 2011; Gaspar-Maia et al., 2011). Especially, tissue-specific genes are sporadically expressed at low level, whereas they were not expected in ESCs (Efroni et al., 2008).
Stem cells cannot be defined as a cell type characterized by stable phenotypes. Pluripotency would be more a state of dynamic heterogeneity of a population driven by transcriptional noise than a discrete state dependent on the fixed expression of a small set of genes (Kalmar et al., 2009). Moreover, the function of the gene regulatory network centered on Nanog might be to generate this dynamic heterogeneity (Kalmar et al., 2009). Works that have tried to “deconstruct” expression heterogeneity in pluripotent stem cells found that genes involved in metabolic or other pathways common to all cells exhibit less variability than genes involved in development and signaling pathways (Kumar et al., 2014). When thousands of ESCs were analyzed using powerful microfluidics devices, thousands of genes were observed as highly variable from cell to cell, especially those linked to metabolism and transcriptional regulation, as well as the targets of the pluripotency regulators (Klein et al., 2015). These regulators themselves together with lineage-specific transcription factors and epigenetic regulators are also among the highly variable genes. This study revealed the intriguing observation that ESC populations cannot be simply divided into two distinct subpopulations of naive and primed states with higher and lower levels of pluripotency factors, respectively (for reviews on the different pluripotent states, see Ware, 2017; Weinberger et al., 2016). Some of these factors indeed fluctuate in a coordinate fashion, but not all of them. There is a continuum of states from high to low pluripotency (Klein et al., 2015).
Moreover, subsequent single-cell genomics studies were performed to decipher the different pluripotent states that can be found in ESC populations and how individual pluripotent cells function. Cultivation in media favoring or not maintenance of the naive state suggested that the levels of overall intercellular gene expression heterogeneity are comparable in both cases (Kolodziejczyk et al., 2015; Messmer et al., 2019). Only specific subsets of genes uniquely defines each pluripotent state, with pluripotency genes being remarkably more variable in conditions allowing priming and more homogeneously expressed in conditions favoring the naive state (Kolodziejczyk et al., 2015). This was also confirmed in other works showing higher heterogeneity in cellular hierarchy (a mix of naive, primed, and differentiated cells), higher bivalency in the epigenetic status, and higher variability in gene expression among ESCs grown in conditions allowing priming (Guo et al., 2016). The same phenomenon is observed for induced pluripotent stem cell populations that contain various states from naive to late primed, with genes and pathways that define differences in pluripotent cell states (Nguyen et al., 2018). In that case, the primed subpopulations were found to harbor higher transcriptional heterogeneity compared with the remaining cells, in coherence with other single-cell studies showing that the transition from pluripotency to lineage commitment phase is characterized by high gene expression variability (Semrau et al., 2017).
Thus, pluripotency rather seems to be a statistical property of stem cell populations (MacArthur and Lemischka, 2013). Functional pluripotency spontaneously emerges from the dynamic variability intrinsic to the pluripotent state (MacArthur and Lemischka, 2013). The whole-genome expression in ESCs creates early, stochastic, and reversible commitments toward differentiation (Hough et al., 2009; Trott et al., 2012). This phenomenon is probabilistic because it depends on the stochastic expression of some pluripotency factors such as Nanog (MacArthur et al., 2012): cells that “by chance” express less Nanog, for instance, differentiate more easily (Kalmar et al., 2009), which is related to more frequent stochastic and reversible commitments to a cell type (MacArthur et al., 2012).
In adult stem cells, as exemplified by hematopoietic stem cells (HSCs), different regulators of differentiation vary independently and stochastically in multipotent cells, what has been linked to random priming toward different cell types (Giladi et al., 2018; Moignard et al., 2015; Pina et al., 2012). For instance, the erythroid lineage is primed while many markers of other lineages are also expressed (Pina et al., 2012). During cell fate specification of hematopoietic multipotent progenitor cells, mixed-lineage intermediates with concurrent expression of factors finally associated with different lineages seem to be obligatory (Olsson et al., 2016). Thus there is no coordination in the HSC expression pattern. Transcriptional heterogeneity in stem cells is thus considered as an advantage for diversifying phenotypes in populations that require diverse potentialities (Torres-Padilla and Chambers, 2014).
On the opposite, a large-scale repression of gene expression occurs during differentiation with expression profiles becoming more discontinuous and stable in differentiated cells (Capp and Laforge, 2020). Differentiation would be above all the suppression of this widespread stochastic gene expression (SGE) (Efroni et al., 2009), which is linked to transition of the chromatin from a dynamic and open state to a more stable and closed state (Ram and Meshorer, 2009). Multiscale analyses revealed that differentiation starts with the release of previous constraints that maintained SGE at relative lower levels in progenitors cells, followed by peak of variability that occurs before a reduction of SGE at lower levels than initially because new constraints are applied (Moussy et al., 2017; Richard et al., 2016). From initial widespread and highly variable expression when growth conditions ensuring the maintenance of the stem state are released, cellular entropy progressively decreases from this transient unstable state with the highest SGE to the final stable differentiated state where gene expression profiles are more homogeneous, coordinated, and restricted (Gao et al., 2020; Richard et al., 2016). While a recent study suggested that the number of expression genes is a better indicator than entropy-based metrics in describing developmental potential, it confirmed the global reduction in chromatin accessibility and/or plasticity during lineage commitment by quantitatively linking it to single-cell gene counts (Gulati et al., 2020). Differentiating cells transit from a looser chromatin that permits wider gene expression to a restricted chromatin accessibility and reduced transcriptional diversity. As in microbial cells, specialization is also associated with decrease in cellular stochasticity in metazoan organisms, but instead of leading to optimal proliferation, full differentiation finally leads to proliferation arrest and integration in a tissue through the establishment of a complete cell-cell interaction network. In that case, the relationships are high stochasticity/generalist/proliferation for stem/progenitor cells and low stochasticity/specialist/low proliferation for differentiated cells.
Finally, it is worth noting that stochastic dedifferentiation events also occur in metazoan when a tissue is injured, for instance, or even in normal tissue from differentiated cells during rejuvenation (Chaffer et al., 2011; Stange et al., 2013; Tata et al., 2013). It then allows production of specialist cells as during development, a process that is associated with proliferation arrest (quiescence) because cells contribute to the functioning of the multicellular organism through a collaborative behavior.
Cancer Cells: Cancer Stem Cells (High Stochasticity) versus Other Cells (Low Stochasticity)
In cancer, a subset of cells called CSCs (also known as “cancer-initiating cells” or “cancer stem-like cells”) is suspected to maintain tumor growth and confer therapeutic resistance (Capp, 2019). These cells possess key characteristics of normal stem cells, namely, self-renewal, unlimited proliferation, but infrequent divisions. Also, they are intrinsically associated with increased phenotypic instability and tumor plasticity as demonstrated in glioblastoma (Dirkse et al., 2019). It seems that the CSC state could be acquired by any cell at any time as dedifferentiation is common in the cancerous population (Rios et al., 2019). Such experimental evidences led to stochastic models of CSC to explain how each cell of a tumor could act as CSC (Beck and Blanpain, 2013; Nguyen et al., 2012). Cancer stem-like cells can arise de novo from non-stem-like cells at a low but significant rate (Gupta et al., 2011). Variations in this ability would be due to intrinsic stochastic variations, especially in terms of gene expression, and environmental conditions (Beck and Blanpain, 2013; Nguyen et al., 2012). CSCs would represent a less differentiated subpopulation with generalist strategy with increased variability in tumor, low proliferation, and high plasticity and evolvability (Csermely et al., 2015) allowing cell adaptation to new environmental conditions, especially in case of therapeutic treatment.
In that perspective, the process of epithelial-to-mesenchymal transition (EMT), whose regulation has been largely deciphered (Lu et al., 2013; Zhang et al., 2014), is worth to consider. This transition plays crucial roles in embryonic development, wound healing, and tumorigenesis. EMT generates mesenchymal-like cells and a variety of intermediate cell states between the epithelial and the mesenchymal state (Dongre and Weinberg, 2019). Especially, hybrid EMT states can be observed in lung cancers and constitute a source of tumor heterogeneity (Udyavar et al., 2017). It also contributes to acquire stemness and cell plasticity (Jolly et al., 2014), and EMT cells can function as CSCs (Dongre and Weinberg, 2019). Moreover, whereas EMT was supposed to be a rare event among cancer cells, this event frequently occurs in breast cancers (Rios et al., 2019), suggesting that the differentiation state of tumor cells is inherently unstable or plastic.
Energy metabolism is another critical aspect that impacts epigenetics and plasticity in cancer cells (Kinnaird et al., 2016). Many connections exist between pluripotency and metabolic activity (Paldi, 2013; Perestrelo et al., 2018). The general trend is that ESC differentiation is associated to a metabolic switch from a glycolytic type to an oxidative type (Yanes et al., 2010): stimulation of glycolysis favors dedifferentiation of differentiated cells into pluripotent cells, whereas inhibition of glycolysis or stimulation of oxidative phosphorylation induces differentiation (Folmes et al., 2012; Zhu et al., 2010). In cancer, some works suggested that CSCs preferentially use glycolysis, whereas others reported a propensity for mitochondrial oxidative phosphorylation, suggesting a possible metabolic plasticity (De Francesco et al., 2018; Snyder et al., 2018). Cancer cells actually seem to possess a hybrid metabolic state where cells can use both glycolysis and oxidative phosphorylation (Jia et al., 2019). This would allow cancer cells to adapt to various microenvironments (Yu et al., 2017). The relationship between this hybrid state and stem-like properties is not deciphered yet, but one might expect that it contributes to metabolic plasticity, and possibly to epigenetic plasticity of CSCs.
Disruption of normal epigenetic marks is now considered as the primary source of the CSC behavior. Thus epigenetic events might initiate cancer, as genetic events do (Flavahan et al., 2017). A common explanation evokes that genetic alterations initiating cancer would induce a “tumor reprogramming” that would reset the epigenetic status and gene expression in initially healthy cells (Vicente-Duenas et al., 2015). Oncogenes would cause “developmental reprogramming” of the epigenome (Sanchez-Garcia, 2015) that would allow them to aberrantly differentiate and pathologically proliferate. The developmental epigenetic status is indeed lost in oncogenesis, and cells revert to a “pseudoprimitive” state that combines regulatory DNA features of embryonic stem cells and of other developing lineages (Stergachis et al., 2013). Stochasticity of gene expression appears to be increased in cancer cells at higher levels than normal stem cells because of a less organized and less stable chromatin structure (Jenkinson et al., 2017). Diverging chromatin states and transcriptional heterogeneity are produced by corrupted coordination of epigenetic modifications as exemplified in chronic lymphocytic leukemia (Pastore et al., 2019). Moreover, this increased stochasticity in cancer cells would facilitate acquisition of novel states as suggested in complex dynamical systems theory (Huang et al., 2009; Li et al., 2016). Cancer cell populations interconvert between these phenotypic states on a stochastic basis (Gupta et al., 2011).
Also, CSCs have the ability to produce cells capable of differentiation, but only partially because differentiation appears to be stopped at intermediate stages. These more differentiated cells harbors strongly proliferate and acquire “specialized” functions that allow full exploitation of the available nutritional resources and co-evolution with surrounding cells, either other tumors cells that acquire other specialized functions that enable intratumoral cooperation or cells from the tumoral microenvironment that can also collaborate with tumors cells for mutual benefit. In this pathological system, cooperation occurs for the benefit of the proliferation and survival of the cancer cells, whereas cell-cell cooperation in metazoans occurs for the benefit of the whole organism. The main difference is that they are no more integrated in a normal cell-cell interaction network that is the result of developmental processes.
The now well-established intratumoral phenotypic heterogeneity shows that from initially plastic CSCs, cells with more stable phenotypes can be generated. However, they keep the ability to easily revert to a more variable stem state on a stochastic basis depending on intrinsic and extrinsic factors (Gupta et al., 2011). Finally, a generalist subpopulation with higher stochasticity and weak proliferation (the CSCs) and another specialist population with weak stochasticity and strong proliferation (more differentiated cells) are observed in cancer. There is a clear analogy with microbial populations. On the contrary, in normal metazoan organisms, subpopulations that are specialist with weak stochasticity stop proliferating because they are progressively integrated in a functional tissue. In all cases, generalists are highly plastic and the specialist state is fully reversible, the transitions between the generalist and specialist states being stochastic.
Significance for Oncogenesis: Return of Metazoan Cells to Unicellular Behavior
A Renewed Atavism Theory of Cancer Based on Cellular Stochasticity
The atavism theory of cancer stipulates that cancer is due to a reversion to phylogenetically prior capabilities, namely, an accidental reactivation of a highly conserved survival program encrypted in every eukaryotic cell (and hence in every metazoans) (Davies and Lineweaver, 2011). During the last years, the atavism model has received an increasing attention both because it appears conceptually and chronologically appealing and also because it seems now empirically supported by molecular evidence. For instance, multicellular organisms indeed derive from unicellular ones, and selfish cells responsible for malignancies also appeared during this major transition in the tree of life (see Aktipis et al., 2015; Domazet-Loso et al., 2007; Grosberg and Strathmann, 2007; Michod and Roze, 2001). By suggesting that cancer is a form of ancestral legacy among metazoans, the atavism theory of cancer could provide an explanation to the commonality of traits displayed by malignant cells from different organs and/or species (Thomas et al., 2017). Assuming that ancestral unicellular organisms evolve under adverse and stressful conditions, the atavism theory also provides a plausible explanation to the fact that malignant cells may well proliferate in anoxic/hypoxic and/or unstable conditions (Gravenmier et al., 2018), and also why they can sometimes survive to aggressive treatments like chemotherapeutic drugs and radiation (Vincent, 2016). Finally, in accordance with the hypothesis that cancer derives from an inappropriate re-expression of ancestral toolkits in first unicellular organisms, Trigos et al. (2017) recently showed that malignant cells both overexpress genes and processes with a unicellular cell origin, and underexpress the multicellular cell pathways that have been selected later on to promote cellular cooperation in metazoans.
However, instead of invoking an ancestral program that would be accidentally reactivated in malignant cells, would it be possible that cancer cells could simply resemble microbial cells by the way they exploit cellular stochasticity to generate specialized cells with optimal capacity to exploit nutritional resources for proliferation, while keeping a subpopulation in a more plastic and generalist state? Contrary to the atavism model, tumor adaptations may also result from a somatic evolution occurring, each time (except in the case of transmissible cancers, Ujvari et al., 2016), under strong convergent selective pressures because the microenvironments in which this evolution occur are governed by the same ecological constraints (i.e., the multicellular body). Given that we detect only “successful” tumors (i.e., those that select adaptations allowing their persistence and growth), we propose that selection would retain malignant cells that adopt a microbial cell lifestyle because this strategy, if heritable, could be the most adapted given the new environmental constraints (that are no more the stabilizing constraints that were present in the healthy tissue). Akin to the atavism model, this hypothesis would account for several cancer characteristics without the need to invoke an ancestral legacy. For instance, the genomic instability, which is one of the best-known hallmarks of cancer, could derive from the selection of a unicellular lifestyle (e.g., Yerlici and Landweber, 2014) rather than an atavistic reexpression per se. Alternatively, one could argue that an ancestral program, if finely tuned and adaptive, is unlikely to generate the large amount of aberrant, even non-viable, malignant cells that are commonly produced during the tumorigenesis, while this fact is not surprising in the context of our hypothesis.
The two aforementioned hypotheses are, in fact, not mutually exclusive because the genetic basis yielding by selection (rather than by an accidental reactivation of a program) to a microbial lifestyle in malignant cells may at least partially rely on genes selected before multicellularity evolved. Said differently ancestral genes would allow the cells' evolvability through a microbial lifestyle without strictly corresponding to specific adaptations of ancestral unicellular organisms. At present it appears challenging to determine which scenario and/or their relative contributions, if both occur, is more likely to explain tumorigenesis, because both seem to be capable of accounting for malignant adaptive traits. Also, we cannot exclude that certain cancers could mostly be governed by atavism sensu stricto, whereas others would rely on the selection in malignant cells for a microbial lifestyle, relying on ancestral genes and/or de novo mutations (e.g., Thomas et al., 2017).
Disruption of Tissue Equilibrium at the Origin of Cell of a Unicellular-like Behavior and Speciation Process
Recently, Gatenby et al. proposed a novel hypothesis for carcinogenesis that integrates genetic and non-genetic drivers of somatic evolution (Gatenby et al., 2020). Although the initial genetic state of a cancer cell is the result of mutations that occurred throughout the lifetime of the host, they propose that this tumorigenesis starts only when cells carrying these oncogenic mutations are in an environment promoting their proliferation (Gatenby et al., 2020). Indeed, most of the time cells, even when they harbor oncogenic mutations, remain under control by local tissue constraints (Martincorena et al., 2015; Yizhak et al., 2019; Yokoyama et al., 2019), being also involved in the cooperative functioning that governs the multicellular host as the unit of natural selection. Somatic evolution is likely to occur when those cells become free from these host constraints, because they can have their own Darwinian dynamics. This can happen following various phenomena, e.g., of injury, inflammation, or infection resulting in dysfunctional local controls. According to Gatenby et al. (Gatenby et al., 2020), the mutations accumulated over the lifetime of the host can subsequently serve as the “‘genetic heritage” of the malignant cell, viewed as a novel evolutionary unit of selection. Said differently, the jump to malignant proliferating cells yielding to cancer is equivalent to a speciation event that must be preceded by a set of mutations representing opportunities for normal cells to become self-defined fitness functions on which natural selection can act in an altered environment.
We agree with this model, but we also propose that for cells becoming insensitive to host controls, self-defined fitness functions allowing them to evolve may also rely on the exploitation of a heritable cellular stochasticity to resemble microbial cells. Based on this idea, a more radical model envisages that cellular stochasticity is initially globally increased because of a disruption of tissue equilibrium (Capp, 2005, 2012a, 2017; Capp and Bataille, 2018, 2020). Several studies have shown that disruption of cell-cell interactions can enhance SGE and phenotypic heterogeneity among differentiating cells. In the rat pituitary tissue, direct cell contacts through gap junctions spatially coordinate prolactin gene expression, whereas enzymatic digestion of extracellular proteins or pharmacological inhibition of gap junctions reduced transcriptional coordination between cells (Featherstone et al., 2016). In Drosophila, loss of epidermal growth factor receptor (EGFR) signaling prevents eye cells from differentiating through a prolonged noisy expression of a transcription factor important in the balance between multipotency and differentiation, suggesting that its dynamic heterogeneity is a necessary element of cell-state transitions in the eye, and that cell states are stabilized through noise reduction by EGFR signaling (Pelaez et al., 2015). A similar mechanism was already described in Caenorhabditis elegans embryos where strong signaling is essential to maintain low expression variability and to ensure reliable neuroblast development (Ji et al., 2013). In embryos that do not have some of the four Frizzled receptors of the Wnt pathway, partial penetration migration defects are observed. The authors showed that this phenotypic heterogeneity has its origin in the increased variability of mab-5 expression in the absence of certain receptors. Thus the transcription of mab-5 appears to be inherently highly stochastic, with a modulation of this variability obtained by Wnt signaling: strong signaling reduces its SGE and is needed for proper cell segregation. An example of such phenomenon in cancer could be EMT because it leads to loss of cell-cell contact and increased cellular plasticity, which makes cells become motile and invasive. It is entirely possible that EMT cells increase cellular plasticity and stochasticity by losing cell-cell contact, although it can be also considered as a gain of a specific cellular function.
Thus, in what can be called the TiDiS theory, increased cellular stochasticity and phenotypic instability (that would correspond to a CSC behavior, Capp, 2019) might originate from an initial loss of environmental control by the normal tissue structure, which could be considered as the sole required event to start cancerous transformation (Capp, 2005, 2012a, 2017; Capp and Bataille, 2018, 2020). If genetic alterations are already present, they would accelerate the process, but it can be considered that the process can start without driver mutations. Multiple examples showed that oncogenic mutations are not sufficient to start transformation (Martincorena et al., 2015; Yizhak et al., 2019; Yokoyama et al., 2019), that oncogenesis can initiate from disruption of the micro-environment (Kode et al., 2014; Maffini et al., 2004; Raaijmakers et al., 2010; Walkley et al., 2007), and that cancer cells harboring multiple mutations can be controlled and reverted by a healthy tissue environment (Booth et al., 2011; Bussard et al., 2010; Hochedlinger et al., 2004; Rubin, 2006; for review, see Solary and Lapane, 2020).
Therefore, the unicellular-like behavior of cancer cells and the speciation process would start from cells that do not fully differentiate (from normal stem cells) or dedifferentiate (from differentiated cells) and remains proliferative, because there are no longer in the tumor the “stabilizing” constraints present in the healthy tissue. This would be the original cause for the loss of the association weak stochasticity/specialist/no proliferation and thus the loss of the cell behavior characteristic of multicellularity. Therefore, cellular behavior would return to that of unicellular cells because cell phenotypes are no more stabilized and cells are no more integrated into a multicellular organism with multiple collaborative differentiated cell types. They would start a speciation process that ultimately leads to new collaborative behavior that is pathological because it only serves the development of the tumor, and that is characterized by the association weak stochasticity/specialist/proliferation found for microorganisms.
Conclusion/Perspectives
Our objective here was to propose an alternative viewpoint to the atavism theory, without excluding the possibility that the two hypotheses, instead of being mutually exclusive, potentially represent the two extremes of a continuum in which the diversity of cancers can emerge. Discriminating between our hypothesis and the atavism one sensu stricto will require in the future the development of methodologies that allow to determine whether cancer adaptations result from the reactivation of ancient programs or from the selection of a microbial lifestyle relying on the stochastic activation of ancestral genes and/or novel mutations.
We propose looking with phylogenetic/phylogenomic analyses for characters that are homologous and/or convergent in microbes and unicellular and multicellular organisms, and are also activated in malignant cells. Reciprocally, we propose to explore the proximate/molecular determinants of adaptive traits in malignant cells from various metazoan species, and then comparing them to those acting in the case of microbes. For instance, the detection in microbial genomes of areas displaying strong similarities with those specifically expressed in malignant cells at key stages of the tumorigenesis would be a crucial information. The atavism scenario predicts that the biological function(s) of these areas in microbes should possess a form of “oncogenic value” when transposed in a cancer metazoan context (e.g., resistance to adverse environmental conditions, proliferation, bet hedging strategy, …). Conversely, our hypothesis does not predict such a correspondence in case of similarities. Comparing the proximate determinants of bet-hedging in microbes that regularly express this strategy in case of adverse conditions to those acting in the malignant cells is another promising direction. Converse to our hypothesis, the atavism hypothesis suggests that exacerbated mutation rates in malignant cells should derive from the same precursors than in microbes, as a programmatic adaptation in case of environmental stress.
Traits others than bet-hedging also appear promising to compare, for instance, the motility acquisition in archaic eukaryotes (Friedl, 2004) with the re-acquisition of motility in malignant cells. Traits like pinocytosis or phagocytosis, which are shared by both protozoans and malignant cells (see Coopman et al., 1998; Lewis, 1937) would be promising to explore with this approach. It would be also relevant to explore if the stochasticity expressed by malignant cells is reinforced by mutations associated with tumor suppressor genes or to other parts of the genomes involved in natural cancer defense. Because these protective mechanisms appeared and evolved with multicellular organisms, they are presumably posterior to the unicellular period. Therefore, it could help to discriminate between the two theories.
Finally, further studies will be also necessary to study the extent to which discriminating between our theory and the atavistic model is relevant for elaborating novel therapies. In lines with our model, an innovative therapeutic strategy would consist in returning to the “normal” relationship for cells of multicellular organisms: low stochasticity/specialist/no proliferation. This would be achieved through mimicking the “normal” microenvironment (Capp, 2012b; Capp and Bataille, 2018, 2020; DeGregory, 2018; Kenny and Bissell, 2003) and allowing cells to interact with this “therapeutic” micro-environment to stop their behavior that resembles the one of unicellular cells. Only the restoration of relationships with the microenvironment resembling the ones that occur in healthy tissues would revert the cancer cells and stop the speciation process that in other cases can only result in propagation in the surrounding tissues.
Limitations of the Study
Additional articles may have been published on the topic since submission of the manuscript.
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Jean-Pascal Capp, capp@insa-toulouse.fr.
Materials Availability
Not applicable as this is a perspective article.
Data and Code Availability
Not applicable as this is a perspective article.
Acknowledgments
This work was supported by INRAE, MICA department (“LimitNoise” project to J.-P.C.), an ANR TRANSCAN (ANR-18-CE35-0009 to F.T.), and the Rotary Club Les Sables d’Olonne (to F.T.).
Author Contributions
J.-P.C. and F.T. formulated the hypotheses and wrote the manuscript.
References
- Ackermann M. A functional perspective on phenotypic heterogeneity in microorganisms. Nat. Rev. Microbiol. 2015;13:497–508. doi: 10.1038/nrmicro3491. [DOI] [PubMed] [Google Scholar]
- Aktipis C.A., Boddy A.M., Jansen G., Hibner U., Hochberg M.E., Maley C.C., Wilkinson G.S. Cancer across the tree of life: cooperation and cheating in multicellularity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015;370:20140219. doi: 10.1098/rstb.2014.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck B., Blanpain C. Unravelling cancer stem cell potential. Nat. Rev. Cancer. 2013;13:727–738. doi: 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- Booth B.W., Boulanger C.A., Anderson L.H., Smith G.H. The normal mammary microenvironment suppresses the tumorigenic phenotype of mouse mammary tumor virus-neu-transformed mammary tumor cells. Oncogene. 2011;30:679–689. doi: 10.1038/onc.2010.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock A., Chang H., Huang S. Non-genetic heterogeneity--a mutation-independent driving force for the somatic evolution of tumours. Nat. Rev. Genet. 2009;10:336–342. doi: 10.1038/nrg2556. [DOI] [PubMed] [Google Scholar]
- Bussard K.M., Boulanger C.A., Booth B.W., Bruno R.D., Smith G.H. Reprogramming human cancer cells in the mouse mammary gland. Cancer Res. 2010;70:6336–6343. doi: 10.1158/0008-5472.CAN-10-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capp J.P. Stochastic gene expression, disruption of tissue averaging effects and cancer as a disease of development. Bioessays. 2005;27:1277–1285. doi: 10.1002/bies.20326. [DOI] [PubMed] [Google Scholar]
- Capp J.P. Belin-Pour la science; 2012. Nouveau regard sur le cancer. Pour une révolution des traitements. [Google Scholar]
- Capp J.P. Stochastic gene expression stabilization as a new therapeutic strategy for cancer. Bioessays. 2012;34:170–173. doi: 10.1002/bies.201100149. [DOI] [PubMed] [Google Scholar]
- Capp J.P. Tissue disruption increases stochastic gene expression thus producing tumors: cancer initiation without driver mutation. Int. J. Cancer. 2017;140:2408–2413. doi: 10.1002/ijc.30596. [DOI] [PubMed] [Google Scholar]
- Capp J.P. Cancer stem cells: from historical roots to a new perspective. J. Oncol. 2019;2019:5189232. doi: 10.1155/2019/5189232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capp J.P., Bataille R. Multiple myeloma exemplifies a model of cancer based on tissue disruption as the initiator event. Front Oncol. 2018;8:355. doi: 10.3389/fonc.2018.00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capp J.P., Bataille R. Multiple myeloma as a bone disease? The tissue disruption-induced cell stochasticity (TiDiS) theory. Cancers (Basel) 2020;12:2158. doi: 10.3390/cancers12082158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capp J.P., Laforge B. A Darwinian and physical look at stem cell biology helps understanding the role of stochasticity in development. Front. Cell Dev. Biol. 2020;8:659. doi: 10.3389/fcell.2020.00659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerulus B., New A.M., Pougach K., Verstrepen K.J. Noise and epigenetic inheritance of single-cell division times influence population fitness. Curr. Biol. 2016;26:1138–1147. doi: 10.1016/j.cub.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer C.L., Brueckmann I., Scheel C., Kaestli A.J., Wiggins P.A., Rodrigues L.O., Brooks M., Reinhardt F., Su Y., Polyak K. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. U S A. 2011;108:7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coopman P.J., Do M.T., Thompson E.W., Mueller S.C. Phagocytosis of cross-linked gelatin matrix by human breast carcinoma cells correlates with their invasive capacity. Clin. Cancer Res. 1998;4:507–515. [PubMed] [Google Scholar]
- Csermely P., Hodsagi J., Korcsmaros T., Modos D., Perez-Lopez A.R., Szalay K., Veres D.V., Lenti K., Wu L.Y., Zhang X.S. Cancer stem cells display extremely large evolvability: alternating plastic and rigid networks as a potential Mechanism: network models, novel therapeutic target strategies, and the contributions of hypoxia, inflammation and cellular senescence. Semin. Cancer Biol. 2015;30:42–51. doi: 10.1016/j.semcancer.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Davies P.C., Lineweaver C.H. Cancer tumors as Metazoa 1.0: tapping genes of ancient ancestors. Phys. Biol. 2011;8:015001. doi: 10.1088/1478-3975/8/1/015001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Francesco E.M., Sotgia F., Lisanti M.P. Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. Biochem. J. 2018;475:1611–1634. doi: 10.1042/BCJ20170164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGregory J. Harvard University Press; 2018. Adaptive Oncogenesis: A New Understanding of How Cancer Evolves inside Us. [Google Scholar]
- Dirkse A., Golebiewska A., Buder T., Nazarov P.V., Muller A., Poovathingal S., Brons N.H.C., Leite S., Sauvageot N., Sarkisjan D. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019;10:1787. doi: 10.1038/s41467-019-09853-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domazet-Loso T., Brajkovic J., Tautz D. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends Genet. 2007;23:533–539. doi: 10.1016/j.tig.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Dongre A., Weinberg R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019;20:69–84. doi: 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- Efroni S., Duttagupta R., Cheng J., Dehghani H., Hoeppner D.J., Dash C., Bazett-Jones D.P., Le Grice S., McKay R.D., Buetow K.H. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell. 2008;2:437–447. doi: 10.1016/j.stem.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efroni S., Melcer S., Nissim-Rafinia M., Meshorer E. Stem cells do play with dice: a statistical physics view of transcription. Cell Cycle. 2009;8:43–48. doi: 10.4161/cc.8.1.7216. [DOI] [PubMed] [Google Scholar]
- Egeblad M., Nakasone E.S., Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev. Cell. 2010;18:884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone K., Hey K., Momiji H., McNamara A.V., Patist A.L., Woodburn J., Spiller D.G., Christian H.C., McNeilly A.S., Mullins J.J. Spatially coordinated dynamic gene transcription in living pituitary tissue. Elife. 2016;5:e08494. doi: 10.7554/eLife.08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher C.L., Fisher A.G. Chromatin states in pluripotent, differentiated, and reprogrammed cells. Curr. Opin. Genet. Dev. 2011;21:140–146. doi: 10.1016/j.gde.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Flavahan W.A., Gaskell E., Bernstein B.E. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357:eaal2380. doi: 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes C.D., Dzeja P.P., Nelson T.J., Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012;11:596–606. doi: 10.1016/j.stem.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P. Prespecification and plasticity: shifting mechanisms of cell migration. Curr. Opin. Cell Biol. 2004;16:14–23. doi: 10.1016/j.ceb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Gao N.P., Gandrillon O., Páldi A., Herbach U., Gunawan R. Universality of cell differentiation trajectories revealed by a reconstruction of transcriptional uncertainty landscapes from single-cell transcriptomic data. bioRxiv. 2020 doi: 10.1101/2020.04.23.056069. [DOI] [Google Scholar]
- Gaspar-Maia A., Alajem A., Meshorer E., Ramalho-Santos M. Open chromatin in pluripotency and reprogramming. Nat. Rev. Mol. Cell Biol. 2011;12:36–47. doi: 10.1038/nrm3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby R.A., Avdieiev S., Tsai K.Y., Brown J.S. Integrating genetic and nongenetic drivers of somatic evolution during carcinogenesis: the biplane model. Evol. Appl. 2020;00:1–9. doi: 10.1111/eva.12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giladi A., Paul F., Herzog Y., Lubling Y., Weiner A., Yofe I., Jaitin D., Cabezas-Wallscheid N., Dress R., Ginhoux F. Single-cell characterization of haematopoietic progenitors and their trajectories in homeostasis and perturbed haematopoiesis. Nat. Cell Biol. 2018;20:836–846. doi: 10.1038/s41556-018-0121-4. [DOI] [PubMed] [Google Scholar]
- Gravenmier C.A., Siddique M., Gatenby R.A. Adaptation to stochastic temporal variations in intratumoral blood flow: the Warburg effect as a bet hedging strategy. Bull. Math. Biol. 2018;80:954–970. doi: 10.1007/s11538-017-0261-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosberg R.K., Strathmann R.R. The evolution of multicellularity: a minor major transition? Annu. Rev. Ecol. Evol. Syst. 2007;38:621–654. [Google Scholar]
- Gulati G.S., Sikandar S.S., Wesche D.J., Manjunath A., Bharadwaj A., Berger M.J., Ilagan F., Kuo A.H., Hsieh R.W., Cai S. Single-cell transcriptional diversity is a hallmark of developmental potential. Science. 2020;367:405–411. doi: 10.1126/science.aax0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G., Pinello L., Han X., Lai S., Shen L., Lin T.W., Zou K., Yuan G.C., Orkin S.H. Serum-based culture conditions provoke gene expression variability in mouse embryonic stem cells as revealed by single-cell analysis. Cell Rep. 2016;14:956–965. doi: 10.1016/j.celrep.2015.12.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P.B., Fillmore C.M., Jiang G., Shapira S.D., Tao K., Kuperwasser C., Lander E.S. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- Hochedlinger K., Blelloch R., Brennan C., Yamada Y., Kim M., Chin L., Jaenisch R. Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev. 2004;18:1875–1885. doi: 10.1101/gad.1213504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough S.R., Laslett A.L., Grimmond S.B., Kolle G., Pera M.F. A continuum of cell states spans pluripotency and lineage commitment in human embryonic stem cells. PLoS One. 2009;4:e7708. doi: 10.1371/journal.pone.0007708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S., Ernberg I., Kauffman S. Cancer attractors: a systems view of tumors from a gene network dynamics and developmental perspective. Semin. Cell Dev. Biol. 2009;20:869–876. doi: 10.1016/j.semcdb.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson G., Pujadas E., Goutsias J., Feinberg A.P. Potential energy landscapes identify the information-theoretic nature of the epigenome. Nat. Genet. 2017;49:719–729. doi: 10.1038/ng.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji N., Middelkoop T.C., Mentink R.A., Betist M.C., Tonegawa S., Mooijman D., Korswagen H.C., van Oudenaarden A. Feedback control of gene expression variability in the Caenorhabditis elegans Wnt pathway. Cell. 2013;155:869–880. doi: 10.1016/j.cell.2013.09.060. [DOI] [PubMed] [Google Scholar]
- Jia D., Lu M., Jung K.H., Park J.H., Yu L., Onuchic J.N., Kaipparettu B.A., Levine H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. U S A. 2019;116:3909–3918. doi: 10.1073/pnas.1816391116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly M.K., Huang B., Lu M., Mani S.A., Levine H., Ben-Jacob E. Towards elucidating the connection between epithelial-mesenchymal transitions and stemness. J. R. Soc. Interface. 2014;11:20140962. doi: 10.1098/rsif.2014.0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmar T., Lim C., Hayward P., Munoz-Descalzo S., Nichols J., Garcia-Ojalvo J., Martinez Arias A. Regulated fluctuations in nanog expression mediate cell fate decisions in embryonic stem cells. PLoS Biol. 2009;7:e1000149. doi: 10.1371/journal.pbio.1000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny P.A., Bissell M.J. Tumor reversion: correction of malignant behavior by microenvironmental cues. Int. J. Cancer. 2003;107:688–695. doi: 10.1002/ijc.11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnaird A., Zhao S., Wellen K.E., Michelakis E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer. 2016;16:694–707. doi: 10.1038/nrc.2016.82. [DOI] [PubMed] [Google Scholar]
- Klein A.M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V., Peshkin L., Weitz D.A., Kirschner M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kode A., Manavalan J.S., Mosialou I., Bhagat G., Rathinam C.V., Luo N., Khiabanian H., Lee A., Murty V.V., Friedman R. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature. 2014;506:240–244. doi: 10.1038/nature12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziejczyk A.A., Kim J.K., Tsang J.C., Ilicic T., Henriksson J., Natarajan K.N., Tuck A.C., Gao X., Buhler M., Liu P. Single cell RNA-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell. 2015;17:471–485. doi: 10.1016/j.stem.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotte O., Volkmer B., Radzikowski J.L., Heinemann M. Phenotypic bistability in Escherichia coli's central carbon metabolism. Mol. Syst. Biol. 2014;10:736. doi: 10.15252/msb.20135022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R.M., Cahan P., Shalek A.K., Satija R., DaleyKeyser A.J., Li H., Zhang J., Pardee K., Gennert D., Trombetta J.J. Deconstructing transcriptional heterogeneity in pluripotent stem cells. Nature. 2014;516:56–61. doi: 10.1038/nature13920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G., Hall R.R., 3rd, Ahmed A.U. Cancer stem cells: cellular plasticity, niche, and its clinical relevance. J. Stem Cell Res. Ther. 2016;6:363. doi: 10.4172/2157-7633.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S.F., Ziv N., Siegal M.L. Bet hedging in yeast by heterogeneous, age-correlated expression of a stress protectant. PLoS Biol. 2012;10:e1001325. doi: 10.1371/journal.pbio.1001325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis W.H. Pinocytosis by malignant cells. Cancer Res. 1937;29:666–679. [Google Scholar]
- Li Q., Wennborg A., Aurell E., Dekel E., Zou J.Z., Xu Y., Huang S., Ernberg I. Dynamics inside the cancer cell attractor reveal cell heterogeneity, limits of stability, and escape. Proc. Natl. Acad. Sci. U S A. 2016;113:2672–2677. doi: 10.1073/pnas.1519210113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M., Jolly M.K., Levine H., Onuchic J.N., Ben-Jacob E. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl. Acad. Sci. U S A. 2013;110:18144–18149. doi: 10.1073/pnas.1318192110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur B.D., Lemischka I.R. Statistical mechanics of pluripotency. Cell. 2013;154:484–489. doi: 10.1016/j.cell.2013.07.024. [DOI] [PubMed] [Google Scholar]
- MacArthur B.D., Sevilla A., Lenz M., Muller F.J., Schuldt B.M., Schuppert A.A., Ridden S.J., Stumpf P.S., Fidalgo M., Ma'ayan A. Nanog-dependent feedback loops regulate murine embryonic stem cell heterogeneity. Nat. Cell Biol. 2012;14:1139–1147. doi: 10.1038/ncb2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffini M.V., Soto A.M., Calabro J.M., Ucci A.A., Sonnenschein C. The stroma as a crucial target in rat mammary gland carcinogenesis. J. Cell Sci. 2004;117:1495–1502. doi: 10.1242/jcs.01000. [DOI] [PubMed] [Google Scholar]
- Martincorena I., Roshan A., Gerstung M., Ellis P., Van Loo P., McLaren S., Wedge D.C., Fullam A., Alexandrov L.B., Tubio J.M. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshorer E., Yellajoshula D., George E., Scambler P.J., Brown D.T., Misteli T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell. 2006;10:105–116. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messmer T., von Meyenn F., Savino A., Santos F., Mohammed H., Lun A.T.L., Marioni J.C., Reik W. Transcriptional heterogeneity in naive and primed human pluripotent stem cells at single-cell resolution. Cell Rep. 2019;26:815–824 e814. doi: 10.1016/j.celrep.2018.12.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michod R.E., Roze D. Cooperation and conflict in the evolution of multicellularity. Heredity (Edinb) 2001;86:1–7. doi: 10.1046/j.1365-2540.2001.00808.x. [DOI] [PubMed] [Google Scholar]
- Moignard V., Woodhouse S., Haghverdi L., Lilly A.J., Tanaka Y., Wilkinson A.C., Buettner F., Macaulay I.C., Jawaid W., Diamanti E. Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nat. Biotechnol. 2015;33:269–276. doi: 10.1038/nbt.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussy A., Cosette J., Parmentier R., da Silva C., Corre G., Richard A., Gandrillon O., Stockholm D., Paldi A. Integrated time-lapse and single-cell transcription studies highlight the variable and dynamic nature of human hematopoietic cell fate commitment. PLoS Biol. 2017;15:e2001867. doi: 10.1371/journal.pbio.2001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New A.M., Cerulus B., Govers S.K., Perez-Samper G., Zhu B., Boogmans S., Xavier J.B., Verstrepen K.J. Different levels of catabolite repression optimize growth in stable and variable environments. PLoS Biol. 2014;12:e1001764. doi: 10.1371/journal.pbio.1001764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.V., Vanner R., Dirks P., Eaves C.J. Cancer stem cells: an evolving concept. Nat. Rev. Cancer. 2012;12:133–143. doi: 10.1038/nrc3184. [DOI] [PubMed] [Google Scholar]
- Nguyen Q.H., Lukowski S.W., Chiu H.S., Senabouth A., Bruxner T.J.C., Christ A.N., Palpant N.J., Powell J.E. Single-cell RNA-seq of human induced pluripotent stem cells reveals cellular heterogeneity and cell state transitions between subpopulations. Genome Res. 2018;28:1053–1066. doi: 10.1101/gr.223925.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A., Venkatasubramanian M., Chaudhri V.K., Aronow B.J., Salomonis N., Singh H., Grimes H.L. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature. 2016;537:698–702. doi: 10.1038/nature19348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paldi A. Effects of the in vitro manipulation of stem cells: epigenetic mechanisms as mediators of induced metabolic fluctuations. Epigenomics. 2013;5:429–437. doi: 10.2217/epi.13.35. [DOI] [PubMed] [Google Scholar]
- Pastore A., Gaiti F., Lu S.X., Brand R.M., Kulm S., Chaligne R., Gu H., Huang K.Y., Stamenova E.K., Beguelin W. Corrupted coordination of epigenetic modifications leads to diverging chromatin states and transcriptional heterogeneity in CLL. Nat. Commun. 2019;10:1874. doi: 10.1038/s41467-019-09645-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelaez N., Gavalda-Miralles A., Wang B., Navarro H.T., Gudjonson H., Rebay I., Dinner A.R., Katsaggelos A.K., Amaral L.A., Carthew R.W. Dynamics and heterogeneity of a fate determinant during transition towards cell differentiation. Elife. 2015;4:e08924. doi: 10.7554/eLife.08924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perestrelo T., Correia M., Ramalho-Santos J., Wirtz D. Metabolic and mechanical cues regulating pluripotent stem cell fate. Trends Cell Biol. 2018;28:1014–1029. doi: 10.1016/j.tcb.2018.09.005. [DOI] [PubMed] [Google Scholar]
- Pina C., Fugazza C., Tipping A.J., Brown J., Soneji S., Teles J., Peterson C., Enver T. Inferring rules of lineage commitment in haematopoiesis. Nat. Cell Biol. 2012;14:287–294. doi: 10.1038/ncb2442. [DOI] [PubMed] [Google Scholar]
- Raaijmakers M.H., Mukherjee S., Guo S., Zhang S., Kobayashi T., Schoonmaker J.A., Ebert B.L., Al-Shahrour F., Hasserjian R.P., Scadden E.O. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464:852–857. doi: 10.1038/nature08851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram E.V., Meshorer E. Transcriptional competence in pluripotency. Genes Dev. 2009;23:2793–2798. doi: 10.1101/gad.1881609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard A., Boullu L., Herbach U., Bonnafoux A., Morin V., Vallin E., Guillemin A., Papili Gao N., Gunawan R., Cosette J. Single-cell-based analysis highlights a surge in cell-to-cell molecular variability preceding irreversible commitment in a differentiation process. PLoS Biol. 2016;14:e1002585. doi: 10.1371/journal.pbio.1002585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios A.C., Capaldo B.D., Vaillant F., Pal B., van Ineveld R., Dawson C.A., Chen Y., Nolan E., Fu N.Y., Group D. Intraclonal plasticity in mammary tumors revealed through large-scale single-cell resolution 3D imaging. Cancer Cell. 2019;35:618–632 e616. doi: 10.1016/j.ccell.2019.02.010. [DOI] [PubMed] [Google Scholar]
- Rubin H. What keeps cells in tissues behaving normally in the face of myriad mutations? Bioessays. 2006;28:515–524. doi: 10.1002/bies.20403. [DOI] [PubMed] [Google Scholar]
- Sanchez-Garcia I. How tumour cell identity is established? Semin. Cancer Biol. 2015;32:1–2. doi: 10.1016/j.semcancer.2015.04.004. [DOI] [PubMed] [Google Scholar]
- Semrau S., Goldmann J.E., Soumillon M., Mikkelsen T.S., Jaenisch R., van Oudenaarden A. Dynamics of lineage commitment revealed by single-cell transcriptomics of differentiating embryonic stem cells. Nat. Commun. 2017;8:1096. doi: 10.1038/s41467-017-01076-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegal M.L. Shifting sugars and shifting paradigms. PLoS Biol. 2015;13:e1002068. doi: 10.1371/journal.pbio.1002068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder V., Reed-Newman T.C., Arnold L., Thomas S.M., Anant S. Cancer stem cell metabolism and potential therapeutic targets. Front. Oncol. 2018;8:203. doi: 10.3389/fonc.2018.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solary E., Lapane L. The role of host environment in cancer evolution. Evol. Appl. 2020;13:1756–1770. [Google Scholar]
- Solopova A., van Gestel J., Weissing F.J., Bachmann H., Teusink B., Kok J., Kuipers O.P. Bet-hedging during bacterial diauxic shift. Proc. Natl. Acad. Sci. U S A. 2014;111:7427–7432. doi: 10.1073/pnas.1320063111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivakov M., Fisher A.G. Epigenetic signatures of stem-cell identity. Nat. Rev. Genet. 2007;8:263–271. doi: 10.1038/nrg2046. [DOI] [PubMed] [Google Scholar]
- Sprouffske K., Merlo L.M., Gerrish P.J., Maley C.C., Sniegowski P.D. Cancer in light of experimental evolution. Curr. Biol. 2012;22:R762–R771. doi: 10.1016/j.cub.2012.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stange D.E., Koo B.K., Huch M., Sibbel G., Basak O., Lyubimova A., Kujala P., Bartfeld S., Koster J., Geahlen J.H. Differentiated Troy+ chief cells act as reserve stem cells to generate all lineages of the stomach epithelium. Cell. 2013;155:357–368. doi: 10.1016/j.cell.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergachis A.B., Neph S., Reynolds A., Humbert R., Miller B., Paige S.L., Vernot B., Cheng J.B., Thurman R.E., Sandstrom R. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell. 2013;154:888–903. doi: 10.1016/j.cell.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tata P.R., Mou H., Pardo-Saganta A., Zhao R., Prabhu M., Law B.M., Vinarsky V., Cho J.L., Breton S., Sahay A. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013;503:218–223. doi: 10.1038/nature12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas F., Ujvari B., Renaud F., Vincent M. Cancer adaptations: atavism, de novo selection, or something in between? Bioessays. 2017;39 doi: 10.1002/bies.201700039. [DOI] [PubMed] [Google Scholar]
- Torres-Padilla M.E., Chambers I. Transcription factor heterogeneity in pluripotent stem cells: a stochastic advantage. Development. 2014;141:2173–2181. doi: 10.1242/dev.102624. [DOI] [PubMed] [Google Scholar]
- Trigos A.S., Pearson R.B., Papenfuss A.T., Goode D.L. Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors. Proc. Natl. Acad. Sci. U S A. 2017;114:6406–6411. doi: 10.1073/pnas.1617743114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigos A.S., Pearson R.B., Papenfuss A.T., Goode D.L. How the evolution of multicellularity set the stage for cancer. Br. J. Cancer. 2018;118:145–152. doi: 10.1038/bjc.2017.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott J., Hayashi K., Surani A., Babu M.M., Martinez-Arias A. Dissecting ensemble networks in ES cell populations reveals micro-heterogeneity underlying pluripotency. Mol. Biosyst. 2012;8:744–752. doi: 10.1039/c1mb05398a. [DOI] [PubMed] [Google Scholar]
- Udyavar A.R., Wooten D.J., Hoeksema M., Bansal M., Califano A., Estrada L., Schnell S., Irish J.M., Massion P.P., Quaranta V. Novel hybrid phenotype revealed in small cell lung cancer by a transcription factor network model that can explain tumor heterogeneity. Cancer Res. 2017;77:1063–1074. doi: 10.1158/0008-5472.CAN-16-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujvari B., Gatenby R.A., Thomas F. The evolutionary ecology of transmissible cancers. Infect. Genet. Evol. 2016;39:293–303. doi: 10.1016/j.meegid.2016.02.005. [DOI] [PubMed] [Google Scholar]
- van Dijk D., Dhar R., Missarova A.M., Espinar L., Blevins W.R., Lehner B., Carey L.B. Slow-growing cells within isogenic populations have increased RNA polymerase error rates and DNA damage. Nat. Commun. 2015;6:7972. doi: 10.1038/ncomms8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturelli O.S., Zuleta I., Murray R.M., El-Samad H. Population diversification in a yeast metabolic program promotes anticipation of environmental shifts. PLoS Biol. 2015;13:e1002042. doi: 10.1371/journal.pbio.1002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Duenas C., Hauer J., Ruiz-Roca L., Ingenhag D., Rodriguez-Meira A., Auer F., Borkhardt A., Sanchez-Garcia I. Tumoral stem cell reprogramming as a driver of cancer: theory, biological models, implications in cancer therapy. Semin. Cancer Biol. 2015;32:3–9. doi: 10.1016/j.semcancer.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Vincent M. Cancer: a de-repression of a default survival program common to all cells?: a life-history perspective on the nature of cancer. Bioessays. 2012;34:72–82. doi: 10.1002/bies.201100049. [DOI] [PubMed] [Google Scholar]
- Vincent M. Resistance to cancer chemotherapy as an atavism? Bioessays. 2016;38:1065. doi: 10.1002/bies.201600166. [DOI] [PubMed] [Google Scholar]
- Walkley C.R., Olsen G.H., Dworkin S., Fabb S.A., Swann J., McArthur G.A., Westmoreland S.V., Chambon P., Scadden D.T., Purton L.E. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129:1097–1110. doi: 10.1016/j.cell.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Atolia E., Hua B., Savir Y., Escalante-Chong R., Springer M. Natural variation in preparation for nutrient depletion reveals a cost-benefit tradeoff. PLoS Biol. 2015;13:e1002041. doi: 10.1371/journal.pbio.1002041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware C.B. Concise review: lessons from naive human pluripotent cells. Stem Cells. 2017;35:35–41. doi: 10.1002/stem.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger L., Ayyash M., Novershtern N., Hanna J.H. Dynamic stem cell states: naive to primed pluripotency in rodents and humans. Nat. Rev. Mol. Cell Biol. 2016;17:155–169. doi: 10.1038/nrm.2015.28. [DOI] [PubMed] [Google Scholar]
- Yanes O., Clark J., Wong D.M., Patti G.J., Sanchez-Ruiz A., Benton H.P., Trauger S.A., Desponts C., Ding S., Siuzdak G. Metabolic oxidation regulates embryonic stem cell differentiation. Nat. Chem. Biol. 2010;6:411–417. doi: 10.1038/nchembio.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yerlici V.T., Landweber L.F. Programmed genome rearrangements in the ciliate oxytricha. Microbiol. Spectr. 2014;2:10. doi: 10.1128/microbiolspec.MDNA3-0025-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhak K., Aguet F., Kim J., Hess J.M., Kubler K., Grimsby J., Frazer R., Zhang H., Haradhvala N.J., Rosebrock D. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 2019;364:eaaw0726. doi: 10.1126/science.aaw0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A., Kakiuchi N., Yoshizato T., Nannya Y., Suzuki H., Takeuchi Y., Shiozawa Y., Sato Y., Aoki K., Kim S.K. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. 2019;565:312–317. doi: 10.1038/s41586-018-0811-x. [DOI] [PubMed] [Google Scholar]
- Yu L., Lu M., Jia D., Ma J., Ben-Jacob E., Levine H., Kaipparettu B.A., Onuchic J.N. Modeling the genetic regulation of cancer metabolism: interplay between glycolysis and oxidative phosphorylation. Cancer Res. 2017;77:1564–1574. doi: 10.1158/0008-5472.CAN-16-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Tian X.J., Zhang H., Teng Y., Li R., Bai F., Elankumaran S., Xing J. TGF-beta-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci. Signal. 2014;7:ra91. doi: 10.1126/scisignal.2005304. [DOI] [PubMed] [Google Scholar]
- Zhu S., Li W., Zhou H., Wei W., Ambasudhan R., Lin T., Kim J., Zhang K., Ding S. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell. 2010;7:651–655. doi: 10.1016/j.stem.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable as this is a perspective article.