

Abstract
Chaperone networks are required for the shearing and generation of transmissible propagons from pre‐existing prion aggregates. However, other cellular networks needed for maintaining yeast prions are largely uncharacterized. Here, we establish a novel role for actin networks in prion maintenance. The [PIN +] prion, also known as [RNQ +], exists as stable variants dependent upon the chaperone machinery for the transmission of propagons to daughter cells during cell division and cytoplasmic transfer. Loss of the Hsp104 molecular chaperone leads to the growth of prion particles until they are too large to be transmitted. Here, we isolated a unique [PIN +] variant, which is unstable in actin mutants. This prion loss is observed over many generations, and coincides with the detection of both high molecular weight species of Rnq1 and large visible aggregates that are asymmetrically retained during cell division. Our data suggest that the irregular actin networks found in these mutants may influence propagon number by slowly permitting aggregate growth over time, resulting in the generation of nontransmissible large aggregates. Thus, we show the potential contribution of cytoskeletal networks in the transmission of prion propagons, which parallels models that have been proposed for cell‐to‐cell transmission of small amyloids in neurodegenerative protein aggregation diseases.
Keywords: [PIN+], [RNQ+], actin cytoskeleton, protein aggregates, Rnq1, yeast prion variant
This study establishes a novel role for actin networks in prion maintenance. A unique variant of the [PIN +] prion is unstable in actin mutants carrying mutations within a binding site for Fimbrin, a conserved actin bundling protein. Our data suggest that irregular actin networks can influence propagon number by slowly permitting aggregate growth over time, resulting in the generation of non‐transmissible propagons.
1. INTRODUCTION
Several neurodegenerative and systemic disorders such as prion disease, Huntington's disease, and Transthyretin amyloidosis are considered protein misfolding disorders. In each of these disorders, one specific protein population can misfold and assemble into large inclusions that are considered hallmarks of these diseases. In many cases, the misfolded protein is associated with different types of protein aggregates. There are small oligomeric complexes and high molecular weight species that are commonly observed as fibrils or inclusions. The small oligomeric complexes tend to be toxic, possibly due to their ability to diffuse freely (Lambert et al., 1998; Glabe, 2006; Winner et al., 2011), whereas large fibrils or high molecular weight aggregates possibly sequester smaller toxic oligomers dampening toxicity (Arrasate et al., 2004).
While a single disease is associated with the misfolding and aggregation of a specific protein, a range of symptoms and disease on‐set are observed in patients (reviewed in Igel‐Egalon et al., 2018). The difference in disease presentation is associated with different physical protein conformations, commonly referred to as strains in mammalian systems. In the case of prion disease, different protein conformations among strains exhibit distinct glycosylation patterns, protease resistance, and protein sedimentation profiles (Bessen and Marsh, 1992; Collinge et al., 1996; Parchi et al., 1996; Khalili‐Shirazi et al., 2005). These differences account for the differences in clinical symptoms and incubation times among prion disease patients, and in relative abilities to jump species barriers.
Recently the link between aggregate size and strain conformation has become clearer. Work by Morales and coworkers (2016) has found that a certain prion strain associated with longer incubation times also had extremely large aggregates as shown by sedimentation studies. This correlation suggests the proportion of protein associated with large molecular weight species could play an important role in phenotypes associated with specific strains. Therefore, it can be envisioned that shifting protein populations from the oligomeric state to large aggregates can lead to less detrimental phenotypes. However, the cellular mechanisms involved in maintaining these different protein populations are poorly understood.
The study of prions in yeast has greatly contributed to our understanding of protein aggregate populations and prion strains. In yeast, several proteins have been shown to form amyloids called prions (Cox, 1965; Lacroute, 1971; Wickner, 1994; Derkatch et al., 1997; Sondheimer and Lindquist, 2000; Volkov et al., 2002; Du et al., 2008; Alberti et al., 2009; Patel et al., 2009; Rogoza et al., 2010; Halfmann et al., 2012; Suzuki et al., 2012). Similar to human amyloids, these yeast proteins can misfold, and form both small oligomers and high molecular weight aggregates. The transmission of most yeast prions to daughters is dependent upon chaperone networks. Hsp104, along with Hsp70 and Hsp40 chaperones, works to shear larger aggregates into smaller transmissible seeds or propagons (Shorter and Lindquist, 2004, 2008; Aron et al., 2007; Higurashi et al., 2008; Winkler et al., 2012).
Yeast prions can also adopt alternative protein conformations with distinct biochemical characteristics and phenotypes (Wickner, 1994; Glover et al., 1997; King et al., 1997; Sondheimer and Lindquist, 2000; Schlumpberger et al., 2001; Bradley et al., 2002; Bradley and Liebman, 2003). However, due to nomenclature conventions within the yeast field, prion strains are often referred to as variants in yeast. Two yeast prions, [PSI +] and [PIN +], have contributed most substantively to advancing our understanding of prion variants. [PSI +] is the misfolded aggregating form of the Sup35 protein, and the [PIN +] prion (also referred to as the [RNQ+] prion) is the misfolded aggregating form of the Rnq1 protein. In the case of [PIN +], distinct variants have a range of visual fluorescent aggregate phenotypes (Bradley and Liebman, 2003; Huang et al., 2013), and differential abilities to promote the formation of the [PSI +] prion (Derkatch et al., 1997; Derkatch et al., 2001; Osherovich and Weissman, 2001; Bradley et al., 2002). It has been shown that during the prion formation process, a single cell can harbor more than one conformation subtype. The transmission of specific subtypes to daughter cells leads to the propagation of certain variants, some of which are more stable than others (Sharma and Liebman, 2012; Huang et al., 2013). While the cellular mechanisms that lead to the destabilization of specific subtypes are not well defined, some mechanisms have been uncovered via treatment with external stresses. For example, transient heat shock leads to loss of the weak [PSI +] variant from cell populations (Bailleul et al., 1999; Wegrzyn et al., 2001; Klaips et al., 2014) as a result of asymmetrical retention of Hsp104 (Klaips et al., 2014). Despite our understanding of how molecular chaperones influence prion stability, other cellular mechanisms involved in prion propagation are poorly understood.
In this study, we exploited a unique variant of the [PIN +] prion to show that actin cytoskeletal networks influence prion stability. This prion variant, while stable in different wildtype genetic backgrounds, is lost over many generations in actin point mutants, which have irregular actin networks. This instability over time correlates with the observation of large transient fluorescent foci and the detection of higher molecular weight aggregates over time. Taken together, results suggest that normal actin cytoskeletal networks play an important role in maintaining a population of small propagons that are transmissible. Our study uncovers a role for the actin cytoskeletal network in the maintenance of prion transmission over many generations.
2. RESULTS
2.1. High [PIN +] is mitotically stable, routinely cytoduced with high fidelity, but has aggregation profiles influenced by genetic background
In order to understand what influences the stability of [PIN +], we needed to establish a baseline of the frequency of prion loss and transmission fidelity. We confirmed that several variants of [PIN +], including high [PIN +], were maintained over several hundred generations (Figure S1). Next, we used a process called cytoduction, which transfers cytoplasmic, but not nuclear, material from a donor to a recipient cell (see Experimental Procedures; Conde and Fink, 1976). The donor strain was wildtype C10B‐H49 high [PIN +] (represented by cell a, Figure 1a), and the recipient strain was wildtype BY4741 [pin −]. Throughout the remainder of this manuscript, C10B‐H49 and BY4741 will be used to refer to the wildtype genotype of their respective strain backgrounds, whereas any variation from this genotype will be indicated by the strain background preceded by a mutant allele designation (Table S1). Analysis of cell populations containing high [PIN +] in the C10B‐H49 genetic background showed that the majority of cells contain multiple Rnq1‐GFP fluorescent foci that are highly mobile. This phenotype is commonly called multiple‐dot (m.d.) [PIN +] (Figure 1a,b; cell a; Figure S2; Bradley and Liebman, 2003). It should be noted that terms such as “high” versus “m.d.” both refer to the identical [PIN +] variant, and the different names reflect the historical characterization of [PIN +] variants by their relative ability to promote formation of the [PSI +] prion (such as low or high [PIN+]; Bradley et al., 2002) versus their visual fluorescent aggregate phenotypes (such as single or multiple dots; Bradley and Liebman, 2003).
FIGURE 1.
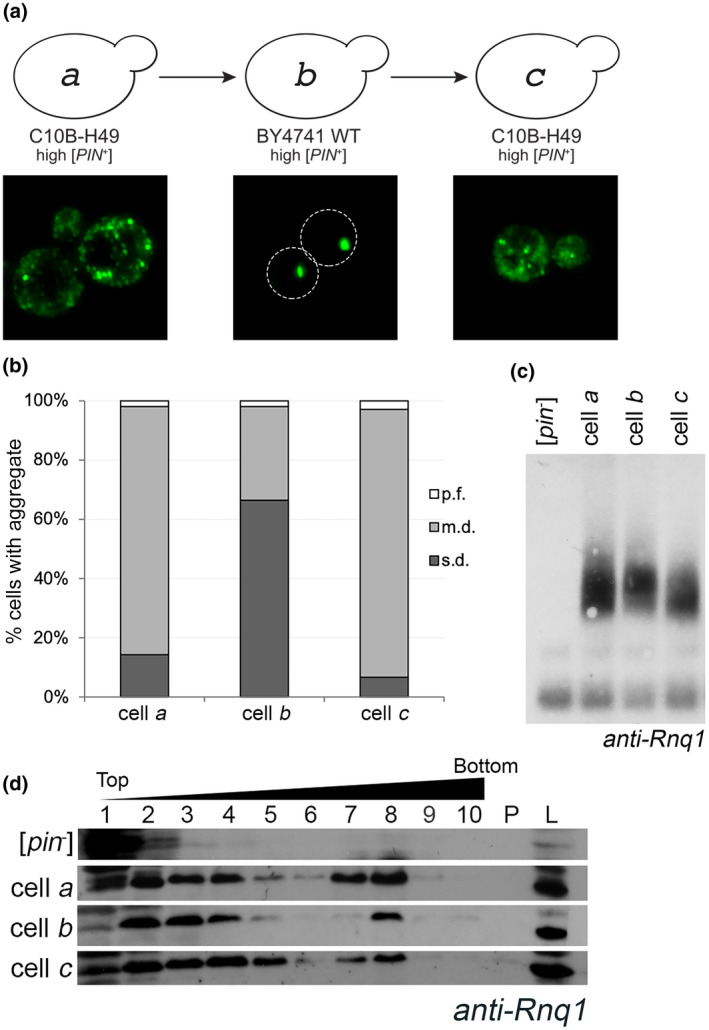
The aggregation phenotype of high [PIN +] in the C10B‐H49 background is maintained after it is transferred through the BY4741 genetic background. (a) Diagram of cytoduction, with representative fluorescent micrographs below. All cytoductions involved transfer of prions from a [PIN +] donor to a [pin −] recipient. Diagram and fluorescent images depict the prion variant after cytoduction. A high [PIN +] C10B‐H49 donor (represented by cell a) was used to introduce the prion into a [pin −] BY4741 background. The resulting cytoductant is represented by cell b. Cell b (high [PIN +] BY4741) was used to introduce the prion into a [pin −] version of C10B‐H49. The cytoductant is represented by cell c. Transient overexpression of Rnq1‐GFP allows for the detection of aggregates within the cytoductants. Representative images are deconvolved maximum image projections of several z‐stacks. (b) Cells containing multiple dots (m.d.), single dot (s.d.), or petite foci (p.f.) were assessed in four independent cultures from each strain. A minimum of 300 cells per culture was scored and means of each population are shown. The number of m.d.‐containing cells in BY4741 recipient strains were significantly different compared to C10B‐H49 donor and recipient cells, as determined by Chi‐square test of independence (p < .0001). (c) Fresh lysates from the indicated strains, including C10B‐H49 [pin −] in the first lane, were run on an SDD‐AGE and analyzed by Western blot using anti‐Rnq1 antibody. Due to several cross‐reacting bands between 20 and 120 kDa using this polyclonal antibody, SDD‐AGE is able to confirm presence or absence of higher molecular weight oligomers, but does not have adequate resolution to affirm presence of the Rnq1 monomer. More definitive evidence for the presence of Rnq1 monomer in [pin −] strains is provided via SDS‐PAGE (Figure S5). (d) Lysates from the indicated strains, including 74D‐694 [pin −] as the top row, were loaded onto discontinuous sucrose (10%, 40%, and 60% sucrose) gradients and fractionated. 10 fractions were collected. Fractions 7 and 8 usually are the interphase between the 40% and 60% sucrose. Protein pelleted at the bottom of the gradient (P) and whole‐cell lysates (L) are shown [Colour figure can be viewed at wileyonlinelibrary.com]
Upon introducing high [PIN +] from the C10B‐H49 donor strain into a BY4741 [pin −] recipient by cytoduction, we observed changes in Rnq1‐GFP aggregate phenotype. Within the BY4741‐cytoduced population, 65.8% of the cells exhibited a single static fluorescent focus, commonly called single dot (s.d.) [PIN +] (represented by cell b, Figure 1a,b; Figure S3). Analysis of replicate cultures confirmed these differences in fluorescent aggregate phenotypes. Bradley and Liebman (2003) reported that a similar cytoduction maintained a multiple‐dot phenotype. However, authors did not appear to quantify the number of cells within a population that had multiple dots versus any other aggregate phenotype. We found quantifications of populations extremely important, designating the population phenotype as m.d. or s.d. [PIN +] according to that which was present in the majority (>50%) of cells. It was also observed that a small subset of cells in both backgrounds exhibited previously described petite foci (p.f.), which are very small foci detectable at 630× magnification (Huang et al., 2013). The systematic characterization of cell populations within a culture, with respect to these aggregate phenotypes, provided a general “aggregate profile” for each genetic background (Figure 1b).
To confirm that this altered aggregation profile did not represent a permanent change of the prion, we cytoduced the prion from BY4741 back into a [pin −] version of C10B‐H49. The C10B‐H49 cytoductants had an aggregation profile comparable to the original C10B‐H49 donor strain (represented by cells c and a, respectively, Figure 1a,b). In fact, comparison of time‐lapse movies of the predominant cell type in these three strains showed that both C10B‐H49 cells a and c had similar highly mobile m.d. aggregates, unlike the predominant cell type in BY4741 cell b with static s.d. aggregates (Figures S2–S4). These data suggest that while the aggregation profile is different in the BY4741 background, the profile reverted back to the m.d. aggregation phenotype. Presence of the [PIN +] prion was further validated biochemically, by the absence of an Rnq1 band in unboiled lysates (Figure S5), and by the detection of similarly sized SDS‐resistant oligomers and large molecular weight species (Figure 1c,d). [PIN +] was also detected functionally by the ability to promote [PSI +] formation (Figure S6). Taken together, these data are consistent with prior observations demonstrating that the process of cytoduction does not typically alter prion variants (Bateman and Wickner, 2013). Thus, throughout the remainder of this manuscript, we will refer to the [PIN +] variant in the BY4741 and C10B‐H49 genetic backgrounds (represented by cells a‐c) as high [PIN +], but refer to these visual differences as the s.d. and m.d. aggregate profiles or phenotypes.
2.2. A unique [PIN +] variant: µdot [PIN +]
We serendipitously isolated a unique [PIN +] variant in a separate study looking at factors influencing prion formation. We previously found that decreased [PSI +] formation was observed in several single gene knockout strains that influenced actin organization, including deletion of the Fimbrin actin bundling homolog SAC6 (Manogaran et al., 2011). As part of this study, we cytoduced high [PIN +] into four [pin −] recipient temperature‐sensitive actin mutants that had been constructed in the BY4741 genetic background, including act1‐120, which contains mutations within the Sac6‐binding region of actin (Figure S7a; Holtzman et al., 1994; Honts et al., 1994; Amberg et al., 1995; Miao et al., 2016). All actin mutants showed severe growth defects at restrictive temperature, limited growth defects at permissive temperature, and larger cell size distribution at permissive temperatures (Figure S7b,c; Wertman et al., 1992). While only two of the mutants, act1‐122 and act1‐120, exhibited either depolarized or partially polarized actin patches within a majority of cells (Figure S7d), all four actin mutants had comparable Rnq1‐GFP aggregate profiles associated with library [PIN +], a [PIN +] variant endogenously found in BY4741 strains (Figure S7e; Manogaran et al., 2010).
When high [PIN +] from C10B‐H49 strains was cytoduced into [pin −] recipient actin mutants, the act1‐120, act1‐101, and act1‐129 mutants all displayed the s.d. aggregate profile (Figure 2a, represented by cell d, act1 mutant), similar to wildtype BY4741 (cell b). In contrast, act1‐122 BY4741 mutants exhibited an entirely different aggregation phenotype (Figure 2a, cell d; act1‐122). At first glance, the cells appeared to have diffuse cytoplasmic fluorescence (Figure 2b). However, upon closer inspection, the cells contained many small mobile foci, visible by time‐lapse microscopy (Figure S8). The small foci are considerably smaller than the petite foci (p.f.) previously observed by Huang et al. (2013). In this paper, we will call this new aggregation pattern of Rnq1‐GFP “micro dot” (µ.d.).
FIGURE 2.
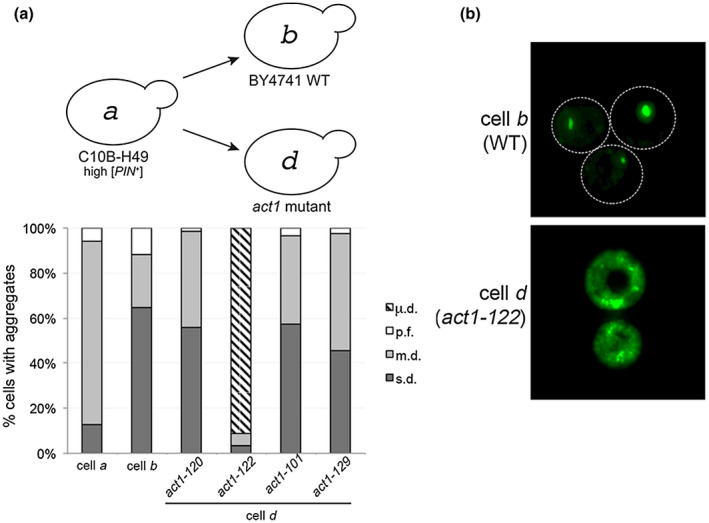
New aggregation phenotype, µ.d., found in act‐122 cytoductant. (a) High [PIN +] was cytoduced from wildtype C10B‐H49 (cell a) into [pin −] BY4741 wildtype or actin mutant recipient strains. The diagram depicts the prion variant after cytoduction. The type of aggregates found in each strain was counted as described in Figure 1b. Each bar represents the mean of four independent cultures and a minimum of 300 cells per culture. (b) Fluorescent micrographs of [PIN +] in wildtype BY4741 and act1‐122 BY4741 strains. While representative images are deconvolved maximum image projections of several z‐stacks, additional contrast was added to cell d to demonstrate the visual differences between both strains (see Figure S8 for raw time‐lapse images) [Colour figure can be viewed at wileyonlinelibrary.com]
We expected that if [PIN +] were cytoduced from act1‐122 BY4741 back into a wildtype C10B‐H49 [pin −] recipient strain, the visual aggregates would revert back to the m.d. aggregation profile of high [PIN +] in that background, similar to our previous observations in Figure 1. However, the C10B‐H49 cytoductants retained the µ.d. aggregation phenotype (represented by cell e, Figure 3a,b). Similar to the µ.d. aggregates in the act1‐122 BY4741 mutants, initial inspection suggested that C10B‐H49 cells exhibited diffuse fluorescence (Figure 3a,b, cell e), but time‐lapse microscopy showed that these cells also contained many small mobile foci (Figure S9). Additionally, population analysis reveals that the µ.d. aggregate phenotype is present in a relatively comparable proportion of cells in act1‐122 BY4741 and these C10B‐H49 strains (represented by cells d and e; Figure 3c). The [PIN +] status of these strains was verified by the presence of SDS‐resistant oligomers (Figure 3d).
FIGURE 3.
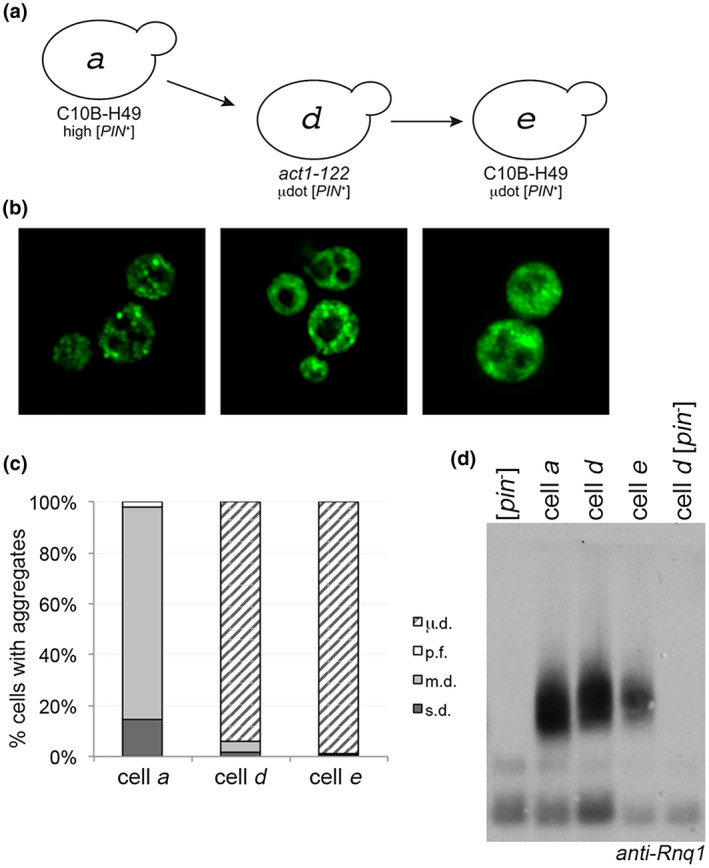
Microdot phenotype does not revert to multiple dots when introduced into the C10B‐H49 background. (a) µdot [PIN +] in the act1‐122 strain (cell d) was cytoduced into the C10B‐H49 background (cell e). (b) Fluorescent images of cells a, d, and e. (c) Similar to Figures 1b and 2c, each bar represents the mean of at least four independent cultures and a minimum of 300 cells per culture. (d) Western blot of lysates of the indicated strains, including wildtype BY4741 [pin −] in the first lane, run on SDD‐AGE gels. Anti‐Rnq1 antibody was used to detect Rnq1 oligomers [Colour figure can be viewed at wileyonlinelibrary.com]
[PIN +] status was also verified functionally by the ability of all strains to promote [PSI +] formation (Figure S6c). Notably, induction frequencies of strains within the same C10B‐H49 genetic background, but with m.d. (cell c; S6C) versus µ.d. (cell e) aggregation phenotypes, are significantly different. Moreover, the ability to retain the µ.d. aggregation profile between the act1‐122 BY4741 and the wildtype C10B‐H49 strains (cells d to e) is in stark contrast to the cytoduction of high [PIN +] from the wildtype BY4741 genetic background into a C10B‐H49 [pin −] recipient strain (Figure 1a,b, cell b to cell c), where reversion from the s.d. to m.d. aggregation profile was observed (Figure 1b). It should also be noted that DNA sequencing showed that RNQ1 gene sequences were identical between strains, eliminating any concerns that the different aggregation profiles were due to RNQ1 polymorphisms (data not shown). Collectively, these data demonstrate that the µ.d. aggregation phenotype is associated with a prion variant that is unique from high [PIN +], and to distinguish the prion variant from the aggregation phenotype, we refer to the variant as µdot [PIN +].
We considered the possibility that the act1‐122 mutation would reproducibly change high [PIN +] into µdot [PIN +]. Therefore, we replicated the original cytoduction experiment, transferring high [PIN +] from C10B‐H49 into act1‐122 BY4741 [pin −] recipient strains (e.g., repeating cell a to cell d cytoductions). Analysis of several independent cytoductions showed that act1‐122 BY4741 strains can acquire aggregation profiles that were phenotypically indistinguishable from wildtype BY4741 cytoduced with high [PIN +] (Figure S10). Therefore, our data suggest that the µdot [PIN +] variant arose due to a rare event coincident with cytoduction.
2.3. Fluorescent aggregates of µdot [PIN +] exhibit transient larger aggregates
Our initial observations and aggregate profile determinations of µdot [PIN +] revealed presence of a very small subpopulation of cells with intensely bright foci similar to single‐ or multiple‐dot foci in both act1‐122 BY4741 and in C10B‐H49 (Figure 3c, s.d. and m.d. categories for cells d and e), although such cells were slightly less frequent in C10B‐H49 (Figure 3c, cell e). Using 3D time‐lapse microscopy, we followed individual cells of this type from both strains, and observed rapid dispersal of these bright foci to a µ.d. aggregate phenotype (Figure 4a). Prolonged time‐lapse microscopy was performed on multiple cells from four independent cultures of act1‐122 BY4741 µdot [PIN +] strains. These time‐lapse videos show that the larger aggregates coalesce and disperse several times (Figure 4b). The duration in which large aggregates were detected was highly variable, yet the duration often increased with each successive appearance. In addition, passage of these large aggregates to the daughter cells was never observed in any of the four time‐lapse analyses (data not shown).
FIGURE 4.
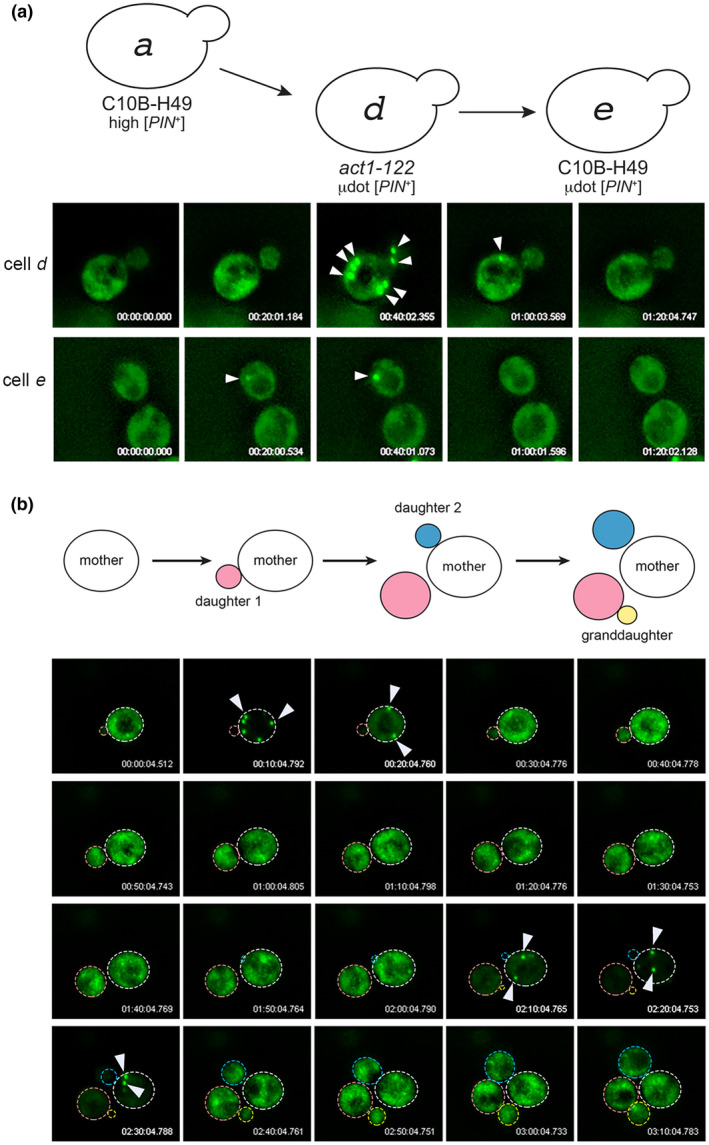
Cells containing µdot [PIN+] show transient large aggregates. (a) 3D time‐lapse imaging of cell d (act1‐122) or cell e (wildtype C10B‐H49) containing µdot [PIN+]. The appearance and disappearance of large aggregates are indicated with arrowheads. (b) A 3D time‐lapse of a single dividing cell shows the appearance and disappearance of large aggregates over several cell divisions. All 3D time‐lapse imaging was done without addition of copper sulfate, such that low Rnq1‐GFP fluorescence reflects trace amounts of copper sulfate in the synthetic complete media (see Experimental Procedures) [Colour figure can be viewed at wileyonlinelibrary.com]
2.4. Actin mutants can render µdot [PIN +] unstable
The cytoduction of high [PIN +] from BY4741 (cell b) into a C10B‐H49 [pin −] recipient strain (cell c) faithfully transmitted the prion (100%, Table 1), whereas our initial efforts to cytoduce µdot [PIN +] from the act1‐122 BY4741 mutant (cell d) into a C10B‐H49 [pin −] recipient strain (cell e) were variable (3.85%–100%,Table 1). Some C10B‐H49 cytoductants faithfully received µdot [PIN +] and other cytoductants were [pin −]. Interestingly, we were able to transmit µdot [PIN +] faithfully through a variation of the cytoduction technique called plasmiduction (see Experimental Procedures; Natsoulis et al., 1994; Vitrenko et al., 2007), where cytoplasmic transfer is monitored through the transfer of a plasmid. We used a plasmid containing RNQ1‐GFP driven by a copper‐inducible promoter to facilitate immediate screening for [PIN +] cytoductants, but copper was not added during the cytoduction procedure. We found that all C10B‐H49 cytoductants possessing the plasmid also contained µdot [PIN +] (Table 1). We suspect that a low level of Rnq1‐GFP gene expression by the copper‐inducible promoter, resulting from low levels of copper found intrinsically in the synthetic media (see Experimental Procedures), may have contributed to maintenance of µdot [PIN +] through plasmiduction.
TABLE 1.
Cytoduction of μdot [PIN +] into C10B‐H49 cells was variable
| Cytoductions | ||
|---|---|---|
| Donor | Recipient | Percent cytoductants that retained [PIN +] |
|
cell b BY4741 high [PIN +] |
cell c C10B‐H49 |
100% (16/16) |
|
cell d BY4741 μdot [PIN +] − |
cell e C10B‐H49 |
95.6% (22/23) |
|
cell d BY4741 μdot [PIN +] |
cell e C10B‐H49 |
100% (23/23) |
|
cell d BY4741 μdot [PIN +] |
cell e C10B‐H49 |
3.85% (1/26) |
|
cell d BY4741 μdot [PIN +] |
cell e C10B‐H49 |
95.6% (22/23) |
| Plasmiductions | ||
|---|---|---|
|
cell b BY4741 high [PIN +] + RNQ1‐GFP |
cell c C10B‐H49 |
100% (32/32) |
|
cell d BY4741 μdot [PIN +] + RNQ1‐GFP |
cell e C10B‐H49 |
100% (8/8) |
|
cell d BY4741 μdot [PIN +] + RNQ1‐GFP |
cell e C10B‐H49 |
100% (8/8) |
|
cell d BY4741 μdot [PIN +] + RNQ1‐GFP |
cell e C10B‐H49 |
100% (8/8) |
|
cell d BY4741 μdot [PIN +] + RNQ1‐GFP |
cell e C10B‐H49 |
100% (8/8) |
Cytoduction or plasmiduction were used to introduce µdot [PIN +] into [pin −] recipients with the C10B‐H49 genetic background. One wildtype control (M362, high [PIN +] cell b) and four act1‐122 strains (M363‐M365, M378; µdot [PIN +] cell d) were used as prion donors [pin −] C10B‐H49 recipient cells to generate cells c and e, respectively (see Experimental Procedures). Individual cytoductants were mated to tester strains containing RNQ1‐GFP plasmids and scored for the presence of aggregates in cytoductant cell populations. The percentage of [PIN +] cytoductants is shown with total number of cytoductants screened in parentheses. Since plasmiductants already contained the RNQ1‐GFP plasmid, plasmiductants were directly scored for the presence of Rnq1‐GFP aggregates in cell populations. Each line represents a single mating between donor and recipient cells.
Based on the variability of µdot [PIN +] transfer through cytoduction, we postulated that µdot [PIN +] in act1‐122 BY4741 strains may be unstable. Therefore, we performed mitotic stability experiments in the absence of the plasmid containing RNQ1‐GFP. The act1‐122 BY4741 µdot [PIN+] strain was subcloned from frozen glycerol stocks on rich media and grown up to 100 generations. Wildtype BY4741 strains containing high [PIN+] were tested in parallel as a control. To check for the presence of [PIN +] after 100 generations, single colonies were picked and mated with a [pin −] tester containing the copper‐inducible RNQ1‐GFP plasmid. Rnq1‐GFP was transiently induced in individual diploid colonies, and each colony was individually scored for the presence of Rnq1‐GFP aggregates within the cell population. We found that all wildtype BY4741 high [PIN +] colonies (cell b) contained [PIN +], even after 100 generations (Figure 5a,b). However, act1‐122 BY4741 µdot [PIN +]‐containing strains (cell d) showed a dramatic decrease in the number of colonies that were [PIN +] by generation 40. Moreover, prion loss seemed to continue as the cells underwent further division (Figure 5b).
FIGURE 5.
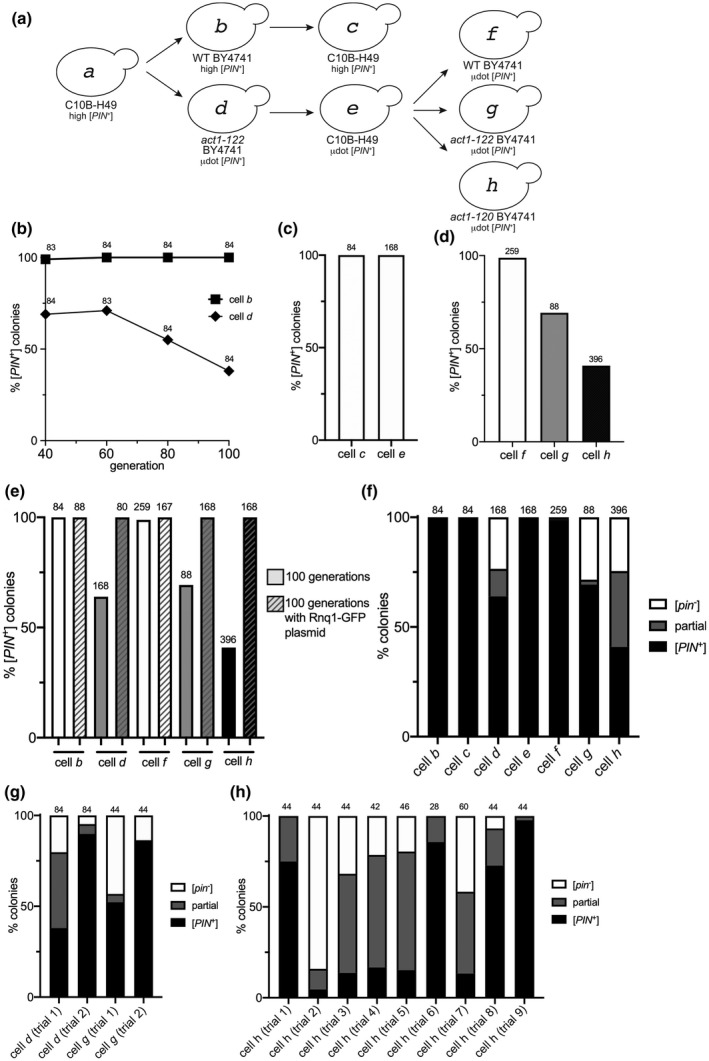
µdot [PIN+] is mitotically unstable in BY4741 strains containing the act1‐122 and act1‐120 mutations. (a) µdot [PIN+] from cell e was cytoduced into wildtype BY4741 (cell f), act1‐122 BY4741 (cell g), or act1‐120 BY4741 (cell h). Cytoductions involving cells a through e are described on the previous figures. (b) Strains represented by cells b and d were propagated for the indicated generations. Individual colonies were mated to a Rnq1‐GFP tester strain and each diploid colony was scored for the complete loss of [PIN +] by the detection of diffuse fluorescence in all of the cells within the population. The number of colonies scored (n) is indicated above each data point. In generations 60–100, the act1‐122 BY4741 strain (cell d) lost the [PIN +] prion in a linear manner (R 2 = 1), whereas loss was not observed in wild type (slope = 0; R 2 = 1). The slope of [PIN +] loss in act1‐122 BY4741 is significantly different from wild type (p < 0.005). (c) The percent of colonies that lost the prion in C10B‐H49 strains containing either high [PIN +] (cell c) or µdot [PIN+] (cell e) after 100 generations of growth. (d) Percent of colonies that lost µdot [PIN+] in wildtype (cell f), act1‐122 (cell g), or act1‐120 (cell h) strains. (e) Comparison of prion loss for all BY4741 strains. All strains were propagated for 100 generations, both with and without the RNQ1‐GFP plasmid (p3034), and individual colonies were scored for [PIN+] loss. (f) A summary of prion loss of all strains (cells b through h). Colonies composed only of cells containing diffuse fluorescence ([pin −], white box), colonies composed of a mix of cells with either diffuse or fluorescent aggregates (partial, gray box), and colonies where all cells have aggregates ([PIN +]; black box) are shown. (g and h) Prion loss observed in individual trials (replicates using independent lab strains, Table S1) of act1‐122 BY4741 (cell d or g) and act1‐120 BY4741 (cell h) strains containing µdot [PIN+]. For all charts, values provided above individual bars or graph data points indicate the total number of colonies assayed.
Given the instability of µdot [PIN +] in act1‐122 BY4741, we asked whether the same µdot [PIN +] variant would also be unstable once we had cytoduced the prion back into [pin −] recipient with the C10B‐H49 genetic background. Using µdot [PIN +] C10B‐H49 cytoductants (cell e) obtained via plasmiduction, plasmids were lost from the cytoductants by subcloning strains on nonselective rich media, and then strains were successively subcloned on rich media for an additional 100 generations. µdot [PIN +] appeared to be faithfully maintained throughout these 100 generations, similar to high [PIN +] in the same wildtype genetic background (cells c and e, Figure 5c).
Given the stability of µdot [PIN +] observed in the C10B‐H49 genetic background one could draw two possible conclusions: (1) prion stability is dependent upon genetic context (i.e., presence of the act1‐122 mutation, BY4741 genetic background, or both), or (2) that this subsequent cytoduction (from cell d to cell e) had imposed a selection for stable prion conformers. To differentiate between these possibilities, we completed one additional set of cytoductions back into [pin −] recipients of wildtype BY4741 and act1‐122 BY4741 (Figure 5a; from cell e to cells f and g). We also decided to include a related yet independent actin [pin −] recipient mutant strain, act1‐120 BY4741 (cell h), which contains two point mutations within the Sac6‐binding region of actin (E99A/E100A). Based upon our observations in Table 1, these cytoductions were completed in the presence of the copper‐inducible RNQ1‐GFP plasmid (without supplemented copper) to enhance successful transmission of µdot [PIN +] to the recipient cell, and facilitate immediate confirmation of [PIN +] status. RNQ1‐GFP plasmids were lost from the cytoductants, as described above, and cytoductants were successively subcloned on rich media through 100 generations. Similar to C10B‐H49 (cell e), all of the wildtype BY4741 colonies (cell f) had faithfully maintained µdot [PIN +] throughout the 100 generations, whereas both act1‐122 and act1‐120 BY4741 with µdot [PIN +] were subject to prion loss (cells g and h; Figure 5d). These data support the concept that integrity of the actin network plays an important role in propagating [PIN +]. It is important to note that other [PIN +] variants such as high [PIN +] and library [PIN +] are stably maintained in these actin mutants (Figure S11), indicating that this unique prion variant is able to uncover the role of actin in prion stability.
Since the presence of the copper‐inducible RNQ1‐GFP plasmid provided faithful transmission of µdot [PIN +] through plasmiduction (Table 1), we postulated that the presence of the same plasmid may stabilize the prion in act1‐122 and act1‐120 BY4741 µdot [PIN +] strains through mitosis. We found that the simple presence of the RNQ1‐GFP plasmid, without any additional copper supplementation, enabled µdot [PIN +] to be faithfully maintained for 100 generations (Figure 5e). Similar to the cytoduction experiments (Table 1), the ability to rescue µdot [PIN +] instability with addition of even modest levels of Rnq1‐GFP suggests that the level of endogenous Rnq1 protein alone is not sufficient to maintain µdot [PIN +] through mitosis in BY4741 act1‐122 or act1‐120 mutants.
In addition to noting the impact of Rnq1‐GFP on maintenance of µdot [PIN +] in these act1 mutant alleles, we were interested in learning more about the progression of [PIN+] loss. Closer inspection of the visual aggregate profiles among the 100 generation act1‐122 and act1‐120 strains relative to wildtype genetic backgrounds showed that some colonies were completely comprised of diffuse cell populations, indicating that all the cells in the colony were [pin −], whereas other colonies displayed a mixed population of cells with some containing aggregates and others exhibiting diffuse fluorescence (partial, Figure 5f). It seems likely that those colonies that have a portion of cells with aggregates are in the process of losing the prion from the population.
Several independent act1‐122 and act1‐120 strains were checked for µdot [PIN +] stability spanning 100 generations. Some strains exhibited very little prion loss, whereas others showed more accelerated prion loss (Figure 5g,h). The variability in loss among strains suggests that the rate of prion loss, and possibly what triggers loss, is influenced by varied biological or environmental factors within independent derived lineages. Taken together, our data suggest that BY4741 strains with act1‐122 or act1‐120 mutations can render µdot [PIN +] unstable.
2.5. Prion aggregates become larger over time in act1‐122 mutants
Based on work with Hsp104, prion loss can occur through growth of the aggregate, leading to poor transmission. Therefore, we were interested in whether µdot [PIN +] loss in act1‐122 BY4741 was due to an increase in Rnq1 aggregate size. In act1‐122 BY4741 µdot [PIN +] strains grown for 60 generations, Rnq1 was found to fractionate to the pellet, indicative of extremely large molecular weight complexes (cell d, Figure 6). Fractionation of Rnq1 to the pellet was not observed in either wildtype C10B‐H49 or BY4741 strains containing high [PIN +] (Figure 1d), or wildtype C10B‐H49 strains containing µdot [PIN +] grown for 60 generations (cell e; Figure 6a). To further confirm that Rnq1 aggregates become larger over time in act1‐122 BY4741 mutants with µdot [PIN +], we looked at Rnq1 fractionation profiles at 20 and 60 generations in the act1‐122 BY4741 that had been newly cytoduced with µdot [PIN +] (cell g, Figure 6c). Rnq1 was not readily detectable in pellet fractions at 20 generations, but was clearly evident by 60 generations, consistent with Rnq1 aggregates growing in size over time.
FIGURE 6.
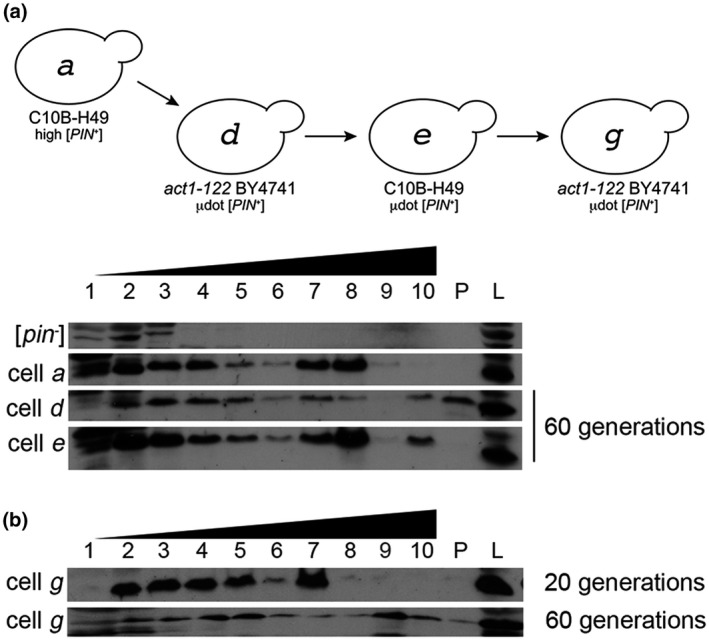
µdot [PIN+] aggregates increase in size over time in act1‐122 mutants. (a) Cell lysates from cells a, d, and e were fractionated on a discontinuous (10%, 40%, 60%) sucrose gradient. Cells d and e were the same strains shown in Figure 5c,d that were grown for 60 generations. As in Figure 1, fraction 1 represents the top of the gradient, fraction 10 is the bottom. The lane labeled P shows protein found in the pellet of gradient and L is whole‐cell lysates. (b) Cell g was propagated for either 20 or 60 generations and lysates were run on discontinuous sucrose gradients. Each gel image is representative of biological replicate trials.
3. DISCUSSION
In this study, we obtained a variant of [PIN +] coincident with cytoduction that had different phenotypic properties from the original high [PIN+] variant. While able to be transmitted through cytoduction, this new µdot [PIN +] variant was lost over many generations in both act1‐122 and act1‐120 BY4741 mutant backgrounds (Figure 5). This loss was correlated with the presence of large visual Rnq1‐GFP foci (Figure 4) as well as the production of extremely large molecular weight species (Figure 6). Discovery of this new variant has provided valuable insight into the role of actin networks in prion propagation, specifically the ability of the actin cytoskeleton to influence the availability of transmissible aggregates for prolonged propagation of prions over many generations.
3.1. µdot [PIN +]: unknown origin and stochastic loss
The mechanism by which we obtained the µdot [PIN +] variant is unclear. Most known prion variants are reproducibly propagated and cytoduced as the same conformer. Cells containing newly formed prion aggregates can yield progeny with multiple variants, but once transmitted over several generations, only one variant is detectable (Bradley et al., 2002; Sharma and Liebman, 2012). Therefore, the detection of an alternate prion variant through cytoduction is unexpected. Two models have been proposed whereby new prion variants may be obtained (Bateman and Wickner, 2013). Either a cloud of different variants is simultaneously propagated and sometimes certain variants dominate through cytoduction (Bateman and Wickner, 2013) or conversion occurs through a rare mistemplating event where a new prion variant forms from a pre‐existing prion (Roberts et al., 2009; Lin et al., 2011).
The observed rate of µdot [PIN +] loss from individual act1‐122 or act1‐120 BY4741 cell lineages was variable (Figure 5). Even though loss appears to be stochastic among cell lineages, the variability could reflect unidentified factors such as aging or environmental stimuli, differences in rate of loss per generation, or perhaps a difference in timing of some triggering event that initiates prion loss. Coincident with the concept of stochastic loss over many generations, we see that Rnq1 is associated with high molecular weight species only after about 60 generations in BY4741 act1‐122 µdot [PIN +] strains (Figure 6). Other studies have observed that higher molecular weight species are associated with some de novo generated [PIN +] variants that were quickly lost after only a few generations (Huang et al., 2013). Therefore, the gradual accumulation of higher molecular weight species over many generations, which seems to specifically occur in the presence of irregular cytoskeletal networks, may also contribute substantially to the stochastic nature of µdot [PIN +] loss in these strains.
3.2. Propagons, actin, and prion stability
It is also possible that with the increased size of Rnq1 aggregates, a smaller number of propagons are available to be transmitted to daughter cells. It has been shown that different [PIN +] variants have different propagon numbers (Sharma and Liebman, 2013). For example, high [PIN +] has four times more propagons as low [PIN +]. It is possible that µdot [PIN +] naturally has a low propagon number. However in the presence of compromised actin networks, the growth of the prion aggregate leaves less Rnq1 protein available for the propagon generation. This idea of an insufficient amount of protein available for the generation of propagons is supported by the fact that when additional Rnq1‐GFP is introduced, µdot [PIN +] is stable in act1‐122 and act1‐120 BY4741 mutant backgrounds even after 100 generations (Figure 5).
It is interesting that loss of µdot [PIN +] is observed in actin mutants but not wildtype cells, suggesting that actin could have a direct role in the transmission of propagons to daughter cells. Several studies have shown a link between actin and prion formation (Ganusova et al., 2006; Manogaran et al., 2011; Speldewinde et al., 2017). For example, loss of the actin bundling protein, Sac6, leads to reduced prion formation (Manogaran et al., 2011). However, this study is the first to show that actin networks can influence the stability of a prion, albeit a certain variant. Since both act1‐122 and act1‐120 mutants contain substitutions within the Sac6 actin‐binding region (Holtzman et al., 1994; Honts et al., 1994; Amberg et al., 1995; Miao et al., 2016), and these mutants display actin patch depolarization (Figure S7), it is possible that actin bundling or actin patch polarization is required for prion stability. It has also been suggested that the actin/myosin network plays a role in depositing excess prion aggregates into amyloid deposition sites near the vacuole (Kumar et al., 2016), but how transport of prion aggregates to these inclusion sites contributes to prion stability is unclear. However, there may be other explanations that imply that actin has an indirect role. Given that actin mutant cells have increased average cell size relative to wildtype cells (Figure S7), we also considered the potential role cell size may play in rendering µdot [PIN +] unstable. However, cell size alone is insufficient to cause prion loss because both library and high [PIN +] are maintained in these strains (Figure S11). Because both act1 mutants exhibiting µdot [PIN +] instability are in the same BY4741 genetic background, determining whether something in the BY4741 background contributes to instability will require further investigation. While our data cannot rule out the influence of cell size, or other contributing factors, these studies seem to suggest a “perfect storm” scenario where the combination of irregular actin networks, accumulation of higher molecular weight aggregates, possible lower propagon number, and cell size compound to render the µdot [PIN +] variant unstable.
We also considered whether µdot [PIN +] loss in these actin mutants may be mediated through Hsp104. A link between Hsp104 and actin networks has been suggested in the retention of heat shocked or oxidatively damaged proteins, using actin networks to move and asymmetrically sequester those proteins in the mother cell (Erjavec and Nystrom, 2007; Tessarz et al., 2009; Liu et al., 2011; Song et al., 2014). A similar mechanism of actin network‐mediated asymmetric retention appears to work on aggregates of the heterologous human HTT103Q amyloid (Liu et al., 2011; Song et al., 2014). Under this model of interaction, irregular actin networks would be expected to reduce asymmetric retention in the mother, and thereby enhance prion transmission to daughter cells, rather than reduce prion transmission as observed here. Additionally, if these actin mutations were interfering with Hsp104 activity, we would expect destabilization of all [PIN +] variants, but we observed that other [PIN +] variants are maintained normally in these strains (Figure S11). However, it has been shown that a size threshold for the transmission of propagons through the budneck exists for the [PSI +] prion (Derdowski et al., 2010). It is possible that the observed increase in µdot [PIN +] aggregate size over many generations (Figure 6) results in prion loss because these large prion propagons are above the size threshold to pass through the bud neck.
These findings parallel models involving cytoskeletal networks that have been proposed for the cell‐to‐cell transmission of small amyloids to other neurons in neurodegenerative protein aggregation diseases. In one model, oligomers and fibrils are proposed to be packaged into exosomes to be released from the cell, and taken up by nearby neurons through actin‐mediated endocytosis (Fevrier et al., 2004; Miranda and Di Paolo, 2018). Another model involving cytoskeletal networks involves tunneling nanotubules, which are normally used to transfer organelles between neurons, but which may also serve as a conduit for transfer of amyloid oligomers or aggregates (Gousset et al., 2009; Victoria and Zurzolo, 2017). It is possible that dysfunction of cytoskeletal networks could limit the transmission or spreading of amyloid in vivo. Regardless of what other factors may contribute to µdot [PIN +] instability, our data provide support that cytoskeletal networks play a role in prion propagation and thereby stability.
4. EXPERIMENTAL PROCEDURES
4.1. Strains and plasmids
Yeast strains in 74D‐694, BY4741, or C10B‐H49 genetic backgrounds were used as indicated (Table S1). BY4741 strains of act1‐120, act1‐122, act1‐101, and act1‐129 were originally made by Wertman et al. (1992), and more recently validated and tagged with NATR selection by the Amberg lab (Viggiano et al., 2010). All act1 mutants were sequenced to confirm the strains contained mutations in the ACT1 gene. Upon transformation with plasmids containing RNQ1‐GFP (p3034 or p3036), these strains were found to contain the “library” variant of [PIN +] (as described by Manogaran et al., 2010). These BY4741 strains were cured of all prions by subcloning at least three times on 5 mM Guanidine–HCl (GuHCl), and verified for the loss of prions by transiently expressing Sup35PrD‐GFP (p3031) or RNQ1‐GFP (p3034 or p3036) fusion proteins and visualizing diffuse fluorescence indicative of the nonprion state. Strains and plasmids used in this study are listed in Tables S1 and S2.
4.2. Cultivation procedures
Yeast strains were grown using standard media and cultivation procedures (Sherman et al., 1986). Complex media containing 2% dextrose (YPD) or synthetic complete media containing the required amino acids and 2% dextrose (SD) was used as indicated. A major component of the synthetic complete media is yeast nitrogen base (BD Difco), which contains trace amounts of copper sulfate, resulting in final concentrations up to 0.25 µM. Strains transformed with plasmids were maintained on synthetic complete media lacking the specific amino acid.
4.3. Fluorescent microscopy
To detect the Rnq1 protein by fluorescent microscopy, strains were transformed with a plasmid containing a copper‐inducible pCUP1‐RNQ1‐GFP (p3034, HIS3). For 3D microscopy, cells were grown for 16–18 hours in plasmid selective media. To induce plasmid expression, 10–50 µM of copper sulfate was added to the media and cultures were grown for an additional 4–6 hours. Cultures were screened for the presence of GFP aggregates using either a 63× (oil immersion, N.A. 1.4) or 100× (oil immersion, N.A. 1.44) objective. µdot [PIN +] was most easily detected with the 100× objective. Z‐stack images containing between 10‐ and 21‐step z‐stacks were captured under a DMI6000 Leica inverted microscope using Leica LASX software. To ensure captured images were able to resolve the aggregate details for print, images were subjected to 3D deconvolution using Autoquant deconvolution algorithms with background removed and intensity rescaled (Media Cybernetics). All images shown are maximum projection unless indicated otherwise. For counting cells with aggregates, a minimum of 300 cells were counted in at least three independent cultures.
Time‐lapse microscopy of cultures was performed according to Sharma et al., (2017). Briefly, cells were grown for 6 hours in synthetic media without added copper, but containing trace concentrations up to 0.25 µM (Cultivation procedures). While this concentration is approximately 1/40–1/200 the concentration routinely added to induce plasmid expression, it proves sufficient to yield detectable Rnq1‐GFP fluorescence. 50 μL of culture was added to 250 μL of fresh selective media, and placed in concanavalin A‐coated Ibidi 1 µ‐slide 8‐well glass bottom slides. 3D images were captured every 10 minutes over 6–12 hours. To adjust for low Rnq1‐GFP fluorescence levels, videos were subjected to deconvolution and maximum projection as indicated above to resolve aggregate dynamics.
4.4. Cytoduction and plasmiduction of prions
Cytoduction is the transfer of cytoplasm from one cell to another, and can be used to transfer prions from a donor strain to a [pin −] recipient strain. The presence of a kar1 mutation permits mating and mixture of cellular contents, but prevents fusion between the nuclei of donor and recipient strain (Conde and Fink, 1976). Cytoduction can be monitored by mitochondrial transfer, but a variation of cytoduction, sometimes referred to as plasmiduction, can be monitored by transfer of plasmid‐born markers from a donor strain to a [pin −] recipient strain (Natsoulis et al., 1994). It has been reported that better cytoplasmic [PIN +] transfer can be achieved using plasmid‐based markers (Vitrenko et al., 2007).
All recipient strains were streaked on 5mM guanidine hydrochloride three times to cure the prion. To verify prion loss, all strains were transformed with copper‐inducible RNQ1‐GFP plasmid (p3034 or p3036), or mated to tester strains containing the RNQ1‐GFP plasmid, and viewed under fluorescent microscopy for diffuse cytoplasmic fluorescence after 4 hours of 50 µM copper induction. Comparable fluorescence assays were completed using Sup35PrD‐GFP (p3031) to confirm that all strains were [psi −].
Cytoduction was performed to transfer prions from some BY4741 strains to a C10B‐H49 recipient strain (Figures 1a, 3a, and Table 1; D101), which was [pin −] [psi −] [rho0] and contained a cycloheximide recessive allele. After mating, cytoductants were selected on YPGlycerol media supplemented with cycloheximide. Cytoductants were verified for growth on selective media, and saved to glycerol stocks. The transfer of prions from cell b to c, or cell d to e, was also repeated through plasmiduction, and all transfer of prions to cells f, g, and h were performed through plasmiduction using the RNQ1‐GFP (HIS3, p3034) plasmid. The presence of [PIN +] among resulting plasmiductants was verified by the presence of fluorescent aggregates (as described above). Confirmed [PIN +] plasmiductants were saved as glycerol stocks (Table S1), simultaneously streaked on rich media to lose the plasmid containing RNQ1‐GFP, and then resaved as glycerol stocks with no plasmid (Table S1).
4.5. Biochemical analysis
Cell lysates were prepared from 50 ml of overnight cultures and lysed according to Sharma et al. (2017). 100 µg of crude lysates was treated with 2% SDS‐Buffer and boiled (for SDS‐PAGE) or unboiled (for SDD‐AGE). For sucrose gradient analysis, 8 mg of freshly prepared protein lysate was loaded on a discontinuous sucrose gradient (10%, 40%, and 60% sucrose in lysis buffer). The discontinuous gradient allows for the capture of large Rnq1 aggregates between the 40% and 60% interface, which was typically in the 7th or 8th fraction (Figure 1d). Gradients were spun at 16,000× g for approximately 130 minutes. 10 fractions were immediately removed from the gradient, and the pellet was resuspended in 1X lysis buffer with protease inhibitor. All fractions were diluted in half and loaded on SDS‐PAGE. All gels were transferred onto PVDF and immunoblotted with anti‐Rnq1 polyclonal antibody (courtesy of Dr. Heather True, Washington University).
4.6. Mitotic stability
All original BY4741 plasmiductants were selected using a HIS3 plasmid that also contains RNQ1‐GFP to facilitate immediate scoring for [PIN +]. The RNQ1‐GFP‐containing plasmid (HIS3, p3034) was lost by subcloning cultures on YPD, and selecting individual colonies that grew on synthetic complete media (SD12) but failed to grow on SD‐His media. These strains were immediately saved to glycerol stocks (See Table S1). [PIN +] stability experiments were initiated upon streaking each strain on YPD fresh from glycerol stocks, pooling at least 20 comparably sized colonies, and streaking this pool on YPD to yield ~100 single colonies representing 40 generations from cryogenic storage. These “40‐generation” single colonies were picked directly into 96‐well plates, including blinded [PIN +] and [pin −] controls, pinned to YPD plates for overnight growth, and mated with D163 tester strain transformed with a plasmid that contains RNQ1‐GFP (LEU2, p3036). Diploids were selected on SD‐Leu‐Ura plates, and pinned to deep well plates containing 750 µl of SD‐Leu‐Ura liquid with 25 µM CuSO4 for overnight growth with shaking at 30C. Individual diploid cultures were screened by fluorescence microscopy for presence of Rnq1‐GFP aggregates versus diffuse fluorescence, indicative of [PIN +] loss. Cell populations of individual colonies were scored by the following: all cells had Rnq1‐GFP aggregates ([PIN +]), only a subpopulation of cells had aggregates (partial), or all cells showed diffuse Rnq1‐GFP fluorescence ([pin −]).
To generate colonies representing 60 generations from cryogenic storage, but also to avoid founder effects, cells from the densely populated region of the above “40‐generation” YPD plate were further subcloned by streaking on a fresh YPD plate to generate ~100 “60‐generation” single colonies. Precisely as described above, “60‐generation” single colonies were picked to 96‐well plates, pinned to YPD, mated to the D163 tester containing p3036, grown on SD‐Leu‐Ura plates, pinned to SD‐Leu‐Ura liquid media with 25 µM CuSO4 for overnight growth at 30°C, and screened for the number of colonies that were [PIN +]. This entire procedure from densely populated region of cell harvest up through microscopic screening was repeated for 80 and 100 generations.
To test mitotic stability in the presence of low‐level Rnq1‐GFP expression, strains representing cells f, g, and h, and retaining the RNQ1‐GFP plasmid (HIS3, p3034) used for plasmiduction, were streaked from cryogenic storage onto SD‐His plates. Stability experiments were carried out similar to above, except that strains were successively subcloned onto SD‐His plates that did not include any supplemented CuSO4, such that the only Rnq1‐GFP expression was generated through the trace copper found in the media. Single colonies, along with desired [PIN +] and [pin −] controls, were then picked directly into 96‐well plates containing SD‐His liquid with 25 µM CuSO4, and grown at 30°C overnight with shaking. Individual cultures were screened by fluorescence microscopy for presence of Rnq1‐GFP aggregates versus diffuse fluorescence indicative of [PIN +] loss.
AUTHOR CONTRIBUTIONS
All authors have made major contributions to the manuscript. J.E.D. and A.L.M contributed to the conceptual design of the study. J.E.D., M.J.O., D.R.L., J.A.R., B.T.W., E.E.D., A.M.K., S.J.M., and A.L.M contributed to the acquisition, analysis, and/or interpretation of the data. J.E.D., D.R.L, and A.L.M have written the manuscript.
Supporting information
Supplementary Material
Video S2
Video S3
Video S4
Video S8
Video S9
Acknowledgments
We thank Dr. Susan Liebman for strains and plasmids, Dr. David Amberg for the actin mutants, and Dr. Heather True for the Rnq1 polyclonal antibodies used in this study. Thank you to Dr. Lisa Petrella for helpful comments on the manuscript. We also thank Dr. Brian Haarer for technical guidance regarding the actin mutants. This work was supported by the National Institutes of Health (NIH) grant GM109336 to ALM, and individual Marquette University Honors Fellowships to M.J.O, J.A.R., E.E.D, and B.T.W.
Dorweiler JE, Oddo MJ, Lyke DR, et al. The actin cytoskeletal network plays a role in yeast prion transmission and contributes to prion stability. Mol Microbiol. 2020;114:480–494. 10.1111/mmi.14528
REFERENCES
- Alberti, S. , Halfmann, R. , King, O. , Kapila, A. and Lindquist, S. (2009) A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell, 137, 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg, D.C. , Basart, E. and Botstein, D. (1995) Defining protein interactions with yeast actin in vivo. Natural Structural Biology, 2, 28–35. [DOI] [PubMed] [Google Scholar]
- Aron, R. , Higurashi, T. , Sahi, C. and Craig, E.A. (2007) J‐protein co‐chaperone Sis1 required for generation of [RNQ+] seeds necessary for prion propagation. EMBO Journal, 26, 3794–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate, M. , Mitra, S. , Schweitzer, E.S. , Segal, M.R. and Finkbeiner, S. (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature, 431, 805–810. [DOI] [PubMed] [Google Scholar]
- Bailleul, P.A. , Newnam, G.P. , Steenbergen, J.N. and Chernoff, Y.O. (1999) Genetic study of interactions between the cytoskeletal assembly protein sla1 and prion‐forming domain of the release factor Sup35 (eRF3) in Saccharomyces cerevisiae . Genetics, 153, 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman, D.A. and Wickner, R.B. (2013) The [PSI+] prion exists as a dynamic cloud of variants. PLoS genetics, 9, e1003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessen, R.A. and Marsh, R.F. (1992) Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. Journal of Virology, 66, 2096–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley, M.E. , Edskes, H.K. , Hong, J.Y. , Wickner, R.B. and Liebman, S.W. (2002) Interactions among prions and prion “strains” in yeast. Proceedings of the National Academy of Sciences of the United States of America, 99(Suppl 4), 16392–16399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley, M.E. and Liebman, S.W. (2003) Destabilizing interactions among [PSI(+)] and [PIN(+)] yeast prion variants. Genetics, 165, 1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge, J. , Sidle, K.C. , Meads, J. , Ironside, J. and Hill, A.F. (1996) Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature, 383, 685–690. [DOI] [PubMed] [Google Scholar]
- Conde, J. and Fink, G.R. (1976) A mutant of Saccharomyces cerevisiae defective for nuclear fusion. Proceedings of the National Academy of Sciences of the United States of America, 73, 3651–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, B.S. (1965) [PSI], a cytoplasmic suppressor of super‐suppressors in yeast. Heredity, 20505–521. [Google Scholar]
- Derdowski, A. , Sindi, S.S. , Klaips, C.L. , DiSalvo, S. and Serio, T.R. (2010) A size threshold limits prion transmission and establishes phenotypic diversity. Science, 330, 680–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkatch, I.L. , Bradley, M.E. , Hong, J.Y. and Liebman, S.W. (2001) Prions affect the appearance of other prions: the story of [PIN(+)]. Cell, 106, 171–182. [DOI] [PubMed] [Google Scholar]
- Derkatch, I.L. , Bradley, M.E. , Zhou, P. , Chernoff, Y.O. and Liebman, S.W. (1997) Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae . Genetics, 147, 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, Z. , Park, K.W. , Yu, H. , Fan, Q. and Li, L. (2008) Newly identified prion linked to the chromatin‐remodeling factor Swi1 in Saccharomyces cerevisiae . Nature genetics, 40, 460–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erjavec, N. and Nystrom, T. (2007) Sir2p‐dependent protein segregation gives rise to a superior reactive oxygen species management in the progeny of Saccharomyces cerevisiae . Proceedings of the National Academy of Sciences of the United States of America, 104, 10877–10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fevrier, B. , Vilette, D. , Archer, F. , Loew, D. , Faigle, W. , Vidal, M. , et al (2004) Cells release prions in association with exosomes. Proceedings of the National Academy of Sciences of the United States of America, 101, 9683–9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganusova, E.E. , Ozolins, L.N. , Bhagat, S. , Newnam, G.P. , Wegrzyn, R.D. , Sherman, M.Y. , et al (2006) Modulation of prion formation, aggregation, and toxicity by the actin cytoskeleton in yeast. Molecular and Cellular Biology, 26, 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabe, C.G. (2006) Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiology of Aging, 27, 570–575. [DOI] [PubMed] [Google Scholar]
- Glover, J.R. , Kowal, A.S. , Schirmer, E.C. , Patino, M.M. , Liu, J.J. and Lindquist, S. (1997) Self‐seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion‐like factor of S. cerevisiae . Cell, 89, 811–819. [DOI] [PubMed] [Google Scholar]
- Gousset, K. , Schiff, E. , Langevin, C. , Marijanovic, Z. , Caputo, A. , Browman, D.T. , et al (2009) Prions hijack tunnelling nanotubes for intercellular spread. Nature Cell Biology, 11, 328–336. [DOI] [PubMed] [Google Scholar]
- Halfmann, R. , Jarosz, D.F. , Jones, S.K. , Chang, A. , Lancaster, A.K. and Lindquist, S. (2012) Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature, 482, 363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higurashi, T. , Hines, J.K. , Sahi, C. , Aron, R. and Craig, E.A. (2008) Specificity of the J‐protein Sis1 in the propagation of 3 yeast prions. Proceedings of the National Academy of Sciences of the United States of America, 105, 16596–16601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman, D.A. , Wertman, K.F. and Drubin, D.G. (1994) Mapping actin surfaces required for functional interactions in vivo. Journal of Cell Biology, 126, 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honts, J.E. , Sandrock, T.S. , Brower, S.M. , O'Dell, J.L. and Adams, A.E. (1994) Actin mutations that show suppression with fimbrin mutations identify a likely fimbrin‐binding site on actin. Journal of Cell Biology, 126, 413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, V.J. , Stein, K.C. and True, H.L. (2013) Spontaneous variants of the [RNQ+] prion in yeast demonstrate the extensive conformational diversity possible with prion proteins. PLoS One, 8, e79582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igel‐Egalon, A. , Béringue, V. , Rezaei, H. , & Sibille, P. (2018). Prion Strains and Transmission Barrier Phenomena. Pathogens, 7, 5 10.3390/pathogens7010005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili‐Shirazi, A. , Summers, L. , Linehan, J. , Mallinson, G. , Anstee, D. , Hawke, S. , et al (2005) PrP glycoforms are associated in a strain‐specific ratio in native PrPSc. Journal of General Virology, 86, 2635–2644. [DOI] [PubMed] [Google Scholar]
- King, C.Y. , Tittmann, P. , Gross, H. , Gebert, R. , Aebi, M. and Wuthrich, K. (1997) Prion‐inducing domain 2–114 of yeast Sup35 protein transforms in vitro into amyloid‐like filaments. Proceedings of the National Academy of Sciences of the United States of America, 94, 6618–6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaips, C.L. , Hochstrasser, M.L. , Langlois, C.R. and Serio, T.R. (2014) Spatial quality control bypasses cell‐based limitations on proteostasis to promote prion curing. eLife, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, R. , Nawroth, P.P. and Tyedmers, J. (2016) Prion aggregates are recruited to the insoluble protein deposit (IPOD) via myosin 2‐based vesicular transport. PLoS genetics, 12, e1006324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroute, F. (1971) Non‐Mendelian mutation allowing ureidosuccinic acid uptake in yeast. Journal of Bacteriology, 106, 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert, M.P. , Barlow, A.K. , Chromy, B.A. , Edwards, C. , Freed, R. , Liosatos, M. , et al (1998) Diffusible, nonfibrillar ligands derived from Abeta1‐42 are potent central nervous system neurotoxins. Proceedings of the National Academy of Sciences of the United States of America, 95, 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, J.Y. , Liao, T.Y. , Lee, H.C. and King, C.Y. (2011) Inter‐allelic prion propagation reveals conformational relationships among a multitude of [PSI] strains. PLoS genetics, 7, e1002297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, B. , Larsson, L. , Franssens, V. , Hao, X. , Hill, S.M. , Andersson, V. , et al (2011) Segregation of protein aggregates involves actin and the polarity machinery. Cell, 147, 959–961. [DOI] [PubMed] [Google Scholar]
- Manogaran, A.L. , Fajardo, V.M. , Reid, R.J. , Rothstein, R. and Liebman, S.W. (2010) Most, but not all, yeast strains in the deletion library contain the [PIN(+)] prion. Yeast, 27, 159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manogaran, A.L. , Hong, J.Y. , Hufana, J. , Tyedmers, J. , Lindquist, S. and Liebman, S.W. (2011) Prion formation and polyglutamine aggregation are controlled by two classes of genes. PLoS genetics, 7, e1001386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, Y. , Han, X. , Zheng, L. , Xie, Y. , Mu, Y. , Yates, J.R., 3rd , et al (2016) Fimbrin phosphorylation by metaphase Cdk1 regulates actin cable dynamics in budding yeast. Nature Communications, 7, 11265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda, A.M. and Di Paolo, G. (2018) Endolysosomal dysfunction and exosome secretion: implications for neurodegenerative disorders. Cell Stress, 2, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales, R. , Hu, P.P. , Duran‐Aniotz, C. , Moda, F. , Diaz‐Espinoza, R. , Chen, B. , et al (2016) Strain‐dependent profile of misfolded prion protein aggregates. Scientific Reports, 6, 20526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsoulis, G. , Winston, F. and Boeke, J.D. (1994) The SPT10 and SPT21 genes of Saccharomyces cerevisiae . Genetics, 136, 93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osherovich, L.Z. and Weissman, J.S. (2001) Multiple Gln/Asn‐rich prion domains confer susceptibility to induction of the yeast [PSI(+)] prion. Cell, 106, 183–194. [DOI] [PubMed] [Google Scholar]
- Parchi, P. , Castellani, R. , Capellari, S. , Ghetti, B. , Young, K. , Chen, S.G. , et al (1996) Molecular basis of phenotypic variability in sporadic Creutzfeldt‐Jakob disease. Annals of Neurology, 39, 767–778. [DOI] [PubMed] [Google Scholar]
- Patel, B.K. , Gavin‐Smyth, J. and Liebman, S.W. (2009) The yeast global transcriptional co‐repressor protein Cyc8 can propagate as a prion. Nature Cell Biology, 11, 344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, B.E. , Duennwald, M.L. , Wang, H. , Chung, C. , Lopreiato, N.P. , Sweeny, E.A. , et al (2009) A synergistic small‐molecule combination directly eradicates diverse prion strain structures. Nature Chemical Biology, 5, 936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogoza, T. , Goginashvili, A. , Rodionova, S. , Ivanov, M. , Viktorovskaya, O. , Rubel, A. , et al (2010) Non‐Mendelian determinant [ISP+] in yeast is a nuclear‐residing prion form of the global transcriptional regulator Sfp1. Proceedings of the National Academy of Sciences of the United States of America, 107, 10573–10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlumpberger, M. , Prusiner, S.B. and Herskowitz, I. (2001) Induction of distinct [URE3] yeast prion strains. Molecular and Cellular Biology, 21, 7035–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, J. and Liebman, S.W. (2012) [PSI(+) ] prion variant establishment in yeast. Molecular Microbiology, 86, 866–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, J. and Liebman, S.W. (2013) Exploring the basis of [PIN(+)] variant differences in [PSI(+)] induction. Journal of Molecular Biology, 425, 3046–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, J. , Wisniewski, B.T. , Paulson, E. , Obaoye, J.O. , Merrill, S.J. and Manogaran, A.L. (2017) De novo [PSI +] prion formation involves multiple pathways to form infectious oligomers. Scientific Reports, 7, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman, F. , Fink, G.R. and Hicks, J.B. (1986) Methods in Yeast Genetics. Plainview, NY: Cold Spring Harbor Lab. [Google Scholar]
- Shorter, J. and Lindquist, S. (2004) Hsp104 catalyzes formation and elimination of self‐replicating Sup35 prion conformers. Science, 304, 1793–1797. [DOI] [PubMed] [Google Scholar]
- Shorter, J. and Lindquist, S. (2008) Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. EMBO Journal, 27, 2712–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondheimer, N. and Lindquist, S. (2000) Rnq1: an epigenetic modifier of protein function in yeast. Molecular Cell, 5, 163–172. [DOI] [PubMed] [Google Scholar]
- Song, J. , Yang, Q. , Yang, J. , Larsson, L. , Hao, X. , Zhu, X. , et al (2014) Essential genetic interactors of SIR2 required for spatial sequestration and asymmetrical inheritance of protein aggregates. PLoS genetics, 10, e1004539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speldewinde, S.H. , Doronina, V.A. , Tuite, M.F. and Grant, C.M. (2017) Disrupting the cortical actin cytoskeleton points to two distinct mechanisms of yeast [PSI+] prion formation. PLoS Genetics, 13, e1006708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, G. , Shimazu, N. and Tanaka, M. (2012) A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science, 336, 355–359. [DOI] [PubMed] [Google Scholar]
- Tessarz, P. , Schwarz, M. , Mogk, A. and Bukau, B. (2009) The yeast AAA+ chaperone Hsp104 is part of a network that links the actin cytoskeleton with the inheritance of damaged proteins. Molecular and Cellular Biology, 29, 3738–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victoria, G.S. and Zurzolo, C. (2017) The spread of prion‐like proteins by lysosomes and tunneling nanotubes: implications for neurodegenerative diseases. Journal of Cell Biology, 216, 2633–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viggiano, S. , Haarer, B. and Amberg, D.C. (2010) Correction/completion of the yeast actin, alanine scan alleles. Genetics, 185, 391–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitrenko, Y.A. , Pavon, M.E. , Stone, S.I. and Liebman, S.W. (2007) Propagation of the [PIN+] prion by fragments of Rnq1 fused to GFP. Current Genetics, 51, 309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov, K.V. , Aksenova, A.Y. , Soom, M.J. , Osipov, K.V. , Svitin, A.V. , Kurischko, C. , et al (2002) Novel non‐Mendelian determinant involved in the control of translation accuracy in Saccharomyces cerevisiae . Genetics, 160, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn, R.D. , Bapat, K. , Newnam, G.P. , Zink, A.D. and Chernoff, Y.O. (2001) Mechanism of prion loss after Hsp104 inactivation in yeast. Molecular and Cellular Biology, 21, 4656–4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertman, K.F. , Drubin, D.G. and Botstein, D. (1992) Systematic mutational analysis of the yeast ACT1 gene. Genetics, 132, 337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner, R.B. (1994) [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae . Science, 264, 566–569. [DOI] [PubMed] [Google Scholar]
- Winkler, J. , Tyedmers, J. , Bukau, B. and Mogk, A. (2012) Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. Journal of Cell Biology, 198, 387–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner, B. , Jappelli, R. , Maji, S.K. , Desplats, P.A. , Boyer, L. , Aigner, S. , et al (2011) In vivo demonstration that alpha‐synuclein oligomers are toxic. Proceedings of the National Academy of Sciences of the United States of America, 108, 4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Video S2
Video S3
Video S4
Video S8
Video S9