

Abstract
Rationale.
The long held speculation that the brain serotonin system mediates some behavioural effects of the psychostimulant cocaine is supported in part by the high affinity of cocaine for the serotonin transporter (SERT) and by reports that the serotonin transporter (SERT), estimated by SERT binding, is increased in brain of human chronic cocaine users. Excessive SERT activity and consequent synaptic serotonin deficiency might cause a behavioural (e.g., mood) abnormality in chronic users of the drug.
Objective and Methods.
Previous studies focused on changes in SERT binding, which might not necessarily reflect changes in SERT protein. Therefore, we compared levels of SERT protein, using a quantitative Western blot procedure, in autopsied brain (striatum, cerebral cortices) of chronic human cocaine users (n=9), who all tested positive for the drug/metabolite in brain, to those in control subjects (n=15) and, as a separate drug of abuse group, in chronic heroin users (n=11).
Results.
We found no significant difference in protein levels of SERT or the serotonin synthesizing enzyme tryptophan hydroxylase-2 amongst the control and drug abuse groups. In the cocaine users no significant correlations were observed between SERT and brain levels of cocaine plus metabolites, or with levels of serotonin or its metabolite 5-hydroxyindoleacetic acid.
Conclusion.
Our postmortem data suggest that a robust increase in striatal/cerebral cortical SERT protein is not a common characteristic of chronic, human cocaine users.
Keywords: Serotonin transporter, Cocaine, Heroin, Postmortem human brain, Western blot
1. Introduction
Efforts to understand the mechanisms of action of the abused psychostimulant cocaine have mostly focused on the ability of the drug to influence activity of the brain dopamine neurotransmitter system via its inhibition of the dopamine transporter (see (Heal et al., 2014; Howell & Negus, 2014)). However, over the decades there has also been continuing interest in establishing whether some behaviours associated with cocaine use might be explained by its actions on brain serotonin releasing neurons (Cox et al., 2011; Cunningham & Anastasio, 2014). This possibility is clinically relevant given that studies in the human are testing the possibility that serotonergic therapeutics (e.g., lorcaserin) might modify cocaine taking behavior or side effects of the stimulant ((Higgins et al., 2013; Higgins et al., 2020); see (Collins et al., 2018)); but there is uncertainty whether preclinical cocaine-serotonin findings will translate to the human (e.g, (Collins & France, 2018) vs. (Banks & Negus, 2017; Pirtle et al., 2019)). Although the function of serotonin in human brain is still not fully understood, clinical pharmacological literature data strongly support the notion that serotonin is involved in aspects of mood ((Pare & Sandler, 1959; Shopsin et al., 1976); see (Ban, 2001) for a review).
The thrust of the argument that brain serotonin might mediate some actions of cocaine stems in part from observations that 1) the stimulant binds to the serotonin transporter (SERT; (Ross & Renyi, 1969; White & Appel, 1981; George, 1989; Hoffman et al., 1991; Ramamoorthy et al., 1993)) with high affinity and locks it in an outward-open inactive state (Coleman et al., 2019), an action that would be expected to cause, acutely, an increase in synaptic levels of the neurotransmitter; and 2) preclinical data associate SERT blockade or its inactivation with some behavioural cocaine actions (e.g. (Sora et al., 1998; Simmler et al., 2017)). Other preclinical studies, albeit not entirely consistent, have suggested that following chronic cocaine exposure the brain SERT upregulates in concentration ((Cunningham et al., 1992; Belej et al., 1996; Banks et al., 2008; Gould et al., 2011); but see (Katz et al., 1993; Simms & Gallagher, 1996; Little et al., 1998; Sawyer et al., 2012); see below for discussion) perhaps to compensate for the acutely increased synaptic serotonin. In this context, should the SERT upregulation be substantial, it could lead to changes in behaviours in the chronic cocaine condition due to a SERT-induced serotonin deficiency (e.g. depressed mood during cocaine abstinence; (Mash et al., 2000)).
To our knowledge, the literature on SERT levels in brain of human chronic cocaine users is limited to only three early reports in which radioligand binding measurements were made by using the first generation non-selective cocaine analog RTI-55/β-CIT. One investigation, a 123I-β-CIT brain imaging study (Jacobsen et al., 2000), reported an increase in whole diencephalon (17%) and brainstem (32%) binding in early abstinent cocaine users, with marked overlap between cocaine and control group ranges. One major limitation of this study was, as authors acknowledged, the potential confound of dopamine transporter binding by the radiotracer in midbrain and thalamus (Sasaki et al., 2012). In contrast, examination of citalopram-sensitive 125I-RTI-55 binding at a saturating concentration of 3.5 nM in autopsied midbrain of cocaine users reported decreased SERT levels in dorsal and median raphe and substantia nigra (−48–34%; (Little et al., 1998)). In another postmortem investigation using the same radioligand at a much lower concentration of 50 pM, a strikingly marked (up to 2–3 fold) increase in binding (by autoradiography) was observed in some striatal and cerebral cortical areas of autopsied cocaine overdose victims, which was attributed to changes in Bmax rather than Kd but with the magnitude of the changes partly dependent on whether the cocaine users died during an excited delirium episode ((Mash et al., 2000); see below in Discussion).
Given the limited information on brain SERT status in human cocaine users, and the possibility that the reports of increased brain SERT binding might not necessarily translate to elevated concentration of the actual SERT protein, we measured, by quantitative Western blotting, concentrations of SERT protein itself in autopsied brain (striatum and cerebral cortex) of human chronic cocaine users, most of whom were suspected of dying from cocaine intoxication. The SERT protein measurement procedure we have previously successfully employed to detect SERT changes (deficiency) in autopsied brain of persons with Parkinson’s disease (Kish et al., 2008) and of users of amphetamine drugs (Kish et al., 2009; Kish et al., 2010a) in which low brain SERT would be expected. We also include for comparison a matched control group, a second drug abuse group (heroin users) and measurements of serotonin and metabolite 5-hyrdroxyindoleacetic acid (5-HIAA) and striatal protein levels of the rate limiting serotonin-synthesizing enzyme, tryptophan hydroxylase-2 (TPH2).
Based on findings in the previous autopsied human brain study of SERT binding (Mash et al., 2000) we hypothesized that SERT protein would be markedly above normal in both striatum and cerebral cortex of the cocaine users of our study.
2. Subjects and Methods
2.1. Subjects
The study was approved by the Research Ethics Board of the Centre for Addiction and Mental Health at Toronto. Postmortem brain from 9 chronic cocaine users (7 males and 2 females), 11 chronic users of heroin (10 males, 1 female), and 24 controls (21 males and 3 females) was obtained from medical examiner offices in the USA and Canada using a standardized protocol. Subject information, drug histories, and brain drug levels for the drug users are summarized in Table 1 and were previously published (Tong et al., 2018). There were no statistically significant differences in age (control, 33.9±2.4 years; cocaine, 35.4±4.8 years; heroin, 36.2±2.5 years; mean±SEM) or postmortem intervals (PMI, interval between death and freezing of the brain; control, 14.1±1.2 hours; cocaine, 17.0±2.4 hours; heroin, 13.4±2.0 hours) between the control and drug users (one-way ANOVA). Genomic DNA was extracted from brain of all 9 cocaine users, 10 of the 11 heroin users and 15 control subjects who were assessed for SERT levels and the SERT gene promoter polymorphism (5HTTLPR) were determined [allele 1 (L)=450 bp; allele 2 (S)=406 bp; see (Kish et al., 2009)]. The control (8LL/4LS/3SS), cocaine (5LL/3LS/1SS) and heroin (2LL/7LS/1SS) users did not differ significantly with respect to distribution of the 5HTTLPR gene promoter polymorphisms (χ2(4,34)=5.3, p=0.26). At autopsy, one half-brain was fixed in formalin for neuropathological analysis, whereas the other half was immediately frozen until dissection for neurochemical analysis. Blood samples and a brain sample at autopsy were obtained from all of the drug users and the control subjects for drug screening. Sequential scalp hair samples for drug analyses could be obtained from five of the nine cocaine users, 19 of the 24 controls, and 10 of the 11 heroin users. Levels of drugs of abuse in blood and other bodily fluids were measured by the local medical examiner whereas drug analyses in brain and hair samples were conducted at the Armed Forces Institute of Pathology (Washington, DC, USA). Subjects for the cocaine group met the following criteria: 1) presence of cocaine or metabolite benzoylecgonine on toxicology screens in blood or (one subject) urine; autopsied brain, and, if available, scalp hair by GC-MS; 2) absence of other drugs of abuse in bodily fluids, with the exception of ethanol (see below), or in brain; 3) evidence from the case records or interview with next of kin of use of cocaine as the primary drug of abuse for at least 1 year prior to death; and 4) absence of neurological illness or, at autopsy, brain pathology unrelated to use of the drug. Two of the cocaine users (#C3 and #C4 in Table 1) had used alcohol recently before death as indicated by the presence of ethanol in blood and/or cocaethylene in brain. Available hair analysis of five of nine cocaine users revealed presence of only cocaine and/or metabolites in four subjects. Known or suspected causes of death of the cocaine users were cocaine intoxication (five), cardiovascular disease with cocaine as a contributing factors (two), carotid artery aneurysm with cocaine as a contributing factor (one) and chest trauma (one). Heroin users met the following criteria: 1) presence of heroin metabolites (6-acetylmorphine, morphine, or morphine glucuronide) on toxicology screens in blood and autopsied brain; 2) absence of other drugs of abuse in bodily fluids with the exception of ethanol (see below) or other opioid drugs (two subjects had blood samples positive for the opioid drug propoxyphene and its metabolite norpropoxyphene); 3) evidence from the case records of primary use of heroin for at least one year prior to death; and 4) absence of evidence of neurological illness or, at autopsy, brain pathology unrelated to use of the drug. Five of the heroin users (Table 1) had used alcohol recently before death as indicated by the presence of ethanol in blood. Hair analysis of the (10 of 11) heroin users revealed presence of only heroin metabolites in eight of the users. The suspected cause of death was heroin intoxication (seven), mixed drug intoxication (two), and cardiovascular disease with heroin as a contributing factors (two). Control subjects (for which brain serotonin markers have been previously reported in (Kish et al., 2009) were neurologically normal and had no evidence of brain pathology on neuropathological examination or history of drug use and tested negative for drugs of abuse in blood, autopsied brain, and in sequential scalp hair samples where available. The cause of death for the controls were cardiovascular disease (n=13), trauma (n=8), renal failure (n=1), drowning (n=1) and leukemia (n=1).
Table 1.
Characteristics and drug use histories of the 9 cocaine (C1-C9) and 11 heroin (H1-H11) usersa.
| Toxicology | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Case | Age (yrs), Sex | PMI (h) | Duration of use (yrs) | Recent drug use pattern | Route of drug administration | Suspected/known cause of death | Hair | Blood drug levelb | Brain drug levelc |
| C1 | 26,M | 18 | 1–2 | Unknown | Oral; smoked | Cocaine intoxication | NE | 424.0 | 179.7 (152.3) |
| C2 | 21,M | 6 | 2–3 | $150/mo, weekend binges | Nasal | CVD/cocaine intoxication | + | ND | 20.7 (14.0) |
| C3* | 26,F | 18 | 8 | Binge/limited only by funds | Oral; smoked | Cocaine intoxication | ND | 0.26 | 12.4 (10.3) |
| C4* | 36,M | 24 | 3 | Binge/limited only by funds | Nasal | Cocaine intoxication | NE | 27.0 | 157.6 (128.2) |
| C5 | 39,M | 26 | 8 | Considered heavy user | Intravenous; nasal | Cocaine intoxication | + | 0.43 | 11.0 (7.6) |
| C6 | 31,M | 22 | >2 | Limited only by funds | Nasal; smoked | Cocaine intoxication | NE | 39.2 | 164.4 (131.7) |
| C7 | 40,M | 9 | >10 | Binge every 2–3 wk | Nasal; smoked | Gunshot wound to chest | + | 12.34 | 40.4 (20.3) |
| C8 | 70,M | 10 | 55 | $60/mo, 1st wk of a month | Smoked | CVD | NE | 19.44 | 15.7 (10.3) |
| C9 | 30,F | 20 | >1 | Unknown | Smoked | CVD/cocaine intoxication | + | 17.16 | 19.4 (15.3) |
| H1 | 36,M | 8 | 10 | $200 per month | Intravenous | CVD/Narcotic intoxication | NE | 1.93 | 1.79 |
| H2* | 43,M | 13 | 27 | Unknown | Intravenous | Narcotic intoxication | + | 0.60 | 1.79 |
| H3 | 34,M | 23 | >1 | Unknown | Intravenous | Narcotic intoxication | + | 0.67 | 1.19 |
| H4* | 34,M | 10.5 | >1 | Unknown | Intravenous | Narcotic intoxication | + | 0.42 | 0.80 |
| H5 | 40,M | 5 | 20 | Daily | Intravenous | Mixed drug intoxication | + | 0.32 | 0.61 |
| H6 | 44,F | 18.5 | 23 | Daily, sometimes 1 g per day | Intravenous | CVD/Narcotic intoxication | + | 0.11 | 0.10 |
| H7 | 43,M | 21 | >1 | Daily | Intravenous | Mixed drug intoxication | + | 1.47 | 1.79 |
| H8* | 35,M | 19.5 | >1 | Unknown | Unknown | Narcotic intoxication | ND | 1.09 | 1.80 |
| H9* | 28,M | 8 | 4 | Daily | Intravenous | Narcotic intoxication | + | 0.39 | 0.46 |
| H10 | 19,M | 9.5 | >1 | Unknown | Unknown | Narcotic intoxication | ND | 0.39 | 0.37 |
| H11* | 42,M | 11 | 10 | Unknown | Intravenous | Narcotic intoxication | + | 0.70 | 0.76 |
M = male; F = female; PMI = postmortem interval; DA = dopamine; CVD = cardiovascular disorder.
Cases with ethanol detected in blood. + Drug hair analyses confirmed; NE = not examined; ND = not detected. For cases C2, C9, H1 and H6, cocaine or heroin toxicity was considered to be a possible contributing factor to the cause of death; high levels of propoxyphene (0.63 and 1.2 mg/L, respectively) and norpropoxyphene (1.26 and 3.8 mg/L, respectively) were also detected in blood of cases H5 and H7 and could have contributed to the death.
Information on the cases including brain drug levels has been published previously in (Tong et al., 2018);
Measured in μM of levels of cocaine plus metabolite benzoylecgonine or the heroin metabolite morphine;
Measured in nmol/g tissue of total levels of cocaine (cocaine plus metabolites benzoylecgonine, ecgonine methyl ester, norcocaine, cocaethylene, with levels of cocaine plus benzoylecgonine only in parenthesis for comparison with drug levels in Mash et al. 2000 investigation) in caudate or heroin (morphine plus 6-acetylmorphine plus morphine glucuronide) in occipital cortex.
Brain regions for neurochemical analysis were dissected as previously described (Kish et al., 1988) using the atlas of (Riley, 1943), including the caudate nucleus and putamen [intermediate (along a dorsoventral gradient) subdivision of the middle (along a dorso-ventral gradient) portion of both nuclei], and Brodmann (BA) classification for frontal (BA10), temporal (BA21) and occipital (BA18) cortices.
2.2. SERT, TPH2, serotonin and 5-HIAA analyses
Levels of SERT and TPH2 immunoreactivity and a “control” protein neurone-specific enolase (NSE) were determined in tissue homogenates by quantitative immunoblotting (see (Kish et al., 2005; Kish et al., 2009) for more details) in the cocaine and heroin users and in 15 of the controls (age, 36±3 years; PMI, 14±2 h) who were matched for age and PMI. In brief, five concentrations of a tissue standard were employed consisting of a pooled human caudate sample (for SERT and TPH2) or frontal cortex (for NSE) running on each gel together with the samples. Protein concentration was determined using the Bio-Rad Protein Assay Kit (Bio-Rad, Hercules, CA, USA) with bovine plasma albumin as the standard. SERT, TPH2 or NSE protein immunoreactivity in each sample was determined by interpolation from the linear standard curve and expressed as microgram tissue standard protein per microgram tissue sample protein. The primary antibodies used for SERT, TPH2 and NSE were from Mab Technologies (Stone Mountain, GA, USA; cat# ST-51–1, against an N terminus 16-aa peptide of human SERT), Sigma-Aldrich (St Louis, MO, USA; cat#T0678, against recombinant rabbit TPH) and Santa Cruz Biotechnology (Santa Cruz, CA, USA; cat#sc-21737, against aa 271–285 of human NSE), respectively. The monoclonal SERT antibody ST-51 (IgG1) has been widely used (Ramsey & DeFelice, 2002; Rajkowska et al., 2017), with specificity in autopsied human brain confirmed by detection of a predominant broad protein band centered at the expected molecular weight of 77 kDa (see Fig. 1) in SERT-rich brain areas of basal ganglia, thalamus and hippocampus (Kish et al., 2005), by expected regional brain distribution of SERT immunoreactivity that is highly significantly correlated with positron emission tomography binding of [11C]DASB in living human brain (Tong et al., 2013), and by marked loss of SERT immunoreactivity in brain of a chronic user of the serotonin “neurotoxin” ecstasy (3,4-Methylenedioxy-methamphetamine (MDMA), (Kish et al., 2010a)). Further, that the 77 kDa protein band corresponds to SERT rather than the more abundant dopamine transporter in the striatum was supported by preferential loss of SERT immunoreactivity in caudate versus putamen in brain of patients with Parkinson’s disease, which is opposite to the preferential loss of dopamine transporter in putamen versus caudate in this degenerative condition (Kish et al., 2008). The monoclonal pan-TPH antibody (IgG3) has been characterized previously (Haycock et al., 2002; Carkaci-Salli et al., 2011), with the isoform TPH2 of 55 kDa selectively expressed in the brain. The coefficients of variation for SERT immunoreactivity were 8.9% and 9.3% within and between blots and 11.8% and 9.3%, respectively, for TPH2 immunoreactivity (Kish et al., 2005; Kish et al., 2008; Kish et al., 2009). Levels of serotonin and metabolite 5-HIAA were determined by HPLC-electrochemical detection (Wilson et al., 1994) in 14 of the controls (age, 34±4 years; PMI, 14±2 h) and in all of the cocaine and heroin users, with the data previously reported for the controls (Kish et al., 2009), for eight of the nine cocaine users (Wilson et al., 1996), and for nine of the 11 users of heroin (Kish et al., 2001).
Figure 1.
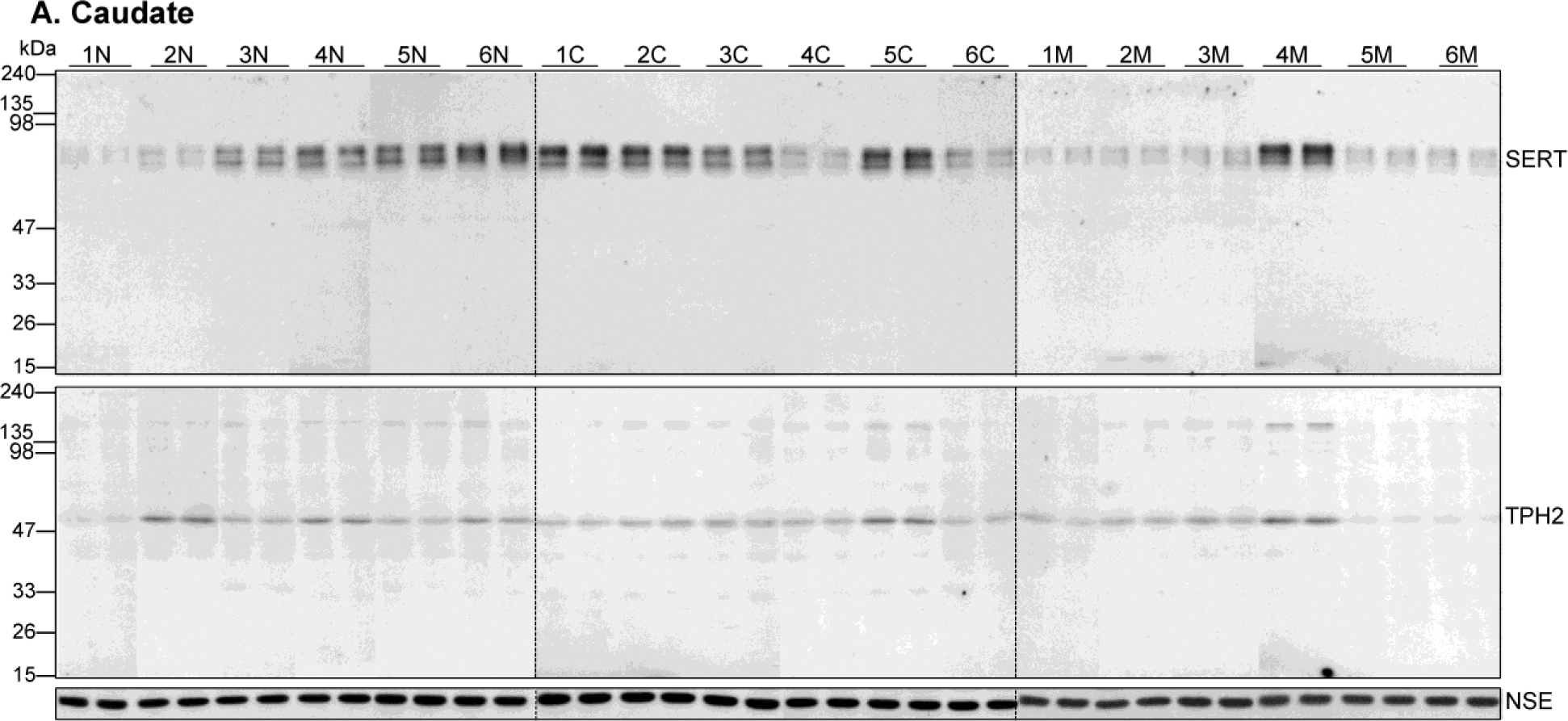
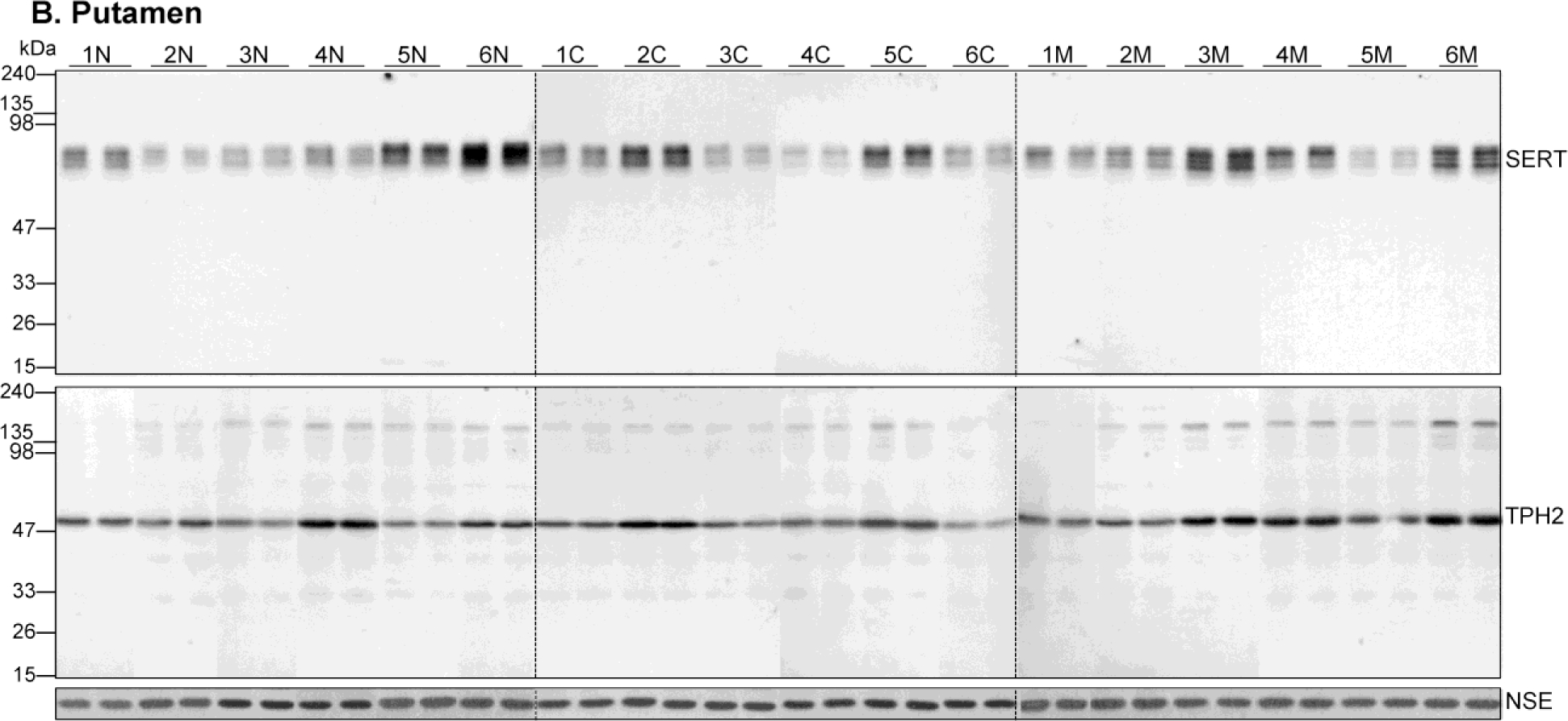
Representative immunoblots of serotonin transporter (SERT), tryptophan hydroxylase-2 (TPH2) and the control protein neuron-specific enolase (NSE) in the caudate nucleus (A) and putamen (B) in six cases each of the control subjects (1N-6N) and users of cocaine (1C-6C) and heroin (1M-6M). Duplicate samples were run for each subjects. Note variable SERT (~77 kDa) and TPH2 (55 kDa) immunoreactivities, but not for NSE (47 kDa), among subjects of both controls and drug users.
2.3. Statistical analyses
Our pre-planned directional hypothesis, based on the previously reported postmortem brain SERT binding studies in the forebrain, was that SERT proteins would be above normal in all examined brain areas of the cocaine users. Statistical analyses were performed by using the StatSoft STATISTICA 7.1 (Tulsa, Oklahoma, USA). Differences in levels of SERT, TPH2, and the other serotonin markers between the controls and drug groups in brain regions examined were conducted using nonparametric Kruskal-Wallis tests (P < 0.05) followed by correction for multiple comparisons (P < 0.05) because many of the data did not pass normality tests (see Fig. 2). Correlations were examined by Pearson product moment correlation or Spearman ranking order correlation as indicated in the text.
Figure 2.
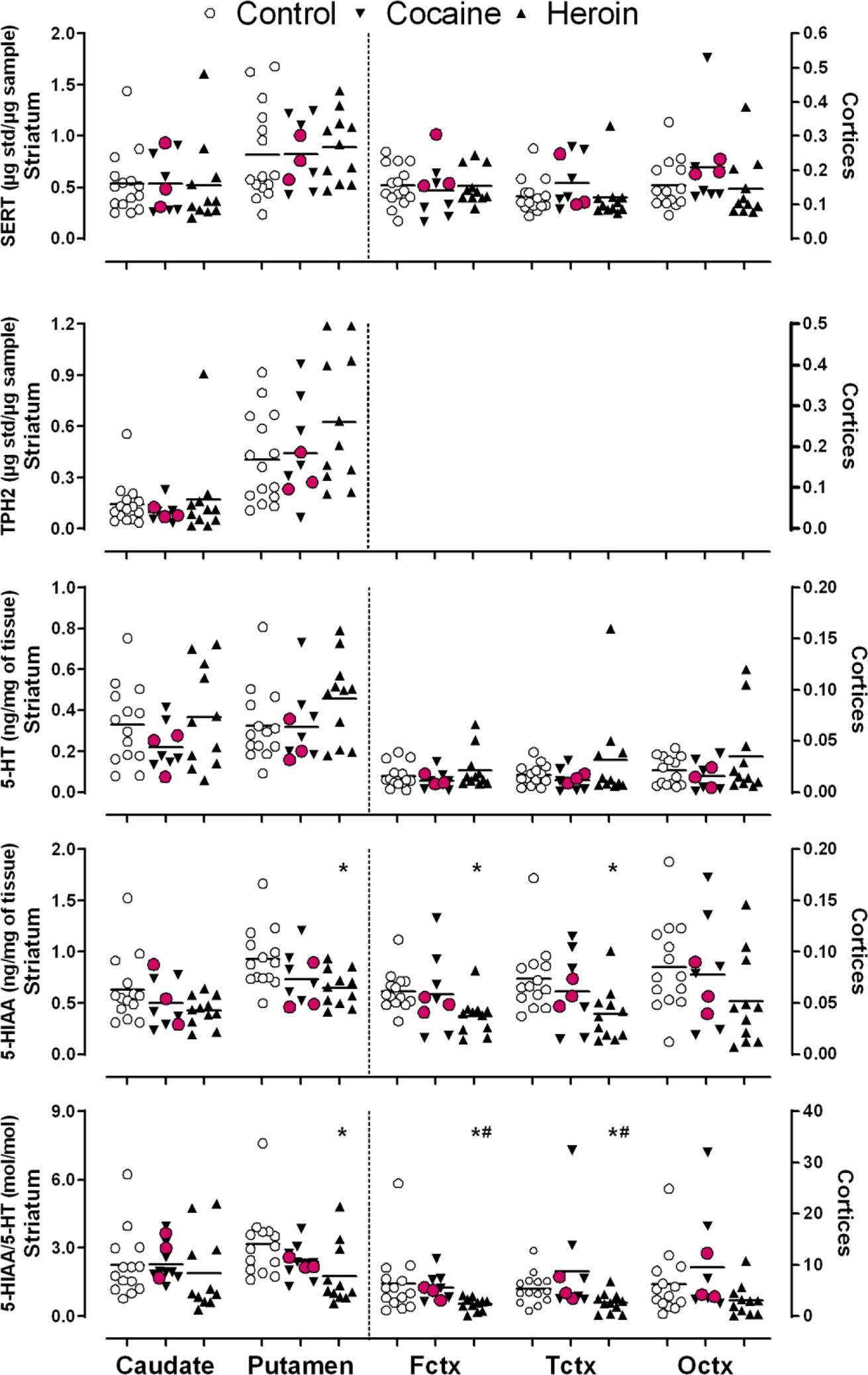
Scatter plots of levels of serotonin transporter (SERT), tryptophan hydroxylase-2 (TPH2), serotonin (5-HT), 5-hydroxyindoleacetic acid (5-HIAA) and the molar ratio of 5-HIAA/5-HT in brain of users of cocaine and heroin and control subjects. Levels of TPH2 were not measured in cerebral cortical areas including frontal (Fctx), temporal (Tctx) and occipital (Octx) cortices because of low levels of the protein that could not be measured reliably. *P < 0.05, drug users vs. healthy controls and #P < 0.05, heroin vs. cocaine users (Kruskal-Wallis tests followed by multiple comparison corrections). Red circles covering down-triangles identify three cocaine users with high levels of cocaine and metabolites in brain. Note much lower levels of the serotonergic markers in cerebral cortices (plotted on right y axis) than in the striatum (plotted on left y axis).
3. Results
As shown in Table 2, protein levels of SERT and TPH2 (the latter examined only in the TPH2-“rich” striatum) and the control protein NSE were normal (P > 0.05) in all examined brain regions of the cocaine and the heroin users (see Fig. 1 for representative immunoblots). An analysis of individual data disclosed extensive overlap between the individual control and drug user SERT and TPH2 values in the brain areas examined (Fig. 2). In agreement with the previous report of a smaller sample size (n = 8–9; Kish et al. 2001), brain concentrations of the serotonin metabolite 5-HIAA and the molar ratio of 5-HIAA vs serotonin, an index of serotonin metabolism, were generally lower in brain of chronic heroin users as compared to those of controls, with significant decrease observed in putamen, frontal and temporal cortices (Table 2 and Fig. 2). However, as previously reported (Wilson et al. 1996), levels of serotonin, 5-HIAA and 5-HIAA/5-HT ratio did not differ between cocaine users and controls (p > 0.05).
Table 2.
Serotonin markers in brain of control subjects and users of cocaine and heroin
| Control | Cocaine | Heroin | P | |
|---|---|---|---|---|
| Caudate | ||||
| Serotonin | 0.33 ± 0.05 (14) | 0.22 ± 0.04 (9) | 0.37 ± 0.08 (11) | 0.32 |
| 5-HIAA | 0.63 ± 0.09 (14) | 0.50 ± 0.08 (9) | 0.42 ± 0.04 (11) | 0.22 |
| 5-HIAA/5-HT (mol/mol) | 2.3 ± 0.4 (14) | 2.3 ± 0.3 (9) | 1.9 ± 0.5 (11) | 0.28 |
| SERT | 0.54 ± 0.08 (15) | 0.54 ± 0.10 (9) | 0.52 ± 0.12 (11) | 0.60 |
| TPH2 | 0.14 ± 0.03 (15) | 0.10 ± 0.02 (9) | 0.17 ± 0.08 (11) | 0.57 |
| NSE | 0.74 ± 0.02 (15) | 0.76 ± 0.04 (9) | 0.72 ± 0.04 (11) | 0.82 |
| Putamen | ||||
| Serotonin | 0.33 ± 0.05 (14) | 0.32 ± 0.06 (9) | 0.46 ± 0.06 (11) | 0.19 |
| 5-HIAA | 0.93 ± 0.08 (14) | 0.73 ± 0.08 (9) | 0.65 ± 0.05 (11)* | 0.021 |
| 5-HIAA/5-HT (mol/mol) | 3.2 ± 0.4 (14) | 2.4 ± 0.3 (9) | 1.8 ± 0.4 (11)* | 0.014 |
| SERT | 0.82 ± 0.12 (15) | 0.82 ± 0.11 (9) | 0.89 ± 0.10 (11) | 0.85 |
| TPH2 | 0.41 ± 0.06 (15) | 0.44 ± 0.10 (9) | 0.63 ± 0.12 (11) | 0.31 |
| NSE | 0.68 ± 0.04 (15) | 0.71 ± 0.03 (9) | 0.73 ± 0.03 (11) | 0.60 |
| Frontal cortex | ||||
| Serotonin | 0.016 ± 0.003 (14) | 0.011 ± 0.003 (9) | 0.022 ± 0.06 (11) | 0.27 |
| 5-HIAA | 0.062 ± 0.005 (14) | 0.058 ± 0.012 (9) | 0.037 ± 0.006 (11)* | 0.008 |
| 5-HIAA/5-HT (mol/mol) | 6.4 ± 1.7 (14) | 5.6 ± 0.9 (9) | 2.4 ± 0.4 (11)*# | 0.010 |
| SERT | 0.16 ± 0.02 (15) | 0.14 ± 0.03 (9) | 0.15 ± 0.02 (11) | 0.77 |
| NSE | 0.79 ± 0.02 (15) | 0.85 ± 0.01 (9) | 0.86 ± 0.04 (11) | 0.14 |
| Temporal cortex | ||||
| Serotonin | 0.017 ± 0.003 (14) | 0.012 ± 0.003 (9) | 0.032 ± 0.014 (11) | 0.34 |
| 5-HIAA | 0.074 ± 0.009 (14) | 0.061 ± 0.012 (9) | 0.039 ± 0.008 (11)* | 0.024 |
| 5-HIAA/5-HT (mol/mol) | 5.4 ± 0.8 (14) | 8.8 ± 3.2 (9) | 2.7 ± 0.6 (11)*# | 0.010 |
| SERT | 0.12 ± 0.01 (15) | 0.16 ± 0.03 (9) | 0.12 ± 0.02 (11) | 0.19 |
| NSE | 0.89 ± 0.03 (15) | 0.89 ± 0.04 (9) | 0.89 ± 0.04 (11) | 0.94 |
| Occipital cortex | ||||
| Serotonin | 0.021 ± 0.004 (14) | 0.015 ± 0.005 (9) | 0.035 ± 0.012 (11) | 0.36 |
| 5-HIAA | 0.085 ± 0.012 (14) | 0.078 ± 0.017 (9) | 0.052 ± 0.013 (11) | 0.11 |
| 5-HIAA/5-HT (mol/mol) | 6.4 ± 1.7 (14) | 9.5 ± 3.3 (9) | 3.1 ± 0.9 (11) | 0.053 |
| SERT | 0.16 ± 0.02 (15) | 0.21 ± 0.04 (9) | 0.15 ± 0.03 (11) | 0.15 |
| NSE | 0.86 ± 0.03 (15) | 0.89 ± 0.04 (9) | 0.96 ± 0.02 (11) | 0.10 |
Data are given as mean ± SEM (n) in ng/mg of wet tissue for serotonin and 5-hydroxyindoleacetic acid (5-HIAA), and in μg tissue standard protein/μg tissue sample protein for serotonin transporter (SERT), tryptophan hydroxylase-2 (TPH2), and neuron specific enolase (NSE). Levels of the serotonin markers in control subjects have been previously reported (Kish et al. 2009); levels of serotonin and 5-HIAA in the caudate and putamen in eight of the nine cocaine users (Wilson et al. 1996) and nine of the eleven heroin users (Kish et al. 2001) were also previously reported.
P < 0.05, drug users vs. healthy controls and
P < 0.05, heroin vs. cocaine users (multiple comparison corrections following Kruskal-Wallis tests, with P < 0.05 in bold).
No significant correlation was observed between levels of the serotonin markers and available drug use parameters of the cocaine and heroin users including duration of use (Pearson) and composite blood and brain drug levels (Spearman). Three cocaine users demonstrated a markedly higher level of cocaine and metabolites in brain than other cocaine users (>150 versus <50 nmol/g tissue); however, the three cocaine users did not have higher mean brain levels of SERT than those with lower brain levels of cocaine plus metabolites (by Mann-Whitney U-test). Indeed, the three cocaine users did not have out-of-range values of any of the serotonin markers (see Fig. 2). Among the drug users, two cocaine users and five heroin users were positive for blood alcohol at the time of autopsy. Comparing the blood alcohol positive to negative drug users (Mann-Whitney U-test), significantly higher levels of SERT in putamen (1.11±0.05 vs 0.73±0.09, p=0.02) and TPH2 in caudate (0.25±0.11 vs 0.08±0.01, p=0.03) were observed, with the difference in caudate SERT (0.71±0.18 vs 0.43±0.06, p=0.18) or putamen TPH2 (0.68±0.14 vs 0.47±0.09, p=0.21) statistically non-significant (see Supplementary Figure).
We found no significant correlation (Pearson) between levels of any serotonin marker and age or PMI of the subjects (all subjects included or in individual groups). We also did not find any significant influence of the 5HTTLPR polymorphism on protein levels of SERT in brain of either healthy controls or drug users (p>0.05, LL vs LS+SS, Mann-Whitney U-test, given very small number of SS amongst the subjects). This differs from contrasting findings in the postmortem brain report of 125I-RTI-55 SERT binding by (Little et al., 1998) but is consistent with other studies showing no influence of the promotor polymorphism on brain SERT binding in vitro and in vivo (Mann et al., 2000; Murthy et al., 2010).
4. Discussion
The main finding of our study is that protein levels of SERT are not markedly increased, as suggested by a previous autopsied brain investigation employing SERT binding, in the examined forebrain regions of the cocaine users of our study. SERT protein was also normal in brain of the heroin users, a drug abuse “control” group, despite decreased brain serotonin metabolism as inferred from decreased levels of 5-HIAA and 5-HIAA/5-HT ratio, consistent with results of an earlier imaging investigation employing the non-specific transporter probe 123I-β-CIT (Cosgrove et al., 2010).
4.1. Limitations
Among the many limitations of postmortem human brain studies is the possibility that postmortem time might have influenced protein levels of SERT. However, there were no statistically significant differences amongst mean postmortem times for the drug and control groups and no statistically significant correlation between postmortem time and brain SERT protein in the groups - and previously, we found similar levels of SERT protein in human biopsied (within 15 minutes of excision) vs. autopsied (mean 14 hours) brain (Kish et al., 2009). Little verified retrospective information could be obtained on long-term (e.g., years) medication history or exposure to other drugs. Thus, the possibility cannot be excluded that other unknown drugs used by the subjects of our study might have influenced brain SERT levels (e.g. by preventing or enhancing a cocaine-induced increase in SERT). In this respect, alcohol use could have been a confounding factor as higher SERT levels were observed in putamen of drug users positive for blood alcohol at autopsy as compared to those negative for alcohol. It should be cautioned that the relationship between blood alcohol and brain SERT protein noted in this report is at most preliminary, given many conflicting reports on serotonergic function and SERT changes in human alcoholism (see (Heinz et al., 2000; Brown et al., 2007; Belmer et al., 2016) for more details). However, a strength of the study is that forensic brain drug and metabolite analyses prove that the drug users must have used cocaine, and used the drug relatively recently. We were also limited by the lack of information in case records on other possible psychiatric (e.g, mood disorders) co-morbidities of the users of cocaine and heroin, which might have contributed to the variability of SERT protein levels (e.g, mood disturbance, see (Meyer, 2007) for review). As to the question whether Western blotting is sufficiently sensitive to detect a SERT change in a human brain condition, in our previous studies we could detect a brain SERT protein change (reduction) in conditions in which a decline would be expected (Parkinson’s disease, (Kish et al., 2008); ecstasy [3,4-methylenedioxymethamphetamine user - single case, (Kish et al., 2010a)) and in human methamphetamine users in which a SERT reduction would not be unexpected (Kish et al., 2009). It is also likely that we would have been able to detect a massive 100–200% increase in SERT protein as suggested by the previous autopsied brain investigation of SERT binding in human cocaine users (Mash et al., 2000). In this respect, a caveat of our study is that levels of total SERT protein in tissue homogenates might not reflect cell surface transporter available for ligand binding given dynamic trafficking/sorting of SERT between plasmalemma membrane and intracellular compartments, e.g., endoplasmic reticulum, Golgi apparatus, synaptic vesicles, and endosomes (see below).
4.2. Comparison with literature findings
To our knowledge, studies of SERT protein in brain of human cocaine users have been scarce, limited to one PET examination and two autoradiography reports. Although there is an extensive experimental animal literature on the influence of repeated (antidepressant) SERT inhibitors on brain SERT binding, this literature is contradictory in terms of whether a change does or does not occur and the direction of the change (e.g., see (Benmansour et al., 1999; Benmansour et al., 2002; Descarries & Riad, 2012; Sawyer et al., 2012)). The variable results might be explained by factors including differences in dosing parameters and withdrawal times, age and specific strain of animals, and brain regions examined.
Analysis of the much more limited pre-clinical literature on repeated cocaine administration on SERT (binding) tentatively suggests that brain SERT upregulation might be a transient change, only observed when animals are either on cocaine or soon after taking the drug (Cunningham et al., 1992; Belej et al., 1996; Banks et al., 2008; Gould et al., 2011) whereas after longer abstinence binding becomes normalized (Katz et al., 1993; Belej et al., 1996). However, this would not seem to be an issue in our investigation as all cocaine users tested positive at autopsy for cocaine or metabolites in brain indicating recent use of the drug. On the other hand, the same animal literature discloses little agreement on the brain areas affected by cocaine (e.g. striatum/caudate SERT binding increased: (Banks et al., 2008); striatum SERT binding normal: (Cunningham et al., 1992; Belej et al., 1996)) and further, that only a relatively small proportion of examined brain areas in these investigations show increased concentration of the transporter. In this regard, in the most regionally exhaustive brain investigation (by autoradiography) a modest (and transient) increase in SERT binding was reported in only five of fifty brain areas/subdivisions assessed in animals sacrificed while on cocaine (Belej et al., 1996). Thus, the pre-clinical animal findings, although limited, suggest that any SERT up-regulation induced by chronic cocaine exposure might not be robust in terms of a global brain effect, and that we might have missed critical areas (if any) affected by the drug in our investigation of a limited number of brain regions.
The strongest evidence for the possibility of a marked cocaine-induced SERT upregulation in human brain is that provided in the postmortem brain autoradiography study of Mash and colleagues (Mash et al., 2000), who examined a total of 14 chronic cocaine users, of which eight subjects died in a state of “excited delirium”, and employed 125I-RTI-55 to measure SERT binding in the presence of benztropine (to block binding of the non-specific ligand to the dopamine transporter). This investigation reported a striking increase (by up to approximately 2–3 fold) in striatum (a region in which we found no difference in SERT protein vs. controls), amygdala, and in some, but not all, cerebral cortical areas, but with the extent of binding increase dependent somewhat on the brain slice location (anterior vs posterior), cerebral cortical subdivision selected and on whether the subjects died in excited delirium (see below). In both our study and that of Mash most, or all, subjects were male, and had similar ages and postmortem times, and the known or suspected cause of death in most subjects was “cocaine toxicity/intoxication”. However, the majority (eight of 14) of drug users in the Mash investigation had preterminal evidence of “excited delirium” before death whereas the case records of our study did not disclose evidence of any pre-terminal bizzare/violent behavior. Levels of cocaine and one or more metabolites (cocaine, benzoylecgonine, econine methylester, norcocaine, cocaethylene; see (Kalasinsky et al., 2000)) were detected in 15 brain regions in all of our nine subjects - and cocaine and metabolite benzoylecgonine were reported in brain of six of the 14 subjects in the Mash investigation for which analyses were conducted. Overall, the cocaine users in current report (Table 1) and the Mash investigation had similar levels of cocaine plus metabolite benzoylecgonine (in nmol/g brain tissue or μM in blood) in brain (69±25 [11–180, median 21] versus 22±9 [5.6–65, median 12]) as well as in blood (60±46 [0–424, median 17] versus 106±86 [1–1220, median 12]). Further, brain and blood drug levels in cocaine users of our study were also similar to the six cocaine overdose subjects of Mash investigation (brain: 25 and 65; blood: 234±197 [6–1220, median 44]), who had higher drug levels than those died in excited delirium (brain: 9.6±1.8 [5.6–14, median 9.4]; blood: 9.8±2.9 [1–26, median 7.5]) and also wider regional extent of brain SERT binding elevation including both anterior and posterior basal ganglia. The detection of cocaine/metabolites in brain of cocaine users of both investigations suggests that all had used the drug “recently” and were therefore subject, just prior to death, to some influence of the drug. The different outcomes between the Mash investigation and our own study, despite some similarities in subject characteristics, could thus be due to methodological differences. Our immunoblotting assay measures total protein levels of SERT in tissue homogenates whereas the autoradiography assay of Mash investigation measures radioligand binding at high affinity sites which may not correspond to all SERT proteins. As a primarily cell surface protein with 12 transmembrane domains, SERT undergoes a variety of post-translational modifications, e.g., glycosylation and phosphorylation, which regulate its association with other proteins, membrane trafficking, and also substrate/ligand binding (Ramamoorthy et al., 1998; Zhong et al., 2012; Rajamanickam et al., 2015; Anderluh et al., 2017). For example, in contrast to 5-HT, MDMA and SSRIs that promote SERT internalization, cocaine is known to promote SERT cell surface localization (Kittler et al., 2010), which might provide an explanation for the differential findings of increased radioligand binding without alteration in total SERT protein. In this scenario, it is noted that the differences in levels of 125I-RTI-55 SERT binding between striatum and cerebral cortical areas in control brains were less than two-fold (see Figures 3 and 4 of (Mash et al., 2000)) whereas the differences in total SERT protein between striatum and cerebral cortices were 3–5 fold (Table 1), suggesting that the striatum might have more SERT protein not available for radioligand binding as compared to cerebral cortex. Alternative explanations for the methodological differences might involve uncertainty in the definition of non-specific binding for an autoradiography assay, in particular given the non-selective profile of 125I-RTI-55 for DAT and SERT and other targets including the so-called SERTsite2 (Rothman et al., 1998) and dominant presence of DAT over SERT in human striatum (Staley et al., 1994). There is also the possibility that a SERT increase is not a common or robust characteristic of cocaine exposure in human users of the drug.
4.3. Conclusion
In conclusion, we find no evidence for a marked increase in SERT protein, as suggested by a previous radioligand binding investigation, in autopsied brain of human, chronic cocaine users. However, our findings, based on a small sample size, are preliminary and require replication. Future investigations might examine SERT or SERT binding (e.g., using now presently available SERT-specific positron emission tomography probes such as 11C-DASB; see (Kish et al., 2010b)) in a much more regionally comprehensive manner should transporter changes be limited to a small number of brain areas. Studies of SERT status in users of drugs of abuse appear to be warranted given the present interest in testing serotonergic agents in these conditions.
Supplementary Material
Funding Information:
This work was supported by a grant from the National Institute of Health, National Institute on Drug Abuse, R01 DA07182 (SJK)
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of interest The authors declare that they have no conflict of interest.
References
- Anderluh A, Hofmaier T, Klotzsch E, et al. (2017) Direct PIP2 binding mediates stable oligomer formation of the serotonin transporter. Nat Commun 8: 14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban TA (2001) Pharmacotherapy of depression: a historical analysis. Journal of neural transmission 108: 707–716. [DOI] [PubMed] [Google Scholar]
- Banks ML, Czoty PW, Gage HD, et al. (2008) Effects of cocaine and MDMA self-administration on serotonin transporter availability in monkeys. Neuropsychopharmacology 33: 219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks ML & Negus SS (2017) Repeated 7-Day Treatment with the 5-HT2C Agonist Lorcaserin or the 5-HT2A Antagonist Pimavanserin Alone or in Combination Fails to Reduce Cocaine vs Food Choice in Male Rhesus Monkeys. Neuropsychopharmacology 42: 1082–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belej T, Manji D, Sioutis S, Barros HM & Nobrega JN (1996) Changes in serotonin and norepinephrine uptake sites after chronic cocaine: pre- vs. post-withdrawal effects. Brain Res 736: 287–296. [DOI] [PubMed] [Google Scholar]
- Belmer A, Patkar OL, Pitman KM & Bartlett SE (2016) Serotonergic Neuroplasticity in Alcohol Addiction. Brain Plast 1: 177–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmansour S, Cecchi M, Morilak DA, et al. (1999) Effects of chronic antidepressant treatments on serotonin transporter function, density, and mRNA level. J Neurosci 19: 10494–10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmansour S, Owens WA, Cecchi M, Morilak DA & Frazer A (2002) Serotonin clearance in vivo is altered to a greater extent by antidepressant-induced downregulation of the serotonin transporter than by acute blockade of this transporter. J Neurosci 22: 6766–6772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AK, George DT, Fujita M, et al. (2007) PET [11C]DASB imaging of serotonin transporters in patients with alcoholism. Alcohol Clin Exp Res 31: 28–32. [DOI] [PubMed] [Google Scholar]
- Carkaci-Salli N, Salli U, Kuntz-Melcavage KL, et al. (2011) TPH2 in the ventral tegmental area of the male rat brain. Brain Res Bull 84: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JA, Yang D, Zhao Z, et al. (2019) Serotonin transporter-ibogaine complexes illuminate mechanisms of inhibition and transport. Nature 569: 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins GT & France CP (2018) Effects of lorcaserin and buspirone, administered alone and as a mixture, on cocaine self-administration in male and female rhesus monkeys. Exp Clin Psychopharmacol 26: 488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins GT, Gerak LR & France CP (2018) The behavioral pharmacology and therapeutic potential of lorcaserin for substance use disorders. Neuropharmacology 142: 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove KP, Tellez-Jacques K, Pittman B, et al. (2010) Dopamine and serotonin transporter availability in chronic heroin users: a [123I]beta-CIT SPECT imaging study. Psychiatry Res 184: 192–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox SM, Benkelfat C, Dagher A, et al. (2011) Effects of lowered serotonin transmission on cocaine-induced striatal dopamine response: PET [11C]raclopride study in humans. Br J Psychiatry 199: 391–397. [DOI] [PubMed] [Google Scholar]
- Cunningham KA & Anastasio NC (2014) Serotonin at the nexus of impulsivity and cue reactivity in cocaine addiction. Neuropharmacology 76 Pt B: 460–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham KA, Paris JM & Goeders NE (1992) Chronic cocaine enhances serotonin autoregulation and serotonin uptake binding. Synapse 11: 112–123. [DOI] [PubMed] [Google Scholar]
- Descarries L & Riad M (2012) Effects of the antidepressant fluoxetine on the subcellular localization of 5-HT1A receptors and SERT. Philos Trans R Soc Lond B Biol Sci 367: 2416–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George FR (1989) Cocaine produces low dose locomotor depressant effects in mice. Psychopharmacology (Berl) 99: 147–150. [DOI] [PubMed] [Google Scholar]
- Gould RW, Gage HD, Banks ML, et al. (2011) Differential effects of cocaine and MDMA self-administration on cortical serotonin transporter availability in monkeys. Neuropharmacology 61: 245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycock JW, Kumer SC, Lewis DA, Vrana KE & Stockmeier CA (2002) A monoclonal antibody to tryptophan hydroxylase: applications and identification of the epitope. J Neurosci Methods 114: 205–212. [DOI] [PubMed] [Google Scholar]
- Heal DJ, Gosden J & Smith SL (2014) Dopamine reuptake transporter (DAT) “inverse agonism”--a novel hypothesis to explain the enigmatic pharmacology of cocaine. Neuropharmacology 87: 19–40. [DOI] [PubMed] [Google Scholar]
- Heinz A, Jones DW, Mazzanti C, et al. (2000) A relationship between serotonin transporter genotype and in vivo protein expression and alcohol neurotoxicity. Biol Psychiatry 47: 643–649. [DOI] [PubMed] [Google Scholar]
- Higgins GA, Fletcher PJ & Shanahan WR (2020) Lorcaserin: A review of its preclinical and clinical pharmacology and therapeutic potential. Pharmacol Ther 205: 107417. [DOI] [PubMed] [Google Scholar]
- Higgins GA, Sellers EM & Fletcher PJ (2013) From obesity to substance abuse: therapeutic opportunities for 5-HT2C receptor agonists. Trends Pharmacol Sci 34: 560–570. [DOI] [PubMed] [Google Scholar]
- Hoffman BJ, Mezey E & Brownstein MJ (1991) Cloning of a serotonin transporter affected by antidepressants. Science 254: 579–580. [DOI] [PubMed] [Google Scholar]
- Howell LL & Negus SS (2014) Monoamine transporter inhibitors and substrates as treatments for stimulant abuse. Adv Pharmacol 69: 129–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen LK, Staley JK, Malison RT, et al. (2000) Elevated central serotonin transporter binding availability in acutely abstinent cocaine-dependent patients. Am J Psychiatry 157: 1134–1140. [DOI] [PubMed] [Google Scholar]
- Kalasinsky KS, Bosy TZ, Schmunk GA, et al. (2000) Regional distribution of cocaine in postmortem brain of chronic human cocaine users. J Forensic Sci 45: 1041–1048. [PubMed] [Google Scholar]
- Katz JL, Griffiths JW, Sharpe LG, De Souza EB & Witkin JM (1993) Cocaine tolerance and cross-tolerance. J Pharmacol Exp Ther 264: 183–192. [PubMed] [Google Scholar]
- Kish SJ, Fitzmaurice PS, Boileau I, et al. (2009) Brain serotonin transporter in human methamphetamine users. Psychopharmacology (Berl) 202: 649–661. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Fitzmaurice PS, Chang LJ, Furukawa Y & Tong J (2010a) Low striatal serotonin transporter protein in a human polydrug MDMA (ecstasy) user: a case study. J Psychopharmacol 24: 281–284. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Furukawa Y, Chang LJ, et al. (2005) Regional distribution of serotonin transporter protein in postmortem human brain: is the cerebellum a SERT-free brain region? Nucl Med Biol 32: 123–128. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Kalasinsky KS, Derkach P, et al. (2001) Striatal dopaminergic and serotonergic markers in human heroin users. Neuropsychopharmacology 24: 561–567. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Lerch J, Furukawa Y, et al. (2010b) Decreased cerebral cortical serotonin transporter binding in ecstasy users: a positron emission tomography/[11C]DASB and structural brain imaging study. Brain 133: 1779–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kish SJ, Shannak K & Hornykiewicz O (1988) Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. Pathophysiologic and clinical implications. N Engl J Med 318: 876–880. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Tong J, Hornykiewicz O, et al. (2008) Preferential loss of serotonin markers in caudate versus putamen in Parkinson’s disease. Brain 131: 120–131. [DOI] [PubMed] [Google Scholar]
- Kittler K, Lau T & Schloss P (2010) Antagonists and substrates differentially regulate serotonin transporter cell surface expression in serotonergic neurons. Eur J Pharmacol 629: 63–67. [DOI] [PubMed] [Google Scholar]
- Little KY, McLaughlin DP, Zhang L, et al. (1998) Cocaine, ethanol, and genotype effects on human midbrain serotonin transporter binding sites and mRNA levels. Am J Psychiatry 155: 207–213. [DOI] [PubMed] [Google Scholar]
- Mann JJ, Huang YY, Underwood MD, et al. (2000) A serotonin transporter gene promoter polymorphism (5-HTTLPR) and prefrontal cortical binding in major depression and suicide. Arch Gen Psychiatry 57: 729–738. [DOI] [PubMed] [Google Scholar]
- Mash DC, Staley JK, Izenwasser S, Basile M & Ruttenber AJ (2000) Serotonin transporters upregulate with chronic cocaine use. J Chem Neuroanat 20: 271–280. [DOI] [PubMed] [Google Scholar]
- Meyer JH (2007) Imaging the serotonin transporter during major depressive disorder and antidepressant treatment. J Psychiatry Neurosci 32: 86–102. [PMC free article] [PubMed] [Google Scholar]
- Murthy NV, Selvaraj S, Cowen PJ, et al. (2010) Serotonin transporter polymorphisms (SLC6A4 insertion/deletion and rs25531) do not affect the availability of 5-HTT to [11C] DASB binding in the living human brain. Neuroimage 52: 50–54. [DOI] [PubMed] [Google Scholar]
- Pare CM & Sandler M (1959) A clinical and biochemical study of a trial of iproniazid in the treatment of depression. J Neurol Neurosurg Psychiatry 22: 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirtle JL, Hickman MD, Boinpelly VC, et al. (2019) The serotonin-2C agonist Lorcaserin delays intravenous choice and modifies the subjective and cardiovascular effects of cocaine: A randomized, controlled human laboratory study. Pharmacol Biochem Behav 180: 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamanickam J, Annamalai B, Rahbek-Clemmensen T, et al. (2015) Akt-mediated regulation of antidepressant-sensitive serotonin transporter function, cell-surface expression and phosphorylation. The Biochemical journal 468: 177–190. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Mahajan G, Legutko B, et al. (2017) Length of axons expressing the serotonin transporter in orbitofrontal cortex is lower with age in depression. Neuroscience 359: 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthy S, Bauman AL, Moore KR, et al. (1993) Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proc Natl Acad Sci U S A 90: 2542–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamoorthy S, Giovanetti E, Qian Y & Blakely RD (1998) Phosphorylation and regulation of antidepressant-sensitive serotonin transporters. J Biol Chem 273: 2458–2466. [DOI] [PubMed] [Google Scholar]
- Ramsey IS & DeFelice LJ (2002) Serotonin transporter function and pharmacology are sensitive to expression level: evidence for an endogenous regulatory factor. J Biol Chem 277: 14475–14482. [DOI] [PubMed] [Google Scholar]
- Riley RA (1943) An atlas of the basal ganglia, brain stem and spinal cord. The Williams & Wilkins Company, Baltimore, MD. [Google Scholar]
- Ross SB & Renyi AL (1969) Inhibition of the uptake of tritiated 5-hydroxytryptamine in brain tissue. Eur J Pharmacol 7: 270–277. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Silverthorn ML, Glowa JR, et al. (1998) Studies of the biogenic amine transporters. VII. Characterization of a novel cocaine binding site identified with [125I]RTI-55 in membranes prepared from human, monkey and guinea pig caudate. Synapse 28: 322–338. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Ito H, Kimura Y, et al. (2012) Quantification of dopamine transporter in human brain using PET with 18F-FE-PE2I. J Nucl Med 53: 1065–1073. [DOI] [PubMed] [Google Scholar]
- Sawyer EK, Mun J, Nye JA, et al. (2012) Neurobiological changes mediating the effects of chronic fluoxetine on cocaine use. Neuropsychopharmacology 37: 1816–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shopsin B, Friedman E & Gershon S (1976) Parachlorophenylalanine reversal of tranylcypromine effects in depressed patients. Arch Gen Psychiatry 33: 811–819. [DOI] [PubMed] [Google Scholar]
- Simmler LD, Anacker AMJ, Levin MH, et al. (2017) Blockade of the 5-HT transporter contributes to the behavioural, neuronal and molecular effects of cocaine. British journal of pharmacology 174: 2716–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms D & Gallagher JP (1996) Modification of serotonin responses in rat dorsolateral septal nucleus neurons by acute and chronic cocaine. J Pharmacol Exp Ther 276: 1292–1303. [PubMed] [Google Scholar]
- Sora I, Wichems C, Takahashi N, et al. (1998) Cocaine reward models: conditioned place preference can be established in dopamine- and in serotonin-transporter knockout mice. Proc Natl Acad Sci U S A 95: 7699–7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley JK, Basile M, Flynn DD & Mash DC (1994) Visualizing dopamine and serotonin transporters in the human brain with the potent cocaine analogue [125I]RTI-55: in vitro binding and autoradiographic characterization. J Neurochem 62: 549–556. [DOI] [PubMed] [Google Scholar]
- Tong J, Fitzmaurice PS, Moszczynska A, et al. (2018) Normal glutathione levels in autopsied brain of chronic users of heroin and of cocaine. Drug Alcohol Depend 190: 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Meyer JH, Furukawa Y, et al. (2013) Distribution of monoamine oxidase proteins in human brain: implications for brain imaging studies. J Cereb Blood Flow Metab 33: 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FJ & Appel JB (1981) A neuropharmacological analysis of the discriminative stimulus properties of fenfluramine. Psychopharmacology (Berl) 73: 110–115. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Levey AI, Bergeron C, et al. (1996) Striatal dopamine, dopamine transporter, and vesicular monoamine transporter in chronic cocaine users. Ann Neurol 40: 428–439. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Nobrega JN, Carroll ME, et al. (1994) Heterogeneous subregional binding patterns of 3H-WIN 35,428 and 3H-GBR 12,935 are differentially regulated by chronic cocaine self-administration. J Neurosci 14: 2966–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Sanchez C & Caron MG (2012) Consideration of allosterism and interacting proteins in the physiological functions of the serotonin transporter. Biochemical pharmacology 83: 435–442. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.