

Abstract
Tea containing abundant catechins is a popular non-alcoholic beverage worldwide. Epigallocatechin-3-gallate (EGCG) is the predominately active substance in catechins, exhibiting a wide range of functional properties including cancer suppression, neuroprotective, metabolic regulation, cardiovascular protection, stress adjustment, and antioxidant in various diseases. Autophagy, a basic cell function, participates in various physiological processes which include clearing away abnormally folded proteins and damaged organelles, and regulating growth. EGCG not only regulates autophagy via increasing Beclin-1 expression and reactive oxygen species generation, but also causing LC3 transition and decreasing p62 expression. EGCG-induced autophagy is involved in the occurrence and development of many human diseases, including cancer, neurological diseases, diabetes, cardiovascular diseases, and injury. Apoptosis is a common cell function in biology and is induced by endoplasmic reticulum stress (ERS) as a cellular stress response which is caused by various internal and external factors. ERS-induced apoptosis of EGCG influences cell survival and death in various diseases via regulating IRE1, ATF6, and PERK signaling pathways, and activating GRP78 and caspase proteins. The present manuscript reviews that the effect of EGCG in autophagy and ERS-induced apoptosis of human diseases.
MeSH Keywords: Apoptosis, Autophagy, Endoplasmic Reticulum Stress
Background
Tea is a popular non-alcoholic beverage and the second most consumed drink after water worldwide [1–3]. Depending on the antioxidant levels and fermentation degree, tea is divided into oolong, black, and green teas [2,4]. Currently, people drink tea in their daily life and the average consumption of tea is about 120 mL per day per person worldwide [5–7]. Green tea as one of the most popular and favorite teas, contains a large amount of catechins. Catechins are the main biologically active components in green tea leaves [8,9]. The catechin group includes major phenolic flavonoids, which are epicatechin, epicatechin-3-gallate, epigallocatechin, and epigallocatechin-3-gallate (EGCG). EGCG has the highest biological activity and is the most common component in total catechins [10,11]. Thus, EGCG is a common and important catechin that has been widely studied in the research of green tea [12,13].
EGCG has the highest concentration in green tea. There are 2 aromatic structures, including 3 carbon bridge structures and a hydroxyl group, in the molecule of EGCG [1,14]. EGCG has health benefits including anti-tumor [15], anti-inflammatory [16], anti-diabetes [17], anti-myocardial infarction [18], anti-cardiac hypertrophy [19], anti-atherosclerosis [20], and antioxidant [21] owing to its abundant phenolic hydroxyl groups. These effects are mainly related to low-density lipoprotein (LDL) cholesterol inhibition, NF-κB inhibition, MPO activity inhibition, decreased levels of glucose and glycated hemoglobin in plasma, decreased inflammatory markers, and reduced reactive oxygen species (ROS) generation [22].
The present manuscript reviews the effect of EGCG in autophagy and endoplasmic reticulum stress (ERS)-induced apoptosis of human diseases narratively. The references are searched for EGCG, autophagy, ERS, apoptosis in PubMed database, and those are selected from 2000–2020.
The Role of EGCG in Autophagy
Autophagy
Autophagy is an important cellular mechanism including cell degradation and recovery, and is highly conserved in all eukaryotes [23]. Autophagy participates in normal physiological processes, which includes clearing away abnormally folded proteins and damaged organelles, and regulating growth [24]. Autophagy includes microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy which function to transport components to lysosomes for degradation and recycling [25]. Microautophagy is directly involved in the phagocytosing and digesting tiny components by lysosomes [26,27], while CMA is regulated and controlled by physiological pressure [28,29]. CMA transfers the cargo into lysosome via the lysosomal membranes [30]. In contrast to microautophagy and CMA, macroautophagy captures cellular components with autophagosomes fusing with lysosome to transport them into the cavity for degradation [31,32]. Macroautophagy is the primary and universal process that cells use to clear away damaged components. Thus, the term macroautophagy is used to represent autophagy in this manuscript.
The Role of EGCG in Autophagy and Human Diseases
Cancer
In the process of tumor progression, autophagy has been found to be downregulated. Autophagy plays a role as a barrier in which a normal cell transforms into a cancerous cell since tumor cells can promote high expression of anti-autophagy genes. EGCG-induced autophagy participates in the development process of various human diseases, including cancer, neurological diseases, diabetes, cardiovascular diseases, injury, and infection. During cancer treatment, EGCG-induced autophagy not only directly affects tumor cells, but also enhances the effect of inhibiting tumor development of targeted drugs, chemotherapy, and combination therapies [33].
EGCG directly kills tumor cells via regulating autophagy with reactive oxygen species (ROS) and light chain 3 (LC3) transition [34,35] in many cancers, including primary effusion lymphoma, breast cancer, oral cancer, mesothelioma, and glioblastoma. In primary effusion lymphoma, EGCG significantly inhibited the growth of BABL-1 and BC-1. EGCG induced autophagy to improve cell death through LC3 transition with increasing Beclin-1 expression and formation of acidic vesicular organelles, and through enhancing ROS generation in primary effusion lymphoma cells [34]. EGCG inhibited cell proliferation since EGCG induced autophagy by enhancing Beclin-1, ATG5, and LC3B and promoted mitochondrial depolarization in breast cancer cells. In vivo, 5, 10, and 20 mg kg−1 EGCG reduced the weight of breast cancer by 20%, 31%, and 34% and only 20 mg kg−1 EGCG significantly decreased glucose, lactic acid, and vascular endothelial growth factor (VEGF) levels [36]. The accumulations of a series of epigenetic and genetic alterations resulted in uncontrolled proliferation division in oral squamous cells. The time-dependent maximal inhibitory concentration of EGCG is 52.3 uM in the SSC-4 cells. EGCG treatment (20 uM) inhibited the proliferation through activating autophagy via upregulating ZEB1, WNT11, IGF1R, FAS, BAK, and BAD genes and inhibiting TP53, MYC, and CASP8 genes in SSC-4 human oral squamous cells [37,38]. The main ROS coming from mitochondria played a crucial role in apoptosis and autophagy. EGCG induced autophagy by increasing the LC3-II expression levels and induced apoptosis via inducing ROS in mesothelioma cell lines, but low concentrations failed to induce cell death [35,39]. However, different concentrations of EGCG have different effects on tumor cells. In primary glioblastoma cell cultures, strong autophagy induction and apoptosis induction was observed after 500 μM EGCG, but the signs of cell death of glioblastoma cells during the observation period of 6 days were not detected under 100 nM EGCG. The data showed that EGCG with a low dose might have chemopreventive effects, but without direct cytotoxicity [40].
Autophagy is utilized by cancer cells to protect themselves from risk factors in order to improve their survival in targeted therapy, chemotherapy and combination therapies [41]. In targeted therapy, EGCG-induced autophagy enhanced the sensitivity of tumor cells to target drugs. Recent studies found that drug resistance is triggered by ROS and reduced by antioxidants in lung cancer cells with gefitinib treatment [42,43]. Drug resistance of gefitinib is related to high LC3 expression and the autophagosomes markers in autophagy [44]. EGCG overcame gefitinib resistance by inhibiting gefitinib-induced autophagy with the decreased expressions of ATG5 and LC3-II/I and the increased p62 expression in A549 cells, and EGCG suppressed tumor growth and increased the survival time in A549 xenograft mouse model [45]. P53 is an important gene which participates in autophagy and apoptosis to promote cell survival. Dual therapy with EGCG and P53 siRNA led to activating pro-apoptotic genes and inhibiting pro-autophagy genes in the Hs578T cell model of TNBC, which suggested that this dual therapy could promote treatment effect [46,47]. The study showed that tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) induced apoptosis in a variety of tumor cells by activating apoptosis markers [48]. TRAIL-induced apoptosis was inhibited by EGCG via manipulating autophagy flux and subsequently reducing the number of death receptors in TRAIL-sensitive HCT116 cells [49].
In chemotherapy, cisplatin as one of the most commonly used chemotherapy drugs for oral squamous cell carcinoma and colorectal cancer, and has drug resistance and serious side effects after treatment. EGCG enhances autophagy and apoptosis with activating AGTs, Beclin-1 and LC3B related pathway, and inhibiting AKT/STAT3 pathway in CAR cells [50–52]. EGCG enhances autophagy effect, which is induced by cisplatin and oxaliplatin with increasing autophagosomes, acidic vesicular organelles, and LC3-II protein in colorectal cancer cells [53]. Clinical effect of doxorubicin (DOX), a significant chemotherapeutic drug for several tumors, is seriously limited because increasing the dose can cause severe cardiotoxicity and lowering the dose impairs treatment effect with drug resistance [54]. In osteosarcoma, EGCG not only could reduce autophagy by decreasing SOX2OT variant 7 to enhance the growth inhibition of DOX, but also could reduce partially Notch3/DLL3 to reduce drug-resistance and the stemness of tumor cells [55]. These results indicate that EGCG may be beneficial in cancer treatment via downregulating death markers and activating autophagy markers. However, autophagy inhibition also contributes to killing tumor cells. In the treatment of hepatocellular carcinoma, EGCG and DOX treatment produce a synergistic effect with increasing cell death by 40 to 60%, increasing apoptosis by 45%, downregulating DOX-induced autophagy and inhibiting the expression of autophagic hallmark microtubule-associated protein LC3 compared with DOX alone [54]. In addition, photothermal-chemotherapy also ablates tumor size and improves the anticancer effect through autophagy flux and inducing the formation of autophagosomes, which is induced by the duo of DOX and EGCG in HeLa tumor models [56].
In combination therapies, low-intensity pulsed electric field (PEF) can improve EGCG to affect tumor cells; ultrasound (US) with tumor cells is the application of physical stimulation in cancer therapy. EGCG combined with low-energy US and low-intensity PEF treatment might cause 20% change in the survival of human pancreatic tumor cells after 72 hours. In addition, 20 μM EGCG increased intracellular ROS levels and LC3-II, and inhibited p-Akt in PANC-1 cells. And 100 μM EGCG increased LC3-II, activated caspase-3 and PARP, and reduced p-Akt in HepG2 cells to result in tumor cell death [57–59].
Neurological diseases
Neurological diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD), produce a huge social problem in the world. Both AD and PD are very popular neurodegenerative diseases worldwide [60,61]. Neurodegenerative diseases are caused by various factors, such as the accumulation of α-synuclein in PD and β-amyloid protein in AD with increased pro-apoptotic proteins, inflammation, and oxidative stress. EGCG has been utilized as the drug for neurodegenerative diseases with extensive biological and pharmacological activities [62,63].
In neurons, autophagy can protect neuronal cells from stress to improve cell survival [64]. EGCG protected neuronal cells against human viruses by inhibiting cytochrome c and Bax translocations, and reducing autophagy with increased LC3-II expression and decreased p62 expression [65]. In addition, EGCG restored autophagy in the mTOR/p70S6K pathway to weaken memory and learning disorders induced by CUMS [66]. EGCG provided the neuroprotective effect in subarachnoid hemorrhage (SAH). EGCG induced normal autophagic flux, regulated Beclin-1 and Atg5 and promptly eliminated damaged mitochondria after SAH. Finally, EGCG increased the neurological scores through inhibiting cell death [67]. Another study found that under EGCG treatment, [Ca2+]m and [Ca2+]i expressions were reduced and oxyhemoglobin-induced mitochondrial dysfunction lessened. Finally, EGCG could rescue autophagy via regulating mRNA expressions of Becn-1, LC3B, and Atg5 after SAH. Therefore, EGCG can protect neuronal cells from or attenuate external damage through the autophagy pathway [68].
Diabetes and its complications
Diabetes is a common metabolic disease characterized by hyperglycemia caused by insulin damage. Clinically, the main and common symptoms are polyuria, blurred vision, and weight loss. Persistent hyperglycemia leads to autonomic dysfunction, foot ulcers, renal failure, and blindness. Flavonoids are beneficial through improving the secretion of insulin, regulating glucose metabolism of hepatocytes, inhibiting the apoptosis of pancreatic β-cells, attenuating oxidative stress, inflammation and insulin resistance, and improving glucose uptake in muscle cells. And EGCG is a member of flavonoids family [69,70].
In GK (Goto-Kakizaki) rats, EGCG enhanced glucose metabolism, inhibited mitochondrial loss and dysfunction, decreased oxidative stress, and reduced autophagy by downregulating the JNK-p53 and ROS-ERK pathways in muscle cells [71]. EGCG-induced autophagy also reduced retinal damage and relieved myocardial mitochondrial dysfunction in chronic complications of diabetes. EGCG protected Müller cells under high glucose from apoptosis through activating autophagy and restoring degradation. EGCG decreased retinal damage and inhibited the reactive gliosis of Müller cells in diabetic retinopathy models [72]. FoxO1 plays as an important transcription factor in insulin signaling. In H9c2 cardiomyoblasts, FoxO1 induced autophagy through the ROS pathway to regulate glucose metabolism, which is inhibited by EGCG [73]. EGCG improved mitochondrial function by increasing autophagy and upstream FoxO factors in the myocardium of diabetic models [74]. These results showed that EGCG would be a potential drug for regulating glucose metabolism and myocardial damage involving diabetes.
Cardiovascular diseases
Ischemic heart disease is a common cardiovascular disease with a high mortality rate. Reperfusion after myocardial ischemia can easily cause injury. EGCG pretreatment reduced autophagy induced by ischemia/reperfusion (I/R), increased cell number, and decreased myocardial infarction area. EGCG protected I/R via inhibiting autophagy through activating PI3K/Akt signals, decreasing Beclin-1, and improving MiR-384 in H9c2 cell lines [75]. Another study found that EGCG post-treatment significantly reduced CK-MB levels and LDH release, decreased myocardial infarct area, reduced apoptosis number, and retained partial heart function in I/R injury models. In addition, EGCG post-treatment decreased I/R injury by rescuing autophagy and reducing apoptosis with reduced cleaved caspase-3, p62, Atg5, Beclin-1, and the ratio of LC3-II/LC3-I, upregulation of PI3K, cathepsin D, endothelial nitric oxide synthase, and Akt [76]. Recent studies have shown autophagy is related to lipid metabolism, which is induced by cholesterol efflux and lipolysis in foam cells. Thence, activating autophagy promoted cholesterol outflow to hinder the formation of advanced atherosclerotic plaques in foam cells. Combined EGCG and oligomeric proanthocyanidins treatment not only activated autophagy, but also stimulated cholesterol efflux via regulating class III PI3K/Beclin-1, implying that is a potential therapeutic method for atherosclerosis [77].
Injury and bacterial infection
The liver, an important metabolic organ, is closely related to autophagy. EGCG increased autophagy through promoting lysosomal acidification and improving the formation of autophagosomes in the liver. EGCG alleviated liver injury via regulating apoptosis and autophagy by inhibiting IL-6/JAKs/STAT3/BNIP3 in ConA-induced hepatitis models [78,79]. EGCG promoted cell survival via upregulating autophagy and shifting the balance of mTOR-AMPK pathways [80]. EGCG treatment inhibited UVB-induced autophagy through reducing autophagosomes and LC3-II, and activating mTOR signals in the age-related macular degeneration. Furthermore, EGCG reduced the toxic effects of UVB on retinal pigment epithelial cells in an autophagic pathway [81].
In bacterial infections, EGCG limited Burkholderia cenocepacia metabolism by enhancing autophagy and inhibiting spread in cystic fibrosis, and promoted cystic fibrosis transmembrane conductance regulator (CFTR) expression [82]. Combined EGCG and 5-AZA treatment could inhibit Legionella infection by rescuing the gene expression of autophagy in infected macrophages [83].
The role of EGCG in endoplasmic reticulum stress (ERS)-induced apoptosis
Endoplasmic reticulum (ER) is a crucial organelle which can synthesize, fold, and secret various proteins in eukaryotic cells. About 30% of cellular proteins are synthesized, folded, processed, and modified to form active functional proteins, including most secreted proteins, membrane-bound proteins, and integrated membrane proteins. The new synthesized proteins encapsulated in vesicles leaves the ER and is transported to Golgi apparatus, then is directed to the inner membrane system or secreted outside the cell. Also, ER participates in some cellular functions, including calcium ion storage, gluconeogenesis, lipid and cholesterol synthesis, and formation of autophagic vesicles. When receiving endogenous or exogenous stimuli including lack of molecular chaperone or cellular energy or Ca2+, disulfide reduction, protein mutations and redox homeostasis, the function of folding protein in ER is disordered. A large number of misfolded or unfolded proteins accumulate in the cavity and cause a series of subsequent reactions called endoplasmic reticulum stress (ERS) [84–86].
ERS-induced apoptosis pathway
ERS is one of common stress responses, which induced apoptosis and interferes with cellular homeostasis. Upon ERS, cells will activate many adaptive functions to respond to changes in protein-folding, called unfolded protein response (UPR). UPR protects cells from stress by improving the ability of synthesizing new proteins and improving protein degradation through autophagy. When ERS is continuous and robust, the amount of the protein in the ER exceeds greatly its fold ability. This pathway clears away damaged cells through apoptosis, suggesting the mechanism controlling cell survival might depend on the intensity and duration of stress stimulation. UPR is mainly activated by 3 signaling pathways: protein kinase RNA like ER kinase (PERK), activating transcription factor-6 (ATF6), and inositol requiring protein 1 (IRE1) [84,86].
The accumulation of unfolded proteins stimulates autophosphorylation and oligomerization of IRE1α in the ER. IRE1α, TRAF2, and ASK1 form a complex to activate JNK which promotes apoptosis or autophagy [87]. After being transferred from ER to the Golgi apparatus, ATF6 is cleaved by Site-1 and Site-2 proteases. The N-terminal cytosolic domain of cleaved ATF6 combined with ERS and cAMP response elements, and is transferred into the nucleus to activate target genes such as X-box binding protein 1 (XBP1), C/EBP homologous protein (CHOP), immunoglobulin heavy chain binding protein (BIP), and 78 kDa glucose regulated protein (GRP78). ATF6 directly regulates apoptosis and autophagy through CHOP and XBP-1 [88]. The function of PERK is that attenuates the translation of mRNA under ERS and prevented synthesized proteins from flowing into the already stressed ER region. The activation of PERK inhibits protein translation through eIF2α phosphorylation and allows the specialized translation of transcripts including the vital sensor ATF4. ATF4 could promote apoptosis by degrading apoptosis protein inhibitors and autophagy by regulating ATG genes under prolonged ERS [84,89]. In addition, ER molecular chaperones including GRP78, BIP, GRP94, and PDI, participate in folding, assembling, and transporting of secreted and membrane proteins. See Table 1.
Table 1.
The role of EGCG in autophagy and human diseases.
| Disease model | Dose | EGCG Effect | References |
|---|---|---|---|
| Cancer (primary effusion lymphoma cells) | 20 μg/mL | EGCG increased LC3 transition, formation of acidic vesicular organelles, and ROS generation | 34 |
| Cancer (4T1 breast cancer cells and breast cancer xenograft) | 20 μM; 20 mg kg−1 |
EGCG enhanced Beclin-1, ATG5, LC3B, and mitochondrial depolarization; EGCG reduced the weight, glucose, lactic acid, and VEGF levels in the breast cancer xenograft |
36 |
| Cancer (SSC-4 human oral squamous cells) | 20 μM | EGCG enhanced ZEB1, WNT11, IGF1R, FAS, BAK, and BAD genes and inhibited TP53, MYC, and CASP8 genes | 38 |
| Cancer (human mesothelioma cells) | 200 μM | EGCG increased the LC3-II expression levels and induced ROS | 39 |
| Cancer (glioblastoma cells) | 100 nM; 500 μM | 500 μM EGCG exhibited strong autophagy and apoptosis induction, but 100 nM had no effect. | 40 |
| Cancer (human lung A549 adenocarcinoma cells and A549 xenograft mouse model) | 80 μM; 200 mg kg−1 |
EGCG inhibited LC-3 II/I ratio and AGT5, and improved p62; EGCG suppressed tumor growth and increased the survival time |
45 |
| Cancer (triple negative breast cancer cells) | 40 nmol | EGCG p53 siRNA, and EGCG activated pro-apoptotic genes and inhibited pro-survival genes, autophagy, and cell network formation | 46 |
| Cancer (HCT116 human colorectal cancer cells) | 20 μM | EGCG decreased p62 and LC3 II/I ratio to active autophagy; Inhibition of autophagy sensitized HCT116 to TRAIL-induced apoptosis on EGCG treatment |
49 |
| Cancer (oral cancer CAR cells) | 50 μM | EGCG activated ATGs, Beclin-1, and LC3B related pathway, and inhibited AKT/STAT3 signal pathway | 52 |
| Cancer (human colorectal cancer cells) | 100 μM | EGCG increased autophagosomes, acidic vesicular organelles, and LC3-II protein | 53 |
| Cancer (hepatoma hep3B cells) | 10 μg/mL | EGCG increased cell death and inhibited LC3 | 54 |
| Cancer (osteosarcoma cells) | 20 μg/mL | EGCG decreased SOX2OT variant 7 and inactivate Notch3/DLL3 signaling | 55 |
| Cancer (mouse HeLa tumor model) | 25 mg kg−1 | EGCG induced autophagic flux and accelerated the formation of autophagosomes | 56 |
| Cancer (human pancreatic cancer PANC-1 cells and HepG2 cells) | 20, 100 μM | 20 μM EGCG increased LC3-II and reduced p-Akt in PANC-1 cells; 100 μM EGCG increased LC3-II, activated caspase-3, and PARP, and reduced p-Akt in HepG2 cells |
59 |
| Neurological diseases (primary neuron cells) | 10 μM | EGCG increased LC3-II expression levels and decreased p62 levels | 65 |
| Neurological diseases (CUMS rats) | 25 mg kg−1 | EGCG decreased p62 via mTOR/p70S6K pathway and decreased AβP1–42 levels | 66 |
| Neurological diseases (PC12 cells) | 50 μM | EGCG inhibited Ca2+ influx, protected mitochondrial function, and downregulated Beclin-1 and AGT5 | 67 |
| Neurological diseases (PC12 cells) | 50 μM | EGCG increased LC3B and downregulated Beclin-1 | 68 |
| Diabetes (diabetic GK rats) | 100 mg kg−1 | EGCG reduced Beclin-1 and DRP1, and reversed the phosphorylation of JNK | 71 |
| Diabetes (retinal Müller cells) | 20 μM | EGCG increased LC3-II and beclin-1, decreased P62 and improved lysosomal activity | 72 |
| Diabetes (H9c2 cardiomyoblasts) | 20 μM | EGCG restored ROS production and suppressed cytoplasmic acetylation of FoxO1 | 73 |
| Diabetes (diabetic GK rats) | 100 mg kg−1 | EGCG enhanced autophagy signaling molecules and the FoxOs abundance | 74 |
| Cardiovascular Diseases (H9c2 cardiomyocytes) | 25 μM | EGCG inhibited autophagy through activating PI3K/Akt signals, increased miR-384 and attenuated Beclin-1 levels | 75 |
| Cardiovascular Diseases (Sprague-Dawley rats) | 10 mg kg−1 | EGCG decreased LVEDP, CK-MB, LDH, C3-II/LC3-I ratio, Beclin-1, Atg5 and p62, and increased LVSP | 76 |
| Cardiovascular Diseases (human monocytic THP 1 cell) | 80 μM | EGCG activated autophagy via upregulating LC3B and AGT5 and stimulated cholesterol efflux via regulating class III PI3K/Beclin-1 | 77 |
| Injury (Balb/c mice) | 30 mg kg−1 | EGCG inhibited the production of TNF-α, IL-6, IFN-γ, and IL-1β, and downregulated hepatocyte apoptosis and autophagy via IL-6/JAKs/STAT3/BNIP3 pathway | 78 |
| Injury (human retinal pigment epithelial cells) | 50 μM | EGCG inhibited UVB-induced autophagy through reducing autophagosomes and LC3-II, and activating mTOR signals | 81 |
| Bacterial Infection (cystic fibrosis) | 25 μg/mL | EGCG improved B. cenocepacia clearance by enhancing autophagy and macrophage survival, and inhibiting spread in cystic fibrosis, and promoted CFTR | 82 |
| Bacterial Infection (infected macrophages) | 50 μM | EGCG inhibited Legionella infection by rescuing autophagy genes (Atg5–Atg12 protein complex, LC3) in infected macrophages | 83 |
EGCG – epigallocatechin-3-gallate; ROS – reactive oxygen species; VEGF – vascular endothelial growth factor; B. cenocepaci – Burkholderia cenocepacia CFRT – cystic fibrosis transmembrane conductance regulator.
The Role of EGCG in ERS-Induced Apoptosis and Human Diseases
Cancer
Apoptosis is a common cell function in biology and is induced by ERS as a cellular stress response which is caused by various internal and external factors. EGCG directly inhibits tumor cell growth via ERS-induced apoptosis in different cancers. In colorectal cancer cells, EGCG induced ERS in HT-29 cells by upregulating BiP, PERK, phosphorylation eIF2α, ATF4, and IRE1α. And apoptosis was induced by increased the activity of caspase-3/7 after EGCG treatment [90,91]. EGCG efficiently inhibited glucosidase II, which participates in quality control and glycoprotein processing in the ER of rat liver microsomes. EGCG interfered with protein processing owing to inhibiting glucosidase II in liver microsomes of hepatoma cells, and ERS induced incomplete UPR with main pro-apoptotic components, including increased eIF2α phosphorylation, cleavage of procaspase-12, induction of CHOP/GADD153, and depletion of ER calcium [92,93]. GRP78 can trigger UPR via activating XBP1 and CHOP in the endoplasmic reticulum to restore cell homeostasis. MMe cells with EGCG treatment improved GRP78 expression in the endoplasmic reticulum, and induced EDEM, CHOP, XBP1, and ATF4 expressions, and increased the activity of caspase-3 and caspase-8. GRP78 accumulation converted UPR of MMe cells into pro-apoptotic ERS [94,95]. Furthermore EGCG only induced ERS related apoptosis in tumor cells and was not effective for normal cells. EGCG promoted ROS production and induced apoptosis via p38 MAPK phosphorylation, caspase-8 activation, and proteolytic cleavage of Bid and JNK pathway activation in glioblastoma cells. However, EGCG did not promote apoptosis in astrocytes [96].
EGCG not only has a direct effect on tumors through ERS-induced apoptosis, but also promotes the sensitivity of tumor cells to therapeutic drugs or methods. siRNA downregulation of GRP78 and EGCG treatment improved apoptosis induced by celecoxib, thereby activating CHOP, caspase-4, GRP78, and IRE-1α in urothelial carcinoma cells [97]. During chemotherapy, EGCG enhanced chrysin-induced apoptosis by overcoming GRP78 expression and enhancing caspase-7 and poly polymerase cleavage in human hepatoma cells [98]. EGCG alone wasn’t beneficial for survival, but significantly promoted the current treatment effect of temozolomide by reducing GRP78 expression and increasing CHOP expression. And life span extension was significantly higher under combination therapy compared with temozolomide alone [99]. EGCG enhanced the activation of ERS-induced apoptosis markers including PARP cleavage, caspase-7 and JNK phosphorylation in breast cancer with vinblastine and Taxol treatment. Inhibition of caspase-7 and JNK eliminated the sensitivity to vinblastine and Taxol in breast cancer with EGCG treatment [100].
However, another research reported that EGCG can antagonize chemotherapy drugs to prevent tumor cell apoptosis. EGCG effectively prevented tumor cell death mediated by BZM in glioblastoma and multiple myeloma. EGCG could only significantly antagonize the function of boronic acid proteasome inhibitors, but not the function of non-boronic acid proteasome inhibitors. Since EGCG directly reacted with BZM to block the effect of BZM, BZM could not trigger tumor cell death through activation of ERS and caspase-7. These results indicated that drinking tea would be banned during BZM therapy owing to antagonistic effect of EGCG on BZM [101,102].
Neurological diseases
Previous studies have found that EGCG protects central nervous system from diseases with its anti-inflammatory and antioxidant properties. Recent studies showed that EGCG reduced ERS-induced apoptosis by downregulating cleaved caspase-3 and caspase-12, CHOP, and GRP78 in a dose-dependent manner in SH-SY5Y Cells. Simultaneously EGCG inhibited neuronal apoptosis with reducing ER abnormal ultrastructural swelling and downregulating ERS-associated proteins in APP/PS1 transgenic mice. In conclusion, EGCG decreased the neurotoxicity via decreasing ERS related apoptosis in the AD [103,104]. In transient focal cerebral ischemia rat models, EGCG treatment after ischemia inhibited ERS with decreasing the expression of glucose-regulated caspase-12, CHOP, and GRP78, and improved the neurological status via inhibiting TRPC6 proteolysis and activating CREB in the MEK/ERK pathway [105]. In familial amyloidotic polyneuropathy mice models, EGCG inhibited 50% deposition of transthyretin toxic that aggregates along peripheral nervous system and gastrointestinal tract system. In addition, EGCG significantly reduced the expression of the markers related non-fibrillar transthyretin deposition, including BiP, the phosphorylated eIF2a, protein oxidation marker-3-nitrotyrosine and death receptor Fas [106].
Diabetes and its complications
The β cells of Langerhans islets absorb glucose by GLUT2, which results in the secretion of insulin to maintain glucose homeostasis. EGCG promoted the secretion of insulin and glucose tolerance, reduced the number of Langerhans pathological islets and ERS markers of the islet, increased area and number of islets, and increased the pancreatic endocrine area of db/db mice [107,108]. A-type-EGCG-dimer is beneficial for the body and affect glucose metabolism in the liver. A-type EGCG dimer prevented insulin resistance and hyperglycemia by inhibiting ERS-induced apoptosis, decreasing the levels of G6Pase and PEPCK, and the activations of ATF4, p-JNK, p-IRE1, and p-PERK in rat liver [109]. Podocytes participate in maintaining the integrity of the glomerular filtration barrier and preventing the production of proteinuria. Podocyte injury can affect the development and prognosis of diabetic nephropathy. EGCG attenuated apoptosis of glucose-induced podocyte through inhibiting ERS with attenuating the expressions of caspase-12, p-PERK, and GRP78 in mouse podocytes [110]. It has been found that activating nuclear factor erythroid 2 related factor 2 (NRF2) can attenuate diabetic testicular damage in rodents. EGCG reduced apoptosis of testicular cell by ERS, oxidative damage, and inflammation, and activated the expression of NRF2 in diabetic mice [111].
Cardiovascular diseases
Endothelial dysfunction is a common cause of cardiovascular diseases, which is affected by many factors including oxidative stress, renin-angiotensin system, oxidized low density lipoprotein, and homocysteine. ERS related NOD-like receptor pyrin domain containing-3 (NLRP3) and thioredoxin-interacting protein (TXNIP) signals are important factors in the endothelial dysfunction and induce inflammation and cell death by producing IL-1β. EGCG is a member of flavonoids groups and has beneficial effects on cardiovascular diseases. One study showed that EGCG inhibited ROS levels and the activation of NLRP3 and TXNIP inflammasome, resulting in decreased IL-1β expression. Simultaneously EGCG reduced cell apoptosis by inhibiting the activity of caspase-3 and restoring mitochondrial membrane potential. EGCG inhibited the activation of NLRP3 and TXNIP inflammasome by regulating the activity of AMPK to protecting endothelial cells from inflammatory and apoptosis [112,113].
Adverse reactions and injury
Cisplatin (CP) is widely used in the chemotherapy for various cancers and is limited high-dose treatment due to its serious adverse reactions, especially nephrotoxicity. CP causes acute renal injury via renal tubular dysfunction in many patients. In CP-induced nephrotoxicity mice, EGCG reduced immunohistochemical damage and biochemical factors, and decreased the expression of phosphorylated ERK, GRP78, and caspase-12. EGCG reduced renal apoptosis by inhibiting ERS [114–116]. Partial bladder outlet obstruction (pBOO) caused by many etiologies, including benign prostatic hyperplasia, cystocele, posterior urethral valves, bladder stones, and urethral stricture, is a common disease of the urinary tract. After the female Sprague-Dawley rats received EGCG treatment, bladder injury and dysfunction was significantly promoted by inhibiting inflammation and ERS-related apoptosis with increased the expression of caspase-12, CHOP, and cyclooxygenase-2 at 48 hours and 30 days [117,118]. ERS-induced apoptosis closely correlated with the progression of age-related macular degeneration. ERS caused the accumulation of misfolded proteins and activated UPR to promote cell survival. EGCG inhibited ERS-mediated apoptosis via downregulating cleaved caspase-12, cleaved caspase-3, cleaved PARP, IRE1α, ERO1α, PERK, CHOP, GRP78, and phosphorylation at ser9 of GSK3β, and upregulating the expression of phosphorylation ser380 of PTEN and ser473 of AKT in retinal pigment epithelial cells [119,120]. See Table 2.
Table 2.
The role of EGCG in ERS-induced apoptosis and human diseases.
| Disease model | Dose | EGCG Effect | References |
|---|---|---|---|
| Cancer (colorectal cancer cells) | 125, 250, 500, 1000 μM | EGCG upregulated BiP, PERK, phosphorylation eIF2α, ATF4, and IRE1α and increased caspase 3/7 activity | 90 |
| Cancer (mouse hepatoma cells) | 100 μM | EGCG inhibited glucosidase II, increased eIF2α phosphorylation, cleavage of procaspase-12, induction of CHOP/GADD153, and depletion of ER calcium | 92 |
| Cancer (MMe cells) | 5, 10, 50, 100 μM | EGCG improved GRP78, induced EDEM, CHOP, XBP1, ATF4 expressions, and increased the activity of caspase 3 and 8 | 94 |
| Cancer (human glioblastoma T98G and U87MG cells) | 50 μM | EGCG promoted ROS production, induced p38 MAPK phosphorylation, caspase-8 activation, proteolytic cleavage of Bid, and activated JNK pathway | 96 |
| Cancer (human urothelial carcinoma cells) | 10, 20, 33.3, 40 μM | EGCG improved apoptosis by activating CHOP, caspase 4, GRP78, and IRE-1α | 97 |
| Cancer (hepatoma cells) | 20 μM | EGCG overcame chrysin-induced GRP78 expression and potentiated the activation of caspase-7 by chrysin. | 98 |
| Cancer (glioblastoma cells) | 20 μM | EGCG promoted the current treatment effect of temozolomide by reducing GRP78 and upregulating CHOP | 99 |
| Cancer (breast cancer cells) | 10 μM EGCG | EGCG induced PARP cleavage, caspase 7 activation and JNK phosphorylation in breast cancer with vinblastine and Taxol treatment | 100 |
| Cancer (multiple myeloma cells and glioblastoma cells) | 10, 20 μM | EGCG directly reacted with BZM to block the effect of BZM and EGCG prevented proteasome inhibition and ER stress induction | 101 |
| Neurological Diseases (SH-SY5Y cells) | 5, 10, 20 μM | EGCG reduced ERS-induced apoptosis by downregulating cleaved caspase 3 and caspase 12, CHOP, GRP78 | 103 |
| Neurological Diseases (neuronal cells) | 25 μM | EGCG inhibited ERS with decreasing caspase-12, CHOP, and GRP78, and improved the neurological status via inhibiting TRPC6 proteolysis and activating CREB via the MEK/ERK pathway | 105 |
| Neurological Diseases (familial amyloidotic polyneuropathy mice models) | 100 mg kg−1 | EGCG reduced BiP, the phosphorylated eIF2α, protein oxidation marker-3-nitrotyrosine and death receptor Fas | 106 |
| Diabetes (db/db mice) | 10 g kg−1 | EGCG promoted the secretion of insulin and glucose tolerance, reduced the number of Langerhans pathological islets and ERS markers of the islet, increased area and number of islets, and increased the pancreatic endocrine area | 108 |
| Diabetes (Wistar rats) | 200 mg kg−1 | A-type EGCG dimer prevented insulin resistance and hyperglycemia by inhibiting ERS-induced apoptosis, decreasing the levels of G6Pase and PEPCK, and the activities of ATF4, p-JNK, p-IRE1 and p-PERK | 109 |
| Diabetes (podocytes) | 20 μM | EGCG attenuated apoptosis of glucose-induced podocyte through inhibiting ERS with attenuating the expressions of caspase-12, p-PERK and GRP78 | 110 |
| Diabetes (diabetic mice) | 100 mg kg−1 | EGCG reduced apoptosis of testicular cell by ERS, oxidative damage, and inflammation, and activated the expression of NRF2 | 110 |
| Cardiovascular Diseases (endothelial cells) | 10 μM | EGCG enhanced AMPK phosphorylation, suppressed ROS production, TXNIP induction, NLRP3 inflammasome activation and attenuated mitochondrial cell apoptosis. | 112 |
| Adverse Reactions (C57/BL6 mice) | 100 mg kg−1 | EGCG attenuated the CP-induced renal dysfunction, kidney tubular damage, and decreased the expression of phosphorylated ERK, GRP78, and caspase 12 | 114 |
| Injury (Sprague-Dawley rats) | 4.5 mg kg−1 | EGCG improved pBOO-induced histologic changes, bladder dysfunction, and the overexpression of cyclooxygenase-2, CHOP, and caspase-12 | 117 |
| Injury (primary retinal pigment epithelial cells) | 10 μM | EGCG inhibited ERS-mediated apoptosis via downregulating cleaved caspase-12, cleaved caspase-3, cleaved PARP, IRE1α, ERO1α, PERK, CHOP, GRP78, and phosphorylation at ser9 of GSK3β, and upregulating the expression of phosphorylation ser380 of PTEN and ser473 of AKT | 119 |
EGCG – epigallocatechin-3-gallate; ROS – reactive oxygen species; VEGF – vascular endothelial growth factor; CFRT – cystic fibrosis transmembrane conductance regulator.
Conclusions
Autophagy and apoptosis closely correlated with the progression of human diseases. As EGCG is associated with apoptosis and autophagy, EGCG exhibits a wide range of functional properties, including cancer prevention, neuroprotection, anti-cardiovascular effect, and anti-diabetic effect (Figure 1). EGCG not only regulates autophagy via increasing Beclin-1 expression and ROS generation, but also causing LC3 transition and decreasing p62 expression. ERS-induced apoptosis of EGCG influences cell survival and death in various diseases via regulating IRE1, ATF6, and PERK signaling pathways, and activating GRP78 and caspase proteins. EGCG-induced autophagy and ERS-induced apoptosis of EGCG is involved in human diseases, including cancer, neurological diseases, diabetes, cardiovascular diseases, and injury.
Figure 1.
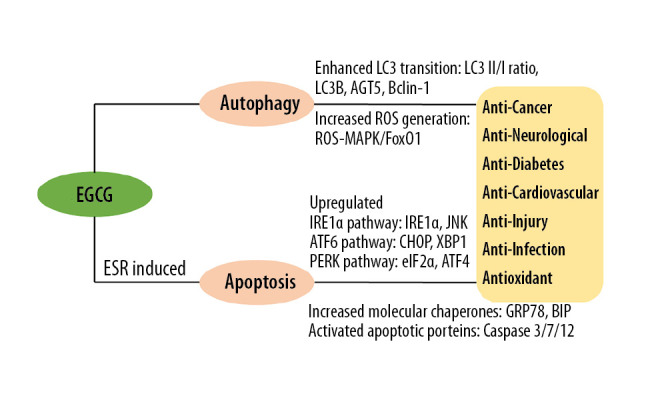
Mechanism of EGCG in autophagy and ERS-induced apoptosis in human disease. EGCG – epigallocatechin-3-gallate; ERS – endoplasmic reticulum stress; ROS – reactive oxygen species.
As natural compounds can interact with the complex network of interlinked physiological processes, these findings of EGCG obtained from a number of experimental models and cell lines cannot be directly inferred into humans. Humans and other organisms have conservative mechanisms, and these mechanisms were inherited in the course of biological evolution. But the models and cell lines of EGCG are not perfectly matched owing to human complex physiology [3,121]. In addition, EGCG metabolism in the intestine and the circulatory systems of these models were unique in different studies [122]. For example, there are different effects of different concentration EGCG in different model organisms for cancer therapy in the manuscript. Future studies should focus on humans to determine whether the same benefits in the models can be replicated. Clinical trials will be conducted to determine the optimal dosage of EGCG to achieve the maximum health benefits of humans, which will be the novel and potential leap in basic and clinical research. Researchers need to make more efforts to convert large amounts of preclinical data into effective human treatment methods.
Footnotes
Source of support: Departmental sources
References
- 1.Sharifi-Rad M, Pezzani R, Redaelli M, et al. Preclinical pharmacological activities of epigallocatechin-3-gallate in signaling pathways: An update on cancer. Molecules. 2020;25:E467. doi: 10.3390/molecules25030467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Graham HN. Green tea composition, consumption, and polyphenol chemistry. Prev Med. 1992;21:334–50. doi: 10.1016/0091-7435(92)90041-f. [DOI] [PubMed] [Google Scholar]
- 3.Prasanth MI, Sivamaruthi BS, Chaiyasut C, Tencomnao T. A review of the role of green tea (camellia sinensis) in antiphotoaging, stress resistance, neuroprotection, and autophagy. Nutrients. 2019;11:E474. doi: 10.3390/nu11020474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan EW, Soh EY, Tie PP, Law YP. Antioxidant and antibacterial properties of green, black, and herbal teas of Camellia sinensis. Pharmacognosy Res. 2011;3:266–72. doi: 10.4103/0974-8490.89748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vuong QV. Epidemiological evidence linking tea consumption to human health: A review. Crit Rev Food Sci Nutr. 2014;54:523–36. doi: 10.1080/10408398.2011.594184. [DOI] [PubMed] [Google Scholar]
- 6.Wierzejska R. Tea and health – a review of the current state of knowledge. Przegl Epidemiol. 2014;68:501–6. 595–99. [PubMed] [Google Scholar]
- 7.Hayat K, Iqbal H, Malik U, et al. Tea and its consumption: benefits and risks. Crit Rev Food Sci Nutr. 2015;5:939–54. doi: 10.1080/10408398.2012.678949. [DOI] [PubMed] [Google Scholar]
- 8.Zink A, Traidl-Hoffmann C. Green tea in dermatology – myths and facts. J Dtsch Dermatol Ges. 2015;13:768–75. doi: 10.1111/ddg.12737. [DOI] [PubMed] [Google Scholar]
- 9.Pae M, Wu D. Immunomodulating effects of epigallocatechin-3-gallate from green tea: Mechanisms and applications. Food Funct. 2013;4:1287–303. doi: 10.1039/c3fo60076a. [DOI] [PubMed] [Google Scholar]
- 10.Sang S, Lambert JD, Ho CT, Yang CS. The chemistry and biotransformation of tea constituents. Pharmacol Res. 2011;64:87–99. doi: 10.1016/j.phrs.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 11.Min KJ, Kwon TK. Anticancer effects and molecular mechanisms of epigallocatechin-3-gallate. Integr Med Res. 2014;3:16–24. doi: 10.1016/j.imr.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagle DG, Ferreira D, Zhou YD. Epigallocatechin-3-gallate (EGCG): Chemical and biomedical perspectives. Phytochemistry. 2006;67:1849–55. doi: 10.1016/j.phytochem.2006.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mereles D, Hunstein W. Epigallocatechin-3-gallate (EGCG) for clinical trials: More pitfalls than promises? Int J Mol Sci. 2011;12:5592–603. doi: 10.3390/ijms12095592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Botten D, Fugallo G, Fraternali F, Molteni C. Structural properties of green tea catechins. J Phys Chem B. 2015;119:12860–67. doi: 10.1021/acs.jpcb.5b08737. [DOI] [PubMed] [Google Scholar]
- 15.Fujiki H, Watanabe T, Sueoka E, et al. Cancer prevention with green tea and its principal constituent, EGCG: from early investigations to current focus on human cancer stem cells. Mol Cells. 2018;41:73–82. doi: 10.14348/molcells.2018.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumazoe M, Nakamura Y, Yamashita M, et al. Green tea polyphenol epigallocatechin-3-gallate suppresses toll-like receptor 4 expression via up-regulation of E3 ubiquitin-protein ligase RNF216. J Biol Chem. 2017;292:4077–88. doi: 10.1074/jbc.M116.755959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li T, Liu J, Zhang X, Ji G. Antidiabetic activity of lipophilic (−)-epigallocatechin-3-gallate derivative under its role of α-glucosidase inhibition. Biomed Pharmacother. 2007;61:91–96. doi: 10.1016/j.biopha.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Othman AI, Elkomy MM, El-Missiry MA, Dardor M. Epigallocatechin-3-gallate prevents cardiac apoptosis by modulating the intrinsic apoptotic pathway in isoproterenol-induced myocardial infarction. Eur J Pharmacol. 2017;794:27–36. doi: 10.1016/j.ejphar.2016.11.014. [DOI] [PubMed] [Google Scholar]
- 19.Sheng R, Gu ZL, Xie ML, et al. EGCG inhibits proliferation of cardiac fibroblasts in rats with cardiac hypertrophy. Planta Med. 2009;75:113–20. doi: 10.1055/s-0028-1088387. [DOI] [PubMed] [Google Scholar]
- 20.Xu X, Pan J, Zhou X. Amelioration of lipid profile and level of antioxidant activities by epigallocatechin-gallate in a rat model of atherogenesis. Heart Lung Circ. 2014;23:1194–201. doi: 10.1016/j.hlc.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 21.Campanella L, Bonanni A, Tomassetti M. Determination of the antioxidant capacity of samples of different types of tea, or of beverages based on tea or other herbal products, using a superoxide dismutase biosensor. J Pharm Biomed Anal. 2003;32:725–36. doi: 10.1016/s0731-7085(03)00180-8. [DOI] [PubMed] [Google Scholar]
- 22.Eng QY, Thanikachalam PV, Ramamurthy S. Molecular understanding of Epigallocatechin gallate (EGCG) in cardiovascular and metabolic diseases. J Ethnopharmacol. 2018;210:296–310. doi: 10.1016/j.jep.2017.08.035. [DOI] [PubMed] [Google Scholar]
- 23.Marshall RS, Vierstra RD. Autophagy: The master of bulk and selective recycling. Annu Rev Plant Biol. 2018;69:173–208. doi: 10.1146/annurev-arplant-042817-040606. [DOI] [PubMed] [Google Scholar]
- 24.Ravanan P, Srikumar IF, Talwar P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017;188:53–67. doi: 10.1016/j.lfs.2017.08.029. [DOI] [PubMed] [Google Scholar]
- 25.Yang Z, Klionsky DJ. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagar R. Autophagy: A brief overview in perspective of dermatology. Indian J Dermatol Venereol Leprol. 2017;83:290–97. doi: 10.4103/0378-6323.196320. [DOI] [PubMed] [Google Scholar]
- 27.Paolini A, Omairi S, Mitchell R, et al. Attenuation of autophagy impacts on muscle fibre development, starvation induced stress and fibre regeneration following acute injury. Sci Rep. 2018;8:9062. doi: 10.1038/s41598-018-27429-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campbell P, Morris H, Schapira A. Chaperone-mediated autophagy as a therapeutic target for Parkinson disease. Expert Opin Ther Targets. 2018;22:823–32. doi: 10.1080/14728222.2018.1517156. [DOI] [PubMed] [Google Scholar]
- 29.Orenstein SJ, Cuervo AM. Chaperone-mediated autophagy: Molecular mechanisms and physiological relevance. Semin Cell Dev Biol. 2010;21:719–26. doi: 10.1016/j.semcdb.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kunz JB, Schwarz H, Mayer A. Determination of four sequential stages during microautophagy in vitro. J Biol Chem. 2004;279:9987–96. doi: 10.1074/jbc.M307905200. [DOI] [PubMed] [Google Scholar]
- 31.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;(Suppl 2):1542–52. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parzych KR, Klionsky DJ. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20:460–73. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khandia R, Dadar M, Munjal A, et al. A comprehensive review of autophagy and its various roles in infectious, non-infectious, and lifestyle diseases: Current knowledge and prospects for disease prevention, novel drug design, and therapy. Cells. 2019;8:E674. doi: 10.3390/cells8070674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsai CY, Chen CY, Chiou YH, et al. Epigallocatechin-3-gallate suppresses human herpesvirus 8 replication and induces ROS leading to apoptosis and autophagy in primary effusion lymphoma cells. Int J Mol Sci. 2017;19:E16. doi: 10.3390/ijms19010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Min NY, Kim JH, Choi JH, et al. Selective death of cancer cells by preferential induction of reactive oxygen species in response to (−)-epigallocatechin-3-gallate. Biochem Biophys Res Commun. 2012;421:91–97. doi: 10.1016/j.bbrc.2012.03.120. [DOI] [PubMed] [Google Scholar]
- 36.Wei R, Mao L, Xu P, et al. Suppressing glucose metabolism with epigallocatechin-3-gallate (EGCG) reduces breast cancer cell growth in preclinical models. Food Funct. 2018;9:5682–96. doi: 10.1039/c8fo01397g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gualtero DF, Suarez Castillo A. Biomarkers in saliva for the detection of oral squamous cell carcinoma and their potential use for early diagnosis: A systematic review. Acta Odontol Scand. 2016;74:170–77. doi: 10.3109/00016357.2015.1110249. [DOI] [PubMed] [Google Scholar]
- 38.Irimie AI, Braicu C, Zanoaga O, et al. Epigallocatechin-3-gallate suppresses cell proliferation and promotes apoptosis and autophagy in oral cancer SSC-4 cells. Onco Targets Ther. 2015;8:461–70. doi: 10.2147/OTT.S78358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Satoh M, Takemura Y, Hamada H, et al. EGCG induces human mesothelioma cell death by inducing reactive oxygen species and autophagy. Cancer Cell Int. 2013;13:19. doi: 10.1186/1475-2867-13-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grube S, Ewald C, Kögler C, et al. Achievable central nervous system concentrations of the green tea catechin EGCG induce stress in glioblastoma cells in vitro. Nutr Cancer. 2018;70:1145–58. doi: 10.1080/01635581.2018.1495239. [DOI] [PubMed] [Google Scholar]
- 41.Kardideh B, Samimi Z, Norooznezhad F, et al. Autophagy, cancer and angiogenesis: Where is the link? Cell Biosci. 2019;9:65. doi: 10.1186/s13578-019-0327-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aljohani H, Koncar RF, Zarzour A, et al. ROS1 amplification mediates resistance to gefitinib in glioblastoma cells. Oncotarget. 2015;6:20388–95. doi: 10.18632/oncotarget.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okon IS, Coughlan KA, Zhang M, et al. Gefitinib-mediated reactive oxygen species (ROS) instigates mitochondrial dysfunction and drug resistance in lung cancer cells. J Biol Chem. 2015;290:9101–10. doi: 10.1074/jbc.M114.631580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao L, Yang G, Shi Y, et al. Co-delivery of Gefitinib and chloroquine by chitosan nanoparticles for overcoming the drug acquired resistance. J Nanobiotechnology. 2015;13:57. doi: 10.1186/s12951-015-0121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meng J, Chang C, Chen Y, et al. EGCG overcomes gefitinib resistance by inhibiting autophagy and augmenting cell death through targeting ERK phosphorylation in NSCLC. Onco Targets Ther. 2019;12:6033–43. doi: 10.2147/OTT.S209441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Braicu C, Pileczki V, Pop L, et al. Dual targeted therapy with p53 siRNA and epigallocatechin gallate in a triple negative breast cancer cell model. PLoS One. 2015;10:e0120936. doi: 10.1371/journal.pone.0120936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crighton D, Wilkinson S, O’Prey J, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 48.Abdulghani J, El-Deiry WS. TRAIL receptor signaling and therapeutics. Expert Opin Ther Targets. 2010;14:1091–108. doi: 10.1517/14728222.2010.519701. [DOI] [PubMed] [Google Scholar]
- 49.Kim SW, Moon JH, Park SY. Activation of autophagic flux by epigallocatechin gallate mitigates TRAIL-induced tumor cell apoptosis via down-regulation of death receptors. Oncotarget. 2016;7:65660–68. doi: 10.18632/oncotarget.11597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pendleton KP, Grandis JR. Cisplatin-based chemotherapy options for recurrent and/or metastatic squamous cell cancer of the head and neck. Clin Med Insights Ther. 2013;2013 doi: 10.4137/CMT.S10409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu CC, Yang JS, Chiang JH, et al. Cell death caused by quinazolinone HMJ-38 challenge in oral carcinoma CAL 27 cells: Dissections of endoplasmic reticulum stress, mitochondrial dysfunction and tumor xenografts. Biochim Biophys Acta. 2014;1840:2310–20. doi: 10.1016/j.bbagen.2014.02.022. [DOI] [PubMed] [Google Scholar]
- 52.Yuan CH, Horng CT, Lee CF, et al. Epigallocatechin gallate sensitizes cisplatin-resistant oral cancer CAR cell apoptosis and autophagy through stimulating AKT/STAT3 pathway and suppressing multidrug resistance 1 signaling. Environ Toxicol. 2017;32:845–55. doi: 10.1002/tox.22284. [DOI] [PubMed] [Google Scholar]
- 53.Hu F, Wei F, Wang Y, et al. EGCG synergizes the therapeutic effect of cisplatin and oxaliplatin through autophagic pathway in human colorectal cancer cells. J Pharmacol Sci. 2015;128:27–34. doi: 10.1016/j.jphs.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 54.Chen L, Ye HL, Zhang G, et al. Autophagy inhibition contributes to the synergistic interaction between EGCG and doxorubicin to kill the hepatoma Hep3B cells. PLoS One. 2014;9:e85771. doi: 10.1371/journal.pone.0085771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang W, Chen D, Zhu K. SOX2OT variant 7 contributes to the synergistic interaction between EGCG and Doxorubicin to kill osteosarcoma via autophagy and stemness inhibition. J Exp Clin Cancer Res. 2018;37:37. doi: 10.1186/s13046-018-0689-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen X, Tong R, Liu B, et al. Duo of (−)-epigallocatechin-3-gallate and doxorubicin loaded by polydopamine coating ZIF-8 in the regulation of autophagy for chemo-photothermal synergistic therapy. Biomater Sci. 2020;8(5):1380–93. doi: 10.1039/c9bm01614g. [DOI] [PubMed] [Google Scholar]
- 57.Hsieh CH, Lu CH, Chen WT, et al. Application of non-invasive low strength pulsed electric field to EGCG treatment synergistically enhanced the inhibition effect on PANC-1 cells. PLoS One. 2017;12:e0188885. doi: 10.1371/journal.pone.0188885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou YF. High intensity focused ultrasound in clinical tumor ablation. World J Clin Oncol. 2011;2:8–27. doi: 10.5306/wjco.v2.i1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsieh CH, Lu CH, Kuo YY, et al. Studies on the non-invasive anticancer remedy of the triple combination of epigallocatechin gallate, pulsed electric field, and ultrasound. PLoS One. 2018;13:e0201920. doi: 10.1371/journal.pone.0201920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–22. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rocha EM, De Miranda B, Sanders LH. Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis. 2018;109:249–57. doi: 10.1016/j.nbd.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 62.Lane DJR, Ayton S, Bush AI. Iron and Alzheimer’s diseases: an update on emerging mechanisms. J Alzheimers Dis. 2018;64:S379–S395. doi: 10.3233/JAD-179944. [DOI] [PubMed] [Google Scholar]
- 63.Singh NA, Mandal AK, Khan ZA. Potential neuroprotective properties of epigallocatechin-3-gallate (EGCG) Nutr J. 2016;15:60. doi: 10.1186/s12937-016-0179-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cherra SJ, 3rd, Chu CT. Autophagy in neuroprotection and neurodegeneration: a question of balance. Future Neurol. 2008;3:309–323. doi: 10.2217/14796708.3.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee JH, Moon JH, Kim SW, et al. EGCG-mediated autophagy flux has a neuroprotection effect via a class III histone deacetylase in primary neuron cells. Oncotarget. 2015;6:9701–17. doi: 10.18632/oncotarget.3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gu HF, Nie YX, Tong QZ, et al. Epigallocatechin-3-gallate attenuates impairment of learning and memory in chronic unpredictable mild stress-treated rats by restoring hippocampal autophagic flux. PLoS One. 2014;9:e112683. doi: 10.1371/journal.pone.0112683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen Y, Chen J, Sun X, et al. Evaluation of the neuroprotective effect of EGCG: a potential mechanism of mitochondrial dysfunction and mitochondrial dynamics after subarachnoid hemorrhage. Food Funct. 2018;9:6349–59. doi: 10.1039/c8fo01497c. [DOI] [PubMed] [Google Scholar]
- 68.Chen Y, Huang L, Zhang H, et al. Reduction in autophagy by (−)-epigallocatechin-3-gallate (EGCG): A potential mechanism of prevention of mitochondrial dysfunction after subarachnoid hemorrhage. Mol Neurobiol. 2017;54:392–405. doi: 10.1007/s12035-015-9629-9. [DOI] [PubMed] [Google Scholar]
- 69.Babu PV, Liu D, Gilbert ER. Recent advances in understanding the anti-diabetic actions of dietary flavonoids. J Nutr Biochem. 2013;24:1777–89. doi: 10.1016/j.jnutbio.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karaa A, Goldstein A. The spectrum of clinical presentation, diagnosis, and management of mitochondrial forms of diabetes. Pediatr Diabetes. 2015;16:1–9. doi: 10.1111/pedi.12223. [DOI] [PubMed] [Google Scholar]
- 71.Yan J, Feng Z, Liu J, et al. Enhanced autophagy plays a cardinal role in mitochondrial dysfunction in type 2 diabetic Goto-Kakizaki (GK) rats: Ameliorating effects of (−)-epigallocatechin-3-gallate. J Nutr Biochem. 2012;23:716–24. doi: 10.1016/j.jnutbio.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 72.Wang L, Sun X, Zhu M, et al. Epigallocatechin-3-gallate stimulates autophagy and reduces apoptosis levels in retinal Müller cells under high-glucose conditions. Exp Cell Res. 2019;380:149–58. doi: 10.1016/j.yexcr.2019.04.014. [DOI] [PubMed] [Google Scholar]
- 73.Liu J, Tang Y, Feng Z, et al. Acetylated FoxO1 mediates high-glucose induced autophagy in H9c2 cardiomyoblasts: Regulation by a polyphenol -(−)-epigallocatechin-3-gallate. Metabolism. 2014;63(10):1314–23. doi: 10.1016/j.metabol.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 74.Liu J, Tang Y, Feng Z, et al. (−)-Epigallocatechin-3-gallate attenuated myocardial mitochondrial dysfunction and autophagy in diabetic Goto-Kakizaki rats. Free Radic Res. 2014;48(8):898–906. doi: 10.3109/10715762.2014.920955. [DOI] [PubMed] [Google Scholar]
- 75.Zhang C, Liang R, Gan X, et al. MicroRNA-384-5p/Beclin-1 As potential indicators for epigallocatechin gallate against cardiomyocytes ischemia reperfusion injury by inhibiting autophagy via PI3K/Akt pathway. Drug Des Devel Ther. 2019;13:3607–23. doi: 10.2147/DDDT.S219074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xuan F, Jian J. Epigallocatechin gallate exerts protective effects against myocardial ischemia/reperfusion injury through the PI3K/Akt pathway-mediated inhibition of apoptosis and the restoration of the autophagic flux. Int J Mol Med. 2016;38:328–36. doi: 10.3892/ijmm.2016.2615. [DOI] [PubMed] [Google Scholar]
- 77.Jamuna S, Ashokkumar R, Sakeena Sadullah MS, Devaraj SN. Oligomeric proanthocyanidins and epigallocatechin gallate aggravate autophagy of foam cells through the activation of Class III PI3K/Beclin1-complex mediated cholesterol efflux. Biofactors. 2019;45:763–73. doi: 10.1002/biof.1537. [DOI] [PubMed] [Google Scholar]
- 78.Li S, Xia Y, Chen K, et al. Epigallocatechin-3-gallate attenuates apoptosis and autophagy in concanavalin A – induced hepatitis by inhibiting BNIP3. Drug Des Devel Ther. 2016;10:631–47. doi: 10.2147/DDDT.S99420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou J, Farah BL, Sinha RA, et al. Epigallocatechin-3-gallate (EGCG), a green tea polyphenol, stimulates hepatic autophagy and lipid clearance. PLoS One. 2014;9:e87161. doi: 10.1371/journal.pone.0087161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Holczer M, Besze B, Zámbó V, et al. Epigallocatechin-3-gallate (EGCG) promotes autophagy-dependent survival via influencing the balance of mTOR-AMPK pathways upon endoplasmic reticulum stress. Oxid Med Cell Longev. 2018;2018 doi: 10.1155/2018/6721530. 6721530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li CP, Yao J, Tao ZF, et al. Epigallocatechin-gallate (EGCG) regulates autophagy in human retinal pigment epithelial cells: A potential role for reducing UVB light-induced retinal damage. Biochem Biophys Res Commun. 2013;438:739–45. doi: 10.1016/j.bbrc.2013.07.097. [DOI] [PubMed] [Google Scholar]
- 82.Caution K, Pan A, Krause K, et al. Methylomic correlates of autophagy activity in cystic fibrosis. J Cyst Fibros. 2019;18:491–500. doi: 10.1016/j.jcf.2019.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Abd E, Maksoud AI, Elebeedy D, Abass NH, et al. Methylomic changes of autophagy-related genes by legionella effector Lpg2936 in infected macrophages. Front Cell Dev Biol. 2020;7:390. doi: 10.3389/fcell.2019.00390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Song S, Tan J, Miao Y, et al. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J Cell Physiol. 2017;232:2977–84. doi: 10.1002/jcp.25785. [DOI] [PubMed] [Google Scholar]
- 85.Fernández A, Ordóñez R, Reiter RJ, et al. Melatonin and endoplasmic reticulum stress: relation to autophagy and apoptosis. J Pineal Res. 2015;59:292–307. doi: 10.1111/jpi.12264. [DOI] [PubMed] [Google Scholar]
- 86.Bettigole SE, Glimcher LH. Endoplasmic reticulum stress in immunity. Annu Rev Immunol. 2015;33:107–38. doi: 10.1146/annurev-immunol-032414-112116. [DOI] [PubMed] [Google Scholar]
- 87.Park S, Lim Y, Lee D, et al. Modulation of protein synthesis by eIF2α phosphorylation protects cell from heat stress-mediated apoptosis. Cells. 2018;7:E254. doi: 10.3390/cells7120254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–94. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–70. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Md Nesran ZN, Shafie NH, Ishak AH, et al. Induction of endoplasmic reticulum stress pathway by green tea epigallocatechin-3-gallate (EGCG) in colorectal cancer cells: Activation of PERK/p-eIF2α/ATF4 and IRE1α. Biomed Res Int. 2019;2019 doi: 10.1155/2019/3480569. 3480569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wong RS. Apoptosis in cancer: From pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87. doi: 10.1186/1756-9966-30-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Magyar JE, Gamberucci A, Konta L, et al. Endoplasmic reticulum stress underlying the pro-apoptotic effect of epigallocatechin gallate in mouse hepatoma cells. Int J Biochem Cell Biol. 2009;41:694–700. doi: 10.1016/j.biocel.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 93.Gamberucci A, Konta L, Colucci A, et al. Green tea flavonols inhibit glucosidase II. Biochem Pharmacol. 2006;72:640–46. doi: 10.1016/j.bcp.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 94.Martinotti S, Ranzato E, Burlando B. (−)- Epigallocatechin-3-gallate induces GRP78 accumulation in the ER and shifts mesothelioma constitutive UPR into proapoptotic ER stress. J Cell Physiol. 2018;233:7082–90. doi: 10.1002/jcp.26631. [DOI] [PubMed] [Google Scholar]
- 95.Korennykh A, Walter P. Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol. 2012;28:251–77. doi: 10.1146/annurev-cellbio-101011-155826. [DOI] [PubMed] [Google Scholar]
- 96.Das A, Banik NL, Ray SK. Flavonoids activated caspases for apoptosis in human glioblastoma T98G and U87MG cells but not in human normal astrocytes. Cancer. 2010;116:164–76. doi: 10.1002/cncr.24699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huang KH, Kuo KL, Chen SC, et al. Down-regulation of glucose-regulated protein (GRP) 78 potentiates cytotoxic effect of celecoxib in human urothelial carcinoma cells. PLoS One. 2012;7:e33615. doi: 10.1371/journal.pone.0033615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sun X, Huo X, Luo T, et al. The anticancer flavonoid chrysin induces the unfolded protein response in hepatoma cells. J Cell Mol Med. 2011;15:2389–98. doi: 10.1111/j.1582-4934.2010.01244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen TC, Wang W, Golden EB, et al. Green tea epigallocatechin gallate enhances therapeutic efficacy of temozolomide in orthotopic mouse glioblastoma models. Cancer Lett. 2011;302:100–8. doi: 10.1016/j.canlet.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 100.Wang J, Yin Y, Hua H, et al. Blockade of GRP78 sensitizes breast cancer cells to microtubules-interfering agents that induce the unfolded protein response. J Cell Mol Med. 2009;3:3888–97. doi: 10.1111/j.1582-4934.2009.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Golden EB, Lam PY, Kardosh A, et al. Green tea polyphenols block the anticancer effects of bortezomib and other boronic acid-based proteasome inhibitors. Blood. 2009;113:5927–37. doi: 10.1182/blood-2008-07-171389. [DOI] [PubMed] [Google Scholar]
- 102.Ludwig H, Khayat D, Giaccone G, Facon T. Proteasome inhibition and its clinical prospects in the treatment of hematologic and solid malignancies. Cancer. 2005;104:1794–807. doi: 10.1002/cncr.21414. [DOI] [PubMed] [Google Scholar]
- 103.Du K, Liu M, Zhong X, et al. Epigallocatechin gallate reduces amyloid β-induced neurotoxicity via inhibiting endoplasmic reticulum stress-mediated apoptosis. Mol Nutr Food Res. 2018;62:e1700890. doi: 10.1002/mnfr.201700890. [DOI] [PubMed] [Google Scholar]
- 104.He Q, Bao L, Zimering J, et al. The protective role of (−)-epigallocatechin-3-gallate in thrombin-induced neuronal cell apoptosis and JNK-MAPK activation. Neuroreport. 2015;26:416–23. doi: 10.1097/WNR.0000000000000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yao C, Zhang J, Liu G, et al. Neuroprotection by (−)-epigallocatechin-3-gallate in a rat model of stroke is mediated through inhibition of endoplasmic reticulum stress. Mol Med Rep. 2014;9:69–76. doi: 10.3892/mmr.2013.1778. [DOI] [PubMed] [Google Scholar]
- 106.Ferreira N, Saraiva MJ, Almeida MR. Epigallocatechin-3-gallate as a potential therapeutic drug for TTR-related amyloidosis: “In vivo” evidence from FAP mice models. PLoS One. 2012;7:e29933. doi: 10.1371/journal.pone.0029933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hanhineva K, Törrönen R, Bondia-Pons I, et al. Impact of dietary polyphenols on carbohydrate metabolism. Int J Mol Sci. 2010;11:1365–402. doi: 10.3390/ijms11041365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ortsäter H, Grankvist N, Wolfram S, et al. Diet supplementation with green tea extract epigallocatechin gallate prevents progression to glucose intolerance in db/db mice. Nutr Metab (Lond) 2012;9:11. doi: 10.1186/1743-7075-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu CM, Ma JQ, Sun JM, et al. Association of changes in ER stress-mediated signaling pathway with lead-induced insulin resistance and apoptosis in rats and their prevention by A-type dimeric epigallocatechin-3-gallate. Food Chem Toxicol. 2017;110:325–32. doi: 10.1016/j.fct.2017.10.040. [DOI] [PubMed] [Google Scholar]
- 110.Xiang C, Xiao X, Jiang B, et al. Epigallocatechin-3-gallate protects from high glucose induced podocyte apoptosis via suppressing endoplasmic reticulum stress. Mol Med Rep. 2017;16:6142–47. doi: 10.3892/mmr.2017.7388. [DOI] [PubMed] [Google Scholar]
- 111.Pan C, Zhou S, Wu J, et al. NRF2 Plays a critical role in both self and EGCG protection against diabetic testicular damage. Oxid Med Cell Longev. 2017;2017 doi: 10.1155/2017/3172692. 3172692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wu J, Xu X, Li Y, et al. Quercetin, luteolin and epigallocatechin gallate alleviate TXNIP and NLRP3-mediated inflammation and apoptosis with regulation of AMPK in endothelial cells. Eur J Pharmacol. 2014;745:59–68. doi: 10.1016/j.ejphar.2014.09.046. [DOI] [PubMed] [Google Scholar]
- 113.Calles-Escandon J, Cipolla M. Diabetes and endothelial dysfunction: A clinical perspective. Endocr Rev. 2001;22:36–52. doi: 10.1210/edrv.22.1.0417. [DOI] [PubMed] [Google Scholar]
- 114.Chen B, Liu G, Zou P, et al. Epigallocatechin-3-gallate protects against cisplatin-induced nephrotoxicity by inhibiting endoplasmic reticulum stress-induced apoptosis. Exp Biol Med (Maywood) 2015;240:1513–19. doi: 10.1177/1535370215573394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Luke DR, Vadiei K, Lopez-Berestein G. Role of vascular congestion in cisplatin-induced acute renal failure in the rat. Nephrol Dial Transplant. 1992;7:1–7. [PubMed] [Google Scholar]
- 116.Bottone MG, Soldani C, Veneroni P, et al. Cell proliferation, apoptosis and mitochondrial damage in rat B50 neuronal cells after cisplatin treatment. Cell Prolif. 2008;41:506–20. doi: 10.1111/j.1365-2184.2008.00530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hsieh JT, Kuo KL, Liu SH, et al. Epigallocatechin gallate attenuates partial bladder outlet obstruction-induced bladder injury via suppression of endoplasmic reticulum stress-related apoptosis – in vivo study. Urology. 2016;91:e1–9. doi: 10.1016/j.urology.2015.12.020. [DOI] [PubMed] [Google Scholar]
- 118.Steers WD, De Groat WC. Effect of bladder outlet obstruction on micturition reflex pathways in the rat. J Urol. 1988;140:864–71. doi: 10.1016/s0022-5347(17)41846-5. [DOI] [PubMed] [Google Scholar]
- 119.Karthikeyan B, Harini L, Krishnakumar V, et al. Insights on the involvement of (−)-epigallocatechin gallate in ER stress-mediated apoptosis in age-related macular degeneration. Apoptosis. 2017;22:72–85. doi: 10.1007/s10495-016-1318-2. [DOI] [PubMed] [Google Scholar]
- 120.Fritsche LG, Fariss RN, Stambolian D, et al. Age-related macular degeneration: Genetics and biology coming together. Annu Rev Genomics Hum Genet. 2014;15:151–71. doi: 10.1146/annurev-genom-090413-025610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Neuhaus CP. Humans as model organisms. Ethics Hum Res. 2019;4:35–37. doi: 10.1002/eahr.500011. [DOI] [PubMed] [Google Scholar]
- 122.Peluso I, Serafini M. Antioxidants from black and green tea: From dietary modulation of oxidative stress to pharmacological mechanisms. Br J Pharmacol. 2017;174:1195–208. doi: 10.1111/bph.13649. [DOI] [PMC free article] [PubMed] [Google Scholar]