

Abstract
Background
Isolated hypogonadotropic hypogonadism (IHH) and Kallmann syndrome (KS) are rare genetic diseases that cause male infertility. The chromodomain helicase DNA‐binding protein 7 (CHD7) gene is commonly associated with KS and IHH. We speculated that CHD7 variants may be associated with male infertility.
Methods
Two hundred males with azoospermia and 120 with oligozoospermia were recruited. The patients underwent clinical examination and reproductive hormone testing. A panel of genes including CHD7 and others related to spermatogenic failure was sequenced by targeted‐gene exome sequencing.
Results
Three patients with severe oligozoospermia had CHD7 variants (a detection rate of 0.94% (3/320)). After prediction software analysis, two of the variants c.3464G>A (p.R1155H) and c.4516G>A (p.G1506S) were predicted to be likely pathogenic. Although predicted to be benign, the variants of c.2824A>G (p.T942A) located in the chromodomain 2 could not be excluded as disease causing. The patients with variants had small testicular volumes. In particular, the testes of the patient with a p.G1506S variant varied in size (left, 8 ml; right, 4.5 ml). Two patients (patients 31 and 120) had low E2 levels and two (patients 83 and 120) had low T levels. Ultimately, these variants were classified as “variants of unknown significant” that may be associated with male infertility.
Conclusions
There may be a relationship between the CHD7 gene missense variants and male infertility. These variants are easier to find in patients with azoospermia and severe oligospermia whose testosterone levels are decreased.
Keywords: chromodomain helicase DNA‐binding protein 7, male infertility, missense variants, targeted‐gene exome sequencing
Chromodomain helicase DNA‐binding protein 7 (CHD7) is one of the reported more commonly genes related to KS and IHH. We speculated that CHD7 variants may have an association with male infertility. These variants are easier to find in patients with azoospermia and severe oligospermia whose testosterone levels are decreased.
1. INTRODUCTION
Infertility is defined as unprotected sexual intercourse for more than one year without pregnancy (Zegers‐Hochschild et al., 2009). About 7% of men suffer from infertility problems, and male infertility accounts for about half of infertile couples (Krausz & Riera‐Escamilla, 2018; Winters & Walsh, 2014). Infertile males often exhibit abnormalities in semen quality, such as azoospermia and oligozoospermia. Male infertility can be divided into three categories: acquired, congenital, and unexplained. At least 15 to 30% of male infertility may be caused by genetic factors (Krausz et al., 2006; Krausz, Forti, & McElreavey, 2003). Congenital male infertility can result from genetic factors or a developmental syndrome (Skakkebaek, Rajpert‐De Meyts, & Main, 2001). Although many studies in the last decades have tried to find the cause of male infertility, most infertile men have been classified as having unexplained infertility, most likely due to genetic mutations associated with spermatogenic failure. In recent years, more than 600 genes associated with male infertility have been reported in the Jackson Laboratory's Mouse Genome Informatics database (http://www.informatics.jax.org/), and 2,300 testicular‐specific genes have been found in humans (Schultz, Hamra, & Garbers, 2003). With the development of high‐throughput sequencing technology, the number of genes associated with male infertility has increased in recent years. Although the number of genes is very large, research into these genes is at an early stage, and gene analysis has not been widely used in clinical diagnosis. Clinical genetic diagnosis remains focused on karyotype analysis, detection of azoospermia factor (AZF) microdeletions, and detection of mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene in obstructive azoospermia.
Isolated hypogonadotropic hypogonadism (IHH) is a rare genetic disease that causes male infertility, manifesting as hypogonadotropic hypogonadism with or without anosmia or hyposmia (Boehm et al., 2015). IHH accompanied by anosmia is called Kallmann syndrome (KS); if IHH is not accompanied by olfactory dysfunction, it is called normosmic IHH (nIHH). IHH is a genetically heterogeneous disease. In addition to infertility, it can present with various developmental abnormalities related to puberty. These symptoms are caused by dysfunctions in the production, secretion, and action of hypothalamic gonadotropin‐releasing hormone (GnRH) (Zhou et al., 2018). Recently, more than 30 genes have been reported to be involved in IHH, including ANOS1, FGFR1/FGF8, PROK2/PROKR2, and CHD7, all of which regulate the development and migration of GnRH neurons during embryonic development, as well as the synthesis, secretion, and action of GnRH (Kim, 2015).
Chromodomain helicase DNA‐binding protein 7 (CHD7) gene (MIM 608892) is one of nine members of the CHD protein family, which have the common function of hydrolyzing ATP and changing the structure of the nucleosome (Marfella & Imbalzano, 2007). The CHD7 gene is located on chromosome 8q12 and has 38 exons and a size of 188 kb (Vissers et al., 2004). It encodes 2,997 amino acids and the CHD7 protein has five nuclear localization signals (Schnetz et al., 2009). Mutations in CHD7 can lead to a syndrome involving multiple organ disorders, called CHARGE syndrome (coloboma, heart defect, atresia choanae, growth and developmental retardation, genital hypoplasia, ear anomalies/deafness; MIM 214800) (Vissers et al., 2004). In addition to CHARGE syndrome, CHD7 mutations are commonly reported in KS and IHH.
Because IHH usually leads to male infertility, we designed a gene panel that included IHH‐related genes and others related to spermatogenic failure. We sequenced 200 patients with azoospermia and 120 patients with oligozoospermia using the gene panel to determine relationships between male infertility and genetic mutations. In this study, we detected three CHD7 missense variants in patients with severe oligozoospermia. We describe here the relationships between CHD7 variants and male infertility.
2. MATERIAL AND METHODS
2.1. Ethical Compliance
All patients agreed to participate in the research and signed informed consent. This study was approved by the Ethics Committee of the First Hospital of Jilin University.
2.2. Patients
Three hundred and twenty patients with failure of spermatogenesis (200 azoospermic and 120 oligozoospermic), ranging from 21 to 39 years, were recruited by the Center for Reproductive Medicine, The First Hospital of Jilin University. Semen samples of the patients were obtained after a 3‐ to 7‐day period of ejaculatory abstinence, and semen analyses were performed three times within an interval of three months according to the World Health Organization guidelines (5th). Absence of spermatozoa in the semen ejaculate, if detected three times, was considered azoospermia. A sperm concentration of <15 × 106/ml was considered oligozoospermia. These patients all excluded abnormal female factors. Patients with known causes of male infertility were excluded, including those with obstructive causes, infectious factors, genitourinary injury, chromosomal abnormalities, and AZF microdeletions. Family genetic history and personal habits were collected by using a questionnaire survey. Physical examinations were conducted by the experienced andrologists. Family history included whether there is a history of genetic disease, hypertension, diabetes, and other special diseases. Physical examinations included examination of male sexual characteristics such as penis length, testicle size and texture, beard, hair distribution, and laryngeal knots. The patients’ medical histories were unremarkable for infertility risk factors. The patients were tested for reproductive hormone levels including luteinizing hormone (LH), follicle stimulating hormone (FSH), testosterone (T), and estradiol (E2) by electrochemiluminescent immunoassays to assess endocrine status.
2.3. Targeted‐gene exome sequencing
An in‐house targeted gene panel (Beijing Medriv Academy of Genetics and Reproduction, Beijing, P. R. China), which included CHD7, was used for sequencing on the Illumina MiSeq platform (Illumina Inc., San Diego, CA, USA). The selected targeted next‐generation sequencing and data analysis were performed as described previously (Yang et al., 2018). Briefly, DNA from peripheral blood samples was first extracted using a DNA extraction kit (Beijing Tiangen Biotech Co., Ltd., Beijing, China) and then sequenced using the target gene panel on the Illumina MiSeq platform. The gene panel contained 52 genes involved in spermatogenesis failure, including CHD7. Fifty‐two genes contained in the panel are listed in Table S1. After sequencing, low‐quality base sequences and adaptor sequences were removed, and sequence alignment was performed using the hg19 human reference sequence using Burrows‐Wheeler aligner software. Duplicated reads from library and PCR preparation were removed using Picard tools. Variants with minor allele frequencies >1% in the databases, intronic variants, and synonymous variants unlikely to be deleterious were excluded. Nonsynonymous variants and splice site variants were retained.
The target gene variants were named by reference to the CHD7 gene sequence in the National Center for Biotechnology Information database (NM_017780.4). The prediction of the effect of the identified variants on protein function was based on three software programs: PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), and Mutation Taster (http://www.mutationtaster.org/). The results were further confirmed by conventional PCR and Sanger sequencing (ABI 3730XL, BGI Genomics, Shenzhen, China). The resulting PDB files were then opened using SPDBV_4.10_PC software to view the three‐dimensional protein model (Pitteloud et al., 2006).
3. RESULTS
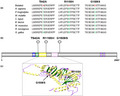
In this study, 320 infertile men were tested using an in‐house targeted gene panel that contained 52 genes associated with spermatogenic failure to find the cause of azoospermia or oligozoospermia. Three patients were found to carry variants in the CHD7 gene. All variants were verified by Sanger sequencing, and the mutated sequences were highly conserved (Figures 1 and 2a). P31 carried a c.3464G>A (p.R1155H) variant, P83 carried a c.2824A>G (p.T942A) variant, and P120 carried a c.4516G>A (p.G1506S) variant. These three variants have been reported in the dbSNP135 database (Table 1). According to SIFT, PolyPhen‐2.0 and Variant Taster software, two variants (p.R1155H and p.G1506S) were predicted to be pathogenic. The p.T942A missense variant was considered disease causing according to Mutation Taster analysis, but considered benign according to SIFT and PolyPhen‐2.0 analysis (Table 2). These three variants have also been described in the gnomAD database; the Allele frequency (AF) in East Asian was 0 for p.R1155H, 0.0013 for p.T942A, and 0.0006 for p.G1506S. The variant of p.T942A was located in the functional domain of chromodomain 2 and p.R1155H in the helicase N domain. The variant of p.G1506S was located near the helicase C domain (Figure 2b,c).
Figure 1.
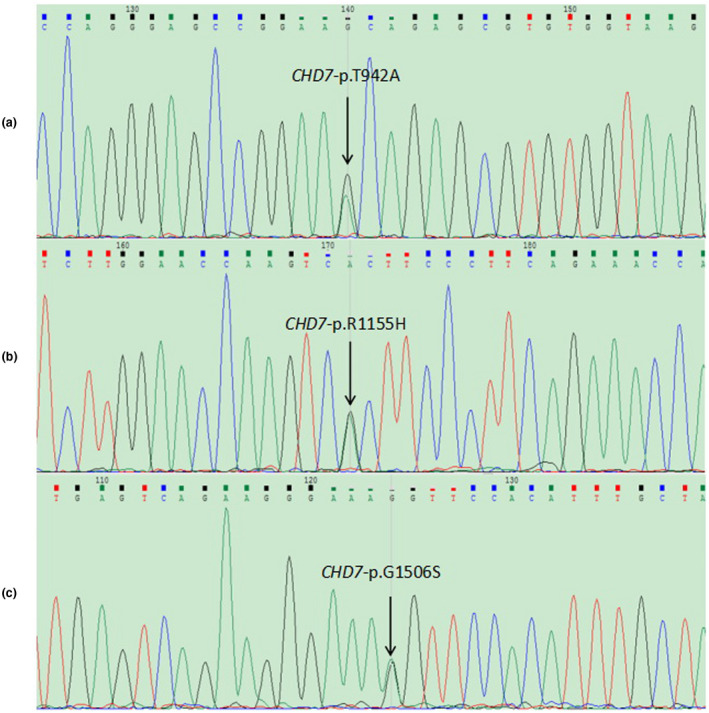
Sanger sequencing of CHD7 gene variants identified in patients. (a) c.2824A>G (p.T942A) variant in P83. (b) c.3464G>A (p.R1155H) variant in P31. (c) c.4516G>A (p.G1506S) variant in P120
Figure 2.
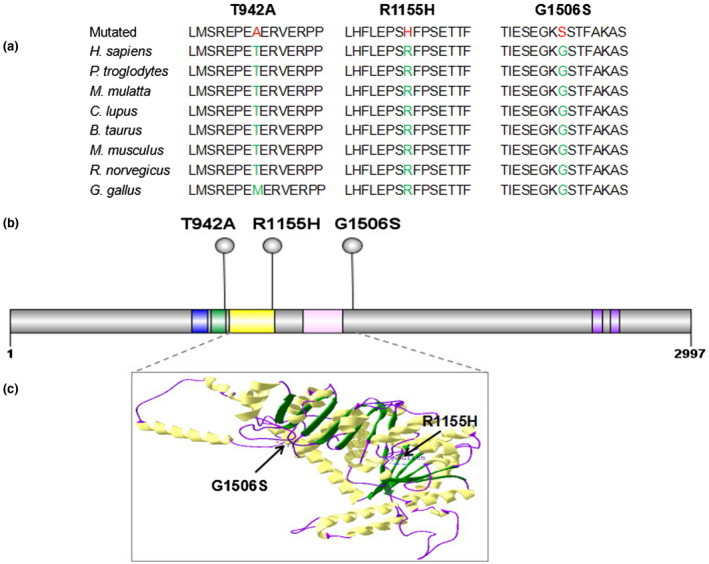
Conservation and locations of the CHD7 gene variants. (a) The conservation of variants (p.T942A, p.R1155H, and p.G1506S) was analyzed across different species. (b) Schematic of the CHD7 protein (amino acid 1 to 2,997) and locations of identified variants. The functional domains of CHD7 are shown as follows: blue, chromodomain 1; green, chromodomain 2; yellow, helicase N; pink, helicase C; purple, BRK. (c) Structural model of CHD7 helicase domains are shown as follows: yellow, α‐helix; green, β‐sheet; purple, the loops. The location of pathogenic variants (p.R1155H and p.G1506S) is indicated by arrows
Table 1.
CHD7 variants identified in patients with idiopathic male infertility
| Variant no. | Exon | Sequence variant | Amino acid change | Chromosomal locution | Reported | rs number | Sample ID |
|---|---|---|---|---|---|---|---|
| 1 | 14 | c.3464G>A | p.R1155H | chr8:61741307 | Yes | rs762669262 | P31 |
| 2 | 10 | c.2824A>G | p.T942A | chr8:61734475 | Yes | rs370194460 | P83 |
| 3 | 19 | c.4516G>A | p.G1506S | chr8:61750797 | Yes | rs750258756 | P120 |
Table 2.
List of missense variants of CHD7 predicted to be functionally significant by bioinformatics analysis
| Variant no. | Nucleotide change | Amino acid change | Allele frequency in East Asian (gnomAD) | SIFT | PolyPhen‐2.0 | Mutationtaster | |||
|---|---|---|---|---|---|---|---|---|---|
| Score | Prediction | Score | Prediction | Score | Prediction | ||||
| 1 | c.G3464A | p.R1155H | 0 | 0.04 | Deleterious | 0.930 | Probably damaging | / | Disease causing |
| 2 | c.A2824G | p.T942A | 0.0013 | 0.8 | Tolerated | 0.004 | Benign | / | Disease causing |
| 3 | c.G4516A | p.G1506S | 0.0006 | 0.12 | Tolerated | 0.926 | Probably damaging | / | Disease causing |
The CHD7 gene variants have been reported to be associated with CHARGE syndrome and IHH syndrome, but none of the patients with three variants we detected showed the main manifestations of either CHARGE or IHH syndromes. Clinical data about the patients are given in Table 3. Semen analysis showed severe oligozoospermia and physical examination exhibited normal male sexual characteristics of all the patients except a smaller testicular volumes. Scrotal colour Doppler ultrasonography revealed small testicular volumes (<15 ml). In particular, testes of P120 were the smallest and varied in size (left, 8 ml; right, 4.5 ml). The hormonal profile of P31 showed low E2 level, and that of P83 showed low T level; P120 had low levels of both T and E2. These three variants underwent an olfactory test to rule out the possibility of KS syndrome. Unable to distinguish odors such as alcohol, white vinegar, water and shampoo, it was intended to diagnose KS syndrome. Our patients can distinguish these odors normally. Ultimately, the three variants detected in this study were classified as “variants of unknown significant” that may be associated with male infertility.
Table 3.
Clinical information and hormone profile of patients with male infertility with CHD7 missense variants
| Sample ID | Age (years) | Height (cm) | Weight (kg) | Infetile years | Sperm count | FSH | LH | T | E2 | Left testicular volume (ml) | Right testicular volume (ml) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (mill./ml) | (mIU/ml) | (mIU/ml) | (nmol/L) | (pg/ml) | |||||||
| P31 | 30 | 176 | 75 | 7 | 4.72 | 3.7 | 2.3 | 11.1 | 17.46↓ | 12 | 12 |
| P83 | 29 | 175 | 76 | 2 | 2.00 | 8.3 | 4.9 | 6.4↓ | 34.39 | 12 | 12 |
| P120 | 24 | 172 | 90 | 1 | 2.16 | 9 | 5.2 | 8.8↓ | 23.85↓ | 8 | 4.5 |
FSH: Follicle‐stimulating hormone (1.5‑12.4 mIU/ml); LH: Luteinizing hormone (1.7–8.6 mIU/ml); T: Testosterone (9.9–27.8 nmol/L); E2: Estradiol (25.8–60.7 pg/ml); ↑: elevated; ↓: decreased.
4. DISCUSSION
In this study, three patients with severe oligozoospermia were found to carry CHD7 variants, a detection rate of 0.94% (3/320). The relationship between CHD7 variants and male infertility has not been studied previously. The three variants of unknown significant of CHD7 gene were discovered accidentally by gene panel detection in patients with spermatogenic failure. Therefore the situation was discussed in this paper. In the previous studies, 70% to 90% of patients with CHRGE syndrome carried pathogenic heterozygous mutations in CHD7 gene (Bergman et al., 2012; Janssen et al., 2012; Zentner, Layman, Martin, & Scacheri, 2010). In addition to FGFR1, CHD7 is the second most important disease‐causing gene associated with IHH or KS (Pitteloud et al., 2006). At present, the genetic model of CHD7 has not been well studied, but it may involve autosomal dominant inheritance. The incidence of CHD7 is about 6% in patients with IHH (Dode & Hardelin, 2010). According to the report of Marcos et al., the incidence of CHD7 mutation in patients with KS was 6.2%, whether or not these patients had some symptoms of CHARGE syndrome (Marcos et al., 2014). The detection rate of CHD7 variants in this study was much lower than in previous reports; however, finding CHD7 variants in patients with male infertility has important implications for the genetic counseling and fertility guidance for these patients.
Generally, patients with IHH have lower levels of sex hormones. Levels of FSH and LH are low or normal in patients with IHH although with the help of the negative feedback modulation of the hypothalamic‐pituitary‐gonadal axis. It usually indicates hypogonadism. These findings indicate defects in hypothalamic or pituitary function (Crowley, Filicori, Spratt, & Santoro, 1985). IHH or KS usually causes an irreversible delay in puberty in women, who present with hypoestrogenic amenorrhea at the age of 17; men with IHH or KS have low testosterone at age 18 (Bhagavath et al., 2006). The three patients with CHD7 variants in this study presented with severe oligozoospermia but not azoospermia, which may be related to their hormone levels. The levels of testosterone were low in two of the patients (P83 and P120), but FSH and LH levels were normal in all three patients. In addition, the testicular volume of these patients were smaller than that of normal men, especially the P120 testis were the smallest and vary in size on both sides. This indicated that the patients did not show symptoms of IHH or that the symptoms were mild. To induce pubertal development or maintain sex hormone levels in adult males, most IHH patients need long‐term testosterone replacement therapy (TRT) (Han & Bouloux, 2010). More than 10% of HH patients may achieve reversal of hypogonadism, and some patients can achieve normal sperm counts (Raivio et al., 2007). A patient with KS with a truncated CHD7 mutation showed that hypogonadotropic dysfunction could be reversed by TRT (Laitinen et al., 2012). Therefore, our patients with low testosterone may also benefit from TRT treatment. However, it is important to inform the patients about the risk of hereditary CHD7 defects in offspring before treatment.
Mutations in CHD7 can cause CHARGE syndrome, which is characterized by multiple organ defects, nIHH, and KS. Different types of CHD7 mutations lead to a variety of clinical phenotypes. In general, CHARGE syndrome is caused by very harmful protein‐truncating mutations, whereas nIHH is caused by less harmful missense mutations (Marcos et al., 2014). All variants detected in our patients were missense variants in highly conserved amino acid sites, and two of them occurred in functional domains of CHD7 (p.T942A variant located in the chromodomain 2 domain and p.R1155H in the helicase N domain). These domains play an important role in the chromatin remodeling activity of the CHD7 protein. Although the p.G1506S variant was not located in a catalytically active domain, bioinformatics analysis showed that the mutation could affect the function of protein. Therefore, the three variants detected in this study were classified as “variants of unknown significant” that may be associated with male infertility.
Wen et al. reported that a male patient with KS with some symptoms of CHARGE syndrome carried a p.E2191K mutation in CHD7. In pedigree analysis, the patient's mother and younger brother were found to have the same mutation without any clinical symptoms (Wen, Pan, Xu, Wang, & Hu, 2018). In another study, six patients with IHH had rare mutations of CHD7. These mutations were inherited from one of the parents, but none of the parents manifested any characteristics of CHARGE syndrome or IHH (Goncalves et al., 2019). Our patients showed only severe oligozoospermia without characteristics of CHARGE syndrome or olfactory abnormalities. Thus, CHD7 variants may have incomplete penetrance. These phenotypic differences may be related to modified genes or to the impact of environmental factors.
5. CONCLUSIONS
Our results suggest that there may be a relationship between the CHD7 gene missense variants and male infertility. These variants are easier to find in patients with azoospermia and severe oligospermia whose testosterone levels are decreased. Although variants in CHD7 are commonly sporadic, genetic counseling of these patients is important because of potential autosomal dominant inheritance and reports of incomplete penetrance.
CONFLICT OF INTERESTS
The authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
Ruizhi Liu and Leilei Li conceived and designed this study. Ruixue Wang and Yang Yu completed the experiments. Hongguo Zhang and Yuting Jiang conducted the data analysis. Yuting Jiang and Xiao Yang collected the blood samples and the clinical data. Leilei Li finished the article. Ruizhi Liu revised the manuscript and made valuable suggestions.
Supporting information
Table S1
ACKNOWLEDGMENT
This research was generously supported by a grant from the Finance Department Health Special Project of Jilin Province, China (JLSCZD2019‐022).
Li L, Wang R, Yu Y, et al. CHD7 missense variants and clinical characteristics of Chinese males with infertility. Mol Genet Genomic Med. 2020;8:e1372 10.1002/mgg3.1372
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Bergman, J. E. H. , Janssen, N. , van der Sloot, A. M. , de Walle, H. E. K. , Schoots, J. , Rendtorff, N. D. , … Hofstra, R. M. W. (2012). A novel classification system to predict the pathogenic effects of CHD7 missense variants in CHARGE syndrome. Human Mutation, 33(8), 1251–1260. 10.1002/humu.22106 [DOI] [PubMed] [Google Scholar]
- Bhagavath, B. , Podolsky, R. H. , Ozata, M. , Bolu, E. , Bick, D. P. , Kulharya, A. , … Layman, L. C. (2006). Clinical and molecular characterization of a large sample of patients with hypogonadotropic hypogonadism. Fertility and Sterility, 85(3), 706–713. 10.1016/j.fertnstert.2005.08.044 [DOI] [PubMed] [Google Scholar]
- Boehm, U. , Bouloux, P.‐M. , Dattani, M. T. , de Roux, N. , Dodé, C. , Dunkel, L. , … Young, J. (2015). Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism—Pathogenesis, diagnosis and treatment. Nature Reviews Endocrinology, 11(9), 547–564. 10.1038/nrendo.2015.112 [DOI] [PubMed] [Google Scholar]
- Crowley, W. F., Jr. , Filicori, M. , Spratt, D. I. , & Santoro, N. F. (1985). The physiology of gonadotropin‐releasing hormone (GnRH) secretion in men and women. Recent Progress in Hormone Research, 41, 473–531. 10.1016/b978-0-12-571141-8.50015-9 [DOI] [PubMed] [Google Scholar]
- Dode, C. , & Hardelin, J. P. (2010). Clinical genetics of Kallmann syndrome. Annales d'Endocrinologie, 71(3), 149–157. 10.1016/j.ando.2010.02.005 [DOI] [PubMed] [Google Scholar]
- Goncalves, C. I. , Patriarca, F. M. , Aragues, J. M. , Carvalho, D. , Fonseca, F. , Martins, S. , … Lemos, M. C. (2019). High frequency of CHD7 variants in congenital hypogonadotropic hypogonadism. Scientific Reports, 9(1), 1597 10.1038/s41598-018-38178-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, T. S. , & Bouloux, P. M. (2010). What is the optimal therapy for young males with hypogonadotropic hypogonadism? Clinical Endocrinology, 72(6), 731–737. 10.1111/j.1365-2265.2009.03746.x [DOI] [PubMed] [Google Scholar]
- Janssen, N. , Bergman, J. E. , Swertz, M. A. , Tranebjaerg, L. , Lodahl, M. , Schoots, J. , … Hoefsloot, L. H. (2012). Variant update on the CHD7 gene involved in CHARGE syndrome. Human Mutation, 33(8), 1149–1160. 10.1002/humu.22086 [DOI] [PubMed] [Google Scholar]
- Kim, S. H. (2015). Congenital hypogonadotropic hypogonadism and Kallmann syndrome: Past, present, and future. Endocrinology and Metabolism, 30(4), 456–466. 10.3803/EnM.2015.30.4.456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausz, C. , Degl'Innocenti, S. , Nuti, F. , Morelli, A. , Felici, F. , Sansone, M. , … Forti, G. (2006). Natural transmission of USP9Y gene variants: A new perspective on the role of AZFa genes in male fertility. Human Molecular Genetics, 15(18), 2673–2681. 10.1093/hmg/ddl198 [DOI] [PubMed] [Google Scholar]
- Krausz, C. , Forti, G. , & McElreavey, K. (2003). The Y chromosome and male fertility and infertility. International Journal of Andrology, 26(2), 70–75. 10.1046/j.1365-2605.2003.00402.x [DOI] [PubMed] [Google Scholar]
- Krausz, C. , & Riera‐Escamilla, A. (2018). Genetics of male infertility. Nature Reviews Urology, 15(6), 369–384. 10.1038/s41585-018-0003-3 [DOI] [PubMed] [Google Scholar]
- Laitinen, E. M. , Tommiska, J. , Sane, T. , Vaaralahti, K. , Toppari, J. , & Raivio, T. (2012). Reversible congenital hypogonadotropic hypogonadism in patients with CHD7, FGFR1 or GNRHR variants. PLoS One, 7(6), e39450 10.1371/journal.pone.0039450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos, S. , Sarfati, J. , Leroy, C. , Fouveaut, C. , Parent, P. , Metz, C. , … Dode, C. (2014). The prevalence of CHD7 missense versus truncating variants is higher in patients with Kallmann syndrome than in typical CHARGE patients. Journal of Clinical Endocrinology and Metabolism, 99(10), E2138–E2143. 10.1210/jc.2014-2110 [DOI] [PubMed] [Google Scholar]
- Marfella, C. G. , & Imbalzano, A. N. (2007). The Chd family of chromatin remodelers. Mutation Research, 618(1–2), 30–40. 10.1016/j.mrfmmm.2006.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteloud, N. , Acierno, J. S. Jr , Meysing, A. , Eliseenkova, A. V. , Ma, J. , Ibrahimi, O. A. , … Crowley, W. F., Jr. (2006). Variants in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proceedings of the National Academy of Sciences of the United States of America, 103(16), 6281–6286. 10.1073/pnas.0600962103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivio, T. , Falardeau, J. , Dwyer, A. , Quinton, R. , Hayes, F. J. , Hughes, V. A. , … Pitteloud, N. (2007). Reversal of idiopathic hypogonadotropic hypogonadism. New England Journal of Medicine, 357(9), 863–873. 10.1056/NEJMoa066494 [DOI] [PubMed] [Google Scholar]
- Schnetz, M. P. , Bartels, C. F. , Shastri, K. , Balasubramanian, D. , Zentner, G. E. , Balaji, R. , … Scacheri, P. C. (2009). Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Research, 19(4), 590–601. 10.1101/gr.086983.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, N. , Hamra, F. K. , & Garbers, D. L. (2003). A multitude of genes expressed solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proceedings of the National Academy of Sciences of the United States of America, 100(21), 12201–12206. 10.1073/pnas.1635054100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skakkebaek, N. E. , Rajpert‐De Meyts, E. , & Main, K. M. (2001). Testicular dysgenesis syndrome: An increasingly common developmental disorder with environmental aspects. Human Reproduction, 16(5), 972–978. 10.1093/humrep/16.5.972 [DOI] [PubMed] [Google Scholar]
- Vissers, L. E. , van Ravenswaaij, C. M. , Admiraal, R. , Hurst, J. A. , de Vries, B. B. , Janssen, I. M. , … van Kessel, A. G. (2004). Variants in a new member of the chromodomain gene family cause CHARGE syndrome. Nature Genetics, 36(9), 955–957. 10.1038/ng1407 [DOI] [PubMed] [Google Scholar]
- Wen, J. , Pan, L. , Xu, X. , Wang, J. , & Hu, C. (2018). Clinical data and genetic variant in Kallmann syndrome with CHARGE syndrome: Case report and pedigree analysis. Medicine (Baltimore), 97(27), e11284 10.1097/MD.0000000000011284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters, B. R. , & Walsh, T. J. (2014). The epidemiology of male infertility. Urologic Clinics of North America, 41(1), 195–204. 10.1016/j.ucl.2013.08.006 [DOI] [PubMed] [Google Scholar]
- Yang, X. , Zhu, D. , Zhang, H. , Jiang, Y. , Hu, X. , Geng, D. , … Liu, R. (2018). Associations between DNAH1 gene polymorphisms and male infertility: A retrospective study. Medicine (Baltimore), 97(49), e13493 10.1097/md.0000000000013493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegers‐Hochschild, F. , Adamson, G. D. , de Mouzon, J. , Ishihara, O. , Mansour, R. , Nygren, K. , … van der Poel, S. (2009). The International Committee for Monitoring Assisted Reproductive Technology (ICMART) and the World Health Organization (WHO) revised glossary on ART terminology, 2009. Human Reproduction, 24(11), 2683–2687. 10.1093/humrep/dep343 [DOI] [PubMed] [Google Scholar]
- Zentner, G. E. , Layman, W. S. , Martin, D. M. , & Scacheri, P. C. (2010). Molecular and phenotypic aspects of CHD7 variant in CHARGE syndrome. American Journal of Medical Genetics. Part A, 152A(3), 674–686. 10.1002/ajmg.a.33323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, C. , Niu, Y. , Xu, H. , Li, Z. , Wang, T. , Yang, W. , … Liu, J. (2018). Variant profiles and clinical characteristics of Chinese males with isolated hypogonadotropic hypogonadism. Fertility and Sterility, 110(3), 486–495.e5. 10.1016/j.fertnstert.2018.04.010 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.