

Abstract
Ca2+ channels with a CaV1.3 pore-forming α1 subunit have been implicated in both neurodegenerative and neuropsychiatric disorders, motivating the development of selective and potent inhibitors of CaV1.3 vs CaV1.2 channels, the calcium channels implicated in hypertensive disorders. We have previously identified pyrimidine-2,4,6-triones (PYTs) that preferentially inhibit CaV1.3 channels, but the structural determinants of their interaction with the channel have not been identified, impeding their development into drugs. Using a combination of biochemical, computational, and molecular biological approaches, it was found that PYTs bind to the dihydropyridine (DHP) binding pocket of the CaV1.3 subunit, establishing them as negative allosteric modulators of channel gating. Site-directed mutagenesis, based on homology models of CaV1.3 and CaV1.2 channels, revealed that a single amino acid residue within the DHP binding pocket (M1078) is responsible for the selectivity of PYTs for CaV1.3 over CaV1.2. In addition to providing direction for chemical optimization, these results suggest that, like dihydropyridines, PYTs have pharmacological features that could make them of broad clinical utility.
Keywords: CaV1.3; L-type calcium channels; pyrimidine-2,4,6-triones; Parkinson’s disease; dihydropyridines; homology model; site-specific mutagenesis
Graphical Abstract
Plasma membrane, voltage-dependent Ca2+ channels regulate a wide range of cellular functions throughout the body. These channels are heteromeric proteins, consisting of a pore-forming α1 subunit and auxiliary subunits. These subunits dictate channel gating, trafficking, localization, and coupling with intracellular signaling cascades.1 Because of its centrality to channel function and pharmacology, Ca2+ channels are divided into classes based on their pore-forming α1 subunit. One these classes – L-type – has a pore-forming CaV1 α1 subunit; four genes have been cloned (CACNA1A-D).2 In the brain, channels with a CaV1.2 (CACNA1C) subunit predominate, consisting of roughly 90% of all L-type Ca2+ channels, with CaV1.3 (CACNA1D) channels making up almost all of the remainder.3
Although CaV1 channels are commonly considered ‘high-voltage activated’ because their open probability increases only at membrane potentials above spike threshold in neurons, CaV1.3 channels have an activation voltage-dependence that is very similar to that of ‘low threshold’ Ca2+ channels that open at membrane potentials below or near spike threshold.4 As a consequence, the functional roles played by CaV13 channels appear to be distinct from those of high-threshold CaV1.2 channels. For example, in the brain CaV1.3 channels have been implicated in the genesis of autonomous pacemaking and integration of synaptic events underlying plasticity.5, 6, 7, 8 They also have been linked to a wide variety of psychomotor disorders, including Parkinson’s disease (PD), autism spectrum disorder (ASD), bipolar disorder, and drug abuse.6, 9, 10, 11 CaV1.2 channels, on the other hand, also are abundant in peripheral organs, including the heart, and are the target of a variety of antihypertensive agents.12 Poor selectivity of compounds for CaV 1.3 over CaV1.2 channels leads to undesirable cardiovascular side effects.3
Despite the growing body of evidence linking CaV1.3 Ca2+ channels to human disease, a potent and selective inhibitor of CaV1.3 channels that might be suitable for clinical use has not been developed. At present, the only class-specific inhibitors of CaV1.3 channels approved for human use are 1,4-dihydropyridines (DHPs); however, DHPs are potent inhibitors of both CaV1.2 and CaV1.3 channels, with most members of this drug class exhibiting a 5–10-fold higher affinity for CaV1.2 channels.3 A key feature of DHPs is that they are negative allosteric modulators (NAMs) of Cav1 channel gating, rather than being pore-blockers.1 This confers voltage-dependence to their action, as their affinity is several orders of magnitude lower at membrane potentials at which most neurons rest, increasing into the nanomolar range with depolarization and activity. This ‘use-dependence’ is undoubtedly critical to their clinical efficacy and minimal side-effect profile.
Given the clinical and scientific rationale for a CaV1.3-selective inhibitor, some years ago our group undertook a compound screening effort. Starting from a conventional Ca2+ fluorescence-based counter-screen against CaV1.2 and CaV1.3 channels, a pyrimidine-2,4,6-trione (PYT) was found to show a slight preference for CaV1.3 channels over CaV1.2 channels. Multiple cycles of lead optimization of the initial hit compound eventually led to 1-(3-chlorophenethyl)-3-cyclopentylpyrimidine-2,4,6-trione (cp-PYT), which was found to be highly selective for inhibition of CaV1.3 channels over CaV1.2 channels.13 However, it was unclear where on the channel cp-PYT bound. The fact that the auxiliary subunits of the CaV12 and CaV1.3 channels used in the assay were the same clearly implicated the pore-forming a1 subunit. But it was unclear where on the a1 subunit that binding took place.
Subsequent to our initial report, two papers appeared that raised questions about the potential value of cp-PYT (and its derivatives) as CaV1.3 channel inhibitors. In the first paper, Ortner et al.14 reported that micromolar concentrations of cp-PYT slowed the deactivation of tail currents, essentially enhancing – not inhibiting – currents through CaV1.3 channels. In the second paper, Huang et al.15 reported that cp-PYT did inhibit CaV1.3 channels, but that the magnitude and selectivity of the inhibition was dependent on the auxiliary β subunit, not the α1 subunit.
The present studies were designed to determine the site of binding of cp-PYT to CaV1.3 channels, which should allow future rational design of more potent and selective NAMs of CaV1.3, thereby minimizing the undesirable cardiovascular effects of CaV1.2 binding. In addition, these studies clarified the above-noted literature discrepancies as well. To this end, a combination of approaches, including site-directed mutagenesis, equilibrium radiolabeled binding studies, and computer modeling, were used to define the cp-PYT binding site on heterologous CaV1.3 channels. In addition, the effects of cp-PYT on natively expressed CaV1.3 channels in substantia nigra dopaminergic neurons were determined. These studies revealed that cp-PYT binds to the same region of the α1 subunit as DHPs. Homology modeling and site-directed mutagenesis studies demonstrated that the selectivity of cp-PYT for CaV1.3 channels at this site was dependent on a single amino acid residue. As with DHPs, cp-PYT binding to this site and inhibition of channel opening was voltage-dependent, making cp-PYT a NAM, like DHPs. From these studies, it was realized that the previously reported study that failed to see cp-PYT inhibition14 used a variant of the CaV1.3 subunit lacking this key single amino acid residue, thereby abolishing the activity of cp-PYT. In addition to this high affinity binding site, a lower affinity site was found. Binding of cp-PYT to this lower affinity site led to slowing of tail current kinetics and could confer a dependence upon the α1 subunit used to form the channel, as noted by earlier investigators.15 Collectively, these studies not only provide a roadmap for the development of more potent and selective CaV1.3 channel NAMs that could be of clinical utility, they also provide explanations for the discrepancies with previous studies.
RESULTS
Site-directed mutagenesis of the DHP binding site disrupted PYT inhibition
CaV1 channels are known to be inhibited by three drug classes: 1,4-dihydropyridines (DHPs), phenylalkylamines, and benzothiazepine. These three classes have overlapping and allosterically-coupled binding sites16, 17 The best characterized of these sites is the DHP binding site. As a first step toward determining if PYTs interact with this site, a known key residue in this binding pocket was mutated, and the effects of this mutation on the ability of cp-PYT to inhibit channel currents were assessed. In the CaV1.2 subunit, T1066 is critical to DHP binding and channel inhibition.18, 19, 20 Mutating this amino acid to tyrosine disrupts DHP binding.19 The homologous amino acid in the CaV1.3 subunit is T1081 (Fig. 1a,b). Therefore, to determine the effect on DHP and PYT the corresponding threonine in CaV1.3 was mutated. This site in the Cav1.2 subunit is well established to determine DHP sensitivity.3 Mutating this threonine residue to a tyrosine (T1081Y) had no effect on CaV1.3 channel gating or current when expressed in HEK293 cells with auxiliary β3 and α2δ subunits (data not shown). However, this substitution significantly reduced the inhibition of evoked currents by a saturating concentration of the DHP isradipine (5 μM) (Fig. 1c,d).21 This mutation also significantly diminished the inhibition of evoked currents by cp-PYT at a nominally saturating concentration (100 μM) (Fig. 1c,d).
Figure 1. cp-PYT and isradipine are sensitive to the Cav1.3-T1081Y mutation.

a. Schematic diagram of the α1-subunit for neuronal L-type calcium channels. The third domain is highlighted, showing the amino acids’ sequence of the S5 segment that confers 1,4-dihydropyridine (DHP) sensitivity: CaV1.3-T1081 and CaV1.2-T1066. b. T1081 on the CaV1.3 subunit was mutated to Y, which should render the channels insensitive to DHPs. c. Both 5 μM isradipine (blue) and 100 μM cp-PYT (red) failed to significantly inhibit currents in CaV1.3-T1081Y channels, suggesting a common binding site. d. Box plot summarizing the percent inhibition observed after application of either 5 μM isradipine (blue) or 100 μM cp-PYT (red) to wild-type CaV1.3 (isradipine: n = 5, median = 90%; cp-PYT: n = 7, 60%) and CaV1.3-T1081Y (isradipine: n = 5, median = 1%; cp-PYT: n = 5, median = 2%) Ca2+ channels. Both the isradipine and cp-PYT effects were statistically significant (Mann-Whitney Rank Sum Test, **p < 0.001). e. SKP004D11, a potent and CaV1.3 selective inhibitor was tritiated in order to label the cp-PYT binding site on CaV13 L-type calcium channels. The specific binding of 3H2-SKP004D11 was determined with a displacement assay using non-labeled SKP004D11 (n = 6, median specific binding = 59%). f. 3H2-SKP004D11 specific binding was displaced from CaV1.3 LTCCs in dissociated HEK293 membranes in a dose-dependent manner that was fit with a Langmuir isotherm (Ki = ~60 nM; n = 4; means are plotted). g. Scatchard plot of the 3H2-SKP004D11 displacement data from CaV1.3 channels in dissociated membrane preparations (red; mean values from four independent experiments). The Kd of SKP004D11 for CaV1.3 LTCCs was derived from the negative-inverse of the slope from the Scatchard plot (n = 4, median = 40 nM). h. Isradipine displaced 3H2-SKP004D11 (Ki = 68 nM). This was determined by a least-squares fit of a Langmuir isotherm and employment of the Cheng-Prusoff equation. For all binding experiments the 3H2-SKP004D11 concentration was 3.0 nM and membrane protein concentrations for stably expressed CaV1.3 LTCCs in HEK-293 cells were between 0.1 and 0.4 mg/mL (see Experimental Section: Binding Experiments). i. Isradipine binding to HEK293 membranes is dependent on the presence of Cav1.3 channel protein.
The ability of the T1081Y mutation to diminish cp-PYT inhibition of CaV1.3 channel currents suggests that PYTs bind at the DHP site. To support this hypothesis, a tritiated version of one of our most potent PYT compounds (3H2-D11)13 was synthesized (Supporting Information Fig. S1) and equilibrium binding experiments were conducted with membrane fractions from HEK293 cells expressing functional CaV1.3 channels (Fig. 1e).13 The binding affinity of (3H2-D11) in membrane fractions was several-fold higher than that seen in intact cells (Fig. 1f,g), potentially because binding was voltage-dependent, as was the case for DHPs (membrane fractions were nominally at 0 mV in contrast to intact cells). Importantly, isradipine displaced (3H2-D11) binding (Fig. 1h), suggesting that PYTs and DHPs bind to physically close sites. Isradipine failed to displace a small fraction of the (3H2-D11) binding (~20%), suggesting that there was another minor PYT binding site. Our experimental system was calibrated prior to binding experiments using isradipine; a linear dependence of channel protein concentration with the specific binding of tritiated isradipine was observed (Fig. 1i).
Structural determinants of PYT binding to the CaV1.3 subunit
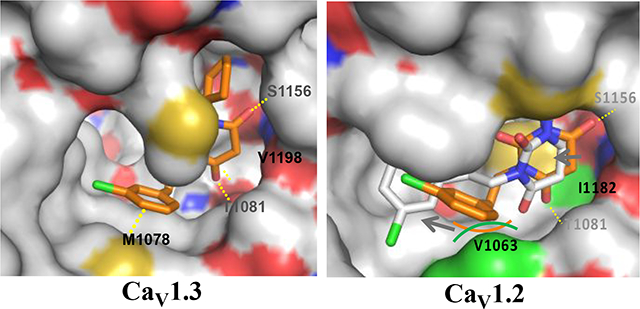
To explore the structural determinants of PYT binding to the DHP site,22 the recently published structural data for the CaV1.1 channel23 was used to construct homology models of CaV1.2 and CaV1.3 channels.24 The mapping of protein sequences in 3-D space was achieved using the Homology Modeling module, and the energy minimization of the produced structures was performed using the Prime module (OPLS3e force field), in Schrödinger software. Compound docking studies were then performed using the Induced Fit Docking module in the Schrödinger software. In these studies, both isradipine and cp-PYT were found to fit into the DHP binding pocket ringed by M1078, T1081, T1082, Q1085, F1154, S1156, Y1194, V1198, M1212, I1205, F1209, I1489, Y1492, and M1493 (Fig. 2a, b). Of those surrounding amino acids, Q1085, F1154, V1198, M1212, and M1493 are known to affect DHP binding.18–20 As shown in Fig. 2a, the benzooxadiazole oxygen of the isradipine faces S1156, and one ester on the pyridine ring of isradipine interacts with the polar residue T1082. In the case of cp-PYT, two of the three carbonyl groups interact with polar residues T1081, Q1085, and S1156 (Fig. 2b). However, the prominent interaction of cp-PYT is the hydrophobic interaction between the side chains of cp-PYT (cyclopentyl and 3-chlorophenethyl groups) and the hydrophobic residues of Cav1.3 (M1078, F1154, V1198, I1205, F1206, F1209 M1493, and F1497). In contrast to isradipine, cp-PYT binding to CaV1.3 is influenced by the interaction with M1078 and V1198 in the DHP binding pocket of the CaV1.3 model (Fig. 2b). M1078 and V1198 of CaV1.3 are the only residues that differ from CaV1.2 in that binding site.
Figure 2. Homology model of CaV1.3 and docking of cp-PYT reveals that cp-PYT uniquely engages Met1078 in CaV1.3-LTCCs.

Proposed docking poses of isradipine (a) and cp-PYT (b) in the CaV1.3 structural model. Isradipine is represented in purple and cp-PYT is represented in orange, while CaV1.3 is represented by green ribbon diagrams. Key binding site residues and interactions with ligands are shown with dark green sticks and with yellow dash lines. Sequence numbers are based on rat CaV1.3 and CaV1.2 subunits. c. Schematic diagram of the CaV1 α1-subunit. The consensus amino acid sequence of the S5 segment of the third (blue) and the S6 segment of the fourth (green) transmembrane domains of nLTCCs are shown. The docking model predicts that methionine-1078 and valine-1198 (red) of the CaV1.3 α1-subunit underlie cp-PYT selective inhibition. d. Voltage-clamp currents evoked in HEK293 cells expressing CaV1.3 (left) or CaV1.2 (right) channels; CaV1.3 channels have a pore-forming α1 subunit with an M1078V mutation, whereas CaV1.2 channels had a pore-forming a1 subunit with an V1063M mutation. Current records are shown before and after bath application of cp-PYT (100 μM). Voltage protocol is shown at the bottom. e. Population data of the inhibition observed by application of either 100 μM cp-PYT (left) or 5 μM isradipine (right) to CaV1.3 (black, cp-PYT: n = 6, 60%, isradipine: n = 5, 90%), CaV1.3-M1078V (blue, cp-PYT: n = 6, 4%, isradipine: n = 5, 78%), and CaV1.3-V1198I (green, cp-PYT: n = 6, 45%, isradipine: n = 6, 94%) L-type calcium channels (LTCCs). The inhibition of CaV1.3 as compared to CaV1.3-M1078V LTCCs by application of 100 μM cp-PYT was deemed significant by the Mann-Whitney Rank Sum Test (**p < 0.001). f. Population data of the inhibition observed by application of either 100 μM cp-PYT (left) or 5 μM isradipine (right) to CaV1.2 (black, cp-PYT: n = 8, 21%, isradipine: n = 6, 78%), CaV1.2-V1063M (blue, cp-PYT: n = 6, 47%, isradipine: n = 6, 92%), CaV1.2-I1183V (green, cp-PYT: n = 8, 12%, isradipine: n = 8, 95%), and CaV1.2-V1063M+I1183V (gray, cp-PYT: n = 10, 32%, isradipine: n = 10, 93%) L-type calcium channels. The inhibition of CaV1.2 as compared to CaV1.2-V1063M and CaV1.2-V1063M+I1183V by application of 100 μM cp-PYT was deemed significant by the Mann-Whitney Rank Sum Test (*p < 0.05).
To test whether these two amino acids account for the selective inhibition of CaV1.3 channels, two sets of site-directed mutagenesis experiments were performed. First, the CaV1.3 subunit was mutated to mimic the CaV1.2 subunit by 1) replacing Met at 1078 in domain III-S5 of the CaV1.3 subunit with a Val, which is found at corresponding site 1063 in the CaV1.2 subunit and 2) replacing Val at 1198 in domain IV-S6 of the CaV1.3 subunit with Ile found at corresponding site 1183 of the CaV1.2 subunit (Fig. 2c). Second, analogous mutations were made in the CaV1.2 subunit to make them resemble the CaV1.3 subunit, namely, V1063 in the CaV1.2 subunit was replaced with a Met, and I1183 in the CaV1.2 subunit was replaced with a Val. The ability of isradipine and PYTs to inhibit these mutated channels was assessed by expressing the channels in HEK293 cells and performing patch clamp experiments with Ba2+ as the charge carrier. All of the channel mutants were fully functional and yielded robust patch clamp currents (Supporting Information Fig. S2). The ability of cp-PYT to inhibit CaV1.3 channel currents was dramatically reduced by the M1078V mutation (Fig. 2d,e), i.e., giving it CaV1.2 binding site character. In contrast, it was not significantly altered by the V1198I mutation (Fig. 2e). None of these mutations affected inhibition by isradipine at saturating concentrations (Fig. 2e). Conversely, the V1063M mutation in the Cav1.2 channel significantly enhanced cp-PYT inhibition, i.e., giving CaV1.2 more CaV1.3 binding site character, but the I1183V mutation did not (Fig. 2d,f). Again, none of these mutations significantly altered the ability of saturating concentrations of isradipine to inhibit channel currents (Fig. 2f). These data strongly suggest that M1078 in the CaV1.3 subunit is critical to the selective inhibition of channel currents by cp-PYTs.
Why is M1078 in the CaV1.3 subunit so important? Our previous work13 suggested that the relative size of the accessible side chains of M1078 (and V1063 in the CaV1.2 subunit) might be important for PYT binding. To determine if this is the case for cp-PYT, two sets of experiments were performed. First, M1078 in the CaV1.3 subunit was replaced with amino acids having progressively less sterically hindering side chains (Fig. 3a,b). Second, V1063 in the CaV1.2 subunit was exchanged with amino acids with progressively larger side chains (Fig. 3c). These experiments revealed a correlation between the size of the amino acid side chain at either position 1078 in CaV1.3 or 1063 in CaV1.2 LTCCs and cp-PYT inhibition (Fig. 3d). In contrast, these mutations had no effect on the inhibition of currents by isradipine (Fig. 3e).
Figure 3. Modulating the steric demand of a single amino acid residue directs pyrimidine-2,4,6-trione sensitivity.

a. Schematic drawing of amino acids placed into the α1-subunit of CaV1.3 at position 1078 or CaV1.2 at position 1063. Amino acids are shown by size of accessible surface area. The native amino acid at these aligned positions is methionine for CaV1.3 (black) and valine for CaV1.2 (red). b. Resultant averaged current traces of CaV1.3 L-type calcium channels, evoked by a −80 mV to 0 mV voltage step for 100 msec, before (gray) and after 100 μM cp-PYT (blue) application. Each pairing of traces represents mutations at CaV1.3–1078 that express amino acids of descending size of accessible amino acid surface area. c. Resultant averaged current traces of CaV1.2 L-type calcium channels, evoked by a −80 mV to 0 mV voltage step for 100 msec, before (gray) and after (blue) 100 μM cp-PYT application. Each pairing of traces represents mutations at CaV1.2–1063 that express amino acids of ascending size of accessible amino acid surface area. All horizontal scale bars represent 50 msec. d. Population data of the inhibition observed by 100 μM cp-PYT application on L-type calcium channels with differing sizes of accessible amino acid surface area at either CaV1.3–1078 (black symbols): CaV1.3-M1078K (n = 5, median = 58%), Cay1.3-M1078I (n = 6, median = 37%), Cay1.3-M1078L (n = 5, median = 31%), Cay1.3-M1078V (n = 8, median = 4%), and CaV1.3-M1078A (n = 4, median =17%); or at CaV1.2–1063 (red symbols): CaV1.2-V1063I (n = 5, median = 48%), CaV1.2-V1063L (n = 4, median = 35%), CaV1.2-V1063M (n = 6, median = 47%), and CaV1.2-V1063K (n = 4, 40%) relative to the inhibition of 100 μM cp-PYT on wild-type CaV1.3 (black symbols) (n = 5, median = 60%) or CaV1.2 (red symbols) (n = 6, median = 21%) LTCCs. Open black circles (CaV1.3) or red circles (CaV1.2) represent individual experiments, while filled circles represent median responses. Linear regression analysis of the median cp-PYT responses led to a linear relationship of the % inhibition and size of accessible side chain (n = 11, r2 = 0.57). e. Population data of the inhibition observed by 5 μM isradipine for channels with differing sizes of accessible amino acid surface area at either CaV1.3–1078 (black symbols): CaV1.3-M1078K (n = 4, median = 88%), CaV1.3-M1078I (n = 5, median = 85%), CaV1.3-M1078L (n = 6, median = 69%), CaV1.3-M1078V (n = 6, median = 78%), and CaV1.3-M1078A (n = 4, median = 84%); or at CaV1.2–1063 (red): CaV1.2-V1063I (n = 5, median = 91%), CaV1.2-V1063L (n = 6, median = 95%), CaV1.2-V1063M (n = 6, median = 92%), and CaV1.2-V1063K (n = 4, median = 72%) relative to the inhibition of 5 μM isradipine on wild-type CaV1.3 (black symbols) (n = 7, median = 90%) or CaV1.2 (red symbols) (n = 5, median = 78%) LTCCs. Open circles black (CaV1.3) or red circles (CaV1.2) represent individual experiments, while filled circles represent medians. Linear regression analysis of the median isradipine responses did not result in a linear relationship of the % inhibition and size of accessible side chain (n = 11, r2 = 0.04).
PYT inhibition of CaV1.3 channels is voltage-dependent
A key feature of DHP inhibition of CaV1 channels is its voltage-dependence. At depolarized membrane potentials, DHPs are several orders of magnitude more potent than at hyperpolarized membrane potentials.5, 25 Although the determinants of this voltage-dependence have not been completely resolved, conformational changes in the shape of the DHP binding pocket are likely to be responsible for the shift in affinity. Given that PYTs bind in the DHP pocket, their inhibition of CaV1.3 channels could be voltage-dependent as well. To test this hypothesis, CaV1.3 channel currents in HEK293 cells were studied using voltage-clamp techniques. Cells were held at either −80 mV or −50 mV prior to stepping to 0 mV to open channels. At relatively hyperpolarized holding potential (−80 mV), isradipine (5 μM) modestly inhibited peak currents, but this inhibition was dramatically increased by holding at −50 mV, as expected from previous studies (Fig. 4a). As predicted by the hypothesis that cp-PYTs interact with the DHP pocket, the ability of cp-PYT (10 μM) to inhibit CaV1.3 channel currents also was voltage-dependent and increased significantly at depolarized holding potentials (Fig. 4b–d).
Figure 4. cp-PYT inhibition of CaV1.3 channel currents is voltage-dependent.

a. Exemplary current traces evoked in HEK293 cells expressing CaV1.3 channels before (control, black) and after application of isradipine (100 nM, blue) from either a holding potential (VH) = − 80 mV (left) or VH = −50 mV (right). The test step voltage was 0 mV. b, c. Examples of current traces before (black) and after cp-PYT (10 μM, red) application from either VH = − 80 mV (b, left) or VH= −50 mV (c, left) (test step was to 0 mV). Time course of cp-PYT and Cd2+ inhibition of CaV1.3 peak currents from either VH = −80 mV (b, right) or VH = −50mV (c, right). d. Box plot summary of percentage inhibition of CaV1.3 peak currents by isradipine and cp-PYT at either VH = −80 mV or −50 mV (test step to 0 mV). Isradipine (VH: −50 mV, n = 6, median = 92% vs. VH: − 80 mV, n = 5, median = 43%; Mann-Whitney test, P = 0.0043). cp-PYT (VH: −50 mV, n = 6, median = 46% vs. VH: - 80 mV, n = 5, median = 26%; Mann-Whitney test, P = 0.0058).
PYT binding to a low affinity site slows channel deactivation
As shown above, isradipine failed to displace a portion of the PYT (i.e., D11) binding (Fig. 1h), suggesting that there was a second low affinity PYT binding site on the CaV1.3 channel. To test this hypothesis, the effects of low (10 μM) and high (50 μM) concentrations of cp-PYT on channel functioning were studied. At low concentration, cp-PYT decreased the CaV1.3 channel currents evoked by a voltage step to 0 mV in HEK 293 cells, but did not alter tail current kinetics or channel deactivation rates when the voltage was stepped back to −80 mV (Fig. 5a–c). However, at high cp-PYT concentration, not only was the current evoked by a depolarizing step diminished, but tail current kinetics slowed (Fig. 5d–f). These results support the proposition that slowing of deactivation is dose-dependent. To determine if this slowing of deactivation kinetics relies on binding to the DHP site, these experiments were repeated in HEK293 cells expressing the M1078V mutation, which disrupted cp-PYT binding to the DHP pocket. In these mutant channels, the effect of cp-PYT (100 μM) on the peak current was lost (Fig. 2d), but the slowing of tail currents persisted (Fig. 5g,h), suggesting that the second binding site was elsewhere on the channel. Together, these data show that deactivation kinetics are slowed only at high concentrations of cp-PYT, and this slowing is independent of the DHP binding site, clearly suggesting that there was a second, low affinity binding site that was responsible for the cp-PYT effects on channel deactivation.
Figure 5. High micromolar concentrations of cp-PYT slowed tail currents independent of the effect on peak current.

a. CaV1.3 channel currents evoked in HEK293 cells by a voltage step to 0 mv (holding at −80 mV) before and after application of cp-PYT (10 μM). b. Semilog plots of absolute tail currents from a. c. Summary of the principal tail current time constant before (black) and after (red) compound application. There were no changes (n = 4, paired t-test, p = 0.86). d. Exemplary current traces evoked by the same protocol before and after application of cp-PYT at high concentration (50 μM). e. Semilog plot of absolute tail currents from CaV1.3 channels before (black) and after (red) cp-PYT (50 μM) application (e) cp-PYT concentration. f. Plot of the principal tail time constants before and after cp-PYT application. Note the slowing of the tail current kinetics (n = 3, paired t-test, p = 0.04). g. Semilog plot of absolute tail currents from CaV1.3-M1078V mutant channels from before (black) and after (red) cp-PYT (50 μM) application. h. Plot of the principal tail time constant before and after cp-PYT application (n = 4, *p<0.05, Mann-Whitney).
PYTs diminish Ca2+ currents and mitochondrial oxidant stress in dopaminergic neurons
As outlined above, native CaV1.3 channels are heterogeneous. They can differ not only in auxiliary subunits, but also in the alternative splicing of the CaV1.3 subunit itself.26, 27, 28 Moreover, channel proteins are post-translationally modified, most notably by phosphorylation.29 To assess the ability of cp-PYT to inhibit native CaV1.3 channels, dopaminergic neurons in the substantia nigra (SN) of mice were studied. These neurons robustly express CaV1.3 Ca2+ channels that give rise to large oscillations in cytosolic Ca2+ concentration and mitochondrial oxidant stress.5, 30, 31 As a first step in this analysis, SN dopaminergic neurons were acutely isolated from brain slices obtained from young adult mice to allow channels to be voltage-clamped (Fig. 6a, left). Cell-attached recordings from these cells revealed that they exhibited the autonomous activity typical of this cell type (Fig. 6a, right). In whole cell recordings with Ba2+ as the charge carrier, voltage ramps from −70 mV to +30 mV yielded currents with typical voltage dependence.32 Application of a low concentration (5 μM) of cp-PYT produced a modest inhibition of currents that was increased by elevating the concentration (50 μM) (Fig. 6b). Because several channel types contribute to these whole cell currents,32, 33 the experiment was redone in SN dopaminergic neurons from a knockout mouse in which the gene coding for the pore-forming subunit of the CaV1.3 channel (CACNAD1) had been deleted.34 In these cells, cp-PYT (50 μM) had no effect (Fig. 6c,d), indicating that cp-PYT was selectively inhibiting CaV1.3 channels.
Figure 6. cp-PYT inhibits Cav1.3 calcium channels in adult SN dopaminergic (DA) neurons from wild-type mice.

a. Left, TH-positive SN DA neuron (scale bar, 10 μM). Right, cell-attached firing (2–4 Hz) from a typical TH-positive, dissociated SN DA neuron. b. Voltage clamp recordings of an adult (P25) SN dopaminergic neuron from an adult WT mouse showing inhibition by cp-PYT (5 μM) (gray) and (50 μM) (red) application. c. Voltage clamp recordings of an adult (P25) SN dopaminergic neuron from an adult Cav1.3-KO mouse showing a lack of inhibition by cp-PYT (50 μM) application (red). d. Box-plot summarizing a dose-dependent effect of cp-PYT (gray, 5 μM) and (red, 50 μM) application on the maximum calcium permeability of SN dopaminergic neurons from adult WT mice as compared to the maximum calcium permeability of SN DA neurons from a CaV1.3-KO mouse (black, n = 6, p<0.001, Mann-Whitney for 50 μM). e. Left, Drawing of an SN dopaminergic neuron derived from a z-stack obtained with two photon laser scanning microscopy following filling of the cell with Alexa568 (20 μM) and Fluo-4 (200 μM) through a whole-cell recording glass pipette. Calcium imaging was performed along the dendrite, ~100 pm from the soma (location indicated by arrow). Somatic voltage recordings before (left, black) and after (right, red) application of cp-PYT (20 μM). 2PLSM measurements of Fluo-4 fluorescence normalized by the baseline Fluo-4 fluorescence (F0) before (black) and after (red) cp-PYT application. f. Comparison of single calcium transients revealed a marked reduction in amplitude following cp-PYT administration; hashed line denotes the point at which the spike occurred. g. Summary showing that cp-PYT significantly reduced the amplitude of spike-associated dendritic Ca2+ transients (p< 0.01; n = 6, Mann-Whitney). h. Top, schematic of the hypothesized signaling between Cav1.3 channels and mitochondria. Bottom, a high magnification image of a SN dopaminergic neuron from a transgenic TH-mito-roGFP mouse showing cytoplasmic, but not nuclear, labeling. i. Mito-roGFP measurements from a SN dopaminergic neuron revealed a high basal oxidation (left, black trace). Isradipine (right, black trace) and cp-PYT (right, red trace) treatment diminished mitochondrial oxidation. j. Box-plots summarizing mean redox measurements in control (black, n = 9) and following isradipine treatment (black, n = 8) or cp-PYT treatment (red, n = 6); mitochondrial oxidant stress was significantly diminished by both isradipine and cp-PYT treatment (*p<0.05, Mann-Whitney).
As the majority of plasma membrane CaV1.3 channels in SN dopaminergic neurons are positioned in dendrites, two photon laser scanning microscopy (2PLSM) and whole cell patch clamping were used in ex vivo brain slices from young adult mice to assess the impact of cp-PYT on dendritic Ca2+ channels. After dialysis of the Ca2+ dye Fluo-4 (100 μM) with the patch electrode, distal dendrites were imaged while SN dopaminergic neurons were pacemaking (Fig. 6e). Application of cp-PYT (50 μM) to the slice had no effect on pacemaking, but significantly reduced the dendritic Ca2+ oscillations, consistent with an inhibition of CaV1.3 channel opening (Fig. 6e,g).30 Overlaying the dendritic fluorescence signals from before and after cp-PYT application revealed that the greatest inhibition occurred during the slow, depolarizing phase prior to spike initiation (Fig. 6f), an observation consistent with the ability of CaV1.3 channels to activate at hyperpolarized membrane potentials.4, 21, 30 Ca2+ entry through CaV1 channels in SN dopaminergic neurons triggers ryanodine receptor-dependent release of Ca2+ from intracellular stores and stimulation of mitochondrial oxidative phosphorylation (OXPHOS).35 One by-product of increased OXPHOS is the production of reactive oxygen species (ROS) that can be monitored using a genetically- encoded, mitochondrially-targeted, redox-sensitive variant of green fluorescent protein (mito-roGFP) (Fig. 6h).30, 31,35, 36 In ex vivo brain slices from a mouse expressing mito-roGFP under control of a tyrosine hydroxylase promoter fragment (to limit expression to dopaminergic neurons), application of isradipine (1 μM) diminished mitochondrial oxidant stress (Fig. 6i, j), as reported previously.30, 31 Bath application of cp-PYT (50 μM) also significantly diminished mitochondrial oxidant stress in SN dopaminergic neurons (Fig. 6i, j), consistent with the proposition that it inhibited native CaV1.3 channels linked to mitochondrial OXPHOS.30
DISCUSSION
The rationale for a CaV1.3 channel selective inhibitor
Although they constitute a relatively small percentage (~10%) of brain CaV1 Ca2+ channels, CaV1.3 channels have been implicated in a variety of psychomotor disorders,6, 9, 10, 11 the best characterized of these is Parkinson’s disease (PD). The cardinal motor symptoms of PD are caused by the loss of SN dopaminergic neurons.11 Studies in mouse and human models have implicated CaV1.3 Ca2+ channels in mitochondrial dysfunction thought to contribute to the degeneration of these neurons.11 Buttressing this conclusion, epidemiological studies have shown that the treatment of hypertension with DHP, but not other hypertension medication, significantly lowers the risk of developing PD.37 This combination of experimental and clinical findings led to the Phase 3, disease-modification trial in early stage PD patients with the DHP isradipine (STEADY-PD).37, 38 Isradipine was chosen for this trial because it had the highest relative affinity for CaV1.3 channels among clinically approved DHPs.3 However, because of its cardiovascular effects, isradipine dosing in PD patients was limited and not optimal for inhibition of CaV1.3 channels.37 As the distribution of CaV1.3 channels is much more narrow than that of CaV1.2 channels,3 a CaV1.3-selective inhibitor is expected to be much more effective for slowing progression of PD than a DHP.
There are other clinical reasons to pursue a selective inhibitor of CaV1.3 channels. CaV1.3 channels in ventral tegmental area dopaminergic neurons have been implicated in the synaptic events underlying drug addiction, and administration of either isradipine or cp-PYT has been shown to disrupt the reinstatement of alcohol- and cocaine-seeking in rodents.9 In addition, gain of function mutations in CaV1.3 channels have been implicated in autism spectrum disorder and epilepsy,10, 39 further broadening the therapeutic value of a CaV1.3 selective inhibitor.
From a translational perspective, the fact that cp-PYT is a NAM, rather than a channel pore blocker, is significant. Undoubtedly, the modest side-effect profile of DHPs stems from their being NAMs, because indiscriminate blockage of CaV1.2/CaV1.3 channels would be lethal. Their preferential interaction with channels in depolarized membranes or use-dependence increases their selectivity. SN dopaminergic neurons, and other neurons at risk in PD that have been studied, are spontaneously active and reside primarily at membrane potentials where DHPs and cp-PYTs preferentially bind to the DHP site.
A shared binding site
Our studies support the conclusion that DHPs and cp-PYT share a common binding pocket. The first piece of evidence supporting this conclusion is that inhibition of CaV1.3 channels by the DHP isradipine and by cp-PYT is dependent upon threonine at 1081, a residue found in the DHP binding pocket common to CaV1.2 and CaV1.3 α1 subunits (Fig. 2a–d).18, 19, 20 Substitution of threonine at 1081 with tyrosine disrupted the ability of isradipine and cp-PYT to inhibit evoked currents. An additional piece of evidence for a shared binding site is that equilibrium membrane binding experiments revealed that isradipine displaced binding of a tritiated PYT (D11), and tritiated D11 binding was displaced by cp-PYT (Fig. 1e–h).
Elucidation of the important residues in the DHP site that determined selective binding of cp-PYT to the CaV1.3 α1 subunit came from our homology models constructed from the CaV1.1 structure.23, 40 Examination of the region near the conserved threonine residue (1081 in CaV1.3 and 1066 in CaV1.2) revealed minor structural differences between CaV1.2 and CaV1.3 subunits (Fig. 2a,b). Docking simulations revealed that in the CaV1.3 α1 subunit, the 3-chlorophenethyl moiety of cp-PYT interacts with methionine-1078, but isradipine does not. Sequence alignment showed that methionine-1078 in the CaV1.3 α1 subunit is not conserved in the CaV1.2 α1-subunit, but is replaced by a valine (at 1063). Another structural difference seen in the homology models is that the valine at 1198 in the IV transmembrane domain of the CaV1.3 subunit is replaced by isoleucine at 1183 in the CaV1.2 subunit. In the homology model, isradipine could be docked into the DHP binding pocket regardless of which amino acid was present. However, valine at 1063 in CaV1.2 created significant steric hinderance to cp-PYT binding but isoleucine 1183 caused only slight repulsion (Supporting Information Fig. S3). To test the homology modeling results, site-directed mutagenesis was performed. Mutation of methionine 1078 to valine in CaV1.3, thereby giving it CaV1.2 character, had no effect on the inhibition produced by isradipine, but dramatically attenuated the effects of cp-PYT on CaV1.3 channel currents (Fig. 2e) – outcomes remarkably predicted by the homology model. In contrast, mutation of valine at 1198 in CaV1.3 to isoleucine, again giving it CaV1.2 character, had no effect on the ability of isradipine or cp-PYT to inhibit CaV1.3 channel currents (Fig. 2e). The homology model also accurately predicted the consequences of the corresponding mutations to the CaV1.2 subunit. Specifically, mutation of valine at 1063 to methionine, giving it CaV1.3 character, increased the ability of cp-PYT to inhibit evoked currents, whereas mutation of isoleucine at 1183 to valine had no effect on cp-PYT inhibition (Fig. 2f). Neither mutation affected the ability of isradipine to inhibit CaV1.2 channel currents (Fig. 2f). Together, these results provide strong support for the proposition that the selective inhibition of CaV1.3 channels by cp-PYT depends on its interaction with a single amino acid, methionine at 1078, in the DHP binding pocket.
If this selectivity arises from hydrophobic interactions with methionine 1078, as suggested by our previous structure-activity studies,41 then selectivity should be a function of the surface area of the side chains of the residue at position 1078 in CaV1.3. To test this hypothesis, methionine at 1078 in CaV1.3 was mutated to alanine, valine, leucine, isoleucine, or lysine (Fig. 3). Consistent with the homology model predictions, as the surface area of the residue decreased, so did inhibition by cp-PYT. None of these mutations was predicted to affect inhibition by isradipine, and none did. Conversely, mutation of valine at 1063 in the CaV1.2 α1-subunit to isoleucine, leucine, methionine, or lysine revealed that increasing the surface area at this site increased the ability of cp-PYT to inhibit currents, without affecting the efficacy of isradipine.
Reconciliation with previously reported results
Our results not only provide a map for future drug discovery work, they also provide an explanation for previous work that disputed the inhibitory action of cp-PYT on CaV1.3 channels. Ortner et al.14 claimed that cp-PYT was an agonist, rather than an inhibitor, of CaV1.3 channels based on their ability at high micromolar concentrations to slow deactivation of heterologously expressed channels. The cloned α1 subunit used in their experiments, however, had a mutation at methionine 1078, the residue in native channels now shown to be critical for the binding of cp-PYT to the DHP site (Fig. 2). The ‘agonist’ effect on tail current kinetics is attributable to a low affinity binding site we observed elsewhere on the channel, because it was seen even when the DHP site had been mutated (Fig. 5g). It remains to be determined where this site resides, but it is of marginal interest, as in our experiments with native expression systems, as well as those of others,9 there is little evidence that this site significantly mitigates the inhibitory effects of cp-PYT on CaV13 channels. What is less readily explained is the apparent failure of cp-PYT to inhibit CaV13 channels in adrenal chromaffin cells.14
In another series of experiments with heterologously expressed CaV1.3 channels, Huang et al.15 found cp-PYT to be a weak inhibitor of CaV1.3 channels. As virtually all of these experiments were conducted from negative holding potentials, this result is explained by the voltage-dependent interaction of cp-PYT with the DHP binding site. In our work (Figs. 4 and 5), from a holding potential of −80 mV, which was used in the Huang et al. studies, the inhibition of CaV1.3 channel currents by cp-PYT was only modest. But at a more physiologically relevant membrane potential of −50 mV, the inhibition was much more profound. This is precisely the phenomenology expected of a NAM. Lastly, Huang et al. described a β subunit dependence of cp-PYT inhibition. Given the robust effect of β subunits on the voltage-dependence of channel gating, most likely, it reflects the effect of voltage on the structure of the α1-subunit and on accessibility of the DHP binding site. To test this hypothesis, the relationship between channel activation and cp-PYT inhibition should be systematically explored.
Toward a clinically useful drug
Although cp-PYT discriminates between CaV1.3 and CaV1.2 channels, it has limitations. The key to moving this effort forward is our discovery that cp-PYT is a voltage-dependent NAM that interacts with the DHP binding site on CaV1.3 channels. Thus, drug screening efforts need to reliably manipulate membrane voltage for a structure-activity relationship to be ascertained. Recently developed planar patch clamp workstations should allow this effort to move forward on a reasonable timescale. Among the goals of this effort will be to increase the potency of CaV1.3 selective compounds by optimizing the interaction with methionine at 1078, utilizing our homology model for predicted selectivity and efficacy.
CONCLUSIONS
The key findings of our study are that cp-PYT acts as a selective, voltage-dependent, NAM of CaV1.3 Ca2+ channels by binding to the DHP site on the pore-forming α1 subunit and that this interaction depends on a CaV1.3-specific methionine residue at position 1078. The use of homology models developed based on a CaV1.1 structure predicted accurately what should occur upon mutation of various residues in CaV1.3 and CaV1.2, which would lead to CaV1.3 selectivity and that the interaction of cp-PYT with CaV1.3 is hydrophobic and sterically controlled. This selective inhibition is shown to be voltage dependent, as expected for a NAM. The mechanism of action of cp-PYT appears to be inhibition of mitochondrial OXPHOS, thereby reducing mitochondrial stress. In addition to effectively inhibiting heterologously-expressed CaV1.3 channels, cp-PYT also inhibited natively expressed channels in mouse SN dopaminergic neurons. These studies pave the way for development of CaV1.3 channel selective NAMs that could be used in the treatment of a variety of psychomotor disorders, such as Parkinson’s disease, autism spectrum disorder, bipolar disorder, and drug abuse, in which opening of these channels contributes to pathogenesis.
MATERIALS AND METHODS
Chemical Synthesis
The synthesis of cp-PYT was previously described.13
Synthesis of 3[H2]-D11. To a stirred mixture of NiCl2 (3 mg, 0.023 mmol) and (3-chlorophenyl)acetonitrile (40 mg, 0.26 mmol) in methanol (3 mL), NaBT4 (5 mg, 13.1 Ci/mmol) was added portionwise (4 times) over 20 min at room temperature. After gas evolution ceased, NaBH4 (15 mg) was added to the mixture, which was stirred for an additional 1 h. The mixture was diluted with 10 mL of dichloromethane and mixed with (±)-endo-norbornyl isocyanate (43 mg, 1.2 equiv) at room temperature. The reaction mixture was stirred for 1 h, quenched with dichloromethane (20 mL) and brine (30 mL). The organic layer was collected, dried with anhydrous MgSO4, and concentrated under vacuum to afford a yellow oil. The yellow oil was passed through a 4 g silica gel cartridge using an ethyl acetate and hexane mixture (1:1) to give the desired pure urea (20 mg, 26%). To a solution of the urea (15 mg, 0.05 mmol) in dichloromethane (10 mL), malonyl chloride (10 μL, 0.06 mmol) was added with vigorous stirring. After being stirred for an additional 1 h, the solution was washed with brine, dried over anhydrous MgSO4, and concentrated at reduced pressure to a small volume. The resulting mixture was purified using a silica gel cartridge (4 g) to give pure 3[H]-SKP004D11 (9 mg, 7.7 Ci/mol).
Cell Culture
Rat CaV1.3 α1D (GenBank accession number: AF370010) containing all alternative splice sites, rat Cavβ3 (GenBank accession number: M88751), rat Cavα2δ−1 (GenBank accession number: 286488), and rabbit CaV1.2 α1C (GenBank accession number: P15381) cDNAs were used. The potential differences in drug sensitivity caused by alternative splice isoforms of either CaV1.2 or CaV1.3 were not evaluated in these studies. All wild-type constructs were provided by Dr. Diane Lipscombe (Brown University) and Dr. Johannes Hell (University of Iowa). General methods for constructs development and stable transfection of CaV1.3 α1D, CaVβ3, and CaVα2δ−1 into HEK-293 cells for FLIPR screens were described in our earlier study1. Transient transacted cells: tsA201 cells were maintained in D-MEM medium supplemented with 10% fetal bovine serum (Life Technologies) without antibiotics. Cells were trypsinized and plated on poly-D-lysine coated coverslips (BD Bioscience) 24 h before transfection. A mixture of CaV1.3 α1D or mutated CaV1.2/3 α1 C/D, CaVβ3, and CaVα2δ−1 cDNA (in pcDNA3.1 vectors) at a molar ratio 1:1:1 together with 1/40 (w/w) GFP cDNA (Life Technologies) were transfected into tsA201 cells using Mirus TransIT®-LT1 transfection reagent (Mirus®) according to the manufacturer’s protocol. GFP-positively labeled cells were recorded 48 h post-transfection.
Generation of the CaV1.3/CaV1.2 mutations
Mutation constructs were generated using the Agilent® Quik®Change II Site-Directed Mutagenesis Kit (catalog#: 200524–5). Primer design and PCR was performed according to the standard procedures detailed in the instruction manual. In short, rat α1D and rabbit α1C genes were mutated to desired constructs using primers detailed in Table S1, S2. PAGE purified primers were made by IDT. Following the digestion with Dpn I, the PCR mixture was used to transform XL1-Blue supercompetent cells and the sample was plated onto LB + ampicillin (100 μg/mL) plates. The colonies were grown in (5 mL) LB broth containing 100 μg/mL ampicillin and the plasmids were isolated using the Qiagen® QIAprep Spin Miniprep Kit (catalog#: 27104) according the instruction manual. All the mutations were verified by sequencing.
Whole-cell Patch Clamp Electrophysiology
After 48 h of incubation at 37 °C on poly-D-lysine treated coverslips, transiently transfected cells underwent whole-cell patch-clamp electrophysiology. The external solution contained the following (in mM): 140 NaCl, 2 KCl, 2 MgCl2, 1 CaCl2, 15 HEPES, and 10 dextrose at pH 7.4 and an osmolarity of ~305 mOSm/L. Barium currents were resolved using a solution with the following (in mM): 140 NaCl, 2 KCl, 2 MgCl2, 10 BaCl2, 10 HEPES, and 10 dextrose at pH 7.4 and an osmolarity of ~310 mOSm/L. The test compound stock solutions in DMSO (10 mM or just DMSO) were diluted in the barium recording solution to the desired saturating concentration, which was perfused (2 mL/min) into the recording chamber while measuring the evoked barium currents. Barium currents were measured from whole-cell voltage patch-clamp recordings using the Pulse 8.4 software data acquisition system (HEKA, Germany). Signals were low-pass filtered at 1 kHz, digitized (sampled) at 10 kHz and were amplified with an Axopatch 200B patch-clamp amplifier (Axon Instruments). Barium currents were evoked by a depolarizing voltage step from a holding potential of −80 mV to 0 mV for 100 msec with a frequency of 0.05 Hz at room temperature (22 °C −25 °C ). Patch pipettes were pulled from thin-wall borosilicate glass (Sutter Instruments) coated with dental wax and maintained at a resistance of approximately 3–4 mΩ. Internal pipette solutions contained the following (in mM): 180 NMG (N-methyl-D-glucosamine), 40 HEPES, 4 MgCl2, 12 phosphocreatine, 0.1 leupeptin, 2 Na2ATP, 0.5 Na3GTP, 5 BAPTA, pH 7.2–7.3 neutralized by addition of phosphoric acid and an osmolarity of ~280 mOSm/L. Electrophysiological signals were analyzed using Clampfit® 9.2 (Axon Instruments) and IgorPro6® software.
Binding experiments
CaV1.3 membranes from stably expressing HEK-293 cells (~1 X 107 cells/harvest) were washed twice with HEPES buffer (40 mM HEPES, 100 mM NaCl, pH 8.0) and then removed from culture dishes. After harvesting the cells by low speed centrifugation (4,000 X g for 15 min) and resuspension in HEPES buffer with NaCl, cells were lysed with a hand-held Potter homogenizer on ice, and membranes were harvested by high speed centrifugation (100,000 X g for 30 min at 4 °C). These homogenization and centrifugation steps were repeated two additional times. A BSA assay was conducted to determine the protein concentration per sample, and for all of the experiments carried out in this study, 0.4 mg/mL/reaction was used. Reaction mixtures for binding experiments consisted of the following (which were added in this order to a total volume of 1 mL): HEPES buffer, membrane fractions, cold (non-tritiated) compound, and hot (tritiated) compound. The reactions were stopped after 90 min incubation at room temperature and were washed, vacuum filtered, and collected on Whatman GF/C filter paper. After incubation in scintillation fluid for 1 h at room temperature, the amount of bound tritium was determined by scintillation counting. Nonspecific binding of SKP004D11 was determined by displacement of tritiated SKP004D11 by a saturating concentration of ‘cold’ compound. The Kd of SKP004D11 for CaV1.3 LTCCs was derived from a least-squares fit of a Langmuir isotherm (Fig. 1f) and the negative-inverse of the slope from a Scatchard plot (Fig. 1g). Displacement data were fit with a Langmuir isotherm using a least-square method and the Ki estimated using the Cheng-Prusoff equation. For all binding experiments, the 3H-SKP004D11 concentration was 3.0 nM and membrane protein concentrations for stably expressed CaV1.3 LTCCs in HEK-293 cells were between 0.1 and 0.4 mg/mL.
Generation of CaV1.3/CaV1.2 homology models and molecular docking
The structural models of α1 subunit of CaV1.2 and CaV1.3 were built using the recently reported Cryo-EM structure of rabbit CaV1.1 (3.6 A) (PDB code: 5GJW) as a structural template. The missing or overlapping residues, charge, and protonation state of CaV1.1 were fixed using the Protein Preparation Wizard module in Schrodinger 11.9. The sequences of four domains (I-IV) that have voltage sensing segments (S1-S4), central pore forming segments (S5-S5), and P-loop of Cav1.2 (CAC1C_HUMAN) as well as Cav1.3 (CAC1D_HUMAN) were extracted and aligned with the CaV1.1 template using a previously described method23. The sequence alignment was also checked using Clustral omega multiple sequence alignment program. In this model, long extracellular loops that are far from the DHP binding site are excluded because of a lack of structural information. The sequence similarity between CaV1.1 and CaV1.3 is 67% after exclusion of long extracellular loops. The mapping of protein sequences in 3-D space was achieved using the Homology Modeling module, and the energy minimization of produced structures was performed using the Prime module (OPLS3e force field) in Schrödinger software. Docking of ligands with produced proteins was performed with the Induced Fit Docking module in the Schrödinger software package. The protonation, charges, and alternative overlapping atoms were refined using the Protein Preparation Wizard module in Schrödinger software.
Statistics
To avoid errors associated with deviations from normality in the sampling distributions, box plots were used to present data summaries. In box plots, the median is shown as the central bar of the box and the upper and lower interquartiles are presented at the edges of the box; the line from the top and bottom of the box show the distribution limits, excluding outliers (defined as points that are greater than 1.5X the interquartile range from the median). Hypothesis testing relied on the non-parametric, Mann-Whitney U test of significance unless otherwise stated. Differences were considered statistically significant if p < 0.05. Control and experimental groups were interleaved where possible.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Michael J. Fox Foundation, JPB and IDP Foundations, NIH Blueprint Neurotherapeutics program (U01NS080409), the NIH NINDS (P50NS047085), and the Northwestern University Chemistry of Life Processes Institute (Innovation Award) for financial support. We also thank R. Miller for his scientific advice regarding the membrane binding experiments and G. Swanson for the use of equipment and lab space to conduct radioligand binding experiments. S. Kang was supported by National Research Foundation of Korea (NRF) grants funded by MSIT (2018R1A5A2025286 and 2019R1A2C2004142).
ABBREVIATIONS USED
- PD
Parkinson’s disease
- ASD
Autism spectrum disorder
- DHP
dihydropyridine
- NAM
Negative allosteric modulator
- PYT
Pyrimidine-2,4,6-trione
- cp-PYT
1-(3-Chlorophenethyl)-3-cyclopentylpyrimidine-2,4,6-trione
- SN
Substantia nigra
- OXPHOS
Oxidative phosphorylation
- ROS
Reactive oxygen species
Footnotes
The authors declare no competing financial interest
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Synthesis of [3H]-D11, current-voltage relationships of CaV1.3/CaV1.2 mutation constructs in domains III and IV, CaV1.3 and CaV1.2 homology models with cp-PYT docked in to show repulsive effects
REFERENCES
- 1.Catterall WA (2011). Voltage-gated calcium channels. Cold Spring Harb Perspect Biol, 3(8), a003947. doi: 10.1101/cshperspect.a003947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Catterall WA, Striessnig J, Snutch TP, Perez-Reyes E, & International Union of P (2003). International Union of Pharmacology. XL. Compendium of voltage-gated ion channels: calcium channels. Pharmacol Rev, 55(4), 579–581. doi: 10.1124/pr.55.4.8 [DOI] [PubMed] [Google Scholar]
- 3.Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U, … Striessnig J (2009). Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol, 75(2), 407–414. doi: 10.1124/mol.108.049981 [DOI] [PubMed] [Google Scholar]
- 4.Helton TD, Xu W, & Lipscombe D (2005). Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci, 25(44), 10247–10251. doi: 10.1523/JNEUR0SCI.1089-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guzman JN, Sanchez-Padilla J, Chan CS, & Surmeier DJ (2009). Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci, 29(35), 11011–11019. doi: 10.1523/JNEUR0SCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nanou E, & Catterall WA (2018). Calcium Channels, Synaptic Plasticity, and Neuropsychiatric Disease. Neuron, 98(3), 466–481. doi: 10.1016/j.neuron.2018.03.017. [DOI] [PubMed] [Google Scholar]
- 7.Striessnig J, Koschak A, Sinnegger-Brauns MJ, Hetzenauer A, Nguyen NK, Busquet P, … Singewald N (2006). Role of voltage-gated L-type Ca2+ channel isoforms for brain function. Biochem Soc Trans, 34(Pt 5), 903–909. doi: 10.1042/BST0340903. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, … Surmeier DJ (2006). Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron, 50(3), 443–452. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 9.Degoulet M, Stelly CE, Ahn KC, & Morikawa H (2016). L-type Ca(2)(+) channel blockade with antihypertensive medication disrupts VTA synaptic plasticity and drug-associated contextual memory. Mol Psychiatry, 21(3), 394–402. doi: 10.1038/mp.2015.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Striessnig J, Bolz HJ, & Koschak A (2010). Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels. Pflugers Arch, 460(2), 361–374. doi: 10.1007/s00424-010-0800-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Surmeier DJ, Obeso JA, & Halliday GM (2017). Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci, 18(2), 101–113. doi: 10.1038/nrn.2016.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siddiq A; Mukhtar I; Baig SG Hallmarks of calcium channel blockers: a review. World J. Pharm. Pharmaceut. Sci 2019, 8, 1674–1687. [Google Scholar]
- 13.Kang S, Cooper G, Dunne SF, Dusel B, Luan CH, Surmeier DJ, & Silverman RB (2012). CaV1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson’s disease. Nat Commun, 3, 1146. doi: 10.1038/ncomms2149 [DOI] [PubMed] [Google Scholar]
- 14.Ortner NJ, Bock G, Vandael DH, Mauersberger R, Draheim HJ, Gust R, … Striessnig J (2014). Pyrimidine-2,4,6-triones are a new class of voltage-gated L-type Ca2+ channel activators. Nat Commun, 5, 3897. doi: 10.1038/ncomms4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang H, Ng CY, Yu D, Zhai J, Lam Y, & Soong TW (2014). Modest CaV1.342-selective inhibition by compound 8 is beta-subunit dependent. Nat Commun, 5, 4481. doi: 10.1038/ncomms5481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brauns T, Prinz H, Kimball SD, Haugland RP, Striessnig J, & Glossmann H (1997). L-type calcium channels: binding domains for dihydropyridines and benzothiazepines are located in close proximity to each other. Biochemistry, 36(12), 3625–3631. doi: 10.1021/bi9613584 [DOI] [PubMed] [Google Scholar]
- 17.Striessnig J, Goll A, Moosburger K, & Glossmann H (1986). Purified calcium channels have three allosterically coupled drug receptors. FEBS Lett, 197(1–2), 204–210. [DOI] [PubMed] [Google Scholar]
- 18.Catterall WA, & Swanson TM (2015). Structural Basis for Pharmacology of Voltage-Gated Sodium and Calcium Channels. Mol Pharmacol, 88(1), 141–150. doi: 10.1124/mol.114.097659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitterdorfer J, Wang Z, Sinnegger MJ, Hering S, Striessnig J, Grabner M, & Glossmann H (1996). Two amino acid residues in the IIIS5 segment of L-type calcium channels differentially contribute to 1,4-dihydropyridine sensitivity. J Biol Chem, 271(48), 30330–30335. [DOI] [PubMed] [Google Scholar]
- 20.Sinnegger MJ, Wang Z, Grabner M, Hering S, Striessnig J, Glossmann H, & Mitterdorfer J (1997). Nine L-type amino acid residues confer full 1,4-dihydropyridine sensitivity to the neuronal calcium channel alpha1A subunit. Role of L-type Met1188. J Biol Chem, 272(44), 27686–27693. [DOI] [PubMed] [Google Scholar]
- 21.Xu W, & Lipscombe D (2001). Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci, 21(16), 5944–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang L, Gamal El-Din TM, Swanson TM, Pryde DC, Scheuer T, Zheng N, & Catterall WA (2016). Structural basis for inhibition of a voltage-gated Ca(2+) channel by Ca(2+) antagonist drugs. Nature, 537(7618), 117–121. doi: 10.1038/nature19102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M, … Yan N (2016). Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 A resolution. Nature, 537(7619), 191–196. doi: 10.1038/nature19321 [DOI] [PubMed] [Google Scholar]
- 24.Xu L, Li D, Tao L, Yang Y, Li Y, & Hou T (2016). Binding mechanisms of 1,4-dihydropyridine derivatives to L-type calcium channel Cav1.2: a molecular modeling study. Mol Biosyst, 12(2), 379–390. doi: 10.1039/c5mb00781j [DOI] [PubMed] [Google Scholar]
- 25.Bean BP (1984). Nitrendipine block of cardiac calcium channels: high-affinity binding to the inactivated state. Proc Natl Acad Sci U S A, 81(20), 6388–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bock G, Gebhart M, Scharinger A, Jangsangthong W, Busquet P, Poggiani C, … Koschak A (2011). Functional properties of a newly identified C-terminal splice variant of Cav1.3 L-type Ca2+ channels. J Biol Chem, 286(49), 42736–42748. doi: 10.1074/jbc.M111.269951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang H, Yu D, & Soong TW (2013). C-terminal alternative splicing of CaV1.3 channels distinctively modulates their dihydropyridine sensitivity. Mol Pharmacol, 84(4), 643–653. doi: 10.1124/mol.113.087155 [DOI] [PubMed] [Google Scholar]
- 28.Singh A, Gebhart M, Fritsch R, Sinnegger-Brauns MJ, Poggiani C, Hoda JC, … Koschak A (2008). Modulation of voltage- and Ca2+-dependent gating of CaV1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J Biol Chem, 283(30), 20733–20744. doi: 10.1074/jbc.M802254200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liss B, & Striessnig J (2019). The Potential of L-Type Calcium Channels as a Drug Target for Neuroprotective Therapy in Parkinson’s Disease. Annu Rev Pharmacol Toxicol, 59, 263–289. doi: 10.1146/annurev-pharmtox-010818-021214 [DOI] [PubMed] [Google Scholar]
- 30.Guzman JN, Ilijic E, Yang B, Sanchez-Padilla J, Wokosin D, Galtieri D, … Surmeier DJ (2018). Systemic isradipine treatment diminishes calcium-dependent mitochondrial oxidant stress. J Clin Invest, 128(6), 2266–2280. doi: 10.1172/JCI95898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, & Surmeier DJ (2010). Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature, 468(7324), 696–700. doi: 10.1038/nature09536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puopolo M, Raviola E, & Bean BP (2007). Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci, 27(3), 645–656. doi: 10.1523/JNEUROSCI.4341-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cardozo DL, & Bean BP (1995). Voltage-dependent calcium channels in rat midbrain dopamine neurons: modulation by dopamine and GABAB receptors. J Neurophysiol, 74(3), 1137–1148. doi: 10.1152/jn.1995.74.3.1137 [DOI] [PubMed] [Google Scholar]
- 34.Brandt A, Striessnig J, & Moser T (2003). CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci, 23(34), 10832–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez-Padilla J, Guzman JN, Ilijic E, Kondapalli J, Galtieri DJ, Yang B, … Surmeier DJ (2014). Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat Neurosci, 17(6), 832–840. doi: 10.1038/nn.3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldberg JA, Guzman JN, Estep CM, Ilijic E, Kondapalli J, Sanchez-Padilla J, & Surmeier DJ (2012). Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson’s disease. Nat Neurosci, 15(10), 1414–1421. doi: 10.1038/nn.3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Surmeier DJ, Halliday GM, & Simuni T (2017). Calcium, mitochondrial dysfunction and slowing the progression of Parkinson’s disease. Exp Neurol, 298(Pt B), 202–209. doi: 10.1016/j.expneurol.2017.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biglan KM, Oakes D, Lang AE, Hauser RA, Hodgeman K, Greco B, … Parkinson Study Group, S.-P. D. I. I. I. I. (2017). A novel design of a Phase III trial of isradipine in early Parkinson disease (STEADY-PD III). Ann Clin TranslNeurol, 4(6), 360–368. doi: 10.1002/acn3.412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinggera A, Lieb A, Benedetti B, Lampert M, Monteleone S, Liedl KR, … Striessnig J (2015). CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type calcium channels. Biol Psychiatry, 77(9), 816–822. doi: 10.1016/j.biopsych.2014.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Y, Huang G, Wu J, Wu Q, Gao S, Yan Z, … Yan N (2019). Molecular Basis for Ligand Modulation of a Mammalian Voltage-Gated Ca(2+) Channel. Cell, 177(6), 1495–1506 e1412. doi: 10.1016/j.cell.2019.04.043 [DOI] [PubMed] [Google Scholar]
- 41.Kang S, Cooper G, Dunne SF, Luan CH, Surmeier DJ, & Silverman RB (2013). Structure- activity relationship of N,N’-disubstituted pyrimidinetriones as Ca(V)1.3 calcium channel-selective antagonists for Parkinson’s disease. J MedChem, 56(11), 4786–4797. doi: 10.1021/jm4005048 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.