

Abstract
Background and purpose:
Cardiac complications post-stroke are common, and diabetes exacerbates post-stroke cardiac injury. In this study, we tested whether treatment with exosomes harvested from human umbilical cord blood derived CD133+ cells (CD133+Exo) improves cardiac function in type 2 diabetes mellitus (T2DM) stroke mice.
Methods:
Adult (3–4m), male, BKS.Cg-m+/+Leprdb/J (db/db, T2DM) and non-DM (db+) mice were randomized to sham or photothrombotic stroke groups. T2DM-stroke mice were treated with Phosphate-buffered saline (PBS) or CD133+Exo (20μg, i.v) at 3 days after stroke. T2DM sham, and T2DM+CD133+Exo treatment groups were included as controls. Echocardiography was performed, and mice were sacrificed at 28 days after stroke. Cardiomyocyte hypertrophy, myocardial capillary density, interstitial fibrosis and inflammatory factor expression were measured in heart. MicroRNA-126 expression and its target gene expression were measured in the heart.
Results:
T2DM mice exhibit significant cardiac deficits such as decreased left ventricular ejection fraction (LVEF) and shortening fraction (LVSF), increased left ventricular diastolic dimension (LVDD) and reduced heart rate compared to non-DM mice. Stroke in non-DM and T2DM mice significantly decreases LVEF compared to non-DM and T2DM-sham, respectively. Cardiac dysfunction is worse in T2DM-stroke mice compared to non-DM-stroke mice. CD133+Exo treatment of T2DM-stroke mice significantly improves cardiac function identified by increased LVEF and decreased LVDD compared to PBS treated T2DM-stroke mice. In addition, CD133+Exo treatment significantly decreases body weight and blood glucose but does not decrease lesion volume in T2DM-stroke mice. CD133+Exo treatment of T2DM mice significantly decreases body weight and blood glucose but does not improve cardiac function. CD133+Exo treatment in T2DM-stroke mice significantly decreases myocardial cross sectional area, interstitial fibrosis, transforming growth factor beta (TGF-β), numbers of M1 macrophages, and oxidative stress markers 4-HNE (4-Hydroxynonenal) and NADPH oxidase 2 (NOX2) in heart tissue. CD133+Exo treatment increases myocardial capillary density in T2DM-stroke mice as well as upregulates endothelial cell capillary tube formation in-vitro. MiR-126 is highly expressed in CD133+Exo compared to exosomes derived from endothelial cells. Compared to PBS treatment, CD133+Exo treatment significantly increases miR-126 expression in heart and decreases its target gene expression such as Sprouty-related, EVH1 domain-containing protein 1 (Spred-1), vascular cell adhesion protein (VCAM), and monocyte chemoattractant protein 1 (MCP1) in the heart of T2DM-stroke mice.
Conclusions:
CD133+Exo treatment significantly improves cardiac function in T2DM-stroke mice. The cardio-protective effects of CD133+Exo in T2DM-stroke mice may be attributed at least in part to increasing miR-126 expression and decreasing its target protein expression in the heart, increased myocardial capillary density and decreased cardiac inflammatory factor expression.
Keywords: Cardiac function, CD133, Echocardiography, Exosomes, Stroke, Type 2 diabetes mellitus
Introduction
Ischemic stroke patients with diabetes mellitus exhibit distinct clinical patterns and worse neurovascular prognosis compared to non-diabetic stroke patients [1]. Upwards of 30% of stroke patients have diabetes [2, 3]. In addition to neurological injury, cardiac injury is common in stroke patients and is associated with poor stroke outcome and increased risk of death [4–7]. We have previously demonstrated that ischemic stroke in non-diabetic mice induces progressive cardiac dysfunction from acute (3 days) to chronic phase (28 days) after stroke with decreased ejection fraction and shortening fraction compared to non-stroke mice [8, 9]. In addition, stroke induces cardiomyocyte hypertrophy, myocardial interstitial fibrosis, oxidative stress and inflammatory responses compared to non-stroke mice [8]. While these studies demonstrate that stroke induces cardiac deficits even in the absence of primary cardiac disease, common risk factors for stroke and cardiovascular injury such as hypertension, diabetes, and advancing age are known to aggravate cardiac injury irrespective of stroke cause or subtype [10, 11]. Therefore, in diabetic patients, in addition to stroke induced cardiac injury, hyperglycemia induced microvascular and macrovascular damage, high systemic inflammatory status and immune responses may result in worse cardiac function [12]. There is a need to identify treatments that specifically improve cardiac function after stroke in the diabetic population.
Circulating endothelial progenitor cells (EPCs) are involved in the neovasculogenesis and maintenance of vascular homeostasis, and regulate the pathogenesis of diabetic vasculopathy [13]. Treatment of stroke with circulating human EPCs decreases infarct volume, exerts protective effects on the neurovascular unit, and promotes long term neurological functional recovery in mice [14, 15]. The number, function, and mobilization of circulating EPCs are significantly reduced in patients with T2DM and vascular diseases [13, 16]. EPCs, by releasing exosomes, stimulate angiogenesis, enhance recovery in a murine model of hind limb ischemia, as well as protect cardiomyocytes against angiotensin II-induced hypertrophy and apoptosis [17, 18].
Exosomes are small extracellular vesicles (~30–150 nm in diameter) with a bi-lipid membrane that are enriched with nucleic acids like mRNA and microRNAs (miRs) [19]. Exosomes are emerging as novel biomarkers as well as novel therapeutic agents for stroke [20–23]. EPC derived exosomes and exosome-mediated transfer of miRs have been reported to promote EPC treatment induced protective effects on microvasculature in a murine model of sepsis [24]. Human peripheral blood EPC derived exosomes increase the proliferation of cardiac fibroblasts and angiogenesis, by regulating the differentiation of fibroblasts into endothelial cells and decreasing the expression of proteins involved in cardiac fibrosis [25]. CD133 is a marker for hematopoietic stem and progenitor cells, and CD133+/KDR+ identifies EPCs [26, 27]. In this study, we investigate whether exosomes harvested from human umbilical cord blood derived CD133+ cells (CD133+Exo) administered 3 days after stroke in T2DM mice improves cardiac function.
Materials and methods
All experiments were conducted in accordance with the standards and procedures of the American Council on Animal Care and with the approval of Institutional Animal Care and Use Committee of Henry Ford Health System. This manuscript was prepared in accordance with ARRIVE guidelines.
Experimental groups:
Male, adult (3–4m) non-DM (db+) and T2DM (BKS.Cg-m+/+Leprdb/J, Jackson Laboratory) mice were employed and randomized to the following experimental groups: 1) Non-DM (n=10), 2) Non-DM-stroke (n=10), 3) T2DM (n=10), 4) T2DM-stroke+PBS (n=7), 5) T2DM+CD133+Exo (n=7), 5) T2DM-stroke+CD133+Exo (n=8). PBS (200μl) and CD133+Exo (20μg in 200μl PBS) treatments were administered intravenously at 3 days after stroke. The early mortality (3 days after stroke, before treatment) was 25% (5/20) in T2DM mice. There was no mortality after PBS or CD133+Exo treatment. The experimental timeline is provided in Fig.1a.
Fig.1. CD133+Exo treatment reduces body weight and blood glucose in T2DM-stroke mice.
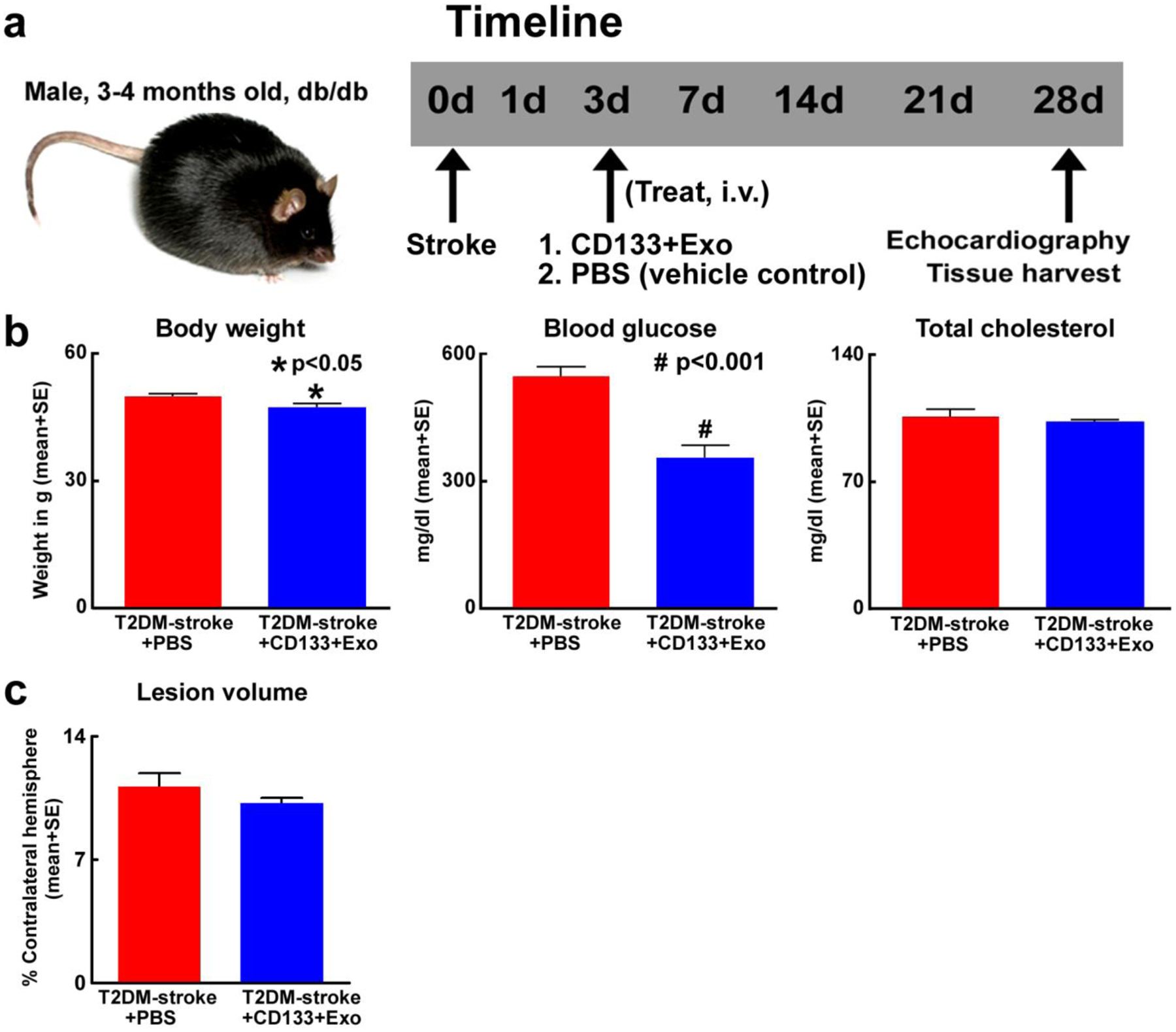
a) Time line of experiments. At 28 days after stroke, CD133+Exo treatment significantly b) decreases body weight and blood glucose without altering cholesterol level and c) does not decrease lesion volume when compared to PBS treated T2DM-stroke mice. T2DM-stroke+PBS: n=7; T2DM-stroke+CD133+Exo: n=8.
Photothrombotic ischemic stroke model:
Diabetic patients have a high prevalence of subcortical infarction [28] associated with the development of cerebral small vessel occlusions [29, 30]. The photothrombotic stroke model is a model of occlusion of small cerebral vessels and generates highly reproducible infarcts in the frontal and parietal cortex without damaging the cardiovascular regions of the brain (insular cortex) and hypothalamus [31–33]. Briefly, mice were anesthetized in a chamber with 3.5% isoflurane and then the head was fixed on the stereotaxic instrument and anesthesia maintained with 1.5% isoflurane in a mixture of 70% N2O and 30% O2 using a facemask. Throughout the surgery, rectal temperature was maintained at 37°C using a feedback regulated water heating system. A skin incision was made on the head to bregma along the median. A roundabout rubber was placed on the skull surface such that the sensorimotor area (1.5 to 3 mm lateral; 0.5 to 1 mm anterior of bregma) was covered by the inner circle. A photosensitive dye (Rose Bengal, 40mg/kg) mixed with saline (4ml/kg) was injected using intraperitoneal injection and 5 minutes later, a cold light illuminator was fixed close to the skull surface of sensorimotor area and turned on. After 20 minutes of cold light exposure, the activated dye induces endothelial damage with platelet activation and thrombosis, resulting in local blood flow interruption [32, 33].
Measurement of physiological parameters:
Fasting blood glucose was tested using a glucose analyzer (AgaMatrix Advanced blood glucose monitoring system) and blood lipids measured using CardioChek Plus analyzer (Fischer Scientific). T2DM mice with fasting blood glucose >300 mg/dl at randomization were included in the study. Fasting blood glucose was also measured before sacrifice to test the effect of treatment on glucose modulation.
Echocardiography:
An investigator blinded to the experimental groups performed echocardiographic measurements in conscious mice. Transthoracic Doppler echocardiography was performed on conscious mice using a Doppler echocardiograph (Acuson C516) equipped with a 15-MHz linear transducer (15L-8) [34, 8]. Mice were trained for 3 consecutive days before echocardiography. With repeated training mice remain calm without developing bradycardia. Briefly, the mouse was picked up by the nape of the neck and held firmly in the palm of one hand in the supine position. After training, the left hemithorax was shaved and a pre-warmed ultrasound transmission gel was applied to the chest. All measurements were digitized by goal-directed, diagnostically driven software and 3 beats were averaged for each measurement including: left ventricular (LV) mass, LV diastolic dimension (LVDD), LV ejection fraction (LVEF), LV shortening fraction (LVSF), cardiac output, diastolic interventricular septum thickness (IVSTd), and diastolic posterior wall thickness (PWTd). LVEF was measured using the formula: LVEF = [(LVAd - LVAs)/LVAs × 100], where LVAd is LV diastolic area and LVAs is LV systolic area.
Immunohistochemical evaluation:
At 28 days after stroke, mice were sacrificed. Heart and brain were isolated and fixed in 4% paraformaldehyde before embedding in paraffin. Seven equally spaced (1 mm) brain coronal sections were processed. Hematoxylin and eosin (H&E) staining was employed for lesion volume measurement. The percentage of the infarction volume compared with the contralateral hemisphere is presented [35]. Heart sections (6μm) were cut and PicroSirius Red (PSR) staining was employed to evaluate fibrosis by measuring interstitial collagen fraction (ICF). Rhodamine-labeled Griffonia simplicifolia lectin staining was used to measure capillary density and assess cardiac microcirculation environment; and FITC-Peanut agglutinin (FITC-PNA) was used to calculate myocardial cross-section area (MCSA) to evaluate cardiomyocyte hypertrophy. For immunostaining, antibodies against mouse ED1 (a marker for monocytes/macrophages; 1:30, BIO-RAD); transforming growth factor (TGF-β; 1:500, Santa Cruz); 4-Hydroxynonenal (4-HNE, 1:1000, Millipore Sigma); NADPH oxidase-2 (NOX2; 1:400, BD Bioscience); monocyte chemoattractant protein 1 (MCP1; 1:100, Abcam) were employed. Negative controls were processed in a similar fashion, but without adding the primary antibody.
Quantification analysis:
An investigator blinded to the experimental groups performed all quantification analysis. Five slides from each heart, with each slide containing 3 fields of view were digitized under a 20× objective (Olympus BX40) using a 3-CCD color video camera (Sony DXC-970MD) interfaced with an MCID image analysis system (Imaging Research). An in-built densitometry function in MCID was used to calculate positive areas of immunolabeling with uniform threshold set above unstained area for all slides. For cells, the number of positive labeled cells in each field of view was counted and averaged to yield a single value for each animal and presented as number of positive cells/unit area.
Cell culture and exosome isolation:
CD133+ cells (SER-CD34-F, Zenbio) were grown in the culture media for one week and media was collected and filtered with 0.22 μM syringe filter to remove any particulate matter. Endothelial cells (EC, ATCC, catalog #CRL-2299) were cultured in Dulbecco modified Eagle medium (DMEM, Life Technologies) with 10% FBS (fetal bovine serum) and 1% antibiotic/antimycotic (Life Technologies) following the manufacturer’s protocols. Cells were cultured up to 3 passages, followed by culture in exosome depleted FBS media (Systembio) for 3 days. The media was collected to isolate exosomes. Exoquick TC (System Biosciences) was added at a ratio of 2 ml Exoquick/10 ml media. The media was stored overnight at 4°C and then centrifuged at 1500g for 30 minutes. Supernatant was removed and the pellet was resuspended in PBS. Protein concentration was determined using BCA Protein Assay Kit (Pierce) following standard protocol. Exosome size characterization was performed using NanoSight NS 300.
Tube formation assay:
Matrigel (Becton Dickinson) was diluted to 75% with SF-DMEM and 100 μl was added per well in a 96 well plate before being incubated at 37°C for 30 minutes. Meanwhile, mouse brain endothelial cells (mBE, CRL-2299, ATCC) were cultured and counted to yield 22500 cells/well and were suspended in SF-DMEM. 100 μl of cell suspension with treatment were added to each well (n=4/group) and allowed to incubate for 3 hours. After 3 hours, the Matrigel wells were digitized under a 4× objective (Olympus BX40) for measurement of total tube length of capillary tube formation. Tracks of endothelial cells organized into networks of cellular cords (tubes) were counted and averaged in four randomly selected microscopic fields.
MiR-126 measurement:
Samples were lysed in Qiazol reagents and the total RNA was isolated using miRNeasy Mini kit (Qiagen). Briefly, miRNAs were reverse transcribed with the miRNA Reverse Transcription kit (Thermo Fisher Scientific) and PCR amplification was performed with the TaqMan miRNA assay kit (hsa-miR-126–3p, Thermo Fisher Scientific, catalog #4427975, which is specific for mature miRNA sequences) according to the manufacturer’s protocols, with U6 snRNA as an internal control [36]. For the qPCR reactions, we used 2 μl of isolated RNA per cDNA reaction, and then used 3 μl of cDNA for each PCR reaction.
PCR:
Total RNA was isolated with TRIzol (Life Technologies), following standard protocol. Following isolation, 2 μg of RNA was used to make cDNA. Quantitative PCR was performed using the SYBR Green real time PCR method on a ViiA7 thermocycler (Applied Biosystems, Foster City, CA) using 3-stage program parameters provided by the manufacturer, as follows; 2 min at 50°C, 10 min at 95°C, and then 40 cycles of 15 seconds at 95°C and 1 min at 60°C. Each sample was tested in triplicate, and analysis of relative gene expression data using the 2−ΔΔCT method. The following primers were used:
MCP1: Fwd: ctgctactcattcac cag caa g; Rev: ctc tct ctt gag cttg gtg aca
VCAM: Fwd: cag gtg gag gtc tac tca ttc c; Rev: ctc cag atg gtc aaa ggg ata c
Spred-1: Fwd: agc agt gcc taa aat gag ctt c; Rev: aag agg gag agg aga agc aag t
Statistical analysis:
One-way Analysis of Variance was employed for the evaluation of functional outcome and histology, respectively. “Contrast/estimate” statement was used to test the group difference. If an overall treatment group effect was detected at p<0.05, pair-wise comparisons were made. All data are presented as mean ± standard error (SE).
Results
CD133+Exo treatment significantly lowers body weight and blood glucose, and improves cardiac function in T2DM stroke mice
Fig.1b shows that compared to PBS treated T2DM-stroke mice, CD133+Exo treated T2DM-stroke mice exhibit significantly lower blood glucose, and modest body weight loss at 28 days after stroke. However, CD133+Exo did not have a cholesterol lowering effect and the total cholesterol was not significantly different between PBS and CD133+Exo treated T2DM stroke mice. CD133+Exo treatment does not decrease lesion volume at 28 days after stroke (Fig.1c) compared to PBS treated T2DM-stroke mice.
To test whether T2DM mice have cardiac dysfunction and whether stroke induces or exacerbates cardiac dysfunction in the T2DM mice, echocardiography was performed at 28 days after stroke in non-DM and T2DM mice. Data in Fig.2a indicate compared to non-DM mice, T2DM mice exhibit significantly reduced LVEF, SF, heart rate, and increased LVDD. In non-DM and T2DM mice, stroke significantly decreases LVEF at 1 month compared to non-DM and T2DM sham mice, respectively. T2DM-stroke mice have significantly reduced LVEF, heart rate and increased LVDD compared to non-DM-stroke mice at 1 month after stroke. Therefore, stroke and T2DM induces significant cardiac dysfunction with worse function in T2DM-stroke mice. In T2DM-stroke mice cardiac dysfunction was evident at 1 month after stroke but not at 3 days after stroke when compared to T2DM sham mice. Therefore, it is unlikely that there were baseline cardiac functional deficits between control and treatment groups at randomization on day 3 after stroke.
Fig.2. Stroke and T2DM induce cardiac dysfunction in mice and CD133+Exo treatment improves cardiac function in T2DM-stroke mice.
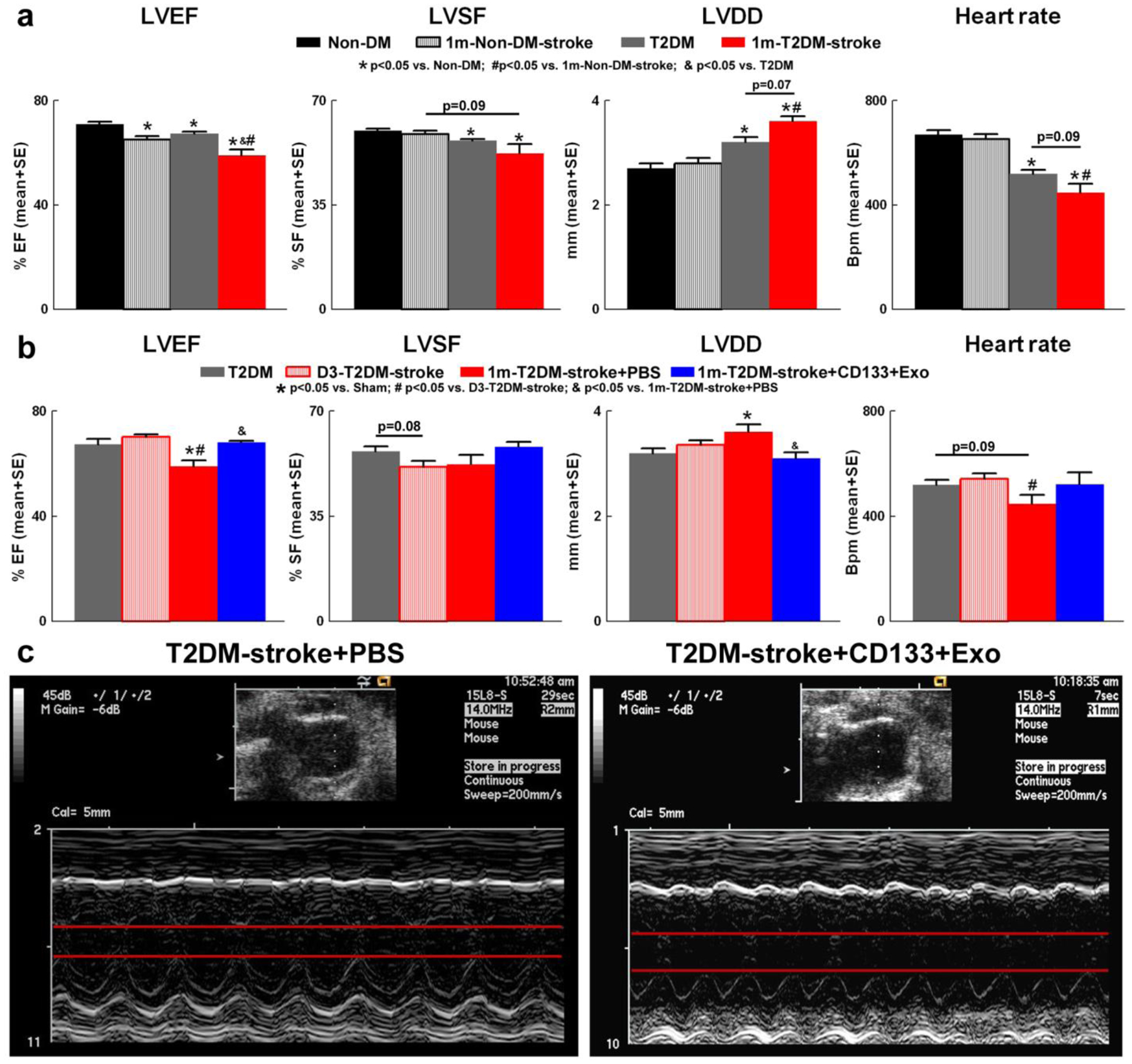
a) Compared to non-DM mice, T2DM mice exhibit significantly reduced left ventricular ejection fraction (LVEF), left ventricular shortening fraction (LVSF), heart rate, and increased left ventricular diastolic dimension (LVDD). In non-DM and T2DM mice, stroke significantly decreases LVEF at 1 month compared to non-DM and T2DM sham mice, respectively. T2DM-stroke mice have significantly reduced LVEF, heart rate and increased LVDD compared to non-DM-stroke mice at 1 month after stroke. b) Stroke in T2DM mice significantly decreases LVEF and increases LVDD at 1 month after stroke but not at 3 days after stroke. CD133+Exo treatment significantly increases LVEF and decreases LVDD but does not significantly increase LVSF compared to PBS treated T2DM-stroke mice. There were no significant differences in heart rate between stroke and control or between PBS and CD133+Exo treated T2DM-stroke mice. c) Representative images of M-mode transthoracic echocardiography measurements in short-axis view. Non-DM: n=10; Non-DM-stroke: n=10; T2DM: n=10; T2DM-stroke+PBS: n=7; T2DM+CD133+Exo: n=7.
To test the therapeutic effects of CD133+Exo treatment on cardiac function, echocardiography was performed at 28 days after stroke in conscious mice. Fig.2b–c shows that compared to PBS treatment, CD133+Exo treatment in T2DM-stroke mice significantly increases LVEF, and decreases LVDD. There were no significant differences between PBS and CD133+Exo treated T2DM-stroke mice in heart rate, cardiac output (T2DM-stroke: 5.4±0.4 ml/min/10g b.wt; +CD133+Exo: 6.1±0.5 ml/min/10g b.wt), LV mass (T2DM-stroke: 23.3±1.3 mg/10g b.wt; +CD133+Exo: 19.2±1.5 mg/10g b.wt), LVSF (T2DM-stroke: 52.2±3.2%; +CD133+Exo: 58.1±1.7%) or LV systolic dimension (T2DM-stroke: 1.7±0.2 mm; +CD133+Exo: 1.3±0.1 mm).
To test whether the cardio-protective effects of CD133+Exo is specific to diabetic cardiac dysfunction or to post-stroke cardiac dysfunction, we evaluated cardiac function at 1 month after CD133+Exo in T2DM sham mice. Fig.3 indicate that CD133+Exo treatment in T2DM mice significantly decreases body weight and blood glucose but does not significantly affect cardiac function evaluated by echocardiography at 1 month after treatment, and the cardio-protective effects of CD133+Exo treatment are most likely specific to post-stroke cardiac dysfunction in diabetic mice.
Fig.3. CD133+Exo treatment reduces body weight and blood glucose but does not improve cardiac function in T2DM mice.

a) Representative images of M-mode transthoracic echocardiography measurements in short-axis view. b) CD133+Exo treatment in T2DM mice does not significantly affect cardiac function evaluated by echocardiography at 1 month after treatment. c) CD133+Exo treatment in T2DM mice significantly decreases body weight and blood glucose compared to baseline values as well as T2DM sham mice at 1 month. T2DM: n=10; T2DM+CD133+Exo: n=7
CD133+Exo treatment significantly decreases cardiac pathological remodeling and increases myocardial capillary density in T2DM stroke mice
Immunohistochemistry was used to evaluate the effects of CD133+Exo treatment on cardiac pathological remodeling after stroke in T2DM mice. Fig.4a shows that CD133+Exo treatment of T2DM stroke mice significantly decreases cardiomyocyte hypertrophy compared to PBS treated T2DM-stroke mice as indicated by decreased myocyte cross sectional area measured using FITC-PNA staining. Fig.4b shows that CD133+Exo treatment significantly decreases interstitial fibrosis compared to control T2DM-stroke mice as indicated by decreased interstitial collagen fraction (ICF) in PSR staining.
Fig.4. CD133+Exo treatment decreases cardiomyocyte hypertrophy and cardiac fibrosis while increasing myocardial capillary density in T2DM-stroke mice.
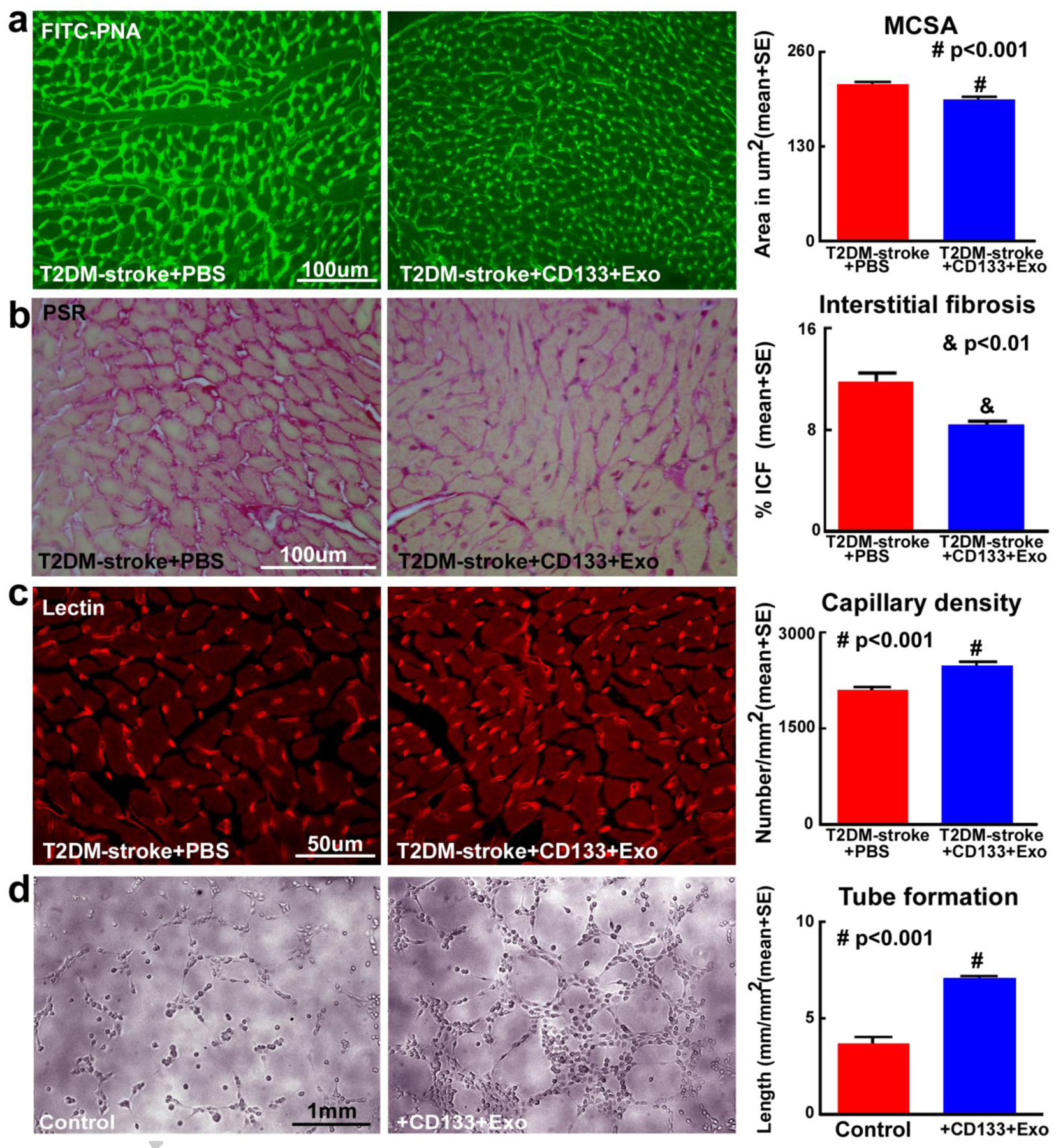
a) CD133+Exo treatment significantly decreases cardiomyocyte cross section area (MCSA) compared to PBS treated T2DM-stroke mice, indicated by FITC-Peanut agglutinin (FITC-PNA) immunostaining and quantification analysis. b) CD133+Exo treatment of T2DM-stroke mice significantly decreases interstitial fibrosis in the heart indicated by decreased interstitial collagen fraction (ICF) in PicroSirius Red (PSR) immunostaining. c) CD133+Exo treatment significantly increases myocardial capillary density compared to PBS treated T2DM-stroke miceindicated by Rhodamine-labeled Griffoniasimplicifolia lectin (lectin) immunostaining and quantification analysis. T2DM-stroke+PBS: n=7; T2DM-stroke+CD133+Exo: n=8. d) In-vitro, CD133+Exo treatment significantly increases capillary tube formation of mouse brain endothelial cells compared to media control.
To test whether CD133+Exo treatment increases myocardial vascular remodeling in T2DM-stroke mice, we measured capillary density in heart using Rhodamine-labeled Griffoniasimplicifolia lectin staining. Fig.4c shows that compared to PBS treated T2DM-stroke control mice, CD133+Exo treatment significantly increases myocardial capillary density. In addition, we employed a capillary tube formation assay in vitro to verify the effects of CD133+Exo on endothelial capillary tube formation/angiogenesis. Fig.4d shows that compared to media control, CD133+Exo significantly increases endothelial cell capillary tube formation.
CD133+Exo treatment significantly decreases cardiac oxidative stress and inflammation in T2DM stroke mice
To test whether CD133+Exo treatment decreases oxidative stress in heart tissue, we measured 4-HNE and NOX2 expression in the heart. Fig.1 shows that compared to PBS treated T2DM-stroke mice, CD133+Exo treatment significantly decreases NOX2 and 4-HNE expression in T2DM-stroke mice. Since TGF-β is a potent contributor to cardiac fibrosis as well as hypertrophy [37, 38], we measured TGF-β expression in cardiac tissue. Fig.6a shows that CD133+Exo treatment significantly decreases cardiac tissue TGF-β expression compared to T2DM-stroke control mice. We also measured cardiac tissue expression of MCP1 and M1 macrophage (ED1) and found that CD133+Exo treatment significantly decreases cardiac tissue expression of MCP1 and ED1 compared to PBS treated T2DM-stroke mice (Fig.6c–d).
Fig.6. CD133+Exo treatment significantly decreases cardiac inflammation, increases miR-126 expression and decreases its target genes in T2DM-stroke mice.
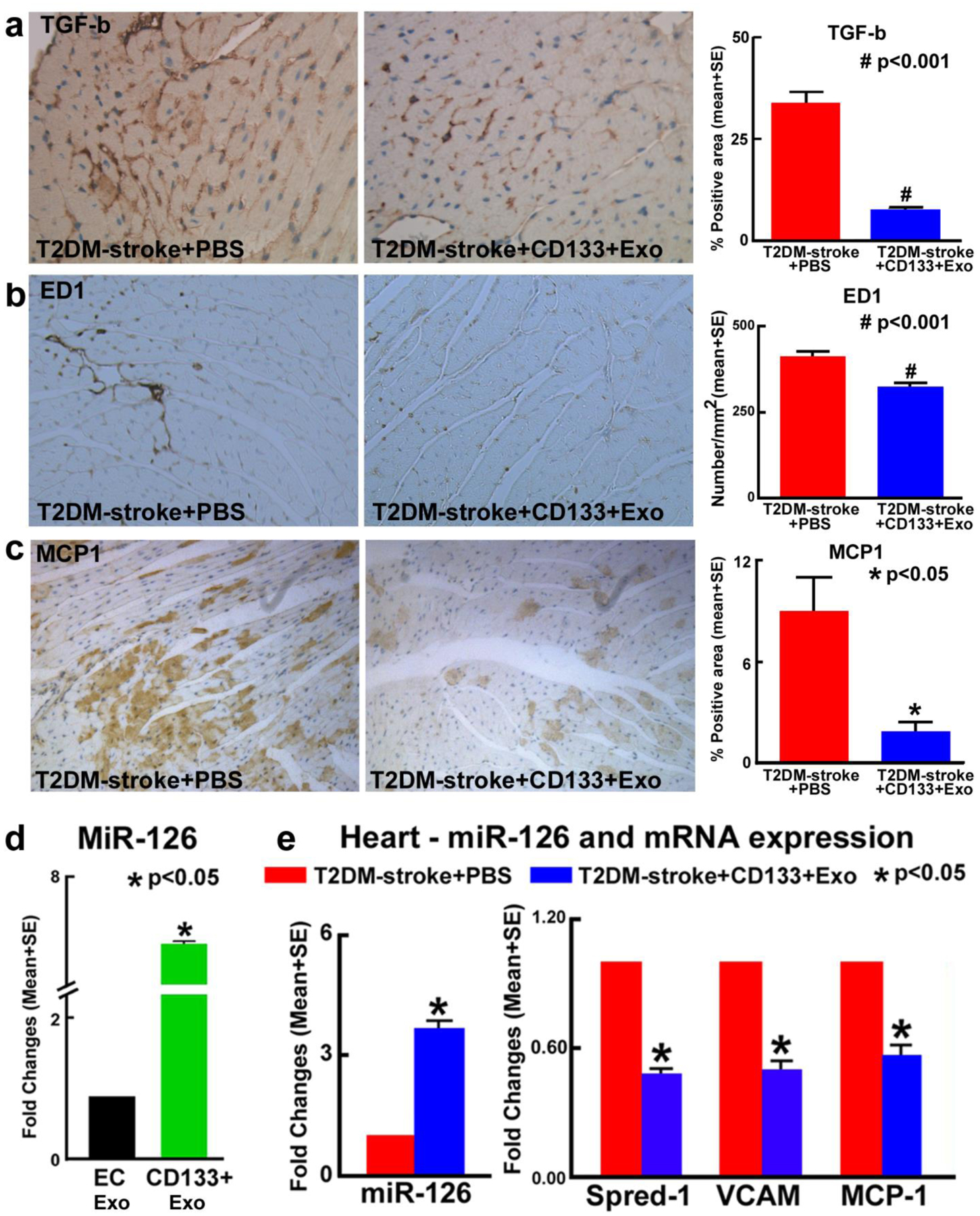
CD133+Exo treatment significantly decreases cardiac tissue inflammatory factor expression such as a) transforming growth factor-β (TGF-β), b) M1 macrophage (ED1) and c) monocyte chemoattractant protein 1 (MCP1) compared to PBS treated T2DM-stroke mice. d) CD133+Exo have higher miR-126 expression compared to EC-Exo. e) CD133+Exo treatment significantly increases cardiac miR-126 expression and decreases its target gene Spred-1, VCAM and MCP-1 expression measured using PCR analysis. T2DM-stroke+PBS: n=7; T2DM-stroke+CD133+Exo: n=8.
CD133+Exo treatment significantly increases miR-126 expression and deceases its target gene expression in the heart of T2DM stroke mice
In our prior studies we have demonstrated the role of miR-126 in mediating cardiac dysfunction after stroke in non-diabetic mice [8]. MiR-126 is down regulated in EPCs derived from diabetic patients, and decreasing miR-126 impairs EPC-mediated functions via its target genes and signaling pathways [39]. Fig.6d shows that miR-126 is highly expressed in CD133+Exo compared to exosomes derived from ECs. To test whether CD133+Exo treatment regulates miR-126 target gene expression, select specific miR-126 target genes such as MCP1, VCAM1 and Spred-1 [40] were evaluated. Fig.6e shows that CD133+Exo treatment significantly decreases the expression of Spred-1, VCAM, and MCP-1 in the heart of T2DM-stroke mice compared to PBS treated T2DM-stroke mice.
Discussion
In this study, we have demonstrated for the first time the therapeutic effects of CD133+Exo on cardiac function after stroke in T2DM mice. Our data show that CD133+Exo treatment administered intravenously at 3 days after stroke does not decrease lesion volume, but improves LVEF, decreases LV dilatation, decreases cardiomyocyte hypertrophy and interstitial fibrosis, increases myocardial capillary density, decreases oxidative stress, increases miR-126 expression and decreases its target gene expression in heart of T2DM-stroke mice.
T2DM and cardiac dysfunction are closely related, and T2DM patients often exhibit increased LV mass, wall thickness, poor myocardial function and arterial stiffness [41]. T2DM (db/db) mice exhibit decreased heart rate, stoke volume and cardiac output with preserved LVEF as well as cardiomyocyte hypertrophy and interstitial fibrosis compared to non-diabetic control mice [42, 43]. In this study, we report that stroke and T2DM induces significant cardiac dysfunction in mice and stroke in T2DM mice induces worse cardiac function compared to non-DM-stroke mice. In addition to cardiac functional deficits, T2DM and stroke also affect myocardial pathological remodeling which refers to cardiac structural changes such as rearrangement of cardiac chamber walls, myocardial interstitial fibrosis, and cardiomyocyte hypertrophy, apoptosis and necrosis [44, 45]. Myocardial interstitial fibrosis is a key pathological step that decreases the systolic and diastolic LV compliance, damages the electrical coupling between cardiomyocytes, decreases capillary density, increases the oxygen diffusion distance of cardiomyocytes, and ultimately affects LV function [46]. Upon examining heart explants from end-stage heart failure patients with and without diabetes obtained at time of heart transplant, it was found that diabetic hearts exhibited significant capillary rarefaction and pericyte loss compared to non-diabetic heart [47]. Hyperglycemic pigs exhibit myocardial capillary rarefaction with concomitant decrease of LVEF compared to wild type pigs, and increasing myocardial capillary density and maturation, and significantly improves LVEF [47]. These studies indicate that diabetes alters myocardial capillary density and therapeutically increasing myocardial capillary density may be associated with improved LVEF. In our study, we found that CD133+Exo treatment significantly decreases interstitial fibrosis and cardiomyocyte hypertrophy and increases myocardial capillary density in T2DM-stroke mice which may contribute to the improved LVEF compared to PBS treated T2DM-stroke mice.
In diabetic patients, lowering blood glucose has been reported to delay the onset/progression of diabetes induced microvascular complications such as nephropathy and retinopathy [48–50]. However, development of cardiovascular disease is robustly associated with vascular injury and inflammation rather than hyperglycemia, and glucose-lowering therapies have limited cardiovascular benefit in diabetic patients [48–50]. We found that treatment of T2DM and T2DM-stroke mice with CD133+Exo significantly decreases blood glucose and body weight compared to T2DM sham or PBS treated T2DM-stroke mice, respectively. While CD133+Exo treatment decreases glucose and body weight in CD133+Exo treated sham mice, it does not improve cardiac function suggesting that the cardio-protective effects of CD133+Exo treatment are most likely specific to post-stroke cardiac dysfunction in diabetic mice and independent of glucose lowering effect.
Inflammatory cytokines and chemokines, reactive oxygen species, oxidative stress, and TGF-β have been implicated in the pathophysiology of cardiac fibrosis [51, 52]. TGF-β mediates cardiac fibrosis by regulating myofibroblast differentiation and migration, as well as collagen secretion [53]. Thus, overexpression of TGF-β increases myocardial stiffness, interstitial cardiac fibrosis and cardiac hypertrophy and induces LV diastolic dysfunction [54–56]. Oxidative stress in cardiomyocytes increases cardiomyocyte death and can cause LV dilation [57]. 4-HNE is produced by activated neutrophils and plays a key role in controlling NOX activity during conditions of increased oxidative stress [58]. Reactive oxygen species (ROS) specifically-derived from NOX2 contribute to several key processes underlying the development of cardiac contractile dysfunction and cardiac pathological remodeling such as myocyte hypertrophy, contractile dysfunction, apoptosis and fibrosis in myocardial infarction, heart failure and doxorubicin-induced cardiotoxicity [59–63]. Increased NOX2 activity in the heart contributes to diabetic cardiomyopathy and cardiac fibrosis [64, 63]. Acute or chronic exposure of cardiomyocytes to high glucose media significantly increases the expression of NOX2 protein and its catalytic subunits while inhibition of NOX2 improves insulin signaling, increases endogenous antioxidant capacity, decreases ER stress, reduces apoptosis and improves cardiomyocyte contractility [63]. Cardiac macrophages assume a proinflammatory phenotype under disease conditions and stimulate myocardial fibroblasts, increase collagen deposition, increase myocardial stiffness thereby leading to cardiac fibrosis and diastolic dysfunction [65, 66]. Our data show that CD133+Exo treatment significantly decreases oxidative stress, TGF-β expression and proinflammatory macrophages which in concert, may contribute to CD133+Exo treatment induced reduction in cardiac fibrosis, cardiomyocyte hypertrophy, and improved cardiac function.
In our prior studies we have demonstrated the role of miR-126 in mediating cardiac dysfunction after stroke in non-diabetic mice [8]. MiR-126 is highly expressed in HUCBCs, ECs and EPCs [36, 67]. We have demonstrated previously that miR-126 mediates HUCBC derived as well as EC-Exo derived neurorestorative effects after stroke in T2DM mice [36, 68]. Our data show that miR-126 is highly expressed in CD133+Exo compared to EC-Exo and CD133+Exo treatment in T2DM-stroke mice significantly increases cardiac miR-126 expression and significantly decreases its target gene expression such as Spred-1, VCAM, and MCP-1 compared to PBS treated T2DM-stroke mice. MiR126 is down regulated in EPCs from diabetic patients, and impairs EPC-mediated function via its target Spred-1 [39]. Increasing miR-126 expression by aerobic training decreases Spred-1 and induces cardiac angiogenesis [69]. Decrease in miR-126 increases Spred-1expression which impairs angiogenic response and is associated decreased activation of right ventricular failure in pulmonary arterial hypertension [70, 71]. Therefore, decreasing Spred-1 via increasing miR-126 may contribute to CD133+Exo treatment induced angiogenic response and increased capillary density in the heart of T2DM-stroke mice. Serum MCP1 level is significantly increased in patients with ischemic stroke and in patients with myocardial infarction compared to control group of patients [72]. MCP1 deficient mice exhibit decreased LV dilatation and improved LV function compared to WT mice subject to myocardial infarction [73]. VCAM-1 is mainly expressed by endothelial cells, smooth muscle cells, and tissue macrophages [74]. VCAM-1 is significantly increased in patients with T2DM and increased levels of soluble VCAM-1 in plasma are independently associated with high risk of mortality from cardiovascular causes [74]. In rats subjected to a model of transient middle cerebral artery occlusion (MCAo) stroke, VCAM-1 was significantly increased in the acute phase after stroke in the ischemic brain and heart [75]. Decrease in VCAM expression may be an indicator of decreased low grade inflammation as well as have implications on macrophage activation. Decreasing miR-126 can consequently increase MCP-1 and VCAM-1 expression promoting infiltration of inflammatory cells into the heart after stroke [8].Therefore, it is likely that CD133+Exo treatment induced increase in miR-126 and decrease in MCP-1 and VCAM1 are involved in decreasing cardiac inflammation after stroke in T2DM mice.
Limitations and future studies:
Investigation of treatments to attenuate cardiac dysfunction after stroke in diabetic population is clinically relevant. Further studies are warranted on direct comparisons on the progression of stroke induced cardiac dysfunction in male and female diabetic animals and the effects of CD133+Exo treatment in attenuating such cardiac deficits. While there is a possibility that in addition to exosomal content such as mRNA and miRs, the effects of the CD133+Exo may be derived from the lipids within the exosome, in our previous studies, there were no significant differences in neurological or cognitive function between liposome treated or PBS treated T2DM-stroke mice [68]. Diabetes is known to aggravate stroke pathology in the ischemic brain [76]. While we have focused our investigation on the effects of CD133+Exo on cardiac function after stroke, the effects of treatment on neurological function and neurovascular remodeling and inflammatory responses within the brain warrant further studies.
Conclusions
In this study, we have demonstrated that CD133+Exo treatment initiated at 3 days after stroke in T2DM mice significantly improves cardiac function and attenuates cardiac pathological remodeling and increases capillary density which maybe attributed at least in part to decreased oxidative stress and inflammatory responses in the heart. Therefore, CD133+Exo warrants further investigation as a novel therapeutic agent for post stroke diabetes induced cardiac dysfunction.
Fig.5. CD133+Exo treatment significantly decreases cardiac oxidative stress in T2DM-stroke mice.
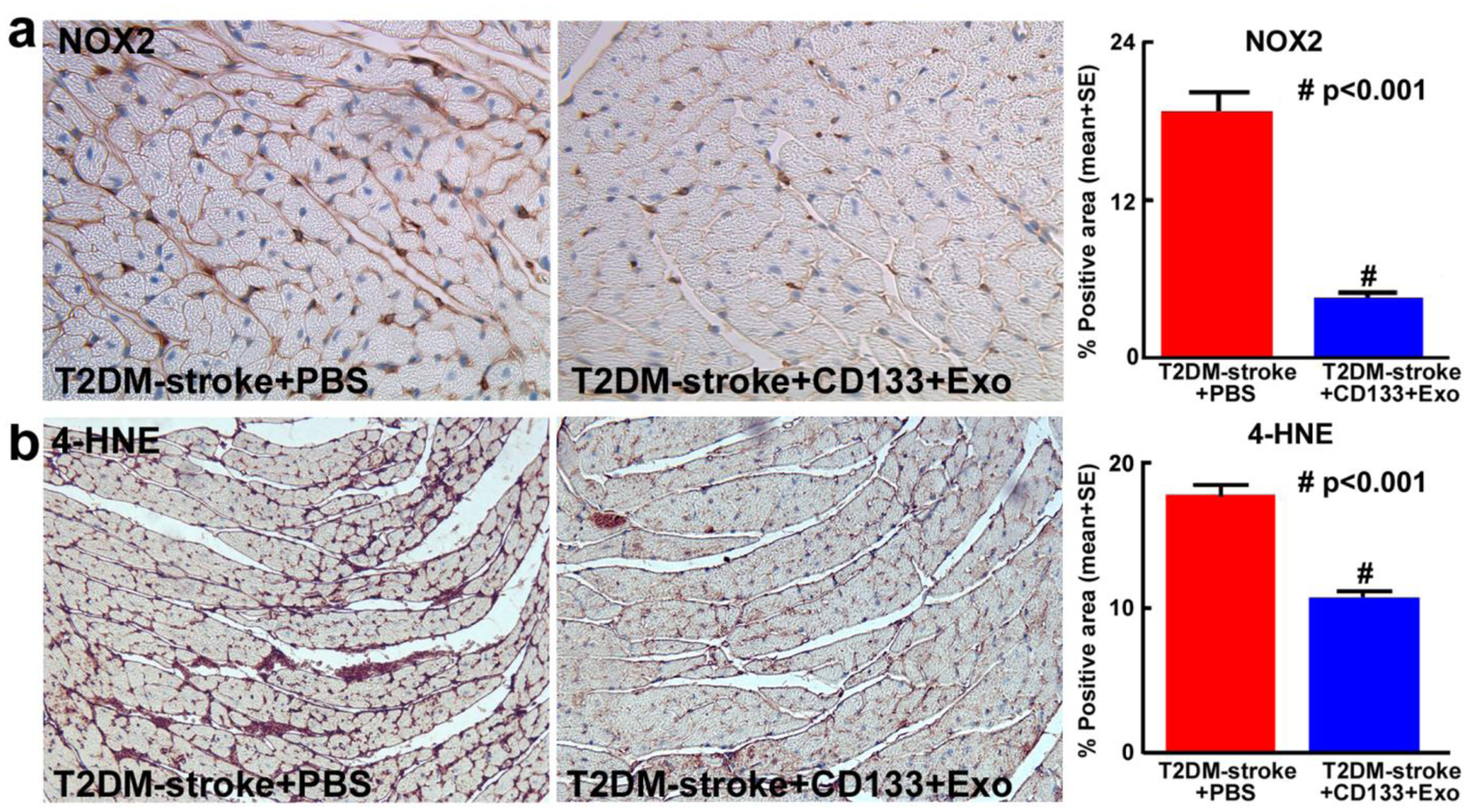
CD133+Exo treatment in T2DM-stroke mice significantly decreases oxidative stress marker a) NADPH oxidase-2 (NOX2) expression and b) 4-Hydroxynonenal (4-HNE) expression compared to PBS treated T2DM-stroke mice. T2DM-stroke+PBS: n=7; T2DM-stroke+CD133+Exo: n=8.
Acknowledgements:
The authors wish to thank Qinge Lu and Sutapa Santra for their technical assistance.
Sources of funding: This work was supported by the National Heart, Lung, and Blood Institute R01HL143432 (JC).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Ethical approval: All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Conflicts of Interest/Disclosures: The authors have no conflicts of interest to disclose. Intellectual Property relating to the topic of this manuscript is subject to patent application (62/586,102) fully owned by Henry Ford Health System.
References
- 1.Nayak AR, Badar SR, Lande N, Kawle AP, Kabra DP, Chandak NH et al. Prediction of Outcome in Diabetic Acute Ischemic Stroke Patients: A Hospital-Based Pilot Study Report. Ann Neurosci. 2016;23(4):199–208. doi: 10.1159/000449480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tziomalos K, Spanou M, Bouziana SD, Papadopoulou M, Giampatzis V, Kostaki S et al. Type 2 diabetes is associated with a worse functional outcome of ischemic stroke. World journal of diabetes. 2014;5(6):939–44. doi: 10.4239/wjd.v5.i6.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP et al. Heart Disease and Stroke Statistics—2019 Update: A Report From the American Heart Association. Circulation. 2019;139(10):e56–e528. doi:doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 4.Ay H, Koroshetz WJ, Benner T, Vangel MG, Melinosky C, Arsava EM et al. Neuroanatomic correlates of stroke-related myocardial injury. Neurology. 2006;66(9):1325–9. doi: 10.1212/01.wnl.0000206077.13705.6d. [DOI] [PubMed] [Google Scholar]
- 5.Oppenheimer SM. Neurogenic cardiac effects of cerebrovascular disease. Curr Opin Neurol. 1994;7(1):20–4. [DOI] [PubMed] [Google Scholar]
- 6.Tokgozoglu SL, Batur MK, Topcuoglu MA, Saribas O, Kes S, Oto A. Effects of stroke localization on cardiac autonomic balance and sudden death. Stroke. 1999;30(7):1307–11. [DOI] [PubMed] [Google Scholar]
- 7.van der Bilt IA, Hasan D, Vandertop WP, Wilde AA, Algra A, Visser FC et al. Impact of cardiac complications on outcome after aneurysmal subarachnoid hemorrhage: a meta-analysis. Neurology. 2009;72(7):635–42. doi: 10.1212/01.wnl.0000342471.07290.07. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Cui C, Yang X, Xu J, Venkat P, Zacharek A et al. MiR-126 Affects Brain-Heart Interaction after Cerebral Ischemic Stroke. Transl Stroke Res. 2017;8(4):374–85. doi: 10.1007/s12975-017-0520-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan T, Chen Z, Chopp M, Venkat P, Zacharek A, Li W et al. Inflammatory responses mediate brain-heart interaction after ischemic stroke in adult mice. J Cereb Blood Flow Metab. 2018:271678×18813317. doi:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verdecchia P, Porcellati C, Reboldi G, Gattobigio R, Borgioni C, Pearson TA et al. Left Ventricular Hypertrophy as an Independent Predictor of Acute Cerebrovascular Events in Essential Hypertension. Circulation. 2001;104(17):2039–44. doi: 10.1161/hc4201.097944. [DOI] [PubMed] [Google Scholar]
- 11.Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22(8):983–8. [DOI] [PubMed] [Google Scholar]
- 12.Brownrigg JRW, Hughes CO, Burleigh D, Karthikesalingam A, Patterson BO, Holt PJ et al. Microvascular disease and risk of cardiovascular events among individuals with type 2 diabetes: a population-level cohort study. The Lancet Diabetes & Endocrinology. 2016;4(7):588–97. doi: 10.1016/S2213-8587(16)30057-2. [DOI] [PubMed] [Google Scholar]
- 13.Fadini GP, Sartore S, Albiero M, Baesso I, Murphy E, Menegolo M et al. Number and Function of Endothelial Progenitor Cells as a Marker of Severity for Diabetic Vasculopathy. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(9):2140–6. doi:doi: 10.1161/01.ATV.0000237750.44469.88. [DOI] [PubMed] [Google Scholar]
- 14.Fan Y, Shen F, Frenzel T, Zhu W, Ye J, Liu J et al. Endothelial progenitor cell transplantation improves long-term stroke outcome in mice. Ann Neurol. 2010;67(4):488–97. doi: 10.1002/ana.21919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarmah D, Kaur H, Saraf J, Pravalika K, Goswami A, Kalia K et al. Getting Closer to an Effective Intervention of Ischemic Stroke: The Big Promise of Stem Cell. Transl Stroke Res. 2018;9(4):356–74. doi: 10.1007/s12975-017-0580-0. [DOI] [PubMed] [Google Scholar]
- 16.Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106(22):2781–6. doi: 10.1161/01.cir.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- 17.Ranghino A, Cantaluppi V, Grange C, Vitillo L, Fop F, Biancone L et al. Endothelial progenitor cell-derived microvesicles improve neovascularization in a murine model of hindlimb ischemia. International journal of immunopathology and pharmacology. 2012;25(1):75–85. doi: 10.1177/039463201202500110. [DOI] [PubMed] [Google Scholar]
- 18.Gu S, Zhang W, Chen J, Ma R, Xiao X, Ma X et al. EPC-derived microvesicles protect cardiomyocytes from Ang II-induced hypertrophy and apoptosis. PloS one. 2014;9(1):e85396–e. doi: 10.1371/journal.pone.0085396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xin H, Li Y, Chopp M. Exosomes/miRNAs as mediating cell-based therapy of stroke. Front Cell Neurosci. 2014;8:377. doi: 10.3389/fncel.2014.00377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen F, Du Y, Esposito E, Liu Y, Guo S, Wang X et al. Effects of Focal Cerebral Ischemia on Exosomal Versus Serum miR126. Transl Stroke Res. 2015;6(6):478–84. doi: 10.1007/s12975-015-0429-3. [DOI] [PubMed] [Google Scholar]
- 21.Chen J, Chopp M. Exosome Therapy for Stroke. Stroke. 2018;49(5):1083–90. doi:doi: 10.1161/STROKEAHA.117.018292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moon GJ, Sung JH, Kim DH, Kim EH, Cho YH, Son JP et al. Application of Mesenchymal Stem Cell-Derived Extracellular Vesicles for Stroke: Biodistribution and MicroRNA Study. Transl Stroke Res. 2019;10(5):509–21. doi: 10.1007/s12975-018-0668-1. [DOI] [PubMed] [Google Scholar]
- 23.Otero-Ortega L, Laso-García F, Gómez-de Frutos M, Fuentes B, Diekhorst L, Díez-Tejedor E et al. Role of Exosomes as a Treatment and Potential Biomarker for Stroke. Transl Stroke Res. 2019;10(3):241–9. doi: 10.1007/s12975-018-0654-7. [DOI] [PubMed] [Google Scholar]
- 24.Zhou Y, Li P, Goodwin AJ, Cook JA, Halushka PV, Chang E et al. Exosomes from Endothelial Progenitor Cells Improve the Outcome of a Murine Model of Sepsis. Molecular therapy : the journal of the American Society of Gene Therapy. 2018;26(5):1375–84. doi: 10.1016/j.ymthe.2018.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ke X, Yang D, Liang J, Wang X, Wu S, Wang X et al. Human Endothelial Progenitor Cell-Derived Exosomes Increase Proliferation and Angiogenesis in Cardiac Fibroblasts by Promoting the Mesenchymal-Endothelial Transition and Reducing High Mobility Group Box 1 Protein B1 Expression. DNA and cell biology. 2017;36(11):1018–28. doi: 10.1089/dna.2017.3836. [DOI] [PubMed] [Google Scholar]
- 26.Thum T, Hoeber S, Froese S, Klink I, Stichtenoth DO, Galuppo P et al. Age-dependent impairment of endothelial progenitor cells is corrected by growth-hormone-mediated increase of insulin-like growth-factor-1. Circulation research. 2007;100(3):434–43. doi: 10.1161/01.RES.0000257912.78915.af. [DOI] [PubMed] [Google Scholar]
- 27.Friedrich EB, Walenta K, Scharlau J, Nickenig G, Werner N. CD34-/CD133+/VEGFR-2+ endothelial progenitor cell subpopulation with potent vasoregenerative capacities. Circ Res. 2006;98(3):e20–5. doi: 10.1161/01.res.0000205765.28940.93. [DOI] [PubMed] [Google Scholar]
- 28.Karapanayiotides T, Piechowski-Jozwiak B, van Melle G, Bogousslavsky J, Devuyst G. Stroke patterns, etiology, and prognosis in patients with diabetes mellitus. Neurology. 2004;62(9):1558–62. [DOI] [PubMed] [Google Scholar]
- 29.Mast H, Koennecke HC, Hartmann A, Stapf C, Marx P. [Association of hypertension and diabetes mellitus with microangiopathic cerebral infarct patterns]. Der Nervenarzt. 1997;68(2):129–34. [DOI] [PubMed] [Google Scholar]
- 30.Mast H, Thompson JL, Lee SH, Mohr JP, Sacco RL. Hypertension and diabetes mellitus as determinants of multiple lacunar infarcts. Stroke. 1995;26(1):30–3. [DOI] [PubMed] [Google Scholar]
- 31.Chen J, Cui C, Yang X, Xu J, Venkat P, Zacharek A et al. MiR-126 Affects Brain-Heart Interaction after Cerebral Ischemic Stroke. Transl Stroke Res. 2017. doi: 10.1007/s12975-017-0520-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Labat-gest V, Tomasi S. Photothrombotic Ischemia: A Minimally Invasive and Reproducible Photochemical Cortical Lesion Model for Mouse Stroke Studies. J Vis Exp. 2013(76):50370. doi: 10.3791/50370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uzdensky AB. Photothrombotic Stroke as a Model of Ischemic Stroke. Transl Stroke Res. 2018;9(5):437–51. doi: 10.1007/s12975-017-0593-8. [DOI] [PubMed] [Google Scholar]
- 34.Yang XP, Liu YH, Rhaleb NE, Kurihara N, Kim HE, Carretero OA. Echocardiographic assessment of cardiac function in conscious and anesthetized mice. The American journal of physiology. 1999;277(5 Pt 2):H1967–74. [DOI] [PubMed] [Google Scholar]
- 35.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10(2):290–3. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 36.Chen J, Ning R, Zacharek A, Cui C, Cui X, Yan T et al. MiR-126 Contributes to Human Umbilical Cord Blood Cell-Induced Neurorestorative Effects After Stroke in Type-2 Diabetic Mice. Stem cells 2016;34(1):102–13. doi: 10.1002/stem.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mewhort HE, Lipon BD, Svystonyuk DA, Teng G, Guzzardi DG, Silva C et al. Monocytes increase human cardiac myofibroblast-mediated extracellular matrix remodeling through TGF-beta1. Am J Physiol Heart Circ Physiol. 2016;310(6):H716–24. doi: 10.1152/ajpheart.00309.2015. [DOI] [PubMed] [Google Scholar]
- 38.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74(2):184–95. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meng S, Cao JT, Zhang B, Zhou Q, Shen CX, Wang CQ. Downregulation of microRNA-126 in endothelial progenitor cells from diabetes patients, impairs their functional properties, via target gene Spred-1. J Mol Cell Cardiol. 2012;53(1):64–72. doi: 10.1016/j.yjmcc.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 40.Small EM, Frost RJA, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation. 2010;121(8):1022–32. doi: 10.1161/CIRCULATIONAHA.109.889048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Devereux RB, Roman MJ, Paranicas M, O’Grady MJ, Lee ET, Welty TK et al. Impact of Diabetes on Cardiac Structure and Function. Circulation. 2000;101(19):2271–6. doi:doi: 10.1161/01.CIR.101.19.2271. [DOI] [PubMed] [Google Scholar]
- 42.Panagia M, Schneider JE, Brown B, Cole MA, Clarke K. Abnormal function and glucose metabolism in the type-2 diabetic db/db mouse heart. Canadian journal of physiology and pharmacology. 2007;85(3–4):289–94. doi: 10.1139/y07-028. [DOI] [PubMed] [Google Scholar]
- 43.Alex L, Russo I, Holoborodko V, Frangogiannis NG. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol. 2018;315(4):H934–h49. doi: 10.1152/ajpheart.00238.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Distefano G, Sciacca P. Molecular pathogenesis of myocardial remodeling and new potential therapeutic targets in chronic heart failure. Italian journal of pediatrics. 2012;38:41. doi: 10.1186/1824-7288-38-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Z, Venkat P, Seyfried D, Chopp M, Yan T, Chen J. Brain-Heart Interaction: Cardiac Complications After Stroke. Circ Res. 2017;121(4):451–68. doi: 10.1161/CIRCRESAHA.117.311170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Donekal S, Venkatesh BA, Liu YC, Liu CY, Yoneyama K, Wu CO et al. Interstitial fibrosis, left ventricular remodeling, and myocardial mechanical behavior in a population-based multiethnic cohort: the Multi-Ethnic Study of Atherosclerosis (MESA) study. Circulation Cardiovascular imaging. 2014;7(2):292–302. doi: 10.1161/circimaging.113.001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hinkel R, Howe A, Renner S, Ng J, Lee S, Klett K et al. Diabetes Mellitus-Induced Microvascular Destabilization in the Myocardium. J Am Coll Cardiol. 2017;69(2):131–43. doi: 10.1016/j.jacc.2016.10.058. [DOI] [PubMed] [Google Scholar]
- 48.Rask-Madsen C, King GL. Vascular complications of diabetes: mechanisms of injury and protective factors. Cell Metab. 2013;17(1):20–33. doi: 10.1016/j.cmet.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohkubo Y, Kishikawa H, Araki E, Miyata T, Isami S, Motoyoshi S et al. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes research and clinical practice. 1995;28(2):103–17. doi: 10.1016/0168-8227(95)01064-k. [DOI] [PubMed] [Google Scholar]
- 50.Giorgino F, Home PD, Tuomilehto J. Glucose Control and Vascular Outcomes in Type 2 Diabetes: Is the Picture Clear? Diabetes Care. 2016;39(Supplement 2):S187–S95. doi: 10.2337/dcS15-3023. [DOI] [PubMed] [Google Scholar]
- 51.Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cellular and molecular life sciences : CMLS. 2014;71(4):549–74. doi: 10.1007/s00018-013-1349-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Leask A Getting to the heart of the matter: new insights into cardiac fibrosis. Circulation research. 2015;116(7):1269–76. doi: 10.1161/CIRCRESAHA.116.305381. [DOI] [PubMed] [Google Scholar]
- 53.Salvarani N, Maguy A, De Simone SA, Miragoli M, Jousset F, Rohr S. TGF-beta1 (Transforming Growth Factor-beta1) Plays a Pivotal Role in Cardiac Myofibroblast Arrhythmogenicity. Circ Arrhythm Electrophysiol. 2017;10(5):e004567. doi: 10.1161/circep.116.004567. [DOI] [PubMed] [Google Scholar]
- 54.Schultz JEJ, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL et al. TGF-β1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002;109(6):787–96. doi: 10.1172/JCI14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51(4):600–6. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosenkranz S, Flesch M, Amann K, Haeuseler C, Kilter H, Seeland U et al. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1). Am J Physiol Heart Circ Physiol. 2002;283(3):H1253–62. doi: 10.1152/ajpheart.00578.2001. [DOI] [PubMed] [Google Scholar]
- 57.Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B et al. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res. 2001;89(3):279–86. [DOI] [PubMed] [Google Scholar]
- 58.Chacko BK, Wall SB, Kramer PA, Ravi S, Mitchell T, Johnson MS et al. Pleiotropic effects of 4-hydroxynonenal on oxidative burst and phagocytosis in neutrophils. Redox biology. 2016;9:57–66. doi: 10.1016/j.redox.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Looi YH, Grieve DJ, Siva A, Walker SJ, Anilkumar N, Cave AC et al. Involvement of Nox2 NADPH oxidase in adverse cardiac remodeling after myocardial infarction. Hypertension. 2008;51(2):319–25. doi: 10.1161/hypertensionaha.107.101980. [DOI] [PubMed] [Google Scholar]
- 60.Zhao Y, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM et al. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer research. 2010;70(22):9287–97. doi: 10.1158/0008-5472.CAN-10-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD et al. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res. 2003;93(9):802–5. doi: 10.1161/01.res.0000099504.30207.f5. [DOI] [PubMed] [Google Scholar]
- 62.Sirker A, Zhang M, Murdoch C, Shah AM. Involvement of NADPH oxidases in cardiac remodelling and heart failure. American journal of nephrology. 2007;27(6):649–60. doi: 10.1159/000109148. [DOI] [PubMed] [Google Scholar]
- 63.Hansen SS, Aasum E, Hafstad AD. The role of NADPH oxidases in diabetic cardiomyopathy. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2018;1864(5, Part B):1908–13. doi: 10.1016/j.bbadis.2017.07.025. [DOI] [PubMed] [Google Scholar]
- 64.Johar S, Cave AC, Narayanapanicker A, Grieve DJ, Shah AM. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2006;20(9):1546–8. doi: 10.1096/fj.05-4642fje. [DOI] [PubMed] [Google Scholar]
- 65.Ma F, Li Y, Jia L, Han Y, Cheng J, Li H et al. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PloS one. 2012;7(5):e35144. doi: 10.1371/journal.pone.0035144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hulsmans M, Sager HB, Roh JD, Valero-Muñoz M, Houstis NE, Iwamoto Y et al. Cardiac macrophages promote diastolic dysfunction. The Journal of Experimental Medicine. 2018;215(2):423–40. doi: 10.1084/jem.20171274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pan Q, Zheng J, Du D, Liao X, Ma C, Yang Y et al. MicroRNA-126 Priming Enhances Functions of Endothelial Progenitor Cells under Physiological and Hypoxic Conditions and Their Therapeutic Efficacy in Cerebral Ischemic Damage. Stem Cells International. 2018;2018:13. doi: 10.1155/2018/2912347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Venkat P, Cui C, Chopp M, Zacharek A, Wang F, Landschoot-Ward J et al. MiR-126 Mediates Brain Endothelial Cell Exosome Treatment-Induced Neurorestorative Effects After Stroke in Type 2 Diabetes Mellitus Mice. Stroke. 2019;50(10):2865–74. doi: 10.1161/strokeaha.119.025371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cantaluppi V, Gatti S, Medica D, Figliolini F, Bruno S, Deregibus MC et al. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney international. 2012;82(4):412–27. doi: 10.1038/ki.2012.105. [DOI] [PubMed] [Google Scholar]
- 70.Potus F, Ruffenach G, Dahou A, Thebault C, Breuils-Bonnet S, Tremblay E et al. Downregulation of MicroRNA-126 Contributes to the Failing Right Ventricle in Pulmonary Arterial Hypertension. Circulation. 2015;132(10):932–43. doi: 10.1161/CIRCULATIONAHA.115.016382. [DOI] [PubMed] [Google Scholar]
- 71.Potus F, Malenfant S, Graydon C, Mainguy V, Tremblay E, Breuils-Bonnet S et al. Impaired angiogenesis and peripheral muscle microcirculation loss contribute to exercise intolerance in pulmonary arterial hypertension. American journal of respiratory and critical care medicine. 2014;190(3):318–28. doi: 10.1164/rccm.201402-0383OC. [DOI] [PubMed] [Google Scholar]
- 72.Arakelyan A, Petrkova J, Hermanova Z, Boyajyan A, Lukl J, Petrek M. Serum levels of the MCP-1 chemokine in patients with ischemic stroke and myocardial infarction. Mediators of inflammation. 2005;2005(3):175–9. doi: 10.1155/MI.2005.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96(8):881–9. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 74.Jager A, van Hinsbergh VW, Kostense PJ, Emeis JJ, Nijpels G, Dekker JM et al. Increased levels of soluble vascular cell adhesion molecule 1 are associated with risk of cardiovascular mortality in type 2 diabetes: the Hoorn study. Diabetes. 2000;49(3):485–91. doi: 10.2337/diabetes.49.3.485. [DOI] [PubMed] [Google Scholar]
- 75.Justicia C, Martín A, Rojas S, Gironella M, Cervera Á, Panés J et al. Anti-VCAM-1 Antibodies did not Protect against Ischemic Damage Either in Rats Or in Mice. J Cereb Blood Flow Metab. 2006;26(3):421–32. doi: 10.1038/sj.jcbfm.9600198. [DOI] [PubMed] [Google Scholar]
- 76.Kim E, Yang J, Park KW, Cho S. Inhibition of VEGF Signaling Reduces Diabetes-Exacerbated Brain Swelling, but Not Infarct Size, in Large Cerebral Infarction in Mice. Transl Stroke Res. 2018;9(5):540–8. doi: 10.1007/s12975-017-0601-z. [DOI] [PMC free article] [PubMed] [Google Scholar]