

Long-chain fatty acids (LCFAs) play an important role in cellular energy metabolism acting both as an important energy source as well as signaling molecules1. LCFA-CoA esters promote their own oxidation by acting as allosteric inhibitors of acetyl-CoA carboxylase (ACC) which reduces the production of malonyl-coenzyme A and relieves inhibition of carnitine palmitoyl-transferase 1, thereby promoting LCFA-CoA transport into the mitochondria for beta-oxidation2-6. Here we report a new level of regulation wherein LCFA-CoA esters themselves allosterically activate AMPK β1 containing isoforms to increase fatty acid oxidation through phosphorylation of ACC. Activation of AMPK by LCFA-CoA esters requires the ADaM (Allosteric Drug and Metabolite) site formed between the α subunit kinase domain and the β subunit. β1-subunit mutations that inhibit AMPK activation by the small molecule activator A-769662, that binds to the ADaM site, also inhibit activation by LCFA-CoAs. Thus, LCFA-CoA metabolites act as direct endogenous AMPK β1-selective activators and promote LCFA oxidation.
Acetyl-CoA carboxylase (ACC) exists as two distinct isoforms, ACC1 and ACC2, that are allosterically activated by citrate and inhibited by LCFA-CoAs5,6. In addition to allosteric regulation, ACC activity is inhibited by covalent modifications involving phosphorylation of ACC1 (Ser80 (Ser79 mice)) and ACC2 (Ser221 (Ser212 mice)) by the AMP-activated protein kinase (AMPK)7-9. AMPK is an evolutionarily conserved metabolic sensor that plays a vital role in regulating energy balance7,10,11. AMPK is expressed in all tissues as a heterotrimer composed of a catalytic α (isoforms 1 or 2) subunit, and β (1 or 2) and γ (1, 2, or 3) regulatory subunits. Canonical activation of AMPK is triggered by energy deficit that causes a rise in the intracellular AMP/ADP:ATP ratio to promote the exchange of ATP for AMP or ADP bound to the regulatory γ-subunit12-16. Binding of AMP and ADP increases kinase activity by inducing a conformational shift that results in enhanced Thr172 phosphorylation within the α-catalytic subunit, while AMP binding additionally results in allosteric activation15,17-22. Evidence suggests that AMPK may also respond to variations in energy substrate availability, and while the mechanisms by which AMPK senses changes in carbohydrate availability have been intensely studied23-30, surprisingly only a few reports have indicated a potential role for lipid moieties to control enzyme activity and the potential mechanisms explaining these observations remain unclear9,31,32.
A structurally diverse class of small molecule synthetic AMPK activators, including A769662, salicylate, SC-4, 991 and PF-06409577 33-38 have been shown to activate AMPK through a distinct mechanism reliant on phosphorylation of Ser108 in the β1-subunit carbohydrate binding module (CBM)36,39. Co-crystallization of AMPK heterotrimers with activators 991 and A769662 derivatives revealed specific interactions with pSer108 and a large hydrophobic pocket created between the kinase domain and β1-CBM35,40. Binding of A769662 was shown to stabilize the interaction between these two subunits, resulting in allosteric activation and protection of AMPKα-pThr172 from dephosphorylation35,40,41. This drug binding site has been termed the ADaM (Allosteric Drug and Metabolite) site based on the speculation that it may also act as a binding site for natural metabolite(s)42.
We initiated our investigation into natural ADaM site ligands by assessing whether LCFAs or their respective metabolically activated CoA (LCFA-CoA) esters (Fig. 1a) directly affected enzyme activity using a TR-FRET SAMS assay format. Studies measuring the activity of AMPKα1β1γ1 isolated from Sf9 cells in the presence of palmitate, palmitoyl-CoA, myristate, or myristoyl-CoA (0.1 to 100 μM) showed that both acyl-CoA esters activated the kinase ~3-fold, whereas their respective unconjugated forms were inactive (Fig. 1b). Importantly, activation was observed at physiological concentrations below the critical micelle concentration for LCFA-CoAs1 with an EC50 value of approximately 1 μM for both palmitoyl-CoA and myristoyl-CoA. To characterize the specificity of AMPK for LCFA-CoA ester chain length, we measured AMPK activity in the presence of acyl-CoAs ranging from C2 (acetyl-CoA) to C18 (oleoyl-CoA) and found that LCFA-CoAs ≥ C12 activated AMPK (Fig. 1c). Palmitoleoyl-CoA and oleoyl-CoA also increased AMPK activity to a similar magnitude, indicating that AMPK also responds to de-saturated forms of LCFA-CoAs (Fig. 1c). We further tested the specificity of AMPK for LCFA-CoAs by showing that several other coenzymes, precursors, and vitamins that share structural features with CoA failed to allosterically activate AMPK (Extended Data Fig. 1a). Moreover, other metabolites involved in LCFA-CoA biosynthesis or catabolism such as malonyl-CoA, free CoA (CoASH), palmitoylcarnitine and pantothenic acid, also failed to stimulate the kinase (Fig. 1d), further illustrating the specificity of AMPK for LCFA-CoAs.
Figure 1. LCFA-CoAs are direct AMPK activators.
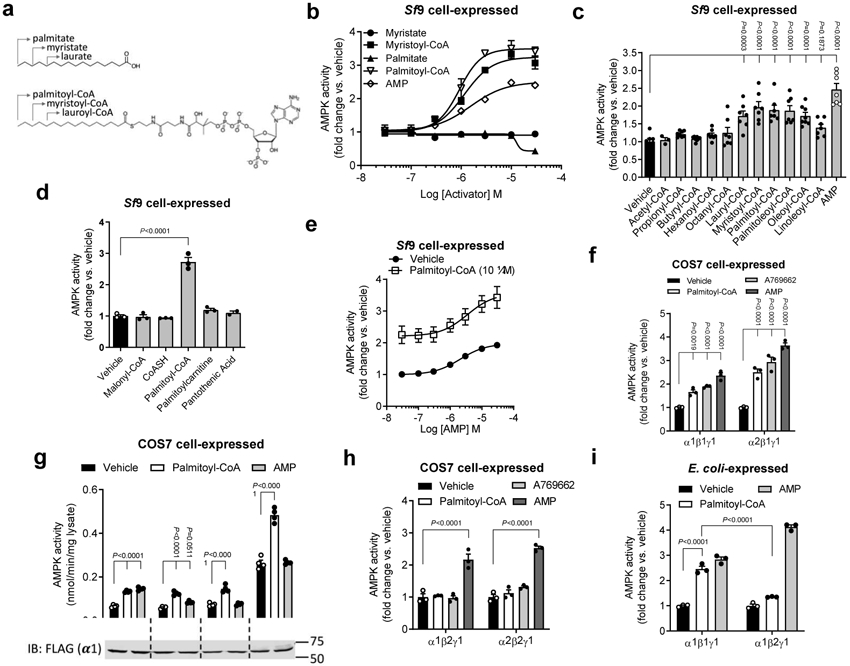
a, Structures of LCFAs (upper) and LCFA-CoAs (lower). b-e, Activities of AMPKα1β1γ1 (Sf9 insect cell-expressed), determined by TR-FRET SAMS assay, in the presence of increasing concentrations of myristate, palmitate, their respective CoA thioester conjugates and AMP (b), acyl-CoA esters (10 μM) with chain lengths ranging from C2 to C18 (c), metabolites or biosynthetic precursors important for palmitoyl-CoA synthesis or catabolism (d) or ± 10 μM palmitoyl-CoA and the indicated concentrations of AMP (e). Data are shown as mean fold change in activity vs. vehicle ± s.e.m. For b, n = 2; for c, n = 7 except for propionyl-, butyryl-, hexanoyl- and oleoyl-CoAs (n =8) and acetyl-CoA (n = 3); for d, n = 3 except for pantothenic acid (n = 2); for e, n = 6. Statistical significance was calculated using one-way ANOVA with Bonferroni’s multiple comparisons test. n represent biological independent experiments. f-h, Activities of AMPK complexes (COS7 cell-expressed; basal activities detailed in Extended Data Table 1), determined by 32P SAMS peptide assay following anti-myc agarose immobilization, of α1 and α2 AMPK ± palmitoyl-CoA (10 μM), A769662 (10 μM) or AMP (100 μM) (f), γ1 mutants ± AMP (100 μM) or palmitoyl-CoA (10 μM) (g) and β1 and β2 AMPK ± palmitoyl-CoA (10 μM), A769662 (10 μM) or AMP (100 μM) (h). For f, h, data are shown as mean fold change in activity vs. vehicle ± s.e.m.; n = 3. For g, data are shown as mean specific activity ± s.e.m.; n = 4. Statistical significance was calculated using two-way ANOVA with Bonferroni’s multiple comparisons test. n represent biological independent experiments. Immunoblot shows relative expression of flag-AMPK in COS7 cells. (i) Activities of purified AMPK complexes α1β1γ1 and α1β2γ1 (E. coli-expressed; basal activities detailed in Extended Data Table 2), determined by 32P SAMS peptide assay, ± palmitoyl-CoA (10 μM) or AMP (100 μM). Data are shown as mean fold change in activity vs. vehicle ± s.e.m.; n = 3. Statistical significance was calculated using two-way ANOVA with Bonferroni’s multiple comparisons test. n represent biological independent experiments.
To establish whether the fatty acid or CoA moieties were individually sufficient to prevent activation by the acyl-CoA conjugate, we pre-incubated AMPKα1β1γ1 complexes with 10 μM palmitoyl-CoA alone or in combination with increasing concentrations of free CoA or palmitate. Palmitoyl-CoA (10 μM) alone increased AMPK activity by ~3-fold but was unaffected by the addition of up to 100 μM free CoA (CoASH) or palmitate (Extended Data Fig. 1b), indicating that neither unconjugated free fatty acids nor free CoA prevented palmitoyl-CoA-dependent activation of AMPK. To determine whether palmitoyl-CoA was mimicking AMP, we measured AMPK activation by palmitoyl-CoA in combination with saturating concentrations of AMP. These studies showed that palmitoyl-CoA activated AMPK to a similar degree in the presence or absence of 100 μM AMP (Fig. 1e), suggesting a distinct mechanism.
To ensure that our observations were not due to artefactual effects specific to our experimental conditions (e.g. TR-FRET assay and Sf9 expressed AMPK) we also investigated palmitoyl-CoA stimulation of AMPK derived from alternate sources, using the 32P radiolabel-based SAMS peptide substrate assay (32P SAMS). Freshly prepared palmitoyl-CoA stimulated the activity of AMPK α1β1γ1 and α2β1γ1 expressed in COS7 mammalian cells and immunoprecipitated on anti-myc agarose via a C-terminal myc-tag on the β1 subunit (Fig. 1f). Interestingly, activation by palmitoyl-CoA, but not A769662 or AMP, was lost when AMPKα1β1γ1 was immobilized on anti-flag agarose or glutathione Sepharose via relevant N-terminal affinity tags on the α subunit (Extended Data Fig. 2a and b). One explanation is that immobilizing AMPK complexes through the α subunit suppresses palmitoyl-CoA binding or allosteric transduction. Activation was also lost when palmitoyl-CoA was prepared in HEPES buffer rather than water, which resulted in ligand aggregation (Extended Data Fig. 2c).
To assess whether palmitoyl-CoA required specific interactions with the γ-regulatory subunit, we treated active AMPK complexes flag-immunoprecipitated from COS7 cells expressing wild type (WT) flag-α1β1γ1, nucleotide site mutants γ1-Asp90Ala (site 1) and γ1-Asp215Ala (site 3), or AMP-insensitive γ1-Arg299Gly mutant. Palmitoyl-CoA (10 μM) increased the activity of all four AMPK complexes, whereas AMP only activated WT AMPK (Fig. 1g). These findings indicate that LCFA-CoAs activate AMPK complexes independently of interactions with the γ-subunit AMP binding sites.
A key defining feature of synthetic ADaM site ligands such as A769662 is their selectivity for AMPK β1 compared to β2 containing complexes35,43. Similar to these pharmacological agents, palmitoyl-CoA was unable to stimulate activities of AMPK β2 complexes, expressed in either COS7 cells (Fig. 1h) or E. coli (Fig. 1i). These data indicate that AMPK activation by LCFA-CoAs occurs at physiologically relevant concentrations, is specific for AMPK β1 containing heterotrimers, requires fatty acyl-CoA chains greater than C12 and is reproducible using distinct assay systems (32P SAMS and TR-FRET) and AMPK protein preparations from multiple sources (insect cell, bacterial and mammalian).
Since palmitoyl-CoA and A769662 share selectivity for β1-AMPK and activated AMPK independent of the γ-subunit AMP binding sites, we reasoned that palmitoyl-CoA might be a natural metabolite capable of activating AMPK via binding to the ADaM site. Consistent with this hypothesis, palmitoyl-CoA was unable to activate AMPK complexes in which the β1-subunit had been N-terminally truncated to remove the CBM (Δ145), a structural modification that disrupts the ADaM site (Fig. 2a). Several studies have indicated that within the ADaM site, Ser108 plays a critical role for allosteric activation36,39. Importantly, mutation of the drug-sensitizing phosphorylation site β1Ser108 to Ala in E. coli-expressed AMPKα1β1γ1 significantly ablated activation by 10 μM palmitoyl-CoA (WT: 2.46 fold activation; β1Ser108Ala: 1.41 fold activation) but not AMP (Fig. 2b), thus highlighting the specificity of the LCFA-CoA activation. Palmitoyl-CoA induced activation of COS7 cell-expressed AMPKα1β1γ1 was also blunted by the β1Ser108Ala mutation (WT: 2.34 fold activation; β1Ser108Ala: 1.84 fold activation) (Fig. 2c), however the inhibitory effect of the mutation was smaller for palmitoyl-CoA compared to CoA esters with shorter acyl chain lengths (Extended Data Fig. 3a). Interestingly, although ADaM site drugs or AMP/ADP are able to prolong AMPK signaling by inducing conformational shifts that protect phosphorylated α-Thr172 from protein phosphatase-dependent dephosphorylation and enzyme deactivation35, palmitoyl-CoA did not attenuate PP2cα-dependent inhibition of AMPK activity (Extended Data Fig. 3b). These findings indicate that the palmitoyl-CoA binding site on AMPK is distinct from the γ-nucleotide binding sites and at least partially overlaps the ADaM site; however, the regulatory effects of its binding may differ from synthetic ligands.
Figure 2. LCFA-CoA activation is mediated through the AMPK ADaM site.

a, Activities of AMPKα1β1γ1 (WT or N-terminal deletions of β1 residues 1-71 (Δ71) or 1-145 (Δ145)) ± palmitoyl-CoA (10 μM) or AMP (100 μM). b, c, Activities of AMPKα1β1γ1 (WT or β1S108A) ± palmitoyl-CoA (10 μM) or AMP (100 μM) (b) or increasing concentration of palmitoyl-CoA (c). For a, b, data are shown as mean fold change in activity vs. vehicle ± s.e.m.; n = 3. For c, data are shown as mean specific activity ± s.e.m.; n = 3. Statistical significance was calculated using two-way ANOVA with Bonferroni’s multiple comparisons test. n represent biological independent experiments. d, In silico modelling of palmitoyl-CoA bound to AMPKα2β1γ1. e, f, Activities of AMPKα1β1γ1 and α2β1γ1 (WT or β1G86E) (e), or AMPK α1β2γ1 and α2β2γ1 (WT or β2E85G) (f) ± palmitoyl-CoA (10 μM), A769662 (10 μM) or AMP (100 μM). Data are shown as mean specific activity ± s.e.m.; n = 3. Statistical significance was calculated using two-way ANOVA with Bonferroni’s multiple comparisons test, or unpaired, 2-tailed Student’s t test (red P values). n represent biological independent experiments. Activities determined by 32P SAMS peptide assay. COS7 cell-expressed AMPK immobilized on anti-myc agarose was used in all experiments unless indicated.
In order to investigate the binding of LCFA-CoA to AMPK we conducted radioligand binding assays. These studies demonstrated that [3H]-palmitoyl-CoA readily bound to both AMPKα1β1γ1 and AMPKα1β2γ1 complexes, an effect that was blocked by the addition of unlabeled palmitoyl-CoA (Extended Data Fig. 3c and d). These findings suggest that although palmitoyl-CoA can effectively bind to both the β1 and β2 subunits, it is only capable of allosterically activating β1-containing complexes.
To further interrogate the binding of LCFA-CoA to AMPK we carried out co-crystallization trials with bacterial AMPKα2β1γ1 in the presence of palmitoyl-CoA, but were unable to obtain diffractable crystals. The CBM in the AMPKα2β1γ1 structure makes extensive crystal contacts so that conformational changes to the CBM upon palmitoyl-CoA binding could inhibit crystallization. In the absence of a co-complex crystal structure we used in silico modelling to test the feasibility of palmitoyl-CoA:AMPKα2β1γ1 interactions at the ADaM site, focusing on potential LCFA-CoA binding pockets involving residues from both α2 and β1 subunits. Palmitoyl-CoA was flexibly docked into a hydrophobic channel at the α2:β1 subunit interface encompassing the ADaM site and a novel pocket located directly below the cyclodextrin binding groove in the β1-CBM (Extended Data Fig. 4). The top 30 ranked palmitoyl-CoA poses were retained for analysis, however in every case the fatty-acyl chain of palmitoyl-CoA adopted a similar orientation to SC4 and A-769662 in the hydrophobic ADaM site and appeared to be optimal for spanning the length of the site (Fig. 2d, Extended Data Fig. 4 and 5 and Supplementary Note). Consistent with the AMPKα1β1γ1 activation data (Fig. 1c), acyl chain lengths C12 – C18 (lauroyl-stearoyl) are able to span the ADaM site and make sufficient hydrophobic interactions to anchor the acyl chain of the LCFA-CoA esters. Acyl chain lengths < C12 are physically unable to span the ADaM site and would participate in fewer interactions than C12 – C18 acyl chains, which may be insufficient to anchor them in the ADaM site. In contrast, acyl chain lengths > C18 would unlikely be accommodated within the hydrophobic ADaM site. While pSer108 makes no direct polar interactions with the acyl chain of palmitoyl-CoA, nor with ADaM site drugs, it does participate in an extensive polar interaction network with α2Thr21, α2Lys29, α2Lys31, β1His109, β1Asn110 and β1Asn111 thereby stabilizing the α2-subunit:β1-CBM interaction (Extended Data Fig. 4d). The majority of the docked poses placed the palmitoyl-CoA adenine ring in various orientations within a hydrophobic pocket lying directly below the cyclodextrin binding groove in the β1-CBM (Fig. 2d and Extended Data Fig. 4a). This pocket is formed by β1 residues Phe82, Trp84, Thr85, Leu93, Tyr125, Phe127, Thr134, Asp136, Pro137, Ser138, Glu139, Asn151 and Ile153, with polar residues forming a border around the hydrophobic pocket interior. Although the di-phosphate moiety is largely solvent exposed it can form interactions with nearby β1-subunit residues Arg83, Thr85, Gly86, Asn111 and Ser138 and α2 subunit residues Asn48, Gln50 and Lys51 (Fig. 2d and Supplementary Note).
AMPK β1 and β2 isoforms share 71% sequence identity and the AMPK β1Ser108 residue is conserved in β2 (Extended Data Figure 6). To understand the β1-isoform functional selectivity displayed by LCFA-CoAs we identified non-conserved residues in close proximity (6.0 Å radius) to the general binding mode of palmitoyl-CoA in our in silico model (Fig. 2d). Sequence and structural alignments of the β-isoforms revealed the largely non-conserved β1 residues as Phe82 (β2Ile81) and Thr85 (β2Ser84) in the adenine ring binding pocket; Gly86 (β2Glu85 in all sequences except D. rerio (Gly) and G. gallus (Asp)) in the di-phosphate moiety binding region; and Thr106 (β2Ile106), Arg107 (β2Lys107) and Asn111 (β2Asp111) in the ADaM site. The side-chain characteristics of β1Phe82 and β1Thr85 are largely conserved in the analogous β2 residues, and only the backbone atoms of β1Thr106 and β1Arg107 contribute to the ADaM site, therefore these residues were considered unlikely to influence β1-isoform selectivity. The presence of negatively charged β2 residues Glu85 and Asp111, however, may result in an unfavorable charge match (i.e. electronic repulsion) with the negatively charged di-phosphate moiety of palmitoyl-CoA, requiring either β2-AMPK or palmitoyl-CoA to adopt a conformation which prevents activation. Alternatively, activation may require conformational rearrangement of the β1-isoform Thr85-Gly86-Gly87-Gly88 loop. Interactions between the side-chains of β2Glu85 and β2Trp83 (hydrogen bond and charge-π interactions) restrict the mobility of the equivalent loop in the β2-isoform (Ser84-Glu85-Gly86-Gly87), potentially preventing activation. Consistent with these structural insights, exchange of β1Gly86 to the analogous β2 residue (Glu85) abolished AMPKα1β1γ1 and α2β1γ1 activation by palmitoyl-CoA (Fig. 2e). Conversely, reciprocal exchange of β2Glu85 to Gly was sufficient to render AMPKα1β2γ1 and α2β2γ1 sensitive to palmitoyl-CoA activation (Fig. 2f). Intriguingly, sensitivity to the β1-selective agonist A769662 followed a similar profile in response to β1/β2 residue exchange at this position, with β1Gly86Glu significantly attenuating, and β2Glu85Gly significantly gaining, activation by A769662, respectively (Fig. 2e and f). Exchange of β1Asn111 to Ala or the analogous β2 residue (Asp111) had no significant effect on AMPKα1β1γ1 activation by palmitoyl-CoA (Extended Data Fig. 7). These data reveal that Ser108 and Gly86 play vital roles in determining the β1-isoform specificity of allosteric activation of AMPK by LCFA-CoAs.
To investigate the potential physiological importance of this regulation we incubated primary mouse hepatocytes (a cell type which expresses predominately AMPK β1) with palmitate and found that within the physiological range of exposures palmitate increased the phosphorylation of ACC S79/212 without altering phosphorylation of AMPK Thr172 (Fig 3a). To directly evaluate the importance of the Co-A derivative in mediating these effects we subsequently treated primary hepatocytes with palmitate (which is taken up, activated to palmitoyl-CoA and metabolized), bromo-palmitate (which is taken up and activated to palmitoyl—CoA but not metabolized) or methyl-palmitate (which is taken up but not activated to palmitoyl-CoA). Consistent with in vitro studies indicating a direct role for palmitoyl-CoA but not palmitate in activating AMPK we found that bromo but not methyl-palmitate increased the phosphorylation of ACC similar to palmitate (Fig 3b). These data support the hypothesis that palmitoyl-CoA is an allosteric activator of AMPK in hepatocytes.
Figure 3. FA-CoAs increase fatty acid oxidation through AMPK phosphorylation of ACC.
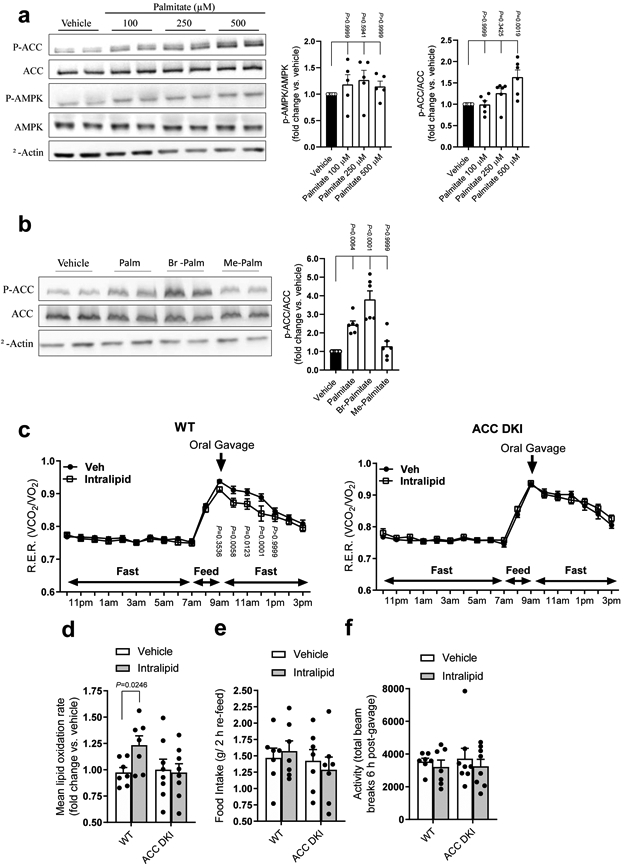
a, Primary mouse hepatocytes from C57Bl6J mice were treated with vehicle or palmitate (100, 250, or 500 μM) followed by western blotting for total AMPKα, phosphorylated-AMPK (p-AMPK, α-Thr172), total ACC, phosphorylated-ACC (p-ACC, Ser79/212) and β-actin. Data are shown as mean fold change in phosphorylation of AMPK (n = 5) or ACC (n = 6) vs. vehicle ± s.e.m. b, Primary mouse hepatocytes from C57Bl6J mice were treated with vehicle or 500 μM palmitate, bromo-palmitate (Br-Palmitate) or methyl-palmitate (Me-Palmitate) followed by western blotting for total ACC, phosphorylated-ACC (p-ACC, Ser79/212) and β-actin. Data are shown as mean fold change in phosphorylation of ACC (n = 6) vs. vehicle ± s.e.m. Statistical significance was calculated using one-way ANOVA with Bonferroni’s multiple comparisons test. n represent biological independent experiments. Representative immunoblots are shown. c, Respiratory Exchange Ratios (R.E.R.) of WT and ACC DKI mice following oral administration of saline (Veh), or Intralipid® (10 ml/kg). Data are shown as mean R.E.R. ± s.e.m. (WT, n = 7; ACC DKI, n = 8). Statistical significance was calculated using two-way ANOVA with Bonferroni’s multiple comparisons test. n represent biological independent experiments. d-f, WT and ACC DKI mice, following oral administration of saline (Vehicle) or Intralipid® (10 ml/kg), were measured for lipid oxidation rates calculated from R.E.R. over 4 h, starting 1 h post-gavage (d), food intake during 2 h re-feed (e) and activity levels 6 h post-gavage (f). Data are shown as mean fold change in lipid oxidation rate vs. vehicle (d), mean food intake (e) and mean total beam breaks (f) ± s.e.m. (WT, n = 7; ACC DKI, n = 8). Statistical significance was calculated using unpaired, 2-tailed Student’s t test with Bonferroni’s multiple comparisons test. n represent biological independent experiments.
In response to synthetic AMPK activators, AMPK regulates fatty acid metabolism in hepatocytes through phosphorylation and inhibition of ACC8,44,45. Therefore, to further characterize the specificity of the response to fatty acids in vivo we treated mice lacking AMPK inhibitory phosphorylation sites on ACC1 (S79A KI) and ACC2 (S212A KI) (ACC DKI mice)8 with vehicle or Intralipid®. ACC DKI mice were resistant to the effects of Intralipid® to enhance fatty acid oxidation (Fig 3c and d). There were no changes in the intake of food (Fig 3e) or physical activity (Fig 3f). These findings indicate a critical role for AMPK phosphorylation of ACC in enhancing fatty acid oxidation in response to acute exposure to long chain fatty acids.
In the present study we demonstrate that LCFA-CoA esters are allosteric activators of AMPK. Our investigations have uncovered a novel mechanism by which LCFA-CoAs directly activate AMPK in a β1 regulatory subunit specific manner. Similar to the synthetic activator A769662, we show that activation by palmitoyl-CoA is attenuated in β2-containing complexes, or by single knock-in mutations of either Ser108 or Gly86 within the β1-CBM. We establish that this interaction is specific for LCFA-CoAs by showing that short-, medium-, and very long-chain acyl-CoAs, free fatty acids or free CoA (CoASH) do not activate AMPK complexes. While the precise molecular binding mode of palmitoyl-CoA remains unknown, the S108A data strongly suggest palmitoyl-CoA is an ADaM site allosteric activator. The importance of Ser108 phosphorylation in maintaining the potency of the ADaM site has been well documented for synthetic ADaM site activators46. In this case we suggest that Ser108 phosphorylation behaves like an ion-pairing reagent for basic residues in the ADaM site, stabilizing the α subunit N lobe/β1-CBM interaction and strengthening the palmitoyl-CoA acyl chain interaction in the binding pocket. In addition, we have identified the non-conserved residue β1Gly86 as a critical palmitoyl-CoA activation determinant.
These studies provide the first evidence that LCFA-CoAs are natural ligands for AMPK, stimulating ACC phosphorylation to promote fatty acid oxidation. Although the functional relationship between adenine nucleotides and LCFA-CoAs, and the consequence of these interactions on structure/function should be further explored, these findings indicate an additional level of regulation in which AMPK senses not only adenine nucleotides, but integrates these signals with changes in the concentration of high-energy LCFA-CoAs to match substrate utilization with availability. Given the AMPK β1 selectivity of the response, it would be anticipated that cell types/tissues where AMPK β1 predominate such as white adipocytes, hepatocytes, the hypothalamus and macrophages would be most affected47,48. In hepatocytes and white adipocytes, it would be expected that fasting or a meal high in lipids, would enhance fatty acid oxidation and suppress fatty acid and cholesterol synthesis through phosphorylation of ACC and HMG-CoA reductase9. In the hypothalamus, fasting induced increases in AMPK activity and ACC phosphorylation are important for stimulating appetite49 and while this effect has been linked to reductions in plasma glucose50, glucose levels are relatively stable compared to the 3–4-fold increase in free fatty acids that occurs with fasting. This suggests that fasting-induced activation of AMPK occurs through LCFA-CoAs and may be vital for increasing appetite; a concept supported by observations that the hypothalamic infusion of fatty acids increases appetite and stimulates hepatic glucose production51. Lastly, in macrophages AMPK is activated by fatty acids and suppresses inflammation48, a finding consistent with observations that fasting induces anti-inflammatory (F4/80+CD11c–) adipose tissue macrophages52. Future studies extending these findings to other metabolic processes known to be regulated by both LCFA-CoAs and AMPK, such as mitochondrial biogenesis and autophagy, are warranted.
Materials and Methods
Dulbecco’s Modified Eagle Media (DMEM), non-essential amino acids, HEPES, phosphate buffered saline (PBS), sodium pyruvate and penicillin/streptomycin were obtained from Invitrogen® (Logon, Utah). Fetal bovine serum (FBS) was obtained from Hyclone® (Grand Island, New York). Br-Palmitate (2-bromohexadecanoic acid, Sigma: 21604), Me-Palmitate (Sigma: P5177–5G) and Na-palmitate (Sigma: P9767–5G) were conjugated to bovine serum albumin (fatty acid free, low endotoxin (Sigma: A8806–1G)). Adenosine monophosphate (AMP), adenosine triphosphate (ATP), acyl-CoA lithium salts, high-performance liquid chromatography (HPLC) grade reagents and solvents, and Sf9 insect cell expressed human, active AMPKα1β1γ1 were acquired from Sigma Chemical Company (St. Louis, MO). Biocoat® type I collagen-coated plates were purchased from Becton Dickinson Labware (Bedford, MA). Phospho-AMPKαThr172 ELISA and antibodies to phospho-AMPKαThr172, AMPKα (total), phospho-ACCSer79, ACC (total) and β-actin (total) were obtained from Cell Signaling Technologies (Beverly, MA). ULight-ACCSer79 peptide (SAMS peptide) and Eu-anti-phospho-ACCSer79 antibody were obtained from PerkinElmer (Waltham, MA).
AMPK activity assay
AMPK enzyme activity was measured using two complementary methods based on either FRET-detection or radiolabeling of the synthetic SAMS peptide substrate. For FRET-based detection, AMPK activity was determined by phosphorylation of the ULight-SAMS peptide. Briefly, 0.5 nM Sf9 insect cell expressed human AMPKα1β1γ1 was pretreated with the indicated activators in 30 μl kinase buffer containing 50 mM HEPES pH 7.5, 1 mM EGTA, 2 mM DTT, 0.01% Tween, in white opaque 96-well microplates at 37°C for 15 min. Reactions were returned to room temperature on an orbital plate shaker for 5 min before a 10 μl addition of a mixture containing 4x ATP (30 μM final) and ULight-SAMS peptide (50 nM final). Plates were briefly centrifuged at 2000 rpm and placed back on the plate shaker at room temperature for 15 min. Reactions were stopped by the addition of 40 μl of detection mix containing 40 mM EDTA and 8 nM Eu-anti-phospho-ACCSer79 antibody. SAMS peptide phosphorylation was determined by TR-FRET (Lm1 Ex = 330 nm, Em = 668 nm (630 nm Co); Lm2 Ex = 330 nm, Em = 620 nm, (570 nm Co). The 668/620 nm fluorescence emission ratio was calibrated to standardized active AMPKα1β1γ1 enzyme with reported activity of 685 nmol/min/mg.
AMPK activity assay using radiolabeled [γ−32P]-ATP was conducted as described previously38. AMPK fragments or heterotrimers were expressed and purified from either E. coli or mammalian cell expression systems as described previously53,54. Specifically, bacterial expression cultures for His-tagged AMPK heterotrimers (WT α1/2 and β1/2 combinations and indicated mutants) were grown in Luria-Bertani broth and induced at 16˚C with 0.25 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), prior to overnight incubation. Cells were lysed using a precooled EmulsiFlex-C5 homogenizer (Avestin) and AMPK purified using Nickel-Sepharose and size exclusion chromatography. AMPK was phosphorylated at α-Thr172 by incubation with CaMKK2 (expressed and purified from E. coli as previously described53 in the presence of 2 mM MgCl2 and 200 μM ATP (1 h, 22˚C), prior to the size exclusion chromatography step. AMPK heterotrimers (WT α1/2 and β1/2 combinations and indicated β or γ1 mutants and β1 truncations) were expressed in mammalian COS7 cells by 48 h transient transfection, with fusion tags on α1/α2 (N-terminal GST or flag), β1/2 (C-terminal myc) and γ1 (N-terminal HA). AMPK complexes were immunoprecipitated from harvested cell lysates using GSH-Sepharose or flag/myc-agarose and beads washed extensively with assay buffer (50 mM HEPES pH 7.4, 1 mM DTT and 0.1% Tween-20) prior to kinase reaction. Activity assays were conducted in the presence of 100 μM SAMS peptide, 5 mM MgCl2 and 200 μM [γ−32P]ATP for 10 min at 30˚C in the presence or absence of ligand (freshly prepared palmitoyl-CoA (prepared in H2O), A769662 or AMP). Phosphotransferase activity was quenched by spotting onto P81 phosphocellulose paper (Whatman, GE Healthcare) followed by repeated washes in 1% phosphoric acid. 32P transfer to the SAMS peptide was quantified by liquid scintillation counting (Perkin Elmer).
PP2Cα protection assay
For the PP2Cα protection assay, AMPK activity was determined under conditions described above for TR-FRET detection with the following exceptions. Following pretreatment with indicated activators +/− 0.5 nM PP2Cα (protein phosphatase-2Cα) in 30 μl kinase buffer containing 50 mM HEPES pH 7.5, 1 mM EGTA, 2 mM DTT, 0.01% Tween, in white opaque 96-well microplates at 37°C for 15 min, reactions were stopped with 40 mM EDTA and AMPK activity determined.
Palmitoyl-CoA binding
AMPK protein used for the palmitoyl-CoA binding assay was purified from E. coli BL21 (DE3) cells. Full length human AMPK α1β1γ1 and AMPK α1β2γ1 complexes were expressed with a His6 tag at the N-terminus of α1. GST-αRIM2: His6-GST-LVPRGS(thrombin cleavage site)-α1(282–374), as described55 . Cells were grown to an OD600 of ~1 at 28°C and induced with 100 μM IPTG at 16°C overnight. Cell pellets were resuspended in 25 mM Tris.HCl, pH 8.0, 300 mM NaCl, 25 mM imidazole, 10% glycerol, and 5 mM β-mercaptoethanol, and lysed by French Press with pressure set to 900 Pa. Supernatants of lysates were loaded onto a 10 ml Ni-NTA column, and eluted with 25 mM Tris.HCl, pH 8.0, 300 mM NaCl, 500 mM imidazole, 10% glycerol, 5 mM β-mercaptoethanol. The eluted protein was further purified by Superdex 200 gel filtration chromatography (GE Healthcare) in 25 mM Tris.HCl, pH 8.0, 300 mM NaCl, 1 mM EDTA, 10% glycerol, 2 mM DTT. AMPK was phosphorylated by incubation with CaMKKβ at 1:0.02 molar ratio in 0.2 mM AMP, 0.2 mM ATP, 2 mM CaCl2, 5 mM MgCl2, 10 mM DTT, and 1 μM calmodulin at room temperature overnight (16 h), followed by further purification through Superdex 200 gel filtration chromatography.
For palmitoyl-CoA binding assay, 4 μM of each AMPK protein were incubated with 0.1 μl palmitoyl [9,10-3H]-CoA (American Radiolabeled Chemicals, Inc., ART 0339, 30–60 Ci/mmol at 1 μCi/μl), each, in 25 mM Tris.HCl, pH 7.4, 100 mM NaCl, and 1 mM MgCl2 for 60 min. Unincorporated [3H]-palmitoyl-CoA was removed by centrifugation through G-50 miniQuick Spin DNA gel filtration spin columns (Roche) pre-equilibrated with the reaction buffer, and the amount of AMPK-bound [3H]-palmitoyl-CoA determined by scintillation counting. Under the conditions used in the radio-ligand-binding assay palmitoyl-CoA reproducibly inhibits AMPK at concentrations above 10 μM, a concentration range at which it forms micelles. For the competition experiment, we therefore reduced the concentration of AMPK to 1 μM and competed with the indicated concentrations of unlabeled palmitoyl-CoA.
Modeling the palmitoyl-CoA interaction with AMPK
All modeling was performed using SYBYL-X 2.1.1 (Certara LP., Princeton, NJ, USA; http://www.certara.com). Flexible docking of palmitoyl-CoA to AMPK was undertaken using Surflex-Dock 2.7 (as implemented in SYBYL-X 2.1.1). The PyMOL Molecular Graphics System 1.8.2.2 (Schrödinger LLC., Cambridge, MA, USA, http://www.pymol.org) was used to construct figures.
The ADaM site ligands (A769662, 991 and SC4), crystallization buffer ligands (imidazole) and all water molecules were removed from the three human AMPKα2β1γ1 full length structures selected for palmitoyl-CoA docking studies (PDB IDs: 4CFF, 5ISO and 6B1U)35,38,56. Ligands in the α2-catalytic subunit ATP site (staurosporine) and the γ1-regulatory subunit AMP sites (i.e AMP) were not removed, since the experimental data indicated that these sites were not involved in palmitoyl-CoA binding. β1Ser108 was phosphorylated in each of the AMPKα2β1γ1structures used for the docking studies. Missing amino acid side-chains were added using a low energy side-chain rotamer library, as implemented within the Biopolymer module of SYBYL-X 2.1.1. Each AMPK protein was then subjected to a short round of optimization to remove any steric conflicts that may have arisen during the addition of the missing side-chains. The MMFF94s molecular mechanics force field and partial atomic charges, along with the conjugate gradient convergence method, were used for the geometry optimization step; termination of the optimization was achieved when the gradient difference of successive steps was < 0.05 kCal/mol.Å or 1000 iterations was reached. Flexible protein side-chain and palmitoyl-CoA docking was performed using Surflex-Dock 2.7; the protomol was generated for each AMPK structure using the multi-channel surface method, a threshold of 0.50 and a bloat value of 3. Although they vary in size and shape for each of the three AMPK structures, the protomols (i.e. a representation of the volume available for ligand occupation, along with energetically favourable locations within this volume for hydrophobic groups, hydrogen bond donors and hydrogen bond acceptors) extend across the interface between the α2- and β1-subunits, fully encompassing two largely hydrophobic pockets (Extended Data Fig. 4). The docking mode used was GeomX with protein flexibility enabled and all other parameters set to default values. The palmitoyl-CoA docked poses were ranked using C-Score57 and the top 30 ranked poses for each of the three AMPK structures were examined for potential interactions with AMPK residues.
Animal experiments
All animal procedures were approved by the McMaster University Animal Ethics Research Board (AUP #:16–12-41). Primary hepatocytes were obtained from 8 week old male C57Bl6J mice by collagenase digestion and suspended in William’s Media E containing 10% FBS and 1% antibiotic-antimicotic and plated in 6- or 12-well collagen coated plates and allowed to adhere for 4–5 h. Cells were then washed with PBS and incubated overnight in the same media. The following morning cells were washed with PBS and serum free media (1% AA, +L-glut) was added. Cells were then incubated in 1% BSA (In Serum Free WME) for 1.5 h. A 50 mM stock solution of Na-Palmitate was made in 50% EtOH 50% H20, and 50 mM stock solutions of Br-Palmitate and Me-Palmitate were made in 100% EtOH. Stock solutions were incubated at 65°C for 10 min and vortexed prior to cell treatment. Cells were treated for 2 h with different vehicle or palmitate conditions (in 1% BSA, serum free WME). At the end of the experiment cells were rapidly washed with cold PBS and snap frozen in liquid N2 in cell lysis buffer. Lysates were stored at −80°C for further analyses.
WT and ACC DKI mice were bred in-house by homozygous pairs derived from the same C57BL6 background. Mice were originally generated by OzGene Pty Ltd. (Perth, Australia) and the targeting strategy previously described47. For the Intralipid® study, 9–10 week-old male mice were maintained under controlled environmental conditions (21°C, 12 h/12 h light-dark cycle) and fed a standard chow diet (Diet 8640, Harlan Teklad, Madison, WI) leading into the study. In order to monitor small changes in R.E.R. an experimental crossover design was utilized, whereby mice of respective genotypes were randomly and evenly divided to receive oral gavage of saline or Intralipid® followed by the opposite treatment 1 week later. Metabolic monitoring was conducted using a Comprehensive Laboratory Animal Monitoring System (Columbus Instruments, Columbus, OH). The experiment was conducted following acclimation to the system for 48 h. Mice were fasted overnight starting at 1900h and given access to food at 0700h for 2 h followed by an oral gavage of vehicle (saline) or Intralipid® (10 ml/kg). Fatty acid oxidation was calculated using the following equation (1.70*VO2 - 1.69*VCO2) as described35.
Immunoblotting
Cell lysates were prepared using 1X lysis buffer containing,20 mM Tris.HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM ethylene glycol tetraacetic acid (EGTA), 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1X phosphatase inhibitor cocktail (Sigma). Total lysate protein concentrations were determined using the BCA Protein Assay (BioRad Laboratories, Hercules, CA). Protein concentrations were adjusted and diluted in 4X LDS (lithium dodecyl sulfate gel sample buffer) containing 50 mM DTT. Proteins were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (4%−12%) Bis/Tris, MOPS running buffer (Invitrogen® Logon, UT). Separated proteins were transferred to polyvinyl difluoride (PVDF) membranes electrophoretically. Nonspecific membrane binding was blocked using 0.2% BSA and membranes were probed with antibodies as indicated.
Data availability
The data that support the findings of this study are available from the corresponding authors upon request. Source data for Figs. 1-3 and Extended Data Figs. 1-7 are available online.
Extended Data
Extended Data Fig. 1. Specificity of AMPK activation by palmitoyl-CoA and effects of co-incubations with free palmitate or coenzyme A.
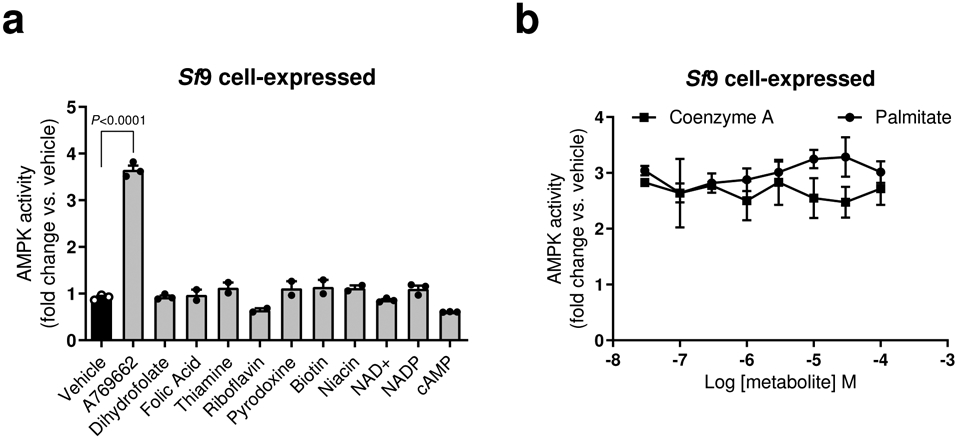
a, b, Activities of AMPKα1β1γ1 (Sf9 insect cell-expressed), determined by TR-FRET SAMS assay, in the presence of coenzymes (100 μM), cofactors (100 μM) and vitamins (100 μM) (a), or following 15 min pre-incubation in the presence of palmitoyl-CoA (10 μM) ± indicated concentrations of free palmitate or coenzyme A (b). Data are shown as mean fold change in AMPK activity vs. vehicle ± s.e.m. For a, n = 3 except for folic acid, thiamine, riboflavin, pyridoxine, biotin and niacin (n = 2); for b, n = 5 (palmitate incubation) or n = 3 (coenzyme A incubation). Statistical significance was calculated using one-way ANOVA with Bonferroni’s multiple comparisons test. n represent biological independent experiments.
Extended Data Fig. 2. Palmitoyl-CoA activation of purified AMPK is sensitive to the method of protein immobilization.
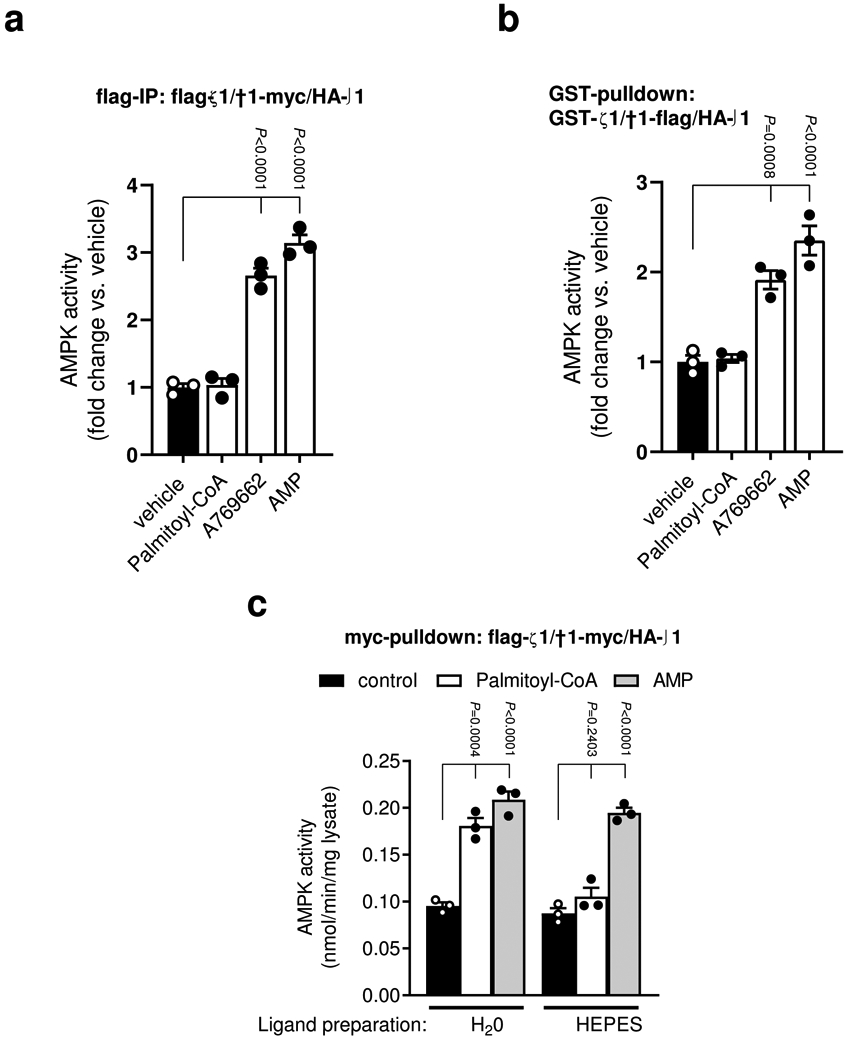
a, b, Activities of AMPKα1β1γ1 (COS7 cell-expressed; fusion tags as indicated) were determined by 32P SAMS peptide assay ± palmitoyl-CoA (10 μM), A769662 (10 μM) or AMP (100 μM), following immobilization on anti-flag agarose (a) or glutathione-Sepharose (b). Data are shown as mean fold change in AMPK activity vs. vehicle ± s.e.m., n = 3. c, Activities of AMPKα1β1γ1 (COS7 cell-expressed; fusion tags as indicated) were determined by 32P SAMS peptide assay ± palmitoyl-CoA (10 μM), prepared in either H2O or 50 mM HEPES, following immobilization on anti-myc agarose. Data are shown as mean specific activity ± s.e.m., n = 3. Statistical significance was calculated using one-way ANOVA with Bonferroni’s multiple comparisons test. n represent biological independent experiments.
Extended Data Fig. 3. Characterization of LCFA-CoA activation of AMPK.
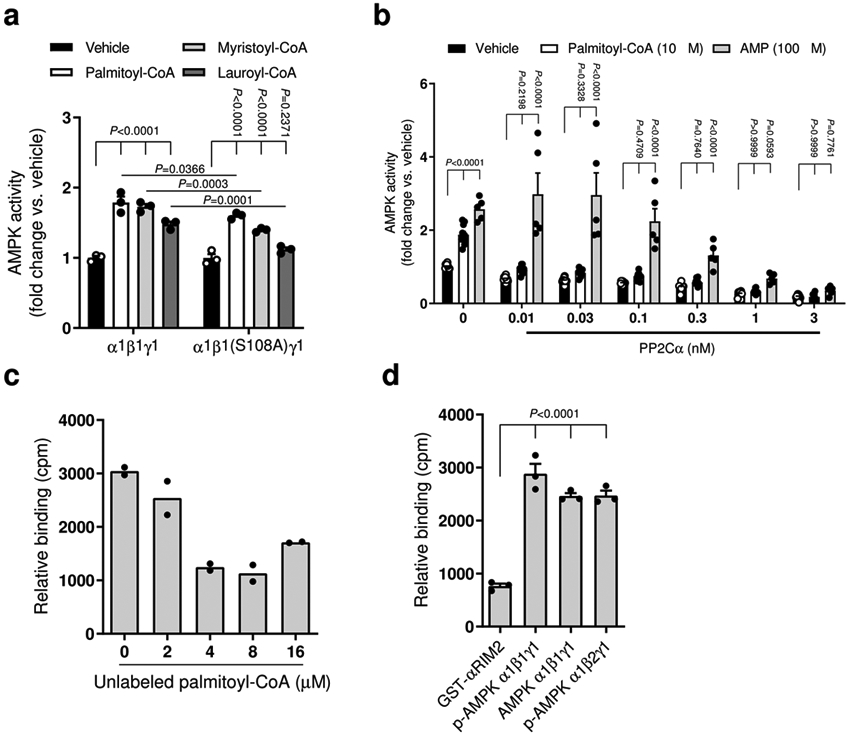
a, Activities of AMPKα1β1γ1 (COS7 cell-expressed, WT and β1S108A mutant) were determined by 32P SAMS assay, following immobilization on anti-myc agarose, ± palmitoyl-CoA, myristoyl-CoA or lauroyl-CoA (10 μM). Data are shown as mean fold change in AMPK activity vs. vehicle ± s.e.m., n=3. Statistical significance was calculated using two-way ANOVA with Bonferroni’s multiple comparisons test. n represent biological independent experiments. b, Activities of AMPKα1β1γ1 (Sf9 insect cell-expressed) were determined by TR-FRET in the presence of the indicated concentration of phosphatase PP2Cα ± AMP (30 μM) or palmitoyl-CoA (10 μM). Data are shown as mean fold change in AMPK activity vs. vehicle ± s.e.m., n=10 except for AMP incubated (n = 5). Statistical significance was calculated using two-way ANOVA with Bonferroni’s multiple comparisons test. n represent biological independent experiments. c, d, Binding of [3H]-palmitoyl-CoA to AMPKα1β1γ1 (E. coli-expressed) ± increasing concentrations of unlabeled palmitoyl-CoA (c), or to various AMPK preparations (d). GST-αRIM2: His6-GST-LVPRGS(thrombin cleavage site)-α1(282–374). Data are shown as mean relative binding ± s.e.m. For c, n = 2; for d, n = 3. Statistical significance was calculated using one-way ANOVA with Bonferroni’s multiple comparisons test. n represent biological independent experiments.
Extended Data Fig. 4. The channel at the interface between AMPK α2- and β1-subunits used for docking palmitoyl-CoA, interactions made by pSer108 and comparison with ADaM site activators.

(a) In the PDB ID:4CFF15 AMPK structure (shown as cartoon), the channel (i.e. the docking protomol, yellow molecular surface) encompasses the ADaM site (located directly above the ATP binding site of the α2-subunit kinase domain) and continues into a pocket located beneath the cyclodextran binding groove of the β1-CBM. In this AMPK structure, the channel is blocked at approximately the α-B helix of the α2-subunit by residues Arg49, Arg53, Pro86 and Thr87 of the α2-subunit and Pro140, Gln154, Lys156, Asp159 and Lys172 of the β1-subunit. Whereas in the (b) 5ISO56 and (c) 6B1U38 AMPK structures, the channel runs the full width across the α2- and β1-subunit interface (i.e. from the start of the ADaM site to the C-interacting helix of the β1-subunit, including the pocket beneath the cyclodextran binding groove of the β1-CBM). Polar interactions made by pSer108 in the (d) palmitoyl-CoA:AMPKα2β1γ1 model, (e) SC4:AMPKα2β1γ1 crystal structure (PDB ID: 6B1U) and (f) A-769662:AMPKα2β1γ1 crystal structure (PDB ID: 4CFF). (g) Overlay of the palmitoyl-CoA:AMPKα2β1γ1 model with the A-769662:AMPKα2β1γ1 and SC4:AMPKα2β1γ1 crystal structures (only A-769962 and SC4 shown for clarity). The same view is shown in panels a-c and d-g, the structures have been aligned via their β1-subunit CBM. In panels a-c, the location of the ATP binding site in the α2-subunit kinase domain is indicated by staurosporine (cyan sticks). Residues from the α2-catalytic subunit are underlined. Polar interactions are indicated by black dashed lines.
Extended Data Fig. 5. Unique palmitoyl-CoA conformation clusters consistent with our experimental data.

Docking into the (a) 4CFF, (b) 5ISO and (c) 6B1U active AMPKα2β1γ1 structures (grey cartoon). Palmitoyl-CoA shown as sticks, with the carbon atoms coloured either yellow or green. (d) Overlay of all docked palmitoyl-CoA poses except for those in Clusters 2, 4 and 5 for the 5ISO AMPK structure. The overlay shows that the different conformational clusters for the 4CFF, 5ISO and 6B1U AMPK structures fall under a general binding mode. Carbon, sulphur, nitrogen, oxygen and phosphorous atoms are coloured grey, yellow, blue, red and orange respectively.
Extended Data Fig. 6. Alignment of AMPK β1 and β2 sequences from diverse species.

Alignments to human AMPKβ1Gly86 and β2Glu85 residues are in bold.
Extended Data Fig. 7. AMPKβ1N111 does not alter sensitivity to palmitoyl-CoA or AMP.
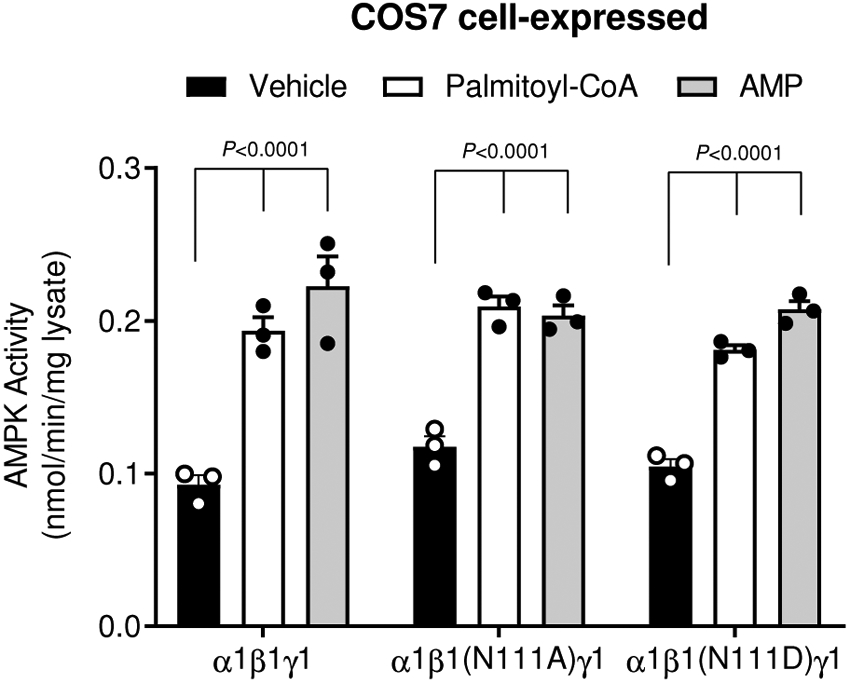
AMPK activity of AMPKα1β1γ1 (WT, β1N111A or β1N111D) was determined by 32P SAMS assay, following immobilization on anti-myc agarose, ± palmitoyl-CoA (10 μM) or AMP (100 μM). Data are shown as mean fold change in activity vs. vehicle, ± s.e.m.; n = 3. Statistical significance was calculated using two-way ANOVA with Bonferroni’s multiple comparisons test. n represent biological independent experiments.
Extended Data Table 1.
Basal activities of AMPK expressed in COS7 mammalian cells
| AMPK complex | immobilization | basal activity (nmol/min/mg lysate) ± s.e.m. |
relates to |
|---|---|---|---|
| α1β1γ1 | myc IP (β-subunit) | 0.1094 ± 2.1 | Fig. 1f |
| α1β2γ1 | myc IP (β-subunit) | 0.1689 ± 17.7 | Fig. 1h |
| α2β1γ1 | myc IP (β-subunit) | 0.238 ± 0.5 | Fig. 1f |
| α2β2γ1 | myc IP (β-subunit) | 0.814 ± 6.7 | Fig. 1h |
| α1β1γl | myc IP (β-subunit) | 0.613 ± 7.5 | Fig. 2a |
| α1β1(N-Δ71)γl | myc IP (β-subunit) | 0.502 ± 4.0 | Fig. 2a |
| α1β1(N-Δ145)γ1 | myc IP (β-subunit) | 0.327 ± 2.5 | Fig. 2a |
| α1β1γ1 | flag IP (α-subunit) | 0.1063 ± 5.9 | ED Fig. 2a |
| α1β1γ1 | GST (α-subunit) | 0.2789 ± 20.2 | ED Fig. 2b |
| α1β1γ1 | myc IP (β-subunit) | 0.1433 ± 5.3 | ED Fig. 3a |
| α1β1(S108A)γl | myc IP (β-subunit) | 0.724 ± 1.4 | ED Fig. 3a |
Extended Data Table 2.
Basal activities of AMPK expressed in E. coli
Supplementary Material
Acknowledgements
This work was supported by the Canadian Institutes of Health Research (201709FDN-CEBA-116200 to GRS), Diabetes Canada (DI-5-17-5302-GS) and project grants from the National Health and Medical Research Council of Australia (NHMRC: 1098459 to JSO, JWS and BEK; 1145265 to JSO and BEK; 1085460 to BEK, SG and GRS; 1138102 to JWS and BEK), Australian Research Council (ARC, DP170101196 to BEK), the Jack Brockhoff Foundation (Grant JBF-4206 to CGL) and the Van Andel Research Institute and the National Institute of Health (NIH R01 GM129436) to K.M. GRS is supported by a Tier 1 Canada Research Chair and a J. Bruce Duncan Chair in Metabolic Diseases. BEK and MWP are NHMRC Fellows, CGL is an NHMRC Early Career Research Fellow. This study was supported in part by the Victorian Government’s Operational Infrastructure Support Program.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Faergeman NJ & Knudsen J Role of long-chain fatty acyl-CoA esters in the regulation of metabolism and in cell signalling. Biochem J 323 ( Pt 1), 1–12 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bortz WM & Lynen F THE INHIBITION OF ACETYL COA CARBOXYLASE BY LONG CHAIN ACYL COA DERIVATIVES. Biochem Z 337, 505–509 (1963). [PubMed] [Google Scholar]
- 3.McGarry JD & Foster DW In support of the roles of malonyl-CoA and carnitine acyltransferase I in the regulation of hepatic fatty acid oxidation and ketogenesis. J Biol Chem 254, 8163–8168 (1979). [PubMed] [Google Scholar]
- 4.McGarry JD, Takabayashi Y & Foster DW The role of malonyl-coa in the coordination of fatty acid synthesis and oxidation in isolated rat hepatocytes. J Biol Chem 253, 8294–8300 (1978). [PubMed] [Google Scholar]
- 5.Trumble GE, Smith MA & Winder WW Purification and characterization of rat skeletal muscle acetyl-CoA carboxylase. Eur J Biochem 231, 192–198, doi: 10.1111/j.1432-1033.1995.tb20686.x (1995). [DOI] [PubMed] [Google Scholar]
- 6.Ogiwara H, Tanabe T, Nikawa J & Numa S Inhibition of rat-liver acetyl-coenzyme-A carboxylase by palmitoyl-coenzyme A. Formation of equimolar enzyme-inhibitor complex. Eur J Biochem 89, 33–41, doi: 10.1111/j.1432-1033.1978.tb20893.x (1978). [DOI] [PubMed] [Google Scholar]
- 7.Steinberg GR & Kemp BE AMPK in Health and Disease. Physiol Rev 89, 1025–1078, doi: 10.1152/physrev.00011.2008 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Fullerton MD et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med 19, 1649–1654, doi: 10.1038/nm.3372 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carling D, Zammit VA & Hardie DG A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett 223, 217–222 (1987). [DOI] [PubMed] [Google Scholar]
- 10.Carling D, Thornton C, Woods A & Sanders MJ AMP-activated protein kinase: new regulation, new roles? Biochem J 445, 11–27, doi: 10.1042/bj20120546 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Viollet B et al. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol 45, 276–295, doi: 10.3109/10409238.2010.488215 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hawley SA et al. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 271, 27879–27887 (1996). [DOI] [PubMed] [Google Scholar]
- 13.Bateman A The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci 22, 12–13 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Chen L et al. AMP-activated protein kinase undergoes nucleotide-dependent conformational changes. Nat Struct Mol Biol 19, 716–718, doi: 10.1038/nsmb.2319 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Xiao B et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449, 496–500, doi: 10.1038/nature06161 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Zhu L et al. Structural insights into the architecture and allostery of full-length AMP-activated protein kinase. Structure 19, 515–522, doi: 10.1016/j.str.2011.01.018 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Carling D, Clarke PR, Zammit VA & Hardie DG Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur J Biochem 186, 129–136 (1989). [DOI] [PubMed] [Google Scholar]
- 18.Cheung PC, Salt IP, Davies SP, Hardie DG & Carling D Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem J 346 Pt 3, 659–669 (2000). [PMC free article] [PubMed] [Google Scholar]
- 19.Davies SP, Helps NR, Cohen PT & Hardie DG 5’-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett 377, 421–425, doi: 10.1016/0014-5793(95)01368-7 (1995). [DOI] [PubMed] [Google Scholar]
- 20.Oakhill JS et al. AMPK is a direct adenylate charge-regulated protein kinase. Science 332, 1433–1435, doi: 10.1126/science.1200094 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA & Carling D Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J 403, 139–148, doi: 10.1042/bj20061520 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suter M et al. Dissecting the role of 5’-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem 281, 32207–32216, doi: 10.1074/jbc.M606357200 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Bieri M et al. AMP-activated protein kinase beta-subunit requires internal motion for optimal carbohydrate binding. Biophys J 102, 305–314, doi: 10.1016/j.bpj.2011.12.012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koay A, Rimmer KA, Mertens HD, Gooley PR & Stapleton D Oligosaccharide recognition and binding to the carbohydrate binding module of AMP-activated protein kinase. FEBS Lett 581, 5055–5059, doi: 10.1016/j.febslet.2007.09.044 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Steinberg GR et al. Reduced glycogen availability is associated with increased AMPKalpha2 activity, nuclear AMPKalpha2 protein abundance, and GLUT4 mRNA expression in contracting human skeletal muscle. Appl Physiol Nutr Metab 31, 302–312, doi: 10.1139/h06-003 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Wojtaszewski JF et al. Regulation of 5’AMP-activated protein kinase activity and substrate utilization in exercising human skeletal muscle. Am J Physiol Endocrinol Metab 284, E813–822, doi: 10.1152/ajpendo.00436.2002 (2003). [DOI] [PubMed] [Google Scholar]
- 27.McBride A, Ghilagaber S, Nikolaev A & Hardie DG The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab 9, 23–34, doi: 10.1016/j.cmet.2008.11.008 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polekhina G et al. Structural basis for glycogen recognition by AMP-activated protein kinase. Structure 13, 1453–1462, doi: 10.1016/j.str.2005.07.008 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Zhang CS et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112–116, doi: 10.1038/nature23275 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li M et al. Transient Receptor Potential V Channels Are Essential for Glucose Sensing by Aldolase and AMPK. Cell Metab 30, 508–524.e512, doi: 10.1016/j.cmet.2019.05.018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watt MJ, Steinberg GR, Chen ZP, Kemp BE & Febbraio MA Fatty acids stimulate AMP-activated protein kinase and enhance fatty acid oxidation in L6 myotubes. J Physiol 574, 139–147, doi: 10.1113/jphysiol.2006.107318 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark H, Carling D & Saggerson D Covalent activation of heart AMP-activated protein kinase in response to physiological concentrations of long-chain fatty acids. Eur J Biochem 271, 2215–2224, doi: 10.1111/j.1432-1033.2004.04151.x (2004). [DOI] [PubMed] [Google Scholar]
- 33.Cool B et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab 3, 403–416, doi: 10.1016/j.cmet.2006.05.005 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Giordanetto F & Karis D Direct AMP-activated protein kinase activators: a review of evidence from the patent literature. Expert Opin Ther Pat 22, 1467–1477, doi: 10.1517/13543776.2012.743994 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Xiao B et al. Structural basis of AMPK regulation by small molecule activators. Nat Commun 4, 3017, doi: 10.1038/ncomms4017 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hawley SA et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 336, 918–922, doi: 10.1126/science.1215327 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cameron KO et al. Discovery and Preclinical Characterization of 6-Chloro-5-[4-(1-hydroxycyclobutyl)phenyl]-1H-indole-3-carboxylic Acid (PF-06409577), a Direct Activator of Adenosine Monophosphate-activated Protein Kinase (AMPK), for the Potential Treatment of Diabetic Nephropathy. J Med Chem. 59, 8068–8081. doi: 80 10.1021/acs.jmedchem.8066b00866. Epub 02016 Aug 00820. (2016). [DOI] [PubMed] [Google Scholar]
- 38.Ngoei KRW et al. Structural Determinants for Small-Molecule Activation of Skeletal Muscle AMPK alpha2beta2gamma1 by the Glucose Importagog SC4. Cell Chem Biol. 25, 728–737.e729. doi: 7 10.1016/j.chembiol.2018.1003.1008. Epub 2018 Apr 1012. (2018). [DOI] [PubMed] [Google Scholar]
- 39.Sanders MJ et al. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem 282, 32539–32548, doi: 10.1074/jbc.M706543200 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Calabrese MF et al. Structural basis for AMPK activation: natural and synthetic ligands regulate kinase activity from opposite poles by different molecular mechanisms. Structure. 22, 1161–1172. doi: 11 10.1016/j.str.2014.1106.1009. Epub 2014 Jul 1124. (2014). [DOI] [PubMed] [Google Scholar]
- 41.Langendorf CG et al. Structural basis of allosteric and synergistic activation of AMPK by furan-2-phosphonic derivative C2 binding. Nat Commun. 7:10912, 10.1038/ncomms10912. (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langendorf CG & Kemp BE Choreography of AMPK activation. Cell Res. 25, 5–6. doi: 10.1038/cr.2014.1163. Epub 2014 Dec 1035. (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scott JW et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol. 15, 1220–1230. doi: 12 10.1016/j.chembiol.2008.1210.1005. (2008). [DOI] [PubMed] [Google Scholar]
- 44.O’Neill HM et al. AMPK phosphorylation of ACC2 is required for skeletal muscle fatty acid oxidation and insulin sensitivity in mice. Diabetologia. 57, 1693–1702. doi: 16 10.1007/s00125-00014-03273-00121. Epub 02014 Jun 00110. (2014). [DOI] [PubMed] [Google Scholar]
- 45.Esquejo RM et al. Activation of Liver AMPK with PF-06409577 Corrects NAFLD and Lowers Cholesterol in Rodent and Primate Preclinical Models. EBioMedicine. 31:122–132., 10.1016/j.ebiom.2018.1004.1009. Epub 2018 Apr 1018. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scott JW et al. Small molecule drug A-769662 and AMP synergistically activate naive AMPK independent of upstream kinase signaling. Chem Biol. 21, 619–627. doi: 6 10.1016/j.chembiol.2014.1003.1006. Epub 2014 Apr 1017. (2014). [DOI] [PubMed] [Google Scholar]
- 47.Dzamko N et al. AMPK beta1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. J Biol Chem. 285, 115–122. doi: 1 10.1074/jbc.M1109.056762. Epub 052009 Nov 056765. (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Galic S et al. Hematopoietic AMPK beta1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J Clin Invest 121, 4903–4915, doi: 10.1172/jci58577 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galic S et al. AMPK signaling to acetyl-CoA carboxylase is required for fasting- and cold-induced appetite but not thermogenesis. Elife 7, doi: 10.7554/eLife.32656 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Claret M et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest 117, 2325–2336, doi: 10.1172/jci31516 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lam TK, Schwartz GJ & Rossetti L Hypothalamic sensing of fatty acids. Nat Neurosci 8, 579–584, doi: 10.1038/nn1456 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Kosteli A et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest 120, 3466–3479, doi: 10.1172/jci42845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oakhill JS et al. beta-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc Natl Acad Sci U S A 107, 19237–19241, doi: 10.1073/pnas.1009705107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dite TA et al. AMP-activated protein kinase selectively inhibited by the type II inhibitor SBI-0206965. J Biol Chem. 293, 8874–8885. doi: 88 10.1074/jbc.RA8118.003547. Epub 002018 Apr 003525. (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li X et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res 25, 50–66, doi: 10.1038/cr.2014.150 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Willows R et al. Phosphorylation of AMPK by upstream kinases is required for activity in mammalian cells. Biochem J. 474, 3059–3073. doi: 30 10.1042/BCJ20170458. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clark RD, Strizhev A, Leonard JM, Blake JF & Matthew JB Consensus scoring for ligand/protein interactions. J Mol Graph Model. 20, 281–295. (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon request. Source data for Figs. 1-3 and Extended Data Figs. 1-7 are available online.