

Abstract
Orthostatic hypotension (OH) is a common clinical manifestation characterized by a significant fall in blood pressure with postural change and is frequently accompanied by debilitating symptoms of orthostatic intolerance. The reported prevalence of OH ranges between 5 to 10% in middle-aged adults with a burden that increases concomitantly with age; in those over 60 years of age, the prevalence is estimated to be over 20%. Unfortunately, the clinical course of OH is not necessarily benign. OH patients are at an increased risk of adverse clinical outcomes including death, falls, cardiovascular and cerebrovascular events, syncope, and impaired quality of life.
The differential diagnosis of OH is broad and includes acute precipitants as well as chronic underlying medical conditions, especially of neurological origin. Appropriate diagnosis relies on a systematic history and physical examination with particular attention to orthostatic vital signs, keeping in mind that ambient conditions during diagnostic testing may affect OH detection due to factors such as diurnal variation.
INTRODUCTION
Orthostatic hypotension (OH) is a clinical manifestation that may or may not be accompanied by symptoms of orthostatic intolerance. It is defined as a sustained reduction of systolic blood pressure (SBP) of at least 20 mmHg or diastolic blood pressure (DBP) of 10 mmHg within three minutes of orthostasis.1 The gravitational shift in blood volume that occurs with active standing is opposed by contraction of skeletal muscle in the legs. Despite this opposition, venous return remains sufficiently compromised to elicit an increase in sympathetic outflow to the heart and blood vessels, and a decrease in parasympathetic vagal activity, which are required to maintain adequate cardiac output. OH occurs when this compensatory autonomic response to standing fails to generate adequate vasoconstriction, resulting in a fall in BP.
The prevalence of OH increases with disorders that affect autonomic nerve transmission and with age, ranging from 5 to 10% in the middle-aged population to over 20% in those older than 60 years.1–3 The reasons for the increased frequency of OH in elderly persons are unclear but likely multifactorial. These may include an increased prevalence of autonomic neurodegenerative disease, polypharmacy with medications known to worsen OH, malnutrition, deconditioning, and age-related physiological changes, such as a blunted baroreceptor response to upright posture and increased rates of axon degeneration that accompany the normal aging process.4,5
Symptomatic OH has a substantial impact on quality of life due to lightheadedness, blurred vision, leg buckling, and syncope that may predispose patients to falls and resultant physical trauma. Additionally, OH is associated with adverse clinical outcomes, such as increased mortality in middle-aged adults, and higher rates of myocardial infarction, heart failure, atrial fibrillation, cerebrovascular events, and cognitive decline in the elderly.6–9 The short- and long-term health complications associated with OH make it a common cause of hospitalization, especially among elderly patients, incurring a significant economic burden on the health care system.10 Thus, appropriate diagnosis of OH and its underlying etiology are critical to alleviate manifest symptoms, improve patient quality of life, and mitigate associated adverse events.
In this review, we describe the differential diagnosis of OH including two broad classification schemata: (a) one based on pathophysiology – neurogenic vs. non-neurogenic, and (b) the other based on temporal changes in orthostatic BP – classical OH, delayed OH (DOH), and initial OH (IOH). This review will also discuss the clinical approach to OH including a detailed history and physical examination with particular emphasis on careful collection of orthostatic vital signs, as well as further investigations that may be useful to help identify the underlying etiology in select settings.
CLINICAL VARIANTS OF ORTHOSTATIC HYPOTENSION
There are three clinical variants of OH that are differentiated on the basis of temporal changes in orthostatic BP: classical OH (Figure 1A), DOH (Figure 1B), and IOH (Figure 1C). Identifying the OH variant at time of initial assessment is clinically relevant as they differ in clinical course and prognosis. For example, one longitudinal study found that more than half of patients initially presenting with DOH developed the classical type over time, and many of whom also developed associated symptoms of neurodegenerative diseases involving the autonomic nervous system.11 Conversely, IOH generally has a benign clinical course without significant neurological morbidity, although it may predispose individuals to syncope.12
Figure 1.
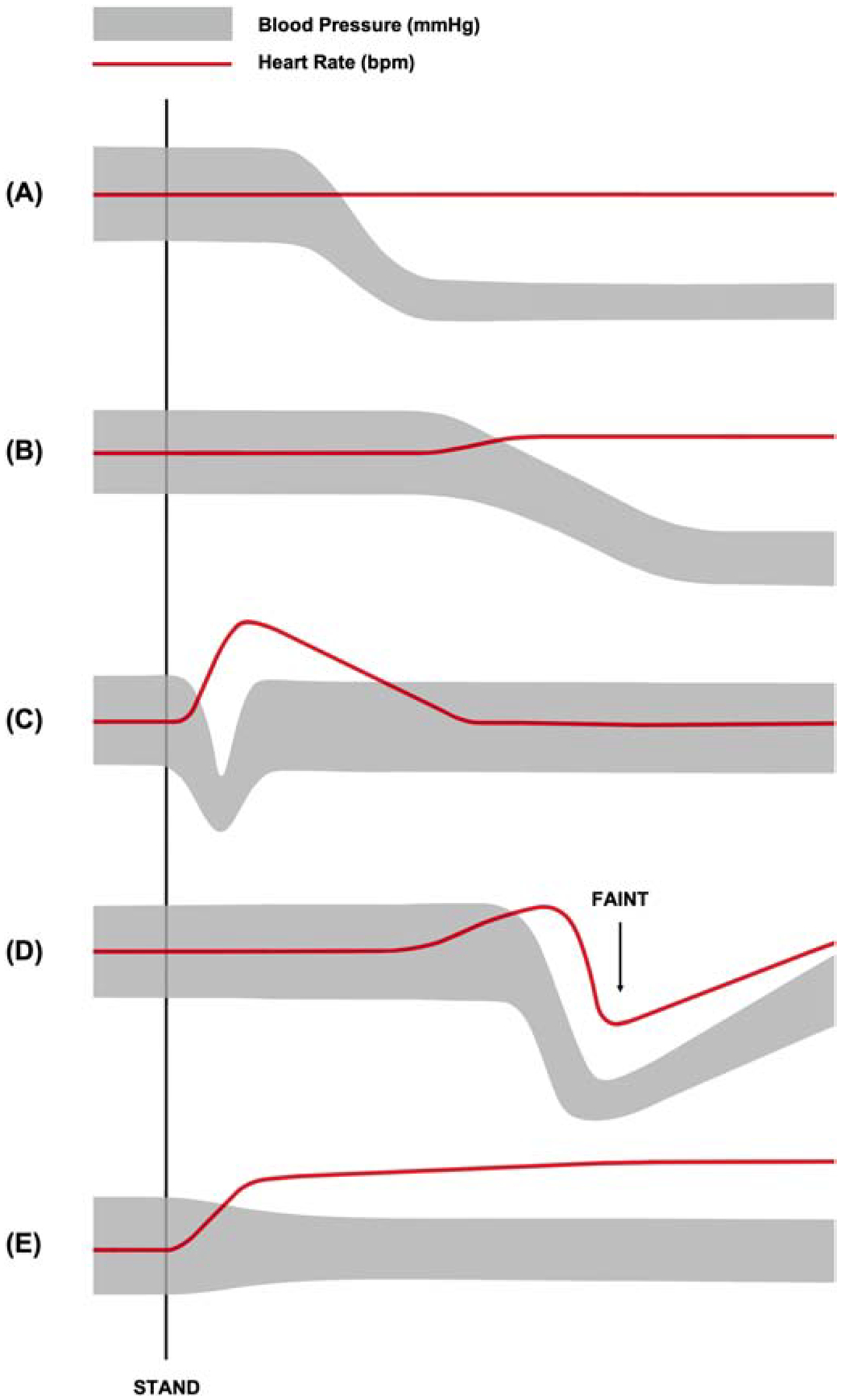
Representative blood pressure and heart rate responses to postural change from supine to active stand in (A) classical orthostatic hypotension, (B) delayed orthostatic hypotension, (C) initial orthostatic hypotension, (D) vasovagal syncope, and (E) postural tachycardia syndrome.
Classical Orthostatic Hypotension
Classical OH is characterized by a sustained reduction in SBP of at least 20 mmHg or DBP of at least 10 mmHg within three minutes of standing or head-up tilt to ≥60° (Table 1).1 In patients with supine hypertension, which occurs in approximately half of patients with nOH, a threshold reduction in BP of 30/15 mmHg may be more appropriate for diagnosis since the magnitude of orthostatic BP decline is measured from the supine position (Table 1).1 Additionally, the majority of classical, non-neurogenic OH cases will exhibit compensatory tachycardia (≥25 bpm) on standing, a response that is severely blunted in nOH (<20 bpm) due to autonomic impairments affecting sympathetic and parasympathetic HR modulation.13
Table 1.
Defining characteristics of orthostatic hypotension (OH) variants, vasovagal syncope (VVS), and postural tachycardia syndrome (POTS).
| Disorder | Hemodynamic Change(s) with Standing | ||
|---|---|---|---|
| Direction and Magnitude | Time of Onset | Duration | |
| Classical OH | ↓ SBP ≥20 mmHg or ↓ DBP ≥10 mmHg | ≤3 min | Sustained |
| Classical OH + Supine HTN | ↓ SBP/DBP ≥30/15 mmHg | ≤3 min | Sustained |
| Delayed OH | ↓ SBP ≥20 mmHg or ↓ DBP ≥10 mmHg | ≥3 min | Sustained |
| Initial OH | ↓ SBP ≥40 mmHg and/or ↓ DBP ≥20 mmHg | ≤15 s | Transient (resolves within 45–60 s of onset) |
| VVS | Preserved BP/HR until syncope prodrome Time of faint: ↓ BP and ↓ HR | Variable | Until faint |
| POTS | ↑ HR ≥30 mmHg No OH | ≤10 min | Sustained |
Abbreviations: SBP, systolic blood pressure; DBP, diastolic blood pressure; HR, heart rate; HTN, hypertension; HR, heart rate
Delayed Orthostatic Hypotension
Patients with postural orthostatic vitals and symptoms characteristic of classical OH, but occur beyond three minutes of standing have DOH (Table 1). Importantly, DOH has been shown to progress to OH in more than half of DOH cases over a 10-year period, many of whom also develop progressive neurodegenerative diseases.11 This suggests that DOH is an early or milder form of adrenergic dysfunction that progresses over time with a poor prognosis.11
Initial Orthostatic Hypotension
IOH is defined as a transient SBP decrease of at least 40 mmHg or DBP decrease of at least 20 mmHg within 15 seconds of standing, typically accompanied by momentary symptoms of cerebral hypoperfusion.1,14 Patients experience complete resolution of symptoms within 45 to 60 seconds of their onset (Table 1).15 Unlike the classical and delayed OH variants, IOH does not appear to be associated with an increased risk of morbidities or mortality.16 It is thought that IOH is merely caused by a delay in vasoconstrictive compensation for gravitational loading during postural change rather than abnormal autonomic physiology.17 This phenomenon is frequently seen in young, otherwise healthy individuals, but generally does not require medical intervention beyond reassurance.14,17
ETIOLOGY AND PREDISPOSING CONDITIONS
OH can be classified on a pathophysiological basis as either neurogenic or nonneurogenic. Non-neurogenic OH is often reversible upon addressing and correcting the underlying etiology. In contrast, neurogenic OH (nOH) is a relatively rare, chronic condition caused by neurodegenerative disorders or peripheral neuropathies associated with autonomic impairments.
Non-Neurogenic Orthostatic Hypotension
Non-neurogenic OH accounts for the majority of OH cases and occurs in the absence of apparent autonomic nervous system disease.18 As previously described, this form of OH typically resolves when the underlying cause is corrected. This can include acute or transitory precipitants of hypotension such as intravascular volume loss, excessive venous pooling, adrenal insufficiency, or the use of certain medications that potentiate hypotension.13
Intravascular volume loss from dehydration, severe anemia, or acute hemorrhage, reduces cardiac preload leading to low cardiac output and subsequent hypotension. Similarly, venous pooling from prolonged static standing, large varicose veins, alcohol-induced vasodilation, or postprandial dilation of splanchnic vessels can significantly diminish venous return.13,19 Finally, the initiation or intensification of deleterious prescription medications may cause or exacerbate hypotension, such as tricyclic antidepressants or antihypertensive medications commonly used for other indications, such as diuretics, nitrates, direct vasodilators, and central sympatholytics.3
Non-neurogenic OH may also occur in the setting of certain chronic conditions. OH is fairly common in heart failure, with a community prevalence of up to 40% in elderly patients and over 80% in those hospitalized for aggravated disease states.20 Systemic BP may be reduced with cardiac pump failure, increasing patient susceptibility to OH. Additionally, some heart failure patients may be volume depleted due to overly aggressive diuresis used to combat chronic neurohormonal activation. Volume depletion can also occur in the case of chronic adrenal insufficiency with reduced mineralocorticoid activity.
Neurogenic Orthostatic Hypotension
Neurogenic OH (nOH) is a relatively rare condition that frequently manifests in patients with primary neurodegenerative disorders due to structural damage of the central and peripheral nervous systems, causing impairment of baroreflex-mediated vasoconstriction.4 Patients with symptoms of primary autonomic dysregulation (e.g. constipation, erectile dysfunction, urinary abnormalities, sweating abnormalities) should be screened for nOH even in the absence of orthostatic symptoms, as patients may be able to tolerate surprisingly low BPs due to adaptive changes in cerebral autoregulatory mechanisms.18,21,22 nOH may also occur secondary to spinal cord injuries or neurologic complications associated with diabetes, amyloidosis, vitamin deficiencies, and select autoimmune diseases.23 While a thorough clinical history is important to identify these neurogenic origins of OH, nOH patients have also been shown to have a greater fall in orthostatic SBP and lesser increase in heart rate (HR) relative to non-neurogenic OH patients. This is due to impaired sympathetic innervation to the heart that makes a proper compensatory tachycardia impossible. Therefore, a ΔHR/ΔSBP ratio lower than 0.5 bpm/mmHg may be diagnostic of nOH.24 While the ΔHR/ΔSBP ratio does not improve diagnostic differentiation between central and peripheral causes of nOH, the relative preservation of sympathetic outflow in patients with central autonomic failure (i.e. multiple system atrophy [MSA]) causes a significantly larger HR increase in these patients compared to those with peripheral autonomic failure (i.e. Lewy body disorders; Parkinson’s disease [PD], Dementia with Lewy Bodies [DLB], and pure autonomic failure [PAF]).
Primary Neurodegenerative Disorders
Synucleinopathies are a group of chronic, progressive neurodegenerative disorders characterized by the abnormal accumulation of fibrillary aggregates rich in the α-synuclein protein throughout the nervous system. These inclusions may be found in the cytoplasm of neurons, neurites, and glia, leading to central and peripheral autonomic failure that may manifest as OH. While synucleinopathies share a common pathological protein, their clinical features and patterns of progressive decline in motor, cognitive, behavioral, and autonomic functions differ immensely based on their distinctive distribution of lesions.25
Lewy bodies are α-synuclein-rich inclusions that develop inside neurons, and associated with both central and peripheral nervous system damage. Alpha-synuclein can also be deposited inside of central oligodendrocytes to form glial cytoplasmic inclusions that are a hallmark of MSA, which is often associated with more rapid disease progression than the Lewy body disorders. However, even with these distinctions, differential diagnosis can be difficult due to significant clinical overlap among synucleinopathies.
The Lewy body synucleinopathies include PD, DLB, and PAF. These three disorders feature overlapping clinical and neuropathologic phenotypes characterized by intraneuronal α-synuclein inclusions.25 In particular, PD and DLB exhibit highly similar clinical characteristics that may only differ in their relative time of onset.
The cardinal feature of PD is parkinsonism (i.e. bradykinesia with rigidity, tremor, or postural instability), but it can be indistinguishable from that of DLB.26 While PD and DLB both have central nervous system features, DLB typically presents with more prominent cognitive problems and fluctuations, as well as visual hallucinations and dementia that coincide with the onset of extrapyramidal symptoms.27 Progressive cognitive decline and dementia typically occur later in the clinical course of PD (if at all) with classical motor symptoms presenting much earlier.27 Autonomic failure and OH are common in both PD and DLB, with the autonomic failure being caused by damage to the peripheral autonomic nerves, similar to what is seen in PAF.28
Interestingly, while the Lewy bodies in PAF are indistinguishable from those in PD, symptoms of autonomic dysfunction are not accompanied by parkinsonism or dementia in PAF, and many patients remain free of neurological morbidities in the long term.29 However, in some cases, initially diagnosed PAF may evolve into other disorders with time, such as MSA. All three Lewy body synucleinopathies can exhibit cardiac sympathetic nerve degeneration, as indicated by impaired [123I]-metaiodobenzylguanidine (123I-MIBG) with single photon emission computed tomography (SPECT) or 6-[18F]fluorodopamine uptake with positron emission tomography (PET), and by low plasma norepinephrine levels.30
In contrast to the Lewy body synucleinopathies where inclusions form inside of neurons, MSA can be considered more of a “central autonomic failure” since the α-synuclein deposition primarily occurs in central oligodendrocytes. Differentiation of MSA from the Lewy body disorders is clinically relevant since MSA has a much more rapid clinical course and considerably poorer prognosis. MSA is further classified into the cerebellar type (MSA-C) or parkinsonian type (MSA-P) based on the symptoms that predominate at the time of diagnosis. MSA-C is primarily characterized by cerebellar or pyramidal (i.e. corticospinal and corticobulbar) dysfunction, including gait and limb ataxia, nystagmus, and dysarthria. Conversely, parkinsonian or extrapyramidal (i.e. brain stem) features predominate in MSA-P, including postural tremor and instability, bradykinesia, and rigidity.26 Autonomic failure is required for a diagnosis of MSA and often presents as urinary incontinence, erectile dysfunction, or OH. Importantly, patients with MSA can initially present with isolated autonomic failure, making it nearly impossible to differentiate MSA from PAF early in the course of disease.28
At initial onset of motor symptoms, the differential diagnosis between MSA-P and PD can be very challenging. A key difference is that patients with MSA experience a more rapid clinical deterioration than those with PD, and also generally respond poorly to therapy with levodopa.28 Additionally, while autonomic deficits are common in PD, their severity and distribution may be used to distinguish PD from MSA-P. PD is predominantly a postganglionic disorder, exhibiting markedly reduced 123I-MIBG uptake on SPECT. In contrast, patients with MSA have central lesions that involve autonomic nuclei, but the efferent postganglionic sympathetic nerves remain intact, as evidenced by the presence of normal or only slightly reduced supine plasma norepinephrine levels, as well as the normal uptake of 123I-MIBG.28
Peripheral Neuropathies
There are a number of systemic disorders and external agents that can cause a peripheral neuropathy and lead to the manifestation of nOH. These include diabetes mellitus, amyloidosis, excessive alcohol use, vitamin deficiencies, immune-mediated injury, infectious diseases, and treatment with chemotherapy agents.23,31 These secondary causes of autonomic failure are often reversible and should be excluded at the outset unless clinical manifestations of primary neurodegenerative disease are evident.
The autonomic neuropathies are a group of disorders in which there is selective damage to the small, lightly myelinated and unmyelinated nerve fibres of the peripheral autonomic nervous system.32 Diabetes mellitus is the most common cause of autonomic neuropathy, which may manifest as HR abnormalities, bladder dysfunction, erectile failure, gastroparesis, constipation, hyperhidrosis, or OH. OH occurs in diabetes largely as a consequence of efferent sympathetic denervation, causing impaired vasoconstriction of the peripheral vascular beds.32
In some cases, OH may be a clue to primary amyloidosis, where insoluble β-fibrillar proteins are deposited in the connective tissue layers surrounding peripheral nerves, as well as in the perineuronal tissues and neural vasculature.32 Although the fibril precursor proteins differ among disparate amyloidoses, they share substantial similarities in their clinical manifestations of neuropathy, the most common being sensorimotor axonal polyneuropathy and carpal tunnel syndrome. Symptoms typically begin with painful paresthesias in the feet and can progress to numbness and motor weakness as larger nerve fibers are affected. The autonomic features of amyloid neuropathy are similar to those of diabetic neuropathy, and include erectile dysfunction, gastrointestinal disturbances, orthostatic symptoms, and OH.
Alcoholic neuropathy is a potential complication of long-term, excessive alcohol consumption characterized by pain and dysesthesias, primarily in the lower extremities, which progressively extend proximally into the arms and legs to cause gait abnormalities accompanied by autonomic dysfunction.33 The exact mechanism behind alcoholic neuropathy is not well understood, but clinical and pathologic findings mainly indicate axonal degeneration with reduced nerve fiber densities.31 While studies have suggested that ethanol or its metabolites have direct neurotoxic effects, alcohol-related thiamine (i.e. vitamin B1) deficiency may also contribute to the pathogenesis of peripheral neuropathy and should be monitored closely in patients with chronic alcoholism.33
While it is rare to see isolated vitamin B1 deficiency in developed nations, vitamin B12 deficiency may be seen in those older than 60 years.34 This may be due to nutritional deficiency, malabsorption, or lack of intrinsic factor. Depletion of vitamin B12 is associated with abnormal erythropoiesis and nervous system demyelination, leading to peripheral neuropathy that manifests as paraesthesia, ataxia, limb weakness, OH, and syncope.34
Finally, several other less frequent causes of peripheral neuropathy may manifest as OH. These include immune-mediated disorders, infectious diseases, and drug toxicity.
Sensory Neuronopathies or Ganglionopathies
In contrast to peripheral neuropathies that primarily affect myelin or nerve axons in a length-dependent pattern, sensory neuronopathies (i.e. ganglionopathies) are characterized by primary degeneration of the dorsal root ganglia and non-length dependent denervation of their axonal projections. The most common cause of ganglionopathy is immune-mediated neuronal destruction, such as in autoimmune autonomic ganglionopathy (AAG). However, autonomic dysfunction, including OH, is highly prevalent in this group of disorders and seemingly unrelated to a specific etiology.35 The underlying mechanism leading to autonomic dysfunction is still unclear, but may be the result of postganglionic nerve fiber impairment or concurrent involvement of the autonomic ganglia.
Exacerbating Medications
Just as medications can mimic the acute non-neurogenic causes of OH, they may also exacerbate the defects in autonomic nerve activity that are characteristic of nOH. Notably, central sympatholytics such as clonidine and methyldopa activate α2-adrenergic receptors to reduce sympathetic outflow to the heart. These agents may be used to blunt tachycardia and hypertension in patients with the hyperadrenergic form of postural tachycardia syndrome (POTS), another autonomic disorder with orthostatic symptoms that may mimic those of OH. Certain antidepressants that influence the autonomic nervous system may also potentiate hypotension. For example, norepinephrine may be depleted with prolonged use of monoamine oxidase inhibitors, and peripheral vasodilation may be caused by α1-adrenergic receptor blockade by tricyclic antidepressants or serotonin antagonists such as trazodone.
Finally, given that the prevalence of both OH and benign prostatic hyperlasia (BPH) increase with age, it is important to consider the hypotensive risk associated with alpha blockers used to treat the lower urinary tract symptoms suggestive of symptomatic BPH. Alpha-1 adrenergic receptor antagonists such as doxazosin, prazosin, and terazosin have become the standard of care for managing BPH, but have the potential to produce OH due to their systemic vasodilatory effects. As such, two clinically “uroselective” alpha blockers, alfuzosin and tamsulosin, have been utilized to specifically target the urologic symptoms of BPH. Of these, tamsulosin has been found to have the lowest potential to reduce BP and cause OH, especially in elderly patients.36 Even so, results from a large, population-based cohort study found that patients who took tamsulosin were significantly more likely to experience severe hypotension requiring hospital admission during the first eight weeks of therapy relative to those taking a 5α-reductase inhibitor for BPH.37 Therefore, while uroselective alpha blockers remain first-line agents in the medical management of BPH, it may be beneficial to attempt therapy with a 5α-reductase inhibitor such as finasteride or dutasteride when drug-induced OH is suspected. While these medications are less effective than alpha blockers in providing immediate symptomatic relief, 5α-reductase inhibitors are associated with a reduced risk of long-term complications and enlarged-prostate-related surgery, slowing of disease progression, and eventual relief of symptoms.38
Medical Mimics
Vasovagal Syncope
Vasovagal syncope (VVS), or neurally-mediated syncope, is a common clinical condition characterized by a transient loss of consciousness due to cerebral hypoperfusion with rapid onset and recovery (Figure 1D).39 VVS is the most common cause of syncope and occurs due to dysfunctional autonomic regulation of postural tone, resulting in a sudden, severe drop in BP and HR after a period of upright posture (Table 1). These episodes of hypotension are accompanied by prodromal symptoms such as lightheadedness, nausea and hot flashes, with syncope ensuing shortly thereafter. Loss of consciousness may also occur in patients with OH, especially under acute aggravating conditions, but its onset is usually more gradual than in VVS and does not follow every episode of hypotension.4 In patients with a history of recurrent syncope, a careful examination of orthostatic vital signs can be helpful. VVS patients typically only exhibit hypotension in the context of syncope prodrome, where it may be accompanied by a relative bradycardia (compared to baseline) and imminent loss of consciousness. Conversely, HR is maintained, or occasionally increased, with postural hypotension in the context of OH and DOH, both of which frequently occur without subsequent syncope.40
Postural Tachycardia Syndrome
POTS is a multifaceted autonomic disorder primarily characterized by chronic symptoms of orthostatic intolerance, similar to those in OH, accompanied by a sustained increase in HR ≥30 bpm within 10 minutes of standing (Figure 1E). Importantly, the diagnostic criteria for POTS requires the orthostatic HR increase to occur in the absence of hypotension (fall in BP ≥20/10 mmHg; Table 1).41 While POTS patients can occasionally have concomitant OH, they must also have excessive orthostatic tachycardia in the setting of preserved BP. If orthostatic tachycardia occurs exclusively in the setting of hypotension, then the diagnosis is OH and not POTS.
Afferent Baroreflex Failure
Afferent baroreflex dysfunction occurs with injury of the baroreceptors, afferent nerves, or their connections in the brainstem.42 Interruption of these inputs to areas of central integration results in unrestrained sympathetic outflow and produces profoundly labile BP, including severe episodes of hypertension and hypotension, as well as OH. Acute baroreflex failure occurs most frequently due to damage of the carotid sinus nerve during surgical removal of neck tumors such as paragangliomas, carotid endarterectomy, or radiation therapy, but may also occur with lesions of the afferent limb (e.g. Guillain-Barré syndrome) or central nervous system.42
Importantly, the pronounced BP fluctuations that accompany afferent baroreflex failure are not necessarily postural, but are rather elicited by a range of exogenous stimuli and endogenous neural perturbations. As such, patients with volatile BP changes that occur irrespective of postural position should be evaluated for afferent baroreflex damage, rather than just disorders related to efferent baroreflex dysfunction (i.e. disorders of autonomic failure).
CLINICAL EVALUATION OF ORTHOSTATIC HYPOTENSION
OH may have subtle and diverse clinical presentations that may be overlooked in the absence of a systematic approach to clinical evaluation. Differential diagnosis relies heavily on the clinical history and comprehensive physical examination, but more sophisticated investigations may be useful in select cases with suspected neurogenic etiology.
History
Obtaining a thorough history is important to identify preexisting conditions and factors that may predispose an individual to orthostatic intolerance. The history should focus on understanding the course of existing neurological, cardiovascular, and endocrine disorders; assessing for signs of autonomic failure; reviewing prescription drug profiles; and determining symptom burden, if any, with emphasis on their onset, frequency, dissolution, and impact on daily functions.
Patients with OH most often present with lightheadedness, blurred vision, or occasionally, syncope.18 Despite significant drops in BP, syncope is not common during episodes of OH, likely due to altered mechanisms of cerebral autoregulation. Some patients may also complain of “coat hanger” pain, platypnea, or other symptoms such as generalized weakness, fatigue, leg buckling, or headache. These patients should be screened for OH, especially in the context of existing neurological deficits. If present, symptoms should only occur upon assuming upright posture, become more severe with ongoing stand, less frequent when seated, and resolve when supine.
Patients reporting a history of rapid-onset, short-lived orthostatic symptoms on standing should be clinically assessed for IOH. Importantly, relying on symptoms alone for OH evaluation without confirmation of positive hemodynamic criteria can lead to unwarranted diagnosis and treatment, thus orthostatic symptoms should not be used to make a diagnosis of OH outside of the context of orthostatic vital signs.44
Physical Examination
Suspected Classical or Delayed Orthostatic Hypotension
Paramount to the physical examination of OH is the proper conduct of orthostatic vital signs. Orthostatic vitals from an active stand test are typically adequate for OH diagnosis, but head-up tilt table testing can aid in confirming a diagnosis of suspected OH when active stand measurements results are borderline. Patients should ideally be supine (not seated) for at least 10 minutes prior to standing in order to establish a reliable baseline.45 SBP, DBP, and HR should be recorded at the end of baseline and subsequently at one, three, five, and ten minutes upon standing.46,47 It is worth noting that a longer duration of upright posture (beyond 10 minutes) may be required to elicit DOH in some patients, and assessment should be guided by clinical history.
In older adults and others who may have difficulties transitioning smoothly from the supine to standing position, it may be preferable to utilize head-up-tilt testing at a minimum 60° angle to objectively evaluate BP changes at the established time points.1,48 Importantly, however, the magnitude of initial orthostatic BP decline is generally smaller with head-up tilt compared to active standing, which may be a consequence of the rapidity of postural change or mechanistic differences in response to passive versus active standing.14
Ambulatory BP monitoring can be useful in patients with suspected OH that do not meet BP criteria in clinic due to the presence of acute factors that contribute to diurnal BP variability.22 It may be difficult to determine whether episodes of hypotension seen throughout a 24-hour period are related to postural change since this is not recorded by the monitoring device. Therefore, it is important to instruct patients to maintain an orthostatic BP diary with measurements taken at different times points over several days to increase diagnostic sensitivity. These time points should include mornings, mealtimes, and upon the onset of orthostatic symptoms.
Suspected Initial Orthostatic Hypotension
Due to the short time course of the BP decrement and recovery in IOH, orthostatic vitals must be assessed in clinic using beat-to-beat monitoring methods, such as finger arterial volume-clamp photoplethysmography, to detect the presence of transient hypotension in those with suspected IOH. Continuous BP and HR measurements should be recorded at the end of a minimum 10-minute supine baseline and for three minutes upon active standing. Importantly, IOH is generally seen with active standing and far less frequently with passive postural change (i.e. head-up tilt testing).
Laboratory Evaluation
Laboratory tests may be ordered to identify overt primary causes of hypotension and disorders causing peripheral neuropathy. These investigations may include blood glucose level to rule out hyperglycemia; complete blood count with hematocrit to rule out severe anemia or infection; an electrolyte panel to rule out abnormalities associated with acute fluid depletion; morning cortisol level to exclude primary adrenal insufficiency; serum immunoelectrophoresis to exclude monoclonal protein associated peripheral neuropathy; and vitamin B12 level to exclude neuropathy from B12 deficiency.49 In selected patients, fractionated catecholamine testing may be useful to confirm the origin of OH. That is, standing plasma norepinephrine levels are typically normal or enhanced in non-neurogenic OH, but significantly reduced in patients with nOH.13 Those with peripheral autonomic failure, as in PD or PAF, may also exhibit low supine plasma norepinephrine levels.50
Autonomic Function Testing
Formal autonomic function testing at a specialized tertiary centre can be useful for differential diagnosis, especially in OH cases with concomitant neurological symptoms. Cardiac parasympathetic function (i.e. cardiovagal function) may be assessed by evaluating heart rate variability with cyclical deep breathing in the supine position. Additionally, the sympathetic cholinergic nervous system can be assessed with sweat tests such as the quantitative sudomotor axon reflex test, while the sympathetic noradrenergic system is evaluated with the cold pressor test, isometric handgrip test, orthostatic challenge, and Valsalva maneuver.50 Patients with nOH are not often unable to mount an adequate sympathetic vasoconstrictor response to Valsalva-induced hypotension, and lack the late phase II BP increase and phase IV BP overshoot responses typical in those with intact autonomic nervous system function (Figure 2).
Figure 2.
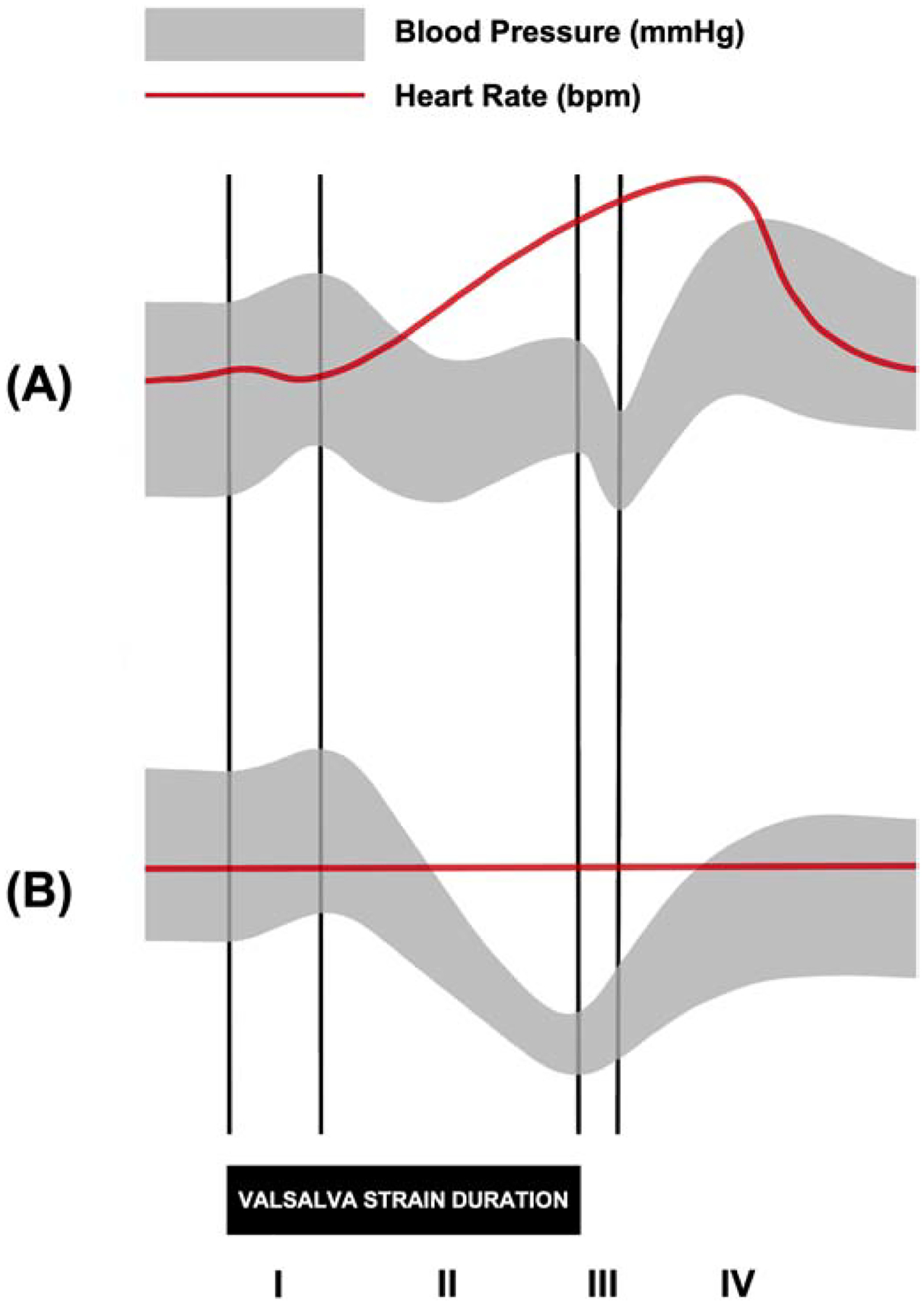
Representative blood pressure (BP) and heart rate (HR) tracings during the Valsalva maneuver in (A) healthy subjects and (B) patients with neurogenic orthostatic hypotension. The Valsalva maneuver is used to evaluate the sympathetic and parasympathetic responses of the baroreflex as an individual forcibly exhales at maximum pressure (~40 mmHg) for 15 seconds. Those with intact autonomic nervous system function are expected to exhibit a specific pattern of physiological responses to the Valsalva maneuver that can be divided into four phases: (I) the mechanical increase in intrathoracic pressure causes a brief increase in BP and decrease in HR; (II, early) the reduction in venous return causes a decrease in stroke volume and BP; (II, late) the baroreflex is activated by the decrease in BP, causing a sympathetically-mediated increase in BP and HR; (III) the pulmonary vascular refills with blood upon termination of the Valsalva maneuver, causing a decrease in BP; (IV) the sudden increase in venous return leads to a compensatory decrease in HR and increase in BP that overshoots baseline BP due to residual sympathetic activity. In patients with impaired autonomic function, as in neurogenic orthostatic hypotension, there is a lack of late phase II BP increase and BP overshoot in phase IV recovery.
Special Considerations in Evaluating Orthostatic Hypotension
Supine Hypertension
Paradoxical supine hypertension occurs in approximately one half of patients with nOH. This condition induces nighttime pressure natriuresis and is often treated with BP lowering medications, both of which can worsen morning OH.51 Prolonged recumbence and overnight volume loss may further aggravate orthostatic symptoms and hypotension in the morning hours. While it can be challenging to manage supine hypertension in the context of OH, there is a risk of end organ damage if severe hypertension is left unaddressed. As previously mentioned, a thresho reduction in BP of 30/15 mmHg may be more appropriate for OH diagnosis in this setting.
Postprandial Hypotension
Postprandial hypotension is characterized by a decline in SBP of at least 20 mmHg within two hours of consuming a meal.52 While postprandial hypotension can be a non-neurogenic cause of OH, the phenomenon occurs in up to 80% of patients with autonomic dysfunction,53 making it an important consideration for those with suspected nOH. One study in patients with PAF found that meal composition significantly impacted the degree of postprandial hypotension, with high glucose meals causing the largest drops in BP followed by high fat and high protein meals, respectively.54 Patients with OH should therefore aim to consume smaller, low carbohydrate meals throughout the day to avoid inducing symptomatic postural hypotension.
CONCLUSION
OH is a clinical manifestation of a diverse collection of neurogenic and non-neurogenic disorders that may be precipitated by acute or chronic factors. The differential diagnosis of OH includes disorders of neurologic, cardiovascular, and endocrine origin, as well as acute intravascular volume depletion, peripheral blood pooling, and offending medication use. Evaluation of suspected OH relies on an understanding of the three temporally differing clinical variants, their presentations in patients with and without existing neurodegenerative disorders, and the rationale for diagnostic testing beyond a comprehensive history and physical examination.
Highlights.
Orthostatic hypotension is associated with increased morbidity and mortality.
There are different forms of orthostatic hypotension, and mimics of orthostatic hypotension.
Understanding these can lead to more accurate diagnoses and better patient care.
FUNDING
SRR is supported by a grant from the Canadian Institutes of Health Research (MOP142426), Ottawa, Canada, and the Vanderbilt Institute for Clinical and Translational Research (VICTR), which is funded by the National Institutes of Health grant 5UL1TR002243 (Bethesda, MD, USA). SRR is a Cardiac Arrhythmia Network of Canada (CANet) funded investigator (London, Ontario, Canada).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
SRR is a consultant to Lundbeck LLC and Theravance Biopharma related to neurogenic orthostatic hypotension, and has received honoraria from the Academy for Continued Healthcare Learning and Medscape for developing continuing medical educational materials about neurogenic orthostatic hypotension. He also serves as DMSB Chair for a Phase 2 study of an irritable bowel syndrome medication for Arena Pharmaceuticals with compensation. SRR is currently the President of the American Autonomic Society without financial compensation.
REFERENCES
- 1.Freeman R, Wieling W, Axelrod FB, Benditt DG, Benarroch E, Biaggioni I, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res. 2011;21(2):69–72. [DOI] [PubMed] [Google Scholar]
- 2.Saedon NI, Pin Tan M, Frith J. The Prevalence of Orthostatic Hypotension: A Systematic Review and Meta-Analysis. J Gerontol A Biol Sci Med Sci. 2020;75(1):117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ricci F, De Caterina R, Fedorowski A. Orthostatic hypotension: Epidemiology, prognosis, and treatment. J Am Coll Cardiol. 2015;66(7):848–60. [DOI] [PubMed] [Google Scholar]
- 4.Freeman R, Abuzinadah AR, Gibbons C, Jones P, Miglis MG, Sinn DI. Orthostatic Hypotension. J Am Coll Cardiol. 2018;72(11):1294–309. [DOI] [PubMed] [Google Scholar]
- 5.Adalbert R, Coleman MP. Review: Axon pathology in age-related neurodegenerative disorders. Neuropathol Appl Neurobiol. 2013;39(2):90–108. [DOI] [PubMed] [Google Scholar]
- 6.Verwoert GC, Mattace-Raso FUS, Hofman A, Heeringa J, Stricker BHC, Breteler MMB, et al. Orthostatic hypotension and risk of cardiovascular disease in elderly people: The Rotterdam study. J Am Geriatr Soc. 2008;56(10):1816–20. [DOI] [PubMed] [Google Scholar]
- 7.Rose KM, Eigenbrodt ML, Biga RL, Couper DJ, Light KC, Sharrett AR, et al. Orthostatic hypotension predicts mortality in middle-aged adults: The Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2006;114(7):630–6. [DOI] [PubMed] [Google Scholar]
- 8.Ricci F, Fedorowski A, Radico F, Romanello M, Tatasciore A, Di Nicola M, et al. Cardiovascular morbidity and mortality related to orthostatic hypotension: A meta-analysis of prospective observational studies. Eur Heart J. 2015;36(25):1609–17. [DOI] [PubMed] [Google Scholar]
- 9.Mehrabian S, Duron E, Labouree F, Rollot F, Bune A, Traykov L, et al. Relationship between orthostatic hypotension and cognitive impairment in the elderly. J Neurol Sci. 2010;299(1–2):45–8. [DOI] [PubMed] [Google Scholar]
- 10.Shibao C, Grijalva CG, Raj SR, Biaggioni I, Griffin MR. Orthostatic Hypotension-Related Hospitalizations in the United States. Am J Med. 2007;120(11):975–80. [DOI] [PubMed] [Google Scholar]
- 11.Gibbons CH, Freeman R. Clinical implications of delayed orthostatic hypotension: A 10-year follow-up study. Neurology. 2015;85(16):1362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Twist DJL, Dinh T, Bouwmans EME, Kroon AA. Initial orthostatic hypotension among patients with unexplained syncope: An overlooked diagnosis? Int J Cardiol. 2018;271:269–73. [DOI] [PubMed] [Google Scholar]
- 13.Kaufmann H, Palma JA. Neurogenic orthostatic hypotension: the very basics. Clin Auton Res. 2017;27(Suppl 1):39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stewart JM, Clarke D. “He’s Dizzy when he Stands Up.” An Introduction to Initial orthostatic Hypotension. J Pediatr. 2011;158:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clarke DA, Medow MS, Taneja I, Ocon AJ, Stewart JM. Initial Orthostatic Hypotension in the Young is Attenuated by Static Handgrip. J Pediatr. 2010;156(6):1019–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finucane C, O’Connell MDL, Donoghue O, Richardson K, Savva GM, Kenny RA. Impaired Orthostatic Blood Pressure Recovery Is Associated with Unexplained and Injurious Falls. J Am Geriatr Soc. 2017;65(3):474–82. [DOI] [PubMed] [Google Scholar]
- 17.Stewart JM. Transient orthostatic hypotension is common in adolescents. J Pediatr. 2002;140(4):418–24. [DOI] [PubMed] [Google Scholar]
- 18.Palma J-A, Kaufmann H. Epidemiology, Diagnosis, and Management of Neurogenic Orthostatic Hypotension. Mov Disord Clin Pract. 2017;4(3):298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Narkiewicz K, Cooley RL, Somers VK. Alcohol potentiates orthostatic hypotension: Implications for alcohol- related syncope. Circulation. 2000;101(4):398–402. [DOI] [PubMed] [Google Scholar]
- 20.Gorelik O, Feldman L, Cohen N. Heart failure and orthostatic hypotension. Heart Fail Rev. 2016;21(5):529–38. [DOI] [PubMed] [Google Scholar]
- 21.Horowitz DR, Kaufmann H. Autoregulatory cerebral vasodilation occurs during orthostatic hypotension in patients with primary autonomic failure. Clin Aut Res. 2001;11(6):363–7. [DOI] [PubMed] [Google Scholar]
- 22.Norcliffe-Kaufmann L, Kaufmann H. Is ambulatory blood pressure monitoring useful in patients with chronic autonomic failure? Clin Auton Res. 2014;24(4):189–92. [DOI] [PubMed] [Google Scholar]
- 23.Fedorowski A, Melander O. Syndromes of orthostatic intolerance: A hidden danger. J Intern Med. 2013;273(4):322–35. [DOI] [PubMed] [Google Scholar]
- 24.Norcliffe-Kaufmann L, Kaufmann H, Palma JA, Shibao CA, Biaggioni I, Peltier AC, et al. Orthostatic heart rate changes in patients with autonomic failure caused by neurodegenerative synucleinopathies. Ann Neurol. 2018;83(3):522–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCann H, Stevens CH, Cartwright H, Halliday GM. α-Synucleinopathy phenotypes. Park Relat Disord. 2014;20(Suppl 1):S62–7. [DOI] [PubMed] [Google Scholar]
- 26.Martí MJ, Tolosa E, Campdelacreu J. Clinical overview of the synucleinopathies. Mov Disord. 2003;18(Suppl 6):S21–27. [DOI] [PubMed] [Google Scholar]
- 27.Sezgin M, Bilgic B, Tinaz S, Emre M. Parkinson’s Disease Dementia and Lewy Body Disease. Semin Neurol. 2019;39(2):274–82. [DOI] [PubMed] [Google Scholar]
- 28.Shibao C, Gamboa A, Diedrich A, Biaggioni I. Management of hypertension in the setting of autonomic failure: A pathophysiological approach. Hypertension. 2005;45(4):469–76. [DOI] [PubMed] [Google Scholar]
- 29.Kaufmann H, Goldstein DS. Pure autonomic failure: A restricted lewy body synucleinopathy or early Parkinson disease? Neurology. 2010;74(7):536–7. [DOI] [PubMed] [Google Scholar]
- 30.Tipre DN, Goldstein DS. Cardiac and extracardiac sympathetic denervation in Parkinson’s disease with orthostatic hypotension and in pure autonomic failure. J Nucl Med. 2005;46:1775–81. [PubMed] [Google Scholar]
- 31.Julian TH, Syeed R, Glascow N, Zis P. Alcohol-induced autonomic dysfunction: a systematic review. Clin Auton Res. 2020;30(1):29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freeman R Autonomic peripheral neuropathy. Lancet. 2005;365(9466):1259–70. [DOI] [PubMed] [Google Scholar]
- 33.Chopra K, Tiwari V. Alcoholic neuropathy: Possible mechanisms and future treatment possibilities. Br J Clin Pharmacol. 2012;73(3):348–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunt A, Harrington D, Robinson S. Vitamin B12 Deficiency. BMJ. 2014;349:g5226. [DOI] [PubMed] [Google Scholar]
- 35.Damasceno A, França MC, Cury H, Nucci A. Autonomic dysfunction in nonparaneoplastic sensory neuronopathy: Beyond sensory abnormalities. J Neurol. 2011;258(2):231–7. [DOI] [PubMed] [Google Scholar]
- 36.Buzelin JM, Fonteyne E, Kontturi M, Witjes WP, Khan A. Comparison of Tamsulosin With Alfuzosin in the Treatment of Patients With Lower Urinary Tract Symptoms Suggestive of Bladder Outlet Obstruction (Symptomatic Benign Prostatic Hyperplasia). The European Tamsulosin Study Group. Br J Urol. 1997;80(4):597–605. [DOI] [PubMed] [Google Scholar]
- 37.Bird ST, Delaney JAC, Brophy JM, Etminan M, Skeldon SC, Hartzema AG. Tamsulosin Treatment for Benign Prostatic Hyperplasia and Risk of Severe Hypotension in Men Aged 40–85 Years in the United States: Risk Window Analyses Using Between and Within Patient Methdology. BMJ. 2013;347:f6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naslund MJ, Miner M. A Review of the Clinical Efficacy and Safety of 5alpha-reductase Inhibitors for the Enlarged Prostate. Clin Ther. 2007;29(1):17–25. [DOI] [PubMed] [Google Scholar]
- 39.Sheldon RS, Grubb BP, Olshansky B, Shen W-KK, Calkins H, Brignole M, et al. 2015 heart rhythm society expert consensus statement on the diagnosis and treatment of postural tachycardia syndrome, inappropriate sinus tachycardia, and vasovagal syncope. Hear Rhythm. 2015;12:e41–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibbons CH, Freeman R. Delayed orthostatic hypotension: A frequent cause of orthostatic intolerance. Neurology. 2006;67(1):28–32. [DOI] [PubMed] [Google Scholar]
- 41.Raj SR, Guzman JC, Harvey P, Richer L, Schondorf R, Seifer C, et al. Canadian Cardiovascular Society Position Statement on Postural Orthostatic Tachycardia Syndrome (POTS) and Related Disorders of Chronic Orthostatic Intolerance. Can J Cardiol. 2020;36(3):357–72. [DOI] [PubMed] [Google Scholar]
- 42.Robertson D, Diedrich A, Chapleau M. Afferent Baroreflex Failure. Auton Neurosci. 2012;172(1–2):1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Norcliffe-Kaufmann L, Axelrod F, Kaufmann H. Afferent baroreflex failure in familial dysautonomia. Neurology. 2010;75(21):1904–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palma JA, Norcliffe-Kaufmann L, Kaufmann H. An orthostatic hypotension mimic: The inebriation-like syndrome in Parkinson disease. Mov Disord. 2016;31(4):598–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cooke J, Carew S, O’Connor M, Costelloe A, Sheehy T, Lyons D. Sitting and standing blood pressure measurements are not accurate for the diagnosis of orthostatic hypotension. QJM. 2009;102(5):355–9. [DOI] [PubMed] [Google Scholar]
- 46.Low PA. Prevalence of orthostatic hypotension. Clin Auton Res. 2008;18(Suppl 1):8–13. [DOI] [PubMed] [Google Scholar]
- 47.Streeten DH, Thomas D, Bell DS. The roles of orthostatic hypotension, orthostatic tachycardia, and subnormal erythrocyte volume in the pathogenesis of the chronic fatigue syndrome. Am J Med Sci. 2000;320:1–8. [DOI] [PubMed] [Google Scholar]
- 48.Aydin AE, Soysal P, Isik AT. Which is preferable for orthostatic hypotension diagnosis in older adults: active standing test or head-up tilt table test? Clin Interv Aging. 2017;12:207–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Freeman R Neurogenic orthostatic hypotension. N Engl J Med. 2008;358(6):615–24. [DOI] [PubMed] [Google Scholar]
- 50.Nwazue VC, Raj SR. Confounders of vasovagal syncope: Orthostatic hypotension. Cardiol Clin. 2013;31(1):89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jordan J, Biaggioni I. Diagnosis and treatment of supine hypertension in autonomic failure patients with orthostatic hypotension. J Clin Hypertens. 2002;4(2):139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trahair LG, Horowitz M, Jones KL. Postprandial hypotension: a systematic review. J Am Med Dir Assoc. 2014;15(6):394–409. [DOI] [PubMed] [Google Scholar]
- 53.Jansen RW. Postprandial hypotension: simple treatment but difficulties with the diagnosis. J Gerontol A Biol Sci Med Sci. 2005;60(10):1268–70. [DOI] [PubMed] [Google Scholar]
- 54.Puvi-Rajasingham S, Mathias CJ. Effect of meal size on post-prandial blood pressure and on postural hypotension in primary autonomic failure. Clin Aut Res. 1996;6(2):111–4. [DOI] [PubMed] [Google Scholar]