

Abstract
The numerous biological roles of NAD+ are organized and coordinated via its compartmentalization within cells. The spatial and temporal partitioning of this intermediary metabolite is intrinsic to understanding the impact of NAD+ on cellular signaling and metabolism. We review evidence supporting the compartmentalization of steady-state NAD+ levels in cells, as well as how the modulation of NAD+ synthesis dynamically regulates signaling by controlling subcellular NAD+ concentrations. We further discuss potential benefits to the cell of compartmentalizing NAD+, and methods for measuring subcellular NAD+ levels.
Subcellular Compartmentalization of NAD+ Synthesis, Catabolism, and Function
Oxidized nicotinamide adenine dinucleotide (NAD+) is a metabolic cofactor that plays essential roles in all domains of life. In over a century of research on NAD+, great strides have been made in understanding the synthesis, degradation, and biological functions of NAD+ (Box 1, Tables 1 and 2). Recent interest in the biology of NAD+ has been driven by a desire to modulate its metabolic and signaling pathways to counteract illnesses stemming from metabolic disorders, tumorigenesis, inflammation, cardiovascular disease, neurodegeneration, and the onset of pathologies associated with advancing age.
Box 1. NAD+ Synthesis and Catabolism.
NAD+ comprises two nucleotides (Figure IA). The steady-state levels of NAD+ are determined by the balance between synthesis and utilization, as described subsequently.
NAD+ Synthesis
The biosynthesis of NAD+ occurs in multiple pathways, including de novo synthesis and salvage pathways [4,105] (Table 1 and Figure IB). De novo synthesis originates with tryptophan, and, through the kynurenine pathway, converges with dedicated NAD+ synthesis pathways through quinolinic acid (QA). Quinolinate phosphoribosyltransferase (QPRT) catalyzes the formation of nicotinic acid mononucleotide (NAMN) from QA, which is subsequently converted to nicotinic acid adenine dinucleotide (NAAD) by one of the three NMNAT enzymes. The final step is the amidation of NAAD by NAD synthetase (NADS), leading to the production of NAD+ (Figure IB) [4,105]. De novo synthesis is generally limited to the liver, kidney, and some immune cells [91,106].
In the mammalian salvage pathways, NAD+ is synthesized from the dietary precursors nicotinic acid (NA), NAM, or NR. In the NA (‘Preiss–Handler’) salvage pathway, NAMN is produced from NA by NAPRT and is ultimately converted to NAD+ by NADS (Figure IB). In the NAM and NR pathways, NMN is produced from NAM by NAMPT or from NR by NMRK1, respectively. Although most tissues heavily rely on NMN and NR pathways, there is variability in vivo for the tissue types that can synthesize NAD+ through the Preiss–Handler pathway [91]. Nonetheless, the final step in both pathways is the synthesis of NAD+ from NMN and ATP by one of the NMNAT enzymes (Figure IB) [4,105].
NAD+ Utilization
NAD+ is a cofactor in cellular redox reactions in key metabolic pathways, where it is reduced to NADH without being catabolized [107]. In other contexts, however, NAD+ is consumed (Table 2). For example, PARP enzymes hydrolyze NAD+ to release ADPR groups that are used for the covalent mono- or poly(ADP-ribosyl)ation of proteins [108,109], DNA [110–113], and RNA [114,115]. Likewise, some sirtuins also catalyze mono(ADP-ribosyl)ation of proteins, whereas others catalyze deacylation using ADPR as an acceptor of acyl groups removed from covalently modified proteins [116,117]. NAD+-degrading enzymes include vertebrate NAD+ glycohydrolases, CD38 [76,77], and SARM1 [30,122] (Table 2). These NADases, which are enzymes that catalyze the hydrolysis of NAD+ to ADP-ribose and nicotinamide, have been identified in bacteria, fungi, and mammals, and some are membrane-anchored [118–121]. NAD+ is also phosphorylated by NAD+ kinase to generate NADP+. NADP+ is further reduced by dehydrogenases to generate NADPH, an important cofactor in biosynthetic pathways and for neutralizing reactive oxygen species generated by metabolic activity [123]. Finally, NAD+ is used for the 5′ capping of mRNAs, which promotes metabolite-directed mRNA turnover in mammals [124].
Table 1.
Key Enzymes in the NAD+ Biosynthetic Pathways
| Enzyme | Activity | Pathway(s) |
|---|---|---|
| Quinolinate phosphoribosyl transferase (QPRT) | Catalyzes the formation of nicotinic acid mononucleotide (NAMN) from quinolinic acid (QA)a | De novo pathway |
| NAD+ synthetase (NADS) | Catalyzes the amidation of NAAD, leading to the production of NAD+ | De novo pathway |
| Nicotinic acid phosphoribosyl transferase (NAPRT) | Catalyzes the formation of NAMN from nicotinic acid (NA); rate-limiting step [125] | NA (‘Preiss–Handler’) salvage pathway |
| Nicotinamide phosphoribosyl transferase (NAMPT) | Catalyzes the production of nicotinamide mononucleotide (NMN) from NA; rate-limiting step [126] | NMN salvage pathway |
| Nicotinamide riboside kinase (NMRK) | Catalyzes the production of NMN from NR; rate-limiting step [87,127] | NR pathway |
| Nicotinamide mononucleotide adenylyltransferase (NMNAT) | Catalyzes the formation of nicotinic acid adenine dinucleotide (NAAD) from NAMN, or of NAD+ from NMN | De novo and salvage pathways |
The de novo NAD+ synthesis pathway originates with tryptophan, and through the kynurenine (Kyn) pathway converges with dedicated NAD+ synthesis pathways via quinolinic acid (QA). The Kyn pathway also feeds other metabolic pathways. Indoleamine-2,3-dioxygenase (IDO) and tryptophan-2,3-dioxygenase (TDO) catalyze the first and rate-limiting step of the Kyn pathway. QPRT is positioned to catalyze the rate-limiting step of the specific de novo NAD+ biosynthesis portion of the pathway, but whether it does so is debated in the literature and may be context-specific [125,128–130].
Table 2.
Major Enzymes That Use or Consume NAD+
| Enzyme | Activities |
|---|---|
| Poly(ADP-ribose) polymerases (PARPs) | Cleave NAD+ to release ADP-ribose (ADPR) groups that are used for the covalent mono- or poly(ADP-ribosyl)ation of proteins, DNA, and RNA |
| Sirtuins | Some sirtuins (e.g., SIRT1) catalyze deacylation of proteins using ADPR as an acceptor of acyl groups (i.e., acetyl, succinyl, malonyl, etc.) Other sirtuins (e.g., SIRT4 and SIRT6) catalyze mono(ADP-ribosyl)ation of proteins |
| CD38 | Ectoenzyme that has both NAD+ glycohydrolase and ADP-ribosyl cyclase activities, which may function in decreasing extracellular NAD+ levels |
| SARM1 | Has both NAD+ glycohydrolase and ADP-ribosyl cyclase activities |
| NAD+ kinase | Phosphorylates NAD+ to generate NADP+; this diminishes the pool of NAD+ available for NAD+-specific enzymes and processes |
NAD+-utilizing (see Glossary) enzymes localize to specific subcellular compartments where they execute specific subcellular functions, leading to compartmentalization of NAD+ synthesis and functions. The classic example of NAD+ compartmentalization is the molecular distinction from its reduced counterpart, NADH, as well as from NADP+ and NADPH. These differences are readily discerned in lysates and in vitro. Additional forms of compartmentalization, nevertheless, are not readily distinguishable in these preparations. These include compartmentalization between the free metabolite and protein-bound fractions of NAD+, as well as the temporal and spatial partitioning of each fraction. In the cell, the abundance of NAD+-dependent dehydrogenase enzymes leads to effective sequestration of most of the NAD+ molecules. NAD+ does not function as a prosthetic group and individual NAD+ and NADH molecules are exchanged; however, the proportion of bound NAD+ has the capacity to buffer local availability of free NAD+.
Free intracellular NAD+ concentrations have been measured directly at 100–120 μM in the nucleus and 50–100 μM in the cytoplasm [1–3]; variations in intracellular concentrations depend on cell type, cellular state, and growth conditions [1]. There are many NAD+-utilizing enzymes that localize to the nucleus or cytoplasm and have Km(NAD+) values that approximate this concentration range [4]. As such, these enzymes are poised for regulation by fluctuations in free NAD+ concentrations. For NAD+-consuming enzymes whose catalytic activity is regulated by signaling pathways, the availability of NAD+ and stimulatory signals can function similarly to two-factor verification, where both are required for efficient catalytic activity of the enzyme. Thus, understanding the regulation of intracellular NAD+ concentrations is intrinsic to understanding its impact on specific enzymatic activities in situ. Notably, intracellular NAD+ is also present in mitochondria, peroxisomes, endoplasmic reticulum, and Golgi [5].
Free NAD+ steady-state levels represent the net overall balance between local synthesis rates and consumption. Although NAD+ molecules are derived from oxidization of NADH, this mechanism has less impact on free NAD+ concentrations in nuclear or cytoplasmic compartments compared to synthetic pathways. This is due to the abundance of NAD+ molecules over NADH molecules in these compartments: the free NAD+/NADH ratio ranges between 400:1 and 700:1 in the nucleus and cytoplasm [1,2,6–9]. Thus, even if NADH levels were to change by tenfold, for example, this would only affect NAD+ levels by 2% or less in these compartments. Mitochondrial NAD+/NADH ratios, however, range between 5:1 and 10:1, highlighting the need to distinctly evaluate free NAD+ in specific compartments to avoid conflation of measurements.
Non-Redundant Roles of NAD+ in Different Cellular Compartments
The concept that NAD+ in different parts of the cell has non-overlapping roles is illustrated by mouse genetic models targeting individual nicotinamide mononucleotide (NMN) adenylyltransferase (NMNAT) enzymes. Many species, including humans, depend on more than one NMNAT enzyme to synthesize NAD+ [10–15] (Table 3). Mammalian NMNAT enzymes have varying expression levels, activities, and subcellular localizations across different tissues [16–18]. NMNAT1 is nuclear, whereas NMNAT2 is associated with the Golgi and acts in the cytoplasm [19–21]. NMNAT3 can localize to the mitochondria or cytoplasm, and its localization and function depend on the cell type [19,22,23].
Table 3.
Homologs of Nicotinamide Mononucleotide Adenylyltransferasea
| Species | NMNAT1 | NMNAT2 | NMNAT3 |
|---|---|---|---|
| H. sapiens | NMNAT1 | NMNAT2 | NMNAT3 |
| M. musculus | Nmnat1 | Nmnat2 | Nmnat3 |
| C. porcellus | Nmnat1 | Nmnat2 | Nmnat3 |
| C. lupus | NMNAT1 | NMNAT2 | NMNAT3 |
| G. gallus | NMNAT1 | NMNAT2 | NMNAT3 |
| B. taurus | NMNAT1 | NMNAT2 | DAA33127 |
| D. rerio | nmnat1 | nmnat2 | nmnat3 |
| X. tropicalis | nmnat1 | nmnat2 | |
| C. elegans | F26H9.4 | W06B3.1 | |
| D. melanogaster | Nmnat | ||
| S. cerevisiae | NMA1/NMA2 |
Data curated from the Ensembl, EggNOG, NCBI Orthologs, and Homologene databases (accessed January 2020).
Distinct Subcellular Requirements for NAD+
Mice genetically ablated for either nuclear or cytoplasmic NMNAT activities are non-viable [24,25], indicating that the functions of the remaining enzymes cannot compensate for the specific loss of NMNAT activity in either the nucleus or cytoplasm, even if NAD+ molecules can traverse between these compartments. Importantly, different Nmnat deletions result in different underlying causes of lethality. Homozygous Nmnat1 mice are embryonic lethal [24]. By contrast, homozygous Nmnat2 mice are born, but are malformed and have distended bladders as well as major muscle innervation and neural development problems [25,26]. Homozygous deletion of Nmnat3 is also lethal; the animals die postnatally from anemia owing to loss of red blood cells, reflecting a cytosolic role for the enzyme in these cells [22]. The distinct timing of when Nmnat knockout mice die – embryonic, perinatal, and postnatal – may additionally suggest that NMNAT activities, and possibly the requirement for various NAD+ pools, are temporally regulated.
The Location of NAD+ Synthesis Matters
Relocalization of NAD+ synthesis to a compartment that has lost its NMNAT activity can be sufficient to rescue specific phenotypes. In cultured dorsal root ganglion neurons from rodents, loss of axonal NMNAT2 results in Wallerian axonal degeneration [27]. Restoration of as little as 25–50% of NMNAT2 activity can prevent degeneration of the axon [27]. Notably, NMNAT activity concurrently prevents the accumulation of precursor NMN that is toxic when NAD+ levels are diminished, and also restores NAD+ levels in these neurons [28–30]. Interestingly, overexpression of NMNAT1 in the nucleus [31] is insufficient to block induced nerve damage, but mislocalization of NMNAT1 to the axonal cytoplasm blocks degeneration [32,33]. Ectopic expression of the NMAT1 fusion protein from the WldS (slow Wallerian degeneration) mutant mouse similarly protects against induced Wallerian degeneration [34,35]. WldS generates a gain-of-function fusion protein with NMNAT activity, and its ability to block axonal degeneration depends on its localization to the axon and its catalytic activity [34,35]. These data indicate that the location of NAD+ synthesis is crucial, and that NMNAT catalytic activity – not a non-enzymatic function [36] – governs this particular phenotype. Interestingly, human disease alleles of NMNAT1 and NMNAT2 have been identified [37–43], suggesting that subcellular NAD+ regulation is similarly compartmentalized and non-redundant in humans.
Together, studies indicate that NAD+ and the pathways of NAD+ biosynthesis serve non-redundant functions in different parts of the cell, and that different pools may be pertinent at different times. The studies also indicate that regulation of NAD+ synthesis can play a large role in directing the local concentrations and compartmentalization of NAD+.
NAD+ Compartmentalization for Regulation
NAD+ compartmentalization plays an important role in regulating biological outcomes, as illustrated in the following examples.
Temporal Regulation of Circadian Rhythms
Modulation of subcellular NAD+ synthesis can regulate the timing of signaling pathways. Mammalian circadian rhythms are coordinated with metabolic activity through controlled expression of NAMPT (Figure 1A) [44–47]. In both the suprachiasmatic nucleus and other circadian tissues, such as liver, NAMPT is directly upregulated by the master regulator CLOCK–BMAL1, a histone acetyl transferase and transcription factor complex [44,45]. Regulation of NAMPT, in turn, results in oscillating NAD+ levels [44,45]. The rhythmic oscillation of NAD+ serves as a feedback ‘timer’ by modulating the activities of NAD+-dependent enzymes, including sirtuins [44–46], helping to establish the periodicity of the cycles. NAMPT oscillations also dictate mitochondrial NAD+ levels and coordinate cellular respiration with awake periods [47]. In these cases, modulation of NAMPT levels drives the rise and fall of NAD+ concentrations that serve to limit the duration of sirtuin activity.
Figure 1. NAD+ Compartmentalization for Regulation.
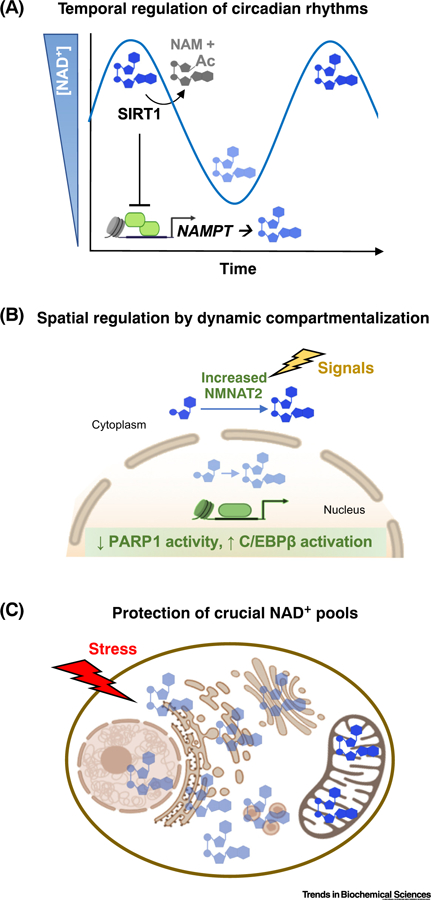
The levels of free NAD+ are tightly controlled both temporally and spatially to precisely modulate signaling in cells. (A) Temporal fluctuations in NAD+ concentrations regulate circadian periodicity rhythms. (B) Spatial partitioning of NAD+ biosynthetic pathways permits signal-dependent initiation of adipogenic differentiation. (C) NAD+ compartmentalization may protect and prioritize support for particularly crucial cellular processes (e.g., in the mitochondria) in response to stress when other subcellular stores are depleted. Abbreviation: Ac, acylation.
Spatial Regulation by Dynamic Compartmentalization
In addition to upregulating specific biosynthetic enzymes to increase local NAD+-dependent activity, these enzymes can be recruited or localized to cellular regions where there may be a high need. For example, NMNAT1 is recruited to genomic loci where PARP1 serves a catalytic-dependent regulatory role (e.g., gene promoters) [48,49], as well as nucleoli [50]. Likewise, NAMPT in HepG2 cells can shuttle between the nucleus and cytoplasm during the S/G2/M phases and G1 phases of the cell cycle, respectively [51].
During the early stages of adipogenesis, a decrease in free nuclear NAD+ concentrations limits PARP1 activity (Figure 1B) [3]. Instead of relocating the NAD+ biosynthetic enzymes, however, adipogenesis-inducing hormones or elevated glucose levels promote an increase in NMNAT2 protein levels, resulting in increased cytoplasmic NAD+ concentrations. During the process, the upregulation of NMNAT2 depletes the availability of the precursor NMN, making it limiting for nuclear NMNAT1. Without sufficient NMN to robustly drive NMNAT1 activity, nuclear NAD+ concentrations simultaneously fall. As a result, the PARP1 activity that depends on nuclear NAD+ is limited, allowing a C/EBPβ adipogenic transcriptional program to drive differentiation of precursor cells into adipocytes [3]. Thus, increased flux of NAD+ biosynthesis in the cytosol can also limit PARP1 activity by competing with and limiting the ability of the cell to synthesize sufficient NAD+ in the nucleus.
Protection of Crucial NAD+ Pools
Compartmentalization affords cells the ability to protect specific pools of NAD+ – particularly life-crucial pools. For example, following genotoxic stress, induction of pseudohypoxic states, or chemically induced NAD+ depletion, mitochondria retain NAD+ levels longer than the nucleus or cytoplasm, and mitochondrial NAD+ levels decrease only after these other compartments have been depleted (Figure 1C) [52–54]. Intriguingly, the continuous turnover of mitochondrial NAD+ by resident consuming enzymes suggests that this pool is regularly replenished. Respiring mitochondria, in particular, depend on NAD+-dependent SIRT3 to prevent buildup of inactivating acylation of mitochondrial enzymes [4]. The prevailing view is that minimal salvaging of NAD+ takes place in mitochondria [53,55,56]. Depletion of the mitochondrial pool of NAD+ only after exhaustion of the cytosolic pool hints at additional regulation between the compartments.
When NAD+ Compartmentalization Goes Awry
Given the importance of NAD+ compartmentalization in key biological systems such as those noted previously, non-physiological alterations in NAD+ compartmentalization are expected to be a driver of disease. Although direct evidence of a role for NAD+ compartmentalization in NAD+-related pathologies (e.g., cancer, diseases of aging) is limited, recent studies are beginning to hint at its importance. For example, the same NMNAT1/ NMNAT2-driven NAD+ compartmentalization that regulates adipogenesis, as noted previously, is also operational in neuroblastoma cells [3]. Many questions remain to be addressed to obtain a complete picture regarding NAD+ compartmentalization in cancer and aging in humans.
NAD+ Compartmentalization in Cancer
Much of the metabolism in highly proliferative cells, including cancerous cells, centers around supporting anabolic and glycolytic pathways in the absence of mitochondrial respiration [57], suggesting that mitochondrial respiration is coordinated with the rest of the cell. Many tumors upregulate the expression of NAD+ biosynthetic enzymes, nicotinate phosphoribosyltransferase (NAPRT) and nicotinamide phosphoribosyltransferase (NAMPT); NAPRT is upregulated via gene amplification and NAMPT is epigenetically upregulated at its promoter [58]. These two enzymes localize predominantly to the cytoplasm and contribute to the cytoplasmic pool of NAD+ [59]. Increased NAD+ in the cytoplasm can promote cell proliferation by supporting glycolysis, as well as serving as a source for NADPH through NAD kinase (NADK). Moreover, the glycolytic enzyme GAPDH shuttles cytoplasmic NAMPT to the nucleus to increase nuclear NAD+ via a direct interaction promoted by cellular stress in transformed epithelial cancer lines [60].
These observations suggest the possibility that changes in subcellular NAD+ levels can be used to communicate between different parts of a cell when mitochondrial function is diminished or when there is a need for increased NAD+ outside the mitochondria. Initial rerouting of NAD+ from mitochondria to the cytoplasm may serve this function because cytoplasmic NAD+ serves as a source for the mitochondrial pool [2,55,61] and alterations in its distribution can affect proliferation [61]. As such, reorganization of subcellular NAD+ levels could represent an early initiating step in tumorigenesis. Because many tumor types broadly depend on upregulated NAD+ for proliferation [58], targeting this pool of NAD+ may have potential as a therapeutic intervention.
NAD+ Compartmentalization in Aging
Declining NAD+ levels have been proposed to underlie the onset of pathologies associated with advanced age, and link age as a risk factor to diseases such as neurodegeneration and cancer. As suggested by preclinical data, lowered NAD+ levels diminish the ability of cells to replenish niches, counteract inflammation and senescence, repair damage, and regulate metabolic pathways [62]. Moreover, lowered NAD+ levels can directly contribute to cellular stress by promoting a reductive environment. Nevertheless, the extent to which decreased NAD+ levels contribute to aging in healthy humans, notably, is still debated [63–67]. Despite evidence for age-dependent decreases of NAD+ in preclinical models, these observations have not always translated directly to humans. Age-related reductions in NAD+ in humans may require particular stressors, be limited to specific cell types or subcellular compartments, or represent modulations of specific pathways. Moreover, alterations in NAD+ compartmentalization generally, or alterations in NAD+ levels in a specific compartment, in the pathologies of aging have not been determined. As such, the complete picture regarding specific cell types, subcellular compartments, and the extent of NAD+ loss in human aging and disease models remains elusive.
On the Outside Looking In: An Extracellular Compartment for NAD+?
In this review we have focused on the synthesis and functions of NAD+ in intracellular compartments. NAD+, however, is also present in the extracellular milieu as extracellular NAD+ (eNAD+) [68], which may be considered as another compartment for NAD+.
Origins and Consumption of eNAD+
Despite of the presence of dimerized extracellular NAMPT (eNAMPT), which can make NMN [69], the enzymes necessary to catalyze the final step of NAD+ synthesis are not known to reside extracellularly [70]. This has raised questions about the provenance of eNAD+. Current thinking is that eNAD+ arises from NAD+ that is synthesized intracellularly and either released upon cell lysis or through pore-forming proteins, such as connexin 43 (Cx43) hemichannels [68,71,72].
More is known about the consumption of eNAD+ than about its origins. The structurally and functionally related NAD+ glycohydrolases CD38 and CD157 are ectoenzymes that hydrolyze eNAD+ to cyclic ADP-ribose (cADPR) and ADPR, which in turn can serve as paracrine second messengers to ultimately mediate intracellular calcium mobilization and signaling [73–75]. Notably, CD38 also targets circulating NAD+ precursors, such as NMN and nicotinamide riboside (NR), for degradation, and the liberated products can be directed to NAD+ biosynthetic pathways inside the cell [76,77]. Studies in Cd38 knockout mice have demonstrated that CD38 is a major consumer of NAD+ in mammals. Thus, CD38 has the potential to govern the biology of eNAD+ by controlling its levels [78].
Biology of eNAD+
Much of our understanding of the biology of eNAD+ comes from studies using Cd38 knockout mice, which have revealed a variety of outcomes that link CD38 to physiological functions, presumably through its ability to degrade eNAD+ and generate cADPR and ADPR as second messengers [73,74]. Defects observed in the knockout mice include impaired neutrophil chemotaxis, defective oxytocin release, and aberrant social behavior, as well as altered tumorigenic potential [73,79,80]. Several recent studies have reported that both intracellular and extracellular NAD+ levels decline with age [81], which occurs with a concurrent increase in CD38 protein levels in various tissues [78,82]. Cd38 knockout mice have twice the levels of NAD+ and are resistant to the age-associated decline in NAD+ [83,84]. By contrast, mice engineered to overexpress CD38 have lower levels of NAD+ and exhibit age-related mitochondrial dysfunction mediated, in part, by regulation of SIRT3 activity [78]. Interestingly, adipose tissue-specific overexpression of NAMPT in mice, which maintains plasma eNAMPT levels and NAD+ synthesis in various tissues, is associated with improved glucose-stimulated insulin secretion, photoreceptor function, and cognitive function [85]. Although the pathway that would lead from NMN synthesis by eNAMPT to increases in eNAD+ is unclear, the parallels between Cd38 knockout and NAMPT overexpression in mice suggest a shared mechanism involving eNAD+.
Adding more complexity, a cell-surface transporter, SLC12A8, was recently reported to mediate the influx of extracellular NMN in the gut [86]. In addition, intracellular nicotinamide riboside kinase (NMRK) has been reported to be rate-limiting for extracellular NMN and NR in NAD+ synthesis [87]. These paradoxical mechanisms, where extracellular NMN is either directly imported or requires dephosphorylation to enter cells as NR, may very well represent mechanisms of compartmentalization in vivo that call for further study. Overall, the available evidence suggests that, although the extracellular space does not function as a separate compartment for NAD+ synthesis, it may represent a separate compartment for NAD+ function. As such, it is likely to be subject to the same temporal and spatial considerations discussed previously for intracellular NAD+.
Methods to Study NAD+ Compartmentalization
The interior of any cell is not a uniform milieu: cells are organized with membrane-bound organelles, macromolecular scaffolding and tracks, and subcompartments with distinct viscosities. The complexities of NAD+ compartmentalization – including being able to distinguish free intracellular fractions from total NAD+ levels – present challenges for monitoring and determining NAD+ concentrations in real time and in vivo. An ideal method would dynamically monitor free NAD+ in an organism with subcellular resolution, be minimally invasive, not compromise the existing pool, and be able to overlook circumstantial differences in oxygen levels and pH in its report of free NAD+. Although each currently available method has its trade-offs, and an all-encompassing perfect solution remains elusive, much progress has been made towards addressing the intricacies of monitoring intracellular NAD+.
Trade-Offs in Spatial and Temporal Resolution
Many commonly applied techniques to monitor NAD+ rely on cellular extracts. Unfortunately, the preparation of extracts compromises intrinsic spatial information from the sample, especially at the subcellular scale, and cannot be used reliably to evaluate free NAD+. These are limitations of colorimetric assays, chromatography, nuclear magnetic resonance, and mass spectrometry (MS). Methods to isolate organelles or subcellular fractions cannot ensure that metabolites remain in their original subcellular compartment (Figure 2). In situ adaptations of these techniques (e.g., MS imaging and gold nanocluster-based probes) are still subject to the challenges of precisely separating out and quantifying free NAD+ from the more abundant protein-bound NAD+ fraction. Available determinations and estimations indicate that the free fraction only comprises 12–25% of total NAD+ [1,2,6]; total NAD+ measurements range from ~300 μM [52,88,89] to as high as 800 μM [90,91] in mammalian cells. The higher abundance of the protein-bound fraction likely overwhelms the detection of free NAD+ in total measurements. In addition, extract-based approaches cannot repeatedly measure from the same cell over time. A minimally invasive in vivo magnetic resonance NAD+ assay has been developed that can distinguish NAD+ from NADH and has been used in physiological settings for patient diagnosis [89,92]. The major trade-offs, however, are that it cannot distinguish free from bound fractions, and the approach is limited for observing fluctuations at the subcellular scale.
Figure 2. Specificity and Resolution when Measuring Intracellular NAD+.

Schematic representations of the specificity and resolution of various methods for measuring intracellular NAD+. (A) Methods are broadly ordered by their ability to both directly and specifically measure the free fraction of intracellular NAD+. Consideration of the entire methodology is taken into account. For example, analytical methods, such as LC-MS/MS, HPLC, and NMR, directly and very specifically measure intracellular NAD+; however, the preparation of biological samples for analysis may conflate free and bound fractions. (B) Methods are organized according to their ability to obtain dynamic measurements from the same locale, as well as by their spatial resolution. Abbreviations: LC, liquid chromatography; MRI, magnetic resonance imaging; MS, mass spectrometry; NMR, nuclear magnetic resonance.
Pitfalls of Indirect Estimations
Indirect projections based on lactate dehydrogenase activity or NADH have historically been used successfully to estimate steady-state levels during homeostasis [6,7]. However, the assumptions necessary to calculate these concentrations are not always valid in all scenarios [8,93]. In particular, observed changes in NADH do not necessarily reflect alterations in free NAD+ levels, and monitoring changes in free NADH levels can overestimate resulting changes in NAD+ concentrations. This is due to the disproportionately high NAD+/NADH ratio in many compartments.
Genetically Encoded Approaches
NAD+, unlike NADH, has no intrinsic fluorescence, and absorbance cannot distinguish its free fraction [7,94]. Currently, the best methods for measuring compartmentalized NAD+ have used genetically encoded sensors (Figure 2) [1,2]. This technique permits full control over the expression of the probe, including restricting its expression to specific cell types, temporally controlling when it is expressed or active, and targeting it to subcellular localization via encoded localization sequences or tethering [95]. In addition, genetically encoded sensors are readily adaptable to in vivo applications [95,96].
An early NAD+ sensor, PARAPLAY, uses the catalytic domain of PARP1. This domain can be auto-modified by poly(ADP-ribose) in an NAD+-dependent manner, and the extent of modification as assessed by Western blotting depends on NAD+ availability [5,56]. Appreciable challenges, nevertheless, include applying this approach to quantitatively measure NAD+ in cells and ensuring that expression of the catalytically active probe minimally affects the local NAD+ pool and its downstream targets. Multiple genetically encoded ratiometric NADH/NAD+ sensors also have been developed and have been invaluable for NADH intracellular measurements [8,9,97,98]. However, because the concentration of free NAD+ exceeds that of free NADH, the current ratiometric NADH/NAD+ sensors become saturated and cannot be used in cells for NAD+ measurements.
To address the need for dedicated NAD+ sensors, two first-generation in situ sensors were developed [1,2]. A semisynthetic sensor called NAD+-Snifit was developed by the laboratory of Johnsson based on their innovative Snifit (snap-tag indicator fluorescent intramolecular tether) system [1,99]. Snifit designs work by an analyte-dependent conformational change to the probe that alters fluorescence resonance energy transfer (FRET). In the case of NAD+-Snifit, binding of free NAD+ induces an intramolecular interaction to increase FRET between a donor associated with a tethered SNAP-tag and an energy acceptor associated with a Halo-tag directly adjacent to the NAD+ binding protein (sulfapyridine). The modularity of the Snifit design permits its successful adaptation for the detection of ions, neurotransmitters, drugs, and other metabolites, including NADP+ and NADPH [100]. Moreover, Snifits can use fluorescent proteins, synthetic dyes, or luciferase to provide resonant energy; readouts can be either fluorescence or bioluminescence. A series of sensitive bioluminescent Snifit sensors for NAD+ – headlined by NAD-cpLuc and NAD-cpLuc2 – were also recently developed as in vitro paper-based assays to facilitate point-of-care readings from biological samples and extracts [101]. These new sensors cover NAD+ concentrations from 10−8 to 10−4 M using a color-change readout of emitted light from blue to red upon NAD+ detection. They represent the most sensitive sensors to date and have multifold dynamic ranges. Although not technically intended for use in situ, they hold great promise for measurements of eNAD+ that may be at much lower concentrations relative to intracellular NAD+.
An independent NAD+ sensor was initiated as a collaboration between the laboratories of Goodman and Cohen [2], with the goal of ultimately facilitating in vivo studies of intracellular NAD+. The fluorescence readout of this sensor is fully genetically encoded and the complete sensor is encoded by a single polypeptide. The sensor combines a permutated NAD+-binding domain modeled after Enterococcus faecalis LigA with the circularly permutated fluorescent protein cpVenus144/145. Binding of local free NAD+ is detected by decreased fluorescence intensity from cpVenus. As a general strategy, circularly permutated fluorescent proteins have aided intracellular sensor designs since the late 1990s, notably the original development of a genetically encoded calcium indicator [102,103].
The development of these sensors has allowed measurement of free subcellular NAD+ for the first time [2]. By achieving subcellular resolution, the sensors established that (i) intracellular NAD+ concentrations are compartmentalized, (ii) subcellular regulation differs by cell type, (iii) local free NAD+ levels match the reported Km(NAD+) values of many NAD+ signaling enzymes, and (iv) lowering NAD+ levels limits the activity of these enzymes in cells [1,2]. Moreover, the new methodologies represent founding tools that can be customized and optimized for the pursuit of new research directions.
Significant technical challenges remain for moving the technologies in vivo and expanding their utility to different physiological compartments. Better evaluation of long-term tolerance, minimizing buffering effects from expression of the probe, and increasing the dynamic range of readouts would all represent important improvements. Advances continue to be made; the developers of SoNAR, originally an NADH/NAD+ sensor, recently re-engineered it as FiNAD to preferentially monitor relative NAD+ levels [104]. The advantages of FiNAD include a large dynamic range (like its predecessor); utility for measurements in bacteria, vertebrates, mammalian cells, and primary human cells; and (like NAD+-Snifit) utility in experiments using high levels of precursor supplementation. However, FiNAD has a Km(NAD+) of ~1.3 mM, limiting its use in mammalian cells. With a Km(NADH) of ~100 μM, crossreactivity with NADH greater than 10 μM further limits the use of FiNAD for mitochondria measurements, as well as in anaerobic or hypoxic cultures [104].
A Call for Additional Designs
Understanding the advantages and limitations of each technology is important. In lieu of a single perfect method for monitoring intracellular NAD+ across all species, compartments, and scenarios, the future will likely include multiple sensors where each serves a specialized purpose. As such, second-generation in situ technologies will need to be developed, and will likely require combinations of both rationale design and unbiased screening [103]. One can imagine a molecular toolbox of customized NAD+ sensors for specific queries. For example, the major differences between fluorescent protein- and NAD+-Snifit-based designs represent distinct advantages and limitations depending on the circumstances. The current fluorescent protein-based sensors are ultimately sensitive to pH and oxygen levels owing to the cpYFP or cpVenus component [2,104]. NAD+-Snifit can utilize pH and oxygen-insensitive FRET pair dyes [1], but reliance on two exogenously applied dyes may spectrally limit options for multiplexing NAD+-Snifit in vivo. In this regard, the utility of each sensor should be evaluated based on its intended application. For example, a sensor designed for measurements in mammalian cells does not need to respond robustly to changes in E. coli [104].
Additional utility will come from different sensors that are sensitive for NAD+ at different ranges, reflecting differences in the expected concentrations in various cellular milieus, cell types, and species. Intracellular measurements for peroxisomes, endoplasmic reticulum, and Golgi, are currently lacking, and the possibility of focal regulation of NAD+ at specific chromatin loci or sites of DNA damage has not been addressed. In addition, expanded palettes for sensors with new wavelengths would be welcomed, especially in the IR range to minimize phototoxicity. Lastly, multiplexing these sensors with detection of NADH, NADP+, and NADPH molecules and the complement of metabolomic data will be crucial to uncover the full role of compartmentalized intracellular NAD+ signaling.
Concluding Remarks
NAD+ is a crucial metabolite and signaling molecule whose detailed functions and biology are incompletely characterized, at best. We are only beginning to elucidate how the temporal and spatial compartmentalization of NAD+ contributes to its numerous biological roles. The emerging view is that NAD+ concentrations are partitioned and dynamically modulated by NAD+ synthesis within the cell. Determining the extent of these mechanisms in vivo and their impact on signaling pathways will be a rich area for future research (see Outstanding Questions). Continued development of new methodologies to measure intracellular NAD+ will be essential to this effort. Because NAD+ is crucial for how organisms in all domains of life organize and extract energy, NAD+ regulation is integral to every discipline and field of study in biology and medicine. Not only are its chemistry and mechanisms part of the history of biochemistry, but its broad biological impact will undoubtedly also continue to bring together expertise across multiple disciplines in the future. The next wave of discoveries in NAD+ biology will be driven by collaborations among scientists with distinct skillsets, including physiologists, enzymologists, cell and molecular biologists, evolutionary biologists, engineers, chemists, physicists, and bioinformaticians.
Outstanding Questions.
What are the regulatory roles for the compartmentalization of NAD+ in vivo? For a full understanding, questions will need to be addressed in cells, as well as at the organ and organismal levels, in both physiological and disease contexts. The application of genetically encoded sensors in model organisms will be crucial for achieving these goals.
How are the roles for NAD+ compartmentalization conserved in different organisms? Different species possess variations in their NAD+ biosynthetic pathways.
How do NAD+ levels in specific compartments interplay with NADH, NADP+, and NADPH levels, as well as with other signaling metabolites?
What is the variation of compartmentalized NAD+ in individual cells?
How do fluctuations in the proportion of protein-bound NAD+ affect free NAD+ concentrations in different parts of the cell?
Can we design methods to precisely regulate NAD+ concentrations in specific cellular compartments?
Figure I. Chemical Structure and Biosynthesis of NAD+.

(A) Chemical structure of oxidized NAD+. NAD+ comprises two nucleotides, one containing an adenine nucleobase and the other containing nicotinamide, joined through their phosphate groups. The location of various chemical moieties contained within NAD+ are indicated: ADP-ribose (ADPR), nicotinamide (NAM), nicotinamide riboside (NR), and nicotinamide mononucleotide (NMN). (B) NAD+ is synthesized de novo in a pathway leading from tryptophan (blue), as well as in ‘salvage’ pathways: (i) the nicotinic acid (NA) (‘Preiss–Handler’) salvage pathway (yellow), (ii) the nicotinamide mononucleotide (NMN) salvage pathway (green), and (iii) the nicotinamide riboside (NR) salvage pathway (green). The abbreviations for the enzymes (gray text) and the intermediates (black plain text) are defined in Table 1.
Highlights.
NAD+ serves an essential role as an electron acceptor (via hydride transfer) in central carbon metabolism. In the absence of intracellular NAD+, cells cannot produce ATP. However, even moderate diminishments in NAD+ levels can limit the signaling activity of NAD+-consuming enzymes.
NAD+ concentrations differ in different parts of the cell, and there are distinct subcellular requirements for NAD+.
Dynamic modulation of subcellular, and possibly extracellular, NAD+ concentrations represent an emerging mechanism for regulating specific NAD+-dependent pathways.
Compartmentalization of NAD+ helps to time responses, communicate cellular status, and protect crucial NAD+ pools.
Genetically encoded sensors represent promising approaches for additional development to generate a molecular toolbox for measuring and studying fluctuations in levels of compartmentalized NAD+.
Acknowledgments
We thank A. Jones, K. Ryu, S. Challa, J. Eller, and M. Stokes for critical comments and suggestions on this work. NAD+-related research in our laboratories is supported by the National Institutes of Health (NIH)/National Institute of General Medical Sciences (NIGMS) grant DP2GM126897 (to X.A.C.), and by the NIH National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant R01 DK069710 and funds from the Cecil H. and Ida Green Center for Reproductive Biology Sciences Endowment (to W.L.K.).
Glossary
- Adipogenesis
the developmental process by which precursor cells develop into adipocytes (fat cells)
- Compartmentalization
separated into isolated or distinct subcellular locales
- Ectoenzyme
an enzyme that is located on, and has catalytic activity at, the surface of a cell, directed towards the exterior of the cell
- Free metabolite
the fraction of a metabolite that is not currently bound to or associated with protein. Typically, this can be considered as the amount of available metabolite
- Multiplexing
combining more than one assay in a simultaneous analysis
- NAD+ biosynthesis
the ordered and regulated series of chemical reactions that lead to the generation of an NAD+ molecule
- NAD+ utilization
the use of NAD+ either as a coenzyme in metabolic reactions or as a substrate for degradation
- Phototoxicity
toxic effects on the cell resulting from exposure to illumination from lasers and high-intensity arc-discharge lamps
- Salvage pathway
a biosynthetic pathway that recycles intermediates from a degradative process
- Subcellular
refers to a defined location, organelle, or structure within a cell. It can also refer to an event or molecule that occurs or resides within the confines of a particular space within a cell
Footnotes
Disclaimer Statement
X.A.C. is an inventor on US patent 10 392 649 covering the NAD+ cpVenus-based sensor described herein. W.L.K. is a founder and consultant for Ribon Therapeutics. He is also an inventor on US patent 9 599 606 covering a set of ADP-ribose detection reagents which have been licensed to and are sold by EMD Millipore.
References
- 1.Sallin O et al. (2018) Semisynthetic biosensors for mapping cellular concentrations of nicotinamide adenine dinucleotides. Elife 7, e32638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cambronne XA et al. (2016) Biosensor reveals multiple sources for mitochondrial NAD+. Science 352, 1474–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryu KW et al. (2018) Metabolic regulation of transcription through compartmentalized NAD+ biosynthesis. Science 360, eaan5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Houtkooper RH et al. (2010) The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr. Rev 31, 194–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dolle C et al. (2010) Visualization of subcellular NAD pools and intra-organellar protein localization by poly-ADP-ribose formation. Cell. Mol. Life Sci 67, 433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williamson DH et al. (1967) The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J 103, 514–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Q et al. (2002) Regulation of corepressor function by nuclear NADH. Science 295, 1895–1897 [DOI] [PubMed] [Google Scholar]
- 8.Hung Yin P. et al. (2011) Imaging cytosolic NADH–NAD+ redox state with a genetically encoded fluorescent biosensor. Cell Metab 14, 545–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao Y et al. (2011) Genetically encoded fluorescent sensors for intracellular NADH detection. Cell Metab 14, 555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emanuelli M et al. (2000) Molecular cloning, chromosomal localization, tissue mRNA levels, bacterial expression, and enzymatic properties of human NMN adenylyltransferase. J. Biol. Chem 276, 406–412 [DOI] [PubMed] [Google Scholar]
- 11.Fernando FS et al. (2002) Human homologue of a gene mutated in the slow Wallerian degeneration (C57BL/Wlds) mouse. Gene 284, 23–29 [DOI] [PubMed] [Google Scholar]
- 12.Raffaelli N et al. (2002) Identification of a novel human nicotinamide mononucleotide adenylyltransferase. Biochem. Biophys. Res. Commun 297, 835–840 [DOI] [PubMed] [Google Scholar]
- 13.Zhang X et al. (2003) Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J. Biol. Chem 278, 13503–13511 [DOI] [PubMed] [Google Scholar]
- 14.Felici R et al. (2013) Insight into molecular and functional properties of NMNAT3 reveals new hints of NAD homeostasis within human mitochondria. PLoS ONE 8, e76938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bockwoldt M et al. (2019) Identification of evolutionary and kinetic drivers of NAD-dependent signaling. Proc. Natl. Acad. Sci. U. S. A 116, 15957–15966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorci L et al. (2007) Initial-rate kinetics of human NMN-adenylyltransferases: substrate and metal ion specificity, inhibition by products and multisubstrate analogues, and isozyme contributions to NAD+ biosynthesis. Biochemistry 46, 4912–4922 [DOI] [PubMed] [Google Scholar]
- 17.Orsomando G et al. (2012) Simultaneous single-sample determination of NMNAT isozyme activities in mouse tissues. PLoS ONE 7, e53271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mori V et al. (2014) Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS ONE 9, e113939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berger F et al. (2005) Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem 280, 36334–36341 [DOI] [PubMed] [Google Scholar]
- 20.Lau C et al. (2010) Isoform-specific targeting and interaction domains in human nicotinamide mononucleotide adenylyltransferases. J. Biol. Chem 285, 18868–18876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayer PR et al. (2010) Expression, localization, and biochemical characterization of nicotinamide mononucleotide adenylyltransferase 2. J. Biol. Chem 285, 40387–40396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hikosaka K et al. (2014) Deficiency of nicotinamide mononucleotide adenylyltransferase 3 (Nmnat3) causes hemolytic anemia by altering the glycolytic flow in mature erythrocytes. J. Biol. Chem 289, 14796–14811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto M et al. (2016) Nmnat3 is dispensable in mitochondrial NAD Level maintenance in vivo. PLoS ONE 11, e0147037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conforti L et al. (2011) Reducing expression of NAD+ synthesizing enzyme NMNAT1 does not affect the rate of Wallerian degeneration. FEBS J 278, 2666–2679 [DOI] [PubMed] [Google Scholar]
- 25.Hicks AN et al. (2012) Nicotinamide mononucleotide adenylyltransferase 2 (Nmnat2) regulates axon integrity in the mouse embryo. PLoS ONE 7, e47869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gilley J and Coleman MP (2010) Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol 8, e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilley J et al. (2013) Rescue of peripheral and CNS axon defects in mice lacking NMNAT2. J. Neurosci 33, 13410–13424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Stefano M et al. (2015) A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ 22, 731–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu HW et al. (2018) Pharmacological bypass of NAD+ salvage pathway protects neurons from chemotherapy-induced degeneration. Proc. Natl. Acad. Sci. U. S. A 115, 10654–10659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Essuman K et al. (2017) The SARM1 Toll/interleukin-1 receptor domain possesses intrinsic NAD+ cleavage activity that promotes pathological axonal degeneration. Neuron 93, 1334–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conforti L et al. (2006) NAD+ and axon degeneration revisited: Nmnat1 cannot substitute for WldS to delay Wallerian degeneration. Cell Death Differ 14, 116–127 [DOI] [PubMed] [Google Scholar]
- 32.Babetto E et al. (2010) Targeting NMNAT1 to axons and synapses Transforms its neuroprotective potency in vivo. J. Neurosci 30, 13291–13304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sasaki Y et al. (2016) NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD+ depletion. Elife 5, e19749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J et al. (2005) A local mechanism mediates NAD-dependent protection of axon degeneration. J. Cell Biol 170, 349–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang MS et al. (2001) The WldS protein protects against axonal degeneration: a model of gene therapy for peripheral neuropathy. Ann. Neurol 50, 773–779 [DOI] [PubMed] [Google Scholar]
- 36.Zhai RG et al. (2008) NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature 452, 887–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiang P-W et al. (2012) Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat. Genet 44, 972–974 [DOI] [PubMed] [Google Scholar]
- 38.Falk MJ et al. (2012) NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet 44, 1040–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koenekoop RK et al. (2012) Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet 44, 1035–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perrault I et al. (2012) Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat. Genet 44, 975–977 [DOI] [PubMed] [Google Scholar]
- 41.Nash BM et al. (2017) NMNAT1 variants cause cone and cone-rod dystrophy. Eur. J. Hum. Genet 26, 428–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huppke P et al. (2019) Homozygous NMNAT2 mutation in sisters with polyneuropathy and erythromelalgia. Exp. Neurol 320, 112958. [DOI] [PubMed] [Google Scholar]
- 43.Lukacs M et al. (2019) Severe biallelic loss-of-function mutations in nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) in two fetuses with fetal akinesia deformation sequence. Exp. Neurol 320, 112961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramsey KM et al. (2009) Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 324, 651–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakahata Y et al. (2009) Circadian control of the NAD+ salvage pathway by CLOCK–SIRT1. Science 324, 654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rutter J (2001) Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 293, 510–514 [DOI] [PubMed] [Google Scholar]
- 47.Peek CB et al. (2013) Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342, 1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang T et al. (2012) Regulation of poly(ADP-ribose) polymerase-1-dependent gene expression through promoter-directed recruitment of a nuclear NAD+ synthase. J. Biol. Chem 287, 12405–12416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang T et al. (2009) Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J. Biol. Chem 284, 20408–20417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song T et al. (2013) The NAD+ synthesis enzyme nicotinamide mononucleotide adenylyltransferase (NMNAT1) regulates ribosomal RNA transcription. J. Biol. Chem 288, 20908–20917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Svoboda P et al. (2019) Nuclear transport of nicotinamide phosphoribosyltransferase is cell cycle-dependent in mammalian cells, and its inhibition slows cell growth. J. Biol. Chem 294, 8676–8689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang H et al. (2007) Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130, 1095–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pittelli M et al. (2010) Inhibition of nicotinamide phosphoribosyltransferase. J. Biol. Chem 285, 34106–34114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gomes Ana P. et al. (2013) Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155, 1624–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davila A et al. (2018) Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. Elife 7, e33246 614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nikiforov A et al. (2011) Pathways and subcellular compartmentation of NAD biosynthesis in human cells. J. Biol. Chem 286, 21767–21778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vander Heiden MG and DeBerardinis RJ (2017) Understanding the intersections between metabolism and cancer biology. Cell 168, 657–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chowdhry S et al. (2019) NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 569, 570–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nikiforov A et al. (2011) Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem 286, 21767–21778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grolla AA et al. (2020) A nicotinamide phosphoribosyltransferase–GAPDH interaction sustains the stress-induced NMN/NAD+ salvage pathway in the nucleus. J. Biol. Chem 295, 3635–3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.VanLinden MR et al. (2015) Subcellular distribution of NAD+ between cytosol and mitochondria determines the metabolic profile of human cells. J. Biol. Chem 290, 27644–27659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Katsyuba E and Auwerx J (2017) Modulating NAD+ metabolism, from bench to bedside. EMBO J 36, 2670–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chaleckis R et al. (2016) Individual variability in human blood metabolites identifies age-related differences. Proc. Natl. Acad. Sci. U. S. A 113, 4252–4259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Elhassan YS et al. (2019) Nicotinamide riboside augments the aged human skeletal muscle NAD+ metabolome and induces transcriptomic and anti-inflammatory signatures. Cell Rep 28, 1717–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Massudi H et al. (2012) Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7, e42357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou CC et al. (2016) Hepatic NAD+ deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br. J. Pharmacol 173, 2352–2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Clement J et al. (2019) The plasma NAD+ metabolome is dysregulated in ‘normal’ aging. Rejuvenation Res 22, 121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haag F et al. (2007) Extracellular NAD and ATP: partners in immune cell modulation. Purinergic Signal 3, 71–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carbone F et al. (2017) Regulation and function of extracellular nicotinamide phosphoribosyltransferase/visfatin. Compr. Physiol 7, 603–621 [DOI] [PubMed] [Google Scholar]
- 70.Nikiforov A et al. (2015) The human NAD metabolome: functions, metabolism and compartmentalization. Crit. Rev. Biochem. Mol. Biol 50, 284–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bruzzone S et al. (2001) Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J 15, 10–12 [DOI] [PubMed] [Google Scholar]
- 72.Fruscione F et al. (2011) Regulation of human mesenchymal stem cell functions by an autocrine loop involving NAD+ release and P2Y11-mediated signaling. Stem Cells Dev 20, 1183–1198 [DOI] [PubMed] [Google Scholar]
- 73.Quarona V et al. (2013) CD38 and CD157: a long journey from activation markers to multifunctional molecules. Cytometry B Clin. Cytom 84, 207–217 [DOI] [PubMed] [Google Scholar]
- 74.Malavasi F et al. (2008) Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev 88, 841–886 [DOI] [PubMed] [Google Scholar]
- 75.Fliegert R et al. (2019) Adenine nucleotides as paracrine mediators and intracellular second messengers in immunity and inflammation. Biochem. Soc. Trans 47, 329–337 [DOI] [PubMed] [Google Scholar]
- 76.Chini EN et al. (2018) The pharmacology of CD38/NADase: an emerging target in cancer and diseases of aging. Trends Pharmacol. Sci 39, 424–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hogan KA et al. (2019) The multi-faceted ecto-enzyme CD38: roles in immunomodulation, cancer, aging, and metabolic diseases. Front. Immunol 10, 1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Camacho-Pereira J et al. (2016) CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab 23, 1127–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bu X et al. (2018) CD38 knockout suppresses tumorigenesis in mice and clonogenic growth of human lung cancer cells. Carcinogenesis 39, 242–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jin D et al. (2007) CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 446, 41–45 [DOI] [PubMed] [Google Scholar]
- 81.Imai S and Guarente L (2014) NAD+ and sirtuins in aging and disease. Trends Cell Biol 24, 464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aksoy P et al. (2006) Regulation of SIRT 1 mediated NAD dependent deacetylation: a novel role for the multifunctional enzyme CD38. Biochem. Biophys. Res. Commun 349, 353–359 [DOI] [PubMed] [Google Scholar]
- 83.Escande C et al. (2013) Flavonoid apigenin is an inhibitor of the NAD+ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 62, 1084–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Barbosa MT et al. (2007) The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J 21, 3629–3639 [DOI] [PubMed] [Google Scholar]
- 85.Yoshida M et al. (2019) Extracellular vesicle-contained eNAMPT delays aging and extends lifespan in mice. Cell Metab 30, 329–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Grozio A et al. (2019) Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab 1, 47–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ratajczak J et al. (2016) NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun 7, 13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Trammell SAJ and Brenner C (2013) Targeted, LCMS-based metabolomics for quantitative measurement of NAD+ metabolites. Comput. Struct. Biotechnol. J 4, e201301012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhu X-H et al. (2015) In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci 112, 2876–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen WW et al. (2016) Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell 166, 1324–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu L et al. (2018) Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab 27, 1067–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lu M et al. (2014) Intracellular redox state revealed by in vivo 31P MRS measurement of NAD+ and NADH contents in brains. Magn. Reson. Med 71, 1959–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sun F et al. (2012) Biochemical issues in estimation of cytosolic free NAD/NADH ratio. PLoS ONE 7, e34525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stringari C et al. (2011) Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proc. Natl. Acad. Sci 108, 13582–13587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bolbat A and Schultz C (2017) Recent developments of genetically encoded optical sensors for cell biology. Biol. Cell 109, 1–23 [DOI] [PubMed] [Google Scholar]
- 96.Chen T-W et al. (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bilan DS et al. (2014) Genetically encoded fluorescent indicator for imaging NAD+/NADH ratio changes in different cellular compartments. Biochim. Biophys. Acta Gen. Subj 1840, 951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhao Y et al. (2015) SoNar, a highly responsive NAD+/NADH sensor, allows high-throughput metabolic screening of antitumor agents. Cell Metab 21, 777–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brun MA et al. (2009) Semisynthetic fluorescent sensor proteins based on self-labeling protein tags. J. Am. Chem. Soc 131, 5873–5884 [DOI] [PubMed] [Google Scholar]
- 100.Farrants H et al. (2017) Rational design and applications of semisynthetic modular biosensors: SNIFITs and LUCIDs. Methods Mol. Biol 1596, 101–117 [DOI] [PubMed] [Google Scholar]
- 101.Yu Q et al. (2019) A biosensor for measuring NAD+ levels at the point of care. Nat. Metab 1, 1219–1225 [DOI] [PubMed] [Google Scholar]
- 102.Baird GS et al. (1999) Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci 96, 11241–11246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sanford L and Palmer A (2017) Recent advances in development of genetically encoded fluorescent sensors. Methods Enzymol 569, 1–49 [DOI] [PubMed] [Google Scholar]
- 104.Zou Y et al. (2020) Illuminating NAD+ metabolism in live cells and in vivo using a genetically encoded fluorescent sensor. Dev. Cell 53, 240–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kulkarni CA and Brookes PS (2019) Cellular compartmentation and the redox/nonredox functions of NAD+. Antioxid. Redox Signal 31, 623–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Minhas PS et al. (2019) Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol 20, 50–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jones DP and Sies H (2015) The redox code. Antioxid. Redox Signal 23, 734–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gibson BA and Kraus WL (2012) New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol 13, 411–424 [DOI] [PubMed] [Google Scholar]
- 109.Gupte R et al. (2017) PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 31, 101–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jankevicius G et al. (2016) The toxin–antitoxin system DarTG catalyzes reversible ADP-ribosylation of DNA. Mol. Cell 64, 1109–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Munnur D and Ahel I (2017) Reversible mono-ADP-ribosylation of DNA breaks. FEBS J 284, 4002–4016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zarkovic G et al. (2018) Characterization of DNA ADP-ribosyltransferase activities of PARP2 and PARP3: new insights into DNA ADP-ribosylation. Nucleic Acids Res 46, 2417–2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Talhaoui I et al. (2016) Poly(ADP-ribose) polymerases covalently modify strand break termini in DNA fragments in vitro. Nucleic Acids Res 44, 9279–9295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Munnur D et al. (2019) Reversible ADP-ribosylation of RNA. Nucleic Acids Res 47, 5658–5669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Munir A et al. (2018) NAD+-dependent synthesis of a 5’-phospho-ADP-ribosylated RNA/DNA cap by RNA 2’-phosphotransferase Tpt1. Nucleic Acids Res 46, 9617–9624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Houtkooper RH et al. (2012) Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol 13, 225–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bheda P et al. (2016) The substrate specificity of sirtuins. Annu. Rev. Biochem 85, 405–429 [DOI] [PubMed] [Google Scholar]
- 118.Cho YS et al. (1998) Auto-ADP-ribosylation of NAD glycohydrolase from Neurospora crassa. Comp. Biochem. Physiol. B Biochem. Mol. Biol 120, 175–181 [DOI] [PubMed] [Google Scholar]
- 119.Ghosh J et al. (2010) Characterization of Streptococcus pyogenes beta-NAD+ glycohydrolase: re-evaluation of enzymatic properties associated with pathogenesis. J. Biol. Chem 285, 5683–5694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim UH et al. (1993) Purification and characterization of NAD glycohydrolase from rabbit erythrocytes. Arch. Biochem. Biophys 305, 147–152 [DOI] [PubMed] [Google Scholar]
- 121.Kim UH et al. (1988) Membrane-associated NAD+ glycohydrolase from rabbit erythrocytes is solubilized by phosphatidylinositol-specific phospholipase C. Biochim. Biophys. Acta 965, 76–81 [DOI] [PubMed] [Google Scholar]
- 122.Carty M and Bowie AG (2019) SARM: From immune regulator to cell executioner. Biochem. Pharmacol 161, 52–62 [DOI] [PubMed] [Google Scholar]
- 123.Tedeschi PM et al. (2016) NAD+ kinase as a therapeutic target in cancer. Clin. Cancer Res 22, 5189–5195 [DOI] [PubMed] [Google Scholar]
- 124.Kiledjian M (2018) Eukaryotic RNA 5′-end NAD+ capping and deNADding. Trends Cell Biol 28, 454–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Audrito V et al. (2020) NAMPT and NAPRT: two metabolic enzymes with key roles in inflammation. Front. Oncol 10, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Revollo JR et al. (2004) The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem 279, 50754–50763 [DOI] [PubMed] [Google Scholar]
- 127.Fletcher RS et al. (2017) Nicotinamide riboside kinases display redundancy in mediating nicotinamide mononucleotide and nicotinamide riboside metabolism in skeletal muscle cells. Mol. Metab 6, 819–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xiao W et al. (2018) NAD(H) and NADP(H) redox couples and cellular energy metabolism. Antioxid. Redox Signal 28, 251–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Castro-Portuguez R and Sutphin GL (2020) Kynurenine pathway, NAD+ synthesis, and mitochondrial function: Targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol 132, 110841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shibata K (2018) Organ co-relationship in tryptophan metabolism and factors that govern the biosynthesis of nicotinamide from tryptophan. J. Nutr. Sci. Vitaminol. (Tokyo) 64, 90–98 [DOI] [PubMed] [Google Scholar]