

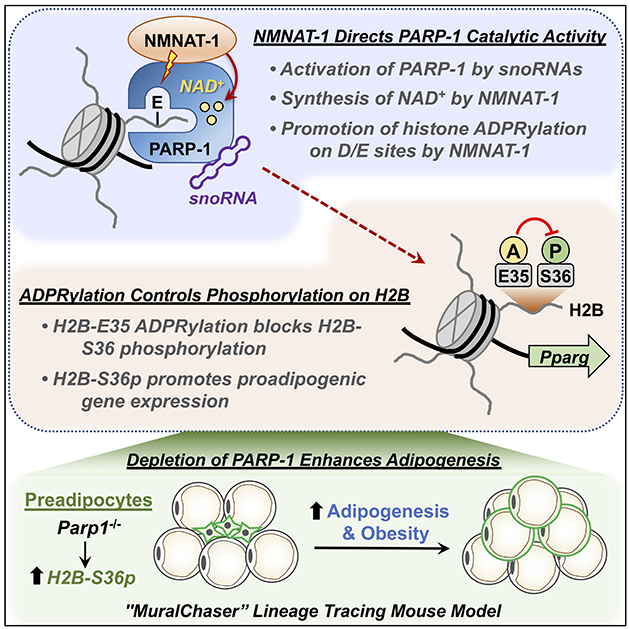
SUMMARY
Although ADP-ribosylation of histones by PARP-1 has been linked to genotoxic stress responses, its role in physiological processes and gene expression has remained elusive. We found that NAD+-dependent ADP-ribosylation of histone H2B-Glu35 by snoRNA-activated PARP-1 inhibits AMP kinase-mediated phosphorylation of adjacent H2B-Ser36, which is required for the proadipogenic gene expression program. The activity of PARP-1 on H2B requires NMNAT-1, a nuclear NAD+ synthase, which directs PARP-1 catalytic activity to Glu and Asp residues. ADP-ribosylation of Glu35 and the subsequent reduction of H2B-Ser36 phosphorylation inhibits the differentiation of adipocyte precursors in cultured cells. Parp1 knockout in preadipocytes in a mouse lineage tracing genetic model increases adipogenesis, leading to obesity. Collectively, our results demonstrate a functional interplay between H2B-Glu35 ADP-ribosylation and H2B-Ser36 phosphorylation that controls adipogenesis.
Keywords: Adipogenesis, ADP-ribosylation, Differentiation, Histones, PARP-1, Phosphorylation, Proliferation, Proteomics, snoRNA
eTOC:
Huang et al. show that the nuclear NAD+ synthase, NMNAT-1, directs PARP-1 catalytic activity to Glu and Asp residues on histones. Physiological ADP-ribosylation of histone H2B-Glu35 by snoRNA-activated PARP-1 with NMNAT-1 inhibits AMPK-mediated phosphorylation of adjacent H2B-Ser36, which is required for proadipogenic gene expression and fat metabolism in vivo.
INTRODUCTION
ADP-ribosylation (ADPRylation) is an NAD+-dependent post-translational modification of proteins, primarily on glutamate (Glu), aspartate (Asp), and serine (Ser) residues, which is mediated by the poly(ADP-ribosyl) transferase (PARP) family of enzymes (Gibson and Kraus, 2012; Ryu et al., 2015). ADPRylation of nuclear proteins has gained considerable attention of late, in part due to development of clinically-approved inhibitors of nuclear PARPs (e.g., PARP-1) as effective cancer therapeutics (Franzese et al., 2019; Kamel et al., 2018). Thousands of substrates of nuclear PARPs have been identified to date (Daniels et al., 2015; Gupte et al., 2017; Mangerich and Altmeyer, 2016), although the biological roles of site-specific ADPRylation have remained elusive. We have recently shown that nuclear NAD+ biosynthesis linked to PARP-1-mediated ADPRylation plays an important role in controlling the cellular events that promote the differentiation of committed preadipocytes into mature adipocytes (Luo et al., 2017; Ryu et al., 2018), demonstrating a key role for nuclear ADPRylation in a fundamental physiological process.
Nuclear proteins, such as core histones, were identified as substrates for ADPRylation decades ago (Burzio et al., 1979; Hilz and Stone, 1976; Jump et al., 1979; Minaga et al., 1979). But, unlike other well characterized histone modifications (e.g., acetylation, methylation, phosphorylation) (Lawrence et al., 2016), the sites of histone ADPRylation are poorly characterized and the functions of histone ADPRylation are unknown. Recent studies have identified serines in core histones as sites of ADPRylation during genotoxic stress, but this modification is less prevalent in physiological states (Larsen et al., 2018; Leidecker et al., 2016; Palazzo et al., 2018). Serine ADPRylation of core histones requires histone poly(ADP-ribosy)lation (PARylation) factor 1 (HPF1), a protein cofactor that promotes the recognition of histone substrates by PARP-1 and directs it to poly(ADP-ribosyl)ate (PARylate) them on Ser residues (Bonfiglio et al., 2017; Gibbs-Seymour et al., 2016; Palazzo et al., 2018). ADPRylation of Glu and Asp residues has also been detected after DNA damage (Gibson et al., 2016; Karch et al., 2017; Rakhimova et al., 2017). Whether an ADPRylation factor like HPF1 exists for Glu and Asp residues and whether such a factor might direct histone ADPRylation on these residues under physiological conditions is a fundamental unanswered question with important biological implications.
The historical focus on PARP-1 and genotoxic stress is based on the potent stimulation of PARP-1 catalytic activity by damaged DNA (e.g., double stranded breaks). One persistent argument against physiological roles of PARP-1 has been the lack of identification of a bona fide activator of PARP-1 catalytic activity that would be present under physiological (i.e., nongenotoxic stress) conditions. Previous studies have suggested that PARP-1 catalytic activity can be stimulated by histones, nucleosomes, and phosphorylation events on PARP-1 (Kraus and Lis, 2003; Krishnakumar and Kraus, 2010), but their role in activating PARP-1 under physiological conditions in vivo has not been demonstrated. Recently, our lab has shown that some snoRNAs are potent activators of PARP-1 catalytic activity in the absence of damaged DNA (Huang et al., 2020; Kim et al., 2019). Here we demonstrate a physiological role for snoRNA-dependent, PARP-1-mediated Glu/Asp ADPRylation events during adipogenesis.
RESULTS
Histones are ADPRylated on Glu and Asp residues in adipocytes in the absence of genotoxic stress
To explore PARP-1-mediated nuclear ADPRylation under physiological conditions in the absence of genotoxic stress, we focused on adipogenesis since recent studies have identified potential roles for PARP-1 and other NAD+-consuming enzymes in adipogenesis (Erener et al., 2012a; Erener et al., 2012b; Luo and Kraus, 2011; Luo et al., 2017; Ryu et al., 2018). For these experiments, we used 3T3-L1 cells, a well-characterized biological model of adipogenesis (Green and Kehinde, 1975). Glu and Asp are the primary sites of ADPRylation in 3T3-L1 cells, as determined by Western blotting for poly(ADP-ribose) (PAR) after treatment of the membrane with hydroxylamine (NH2OH), which chemically cleaves ADPR from Glu and Asp residues, but not other amino acids (Gibson et al., 2017; Zhang et al., 2013) (Figure S1A). To identify PARP-1 protein substrates in 3T3-L1 cells, we employed an NAD+ analog-sensitive PARP (asPARP) approach that we described previously (Gibson and Kraus, 2017; Gibson et al., 2016). In this assay, an asPARP-1 mutant (L877A) catalyzes the ADPRylation of PARP-1-specific substrates using a ‘clickable’ NAD+ analog [i.e., 8-Bu(3-yne)T-NAD+] (Figure 1A). To demonstrate asPARP-1-specific labeling of 3T3-L1 cell proteins, we added purified asPARP-1 to nuclear extracts, initiated the reactions with 8-Bu(3-yne)T-NAD+, clicked to TAMRA fluor, and analyzed the labeled proteins by in-gel fluorescence (Figure S1B). Then, to identify nuclear PARP-1 substrates by mass spectrometry, we expressed the asPARP-1 mutant in 3T3-L1 cells after knockdown of endogenous PARP-1 (Figure S1C) and performed the labeling reaction in isolated nuclei (Figure 1A).
Figure 1. Identification of PARP-1 substrates and specific ADPRylation sites in 3T3-L1 cells.
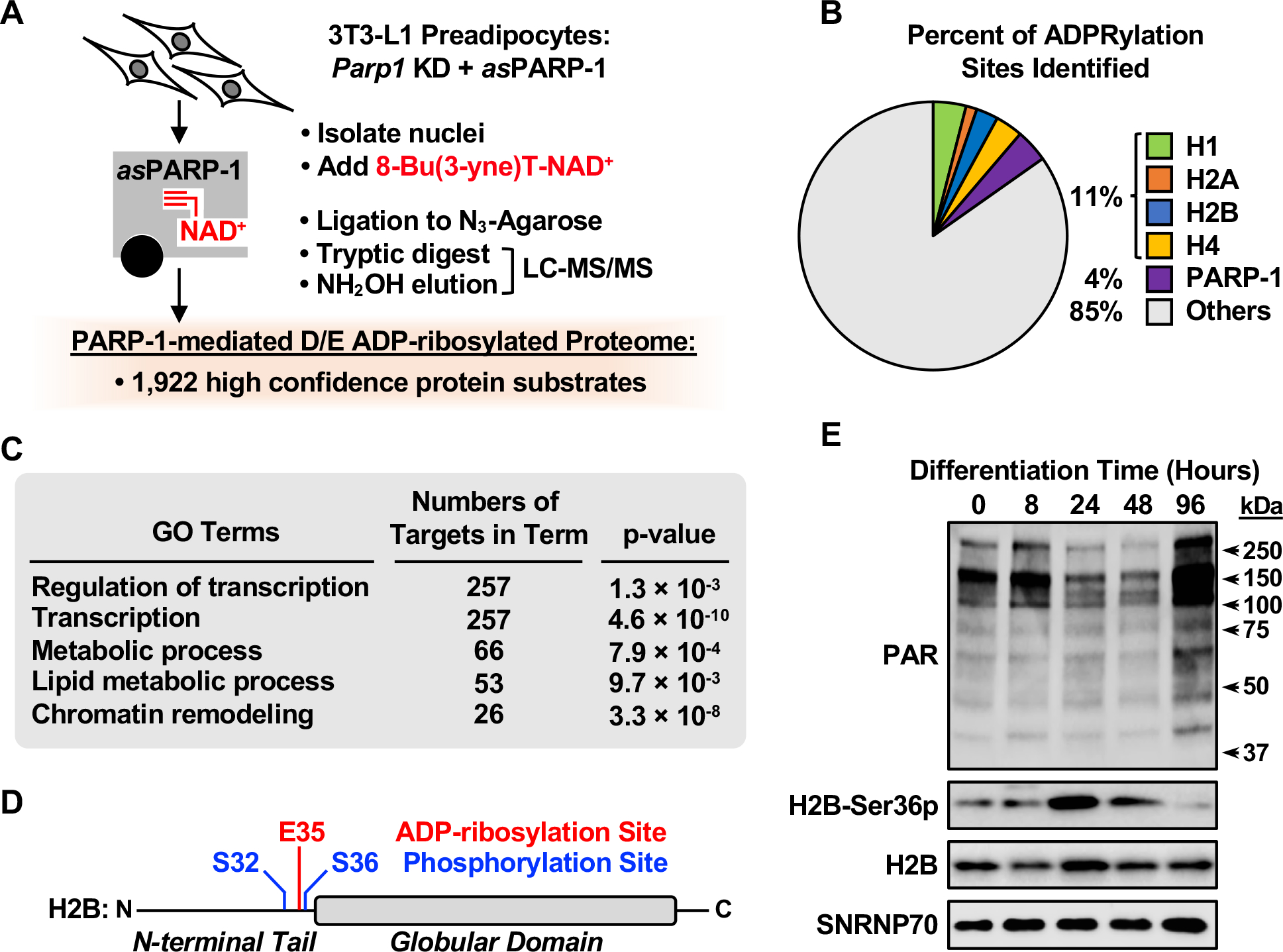
(A) Schematic representation of the identification of PARP-1-specific substrates and Glu/Asp ADPRylation sites in 3T3-L1 preadipocytes using an asPARP approach.
(B) Pie chart showing the percent of PARP-1-mediated ADPRylation sites identified on histones H1, H2A, H2B, and H4, and PARP-1.
(C) Gene ontology terms enriched for the protein targets of PARP-1 mediated ADPRylation.
(D) Schematic diagram showing the PARP-1-mediated Glu35 ADPRylation site determined by LC-MS/MS, as well as two adjacent phosphorylation sites (Ser32 and Ser36) on histone H2B.
(E) Western blots showing the levels of PAR and H2B-Ser36p in nuclear extracts from 3T3-L1 cells during a time course of differentiation with MDI. SNRNP70 was used as a loading control. Molecular weight markers in kilodaltons (kDa) are shown.
[See also Figure S1]
The substrate proteins were enriched by ‘clicking’ to azide-agarose beads using a copper-catalyzed azide-alkyne cycloaddition reaction, followed by trypsin cleavage and mass spectrometry to identify the released peptides (Gibson et al., 2016). Remaining peptides covalently linked to the agarose beads were released by hydroxylamine treatment, which also reduced the complexity of the ADPR modification, creating an adduct on Glu and Asp residues that was readily identifiable by mass spectrometry (Gibson et al., 2016; Zhang et al., 2013) (Figure 1A). Using this approach, we identified 1,922 unique high confidence Glu/Asp ADPRylated protein substrates of PARP-1 (Figures 1A, S1D, and S1E; Table S1). Core and linker histone proteins were enriched in the peptide and site identifications (Figure 1B), with histones H1, H2A, H2B, and H4 all showing multiple Glu or Asp ADPRylation sites (Table S2). Gene ontology (GO) analyses revealed major functions of the PARP-1 substrate proteins in lipid metabolism, chromatin remodeling, and the regulation of transcription (Figure 1C). These results demonstrate that histones are modified on Glu and Asp residues in a physiological context in the absence of genotoxic stress.
We and others have previously shown that ADPRylation sites are enriched near phosphorylation sites across the human proteome (Gibson et al., 2016; Larsen et al., 2018). In this regard, we were intrigued by Glu35 on H2B, which is adjacent to Ser36, a site of both AMPK- and S6K1-mediated phosphorylation (Bungard et al., 2010; Yi et al., 2016) (Figure 1D). Interestingly, the levels of phospho-H2B-Ser36 (H2B-Ser36p) increased dramatically in 3T3-L1 cells 24 to 48 hours after the initiation of signal-induced differentiation using MDI cocktail and then receded, as determined by Western blotting (Figure 1E). This increase occurred during a time frame in which the levels of nuclear NAD+ synthesized by the nuclear NAD+ synthase NMNAT-1 and PARP-1 catalytic activity are at a minimum, that we defined previously (Luo et al., 2017; Ryu et al., 2018) (Figure 1E). Importantly, the levels of H2B-PAR and H2B-Ser36p varied inversely throughout the time course of differentiation (i.e., when one was enriched, the other was depleted) as determined by immunoprecipitation (IP)-Western blotting analyses (Figure S1F). These results suggested to us that ADPRylation might somehow be linked to or inhibit the phosphorylation of H2B-Ser36. Additional analyses indicated that AMPK is the key kinase for H2B-Ser36 phosphorylation during adipogenesis in 3T3-L1 cells (Figure S2, A–C).
Functional interplay between ADPRylation and phosphorylation on H2B
To test the functional link between ADPRylation and H2B-Ser36 phosphorylation directly, we altered the levels of PARP-1 protein and activity in 3T3-L1 cells and then monitored the levels of bulk PAR and H2B-Ser36p by Western blotting. Treatment with the PARP-1-selective inhibitor BYK204165 caused a decrease in the levels of PAR and a dramatic increase in the levels of phospho-AMPKα and H2B-Ser36p, but had no effect on the levels of H2B-Ser32p, a nearby modification (Figures 2A and S3A). This was confirmed by using another PARP inhibitor, Niraparib (Figure S3B). Similarly, knockdown of Parp1 or Nmnat1 using previously characterized shRNAs (Luo et al., 2017; Ryu et al., 2018) caused a decrease in PAR levels and a corresponding increase in H2B-Ser36p levels (Figures 2B and S3C). In contrast, siRNA-mediated depletion of PAR glycohydrolase (PARG), an enzyme that removes PAR chains from PARP-1 substrate proteins, caused an increase in PAR levels and a corresponding reduction in H2B-Ser36p levels (Figures 2C and S3D). H2B-Ser32p was unaffected under similar conditions (Figure S3, E and F). Thus, in all of these perturbation experiments, reductions in PARP-1 activity were associated with increases in the levels of H2B-Ser36p, and vice versa, establishing a clear inverse relationship between ADPRylation and phosphorylation on H2B.
Figure 2. Interplay between ADPRylation and phosphorylation on H2B.
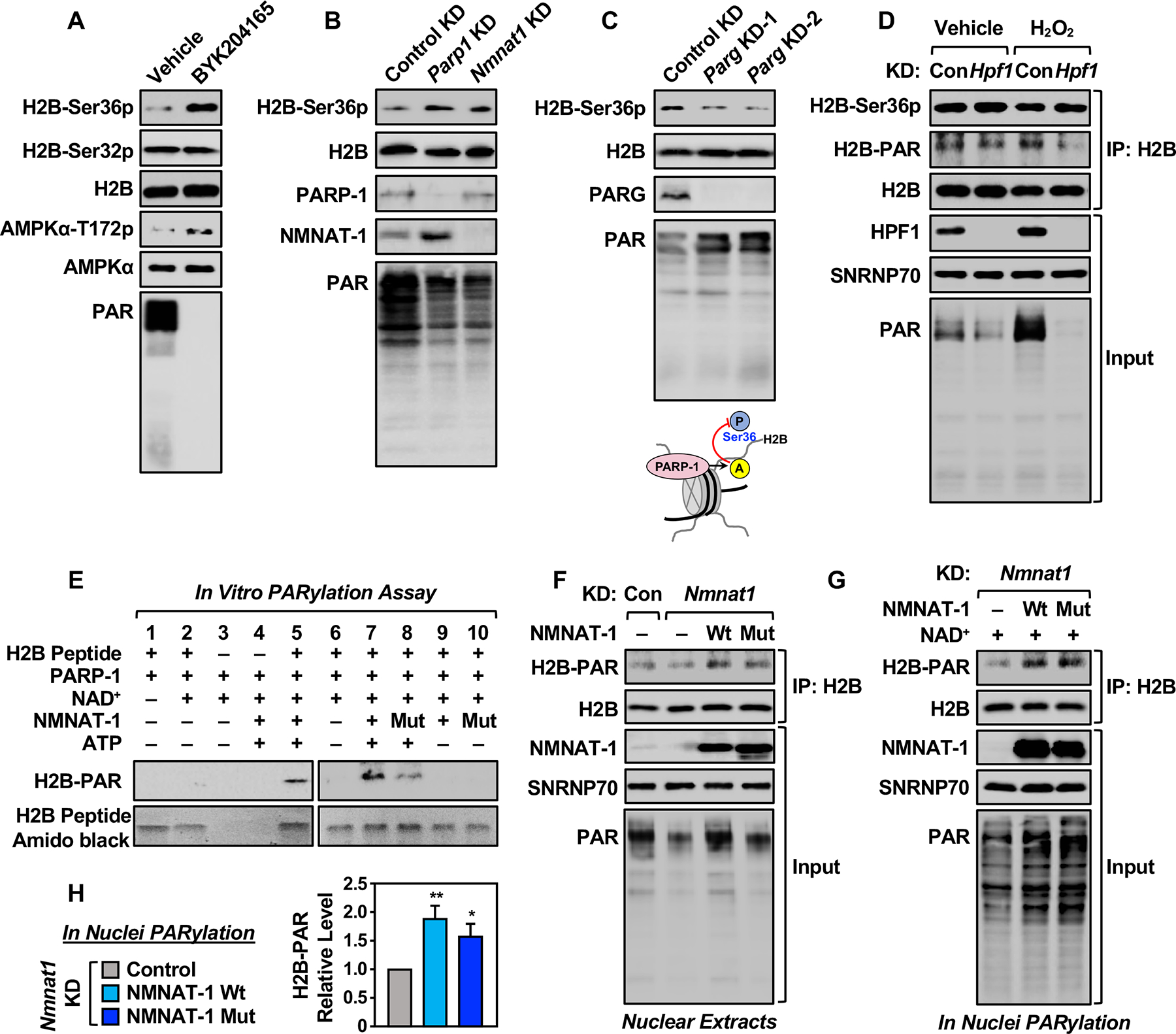
(A) Western blots showing the levels of H2B-Ser36p, H2B-Ser32p, H2B, AMPKα-T172p, AMPKα, and PAR levels in 3T3-L1 cells treated with vehicle or BYK204165 (20 μM).
(B) Western blots showing the levels of H2B-Ser36p, H2B, PARP-1, NMNAT-1, and PAR ± shRNA-mediated knockdown (KD) of Parp1 or Nmnat1 in 3T3-L1 cells.
(C) Western blots showing the levels of H2B-Ser36p, H2B, PARG, and PAR ± siRNA-mediated knockdown (KD) of Parg in 3T3-L1 cells (top panels). Schematic diagram showing that PARP-1 mediated ADPRylation blocks H2B-Ser36p in 3T3-L1 cells (bottom panel).
(D) Western blots showing the levels of H2B-Ser-36p and H2B-PAR on immunoprecipitated nucleosomes (IP with H2B) from 3T3-L1 cells ± siRNA-mediated knockdown (KD) of Hpf1, treated with vehicle or 2 mM H2O2 for 5 min. SNRNP70 was used as a loading control.
(E) In vitro PARylation assays with H2B (10–59) peptide, as well as PARP-1, wild-type or catalytically inactive W170A (Mut) NMNAT-1, NAD+, and ATP as indicated. Note that the addition of exogenous NAD+ abrogates the need for NMNAT-1 catalytic activity. Western blots showing the levels of PARylation on the H2B peptide. Amido black staining was used as a loading control for the peptide.
(F) Western blots showing the levels H2B-PAR immunoprecipitated from Nmnat1 knockdown (KD) 3T3-L1 cells expressing RNAi resistant Wt or Mut NMNAT-1. SNRNP70 was used as a loading control.
(G) Nuclei from Nmnat1 KD 3T3-L1 cells expressing RNAi-resistant Wt or Mut NMNAT-1 were incubated with 100 μM NAD+ to stimulate PARylation prior to immunoprecipitation of H2B. Western blots showing the levels of H2B-PAR. SNRNP70 was used as a loading control.
(H) Quantification of the results shown in (G). Each bar represents the mean + SEM (n = 4). Asterisks indicate significant differences from the Nmnat1 KD group (Student’s t-test; * p < 0.05, and ** p < 0.01).
Next, we asked whether Glu and Asp ADPRylation of histones by PARP-1 during adipogenesis might also require HPF1 or another ADPRylation factor, and if such a factor might influence H2B-Ser36 phosphorylation. Using siRNA-mediated knockdown in 3T3-L1 cells, we observed that HPF1 was required for H2B PARylation in response to genotoxic stress (e.g., treatment with H2O2) (Figure 2D; right two lanes), but not for H2B PARylation in the absence of genotoxic stress (Figure 2D; left two lanes). In addition, the levels of H2B-Ser36p and H2B-Ser32p were largely unaffected by knockdown of Hpf1 or treatment with H2O2 (Figures 2D and S3G). Furthermore, the levels of HPF1 did not vary during adipogenesis (Figure S3H) in spite of dramatic changes in the levels of H2B-Ser36p (Figures 1E and S1F). Together, these results indicate that HPF1 is not a regulator of the interplay between H2B PARylation and H2B-Ser36 phosphorylation during adipogenesis and in the absence of genotoxic stress.
NMNAT-1 directs PARP-1 catalytic activity to histones for Glu and Asp ADPRylation
Given that PARP-1 was unable to PARylate an H2B N-terminal tail peptide containing Glu35 and Ser36 in in a biochemical assay with purified components (Figure 2E, lanes 1–4), we considered the possibility that another ADPRylation factor might support PARP-1-mediated PARylation of H2B. We have previously uncovered an intimate functional relationship between PARP-1 and NMNAT-1, a nuclear enzyme that synthesizes NAD+ from ATP and nicotinamide mononucleotide (NMN) to support PARP-1’s catalytic activity (Ryu et al., 2018; Zhang et al., 2012). Thus, we tested the ability of NMNAT-1 to serve as an ADPRylation factor for PARP-1.
We performed an in vitro PARP-1-mediated ADPRylation assay with an H2B peptide in a reaction containing exogenous NAD+, which was added to abrogate the need for NAD+ synthesis by NMNAT-1. Indeed, under these conditions, we observed that purified wild-type (Wt) NMNAT-1 (Figure S4A) promoted PARP-1-mediated PARylation of the H2B peptide, but only in the presence of ATP (Figure 2E, lanes 5, 7, and 9). ATP hydrolysis by NMNAT-1, however, was not required since similar results were observed with a non-hydrolyzable ATP analog (Figure S4B). These results indicate that NMNAT-1 can direct PARP-1 catalytic activity to H2B in the absence of ongoing NAD+ synthesis. A catalytically inactive NMNAT-1 mutant (W170A; Figure S4, A and C) also supported ATP-dependent PARylation of H2B by PARP-1 when NAD+ was provided in the reaction (Figure 2E, lanes 8 and 10). These results indicate that NMNAT-1 requires ATP, but not its own catalytic activity, to activate PARP-1 catalytic activity toward histone H2B.
The salient features of the NMNAT-1-dependent regulation of PARP-1 catalytic activity with H2B were also observed in cell-based assays. For example, expression of Wt or W170A mutant NMNAT-1 in Nmnat1 knockdown 3T3-L1 cells fully or partially restored H2B PARylation, respectively, in the presence of the endogenous NAD+ in the cells (Figure 2F). In NAD+-depleted nuclei (Luo et al., 2017; Ryu et al., 2018), Wt or W170A mutant NMNAT-1 restored H2B PARylation in the presence of added NAD+, although the activity of the mutant was not as potent as Wt NMNAT-1 (Figure 2, G and H). Not surprisingly given the functional interaction that we observed, both Wt and W170A mutant NMNAT-1 coimmunoprecipitated with PARP-1 from 3T3-L1 cells (Figure 3A), supporting observations from our previous biochemical assays (Zhang et al., 2012). Furthermore, both PARP-1 and NMNAT-1 coimmunoprecipitated with FLAG-H2B ectopically expressed in 3T3-L1 cells, indicating the formation of a PARP-1/histone H2B/NMNAT-1 complex (Figure 3B).
Figure 3. NMNAT-1 directs PARP-1 catalytic activity to histones for Glu and Asp ADPRylation.
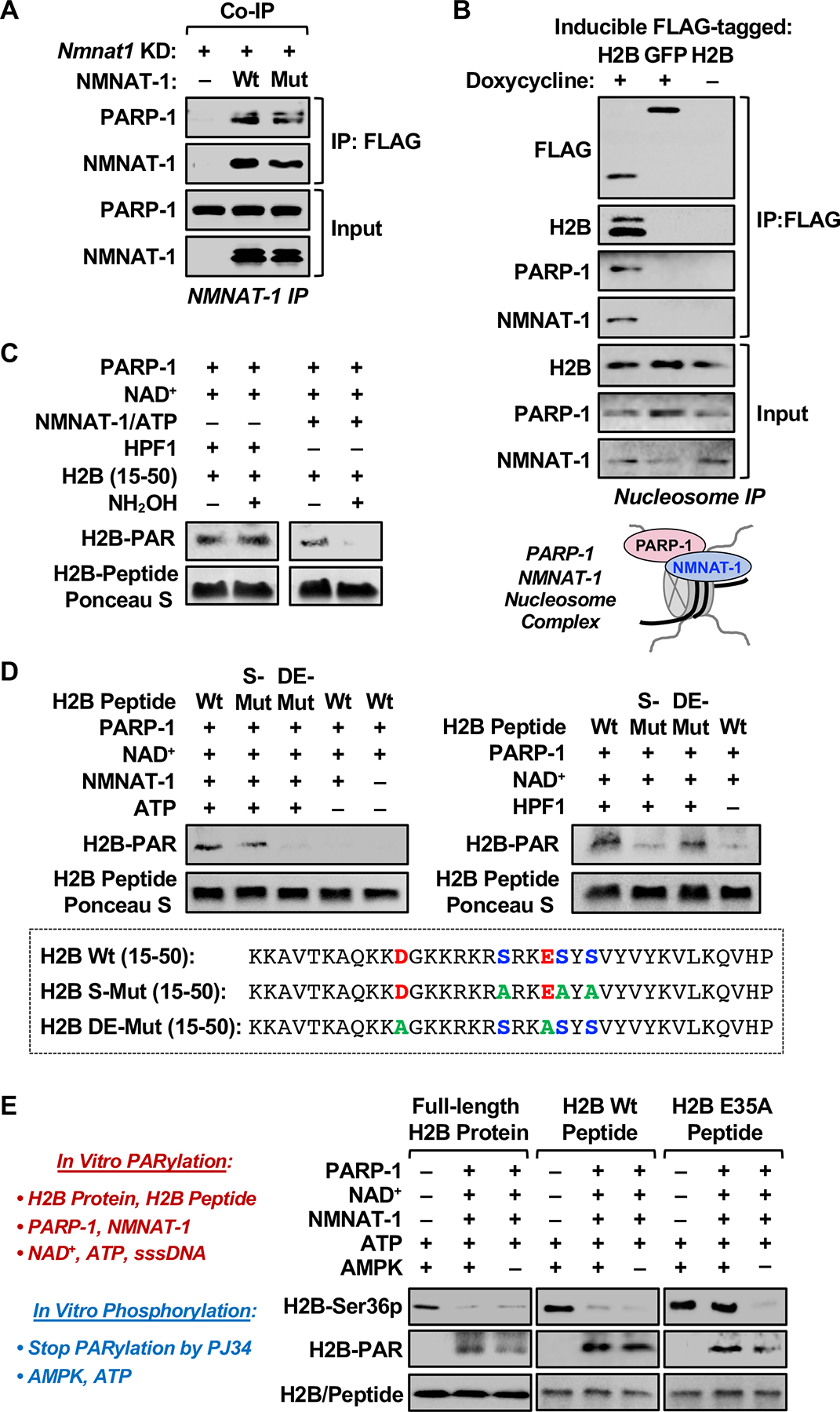
(A) NMNAT-1-interacting proteins were co-immunoprecipitated (co-IP) from nuclear extracts of Nmnat1 KD 3T3-L1 cells expressing FLAG-tagged Wt or Mut NMNAT-1, and analyzed by Western blotting as indicated.
(B) H2B-interacting proteins were immunoprecipitated from nucleosomes isolated from 3T3-L1 cells expressing doxycycline-inducible FLAG-tagged H2B or GFP (used as a control), and analyzed by Western blotting as indicated. Schematic diagram showing the formation of a PARP-1/NMNAT-1/nucleosome ternary complex in 3T3-L1 cells (bottom panel).
(C) In vitro PARylation assays with H2B (15–50) peptide, as well as PARP-1/HPF1 (left panels), PARP-1/NMNAT-1 (right panels), NAD+, and ATP as indicated, with or without subsequent treatment with 1 M hydroxylamine (NH2OH). Western blots showing the levels of PARylated H2B peptide. Ponceau S staining of the peptide was used as a loading control.
(D) In vitro PARylation assays with wild-type, Ser-mutant (S-Mut), and Glu/Asp-mutant (DE-Mut) H2B (15–50) peptides, as well as PARP-1/NMNAT-1 (left panels), PARP-1/HPF1 (right panels), NAD+, and ATP as indicated. Western blots showing the levels of PARylated H2B peptide. Ponceau S staining of the peptide was used as a loading control. The sequences of the H2B peptides are shown in the bottom panel.
(E) In vitro PARylation assays were performed by incubation of full-length H2B protein (left), Wt H2B peptide (10–59) (middle), or E35A H2B peptide (right) with purified PARP-1, NMNAT-1, NAD+, and ATP, followed by phosphorylation assays with the addition of recombinant AMPK complex (combination of α1/β1/γ1 subunits). The PARylation reactions were initiated by the addition of sssDNA and stopped by the addition of PJ34. Western blots showing the levels of H2B-Ser36p and H2B-PAR. Blots of H2B (left bottom panel) or amido black staining of the peptide (middle and right bottom panels) was used as loading controls.
[See also Figure S4]
We then tested the selectivity of PARP-1 toward Glu/Asp or Ser in the presence of NMNAT-1. Using the same reaction conditions noted above, we observed that NH2OH removed essentially all of the PAR signal from the H2B peptide ADPRylated by PARP-1/NMNAT-1, but not by PARP-1/HPF1, indicating Glu/Asp ADPRylation (Figure 3C). Furthermore, mutation of all of the Glu/Asp (D/E) residues in the H2B peptide, but not mutation of all of the Ser (S) residues, blocked PARylation by PARP-1/NMNAT-1, but not by PARP-1/HPF1 (Figure 3D). Moreover, we observed that PARylation by PARP-1/NMNAT-1 occurred with an H2B peptide in which Ser36 was mutated (S36A), but was lost with an H2B peptide in which Glu35 was mutated (E35A) (Figure S4D). Together, these results indicate that the histone ADPRylation activity of PARP-1 with NMNAT-1 is directed toward Glu/Asp residues. More specifically, they confirm that H2B-Glu35 is a site of PARP-1/NMNAT-1-mediated ADPRylation and demonstrate that PARP-1/NMNAT-1 does not PARylate H2B-Ser36. With respect to the latter, H2B-Ser36 is not known to be a site of H2B ADPRylation based on previous mass spectrometry studies focused on histone Ser ADPRylation (Bilan et al., 2017; Bonfiglio et al., 2017; Fontana et al., 2017; Larsen et al., 2018; Leidecker et al., 2016).
To explore in more detail the effects of site-specific histone ADPRylation on histone phosphorylation, we performed in vitro ADPRylation assays on H2B (full-length or N-terminal peptide 10–59) followed by AMPK-mediated phosphorylation. In initial experiments, we found that NMNAT-1-dependent PARylation of H2B inhibited phosphorylation of Ser36 by recombinant AMPK (Figure 3E; left and middle panels). Mutation of Glu35 in the H2B peptide to alanine (Ala) restored phosphorylation of Ser36 by AMPK (Figure 3E; right panel), indicating that PARylation of Glu35 on H2B by PARP-1 inhibits phosphorylation of Ser36. [Note that the H2B-Ser36p antibody recognizes the Glu35Ala (E35A) mutant H2B peptide, but with reduced affinity (Figure S4E). Thus, the effects of mutating Glu35 on H2B-Ser36 phosphorylation may be underestimated]. Importantly, in order-of-addition experiments, unmodified PARP-1 alone, PARylated PARP-1 without further catalytic activity, or PARP-1/NMNAT-1 in the presence of a PARP inhibitor, were unable to block phosphorylation of H2B-Ser36 by AMPK (Figure S4F). The results indicate that direct PARylation of H2B-Glu35 by PARP-1 with NMNAT-1, not PARP-1 binding to H2B or PAR linked to PARP-1, drives the inhibition of AMPK-mediated H2B-Ser36 phosphorylation.
SnoRNAs stimulate PARP-1 activation and PARylation of Histone H2B
We sought to identify physiological activators of PARP-1, as opposed to damaged DNA, that might drive ADPRylation during the differentiation of 3T3-L1 cells. We recently showed that some PARP-1-interacting small nucleolar RNAs (snoRNAs), such as human SNORA37, are potent physiological activators of PARP-1 in vitro and in cells in the absence of DNA damage (Huang et al., 2020; Kim et al., 2019) (Figure S5, A and B). We found that NMNAT-1-dependent, PARP-1-mediated ADPRylation of histone H2B was also observed when PARP-1 was activated by SNORA37, but not other snoRNAs that do not interact with and activate PARP-1 (Kim et al., 2019) (Figure S5, A–C).
We then asked whether PARP-1 activation during adipogenesis might also be mediated by snoRNAs and if these snoRNAs might influence H2B-Ser36 phosphorylation and proadipogenic gene expression. We chose four mouse snoRNAs from 3T3-L1 RNA-seq data (Snora7a, Snord16a, Snord22 and Snora64) that are highly expressed and are present when PARP-1 is most active. In an in vitro autoPARylation assay, PARP-1 was automodified in the presence of in vitro transcribed Snora64, Snora7a, and Snord16a, but was not activated by Snord22 (Figures 4A and S5D). Similar to human SNORA37, mouse Snora64 stimulated PARP-1 to PARylate core histones in nucleosomes in vitro in the presence of NMNAT-1 (Figure S5E). Additionally, we profiled the expression of these snoRNAs by RT-qPCR to determine if their expression patterns during differentiation is consistent with their proposed effects on PARP-1 activity. We observed that the expression patterns are generally consistent with their proposed regulatory functions, especially for Snora64 and Snord16a (Figure S5F). As expected, the expression pattern of the non-stimulatory snoRNA Snor22 showed no correspondence with PARP-1 catalytic activity (Figure S5F).
Figure 4. SnoRNAs are physiological activators of PARP-1 during the differentiation of 3T3-L1 cells.
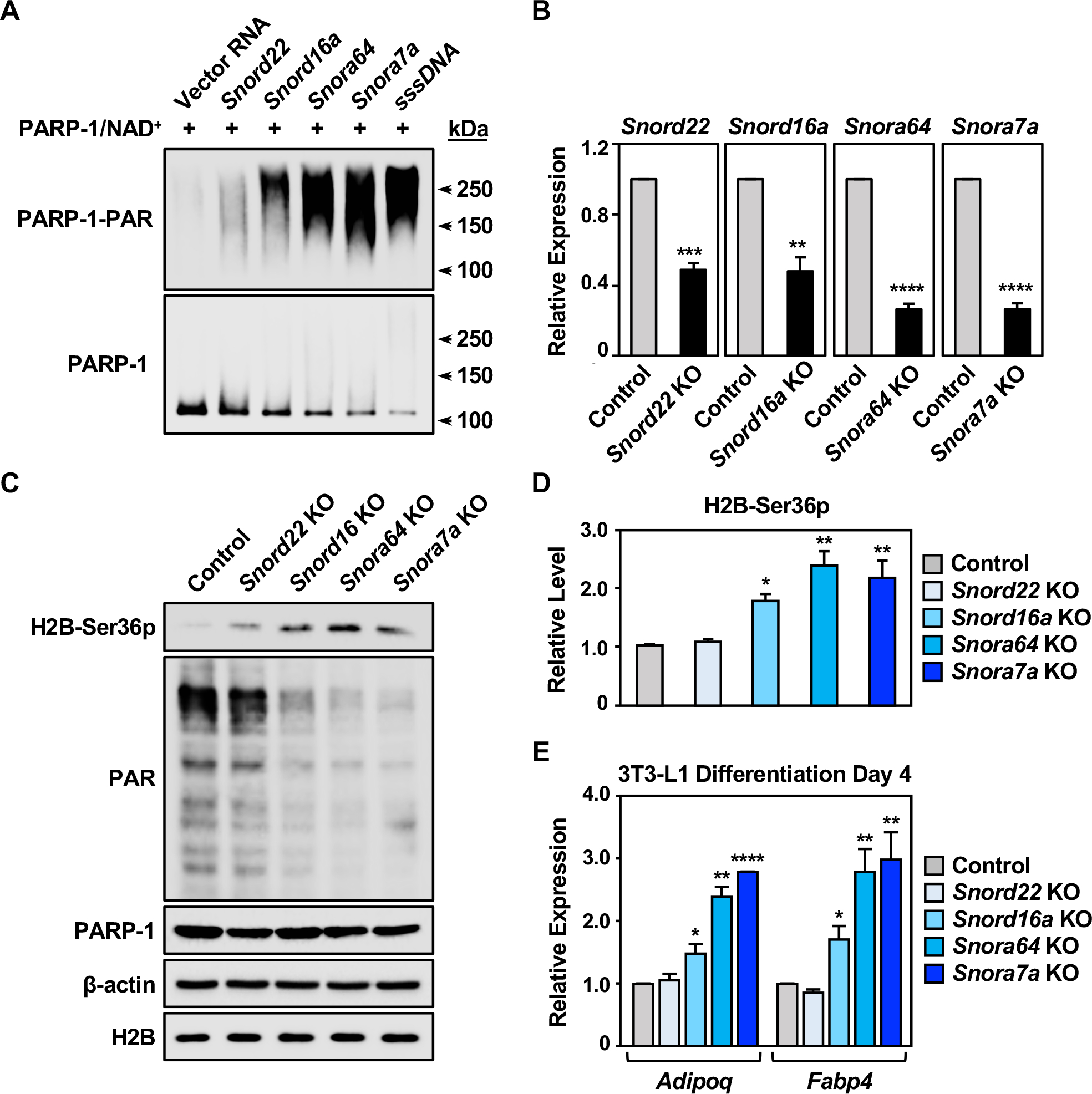
(A) PARP-1 automodification reactions with activation by snoRNAs or sssDNA were analyzed by Western blotting for PAR (upper) and PARP-1 (lower). Molecular weight markers in kDa are shown.
(B) Expression of snoRNAs in 3T3-L1 cells with CRISPR/Cas9-mediated knockout of individual snoRNAs, as determined by RT-qPCR. Each bar represents the mean + SEM (n = 3). Asterisks indicate significant differences from the corresponding Control (Student’s t-test; ** p < 0.01, *** p < 0.001, and **** p < 0.0001).
(C) Western blots showing the levels of H2B-Ser36p, PAR, and PARP-1 in 3T3-L1 cells with CRISPR/Cas9-mediated knockout of individual snoRNAs as indicated. β-actin and H2B were used as loading controls.
(D) Quantification of the H2B-Ser36p levels shown in (C). Each bar represents the mean + SEM (n = 3). Asterisks indicate significant differences from the control (one-way ANOVA followed by Dunnett’s multiple comparisons test; * p<0.05, and ** p < 0.01).
(E) Expression of the adipocyte marker genes Adipoq and Fabp4 in 3T3-L1 cells with CRISPR/Cas9-meditaed knockout of individual snoRNA at differentiation day 4, as determined by RT-qPCR. Each bar represents the mean + SEM (n = 3). Asterisks indicate significant differences from the corresponding control (Student’s t-test; * p < 0.05, ** p < 0.01, and **** p < 0.0001).
[See also Figure S5]
We have previously shown that inhibition of PARP-1 activity promotes differentiation of 3T3-L1 cells (Luo et al., 2017; Ryu et al., 2018). Thus, we examined the role of snoRNA-mediated PARP-1 activation in adipogenesis by generating knockouts of the four snoRNAs noted above in 3T3-L1 cells using CRISPR/Cas9. The snoRNA knockouts in bulk cells induced significant loss of snoRNA expression as determined by RT-qPCR (Figure 4B), but did not affect the expression of their host genes: Snhg1 (Snord22), Rpl4 (Snord16a), Rps2 (Snora64), and Rpl32 (Snora7a) (Figure S5G). We found that knockout of Snord16a, Snora64, or Snora7a caused a significant reduction in the bulk levels of PAR and a dramatic increase in the levels of H2B-Ser36p, supporting our observation of an inverse relationship between ADPRylation and phosphorylation on H2B (Figure 4, C and D). Importantly, knockout of these snoRNAs individually dramatically enhanced the differentiation of 3T3-L1 cells, as determined by the expression of key proadipogenic marker genes (e.g., Adipoq, and Fabp4) at differentiation day 4 (Figure 4E). In contrast, knockout of Snord22, which had no effect on either bulk PAR or H2B-Ser36p levels, did not affect proadipogenic gene expression (Figure 4, C–E). Together, these results demonstrate that snoRNAs are potent physiological activators of PARP-1 that can promote histone H2B ADPRylation during the differentiation of 3T3-L1 cells.
PARylation of Glu35 on H2B by PARP-1 inhibits phosphorylation of Ser36 in cells
Next, we examined the functional interplay between PARylation at Glu35 and phosphorylation at Ser36 on histone H2B in cells. We generated 3T3-L1 cells that constitutively or inducibly (in response to doxycycline; Dox) express FLAG epitope-tagged versions of Wt, Glu35Ala (E35A) mutant, or Ser36Ala (S36A) mutant H2B, of which the latter two cannot be PARylated or phosphorylated, respectively (Figures 5A and S6A). The addition of the FLAG epitope allowed us to distinguish between the endogenous H2B and the ectopically-expressed FLAG-H2B (Figure 5B). We then examined the PARylation and phosphorylation of the Wt and mutant H2B proteins in immunoprecipitated (IP) nucleosomes by Western blotting.
Figure 5. PARylation of Glu35 on H2B by PARP-1 inhibits phosphorylation of Ser36.
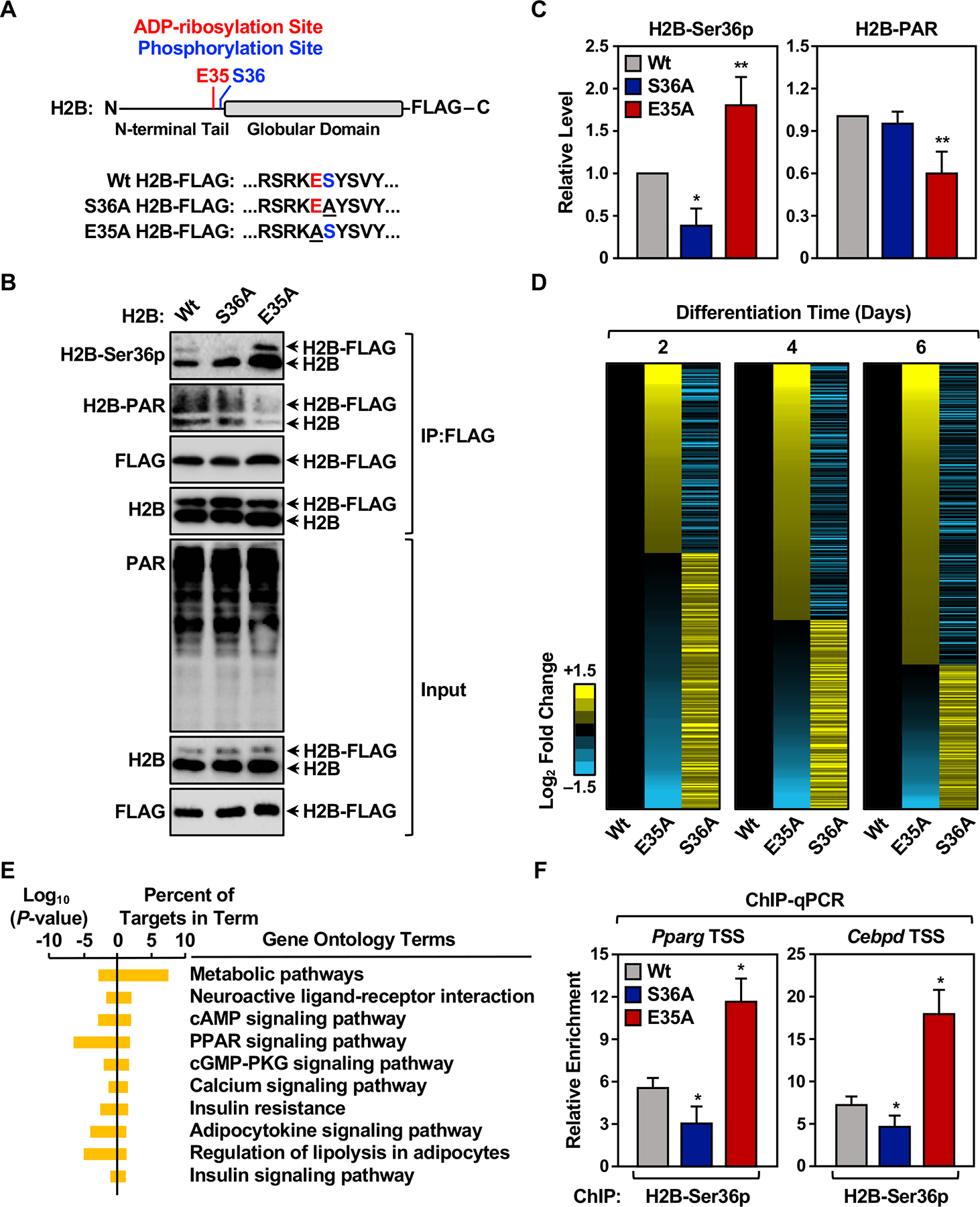
(A) Schematic diagram showing wild-type (Wt), S36A mutant, E35A mutant H2B.
(B) Western blots showing the levels of Ser36p and PAR on endogenous and ectopically-expressed H2B in 3T3-L1 cells expressing FLAG-tagged Wt, S36A mutant, or E35A mutant H2B.
(C) Quantification of the levels of H2B-Ser36p and H2B-PAR on ectopically-expressed H2B shown in (B). Each bar represents the mean + SEM (n = 3). Asterisks indicate significant differences from Wt (one-way ANOVA followed by Dunnett’s multiple comparisons test; * p < 0.05, and ** p < 0.01).
(D) Heat maps showing the results of RNA-seq assays from 3T3-L1 cells expressing Dox-inducible Wt, S36A mutant, or E35A mutant H2B during a time course of differentiation with MDI. Ectopic H2B expression was induced at differentiation day 1. Results represent the fold changes versus the corresponding Wt H2B ectopic expression cell line at days 2, 4 and 6.
(E) Gene ontology terms enriched for the differentially expressed set of genes at day 4.
(F) H2B-Ser36p enrichment was assessed by ChIP-qPCR at the Pparg and Cebpd gene promoters in 3T3-L1 cells expressing Dox-inducible Wt, S36A mutant, or E35A mutant H2B after 2 days of differentiation. Ectopic H2B expression was induced at differentiation day 1. Each bar represents the mean + SEM (n = 3). Asterisks indicate significant differences from Wt (Student’s t-test; * p < 0.05).
[See also Figure S6]
As expected, the E35A mutant exhibited a dramatic impairment of PARylation, which occurred on both the ectopically-expressed FLAG-H2B E35A mutant, as well as the unmutated endogenous H2B (Figure 5, B and C). This result is indicative of inter-histone trans effects within the nucleosome, where failure to PARylate the E35A mutant inhibits PARylation of the unmutated endogenous H2B. This effect is reminiscent of trans effects within or between nucleosomes that have been reported previously for other histone modifications (Martire et al., 2019). The reduction of PARylation observed with E35A mutant was accompanied by a dramatic increase in H2B-Ser36p on both the exogenous and endogenous H2B proteins (Figure 5, B and C). In contrast, the S36A mutant had no effect on PARylation of H2B, but did abrogate phosphorylation of the ectopically expressed S36A mutant, as expected (Figure 5, B and C). Together, these results from cells reaffirm, support, and extend the results from our biochemical assays showing that PARylation of Glu35 on H2B by PARP-1 inhibits phosphorylation of Ser36. Interestingly, H2B-E35 ADPRylation may also impact other histone modifications, such as H3K27 dimethylation (H3K27me2) and H3K79 acetylation (Figure S6B), suggesting a broader epigenomic regulatory role for within chromatin.
Functional interplay between H2B Glu35 PARylation and Ser36 phosphorylation controls adipogenesis
Next, we examined whether the functional interplay between H2B Glu35 PARylation and Ser36 phosphorylation could impact proadipogenic gene expression. We preformed RNA-seq in 3T3-L1 cells ectopically expressing Wt, E35A, or S36A versions of H2B during a time course of differentiation. As shown in the heatmaps in Figure 5D, the E35A and S36A mutants had opposing effects on the expression of a subset of genes at all three differentiation time points tested (Figure 5D). In GO analyses, the regulated genes at day 4 were enriched in terms related to adipogenesis, metabolism, PPAR signaling, and insulin signaling, as expected (Figure 5E). Finally, in chromatin immunoprecipitation (ChIP) assays, we observed an increase in H2B-Ser36p levels at the promoters of proadipogenic genes (i.e., Pparg and Cebpd) in cells expressing the E35A mutant and decrease in H2B-Ser36p levels in cells expressing the S36A mutant (Figure 5F, note that this assay detects H2B-Ser36p on both endogenous and exogenous H2B).
The effects of the H2B mutants on adipogenesis were confirmed by monitoring the expression of key proadipogenic marker genes (e.g., Pparg, Adipoq) by RT-qPCR (Figure 6A), as well as lipid content by staining with Oil Red O (Figure 6B). Additional genomic analyses of H3K27me3, an EZH2-mediated histone modification that represses Wnt signaling during adipogenesis (Yi et al., 2016), confirmed a role for H2B-Glu35 ADP-ribosylation in the transcriptional regulation of adipogenesis (Figure S6, C–E). Together, the results from the gene expression and differentiation assays strongly support the conclusion that H2B Glu35 PARylation and Ser36 phosphorylation act in opposition to control proadipogenic gene expression in adipocytes.
Figure 6. Interplay between H2B Glu35 PARylation and Ser36 phosphorylation controls adipogenesis.
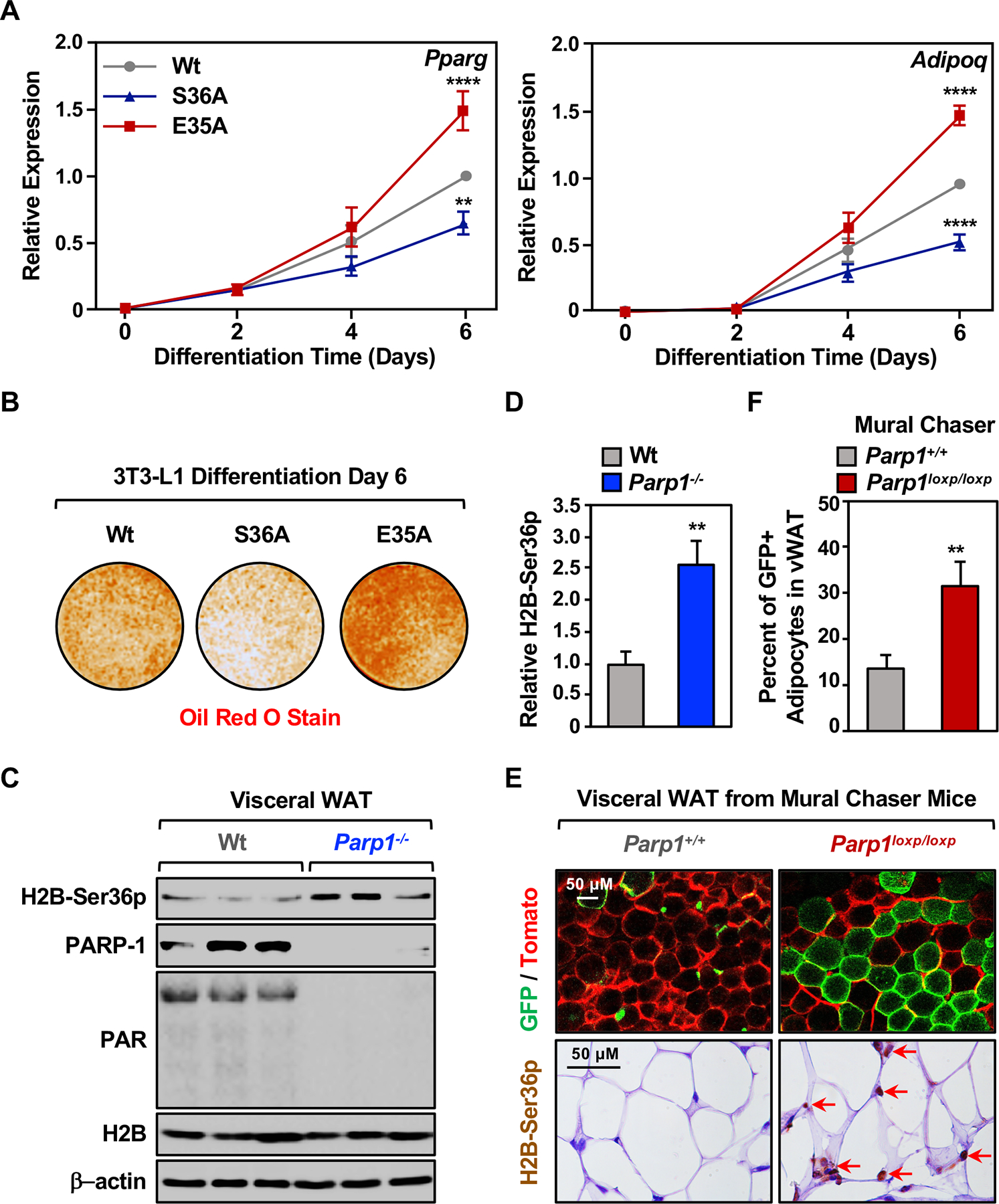
(A) Expression of the adipocyte marker genes Pparg and Adipoq in 3T3-L1 cells expressing Dox-inducible Wt, S36A mutant, or E35A mutant H2B during a time course of differentiation with MDI, as determined by RT-qPCR. Ectopic H2B expression was induced at differentiation day 1. Each point represents the mean + SEM (n = 7). Asterisks indicate significant differences from the corresponding Wt (two-way ANOVA followed by Dunnetts’s multiple comparisons test; ** p < 0.01, and **** p < 0.0001).
(B) Oil red O staining of lipid droplets in 3T3-L1 cells expressing Dox-inducible Wt, S36A mutant, or E35A mutant H2B after 6 days of differentiation with MDI. Ectopic H2B expression was induced at differentiation day 1.
(C) Western blots showing the levels of H2B-Ser36p, PAR, and PARP-1 in visceral white adipose tissue (WAT) from C57BL/6 wild type (Wt) or Parp1 null (Parp1−/−) mice. β-actin was used as a loading control.
(D) Quantification of the H2B-Ser36p levels shown in (C). Each bar represents the mean + SEM (n = 6). Asterisks indicate significant differences from Wt (Student’s t-test; ** p < 0.01).
(E) Imaging of fresh visceral WAT from Dox-inducible Mural Chaser (PdgfrbrtTA; TRE-Cre; Rosa26RmT/mG) Parp1+/+ or Mural Chaser Parp1loxp/loxp mice treated with Dox for 9 days, followed by 4 weeks of high-fat/high sucrose diet. (Top panels) Representative confocal fluorescence images collected at 10x magnification. Membrane-anchored Tomato fluorescent protein is shown as red. Membrane-anchored GFP fluorescent protein is shown as green. Scale bars represent 50 μm. (Bottom panels) Representative images collected at 63x magnification of immunohistochemical staining of H2B-Ser36p in visceral WAT. Scale bars represent 50 μm.
(F) Quantification of the percent of newly formed adipocytes from the Pdgfrβ+ preadipocytes shown in top panels of (E). Each bar represents the mean + SEM (n = 9 in Parp1+/+ group, and n = 6 in Parp1loxp/loxp group). Asterisks indicate significant differences from Parp1+/+ (Student’s t-test; ** p < 0.01).
[See also Figure S7]
To explore this functional interplay in vivo, we used both Parp1 null and conditional mouse genetic models. First, in visceral white adipose tissue (WAT) isolated from Parp1 null mice (de Murcia et al., 1997), we observed an increase in the levels of H2B-Ser36p (Figure 6, C and D), consistent with the effects of Parp1 knockdown in 3T3-L1 cells (Figure 2B). Next, we crossed Parp1loxp/loxp mice (Luo et al., 2017) to Pdgfrb-based lineage tracing mice (‘Mural Chaser’ mice), which allow Dox-inducible deletion of Parp1 and tracking of adipocyte precursors through differentiation (Vishvanath et al., 2016) (Figure S7, A and B). In this system, the cytoplasmic membranes of all cells in the tissue are constitutively labeled with membrane-anchored tdTomato fluorescent protein (mT) (Figure 6E, top left). Treatment with Dox induces expression of Cre recombinase, leading to (1) deletion of the mT gene and expression of membrane-anchored Green fluorescent protein (mG) and (2) deletion of Parp1 in PDGFRβ-positive (PDGFRβ+) adipocyte precursors (Figure S7A). Thus, mG-labeled cells in WAT identify mature adipocytes that arose de novo from an expansion of the adipocyte precursor population (Vishvanath et al., 2016) (Figure S7B). We monitored the frequency of newly formed adipocytes from the PDGFRβ+ precursors by fluorescent imaging of fresh visceral WAT (Figure 6E, top), as well as immunofluorescent (IF) staining for perilipin, which is an adipocyte-specific lipid protein that associates with the surface of lipid droplets, and GFP, which marks newly differentiated adipocytes (Figure S7, C and D).
In mice fed a high-fat/high-sucrose diet for four weeks, we observed a >2-fold increase in GFP positive adipocytes that formed from the PDGFRβ+ precursors upon Parp1 preadipocyte-specific deletion in visceral WAT (Figures 6E, top; S7C, S7D). These results indicate that PARP-1 acts to inhibit adipocyte precursor differentiation in vivo. Importantly, we also observed a dramatic increase in the fraction of mature adipocytes that stained positively for nuclear H2B-Ser36p by immunohistochemistry and immunofluorescent staining in Parp1 preadipocyte-specific deletion Mural Chaser mice (Figure 6E, bottom; Figure S7E). Importantly, H2B-Ser36p was enriched in the GFP+ (i.e., Parp1 knockout) cells (Figure 6E, bottom; Figure S7E). Thus, a number of salient features of our observations in 3T3-L1 cells were also evident in mice in vivo, namely that PARP-1 inhibits H2B-Ser36 phosphorylation and adipogenesis. To further investigate the role of preadipocyte-specific deletion of PARP-1 in metabolic outcomes, we fed the Parp1 preadipocyte-specific deletion Mural Chaser mice with high fat diet for 8 weeks. These mice exhibited a significant increase in body weight (Figure 7A) and fat/mass ratio (Figure 7B) compared to the control mice. These results indicate that preadipocyte-specific deletion of PARP-1 promotes adipogenesis that can lead to obesity.
Figure 7. NMNAT-1-dependent physiological ADPRylation by PARP-1 inhibits H2B-Ser36 phosphorylation and adipogenesis.
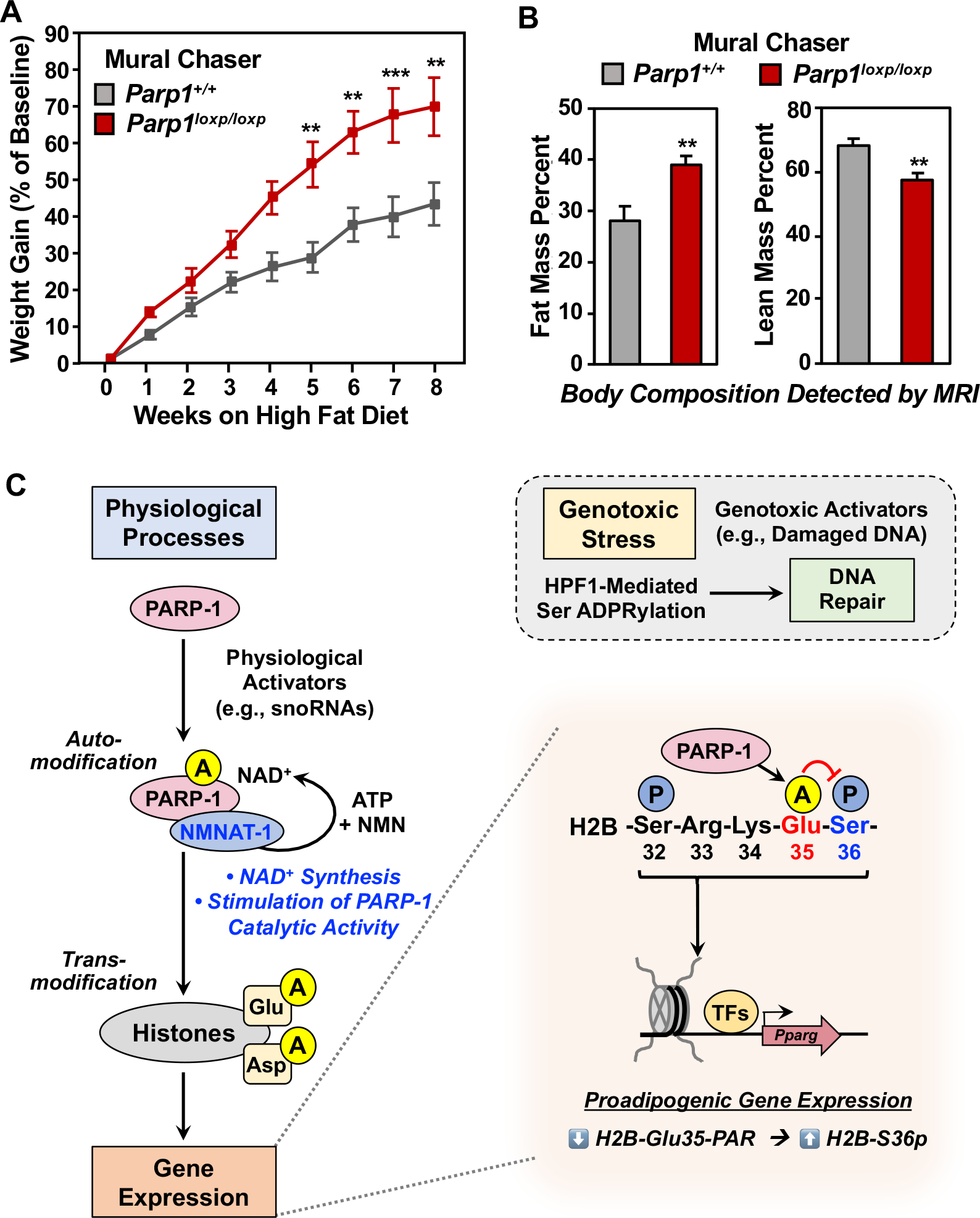
(A) Eight-week old Dox-inducible Mural Chaser Parp1+/+ or Parp1loxp/loxp mice were treated with Dox for 9 days, followed by 8 weeks on a high-fat/high sucrose diet. Body weights of the mice were measured weekly following the start of the high-fat/high sucrose diet. Each point represents the mean + SEM (n = 7 in Parp1+/+ group, and n = 10 in Parp1loxp/loxp group). Asterisks indicate significant differences from Parp1+/+ at individual time point (Student’s t-test; ** p < 0.01, and *** p < 0.01).
(B) Mural Chaser Parp1+/+ or Parp1loxp/loxp mice were treated as described in (A). Fat mass (left panel) and lean mass (right panel) ratio (normalized to body weight) were measured after 8 weeks of high-fat/high sucrose diet feeding. Each bar represents the mean + SEM (n = 7 in Parp1+/+ group, and n = 10 in Parp1loxp/loxp group). Asterisks indicate significant differences from Parp1+/+ (Student’s t-test; ** p < 0.01).
(C) Model for NMNAT-1-dependent histone Glu/Asp ADPRylation by PARP-1 during the physiological process of adipogenesis. See details in the Discussion.
DISCUSSION
The specific biological functions of site-specific histone ADPRylation have remained elusive for decades. As with most other aspects of PARP-1 and ADPRylation, the focus has largely been on events occurring under genotoxic stress conditions. Herein, we have shown that snoRNA-activated PARP-1 PARylates histones on specific Glu and Asp residues under the physiological conditions of adipogenesis in a process that does not require genotoxic stress, damaged DNA, or HPF1, greatly expanding our understanding of the biological potential of histone ADPRylation (Figure 7C).
Mechanisms of physiological histone ADPRylation by PARP-1: Roles of snoRNAs and NMNAT-1
Arguments against physiological effects of PARP-1 (i.e., without genotoxic stress), especially with regard to histone ADPRylation, are centered around the following questions: (1) How is PARP-1 activated in the absence of damaged DNA, the classical activator of PARP-1 catalytic activity? and (2) What directs PARP-1 to histone substrates, which are generally thought to be poor substrates of PARP-1 in the absence of genotoxic stress, which promotes functional interactions between HPF1 and PARP-1? We have addressed both of these questions in the studies described herein, focusing on snoRNAs as endogenous physiological activators of PARP-1 and NMNAT-1 as a factor that directs PARP-1 catalytic activity to Glu and Asp resides on histones (Figure 7C). Given the ubiquitous expression of PARP-1 in mammals, such mechanisms are likely to operate in many tissues.
In a previous study, we identified PARP-1-interacting snoRNAs as potent activators of PARP-1 catalytic activity in the absence of damaged DNA (Huang et al., 2020; Kim et al., 2019). SnoRNAs bind to the zinc-finger DNA binding domain of PARP-1 and are likely to stimulate PARP-1 catalytic activity through an allosteric mechanism, similar to damaged DNA (Kim et al., 2019). Here we show that snoRNAs can activate PARP-1 catalytic activity with histone substrates, establishing snoRNAs as endogenous physiological activators of PARP-1. Notably, one of the snoRNAs that promotes these effects, SNORA7A, has been shown to inhibit adipogenic differentiation of mesenchymal stem cells in humans (Zhang et al., 2017).
Our results also indicate that NMNAT-1, a nuclear NAD+ synthase that synthesizes NAD+ to support PARP-1 catalytic activity in the nucleus (Ryu et al., 2018), also directs PARP-1 catalytic activity to Glu and Asp resides on histones. Interestingly, this function requires the binding of ATP, a substrate for NMNAT-1, but is not dependent on the catalytic activity of NMNAT-1. Previous structural studies of NMNAT proteins have indicated considerable structural flexibility in NMNAT proteins, as well as substrate-induced conformational changes (D’Angelo et al., 2000; Garavaglia et al., 2002; Saridakis et al., 2001). Together, these findings and our results suggest that NMNAT-1 may sense nuclear ATP levels while serving as a PARylation factor for PARP-1, providing an additional level of control.
NMNAT-1 is an ADPRylation specificity factor for PARP-1-mediated Glu/Asp ADPRylation
The concept of an ADPRylation specificity factor was first proposed for HPF1 with PARP-1 (Bonfiglio et al., 2017; Gibbs-Seymour et al., 2016; Palazzo et al., 2018). Although the mechanisms of HPF1’s effects on PARP-1 have been addressed in recent structural studies (Suskiewicz et al., 2020), the functional definition of an ADPRylation specificity factor remains unclear because it encompasses a number of distinct activities. The key observable activity of HPF1 is that Ser residues, which would not otherwise be ADPRylated by PARP-1, are ADPRylated in the presence of HPF1. Other aspects of the functional definition include: (1) a switch from automodification to transmodification by PARP-1 in the presence of HPF1, which functions like a chaperone to bring PARP-1 catalytic activity to trans-substrates, and (2) a switch from Glu/Asp to Ser ADPRylation by PARP-1 in the presence of HPF1 (Bonfiglio et al., 2017; Zhang et al., 2013).
The effects of NMNAT-1 on PARP-1 are similar to, but distinct from, those observed for HPF1, encompassing two of the three activities noted above: (1) Glu/Asp residues, which would not otherwise be ADPRylated by PARP-1, are ADPRylated in the presence of NMNAT-1 and (2) NMNAT-1 redirects PARP-1 from automodification to transmodification, bringing PARP-1 catalytic activity to trans-substrates. NMNAT-1 does not, however, appear to promote a switch from one amino acid substrate to another. While we do not think the latter is an essential component of the definition of a specificity factor, we do think it is important to precisely describe the activities observed in relation to the definition. In summary, both HPF1 and NMNAT-1 share an ability to redirect PARP-1 catalytic activity to specific residues on trans-substrates that would not otherwise be ADPRylated in their absence – HPF1 to Ser and NMNAT-1 to Glu/Asp.
Functional interplay between H2B-Glu35 PARylation and H2B-Ser36 phosphorylation
ADPRylation sites are enriched near phosphorylation sites across the human proteome (Gibson et al., 2016; Larsen et al., 2018). A recent study showed that PARylation and phosphorylation of the same Ser residue in a core histone (i.e., H3-Ser10) are incompatible (Bartlett et al., 2018). In contrast, we found that PARylation of H2B-Glu35 by PARP-1 with NMNAT-1 inhibits phosphorylation of an adjacent residue, H2B-Ser36. Importantly, PARP-1 with NMNAT-1 does not PARylate H2B-Ser36, indicating that direct competition with H2B-Ser36 phosphorylation is not the underlying mechanism. Site-specific ADPRylation may inhibit modification of adjacent residues by steric or charge effects that may restrict access of the modifier enzyme (e.g., AMPK in the case of H2B-Ser36p) or alter the chemistry of the adjacent site (Bartlett et al., 2018; Rothbart and Strahl, 2014; Zhang et al., 2015).
We observed that the levels of H2B-Ser36p increase and are maximal 1 to 2 days after the initiation of differentiation. In multiple assays described herein, H2B-Ser36p correlated with or was required for a set of proadipogenic outcomes. Thus, in this context, we found that ADPRylation of a specific residue on a single core histone protein can antagonize a broader physiological outcome mediated by H2B-Ser36p (Figure 7C). Taken together, these results indicate the potential for highly specific and well-choreographed PARP-1-mediated outcomes, in contrast to the non-specific, broad modification of chromatin proteins observed during genotoxic stress. Furthermore, our results provide a rationale for understanding site-specific ADPRylation of histones as an endpoint of signaling pathways during physiological gene regulation, placing this modification in the same realm as other well characterized site-specific modifications of histone (e.g., acetylation, methylation, phosphorylation) (Lawrence et al., 2016).
Role of PARP-1 in adipogenesis
Previous studies have linked PARP-1 function to adipogenesis and other metabolic phenotypes, but the results have using whole body Parp1 null animals often been inconsistent and contradictory (Asher et al., 2010; Bai et al., 2011; Devalaraja-Narashimha and Padanilam, 2010; Erener et al., 2012a; Erener et al., 2012b; Lehmann et al., 2015; Luo et al., 2017; Ryu et al., 2018). The lineage tracing mice with tissue-specific conditional Parp1 knockout that we used herein have allowed us to unambiguously address questions about the role of PARP-1 in tissue-specific physiological outcomes. Our results demonstrate a definitive role for PARP-1 in repressing the expansion of the population of adipocyte precursors to limit adipogenesis, consistent with previous observations (Devalaraja-Narashimha and Padanilam, 2010; Luo et al., 2017; Ryu et al., 2018). Depletion or inhibition of PARP-1, limiting nuclear NAD+ availability, or mutation of specific sites of ADPRylation on key PARP-1 substrate proteins (e.g., H2B-Glu35, C/EBPβ) releases the PARP-1-mediated repression, allowing adipogenesis to proceed (Devalaraja-Narashimha and Padanilam, 2010; Luo et al., 2017; Ryu et al., 2018). The effects of PARP-1-mediated ADPRylation of H2B are likely occur after mitotic clonal expansion of the preadipocytes, but this needs to be investigated further. Importantly, our results demonstrate that loss of PARP-1 in the population of adipocyte precursors has broader metabolic consequences for the animal, with a significant increase in body weight and fat mass when placed on a high fat diet. These results highlight the importance of examining PARP-1 actions in specific populations of cells. Collectively, our studies reveal the mechanisms underlying physiological site-specific ADPRylation of core histones on Glu and Asp residues by PARP-1.
LIMITATIONS
We attempted to explore the role of H2B-Ser36 phosphorylation on a global scale using ChIP-seq. Unfortunately, the only commercially available H2B-Ser36p antibody does not work for ChIP-seq (Bungard et al., 2010; Yi et al., 2016). Furthermore, the technology for making antibodies to specific sites of ADPRylation does not currently exist. Thus, global ChIP analyses of H2B-Ser36p and H2B-Glu35-PAR will have to wait until suitable reagents are available.
Finally, although we think it is unlikely, we cannot definitively rule out the interesting possibility that an unknown specificity factor functions with PARP-1 to mediate Ser ADPRylation under physiological conditions. Such a possibility might allow for ADPRylation of H2B-Ser36 as a mechanism to block Ser36 phosphorylation. We do think, however, that as other members of the PARP family are explored in more detail, specificity factors with activities similar to HPF1 or NMNAT-1 will be the rule rather than the exception.
STAR ★ METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, W. Lee Kraus (Lee.Kraus@utsouthwestern.edu).
Materials availability
As Lead Contact, W. Lee Kraus is responsible for all reagent and resource requests. Please contact W. Lee Kraus at Lee.Kraus@utsouthwestern.edu with requests and inquiries.
Data and code availability
The RNA-seq and ChIP-seq data sets generated specifically for this study can be accessed from the NCBI’s Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo/) using the superseries accession number GSE136055. The new mass spec data sets generated for these studies are available as supplemental data provided with this manuscript.
EXPERIMENTAL MODELS AND SUBJECT DETAILS Cell culture
3T3-L1 cells (Green and Kehinde, 1975) were obtained from the American Type Cell Culture (ATCC, CL-173) and were regularly verified as mycoplasma-free. They were cultured in DMEM (Cellgro, 10–017-CM) supplemented with 10% fetal bovine serum (Sigma, F8067) and 1% penicillin/streptomycin. Differentiation from preadipocytes into adipocytes was induced in contact-inhibited cells by the addition of MDI cocktail [0.25 mM IBMX (3-isobutyl-1-methylxanthine; Calbiochem, 410957), 1 μM dexamethasone (Sigma, D4902), and 10 μg/mL insulin (Sigma, I-5500)]. The cells were treated with MDI cocktail for 2 days and then cultured in medium containing 10 μg/mL insulin for the indicated time points before collection.
293T cells were obtained from the ATCC (CRL-3216) and were regularly verified mycoplasma-free. They were cultured in DMEM (Cellgro, 10–017-CM) supplemented with 10% fetal bovine serum (Sigma, F8067) and 1% penicillin/streptomycin. MDA-MB-436 cells were purchased from the ATCC (HTB-130) and maintained in RPMI (Sigma-Aldrich, R8758) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
Generation of knockdown, knockout, and ectopic expression cell lines
Stable shRNA-mediated knockdown in 3T3-L1 cells.
Lentiviruses were generated by transfecting the pLKO.1 vectors described in the Method Details, each containing a different shRNA sequence directed against its cognate target (Parp1 or Nmnat1), into 293T cells, together with the expression vectors for the VSV-G envelope protein (pCMV-VSV-G, Addgene plasmid no. 8454), the expression vector for GAG-Pol-Rev (psPAX2, Addgene plasmid no. 12260), and a vector to aid with translation initiation (pAdVAntage, Promega) using Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific, L3000015) according to the manufacturer’s instructions. The viruses were collected in the culture medium, concentrated by using a Lenti-X concentrator (Clontech, 631231), and used to infect 3T3-L1 cells. The infected cells were selected with 2 μg/mL puromycin (Sigma, P9620), expanded, and frozen in aliquots for future use.
Stable ectopic protein expression in 3T3-L1 cells.
Lentiviruses were generated by transfecting the pCDH vectors described in the Method Details into 293T cells. 3T3-L1 cells were infected with the lentiviruses, with subsequent selection using 2 μg/mL puromycin to enrich for cells expressing wild-type, E35A, or S36A histone H2B. Ectopic expression of the cognate proteins was confirmed by Western blotting.
Inducible ectopic protein expression in 3T3-L1 cells.
Lentiviruses were generated by transfecting the pINDUCER20 vectors described in the Method Details into 293T cells. For inducible expression of histone H2B, 3T3-L1 cells were infected with the lentiviruses, with subsequent selection using 800 μg/mL G418 sulfate (Sigma, A1720). The cells were induced with MDI cocktail, treated with 1 μg/mL Dox one day after differentiation, and maintained in medium containing 1 μg/mL Dox for the indicated time points before collection. For inducible expression of PARP-1 or NMNAT-1, 3T3-L1 cells with shRNA-mediated knockdown of Parp1 or Nmnat1 as described above were infected with the lentiviruses, with subsequent selection using 800 μg/mL G418 sulfate, and then were treated with 1 μg/mL Dox for 48 hours. Inducible ectopic expression of the cognate proteins was confirmed by Western blotting.
CRISPR/Cas9-mediated knockout of snoRNAs in 3T3-L1 cells.
Lentiviruses were generated by transfecting the LentiCRISPR v2 vectors described in the Method Details into 293T cells, and used to infect 3T3-L1 cells. The infected cells were selected with 2 μg/mL puromycin (Sigma, P9620), expanded, and frozen in aliquots for future use. The efficiency of snoRNA knockout in bulk cells was verified by RT-qPCR.
Mice used for in vivo experiments
We used various strains of mice for the in vivo experiments described herein. All animal use was performed with oversight from UT Southwestern’s Institutional Animal Care and Use Committee. Mice were maintained on a standard rodent chow diet or specialized diets as specified with 12-hour light and dark cycles.
Parp1-null mice.
Parp1 null (Parp1−/−) mice on a C57BL/6 background were described previously (de Murcia et al., 1997). Eight- to ten-week-old male mice were maintained on a standard rodent chow diet and used for experiments as specified.
Parp1 preadipocyte-specific deletion in Mural Chaser mice.
To generate Parp1 preadipocyte-specific deletion lineage-tracing mice, we used the doxycycline (Dox)-inducible Mural Chaser system (PdgfrbrtTA; TRE-Cre; Rosa26RmT/mG) created by R.G. (Vishvanath et al., 2016). Parp1loxP/loxP mice (Jackson Laboratory, stock no 032650) were generated as described previously (Luo et al., 2017) and crossed with mice carrying TRE-Cre [B6. Cg-Tg(tetO-cre)1Jaw/J; Jackson Laboratory, stock no 006234] and Rosa26RmT/mG [B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J; Jackson Laboratory, stock no 007676] alleles. The lineage-tracing mice were then crossed with Parp1loxP/loxP/TRE-Cre/Rosa26RmT/mG mice to generate preadipocyte-specific deletion of Parp1 in Mural Chaser mice. For experiments, we used six- to eight-week-old male or female lineage-tracing mice (see details below).
Primers used for genotyping.
The following PCR primers were used for genotyping:
Pdgfrb-rtTA forward: 5’-AAGTCATTCCGCTGTGCTCT-3’
Pdgfrb-rtTA reverse: 5’-GTCTCAGAAGTGGGGGCATA-3’
mTmG forward: 5’-CTCTGCTGCCTCCTGGCTTCT-3’
mTmG reverse 1: 5’-CGAGGCGGATCACAAGCAATA-3’
mTmG reverse 2: 5’-TCAATGGGCGGGGGTCGTT-3’
TRE-Cre forward: 5’-ATGCTTCTGTCCGTTTGCC-3’
TRE-Cre reverse: 5’-CAACACCATTTTTTCTGACC-3’
PARP1-Floxed forward: 5’-CTGTGGTCCTCTTGCCTCTG-3’
PARP1-Floxed reverse: 5’-ACTTCCCCAGGGATGGGTTA-3’
METHOD DETAILS
Cell treatments
3T3-L1 cells were grown until confluent and exposed to various treatments for the experiments described herein. For experiments with PARP inhibitor, the cells were treated with 20 μM BYK204165 (Tocris Bioscience, 1104546–89-5), or DMSO vehicle for 2 hours. For activation of AMPKα, the cells were treated with 500 μM 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR, Sigma, A9978) for 1 hour. For inhibition of AMPKα, the cells were treated with 20 μM Dorsomorphin (Sigma, P5499) for 1 hour. For inhibition of phosphatase, the cells were treated with 1 μM Okadaic acid (Sigma, 459620) for 15 minutes. For inhibition of Ca2+/Calmodulin-dependent protein kinase kinase (CaMKK), the cells were treated with 10 μg/mL STO-609 (Sigma, S1318) for 1 hour. For treatment with 2-deoxy-D-glucose (5 mM; Sigma, D8375), the cells were grown until confluent and treated for 24 hours. For induction of DNA damage, the cells were treated with 2 mM H2O2 for 5 minutes.
Antibodies
The custom recombinant poly(ADP-ribose) binding reagent (anti-PAR) was generated and purified in-house (Gibson et al., 2017) (now available from EMD Millipore; catalog no. MABE1031). The custom rabbit polyclonal antiserum against PARP-1 was generated in-house by using a purified recombinant antigen comprising the amino-terminal half of PARP-1 (Kim et al., 2004) (now available from Active Motif; catalog no. 39559). The custom rabbit polyclonal antiserum against NMNAT-1 was generated in-house by using recombinant human and mouse NMNAT-1 (Ryu et al., 2018). The other antibodies used were as follows: Phospho-histone H2B Ser36 (ECM Biosciences, HP4331), Phospho-histone H2B Ser32 (Invitrogen, PA5–40148), Histone H2B (Abcam, ab1790), FLAG (Sigma-Aldrich, F3165), HPF1 (Atlas, HPA043467), PARG (Millipore, MABS61), Phospho-AMPKα Thr172 (Cell Signaling, 2531), AMPKα (Cell Signaling, 2532), Phospho-p70 S6 kinase Thr389 (Cell Signaling, 9205), p70 S6 kinase (Cell Signaling, 9202), SNRNP70 (Abcam, ab83306), β-actin (Cell Signaling, 3700), rabbit IgG (ThermoFisher, 10500C), goat anti-rabbit HRP-conjugated IgG (ThermoFisher, 31460), goat anti-mouse HRP-conjugated IgG (ThermoFisher, 31430), GFP (Abcam, ab13970), Perilipin (Fitzgerald, 20R-PP004), Alexa fluor 488-conjugated goat anti-chicken IgG (Invitrogen, A-11039), Alexa fluor 647-conjugated goat anti-guinea pig IgG (Invitrogen, A-21450), and Alexa fluor 405-conjugated goat anti-rabbit (Abcam, ab175654).
Dot blotting to confirm phospho-histone H2B Ser36 antibody specificity
Peptides representing amino acids 25 through 46 of histone H2B were synthesized containing the amino acid sequences and modifications noted in the figures (Elim Biopharmaceuticals). The peptides were dissolved and diluted serially in H2O to concentrations raging from 62.5 to 1,000 ng/μL. One microliter of each diluted peptide solution was spotted onto a nitrocellulose membrane. The membranes were air dried, blocked with 5% nonfat milk in TBST, incubated with the phospho-histone H2B Ser36 primary antibodies described above in 3% nonfat milk prepared in TBST, and then incubated with anti-rabbit HRP-conjugated IgG. Western blot signals were detected using an ECL detection reagent (Thermo Fisher Scientific, 34077, 34095).
Molecular cloning to generate knockdown and expression vectors
Generation of lentiviral expression vectors for shRNAs targeting Parp1 and Nmnat1.
An shRNA construct targeting mouse Parp1 mRNA was generated by cloning a double-stranded oligonucleotide (5’-GGGCAAGCACAGTGTCAAA-3’) into the pLKO.1 vector (Addgene, plasmid no. 10878). shRNA constructs targeting mouse Nmnat1 mRNA (TRCN0000111435, TRCN0000335596) and control shRNA (SHC002) were purchased from Sigma, and then cloned into the pLKO.1 vector.
Generation of expression vectors for RNAi-resistant wild-type and analog-sensitive mutant (L877A) PARP-1.
cDNA was prepared by extracting total RNA from 3T3-L1 cells using Trizol (Invitrogen, 15596026), followed by reverse transcription using Superscript III reverse transcriptase (Invitrogen, 18080051) and an oligo(dT) primer, according to the manufacturer’s instructions. A Parp1 cDNA was then amplified from the cDNA library. A Parp1 cDNA for an analog-sensitive mutant (L877A) was generated by site-directed mutagenesis using Pfu Turbo DNA polymerase (Agilent, 600250). The PCR products were then cloned into the pINDUCER20 lentiviral doxycycline (Dox)-inducible expression vector (Addgene, plasmid no. 44012) (Meerbrey et al., 2011).
Generation of RNAi-resistant expression vectors for wild-type and catalytic mutant (W170A) NMNAT-1.
cDNA was prepared as described above. An Nmnat1 cDNA was then amplified from the cDNA library. Nmnat1 cDNAs for an RNAi-resistant mutant and a catalytically inactive mutant (W170A) were generated by site-directed mutagenesis using Pfu Turbo DNA polymerase (Agilent, 600250), and then cloned into the pINDUCER20 lentiviral Dox-inducible expression vector.
Generation of lentiviral expression vectors for wild-type, PARylation site mutant, and phosphorylation site mutant histone H2B.
Double-stranded cDNAs encoding carboxyl-terminal FLAG epitope-tagged mouse wild-type, PARylation mutant (E35A), and phosphorylation mutant (S36A) histone H2B were synthesized as gene blocks (Integrated DNA Technologies), and then cloned individually into two different expression vectors using Gibson assembly (NEB, E2621): (1) pCDH-EF1α-MCS-IRES-Puro lentiviral expression vector (System Biosciences, CD532A-2) and (2) pINDUCER20 lentiviral Dox-inducible expression vector.
Generation of in vitro transcription vectors for snoRNAs.
Double-stranded cDNAs encoding mouse Snora7a, Snord16a, Snord22, or Snora64 were synthesized as gene blocks (Integrated DNA Technologies), and then cloned into HindIII- and XbaI-digested pcDNA3.1 (ThermoFisher Scientific) downstream of the T7 promoter. cDNAs encoding individual human snoRNAs were amplified from a cDNA pool by PCR using the snoRNA-specific primer sets listed below and cloned into HindIII and XbaI-digested pcDNA3 (ThermoFisher Scientific) downstream of the T7 promoter, as described previously (Kim et al., 2019).
Cloning primers
SNORA15 Forward: 5’-CGGAATTCGCATGGCCGAATACTGTGTTTTTATC-3’
SNORA15 Reverse: 5’-GCTCTAGAATTTGTATTCACCTTTAATAATGATATGC-3’
SNORA37 Forward: 5’-CGGAATTCTGAGCACTTTCACAGGTCCTCC-3’
SNORA37 Reverse: 5’-GCTCTAGAAATTGTCCCATTGAATGACAGCTGC-3’
SNORA65 Forward: 5’-CGGAATTCTCAGCCACCCGCCACTGCA-3’
SNORA65 Reverse: 5’-GCTCTAGAGCTGTTCCCATGCTTTCGG-3’
Generation of CRISPR/Cas9 lentiviral vectors for sgRNA expression and genome editing.
The CRISPR plasmid LentiCRISPR v2 was obtained from Addgene (plasmid no. 52961). Using the Dharmacon CRISPR Design Tool, optimized single guide RNA (sgRNA) sequences for Snora7a, Snord16a, Snord22, and Snora64 were designed as described below. A non-targeting sequence from the GeCKOv2 Mouse Library Pool A was used as the control (Sanjana et al., 2014). To clone the guide target sequence into LentiCRISPR v2, oligo pairs for each snoRNA were synthesized as described below. Each pair of oligos was annealed, diluted, and then assembled into LentiCRISPR v2 using Golden-Gate sgRNA cloning protocol (Sanjana et al., 2014; Shalem et al., 2014).
sgRNA sequences
Snora7a sgRNA: GCTACCGAGCAAACTGGGAA
Snord16a sgRNA: GTTGCCTGCTGTCAGTATGC
Snord22 sgRNA: GACGTGTGAACATTTCTTCA
Snora64 sgRNA: TCTCCGTACGAAAGTCACAC
Non-targeting sequence: GCGAGGTATTCGGCTCCGCG
Oligo pairs
Snora7a oligo 1: 5’-CACCGGCTACCGAGCAAACTGGGAA-3’
Snora7a oligo 2: 5’-AAACTTCCCAGTTTGCTCGGTAGCC-3’
Snord16a oligo 1: 5’-CACCGGTTGCCTGCTGTCAGTATGC-3’
Snord16a oligo 2: 5’-AAACGCATACTGACAGCAGGCAACC-3’
Snord22 oligo 1: 5’-CACCGGACGTGTGAACATTTCTTCA-3’
Snord22 oligo 2: 5’-AAACTGAAGAAATGTTCACACGTCC-3’
Snora64 oligo 1: 5’-CACCGTCTCCGTACGAAAGTCACAC-3’
Snora64 oligo 2: 5’-AAACGTGTGACTTTCGTACGGAGAC-3’
Non-targeting oligo 1: 5’-CACCGGCGAGGTATTCGGCTCCGCG-3’
Non-targeting oligo 2: 5’-AAACCGCGGAGCCGAATACCTCGCC-3’
Expression and purification of recombinant proteins
Purification of PARP-1 expressed in Sf9 insect cells.
Sf9 insect cells, cultured in SF-II 900 medium (Invitrogen), were transfected with 1 μg of bacmid driving expression of FLAG-tagged wild-type or analog-sensitive (L877A) mouse PARP-1 using Cellfectin transfection reagent (Invitrogen) as described by manufacturer. After three days, the medium was supplemented with 10% FBS, penicillin, and streptomycin, and collected as a baculovirus stock. After multiple rounds of amplification of the stock, the resulting high titer baculovirus was used to infect fresh Sf9 cells to induce expression of PARP-1 protein for two days. The PARP-1 expressing Sf9 cells were then collected by centrifugation, flash frozen in liquid N2, and stored at −80°C.
For purification of PARP-1, the Sf9 cell pellets were thawed on wet ice. The cells were resuspended in FLAG Lysis Buffer (20 mM HEPES pH 7.9, 0.5 M NaCl, 4 mM MgCl2, 0.4 mM EDTA, 20% glycerol, 250 mM nicotinamide, 2 mM β-mercaptoethanol, 2x protease inhibitor cocktail) and lysed by Dounce homogenization (Wheaton). The lysate was clarified by centrifugation (15,000 rpm, 30 min, 4°C), mixed with an equal volume of FLAG Dilution Buffer (20 mM HEPES pH 7.9, 10% glycerol, 0.02% NP-40), sonicated, and then clarified by centrifugation (15,000 rpm, 30 min, 4°C) again. The clarified lysate was mixed with anti-FLAG M2 agarose resin (Sigma, A2220), washed twice with FLAG Wash Buffer #1 (20 mM HEPES pH 7.9, 150 mM NaCl, 2 mM MgCl2, 0.2 mM EDTA, 15 % glycerol, 0.01% NP-40, 100 mM nicotinamide, 0.2 mM β-mercaptoethanol, 1 mM PMSF, 1 μM aprotinin, 100 μM leupeptin), twice with FLAG Wash Buffer #2 (20 mM HEPES pH 7.9, 1 M NaCl, 2 mM MgCl2, 0.2 mM EDTA, 15% glycerol, 0.01% NP-40, 100 mM nicotinamide, 0.2 mM β-mercaptoethanol, 1 mM PMSF, 1 μM aprotinin, 100 μM leupeptin), and twice with FLAG Wash Buffer #3 (20 mM HEPES pH 7.9, 150 mM NaCl, 2 mM MgCl2, 0.2 mM EDTA, 15% glycerol, 0.01% NP-40, 0.2 mM β-mercaptoethanol, 1 mM PMSF). The FLAG-tagged PARP-1 protein was eluted from the anti-FLAG M2 agarose resin with FLAG Wash Buffer #3 containing with 0.5 mg/mL 3x FLAG peptide (Sigma, F4799). The eluted proteins were aliquoted, flash frozen in liquid N2, and stored at −80°C until used. To assess the purity and quality of the purified proteins, 1 μg of purified protein was subjected to SDS–PAGE and stained with Coomassie Brilliant Blue.
Purification of NMNAT-1 and HPF1 expressed in E. coli.
6xHis-tagged mouse wild-type (Wt) or catalytically inactive mutant (W170A) NMNAT-1 (UniProt entry Q9EPA7), and HPF1 (UniProt entry Q8CFE2) was expressed in E. coli strain BL21(DE3) using a pET19b-based bacterial expression vector. The transformed bacteria were grown in LB containing ampicillin at 37 °C until the OD595 reached 0.4–0.6. Recombinant protein expression was induced by the addition of 1 mM IPTG for 2 h at 37 °C. The cells were collected by centrifugation, and the cell pellets were flash-frozen in liquid N2 and stored at −80 °C. The frozen cell pellets were thawed on wet ice and lysed by sonication in Ni-NTA Lysis Buffer (10 mM Tris-HCl pH 7.5, 0.5 M NaCl, 0.1 mM EDTA, 0.1% NP-40, 10% glycerol, 10 mM imidazole, 1 mM PMSF, and 1 mM β-mercaptoethanol). The lysates were clarified by centrifugation at 15,000 rpm using an SS34 rotor (Sorvall) at 4°C for 45 minutes. The supernatant was incubated with 1 mL of Ni-NTA resin equilibrated in Ni-NTA Equilibration Buffer (10 mM Tris-HCl pH 7.5, 0.5 M NaCl, 0.1% NP-40, 10% glycerol, 10 mM imidazole, and 1 mM β-mercaptoethanol) at 4 °C for 2 hours with gentle mixing. The resin was collected by centrifugation at 4 °C for 10 minutes at 1,000 x g, and the supernatant was removed. The resin was washed three times with Ni-NTA Wash Buffer (10 mM Tris-HCl pH 7.5, 1 M NaCl, 0.2% NP-40, 10% glycerol, 10 mM imidazole, and 1 mM PMSF). The recombinant proteins were then eluted using Ni-NTA Elution Buffer (10 mM Tris-HCl pH 7.5, 0.2 M NaCl, 0.1% NP-40, 10% glycerol, 500 mM imidazole, 1 mM PMSF, and 1 mM β-mercaptoethanol). The eluates were collected by centrifugation at 4°C for 10 minutes at 1,000 x g, and dialyzed in Ni-NTA Dialysis Buffer (10 mM Tris-HCl pH 7.5, 0.2 M NaCl, 10% glycerol, 10 mM imidazole, 1 mM PMSF, and 1 mM β-mercaptoethanol). The dialyzed proteins were quantified using a Bradford protein assay (Bio-Rad), aliquoted, flash-frozen in liquid N2, and stored at −80 °C.
Identification of PARP-1 substrates using an analog-sensitive PARP approach
We used an analog-sensitive PARP (asPARP) approach coupled with protein mass spectrometry as described previously (Gibson and Kraus, 2017; Gibson et al., 2016) to identify substrates of PARP-1 ADP-ribosylation activity, as well as the specific amino acid residues modified by PARP-1. First, we verified the system using in vitro assays and in-gel fluorescence. Then, we performed the assay in nuclei from 3T3-L1 cells, followed by mass spectrometry to identify the ADPRylated proteins.
In-gel fluorescence for testing PARP-1 specific modification of nuclear extract proteins.
Nuclear extracts were prepared from 3T3-L1 cell as described previously (Gibson and Kraus, 2017; Gibson et al., 2016). One microgram of purified PARP-1 protein (wild-type or analog-sensitive) was incubated with 50 μg of 3T3-L1 cell nuclear extract and 250 μM of 8-Bu(3-yne)T-NAD+ for 15 minutes at room temperature. The reactions were stopped by methanol:chloroform precipitation. After centrifugation for 10 minutes at maximum speed at room temperature, the protein pellets were dissolved in 1 mL Urea Solubilization Buffer (200 mM HEPES pH 8.0, 8 M urea, 1 M NaCl, and 4% CHAPS). Fifty μL from the 1 mL of labeled and solubilized proteins were combined sequentially in the following order with mixing: 1 μL azido-TAMRA (Click Chemistry Tools), 2 μL of a 50:250 mM CuSO4:THPTA pre-formed catalytic complex, 1 μL of 500 mM aminoguanidine hydrochloride, and 1 μL of 500 mM sodium ascorbate. After incubation in the dark for 2 hours, the fluorescently labeled PARP-1 target proteins were extracted by methanol:chloroform precipitation, run on an SDS-PAGE gel, and visualized using a Bio-Rad Pharos FX Plus Molecular Imager.
In nuclei 8-Bu(3-yne)T-ADP-ribosylation reactions.
Undifferentiated 3T3-L1 with shRNA-mediated depletion of endogenous PARP-1 and inducible ectopic expression of asPARP-1 were harvested in ice-cold PBS and collected by centrifugation. The cells were swollen in Nuclei Isolation Buffer (10 mM HEPES pH 8.0, 2 mM MgCl2, 3 mM CaCl2, 300 mM sucrose, with freshly added 1 mM DTT, 1x protease inhibitors, and 1x phosphatase inhibitors) and the nuclei were released by the addition of 0.65% NP-40 with moderate vortexing. Following collection by centrifugation (1000 x g for 5 minutes at 4°C), the nuclei were resuspended in PARP Reaction Buffer (30 mM Tris•HCl, pH 7.5, 10 mM KCl, 5 mM MgCl2, 5 mM CaCl2, 0.01% NP-40, 0.05 mM EDTA, 20% glycerol, with freshly added 1 mM DTT, protease inhibitors, and phosphatase inhibitors) containing 250 μM 8-Bu(3-yne)T-NAD+ (BIOLOG Life Science Institute, Bremen, Germany) for 30 minutes at 25°C with occasional gentle mixing to allow ADP-ribosylation to occur in the isolated nuclei.
Preparation for LC-MS/MS.
Following in nuclei ADP-ribosylation, the nuclei were collected by centrifugation and resuspended in Urea Solubilization Buffer (200 mM HEPES pH 8.0, 8 M urea, 1 M NaCl, and 4% CHAPS) containing 250 U/mL of Universal nuclease (Pierce/Thermo). The 8-Bu(3-yne)T-ADP(ribosyl)ated nuclear proteins in Urea Solubilization Buffer were combined sequentially in a 2 mL tube in the following order with mixing: 100 μL azido-agarose beads (Click Chemistry Tools), 820 μL water, 40 μL of a 50:250 mM CuSO4:THPTA pre-formed catalytic complex, 20 μL of 500 mM aminoguanidine hydrochloride, and 20 μL of 500 mM sodium ascorbate. After 18 hours of click chemistry reaction time in the dark with slow mixing in a rotating mixer, the beads were collected by centrifugation at room temperature for 1 min. at 1000 RCF in a microcentrifuge and the reaction supernatant was aspirated. The beads were resuspended in 1.8 mL of MilliQ water and were collected by centrifugation at room temperature for 1 min. at 1000 RCF. The beads were then resuspended in 1 mL of SDS Wash Buffer (100 mM Tris•HCl pH 8.0, 1% SDS, 250 mM NaCl, 5 mM EDTA) supplemented with freshly made 1 mM DTT, heated to 70°C for 15 minutes, and then allowed to cool to room temperature.
The beads were collected by centrifugation at room temperature for 5 min. at 1000 x g in a microcentrifuge and the supernatant was aspirated. The beads were then resuspended in 1 mL of SDS Wash Buffer containing 40 mM iodoacetamide and incubated at room temperature for 30 min. in the dark to alkylate the cysteine residues. The beads were then transferred to a 2 mL single-use column (Bio-Rad) and washed as follows: 10 washes of 2 mL each with SDS Wash Buffer, 10 washes of 2 mL each with Urea Wash Buffer (100 mM Tris•HCl, pH 8.0, 8 M urea), and 10 washes of 2 mL each with 20% acetonitrile. Following these extensive washes, the beads were resuspended in 500 μL of Trypsin Digestion Buffer (100 mM Tris•HCl, pH 8.0, 2 mM CaCl2, 10% acetonitrile). Trypsin digestion of bead-bound 8-Bu(3-yne)T-ADP-ribosylated nuclear extract proteins was performed by adding of 1 μg of trypsin (Promega) to the Trypsin Digestion Buffer, with incubation at room temperature overnight with slow mixing on a rotating mixer. The peptides from the trypsin digestion were prepared for LC MS/MS by desalting on a C18 stage tip (Thermo) according to the manufacturer’s protocol and lyophilized for storage at −80°C prior to the LC-MS/MS runs for peptide identification. After the trypsin digestion of the proteins on the beads, which left the peptides covalently linked to the beads through the azide-clicked 8-Bu(3-yne)T-ADP-ribosylation sites, the beads were transferred to a fresh 2 mL single use column (Bio-Rad) and washed as follows: 10 washes of 2 mL each with SDS Wash Buffer, 10 washes of 2 mL each with Urea Wash Buffer, 10 washes of 2 mL each with 20% acetonitrile, and 5 washes of 2 mL each with Peptide Elution Buffer (100 mM HEPES, pH 8.5). The beads were transferred to a microcentrifuge tube and hydroxylamine (Sigma) was added to 0.5 M to elute the glutamate- and aspartate-modified 8-Bu(3-yne)T-ADP-ribosylated peptides from the beads. The eluted peptides were prepared for LC-MS/MS by desalting on a C18 stage tip (Thermo) according to the manufacturer’s protocol and then lyophilized for storage at −80°C prior to LC-MS/MS analysis.
LC-MS/MS analysis.
Following solid-phase extraction cleanup with an Oasis HLB microelution plate (Waters), the trypsin samples were reconstituted in 2% (v/v) acetonitrile (ACN) and 0.1% trifluoroacetic acid in water such that the resulting concentration was 1 μg/μL. Hydroxylamine eluted samples were reconstituted in 6 μL of the same buffer. One microliter of the trypsin samples and 5 μL of the hydroxylamine eluate samples were injected onto an Orbitrap Fusion Lumos mass spectrometer (Thermo Electron) coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system (Dionex). Samples were injected onto a 75 μm i.d., 50-cm long EasySpray column (Thermo) and eluted with a gradient from 1–28% buffer B over 60 minutes. Buffer A contained 2% (v/v) ACN and 0.1% formic acid in water, and Buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water.
The mass spectrometer was operated in positive ion mode with a source voltage of 1.5–2.4 kV and an ion transfer tube temperature of 275 °C. MS scans were acquired at a resolution of 120,000 in the Orbitrap and up to 10 MS/MS spectra were obtained in the ion trap for each full spectrum acquired using higher-energy collisional dissociation (HCD) for ions with charges 2–7. Dynamic exclusion was set for 25 s after an ion was selected for fragmentation. Raw MS data files were analyzed using Proteome Discoverer v2.2 (Thermo), with peptide identification performed using Sequest HT searching against the Mus musculus database from UniProt. Fragment and precursor tolerances of 10 ppm and 0.6 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification, with oxidation of Met and hydroxamic acid addition (+15.0109 Da) to Asp and Glu set as variable modifications. The false-discovery rate (FDR) cutoff was 1% for all proteins and peptides.
Knockdown of PARG and HPF1 by using siRNAs
Commercially available siRNA oligos targeting PARG (Sigma, SASI_Mm01_00167287, SASI_Mm01_00115158) or HPF1 (Thermo Fisher Scientific, s29882, s29883) were transfected at a final concentration of 20 nM using Lipofectamine RNAi MAX reagent (Invitrogen, 13778150) according to the manufacturer’s instructions. All experiments were performed 48 hours after siRNA transfection.
Preparation of cell extracts and Western blotting
3T3-L1 cells were cultured and treated as described above before the preparation of cell extracts.
Preparation of whole cell lysates.
The collected cells were washed with ice-cold PBS and resuspended in Whole Cell Lysis Buffer [50 mM Tris-HCl pH 7.9, 2 mM CaCl2, 0.2% Triton X-100, 100 U/mL micrococcal nuclease (Worthington, 27735), 1x complete protease inhibitor cocktail (Roche, 11697498001), 1x phosphatase inhibitor cocktail (Sigma, P0044, P5726), 250 nM ADP-HPD (Sigma, A0627, a PARG inhibitor), 10 μM PJ34 (Enzo Life Sciences, ALX-270–289, a PARP inhibitor)] and incubated for 15 minutes at room temperature with gentle mixing to lyse the cells and extract the proteins. The lysates were clarified by centrifugation in a microcentrifuge for 5 minutes at 4°C at full speed.
Preparation of nuclear and cytosolic extracts.
The collected cells were washed with ice-cold PBS and resuspended in Isotonic Buffer (10 mM Tris-HCl pH 7.5, 2 mM MgCl2, 3 mM CaCl2, 0.3 M sucrose, with freshly added 1 mM DTT, 1x protease inhibitor cocktail, 1x phosphatase inhibitor cocktail, 250 nM ADP-HPD, and 10 μM PJ34), incubated on ice for 15 minutes, and lysed by the addition of 0.6% IGEPAL CA-630 with gentle vortexing. After centrifugation, the nuclei were collected by centrifugation in a microcentrifuge for 1 minute at 4°C at 1,000 rpm. The supernatant was collected as the cytoplasmic fraction. The pelleted nuclei were resuspended in Nuclear Extraction Buffer (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 25% v/v glycerol, with freshly added 1 mM DTT, 1x protease inhibitor cocktail, 1x phosphatase inhibitor cocktail, 250 nM ADP-HPD, and 10 μM PJ34) and incubated on ice for 30 minutes. The chromatin and soluble nuclear fractions were separated by centrifugation (12,000 x g, 30 min, 4°C). The chromatin fraction was then resuspended in Chromatin Extraction Buffer (10 mM Tris-HCl pH 7.5, 10 mM KCl, 1 mM CaCl2, 1x protease inhibitor cocktail, 1x phosphatase inhibitor cocktail, 250 nM ADP-HPD, and 10 μM PJ34) and then digested with micrococcal nuclease (40 U/ml) at 37°C for 15 min. The reaction was stopped by the addition of 5 mM EGTA, and the soluble chromatin fraction was collected by centrifugation (12,000 x g, 30 min, 4°C).
Determination of protein concentrations and Western blotting.
Protein concentrations were determined using a BCA protein assay (Pierce, 23225). The cell extracts were aliquoted, flash-frozen in liquid N2, and stored at −80 °C. Aliquots of the cell extracts were run on polyacrylamide-SDS gels and transferred to nitrocellulose membranes. After blocking with 5% nonfat milk in TBST, the membranes were incubated with the primary antibodies described above in 3% nonfat milk prepared in TBST, followed by anti-rabbit HRP-conjugated IgG or anti-mouse HRP-conjugated IgG. Western blot signals were detected using an ECL detection reagent (Thermo Fisher Scientific, 34077, 34095).
Immunoprecipitation and analysis
The following protocols were used for nucleosome immunoprecipitation and co-immunoprecipitation and analysis.
Nucleosome immunoprecipitation.
3T3-L1 cells expressing FLAG-tagged wild-type or mutant (E35A, S36A) histone H2B or native 3T3-L1 cells were seeded at ~5×106 cells per 15 cm diameter plate and cultured, differentiated, and treated as described above. Nucleosome isolation was performed as described previously (Jayaram et al., 2016). Briefly, the cells were collected and the nuclei were isolated using Hypotonic Buffer (5 mM HEPES pH 7.5, 10 mM KCl, 5 mM MgCl2, 0.5 mM EDTA, 0.4 mM PMSF, with freshly added 1 mM DTT, 1x protease inhibitor cocktail, 1x phosphatase inhibitor cocktail, 250 nM ADP-HPD, and 10 μM PJ34), followed by centrifugation at 1,350 RCF for 5 minutes in a microcentrifuge. Nuclei were resuspended in Buffer A (15 mM HEPES pH 7.5, 30 mM KCl, 3 mM CaCl2, 3 mM MgCl2, 0.4 mM PMSF, with freshly added 1 mM DTT, 1x protease inhibitor cocktail, 1x phosphatase inhibitor cocktail, 250 nM ADP-HPD, and 10 μM PJ34) and incubated with 40 U/mL micrococcal nuclease at 37°C for 15 minutes. The micrococcal nuclease reaction was stopped by adding 5 mM EDTA, 3 mM EGTA, and 200 mM KCl (final concentrations). After centrifugation at 10,000 x g for 15 minutes at 4°C, the supernatant containing the soluble chromatin (mainly mononucleosomes) was collected, and the concentration was determined using the BCA protein assay. For 3T3-L1 cells expressing FLAG-tagged H2B, 1 mg of chromatin extract protein was incubated with anti-FLAG M2 agarose resin (Sigma, A2220) overnight at 4°C, and then washed 3 times with Wash Buffer (10 mM HEPES pH 7.5, 300 mM KCl, 0.5 mM EDTA, 0.1% Tween 20). For native 3T3-L1 cells, 1 mg of chromatin extract protein was incubated with 4 μL of H2B antibody (Abcam, ab1790) at 4°C overnight, followed by a 2-hour incubation with 100 μL of a 50% slurry of protein A-agarose beads, and then washed 3 times with Wash Buffer. The immunoprecipitated mononucleosomes were recovered from the beads by either eluting with 0.5 mg/mL 3xFLAG peptide in Elution Buffer (20 mM HEPES pH 7.5, 1 mM EDTA, 10% glycerol, 150 mM NaCl, 0.01% NP-40, with freshly added 0.4 mM PMSF, 1 mM DTT, and 1x protease inhibitor cocktail, 1x phosphatase inhibitor cocktail, 250 nM ADP-HPD, and 10 μM PJ34), or heating at 100°C for 5 minutes in 2x SDS-PAGE loading buffer. The immunoprecipitated material was subjected to Western blotting as described above.
Co-immunoprecipitation.
3T3-L1 cells with shRNA-mediated knockdown of Nmnat1 and ectopic inducible expression of FLAG-tagged wild-type or catalytically inactive mutant (W170A) NMNAT-1 were seeded at ~5×106 cells per 15 cm diameter plate and cultured, as described above. For the induction of FLAG-tagged NMNAT-1 expression, the cells were treated with 1 μg/mL of Dox for 48 hours before collection. The cells were collected and extracted as described above. The nuclear extracts were mixed with an equal volume of Dilution Buffer (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 0.2 mM EDTA, with freshly added 1 mM DTT, 1x protease inhibitor cocktail, 1x phosphatase inhibitor cocktail, 250 nM ADP-HPD, and 10 μM PJ34). Aliquots of nuclear extract containing 1 mg of total protein were used for each IP condition. The nuclear extracts were incubated with 50 μL of anti-FLAG M2 agarose resin (Sigma, A2220) overnight at 4°C, and then washed 3 times with Wash Buffer (20 mM HEPES pH 7.6, 150 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA) for 10 minutes at 4°C with constant mixing. The beads were then heated to 100°C for 10 minutes in 2x SDS-PAGE loading solution to release the bound proteins. The immunoprecipitated material was subjected to Western blotting as described above.
In nuclei PARylation reactions
3T3-L1 cells with shRNA-mediated knockdown of Nmnat1 expressing inducible ectopic FLAG-tagged wild-type or catalytically inactive mutant (W170A) NMNAT-1 were seeded at ~5×106 cells per 15 cm diameter plate and cultured as described above. For the induction of FLAG-tagged NMNAT-1 expression, the cells were treated with 1 μg/mL of Dox for 48 hours before collection. In nuclei PARylation reactions were performed as described previously (Luo et al., 2017). Cell pellets were suspended in 1 mL of Buffer A (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, with freshly added 1 mM DTT, 1x protease inhibitor cocktail, and 1x phosphatase inhibitor cocktail) and then lysed by pipetting up and down several times on ice for 15 minutes. The cells were collected by centrifugation at 1,000 x g for 5 minutes at 4°C. After centrifugation, the nuclei were resuspended in 100 μL of Freezing Buffer (50 mM Tris-HCl pH 8.3, 5 mM MgCl2, 40% glycerol, 0.1 mM EDTA, with freshly added 1 mM DTT, 1x protease inhibitor cocktail, and 1x phosphatase inhibitor cocktail).
For in nuclei PARylation reactions, 100 μM NAD+ was added to the reactions containing 3T3-L1 nuclei to enhance PARylation. Briefly, 50 μL of suspended nuclei were incubated with an equal volume of 2x Reaction Buffer (10 mM Tris-HCl pH 8.5, 20 mM KCl, 2 mM DTT, and 2x complete protease inhibitor cocktail) with or without 100 μM NAD+ for 30 minutes at room temperature. The reactions were stopped by adding 33 μL of 4x Stop Buffer (4% SDS and 20 mM EDTA), followed by heating to 100°C for 5 minutes in SDS-PAGE loading buffer. A 5–10 μL aliquot of each reaction was analyzed by Western blotting as described above.
In vitro PARylation assays
In vitro PARylation assays were performed essentially as described previously (Lin et al., 2018; Zhang et al., 2012). The reactions contained 0.1 μM purified PARP-1 and 1 μM wild-type or catalytically inactive mutant (W170A) NMNAT-1. NAD+, ATP, a non-hydrolyzable ATP analog [adenosine 5′-(γ-thio)-triphosphate (ATP-γ-S)], and NMN were included at a final concentration of 100 μM each. H2B peptides were synthesized (Elim Biopharmaceuticals) and included at 3 μg per reaction. The sequences of H2B peptides used for in vitro biochemical assays are listed below. Recombinant H2B proteins (NEB, M2505S) were included at 1 μg per reaction. Assembled recombinant nucleosomes were kindly provided by Dr. Glen Liszczak (UT Southwestern Medical Center), which were used at a final concentration of 500 nM. One hundred ng/μL of sheared salmon sperm DNA (Invitrogen, AM9680) or 200 nM snoRNA was used as an allosteric activator of PARP-1 enzymatic activity. Purified proteins were mixed with the H2B peptides or proteins in PARylation Buffer (20 mM HEPES pH 7.9, 5 mM MgCl2, 5 mM CaCl2, 25 mM KCl2, 0.01% NP-40, 1 mM DTT, 100 ng/μL sheared salmon sperm DNA, or 200 nM snoRNA), before the addition of 100 μM NAD+ to the reaction. The PARylation reactions were incubated at room temperature for 20 minutes and stopped by the addition of one third of a reaction volume of 4x SDS-PAGE Loading Buffer (200 mM Tris•HCl, pH 6.8, 8% SDS, 40% glycerol, 4% β-mercaptoethanol, 50 mM EDTA, 0.08% bromophenol blue), followed by heating to 100°C for 5 minutes. The reaction products were then resolved on a 15% SDS-PAGE gel, transferred to a nitrocellulose membrane, and blotted with an anti-poly-ADP-ribose detection reagent (EMD Millipore, MABE1031).
Sequences of the H2B peptides.
The following H2B peptides were used for in vitro ADPRylation assays.
H2B Wt (10–59): PKKGSKKAVTKAQKKDGKKRKRSRKESYSVYVYKVLKQVHPDTGISSKAM
H2B E35A (10–59): PKKGSKKAVTKAQKKDGKKRKRSRKASYSVYVYKVLKQVHPDTGISSKAM
H2B Wt (15–50): KKAVTKAQKKDGKKRKRSRKESYSVYVYKVLKQVHP
H2B S-Mut (15–50): KKAVTKAQKKDGKKRKRARKEAYAVYVYKVLKQVHP
H2B DE-Mut (15–50): KKAVTKAQKKAGKKRKRSRKASYSVYVYKVLKQVHP
H2B Wt (26–50): GKKRKRSRKESYSVYVYKVLKQVHP
H2B S36A (26–50): GKKRKRSRKEAYSVYVYKVLKQVHP
H2B E35A (26–50): GKKRKRSRKASYSVYVYKVLKQVHP
In vitro phosphorylation assays
In vitro phosphorylation assays were performed as described previously (Greer et al., 2007) with some modifications. Briefly, 0.1 μg of purified AMPK proteins (combination of α1/β1/γ1 subunits; Abcam, 79803) were incubated with 3 μg of H2B peptide in Kinase Reaction Buffer (20 mM HEPES pH 7.5, 5 mM MgCl2, 6.25 mM β-glycerophosphate, 1.25 mM EGTA, 125 μM ATP, and 1 mM DTT) at 30°C for 15 minutes. To determine the effect of PARylation on phosphorylation, we first performed the in vitro PARylation assays as described above. After stopping the PARylation reactions by adding 10 μM PARP inhibitor PJ34, in vitro phosphorylation assays were carried out by adding 0.1 μg recombinant AMPK complex (combination of α1/β1/γ1 subunits, Abcam, ab79803), 6.25 mM β-glycerophosphate, 1.25 mM EGTA, and 125 μM ATP to the reactions with subsequent incubation for 15 minutes at 30 °C. The reactions were then stopped by the addition of one third of a reaction volume of 4x SDS-PAGE Loading Buffer (200 mM Tris•HCl pH 6.8, 8% SDS, 40% glycerol, 4% β-mercaptoethanol, 50 mM EDTA, 0.08% bromophenol blue) followed by heating to 100°C for 5 minutes.
In vitro transcription to generate snoRNAs
To generate snoRNAs in vitro, XbaI-linearized plasmid DNA containing a cDNA of the snoRNA of interest (pcDNA3.1-snoRNA) was gel purified on an agarose gel and transcribed with T7 RNA polymerase (Promega, P207B) according to the manufacturer’s protocol. One μg of the linearized plasmid DNA was incubated with T7 RNA polymerase and NTPs (10 mM each of ATP, CTP, GTP, and UTP) at 37°C for 35 minutes. The template DNA was then removed by treatment with DNase (Promega, M6101) at 37°C for 15 minutes. The reaction was stopped by the addition of 2 μL of 0.2 M EDTA (pH 8.0). The in vitro transcribed snoRNAs were purified using EZ-10 DNAaway RNA Kit (Biobasic, BS88136) and the size of the transcripts were confirmed using 1% denaturing agarose gel electrophoresis with SYBR Gold staining (Invitrogen, S11494).
In vitro PARylation assays with snoRNA-activated PARP-1
Fifty nanograms of purified FLAG epitope-tagged recombinant PARP-1 protein was incubated with 200 nM of in vitro transcribed snoRNA or 100 ng/μL sheared salmon sperm DNA in ADP-ribosylation Buffer (20 mM HEPES pH 7.9, 5 mM MgCl2, 5 mM CaCl2, 25 mM KCl2, and 1 mM DTT) with 100 μM NAD+ at room temperature for 20 minutes. The ADP-ribosylation reactions were stopped by the addition of 4x SDS-PAGE loading buffer followed by heating to100°C for 10 minutes. The reaction products were then resolved on a 8% SDS-PAGE gel, transferred to a nitrocellulose membrane, and blotted with an anti-poly-ADP-ribose detection reagent (EMD Millipore, MABE1031) and PARP-1 antibody.
RNA isolation and RT-qPCR
3T3-L1 cells expressing inducible FLAG-tagged wild-type or mutant (E35A, S36A) histone H2B were seeded in 6-well plates and differentiated with MDI cocktail described above, treated with 1 μg/mL Dox at day 1 after differentiation, and maintained in medium containing 1 μg/mL Dox for the indicated time points before collection. 3T3-L1 cells with CRISPR/Cas9-mediated knockout of individual snoRNAs were seeded in 6-well plates and differentiated with MDI cocktail described above. The cells were collected at the indicated time points and total RNA was isolated using Trizol Reagent (Invitrogen) according to the manufacturer’s protocol. Total RNA was reverse transcribed using oligo (dT) primers and MMLV reverse transcriptase (Promega) to generate cDNA. The cDNA samples were subjected to qPCR using gene-specific primers, as listed below, as described previously (Luo et al., 2014). Briefly, cDNA, 1x SYBR Green PCR master mix, and forward and reverse primers (250 nM) were mixed and subjected to 45 cycles of amplification (95°C for 10 seconds, 60°C for 10 seconds, 72°C for 1 second) following an initial 5 min incubation at 95°C using a Roche LightCycler 480 384-well qPCR system. Melting curve analyses were performed to ensure that only the targeted amplicon was amplified. Target gene expression was normalized to the expression of Tbp mRNA (mouse). Relative expression was determined in comparison to the value of an appropriate control sample. All experiments were performed at least three times with independent biological replicates to ensure reproducibility. All experimental groups that were compared had similar variance as determined by the standard deviation of the biological replicates within each group. Statistical differences between two groups were determined using the Student’s t-test and a statistical significance was defined as p < 0.05.
Primers used for RT-qPCR.
The following primers were used for RT-qPCR.
Adipoq forward: 5’-GACAAGGCCGTTCTCTTCAC-3’
Adipoq reverse: 5’-CAGACTTGGTCTCCCACCTC-3’
Pparg forward: 5’-TGCTGTTATGGGTGAAACTCT-3’
Pparg reverse: 5’-CGCTTGATGTCAAAGGAATGC-3’
Fabp4 forward: 5’-AAGTGGGAGTGGGCTTTGC-3’
Fabp4 reverse: 5’-CCGGATGGTGACCAAATCC-3’
Tbp forward: 5’-TGCTGTTGGTGATTGTTGGT-3’
Tbp reverse: 5’-CTGGCTTGTGTGGGAAAGAT-3’
Snora7a forward: 5’-GCTAGTATTCTTCCAGCTTCGGA-3’
Snora7a reverse: 5’-GCTACCGAGCAAACTGGGAA-3’
Snord16a forward: 5’-ACTCTGTTCACAGCGACAGT-3’
Snord16a reverse: 5’-TGCTCAGTAACAATGTTCGTCAA-3’
Snord22 forward: 5’-TCCTAGCTCCAGAGCCTGAAA-3’
Snord22 reverse: 5’-CAGACAGTTCCTTCTGGAACAA-3’
Snora64 forward: 5’-GGTTTTACCCGTGTGACTTTCG-3’
Snora64 reverse: 5’-GTGGTGCTCCTCTAGGCAAG-3’
Rpl32 forward: 5’-GGCTGCCATCTGTTTTACGG-3’
Rpl32 reverse: 5’-TTTCCGCCAGTTTCGCTTAAT-3’
Rpl4 forward: 5’-AGCACCACGCAAGAAGATTCA-3’
Rpl4 reverse: 5’-TCTCGGATTTGGTTGCCAGT-3’
Snhg1 forward: 5’-TCCTTGTTCGGGGTTTGAGG-3’
Snhg1 reverse: 5’-ACAGCACCCTGACTACAAGC-3’
Rps2 forward: 5’-GACCAGGTTCAAGGCTTTCG-3’
Rps2 reverse: 5’-GCTTGCCAATCTTGTTCCCC-3’
RNA-sequencing
The following methods were used to generate, evaluate for QC, sequence, and analyze the data from RNA-seq libraries.
Generation of RNA-seq libraries.
Two biological replicates of 3T3-L1 cells with inducible ectopic expression of wild-type, E35A, and S36A histone H2B were differentiated and treated as described above. Total RNA was isolated using the RNeasy kit (Qiagen) according to the manufacturer’s instructions. The total RNA was then enriched for polyA+ RNA using Dynabeads Oligo(dT)25 (Invitrogen). The polyA+ RNA was then used to generate strand-specific RNA-seq libraries as described previously (Zhong et al., 2011). The RNA-seq libraries were subjected to QC analyses (i.e., number of PCR cycles required to amplify each library, the final library yield, and the size distribution of final library DNA fragments) and sequenced using an Illumina HiSeq 2000.
Analysis of RNA-seq data.
The raw data were subjected to QC analyses using the FastQC tool. The reads were then mapped to mouse genome (mm10) using the spliced reader aligner TopHat version 2.0.13 (Kim et al., 2013). Transcriptome assembly was performed using cufflinks v.2.2.1 with default parameters (Trapnell et al., 2010). The transcripts were merged into two distinct, non-overlapping sets using cuffmerge, followed by cuffdiff to call the differentially regulated transcripts. The significantly (p <0.05) regulated genes from cells expressing the H2B mutants S36A or E35A were compared to wild-type at the indicated time points.
Chromatin immunoprecipitation-qPCR (ChIP-qPCR)
3T3-L1 cells expressing inducible FLAG-tagged wild-type or mutant (E35A, S36A) histone H2B were seeded at ~5×106 cells per 15 cm diameter plate and differentiated with MDI cocktail described above. They were then treated with 1 μg/mL Dox at day 1 after differentiation and collected at day 2 after differentiation. ChIP was performed as described previously (Krishnakumar et al., 2008), with a few modifications. Briefly, the cells were crosslinked with 1% formaldehyde in PBS for 10 minutes at 37°C and quenched in 125 mM glycine in PBS for 5 minutes at 4°C. The cells were then collected and lysed in Farnham Lysis Buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP-40, with freshly added 1 mM DTT, 1x protease inhibitor cocktail, 1x phosphatase inhibitor cocktail, 250 nM ADP-HPD, and 10 μM PJ34). The crude nuclear pellet was collected by centrifugation, resuspended in Chromatin Lysis Buffer (50 mM Tris-HCl pH 7.9, 1% SDS, 10 mM EDTA, with freshly added 1 mM DTT, 1x protease inhibitor cocktail, 1x phosphatase inhibitor cocktail, 250 nM ADP-HPD, and 10 μM PJ34), and incubated on ice for 10 minutes. The chromatin was sheared at 4°C by sonication using a Bioruptor UC200 to generate chromatin fragments of ~300 bp in length. The soluble chromatin was diluted 1:10 with Dilution Buffer (20 mM Tris-HCl pH 7.9, 0.5% Triton X-100, 2 mM EDTA, 150 mM NaCl, with freshly added 1 mM DTT, 1x protease inhibitor cocktail, 1x phosphatase inhibitor cocktail, 250 nM ADP-HPD and 10 μM PJ34) and pre-cleared with Protein A Dynabeads (Invitrogen, 10002D).
The pre-cleared lysates were incubated with antibodies against the factor of interest (H2B-Ser36p or histone H2B) or with rabbit IgG as a control at 4°C overnight. Twenty μL of Protein A Dynabeads were then added to the sample with continue incubation for an extra 2 hours at 4°C with constant mixing. The immunoprecipitated material was then washed once with Low Salt Wash Buffer (20 mM Tris-HCl pH 7.9, 2 mM EDTA, 125 mM NaCl, 0.05% SDS, 1% Triton X-100, with freshly added 1x protease inhibitor cocktail, and 1x phosphatase inhibitor cocktail), once with High Salt Wash Buffer (20 mM Tris-HCl pH 7.9, 2 mM EDTA, 500 mM NaCl, 0.05% SDS, 1% Triton X-100, with freshly added 1x protease inhibitor cocktail, and 1x phosphatase inhibitor cocktail), once with LiCl Wash Buffer (10 mM Tris-HCl pH 7.9, 1 mM EDTA, 250 mM LiCl, 1% NP-40, 1% sodium deoxycholate, with freshly added 1x protease inhibitor cocktail, and 1x phosphatase inhibitor cocktail), and once with 1x Tris-EDTA (TE). The immunoprecipitated material was eluted in Elution Buffer (100 mM NaHCO3, 1% SDS), de-crosslinked overnight at 65°C, and then digested with proteinase K and RNase H to remove protein and RNA, respectively. The immunoprecipitated genomic DNA was then extracted with phenol:chloroform:isoamyl alcohol and precipitated with ethanol. The ChIPed genomic DNA was subjected to qPCR as described above using the gene specific primers listed below. All experiments were performed on at least three separate biological replicates to ensure reproducibility. Statistical differences between two groups were determined using the Student’s t-test and a statistical significance was defined as p < 0.05.
Primers used for ChIP-qPCR.
The following primers were used for ChIP-qPCR:
Cebpd promoter forward: 5’-CGTACATGGCAGGAGTCGT-3’
Cebpd promoter reverse: 5’-CCGCGGCCTTCTACGAG-3’
Pparg promoter forward: 5’-GGTCCAAAATGTTACTGCTATCCA-3’
Pparg promoter reverse: 5’-GCCTTTATTCTGTCAACTATTCCTT-3’
Chromatin immunoprecipitation-sequencing (ChIP-seq)
Growth of cells.
3T3-L1 cells expressing FLAG-tagged wild-type or E35A mutant histone H2B were seeded at ~5×106 cells per 15 cm diameter plate, and used for H3K27me3 ChIP.
ChIP for H3K27me3.
ChIP was performed as described previously (Murakami et al., 2017)with slight modifications. Briefly, the cells were cross-linked with 1% formaldehyde in PBS for 10 minutes at 37°C and quenched in 125 mM glycine in PBS for 5 minutes at 4°C. Cross-linked cells were then collected by centrifugation and lysed in Farnham Lysis Buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP-40, 1 mM DTT, 1x protease inhibitor cocktail, and 1x phosphatase inhibitor cocktail). The nuclei were collected by brief centrifugation in a microcentrifuge and resuspended in SDS Lysis Buffer (50 mM Tris-HCl pH 7.9, 1% SDS, 10 mM EDTA, 1 mM DTT, 1x protease inhibitor cocktail, and 1x phosphatase inhibitor cocktail) by pipetting and incubation on ice for 10 min. The chromatin was then sheared to ~300 bp DNA fragments by sonication using a Bioruptor Plus sonicator (Diagenode). The soluble chromatin was clarified by centrifugation, diluted 1:10 with ChIP Dilution Buffer (20 mM Tris-HCl pH 7.9, 0.5% Triton X-100, 2 mM EDTA, 150 mM NaCl, 1 mM DTT, 1x protease inhibitor cocktail, and 1x phosphatase inhibitor cocktail) and pre-cleared with protein A Dynabeads (Thermo Fischer, 1002D).
The pre-cleared lysates were incubated with an antibody against H3K27me3 (Active Motif, 39055), or rabbit IgG (as a control) at 4°C overnight with gentle mixing. Protein A Dynabeads were then added to the sample with continue incubation for an extra 2 hours at 4°C with constant mixing. The immunoprecipitated material was then washed with (1) Low Salt Wash Buffer (20 mM Tris-HCl pH 7.9, 2 mM EDTA, 125 mM NaCl, 0.05% SDS, 1% Triton X-100, 1x protease inhibitor cocktail, and 1x phosphatase inhibitor cocktail), (2) High Salt Wash Buffer (20 mM Tris-HCl pH 7.9, 2 mM EDTA, 500 mM NaCl, 0.05% SDS, 1% Triton X-100, 1x protease inhibitor cocktail, and 1x phosphatase inhibitor cocktail), (3) LiCl Wash Buffer (10 mM Tris-HCl pH 7.9, 1 mM EDTA, 250 mM LiCl, 1% NP-40, 1% sodium deoxycholate, 1x protease inhibitor cocktail, and 1x phosphatase inhibitor cocktail), and (4) 1x Tris-EDTA (TE). The immunoprecipitated material was eluted in Elution Buffer (100 mM NaHCO3, 1% SDS), de-crosslinked overnight at 65°C, and then digested with proteinase K and RNase H to remove protein and RNA, respectively. The immunoprecipitated genomic DNA was then extracted with phenol:chloroform:isoamyl alcohol and precipitated with ethanol. The precipitated ChIPed DNA was collected by centrifugation, air dried, and dissolved in DEPC- treated, nuclease-free water.
Generation of ChIP-seq libraries.
ChIP-seq libraries were prepared as previously described with some slight modifications (Murakami et al., 2017). Briefly, the ChIPed DNA or input DNA was purified using AMPure XP beads (Beckman Coulter, A63881). A total of 5–10 ng of ChIPed DNA, or equivalent amounts input DNA, was subjected to end repair using an end-repair mix (Enzymatics, Y9140- LC-L) and 0.1 mM dNTPs. A single dA base was added to the end repaired DNA using the Klenow DNA polymerase (Enzymatics, P7010-HC-L). TruSeq DNA Sample Prep Kit adaptors (custom synthesized by IDT and HPLC purified) were partially annealed to form double-stranded adaptors on one terminus, and annealed to add DNA through double-stranded DNA-DNA ligation using T4 DNA ligase (Enzymatics, L6030-HC-L) in DNA Rapid Ligase Buffer (Enzymatics, B1010). The adaptor-ligated DNA was amplified by PCR using Kapa HiFi Hot Start Ready Mix (KAPA Biosystems, KK2612), and size-selected and purified by electrophoresis on a 1% agarose gel containing SYBR Gold (Invitrogen, S11494). The DNA was excised from the agarose gel and eluted using a QIAquick Gel Extraction Kit (Qiagen, 28706). The quality of the library was assessed using a D1000 ScreenTape (Agilent, 5067–5582) on a 2200 TapeStation (Agilent) and quantified using a Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Q32851). The libraries with unique adaptor barcodes were multiplexed and sequenced on an Illumina HiSeq 2000.
Analysis of ChIP-seq data
Quality control and alignment.
The quality of the ChIP-seq datasets was assessed using the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). The ChIP-seq reads were aligned to the mouse reference genome (mm10) using Bowtie (ver. 0.12.7) with default parameters (Langmead and Salzberg, 2012). Samtools (ver. 0.1.19) (Li and Durbin, 2009) and Picard (ver. 1.127) were used to filter for uniquely mapped reads. Uniquely mapped reads were then converted to bigwig files using BEDTools (Quinlan and Hall, 2010) for visualization in the UCSC genome browser. H3K27me3 peak calling from two replicates per condition was performed using MACS software (ver. 1.4.2) with the default p- value and input condition as a control.
Data visualization and statistics.
For quantitatively assessing the reads distribution in a fixed window around each called peak under various conditions, we generated boxplots for the data. Raw read counts were obtained using the Multicov function in BEDTools and normalized based on library size. Wilcoxon rank sum tests were performed to determine the statistical significance of different comparisons.
Nearest neighboring gene analysis.
The nearest neighboring gene for each identified H3K27me3 peak was determined using Homer custom GTF. The expression of these genes was determined using the FPKM values obtained from the RNA-seq, using custom R scripts.
Oil Red O staining
3T3-L1 cells were seeded in 6-well plates and differentiated with MDI cocktail as described above. After 6 days of differentiation, the cells were rinsed twice with PBS and fixed with 4% paraformaldehyde. The fixed cells were incubated with 60% isopropanol for 5 minutes and then stained with 0.3% oil red-O solution (Sigma, O1391) for 5 minutes.
Imaging of visceral white adipose tissue from Mural Chaser mice
Six- to eight-week-old male and female Parp1 preadipocyte-specific deletion (Parp1loxp/loxp) and the control (Parp1+/+) Mural Chaser mice were maintained on a standard rodent chow diet or chow diet containing 600 mg/kg doxycycline (Dox) (Envigo RMS, Inc.) for 9 days, and then fed a high-fat/high-sucrose diet (45 kcal% fat and 30 kcal% sucrose; Research Diets, D08112601) for four weeks. Fresh visceral white adipose tissue from the mice were collected and directly imaged using a Zeiss LSM 880 Airyscan confocal microscope at the Live Cell Imaging Facility, UT Southwestern Medical Center.
Preparation of tissue lysates from white adipose tissue for Western blotting
Visceral white adipose tissue (~500 mg) from Parp1 null mice, C57BL/6 wild-type mice, Parp1 preadipocyte-specific deletion (Parp1loxp/loxp), and the control (Parp1+/+) Mural Chaser mice was minced and homogenized in Tissue Lysis Buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 10% v/v glycerol, 1% Triton X-100 with freshly added 1 mM DTT, 1x protease inhibitor cocktail, 1x phosphatase inhibitor cocktail, 250 nM ADP-HPD, and 10 μM PJ34), followed by gentle mixing at 4°C for 30 min. After centrifugation (18,000 x g, 20 minutes, 4°C), the fat cake was removed from the top of the liquid layer and the supernatant was collected. Protein concentrations were determined using a BCA protein assay (Pierce, 23225). The tissue lysates were aliquoted, flash-frozen in liquid N2, and stored at −80 °C. Western blotting was performed as described above.
Histological analysis of adipose tissues
Visceral white adipose tissue from Parp1 preadipocyte-specific deletion (Parp1loxp/loxp) and the control (Parp1+/+) Mural Chaser mice were collected and fixed with 10% neutral buffered formalin for 48 hours. Paraffin sections were prepared by the Molecular Pathology Core Facility at UT Southwestern Medical Center.
Indirect immunofluorescence.
The antibodies used for immunofluorescent staining were as follows: chicken anti-GFP (1:500; Abcam, ab13970), guinea pig anti-perilipin (1:1000; Fitzgerald, 20R-PP004), rabbit anti-phospho-histone H2B Ser36 (1:200, ECM Biosciences, HP4331), Alexa fluor 488-conjugated goat anti-chicken IgG (1:400; Invitrogen, A-11039), Alexa fluor 647-conjugated goat anti-guinea pig IgG (1:400; Invitrogen, A-21450), and Alexa fluor 405-conjugated goat anti-rabbit (1:400, Abcam, ab175654). Briefly, paraffin sections were deparaffinized in xylene and rehydrated sequentially in 100%, 95%, 80%, 70%, and 50% ethanol prepared in ddH2O. Antigen retrieval was performed by boiling the sections in 10 mM citrate pH 6.0 for 10 minutes. After cooling to room temperature for 1 hour, the sections were incubated with Fx Signal Enhancer (Invitrogen, I36933) for 30 minutes at room temperature.
The sections were then blocked with 5% normal goat serum (Thermo Fisher Scientific, 50062Z) in PBS at room temperature for 1 hour and then incubated with primary antibodies overnight at 4 °C in PBS containing 5% normal goat serum. After washing twice in PBS, the sections were incubated with secondary antibodies in PBS containing 5% normal goat serum at room temperature for 1 hour. After washing twice with PBS, the sections and coverslips were mounted with Anti-Fade mounting medium with DAPI (Vector Laboratories, H-1200). All images were obtained using a Zeiss LSM 880 Airyscan confocal microscope (Live Cell Imaging Facility, UT Southwestern Medical Center). Ten random 20x magnification fields were photographed from each section from four animals in each group. The frequency of GFP+ and perilipin+ adipocytes from each image was manually quantified under blinded conditions. Statistical differences between groups were determined using the Student’s t-test and statistical significance was defined as p < 0.05.
Immunohistochemistry.
Briefly, paraffin sections were deparaffinized in xylene and rehydrated sequentially in 100%, 95%, 80%, 70%, and 50% ethanol prepared in ddH2O. Antigen retrieval was performed by boiling the sections in 10 mM citrate pH 6.0 for 10 minutes. After cooling to room temperature for 1 hour, the sections were incubated in 3% H2O2 in methanol for 12 minutes. After washing with PBS, the sections were blocked with 5% normal goat serum (Thermo Fisher Scientific, 50062Z) in PBS at room temperature for 1 hour and then incubated with primary antibody Phospho-histone H2B Ser36 (1:500; ECM Biosciences, HP4331) overnight at 4 °C in PBS containing 5% normal goat serum. After washing with PBS, the sections were incubated with biotin-conjugated goat anti-rabbit secondary antibody (Invitrogen, 65–6140) in PBS containing 5% normal goat serum at room temperature for 1 hour. The sections were then incubated with Vectastain ABC reagent (Vector Laboratories, PK-7200) for 30 minutes. The peroxidase was then developed using DAB reagent (Abcam, ab64238). After counterstaining with hematoxylin solution (Sigma, HHS16), the sections were dehydrated and mounted with Permount Mounting Medium (Fisher Scientific, SP15).
Mouse body weight and composition
Eight-week-old male Parp1 preadipocyte-specific deletion (Parp1loxp/loxp) and control (Parp1+/+) Mural Chaser mice were maintained on a standard rodent chow diet or chow diet containing 600 mg/kg doxycycline (Dox) (Envigo RMS, Inc.) for 9 days, and then fed a high-fat/high-sucrose diet (45 kcal% fat and 30 kcal% sucrose; Research Diets, D08112601) for eight weeks. Body weight was monitored weekly. Body composition was measured using an EchoMRI-100 Body Composition Analyzer (UTSW Metabolic Phenotyping Core).
Identification of histone PTMs by mass spectrometry
3T3-L1 cells expressing FLAG-tagged wild-type or E35A mutant histone H2B were seeded at ~5×106 cells per 15 cm diameter plate and cultured as described above. Nucleosome immunoprecipitation was performed as described above. The immunoprecipitated material was electrophoresed on a precast SDS-PAGE gel and the regions containing histone proteins were excised from the gel. The gel pieces were then reduced and alkylated with DTT (20 mM) and iodoacetamide (27.5 mM). A 0.01 μg/μL solution of trypsin in 50 mM triethylammonium bicarbonate (TEAB) was added to completely cover the gel slides and then digested overnight. Following solid-phase extraction cleanup with an Oasis HLB μelution plate (Waters), the resulting peptides were reconstituted in 10 μL of 2% (v/v) acetonitrile (ACN) and 0.1% trifluoroacetic acid in water. Five μL of the solution was injected onto an Orbitrap Fusion Lumos mass spectrometer (Thermo Electron) coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system (Dionex). The samples were injected onto a 75 μm i.d., 75-cm long EasySpray column (Thermo), and eluted with a gradient from 1–28% buffer B over 90 minutes. Buffer A contained 2% (v/v) ACN and 0.1% formic acid in water, and buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water. The mass spectrometer operated in positive ion mode with a source voltage of 1.5–2.4 kV and an ion transfer tube temperature of 275 °C. MS scans were acquired at 120,000 resolution in the Orbitrap and up to 10 MS/MS spectra were obtained in the ion trap for each full spectrum acquired using higher-energy collisional dissociation (HCD) for ions with charges 2–7. Dynamic exclusion was set for 25 s after an ion was selected for fragmentation. Raw MS data files were analyzed using Proteome Discoverer v2.2 (Thermo), with peptide identification performed using Sequest HT searching against the Mus musculus database from UniProt. Fragment and precursor tolerances of 10 ppm and 0.6 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification, and oxidation of Met, acetylation of Lys, mono, di, and trimethylation of Lys and Arg, phosphorylation of Ser, Thr, and Tyr, and ubiquitination of Lys were set as variable modifications. The false-discovery rate (FDR) cutoff was 1% for all proteins and peptides.
QUANTIFICATION AND STATISTICAL ANALYSIS
All sequencing-based genomic experiments were performed a minimum of two times with independent biological samples. Statistical analyses for the genomic experiments were performed using standard genomic statistical tests as described above. All gene specific qPCR-based experiments were performed a minimum of three times with independent biological samples. All Western blotting experiments with quantification were performed a minimum of three times with independent biological samples and analyzed by Image Lab 6.0. Statistical analyses were performed using GraphPad Prism 7. All tests and p-values are provided in the corresponding figures or figure legends. In all figures, the p values are shown as: *, p<0.05; **, p<0.01; ***, p<0.001, ****, p<0.0001.
KEY RESOURCES TABLE
The Key Resources Table is included with the Supplemental Materials.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PARP-1 | Active Motif | Cat# 39559 RRID: AB_2793257 |
| Anti-poly-ADP-ribose binding reagent | Millipore | Cat# MABE1031 RRID: AB_2665467 |
| Phospho-histone H2B Ser36 | ECM Biosciences | Cat# HP4331 |
| Phospho-histone H2B Ser32 | Invitrogen | Cat# PA5–40148 RRID: AB_ 2608946 |
| Histone H2B | Abcam | Cat# ab1790 RRID: AB_302612 |
| FLAG | Sigma-Aldrich | Cat# F3165 RRID: AB_259529 |
| HPF1 | Atlas | Cat# HPA043467 RRID: AB_10793949 |
| PARG | Millipore | Cat# MABS61 RRID: AB_10806473 |
| H3K27me3 | Active Motif | Cat# 39055 RRID: AB_ 2561020 |
| Phospho-AMPKα Thr172 | Cell Signaling | Cat# 2531 RRID: AB_330330 |
| AMPKα | Cell Signaling | Cat# 2532 RRID: AB_330331 |
| Phospho-p70 S6 Kinase Thr389 | Cell Signaling | Cat# 9205 RRID: AB_330944 |
| p70 S6 Kinase | Cell Signaling | Cat# 9202 RRID: AB_331676 |
| GFP | Abcam | Cat# ab13970 RRID: AB_300798 |
| Perilipin | Fitzgerald | Cat# 20R-PP004 RRID: AB_1288416 |
| SNRP70 | Abcam | Cat# ab51266 RRID: AB_882630 |
| β-actin | Cell Signaling | Cat# 4967 RRID: AB_330288 |
| Goat anti-rabbit HRP-conjugated IgG | ThermoFisher | Cat# 31460 RRID: AB_228341 |
| Goat anti-mouse HRP-conjugated IgG | ThermoFisher | Cat# 31430 RRID: AB_10960845 |
| Alexa fluor 488-conjugated goat anti-chicken IgG | Invitrogen | Cat# 3 A-11039 RRID: AB_ 2534096 |
| Alexa fluor 647-conjugated goat anti-guinea pig IgG | Invitrogen | Cat# A-21450 RRID: AB_ 2735091 |
| Alexa fluor 405-conjugated goat anti-rabbit | Abcam | Cat# ab175654 |
| Rabbit IgG | ThermoFisher | Cat# 10500C RRID: AB_2532981 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Histone H2B peptides, wild-type and mutants | Elim Biopharmaceuticals | Peptide ID 8015, 7959, 9351, and 9936 |
| Recombinant human AMPK | Abcam | Cat# ab79803 |
| Mouse Flag-tagged PARP-1, wild-type and mutant | This study | N/A |
| Human Flag-tagged PARP-1 | This study | N/A |
| Mouse His-tagged NMNAT-1, wild-type and mutant | This study | N/A |
| Human His-tagged NMNAT-1, wild-type and mutant | This study | N/A |
| Mouse His-tagged HPF1 | This study | N/A |
| Critical Commercial Assays | ||
| EZ-10 DNAaway RNA Kit | Bio Basic | Cat# BS88133 |
| RNA Screen Tape | Agilent | Cat# 5067–5578 |
| Deposited Data | ||
| 3T3-L1 RNA-seq and ChIP-seq data sets | This study | GEO: GSE136055 |
| Experimental Models: Cell Lines | ||
| 3T3-L1 | ATCC | Cat# CL-173 |
| MDA-MB-436 | ATCC | Cat# HTB-130 |
| 293T | ATCC | Cat# CRL-3216 |
| Recombinant DNA | ||
| pLKO.1-PARP-1 shRNA | This study | N/A |
| pLKO.1-NMNAT-1 shRNA | This study | N/A |
| pINDUCER20-FLAG-PARP-1, wild-type and mutant | This study | N/A |
| pINDUCER20-FLAG- NMNAT-1, wild-type and mutant | This study | N/A |
| pINDUCER20-FLAG-H2B, wild-type and mutants | This study | N/A |
| pINDUCER20-GFP | This study | N/A |
| pCDH-FLAG-H2B, wild-type and mutants | This study | N/A |
| pET19b-His-NMNAT-1, wild-type and mutants | This study | N/A |
| pET19b-His-HPF1 | This study | N/A |
| pcDNA3.1 Snora7a, Snord16a, Snord22, and Snora64 | This study | N/A |
| pcDNA3.1 SNORA15, SNORA37, and SNORA65 | This study | N/A |
| LentiCRISPR v2 sgRNAs for Snora7a, Snord16a, Snord22, Snora64, and non-targeting sgRNA | This study | N/A |
| pLKO.1 | Addgene | Plasmid no. 10878 |
| pINDUCER20 | Addgene | Plasmid no. 44012 |
| pCDH-EF1α-MCS-IRES-Puro | System Biosciences | Cat# CD532A-2 |
| pET19b | EMD Millipore | Cat# 69677 |
| LentiCRISPR v2 | Addgene | Plasmid no. 52961 |
| pCMV-VSV-G | Addgene | Plasmid no. 8454 |
| pAdVAntage | Promega | Cat# TB207 |
| psPAX2 | Addgene | Plasmid no. 12260 |
| Sequence-Based Reagents | ||
| Primers for RT-qPCR | See STAR Methods | N/A |
| Primers for ChIP-qPCR | See STAR Methods | N/A |
| Primers for molecular cloning | See STAR Methods | N/A |
| Software and Algorithms | ||
| FastQC | Babraham Bioinformatics | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Tophat | Trapnell et al., 2009 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks | Trapnell et al., 2014 | http://cole-trapnell-lab.github.io/cufflinks/ |
| DAVID Bioinformatics Resources | LHRI | https://david.ncifcrf.gov/content.jsp?file=citation.htm |
Highlights:
Glu35 on histone H2B is a key site of PARylation and regulation during adipogenesis.
NMNAT-1 promotes snoRNA-activated PARP-1 Glu/Asp PARylation activity.
H2B-Glu35 PARylation inhibits H2B-Ser36 phosphorylation to control adipogenesis.
PARP-1 controls adipogenesis and body weight by regulating adipocyte precursors.
ACKNOWLEDGEMENTS
The authors would like to thank the following: (1) members of the Kraus lab for continued input and feedback on this project, (2) Laura Banaszinski, Rebecca Gupte, Sridevi Challa, and Daeseok Kim for comments and feedback on this manuscript, (3) Tulip Nandu for assisting with the preliminary mass spec data analysis, (4) Andrew Kelleher for assisting with the IHC from mouse tissues, (5) Glen Liszczak for the nucleosomes used in the in vitro PARylation assays, (6) the UT Southwestern Proteomics Core Facility under the direction of Andrew Lemoff, (7) the UT Southwestern Live Cell Imaging Core Facility under the direction of Kate Luby-Phelps, (8) the UT Southwestern Next Generation Sequencing Core under the direction of Ralf Kittler, and (9) the UT Southwestern Histo Pathology Core. This work was supported by a grant from the NIH/NIDDK (R01 DK069710) and funds from the Cecil H. and Ida Green Center for Reproductive Biology Sciences Endowment to W.L.K.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
W.L.K. is a founder and consultant for Ribon Therapeutics, Inc. He is also a co-holder of U.S. Patent 9,599,606 covering the ADP-ribose detection reagents used herein, which have been licensed to and is sold by EMD Millipore.
SUPPLEMENTAL INFORMATION
Supplemental Information includes: (1) a supplemental data document (one table and seven figures) provided as a pdf file and (2) a supplemental table provided as an Excel file.
REFERENCES
- Asher G, Reinke H, Altmeyer M, Gutierrez-Arcelus M, Hottiger MO, and Schibler U (2010). Poly(ADP-ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell 142, 943–953. [DOI] [PubMed] [Google Scholar]
- Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, et al. (2011). PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab 13, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett E, Bonfiglio JJ, Prokhorova E, Colby T, Zobel F, Ahel I, and Matic I (2018). Interplay of histone marks with serine ADP-ribosylation. Cell Rep 24, 3488–3502 e3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilan V, Leutert M, Nanni P, Panse C, and Hottiger MO (2017). Combining higher-energy collision dissociation and electron-transfer/higher-energy collision dissociation fragmentation in a product-dependent manner confidently assigns proteomewide ADP-ribose acceptor sites. Anal Chem 89, 1523–1530. [DOI] [PubMed] [Google Scholar]
- Bonfiglio JJ, Fontana P, Zhang Q, Colby T, Gibbs-Seymour I, Atanassov I, Bartlett E, Zaja R, Ahel I, and Matic I (2017). Serine ADP-ribosylation depends on HPF1. Mol Cell 65, 932–940 e936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, and Berger SL (2010). Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science 329, 1201–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzio LO, Riquelme PT, and Koide SS (1979). ADP ribosylation of rat liver nucleosomal core histones. J Biol Chem 254, 3029–3037. [PubMed] [Google Scholar]
- D’Angelo I, Raffaelli N, Dabusti V, Lorenzi T, Magni G, and Rizzi M (2000). Structure of nicotinamide mononucleotide adenylyltransferase: a key enzyme in NAD(+) biosynthesis. Structure 8, 993–1004. [DOI] [PubMed] [Google Scholar]
- Daniels CM, Ong SE, and Leung AK (2015). The promise of proteomics for the study of ADP-ribosylation. Mol Cell 58, 911–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, et al. (1997). Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A 94, 7303–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devalaraja-Narashimha K, and Padanilam BJ (2010). PARP1 deficiency exacerbates diet-induced obesity in mice. The Journal of endocrinology 205, 243–252. [DOI] [PubMed] [Google Scholar]
- Erener S, Hesse M, Kostadinova R, and Hottiger MO (2012a). Poly(ADP-ribose)polymerase-1 (PARP1) controls adipogenic gene expression and adipocyte function. Mol Endocrinol 26, 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erener S, Mirsaidi A, Hesse M, Tiaden AN, Ellingsgaard H, Kostadinova R, Donath MY, Richards PJ, and Hottiger MO (2012b). ARTD1 deletion causes increased hepatic lipid accumulation in mice fed a high-fat diet and impairs adipocyte function and differentiation. FASEB J 26, 2631–2638. [DOI] [PubMed] [Google Scholar]
- Fontana P, Bonfiglio JJ, Palazzo L, Bartlett E, Matic I, and Ahel I (2017). Serine ADP-ribosylation reversal by the hydrolase ARH3. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzese E, Centonze S, Diana A, Carlino F, Guerrera LP, Di Napoli M, De Vita F, Pignata S, Ciardiello F, and Orditura M (2019). PARP inhibitors in ovarian cancer. Cancer Treat Rev 73, 1–9. [DOI] [PubMed] [Google Scholar]
- Garavaglia S, D’Angelo I, Emanuelli M, Carnevali F, Pierella F, Magni G, and Rizzi M (2002). Structure of human NMN adenylyltransferase. A key nuclear enzyme for NAD homeostasis. J Biol Chem 277, 8524–8530. [DOI] [PubMed] [Google Scholar]
- Gibbs-Seymour I, Fontana P, Rack JGM, and Ahel I (2016). HPF1/C4orf27 is a PARP-1-interacting protein that regulates PARP-1 ADP-ribosylation activity. Mol Cell 62, 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Conrad LB, Huang D, and Kraus WL (2017). Generation and characterization of recombinant antibody-like ADP-ribose binding proteins. Biochemistry 56, 6305–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, and Kraus WL (2012). New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 13, 411–424. [DOI] [PubMed] [Google Scholar]
- Gibson BA, and Kraus WL (2017). Identification of protein substrates of specific PARP enzymes using analog-sensitive PARP mutants and a “clickable” NAD(+) analog. Methods Mol Biol 1608, 111–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H, Schwede F, Yu Y, and Kraus WL (2016). Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science 353, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green H, and Kehinde O (1975). An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell 5, 19–27. [DOI] [PubMed] [Google Scholar]
- Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, and Brunet A (2007). The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem 282, 30107–30119. [DOI] [PubMed] [Google Scholar]
- Gupte R, Liu Z, and Kraus WL (2017). PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 31, 101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilz H, and Stone P (1976). Poly(ADP-ribose) and ADP-ribosylation of proteins. Rev Physiol Biochem Pharmacol 76, 1–58, 177. [DOI] [PubMed] [Google Scholar]
- Huang D, Kim DS, and Kraus WL (2020). Specific binding of snoRNAs to PARP-1 promotes NAD(+)-dependent catalytic activation. Biochemistry 59, 1559–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram H, Hoelper D, Jain SU, Cantone N, Lundgren SM, Poy F, Allis CD, Cummings R, Bellon S, and Lewis PW (2016). S-adenosyl methionine is necessary for inhibition of the methyltransferase G9a by the lysine 9 to methionine mutation on histone H3. Proc Natl Acad Sci U S A 113, 6182–6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jump DB, Butt TR, and Smulson M (1979). Nuclear protein modification and chromatin substructure. 3. Relationship between poly(adenosine diphosphate) ribosylation and different functional forms of chromatin. Biochemistry 18, 983–990. [DOI] [PubMed] [Google Scholar]
- Kamel D, Gray C, Walia JS, and Kumar V (2018). PARP inhibitor drugs in the treatment of breast, ovarian, prostate and pancreatic cancers: An update of clinical trials. Curr Drug Targets 19, 21–37. [DOI] [PubMed] [Google Scholar]
- Karch KR, Langelier MF, Pascal JM, and Garcia BA (2017). The nucleosomal surface is the main target of histone ADP-ribosylation in response to DNA damage. Mol Biosyst 13, 2660–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Camacho CV, Nagari A, Malladi VS, Challa S, and Kraus WL (2019). Activation of PARP-1 by snoRNAs controls ribosome biogenesis and cell growth via the RNA helicase DDX21. Molecular cell 75, 1270–1285 e1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Mauro S, Gevry N, Lis JT, and Kraus WL (2004). NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell 119, 803–814. [DOI] [PubMed] [Google Scholar]
- Kraus WL, and Lis JT (2003). PARP goes transcription. Cell 113, 677–683. [DOI] [PubMed] [Google Scholar]
- Krishnakumar R, Gamble MJ, Frizzell KM, Berrocal JG, Kininis M, and Kraus WL (2008). Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science 319, 819–821. [DOI] [PubMed] [Google Scholar]
- Krishnakumar R, and Kraus WL (2010). The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell 39, 8–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen SC, Hendriks IA, Lyon D, Jensen LJ, and Nielsen ML (2018). Systems-wide analysis of serine ADP-ribosylation reveals widespread occurrence and site-specific overlap with phosphorylation. Cell Rep 24, 2493–2505 e2494. [DOI] [PubMed] [Google Scholar]
- Lawrence M, Daujat S, and Schneider R (2016). Lateral thinking: How histone modifications regulate gene expression. Trends Genet 32, 42–56. [DOI] [PubMed] [Google Scholar]
- Lehmann M, Pirinen E, Mirsaidi A, Kunze FA, Richards PJ, Auwerx J, and Hottiger MO (2015). ARTD1-induced poly-ADP-ribose formation enhances PPARgamma ligand binding and co-factor exchange. Nucleic acids research 43, 129–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidecker O, Bonfiglio JJ, Colby T, Zhang Q, Atanassov I, Zaja R, Palazzo L, Stockum A, Ahel I, and Matic I (2016). Serine is a new target residue for endogenous ADP-ribosylation on histones. Nat Chem Biol 12, 998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KY, Huang D, and Lee Kraus W (2018). Generating Protein-Linked and Protein-Free Mono-, Oligo-, and Poly(ADP-Ribose) In Vitro. Methods Mol Biol 1813, 91–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, and Kraus WL (2011). A one and a two … expanding roles for poly(ADP-ribose) polymerases in metabolism. Cell Metab 13, 353–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Ryu KW, Kim DS, Nandu T, Medina CJ, Gupte R, Gibson BA, Soccio RE, Yu Y, Gupta RK, et al. (2017). PARP-1 controls the adipogenic transcriptional program by PARylating C/EBPbeta and modulating its transcriptional activity. Mol Cell 65, 260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangerich A, and Altmeyer M (2016). Identifying ADP-ribosylation targets by chemical genetics. Transl Cancer Res 5, S1163–S1166. [Google Scholar]
- Martire S, Gogate AA, Whitmill A, Tafessu A, Nguyen J, Teng YC, Tastemel M, and Banaszynski LA (2019). Phosphorylation of histone H3.3 at serine 31 promotes p300 activity and enhancer acetylation. Nat Genet 51, 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, et al. (2011). The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America 108, 3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minaga T, Romaschin AD, Kirsten E, and Kun E (1979). The in vivo distribution of immunoreactive larger than tetrameric polyadenosine diphosphoribose in histone and nonhistone protein fractions of rat liver. J Biol Chem 254, 9663–9668. [PubMed] [Google Scholar]
- Murakami S, Nagari A, and Kraus WL (2017). Dynamic assembly and activation of estrogen receptor alpha enhancers through coregulator switching. Genes & development 31, 1535–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo L, Leidecker O, Prokhorova E, Dauben H, Matic I, and Ahel I (2018). Serine is the major residue for ADP-ribosylation upon DNA damage. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhimova A, Ura S, Hsu DW, Wang HY, Pears CJ, and Lakin ND (2017). Site-specific ADP-ribosylation of histone H2B in response to DNA double strand breaks. Sci Rep 7, 43750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart SB, and Strahl BD (2014). Interpreting the language of histone and DNA modifications. Biochim Biophys Acta 1839, 627–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KW, Kim DS, and Kraus WL (2015). New facets in the regulation of gene expression by ADP-ribosylation and poly(ADP-ribose) polymerases. Chem Rev 115, 2453–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KW, Nandu T, Kim J, Challa S, DeBerardinis RJ, and Kraus WL (2018). Metabolic regulation of transcription through compartmentalized NAD(+) biosynthesis. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saridakis V, Christendat D, Kimber MS, Dharamsi A, Edwards AM, and Pai EF (2001). Insights into ligand binding and catalysis of a central step in NAD+ synthesis: structures of Methanobacterium thermoautotrophicum NMN adenylyltransferase complexes. J Biol Chem 276, 7225–7232. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suskiewicz MJ, Zobel F, Ogden TEH, Fontana P, Ariza A, Yang JC, Zhu K, Bracken L, Hawthorne WJ, Ahel D, et al. (2020). HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation. Nature 579, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishvanath L, MacPherson KA, Hepler C, Wang QA, Shao M, Spurgin SB, Wang MY, Kusminski CM, Morley TS, and Gupta RK (2016). Pdgfrbeta+ mural preadipocytes contribute to adipocyte hyperplasia induced by high-fat-diet feeding and prolonged cold exposure in adult mice. Cell Metab 23, 350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi SA, Um SH, Lee J, Yoo JH, Bang SY, Park EK, Lee MG, Nam KH, Jeon YJ, Park JW, et al. (2016). S6K1 phosphorylation of H2B mediates EZH2 trimethylation of H3: A determinant of early adipogenesis. Mol Cell 62, 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Berrocal JG, Yao J, DuMond ME, Krishnakumar R, Ruhl DD, Ryu KW, Gamble MJ, and Kraus WL (2012). Regulation of poly(ADP-ribose) polymerase-1-dependent gene expression through promoter-directed recruitment of a nuclear NAD+ synthase. The Journal of biological chemistry 287, 12405–12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Cooper S, and Brockdorff N (2015). The interplay of histone modifications -writers that read. EMBO Rep 16, 1467–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang J, Ding M, and Yu Y (2013). Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat Methods 10, 981–984. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xu C, Gu D, Wu M, Yan B, Xu Z, Wang Y, and Liu H (2017). H/ACA box small nucleolar RNA 7A promotes the self-renewal of human umbilical cord mesenchymal stem cells. Stem Cells 35, 222–235. [DOI] [PubMed] [Google Scholar]
- Zhong S, Joung JG, Zheng Y, Chen YR, Liu B, Shao Y, Xiang JZ, Fei Z, and Giovannoni JJ (2011). High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb Protoc 2011, 940–949. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq and ChIP-seq data sets generated specifically for this study can be accessed from the NCBI’s Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo/) using the superseries accession number GSE136055. The new mass spec data sets generated for these studies are available as supplemental data provided with this manuscript.