

Abstract
Objective:
To determine if endothelial dysfunction in a mouse model of diet-induced obesity and in obese humans is mediated by the suppression of endothelial Kir channels.
Approach and Results:
Endothelial dysfunction, observed as reduced dilations to flow, occurred after feeding mice a high-fat, Western diet for eight weeks. The functional downregulation of endothelial Kir2.1 using dominant-negative Kir2.1 construct resulted in substantial reductions in the response to flow in mesenteric arteries of lean mice whereas no effect was observed in arteries of obese mice. Overexpressing WT-Kir2.1 in endothelium of arteries from obese mice resulted in full recovery of the flow response. Exposing freshly isolated endothelial cells to fluid shear during patch clamp electrophysiology revealed that the flow-sensitivity of Kir was virtually abolished in cells from obese mice. Atomic force microscopy revealed that the endothelial glycocalyx was stiffer and the thickness of the glycocalyx layer reduced in arteries from obese mice. We also identified that the length of the glycocalyx is critical to the flow-activation of Kir. Overexpressing Kir2.1 in endothelium of arteries from obese mice restored flow- and heparanase-sensitivity, indicating an important role for heparan sulfates in the flow-activation of Kir. Furthermore, the Kir2.1-dependent component of flow-induced vasodilation was lost in the endothelium of resistance arteries of obese humans obtained from biopsies collected during bariatric surgery.
Conclusions:
We conclude that obesity-induced impairment of flow-induced vasodilation is attributed to the loss of flow-sensitivity of endothelial Kir channels and propose that the latter is mediated by the biophysical alterations of the glycocalyx.
Keywords: Obesity, endothelial dysfunction, K+ channels, shear stress, glycocalyx
Subject codes: Animal Models of Human Disease, Endothelium/Vascular Type/Nitric Oxide, Ion Channels/Membrane Transport, Lipids and Cholesterol
Graphical Abstract
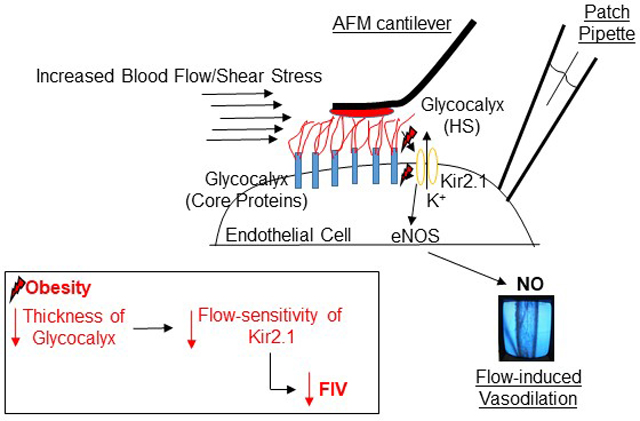
Introduction
Obesity represents a complex and multifactorial metabolic disease with well-established deficits to cardiovascular function. Arteries residing in visceral adipose exhibit impaired vascular responses to endothelium-dependent agonists including intraluminal flow1,2. Obesity-induced endothelial dysfunction precedes and is a predictor for severe disease such as atherosclerosis.
A hallmark of endothelial dysfunction in obesity is impairment of flow-induced vasodilation (FIV), the dilatory response of arteries to flow3. The prevalent thought is that endothelial dysfunction in obesity manifests as the impairment of endothelial NO synthase (eNOS)7 and the loss of NO4, a major vasodilator and anti-inflammatory agent5. Obesity also disrupts the endothelial glycocalyx6,7, a proteoglycan extension of the endothelial layer that is a putative mechanosensing complex8, required for flow-induced NO production9,10.
Our recent studies identified a flow-sensitive inwardly-rectifying K+ channel, Kir2.1, to be a critical mediator of NO-dependent FIV upstream of NO production11. Furthermore, we determined that Kir2.1 is required for eNOS phosphorylation at serine 1177, the site necessary for NO production, that is impaired in obesity12. These findings established that flow-sensitive endothelial Kir channels are critical to endothelial control of vascular tone in response to shear stress. Flow-induced NO production is also dependent on the integrity of the glycocalyx, one of the primary mechanosensors, existing as a direct barrier between the drag force of flowing blood and the vascular endothelium13. Importantly, the integrity of the glycocalyx was shown to be impaired early in obesity6.
In this study, we show that high-fat diet drastically reduces shear-induced activation of Kir and discover that flow sensitivity of Kir requires intact heparan sulfates (HS) of the endothelial glycocalyx. Furthermore, we show that the loss of flow-sensitivity of Kir plays a major role in FIV impairment in high-fat diet induced obesity in mice and in obese humans. We propose that the obesity-induced alterations of the endothelial glycocalyx renders Kir insensitive to shear, ultimately underlying the endothelial dysfunction in the microvasculature.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animals
All studies involving animals were approved by the Animal Care Committee at the University of Illinois at Chicago (UIC). Ten week old male and female mice were randomly divided into diet groups: lean controls maintained on a normal laboratory diet and an obese group fed a high-fat Western diet. Data from male and female mice were combined where indicated. Mice were euthanized by CO2 asphyxiation followed by cervical dislocation, unless otherwise stated, and 1st order mesenteric arteries were isolated from mesenteric adipose.
Analysis of fasted serum lipids and mass spectroscopy
Whole blood was collected following a 6 hour fast. Mice were sedated with isoflurane and decapitated14. Serum samples were assessed for the rodent lipid panel consisting of low density lipoprotein (LDL), high density lipoprotein (HDL), total cholesterol, and triglycerides. Liquid Chromatography-Electrospray Ionization Tandem-Mass Spectrometric (LC-ESI-MS/MS) was performed15.
Transduction of artery endothelium with adenoviral Kir2.1 constructs and FIV
Mesenteric arteries were incubated with adenovirus (AV) containing either dominant-negative (DN) or WT-Kir2.1 constructs for 48 hours under standard culture conditions (10% FBS DMEM medium, 5% CO2, 37°C)16. Endothelial-specific expression was driven by the hVE-Cadherin promoter. FIV was measured as described11,16,17.
Confocal microscopy, flow cytometry, and Western blotting
Excised arteries were opened en face and immunostained18. Images were taken using a Zeiss laser scanning confocal microscope. Single-cell suspensions of digested arteries or human microvascular endothelial cells (HAMECs) were used to detect the expression of endothelial cell (EC) membrane Kir2.1 in CD31+ cells using a BD LSRFortessa flow cytometer. Data was analyzed using Kaluza2.1 software. Western blots on whole mesenteric arcades were performed as described16. HAMECs were exposed to shear stress using a cone-and-plate rheometer for analysis of Akt phosphorylation by Western blotting11.
Perforated whole-cell electrophysiology
ECs were isolated from mesenteric arteries using an established method16,19. Freshly isolated ECs or HAMECs were seeded onto the modified parallel plate flow chamber. Electrophysiology was performed as described16. Flow was administered by gravity perfusion.
Atomic force microscopy
Elastic moduli of HAMECs and en face arteries were measured with an Asylum MFP-3D-Bio atomic force microscope (Santa Barbara, CA)14. A notable modification was the use of a highly flexible cantilever (spring constant of 0.01 N/m versus typically used 0.10 N/m), as was described for the atomic force microscopy (AFM) analysis of the glycocalyx20,21. A 4.5 μm polystyrene bead AFM probe (Novascan, Ames, IA) was used to facilitate even force dispersion. Fifteen to twenty sites were analyzed/artery from one or two vessels per mouse (20–50 independent force-distance measurements/mouse).
Human studies
Human studies were conducted in accordance with the Institutional Review Board at the UIC. Obese human subjects recruited to this study gave informed consent prior to the planned bariatric surgery date. All subjects had a body mass index of ≥ 38 and were between 26–48 years old. Subjects were excluded from study if they had diabetes, cancer, heart disease, a history of smoking, kidney or liver disease, gallbladder disease, or autoimmune/inflammatory disease. Mesenteric adipose biopsies were collected the day of the surgery and arteries immediately cleaned of adipose and incubated with AV, as described above. All subject information was de-identified and stored on a password protected, onsite hard drive. Additional subject clinical measures and demographics are outlined in Supplemental Table 1.
Statistical analyses
Appropriate sample sizes for this study were calculated using a power test set to 0.9 and predicated on our initial findings in male lean vs. obese mice after 8-weeks on respective diets (SFig1). Each data set was tested for normality and equal variance prior to conducting appropriate parametric or nonparametric tests. For parametric data sets, equal variance was not assumed and an unbalanced ANOVA or Student’s t test was performed. The lognormal AFM data were transformed prior to conducting parametric tests22. For all other data sets that exhibited a non-normal distribution, appropriate nonparametric tests were applied. For all tests, significance was set to p < 0.05.
Results
Loss of endothelial Kir2.1 is responsible for impairment of FIV in mesenteric adipose arteries of obese mice.
Arteries in the mesenteric adipose of obese humans and rodents exhibit endothelial dysfunction2,23. To investigate the underlying mechanism, we first fed mice a normal laboratory diet or high-fat Western diet and tested endothelial function at various time points in excised arteries by measuring dilations to flow (Fig.S1), as described11,16,17.
No differences in FIV were detected in arteries from male mice after a 4-week diet (Fig.S1A,D), however, robust endothelial dysfunction was observed after 8 weeks (Fig.S1B,D). This persists after 24 weeks (Fig.S1C,D). Similarly, arteries from obese female mice also had reduced FIV compared to lean controls after 8 weeks on respective diets (Fig.S1E,F). No vascular smooth muscle dysfunction was observed after 8 weeks as measured by the endothelium-independent vasodilator sodium nitroprusside (SNP) (Fig.S2A). In contrast, the 24-week time point showed evidence of vascular smooth muscle dysfunction as evidenced by reduced dilations to SNP (Fig.S2B). Therefore, the role of Kir2.1 in obesity-induced endothelial dysfunction was tested at 8-weeks. Average body weights for lean vs. obese groups before the start of the diet were comparable and, as expected, after the 8-week feeding regimen, obese mice weighed significantly more than lean counterparts (Table S2). However, obesity did not affect the baseline diameter of mesenteric arteries (Table S2).
To test if obesity reduces FIV through impairment of endothelial Kir2.1 channels, functional expression of Kir2.1 in intact mesenteric arteries was downregulated using an endothelial-specific adenoviral construct containing a dominant negative (DN) Kir2.1 (VE-Cad-DN-Kir2.1-AV). Empty adenovirus (AV) was used as control. Notably, Kir2.1 contained an extracellular HA-tag (VE-Cad-WT-Kir2.1-HA-AV), which allowed for the verification of endothelial-specific Kir2.1 expression. Indeed, HA-tag specific fluorescence was observed in intact, but not denuded, en face arteries (Fig. 1A).
Figure 1. The loss of Kir2.1 results in FIV impairment in mesenteric adipose arteries of obese mice.

A) Images of intact and denuded en face mesenteric arteries transduced with the VE-Cadherin-WT-Kir2.1-HA tag (WT-Kir2.1)-adenovirus (AV) and stained for the HA-tag. The magnification is 25x. Ex vivo flow-induced vasodilation (FIV) measurements in arteries from B) lean and C) obese mice incubated with Empty (Em)-AV or VE-Cadherin-dominant negative-Kir2.1-AV (DN-Kir2.1) +/− BaCl2 (Ba) (n = 8). D) Analysis of FIV at a flow of Δ100; * indicates significance when compared to lean Em-AV (p<0.05). E) Ex vivo FIV measurements in arteries isolated from obese mice after incubation with either Em-AV or WT-Kir2.1-AV +/− BaCl2 or LNAME (LN) (n = 8). F) Analysis of the %dilation at Δ100; † and * indicate significance when compared to VE-Cadherin-WT-Kir2.1-HA-AV or Em-AV, respectively. Data from an equal number of male and female mice were combined in B-F.
Downregulation of Kir2.1.
As expected, in lean controls exposed to the Em-AV, the percent dilation to flow reached an average 87.2% in response to the maximum pressure gradient of Δ100 whereas the response in arteries from obese mice was significantly reduced (Fig. 1B, C). Expressing VE-Cad-DN-Kir2.1-AV significantly reduced FIV in arteries from lean mice but had no effect in arteries from obese mice (Fig. 1B–D), suggesting a loss of Kir2.1 contribution to FIV in arteries of obese mice. Successful knockdown of Kir2.1 with VE-Cad-DN-Kir2.1-AV is supported by the lack of an effect of Ba2+, a blocker of Kir channels. No difference was observed with respect to downregulation of Kir2.1 in male vs. female mice (Fig.S2C–D) and so data was combined.
Rescue of FIV with WT Kir2.1.
To test if FIV could be rescued in arteries of obese mice by overexpressing WT-Kir2.1 in endothelium, arteries from obese mice were incubated with VE-Cad-WT-Kir2.1-HA-AV containing a fully functional WT Kir2.1. Overexpressing WT-Kir2.1 in endothelium of visceral arteries of obese mice rescued dilations to flow compared to arteries exposed to Em-AV (Fig. 1E,F) and restored sensitivity to Ba2+. Furthermore, overexpressing WT Kir2.1 in the endothelium of obese mice arteries restored sensitivity to LNAME, suggesting that the overexpression of Kir2.1 works through activation of eNOS.
In addition, we tested the role of the small Ca2+-activated K+ (SK) channel in obesity as these channels contribute to the NO-independent dilations to flow24. In contrast to Kir2.1, the contribution of SK channels to FIV was similar in lean and obese mice as assessed by blocking SK with apamin (Fig.S3).
Kir channels are impaired in endothelial cells of obese mice.
We next tested the functional expression of Kir channels in ECs freshly isolated from lean and obese mice using perforated patch clamp. As expected, we detected well-defined Kir currents with typical inward rectification in freshly isolated ECs that reversed at ~−20 mV in a 60 mM K+ bath. Most importantly, Kir current densities (IKir) were reduced more than 3-fold in ECs isolated from obese mice relative to the IKir detected in ECs from lean counterparts (Fig. 2A–C). EC isolation was verified by the endothelial marker CD31 (Fig. 2A). To determine if decreased Kir2.1 surface expression was an underlying cause of reduced Kir activity, Kir2.1 expression in ECs isolated from mesenteric arteries was assessed by flow cytometry. Kir2.1 expression was analyzed in cells positive for CD31. Kir2.1 surface expression was determined using an antibody that targets an extracellular epitope of the channel. No effect of obesity was observed on endothelial Kir2.1 expression or in the percent of ECs expressing Kir2.1 even after an extended diet of 24 weeks (Fig.S4A–C). In addition, there was no difference in Kir2.1 expression as determined by immunoblotting of intact mesenteric arterial arcades between lean and obese mice (Fig.S4D) further supporting that obesity does not affect Kir2.1 expression. Taken together, these data suggest that obesity impairs Kir activity without affecting membrane expression.
Figure 2. Kir channels are impaired in endothelial cells of obese mice.

Representative patch clamp recordings of IKir in endothelial cells (ECs) from A) lean and B) obese mouse mesenteric arteries. Inset: bright field (BF) and CD31 fluorescence image of a freshly isolated EC. Scale bar=5 μm. C) Group data of Kir current density (CD) at −100 mV (*p <0.05). Representative recordings of IKir in ECs from D) lean and E) obese mice before (Static) and after flow (Shear). D: A picture of the parallel plate flow chamber. Insets show the shear-induced increase in IKir for respective recordings. Shear-induced CD for each cell tested from F) lean and G) obese mice normalized to static CD (n = 12 cells from 6 lean mice and 9 cells from 4 obese mice, *p<0.05). H) Group data comparing shear-induced increases in EC IKir (*p<0.05).
A major feature of Kir channels is that they are activated by shear stress25. To directly test the impact of obesity on the flow-sensitivity of Kir channels, ECs freshly isolated from arteries of lean and obese mice were seeded into a parallel flow chamber designed in our earlier studies to expose cells to well-defined flow during electrophysiological recordings11,16,26. The recordings start prior to the initiation of the flow and then performed continuously during the flow exposure from the same cell. Representative recordings reveal the effects of fluid-shear on IKir in ECs from lean and obese mice: IKir recorded from cells isolated from lean mice show a relatively large current with a significant increase in response to flow whereas IKir recorded from cells isolated from obese mice show smaller current and no increase with flow (Fig. 2D,E). The insets highlight the shear-sensitive components of the current (ISh-ISt). Clearly, the shear-sensitive component is virtually absent in obese mice. Furthermore, in ECs from lean mice, fluid shear induced an increase in IKir in each cell tested (Fig. 2F,H), whereas in ECs from obese mice 8 of 9 cells tested did not respond. (Fig. 2G,H). These findings indicate that a functional impairment renders Kir insensitive to fluid shear.
Mechanism of obesity-induced impairment of Kir channels.
Obesity-induced impairment of Kir cannot be attributed to increased cholesterol:
As expected, feeding mice a high-fat Western diet results in mild dyslipidemia with a small but significant increase in fasted serum LDL (Table S3). Since our previous studies established that Kir channels are suppressed by cholesterol16,26,27, we tested whether obesity also results in an increase in cholesterol in mesenteric arteries via mass spectrometry. However, while serum cholesterol was elevated >2-fold in obese mice no significant difference in arterial cholesterol levels was detected (Fig.S5A,B). Furthermore, we analyzed the tissue levels of several biologically-active lipids and found no or little difference between lean and obese tissues (Fig.S5C–H). These data show that obesity has no significant effect on tissue cholesterol of mesenteric arteries. Additionally, depleting cholesterol from arteries of obese mice using methyl-β-cyclodextrin (MβCD), a treatment that leads to recovery of FIV in severely hypercholesterolemic apoE−/− mice16 (Fig.S5I), had no effect on FIV in obese mice (Fig.S5J,K). These data suggest that mild dyslipidemia that accompanies diet-induced obesity in mice does not induce inhibition of Kir channels via cholesterol.
Obesity-induced biophysical alterations to the glycocalyx impair Kir flow-sensitivity:
Here, we tested whether: 1) obesity alters the biophysical properties of the glycocalyx in resistance arteries, and 2) the glycocalyx regulates the flow-activation of endothelial Kir channels, an effect that is lost with alterations to the glycocalyx.
Obesity alters the biophysical properties of the endothelial glycocalyx and underlying endothelial layer:
To test the biophysical properties of the endothelial glycocalyx of the adipose resistance microvasculature by AFM, we first verified that the arteries maintain both the glycocalyx and the endothelial layer during the en face preparation by staining for HS and for CD31 (Fig.S6A,B). As described previously, using a cantilever with a low spring constant allows for distinguishing the glycocalyx layer based on the shape of the force-distance AFM approach curve: the presence of the glycocalyx manifests as a deviation from the Hertz model, also seen as a break in the slope of the force-distance curve (Fig.S6C,D). Representative traces (Fig. 3A,B) reveal the effects of obesity on the endothelial glycocalyx and underlying endothelial layer, respectively. Obesity increased the elastic moduli of the endothelial glycocalyx (3A) and underlying layer (3B) as well as decreased the length of the glycocalyx (3A). Specifically, the values of the elastic modulus for the glycocalyx of the en face arteries of lean mice range from 440 +/− 100 Pa, the typical range of the glycocalyx elastic moduli of ECs20,21 whereas the glycocalyx of the arteries isolated from obese mice had an elastic modulus ranging from 1100 +/− 140 Pa (Fig. 3D, left column). The glycocalyx also had a decreased length in mesenteric arteries of obese mice averaging 99±9 nm while the glycocalyx in lean mice was an average 176±26 nm (Fig. 3D, right column). Obesity also induced an increase in the elastic modulus of the underlying endothelium (Fig. 3E) which, in addition to the glycocalyx, prompted further investigation into the effects of the endothelium elastic modulus on Kir flow-sensitivity.
Figure 3. Obesity results in increased stiffness and decreased thickness of endothelial glycocalyx.

Representative atomic force microscopy force-distance curves generated from A) the glycocalyx and B) endothelium of en face arteries. Group data comparing the C) elastic moduli of the endothelial glycocalyx (left column), thickness of the glycocalyx (right column), and D) elastic moduli of the endothelial layer in arteries from lean and obese mice. For elastic modulus data sets, each data point represents the measurements taken from an individual artery (n = 8 or 9 arteries from 5 mice/group, *p<0.05).
Does flow-sensitivity of Kir depend on the stiffness of the endothelium or endothelial glycocalyx?
To test this possibility, we seeded HAMECs on hydrogels of different stiffness because the cell elastic modulus is known to increase on stiffer substrates. Specifically, cells were seeded on polyacrylamide gels of 5 kPa and 30 kPa for 24 hours prior to experiments (Fig.S8A). As expected, HAMECs seeded on the stiff gels had a greater cellular elastic modulus than cells seeded on the soft gels (Fig. 4A). The glycocalyx of cells seeded on the stiff gels also had a greater elastic modulus (Fig. 4B) with a significant, positive correlation between cell stiffness and glycocalyx stiffness (Fig.S8B). In contrast, no differences were observed in glycocalyx length between cells seeded on gels of different stiffness (Fig. 4C) allowing us to discriminate the effects of cell/glycocalyx stiffness and glycocalyx length on the flow-activation of Kir. Our data show that no differences in flow-activated Kir currents were observed in HAMECs seeded on the different gels (Fig. 4D,E), indicating that neither elastic moduli are major factors in the flow-sensitivity of Kir.
Figure 4. Flow-sensitivity of Kir channels critically depends on glycocalyx length.

Histograms generated from A, B) endothelial cell and glycocalyx elastic moduli, and C) glycocalyx length of Human Adipose Microvascular Endothelial Cells (HAMECs) seeded on 5 vs. 30 kPa polyacrylamide gels (*p<0.05). D) Representative recordings and E) group data show flow-induced increase in IKir in HAMECs seeded on gels. F) Histograms detailing the length of the glycocalyx following enzymatic removal of heparan sulfates (middle) and hyaluronic acids (right) (*p<0.05). G) Representative recordings and H) group data show flow-induced increase in IKir following treatment with enzymes (*p<0.05). Data represents 4–5 independent experiments. Shear-Static, Sh-St. heparanase III, HEPIII. Hyaluronic acid lyase, HA lyase.
Does flow-sensitivity of Kir depend on glycocalyx length?
To test this possibility, we used HEPIII and HA lyase to degrade different components of the glycocalyx. We found significant effects of the enzymes on glycocalyx length (Fig. 4F) and stiffness (Fig.S8C–E), without affecting the underlying endothelial layer (Fig.S8F–H). We next tested the flow-sensitivity of Kir following treatment with either enzyme. Compared to controls that exhibited robust flow-activation of Kir, each enzyme essentially abolished the Kir response to flow (Fig. 4G,H), indicating the critical role of the glycocalyx in shear-induced Kir activation and suggesting that the length of the glycocalyx is the important parameter.
Importantly, we also tested if the enzymes directly affect the surface expression of Kir channels and thereby reduce channel function independent of perturbations to the glycocalyx. The S.Fig. 7 shows that Kir2.1 expression is unaltered by enzyme treatments. These data further support the proposed mechanism that the enzymes disrupt the physical properties of the glycocalyx which results in the loss of Kir flow-sensitivity.
Overexpression of endothelial Kir2.1 restores sensitivity to HEPIII in ECs from obese mice:
Next, we focused on heparan sulphates (HS), the most abundant class of glycosaminoglycan (GAG)13 in the regulation of Kir activity in freshly isolated ECs from lean and obese mice. First, successful removal of HS by HEPIII was confirmed by staining for HS in en face preparations (Fig.S9A). Freshly isolated cells were exposed to HEPIII and representative recordings from both control and HEPIII-treated, freshly-isolated ECs from lean mice show a well-defined IKir (Fig.S9B) but the current in HEPIII-treated cells was reduced (Fig.S9C). To confirm if HS are required for the flow-sensitivity of Kir channels in freshly-isolated ECs, HEPIII-treated cells were seeded into the flow chamber described above. As expected, and shown in a representative trace highlighting the shear-induced current component ISh-ISt, control ECs from lean mice had a robust increase in IKir in response to flow (Fig. 5A), whereas it did not increase in ECs from lean mice lacking HS indicating that the flow-sensitivity of Kir channels is abolished (Fig.5B).
Figure 5. Overexpressing Kir2.1 in endothelium restores Kir flow- and HEPIII-sensitivity.

Representative shear-induced IKir recordings from A) control and B) heparanse III treated (HEPIII) lean mouse endothelial cells (ECs). Representative IKir recordings from ECs isolated from obese mouse mesenteric arteries exposed to C,D) Empty-adenovirus (Em-AV) or E,F) VE-Cadherin-WT-Kir2.1-AV with and without HEPIII treatment. G) Group data (n = 7–12 cells from 4–8 mice/group, *p<0.05). Data from males and female obese mice were combined. Current Density, CD.
As also shown in Figure 2, the flow-sensitivity of Kir recorded from ECs freshly isolated from obese mice is virtually abolished (Fig. 5C) and as expected, treating ECs from obese mice with HEPIII, had no effect on the flow-activation of Kir (Fig. 5C,D,G). We next tested whether over-expression of Kir2.1 in ECs isolated from arteries of obese mice rescues the flow and HEPIII sensitivity of the currents. We found that this is indeed the case: overexpressing the WT-Kir2.1 restored baseline currents to ~25% of that in lean mice and resulted in a 2-fold increase in the EC-Kir flow-sensitive current as compared to control ECs (Fig. 5E,G). Overexpressing the WT channel also restores HEPIII inhibition of the flow-induced increase in IKir (Fig. 5F,G).
Obesity disrupts glycocalyx-mediated Akt signaling:
We recently showed that flow-induced Akt phosphorylation is dependent on endothelial Kir channels11. Similar to earlier studies28,29, we show here that removal of HA or HS results in reduced Akt phosphorylation in HAMECs exposed to shear stress (Fig.S9D,E). We next investigated, therefore, the role of the flow-induced glycocalyx-mediated Akt activation in mesenteric arteries from lean and obese mice by perfusion of the lumen with HEPIII in the presence and absence of an Akt inhibitor. As expected, Akt inhibition significantly blunts FIV in arteries from lean mice (Fig. 6A). However, after perfusing the vessel lumen with HEPIII to remove the heparan sulfates from the glycocalyx, dilations to flow were blunted and the Akt inhibitor had no further effect (Fig. 6B,G). Sham control vessels that were perfused with buffer without HEPIII responded to flow to the same extent as before the perfusion and retained sensitivity to Akt inhibition (Fig. 6C,G). There was no effect of Akt inhibition or HEPIII on arterial response to flow in obese mice. These data support the hypothesis that the glycocalyx-Kir2.1-Akt signaling axis is impaired in obesity (Fig. 6D–F,H).
Figure 6. Flow-mediated HS-glycocalyx-Akt signaling is impaired in obesity.

The effects of Akt inhibition and heparanse III (HEPIII) on flow-induced vasodilation (FIV) in arteries from A-C,G) lean and D-F,H) obese mice are shown (*p<0.05). Sham arteries received a luminal perfusion of buffer without HEPIII (n=4–10 vessels/treatment from 4–5 mice/group). Akt Inhibitor, Akt Inhib. Both male and female mice were included.
Loss of endothelial Kir2.1 is responsible for impairment of FIV in mesenteric adipose arteries of obese humans.
We tested the role of endothelial Kir2.1 in obese humans to determine if the reduced FIV is due to the loss of Kir channel contributions as we observed in the mouse model of diet-induced obesity. Morbidly obese humans (BMI ≥ 38) undergoing planned bariatric surgery were recruited to this study (for additional subject demographics, please refer to Table S1). Biopsies of mesenteric adipose were collected during surgery and arteries with an average diameter of 153.9 ± 22.8 μm were subsequently isolated and incubated with AV containing constructs for endothelial-specific expression of Kir2.1-DN or WT-Kir2.1. After 48 hours of incubation with AV, arteries were cannulated, pressurized, and pre-constricted as described previously for testing FIV using the pressure gradient method of administering intraluminal flow11,16,17.
Mesenteric arteries from obese subjects incubated with the Em-AV had an average percent dilation to Δ100 flow of 47.7%, which is similar to previous reports on this artery type in obese humans2 and similar to the percent dilations observed in this study for the mesenteric arteries of diet-induced obese mice (48.8%, Fig. 1). Expressing Kir2.1-DN in the endothelium of arteries from obese humans had no effect on FIV (Fig. 6A,C). Notably, overexpressing functional WT Kir2.1 channels in endothelium increased percent dilations to flow, reaching 86.4% at the maximum flow rate of Δ100 (Fig. 6B,D). These data suggest that endothelial Kir2.1 are key components to FIV of human mesenteric arteries and that Kir2.1 channels are dysfunctional in obese human endothelium.
Discussion
Obesity-induced impairment of endothelial function is a key factor in the development of the cardiovascular disease3,4. Here we identify a novel mechanism of endothelial dysfunction in obesity to be a result of reduced function and loss of flow-sensitivity of endothelial Kir2.1 channels. Furthermore, we show that obesity-induced impairment of flow-induced vasodilation can be fully rescued by endothelial-specific overexpression of Kir2.1 in intact arteries. We also provide the first evidence that flow-sensitivity of Kir2.1 critically depends on endothelial HS-glycocalyx, suggesting that flow activation of these channels is the mechanistic link between glycocalyx as a flow mechanosensor and FIV. We propose that obesity-induced alteration of the glycocalyx and changes to its length are critical to the loss of flow-sensitivity of endothelial Kir2.1, which result in FIV impairment via disruption of Akt1/eNOS signaling. Notably, we show that impairment of Kir2.1 channels plays a central role in obesity-induced endothelial dysfunction in both a mouse model of high fat diet induced obesity and in obese humans.
Role of Kir channels in vasodilation:
Based on the unique biophysical properties of Kir channels that are activated by membrane hyperpolarization and by high extracellular K+, it was suggested that Kir channels act as “boosters” of the electrical responses generated by other vascular K+ channels19,30. Our recent studies demonstrated that endothelial Kir are key regulators of flow-induced activation of eNOS11, a major flow-induced signaling pathway that governs a multitude of endothelial responses. Specifically, using a combination of Kir2.1-deficient mice and endothelial-specific viral constructs of Kir2.1 and its dominant-negative mutant, we demonstrated the critical role of endothelial Kir2.1 in FIV of murine and human mesenteric resistance arteries. We also found that flow-induced activation of endothelial Kir2.1 results in the activation of eNOS and NO production and is independent of Ca2+-sensitive K+ channels. Furthermore, we discovered that endothelial Kir2.1 channels are also essential for flow-induced activation of Akt1, a kinase well-known to activate eNOS31. These observations identify endothelial Kir2.1 as an upstream major regulator of flow-induced endothelial signaling. In this study, we show that impairment of endothelial Kir2.1 channels plays a significant role in endothelial dysfunction in obesity.
Obesity, eNOS, and Kir2.1:
The loss of NO bioavailability in obesity is well-established3,4. This loss was attributed to the impairment of eNOS function including disruption of the eNOS/Akt complex, eNOS dimerization32 or in some models a decrease in eNOS expression33.
Our present data suggests a novel paradigm that obesity-induced endothelial dysfunction is a result of the loss of endothelial Kir2.1 channel function. First, downregulation of endothelial Kir2.1 results in a significant decrease in FIV in arteries of lean mice but has no effect on arteries of obese mice indicating that Kir2.1-dependent component of FIV is lost in obese mice. The loss of Kir2.1-component of FIV in mesenteric arteries of obese mice is supported by the finding that Kir currents are drastically decreased and flow sensitivity virtually abolished in mesenteric ECs isolated from obese mice. Second, overexpressing the functional Kir2.1 channel in the endothelium of arteries isolated from obese mice completely rescues FIV in an eNOS-dependent manner. This finding indicates that overexpressing endothelial Kir2.1 recovers endogenous signaling pathways of NO production and supports the notion that Kir channels upstream of eNOS are impaired in obesity. Importantly, FIV was also restored in arteries isolated from mesenteric adipose of obese humans with overexpression of endothelial WT-Kir2.1. This finding in particular supports the diet-induced obese mouse model as a translational approach to investigating mechanisms of obesity-induced impairment of FIV. Our results indicate that dysfunctional endothelial Kir2.1 channels are underlying the reduced FIV and are upstream from eNOS dysfunction in arteries isolated from obese mice and humans.
Impairment of Kir function in high fat diet-induced obesity
Obesity and hypercholesterolemia impair Kir via distinct mechanisms:
Our initial hypothesis was that a high-fat diet inhibits Kir via elevation of cellular cholesterol. This hypothesis was based on our studies establishing the inhibitory effect of cholesterol on Kir channels through direct cholesterol-channel interactions26,34,35. Most recently, we demonstrated that cholesterol-induced suppression of endothelial Kir2.1 is responsible for the loss of flow-induced eNOS activation and impairment of FIV in apoE−/− mice16, a murine model of severe hypercholesterolemia. We found, however, that impairment of endothelial Kir channels in diet-induced obesity cannot be attributed to cholesterol-induced suppression of the channels. In contrast to cholesterol accumulation in vascular tissues of apoE−/− mice, we detected no increase in cholesterol levels in arteries of obese mice using mass spectrometry. Furthermore, unlike the previous findings in the apoE−/− mouse16, depletion of cholesterol did not recover FIV in arteries from obese mice. Thus, our findings indicate that impairment of endothelial Kir is a common factor in hypercholesterolemia- and obesity-induced reductions in FIV but through entirely different mechanisms.
The integrity of the glycocalyx is essential for the flow sensitivity of endothelial Kir:
Previous studies showed that obesity results in increased penetration of red blood cells into the glycocalyx barrier6,7 and that aortas of db/db mice have increased glycocalyx stiffness22. Here, we provide novel and unique insights into the impact of obesity on the biomechanical properties of the glycocalyx in resistance arteries. We show that high-fat diet-induced obesity results not only in increased elastic modulus but also decreased length of the glycocalyx. Furthermore, we show that obesity-induced perturbation of the glycocalyx renders Kir insensitive to flow, resulting in endothelial dysfunction in the microvasculature.
Kir channels were originally proposed to be putative flow sensors based on their presence on the apical membrane and fast response to flow25. However, since Kir channel structure lacks extensive external residues that would “sense” changes in shear forces, we hypothesized that flow-activation of Kir is mediated by the glycocalyx. We identified that glycocalyx length is an important property in regulating the flow-activation of Kir, as evidenced in both ex vivo arteries and ECs treated with HEPIII or HA lyase. In contrast the stiffness of the glycocalyx that was modified by seeding the cells on stiffer vs. softer substrates had no clear association with Kir activity. It is important to note that the glycocalyx elastic modulus of cells seeded on the 30 kPa gels was similar to that observed in the animal model of diet-induced obesity, supporting the in vitro model as a valid approach for testing the role of the glycocalyx elastic modulus on Kir. In contrast, treating the cells with either HEPIII or HA lyase significantly decreased the length but had only a minor effect of glycocalyx stiffness. In the context of biomechanical transduction of shear force, it is logical that a shortened glycocalyx may not be able to transduce shear forces to the downstream signaling events as the torque on the glycocalyx “lever” may be inadequate to do so.
Our findings also demonstrate for the first time that the presence of HS is required for flow-activation of Kir in freshly isolated ECs. It is also noteworthy that overexpression of Kir in ECs isolated from obese mice results in partial recovery of the currents and full recovery of FIV. These data suggest that despite the near complete loss of Kir channel flow-sensitivity in obese mice, the integrity of the HS-glycocalyx is only partially impaired, allowing for the partial rescue of the currents upon introducing the additional channels. Furthermore, even the partial rescue of Kir flow-sensitivity is sufficient to fully restore the FIV in obese mice. It is certainly possible that removal of the glycocalyx may impact other mechanosensors which indirectly results in the inability of Kir to respond to fluid shear. Our study identifies the pathophysiological relevance of the glycocalyx/Kir/Akt axis in obesity.
Implications for obesity in humans
Importantly, we also identified the role of endothelial Kir2.1 in mesenteric arteries of obese humans, an arterial bed known to be highly dysfunctional in obesity. Overexpressing WT-Kir2.1 exclusively in vascular endothelium of arteries from obese subjects restores dilations to flow while expressing the Kir2.1-DN had no effect. Notably, the subjects recruited to this study had cholesterol within the healthy range. An inherent limitation in the obese human study is the lack of lean, healthy controls as these biopsies are obtained during bariatric surgery. However, we showed recently that FIV in subcutaneous adipose arteries obtained from obese patients, known to respond greater to flow than visceral arteries2, maintain intact Kir contribution to FIV11. This is consistent with earlier studies showing that Kir channels are involved in the regulation of vascular tone in healthy human subjects36–38. Taken together, these data indicate that Kir channels are impaired in the vasculature of obese humans.
In conclusion, we propose that obesity-induced alterations of the endothelial glycocalyx interferes with the mechanotransduction mechanism and impairs flow-sensitivity of Kir channels, which in turn result in the loss of flow-induced NO production and impairment of FIV. This study provides a new understanding of (1) the regulation of flow-activated ion channels and (2) mechanisms of obesity-induced endothelial dysfunction. Future studies will focus on determining how HS regulate the flow-activation of Kir as well as the mechanistic relationship between altered physical properties of the glycocalyx and reduced Kir function observed in this study.
Supplementary Material
Figure 7. Kir2.1 does not contribute to FIV in mesenteric adipose arteries of obese humans.

A) The table shows individual subject blood lipids measured by the clinic at the UIC for total cholesterol (Chol.), triglycerides (TriG), high density lipoproteins (HDL), non-HDL (NHDL), and low density lipoproteins (LDL). Flow-induced vasodilation measurements in human arteries exposed to B) VE-Cadherin-dominant negative-Kir2.1-AV (DN-Kir2.1) or C) VE-Cadherin-WT-Kir2.1-HA-AV (WT-Kir2.1). Analysis of the %dilation at a flow of Δ100 for D) DN-Kir2.1 and E) WT-Kir2.1 (*p<0.05). ND – not determined.
Highlights.
Obesity-induced endothelial dysfunction is mediated by a loss in flow-sensitivity of Kir in mouse and human mesenteric arteries
Heparan sulfates of the endothelial glycocalyx are critical regulators of endothelial Kir flow-sensitivity.
A reduced thickness of the glycocalyx in obesity may disrupt the regulation of endothelial Kir channels.
Acknowledgments
We thank Ms. Alema Jackson, BS, Mr. Nicolas Barbera, PhD, Arvind Sridhar, MS, and Wenjing Feng, MD for technical assistance. We thank the Mass Spectrometry Core at National Jewish Health for their contributions.
Sources of Funding
This study was funded by NHLBI R01s HL-073965 (IL and SAP), HL-083298 (IL), HL-141120 (IL, SAP, and ROD), and American Heart Association Postdoctoral Fellowship Award (16POST27000011), 5T32HL007829–24, 5T32HL082547–09, 5T32DK080647–05 (ISF).
Non-standard Abbreviations and Acronyms
- Kir2.1
Inwardly rectifying K+ channel 2.1
- FIV
Flow-induced vasodilation
- NO
Nitric oxide
- eNOS
Endothelial nitric oxide synthase
- ECs
Endothelial cells
- apoE−/−
Apolipoprotein E knockout
- HS
Heparan sulfate
- SNP
Sodium nitroprusside
- DN
Dominant negative
- AV
Adenovirus
- VE-Cad
Vascular endothelial-Cadherin
- Em
Empty
- HA
Hemagglutinin
- SK
Small Ca2+ activated K+ channels
- IKir
Kir current density
- GAGs
Glycosaminoglycans
- MβCD
Methyl-β-cyclodextrin
- LNAME
L-NG-Nitroarginine methyl ester
- LDL
Low density lipoprotein
- HDL
High density lipoprotein
- HAMECs
Human Adipose Microvascular Endothelial Cells
- AFM
Atomic force microscopy
- HEPIII
Heparanase III
Footnotes
Disclosures
None.
References
- 1.Stapleton PA, James ME, Goodwill AG and Frisbee JC. Obesity and vascular dysfunction. Pathophysiology. 2008;15:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grizelj I, Cavka A, Bian JT, Szczurek M, Robinson A, Shinde S, Nguyen V, Braunschweig C, Wang E, Drenjancevic I and Phillips SA. Reduced flow-and acetylcholine-induced dilations in visceral compared to subcutaneous adipose arterioles in human morbid obesity. Microcirculation. 2015;22:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toda N and Okamura T. Obesity impairs vasodilatation and blood flow increase mediated by endothelial nitric oxide: an overview. J Clin Pharmacol. 2013;53:1228–39. [DOI] [PubMed] [Google Scholar]
- 4.Williams IL, Wheatcroft SB, Shah AM and Kearney MT. Obesity, atherosclerosis and the vascular endothelium: mechanisms of reduced nitric oxide bioavailability in obese humans. Int J Obes Relat Metab Disord. 2002;26:754–64. [DOI] [PubMed] [Google Scholar]
- 5.Sharma JN, Al-Omran A and Parvathy SS. Role of nitric oxide in inflammatory diseases. Inflammopharmacology. 2007;15:252–9. [DOI] [PubMed] [Google Scholar]
- 6.Eskens BJ, Leurgans TM, Vink H and Vanteeffelen JW. Early impairment of skeletal muscle endothelial glycocalyx barrier properties in diet-induced obesity in mice. Physiol Rep. 2014;2:e00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee DH, Dane MJ, van den Berg BM, Boels MG, van Teeffelen JW, de Mutsert R, den Heijer M, Rosendaal FR, van der Vlag J, van Zonneveld AJ, Vink H, Rabelink TJ and group NEOs. Deeper penetration of erythrocytes into the endothelial glycocalyx is associated with impaired microvascular perfusion. PLoS One. 2014;9:e96477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tarbell JM and Pahakis MY. Mechanotransduction and the glycocalyx. J Intern Med. 2006;259:339–50. [DOI] [PubMed] [Google Scholar]
- 9.Yen W, Cai B, Yang J, Zhang L, Zeng M, Tarbell JM and Fu BM. Endothelial surface glycocalyx can regulate flow-induced nitric oxide production in microvessels in vivo. PLoS One. 2015;10:e0117133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartosch AMW, Mathews R and Tarbell JM. Endothelial Glycocalyx-Mediated Nitric Oxide Production in Response to Selective AFM Pulling. Biophys J. 2017;113:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahn SJ, Fancher IS, Bian JT, Zhang CX, Schwab S, Gaffin R, Phillips SA and Levitan I. Inwardly rectifying K(+) channels are major contributors to flow-induced vasodilatation in resistance arteries. J Physiol. 2017;595:2339–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Touati S, Meziri F, Devaux S, Berthelot A, Touyz RM and Laurant P. Exercise reverses metabolic syndrome in high-fat diet-induced obese rats. Med Sci Sports Exerc. 2011;43:398–407. [DOI] [PubMed] [Google Scholar]
- 13.Reitsma S, Slaaf DW, Vink H, van Zandvoort MA and oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007;454:345–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Master E, Huang RT, Zhang C, Bogachkov Y, Coles C, Shentu TP, Sheng Y, Fancher IS, Ng C, Christoforidis T, Subbaiah PV, Berdyshev E, Qain Z, Eddington DT, Lee J, Cho M, Fang Y, Minshall RD and Levitan I. Proatherogenic Flow Increases Endothelial Stiffness via Enhanced CD36-Mediated Uptake of Oxidized Low-Density Lipoproteins. Arterioscler Thromb Vasc Biol. 2018;38:64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oh MJ, Zhang C, LeMaster E, Adamos C, Berdyshev E, Bogachkov Y, Kohler EE, Baruah J, Fang Y, Schraufnagel DE, Wary KK and Levitan I. Oxidized LDL signals through Rho-GTPase to induce endothelial cell stiffening and promote capillary formation. J Lipid Res. 2016;57:791–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fancher IS, Ahn SJ, Adamos C, Osborn C, Oh MJ, Fang Y, Reardon CA, Getz GS, Phillips SA and Levitan I. Hypercholesterolemia-Induced Loss of Flow-Induced Vasodilation and Lesion Formation in Apolipoprotein E-Deficient Mice Critically Depend on Inwardly Rectifying K(+) Channels. J Am Heart Assoc. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson AT, Fancher IS, Sudhahar V, Bian JT, Cook MD, Mahmoud AM, Ali MM, Ushio-Fukai M, Brown MD, Fukai T and Phillips SA. Short-term regular aerobic exercise reduces oxidative stress produced by acute in the adipose microvasculature. Am J Physiol Heart Circ Physiol. 2017;312:H896–H906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ko KA, Fujiwara K, Krishnan S and Abe JI. En Face Preparation of Mouse Blood Vessels. J Vis Exp. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sonkusare SK, Dalsgaard T, Bonev AD and Nelson MT. Inward rectifier potassium (Kir2.1) channels as end-stage boosters of endothelium-dependent vasodilators. J Physiol. 2016;594:3271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiesinger A, Peters W, Chappell D, Kentrup D, Reuter S, Pavenstadt H, Oberleithner H and Kumpers P. Nanomechanics of the endothelial glycocalyx in experimental sepsis. PLoS One. 2013;8:e80905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marsh G and Waugh RE. Quantifying the mechanical properties of the endothelial glycocalyx with atomic force microscopy. J Vis Exp. 2013:e50163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Targosz-Korecka M, Jaglarz M, Malek-Zietek KE, Gregorius A, Zakrzewska A, Sitek B, Rajfur Z, Chlopicki S and Szymonski M. AFM-based detection of glycocalyx degradation and endothelial stiffening in the db/db mouse model of diabetes. Sci Rep. 2017;7:15951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prieto D, Contreras C and Sanchez A. Endothelial dysfunction, obesity and insulin resistance. Curr Vasc Pharmacol. 2014;12:412–26. [DOI] [PubMed] [Google Scholar]
- 24.Zhang DX and Gutterman DD. Transient receptor potential channel activation and endothelium-dependent dilation in the systemic circulation. J Cardiovasc Pharmacol. 2011;57:133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olesen SP, Clapham DE and Davies PF. Haemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature. 1988;331:168–70. [DOI] [PubMed] [Google Scholar]
- 26.Fang Y, Mohler ER 3rd, Hsieh E, Osman H, Hashemi SM, Davies PF, Rothblat GH, Wilensky RL and Levitan I. Hypercholesterolemia suppresses inwardly rectifying K+ channels in aortic endothelium in vitro and in vivo. Circ Res. 2006;98:1064–71. [DOI] [PubMed] [Google Scholar]
- 27.Romanenko VG, Fang Y, Byfield F, Travis AJ, Vandenberg CA, Rothblat GH and Levitan I. Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys J. 2004;87:3850–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.dela Paz NG, Melchior B, Shayo FY and Frangos JA. Heparan sulfates mediate the interaction between platelet endothelial cell adhesion molecule-1 (PECAM-1) and the Galphaq/11 subunits of heterotrimeric G proteins. J Biol Chem. 2014;289:7413–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumagai R, Lu X and Kassab GS. Role of glycocalyx in flow-induced production of nitric oxide and reactive oxygen species. Free Radic Biol Med. 2009;47:600–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith PD, Brett SE, Luykenaar KD, Sandow SL, Marrelli SP, Vigmond EJ and Welsh DG. KIR channels function as electrical amplifiers in rat vascular smooth muscle. J Physiol. 2008;586:1147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R and Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–5. [DOI] [PubMed] [Google Scholar]
- 32.Molnar J, Yu S, Mzhavia N, Pau C, Chereshnev I and Dansky HM. Diabetes induces endothelial dysfunction but does not increase neointimal formation in high-fat diet fed C57BL/6J mice. Circ Res. 2005;96:1178–84. [DOI] [PubMed] [Google Scholar]
- 33.Moien-Afshari F, Ghosh S, Elmi S, Rahman MM, Sallam N, Khazaei M, Kieffer TJ, Brownsey RW and Laher I. Exercise restores endothelial function independently of weight loss or hyperglycaemic status in db/db mice. Diabetologia. 2008;51:1327–37. [DOI] [PubMed] [Google Scholar]
- 34.Singh DK, Shentu TP, Enkvetchakul D and Levitan I. Cholesterol regulates prokaryotic Kir channel by direct binding to channel protein. Biochim Biophys Acta. 2011;1808:2527–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenhouse-Dantsker A, Noskov S, Durdagi S, Logothetis DE and Levitan I. Identification of novel cholesterol-binding regions in Kir2 channels. J Biol Chem. 2013;288:31154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hearon CM Jr., Richards JC, Racine ML, Luckasen GJ, Larson DG and Dinenno FA. Amplification of endothelium-dependent vasodilatation in contracting human skeletal muscle: role of KIR channels. J Physiol. 2019;597:1321–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crecelius AR, Luckasen GJ, Larson DG and Dinenno FA. KIR channel activation contributes to onset and steady-state exercise hyperemia in humans. Am J Physiol Heart Circ Physiol. 2014;307:H782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crecelius AR, Richards JC, Luckasen GJ, Larson DG and Dinenno FA. Reactive hyperemia occurs via activation of inwardly rectifying potassium channels and Na+/K+-ATPase in humans. Circ Res. 2013;113:1023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.