

Key Points
Deletion of αv integrins from B cells accelerates TLR-driven autoimmunity.
αv-knockout B cells have increased pathogenic TLR signaling.
αv regulates cytokine production and differentiation of IgG2c plasma cells.
Abstract
Systemic lupus erythematosus (SLE) is defined by loss of B cell tolerance, resulting in production of autoantibodies against nucleic acids and other cellular Ags. Aberrant activation of TLRs by self-derived RNA and DNA is strongly associated with SLE in patients and in mouse models, but the mechanism by which TLR signaling to self-ligands is regulated remains poorly understood. In this study, we show that αv integrin plays a critical role in regulating B cell TLR signaling to self-antigens in mice. We show that deletion of αv from B cells accelerates autoantibody production and autoimmune kidney disease in the Tlr7.1 transgenic mouse model of SLE. Increased autoimmunity was associated with specific expansion of transitional B cells, extrafollicular IgG2c-producing plasma cells, and activation of CD4 and CD8 T cells. Our data show that αv-mediated regulation of TLR signaling in B cells is critical for preventing autoimmunity and indicate that loss of αv promotes escape from tolerance. Thus, we identify a new regulatory pathway in autoimmunity and elucidate upstream signals that adjust B cell activation to prevent development of autoimmunity in a mouse model.
Introduction
A hallmark of systemic lupus erythematosus (SLE) is the production of high levels of class-switched IgG autoantibodies that form pathogenic immune complexes. Appearance of autoantibodies often precedes disease, and loss of B cell tolerance is a critical initiating event for SLE. Although autoantibodies can arise against a wide range of self-antigens, nucleic acids, and associated nuclear Ags, including DNA, RNA, histones, and ribonucleoproteins, dominate the autoantigen repertoire. There is increasing evidence from human genetic studies and mouse models of SLE that recognition of self-derived nucleic acids by TLRs contributes to this loss of tolerance and production of autoantibodies. Polymorphisms and copy number variations in TLR7, which recognizes ssRNA, and in downstream signaling components, such as IRF7, are associated with susceptibility to SLE (1–5). Supporting a causative role for TLR signaling in SLE, overexpression of TLR7, either because of the Y-linked autoimmune accelerator (Yaa) translocation (6–8) or through transgenic (tg) manipulation (9) causes lupus-like autoimmunity in mice.
TLR7 is expressed by conventional and plasmacytoid dendritic cells (pDCs) and B cells, and all these cell types become activated in SLE and contribute to autoimmunity. TLR7 signaling in dendritic cells (DCs) triggers production of inflammatory and immune-stimulatory cytokines, most notably type I IFNs, which are secreted at high levels by pDCs and are associated with severe autoimmunity in human patients and in mouse models (10). In B cells, TLR7 and TLR9 synergize with the BCR (11–13) to promote expansion of B cells specific for nucleic acids and associated Ags and production of autoantibodies (14). TLR signaling also promotes Ag presentation in DCs and B cells, driving T cell–mediated autoimmunity (15, 16). Confirming the importance of TLR signaling in SLE, deletion of Tlr7 and Tlr9 significantly attenuates anti–nucleic acid autoantibody production and autoimmune pathology in a mouse model of SLE (17). Conditional knockout studies reveal that TLR signaling in B cells is critical for many aspects of autoimmunity, including autoantibody production, T cell activation, and glomerular nephritis. Disruption of DC-intrinsic TLR signaling, in contrast, does not prevent autoantibody production or nephritis, although it is required for development of dermatitis and production of type I IFN by pDCs (16). Furthermore, the DC activation observed in this mouse model of autoimmunity is dependent on B cell–intrinsic TLR signaling, suggesting that B cell activation is an essential early step in autoimmune disease (16, 18). However, it remains unclear what mechanisms exist to regulate B cell TLR signaling in response to self-antigens and how loss of these mechanisms in B cells impacts development of autoimmunity.
We have recently identified a regulatory role for the integrin αvβ3 in TLR signaling in B cells (19, 20). We have shown that αvβ3 promotes recruitment of the autophagy component LC3 to TLR-containing endosomes, which, in turn, promotes endosome fusion with lysosomes, terminating TLR signaling. Deletion of either αv or β3 integrins from B cells delays TLR trafficking, resulting in increased signaling in vitro and increased B cell proliferation and Ab production in vivo (19). Disruption of this regulatory mechanism specifically affects responses to Ags associated with TLR ligands, causing expansion of plasma and memory B cells and increased production of high-affinity, IgG2a/c class-switched Abs (20). We have previously reported that αv-knockout mice also develop serum anti-dsDNA autoantibodies as they age, leading us to hypothesize that this regulatory mechanism contributes to B cell tolerance to self-TLR ligands (19). The αv-knockout mice thus provide us with a unique model to alter the strength of B cell TLR signaling and disrupt tolerance. To confirm whether loss of this mechanism can lead to development of autoimmunity, we have crossed B cell–specific αv integrin knockout mice with the Tlr7.1 tg model of autoimmune disease. In this study, we show that deletion of αv from B cells promotes production of autoantibodies to a wide range of autoantigens, including RNA and small nuclear ribonucleoproteins (snRNPs), leading to increased Ab and complement deposition in kidneys. This is associated with a major expansion of IgG2c class-switched plasmablasts, increased inflammatory cytokine production by B cells, and activation of CD4 and CD8 T cells. Together, these data identify αvβ3 as a physiological regulator of autoreactive B cell activation and show that alterations in the strength of TLR signaling in B cells can accelerate autoimmunity.
Materials and Methods
Mice
αv-CD19 (19) and TLR7.1 tg mice (9) have been described previously. All mice were housed under specific pathogen-free conditions at Benaroya Research Institute. All animal experiments were performed under appropriate licenses and institutional review within local and national guidelines for animal care.
Abs and reagents
Anti-mouse Abs used for flow cytometry include the following: CD95 (Jo2), CD138 (281-2), CD4 (RM4-5), CD38 (90), MHC class II (2G9), CD23 (B3B4), CD80 (B7-1), CD44 (IM7), B220 (RA3-6B2), CD11b (M1/70), IgM (R6-60.2), IgD (11-26c.2a), CD19 (1D3), and Mouse BD Fc Block (2.4G2) from BD Biosciences. CD8α (53-6.7), GL7 (GL7), CD86 (GL1), CD21 (7E9), CD69 (H1.2F3), CD25 (PC61), and CD73 (Ty/11.8) were from BioLegend. Anti–NFκ-B (D14E12), anti-LSD1 (C69G12), and HRP-conjugated anti-rabbit IgG were from Cell Signaling Technology. Alkaline phosphatase–conjugated anti-mouse IgG, alkaline phosphatase–conjugated anti-mouse IgG2c, and anti-mouse IgG (H chain + L chain) were from SouthernBiotech. PNA-FITC was from Vector Laboratories, and R848 was from InvivoGen.
Flow cytometry and cell sorting and stimulations
Spleen cells were harvested in PBS/0.5% BSA/2 mM EDTA and depleted of RBCs (ACK lysis buffer; Life Technologies). Single-cell suspensions blocked with Fc Block (BD Biosciences) were stained with a combination of fluorochrome-tagged Abs for surface markers and LIVE/DEAD cell marker (Thermo Fisher Scientific) at 4°C for 30 min. For sorting of spleen marginal zone (MZ), follicular (Fo), and transitional 1 (T1) B cells, RBC-lysed single-cell suspensions were labeled with anti-B220, anti-CD23, and anti-CD21 and anti-CD24 Abs, then sorted with FACSAria II (BD Bioscience). For studies on T cell activation and proliferation, FACS-sorted B cells or total B cells after enrichment (Stem Cell Technologies) were plated on 96-well plates in complete RPMI 1640 (10% FBS, 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin, and 50 μM 2-ME). Magnetically sorted naive CD4 T cells (Miltenyi Biotec) were added to the B cells with or without 2.5 μg/ml anti-CD3 (145-2C11; BD Biosciences). Cells were cultured at the ratio of 1:4 B cells to T cells. CD4 T cell activation was measured at 48 h by flow cytometry using activation specific markers. For measurement of proliferation, cells were pulsed with 1 μCi/well tritiated thymidine ([3H]TdR) for 18 h prior to harvest; incorporation was determined by liquid scintillation spectrometry.
Cytokine array
For analysis of cytokines produced by B cell populations, FACS-sorted MZ, Fo, or T1 B cells were plated in 96-well plates at the density of 100,000 cells per well and stimulated with TLR7 ligand R848 (5 μg/ml). Supernatants were harvested 48 h later and cytokine production measured by LEGENDplex bead-based multianalyte flow assay kit from BioLegend, according to the manufacturer’s protocol. Cytokine production was analyzed by FACSCalibur and quantified using LEGENDplex software (BioLegend).
Western blot
For analysis of NF-κB activation, FACS-sorted MZ, Fo, and T1 B cell subsets were stimulated for indicated times with TLR7 ligand R848 (5 μg/ml). Nuclear extracts were prepared by lysing cells in hypotonic nuclear extraction buffer (1 M HEPES [pH 7.5], 5 M NaCl, 0.5 M EDTA [pH 8], 50% glycerol, 10% IGEPAL, and 10% Triton X-100) for 10 min, followed by centrifugation at 1500 × g for 5 min at 4°C to pellet the nuclei, and nuclei were resuspended in radioimmunoprecipitation assay buffer. Lysates were centrifuged for 10 min at 4°C at 14,000 × g, and supernatant was collected as nuclear fraction. Proteins were quantified by BCA assay (Pierce), separated by electrophoresis using NuPAGE Bis-Tris gels (Invitrogen) and blotted onto PVDF membranes. Nonspecific binding was blocked with 5% BSA in TBST (0.1%), followed by incubation with primary Abs overnight at 4°C. HRP-conjugated secondary Abs were added for 1 h at room temperature and developed using ECL reagents (MilliporeSigma).
ELISA
For detection of anti-RNA Abs, Immulon 2HB Microtiter Plates were first treated with poly-l-lysine and then coated with 10 μg/ml yeast RNA (Sigma-Aldrich) diluted in PBS. Plates were blocked with 2% BSA, 2% FCS, 0.1% Tween 20, and 0.02% sodium azide in PBS, and sera was added in serial dilution and incubated overnight at 4°C. Plates were washed, and bound Abs were detected by adding alkaline phosphatase–conjugated goat anti-mouse IgG or IgG2c (SouthernBiotech) diluted in blocking buffer for 60 min at 37°C. Secondary Abs were detected by using disodium p-nitrophenyl phosphate substrate (Sigma-Aldrich) and absorbance (OD) read at 405 nm. For detection of total Ig in culture, supernatants plates were coated with 10 μg/ml goat anti-mouse IgG (H and L chain specific) (SouthernBiotech) in PBS at 4°C overnight. Plates were blocked as above and incubated with varying dilutions of cell culture supernatants in PBS. After incubation with alkaline phosphatase–conjugated IgG or IgG2c Abs (SouthernBiotech), color was developed as above with disodium p-nitrophenyl phosphate substrate.
Autoantigen array
Autoantigen microarrays were performed at University of Texas Southwestern Medical Center, Microarray Core Facility, Dallas, Texas (https://microarray.swmed.edu/products/product/autoantigen-microarray-panel-i/).
Anti-nuclear Ab
For anti-nuclear Ab (ANA) assays, diluted serum (1/40) was added to fixed Hep-2 ANA slides (MBL Bion), and FITC-conjugated goat anti-mouse IgG was used as detection Ab. Fluorescent images were obtained using Nikon Ti (Eclipse) inverted microscope at original magnification ×40 with constant exposure.
Kidney immunofluorescence staining
Mouse kidneys were embedded in OCT compound and snap frozen over dry ice. Six-micromolar sections were cut on cryostat and mounted on Superfrost Plus Slide and fixed with ice cold acetone for 5 min. After rehydration with PBS, slides were blocked with PBS/5% BSA and stained with IgG-FITC (Alexa 488 IgG, catalog no. A11001; Invitrogen) or C3-FITC (catalog no. 855500; MP Biomedicals). Images were acquired using Nikon Ti (Eclipse) inverted microscope with Ultraview Spinning Disc (CSU-X1) confocal scanner (PerkinElmer).
Results
Deletion of αv from B cells increases splenomegaly in Tlr7.1 tg mice
To determine whether B cell αv integrins affected autoimmunity driven by TLR signaling, we used an established model in which the Tlr7 gene is overexpressed from a bacterial artificial chromosome transgene [Tlr7.1 tg mice (9)], resulting in autoimmunity associated with expansion of autoreactive B cells (9, 21). αv-CD19 mice (19) were crossed with Tlr7.1 tg mice to generate αv-CD19.Tlr7 mice; littermates hemizygous for the αv-flox allele but with the same CD19-Cre and Tlr7.1 tg alleles (Tlr7.tg) served as controls. Tlr7.tg mice develop exacerbated immune dysregulation with age, often requiring euthanasia from 3 mo of age (9, 22). αv-CD19.Tlr7 mice showed increased incidence of sudden death or development of severe autoimmune defects, such as anemia, that required euthanasia compared with littermate controls. This was most pronounced in female mice, resulting in ∼40% mortality by 10 wk of age (Fig. 1A). αv-CD19.Tlr7 mice also had significantly larger spleens than both Tlr7.tg littermates and non-tg controls, and as with mortality, the effects of αv deletion on splenomegaly was most prominent in females (Fig. 1B). Non-tg αv-CD19 mice do not have splenomegaly, indicating that this increase in spleen size was due to a synergistic effect between αv deletion and Tlr7 overexpression. Furthermore, αv-CD19.Tlr7 developed splenomegaly earlier than littermate controls. Almost all of the αv-CD19.Tlr7 mice analyzed had enlarged spleens by 6–8 wk of age, whereas only 30% of Tlr7.tg control mice had significantly enlarged spleens at this age and had not yet developed the severe splenomegaly and widespread autoimmune inflammation reported for this strain at 10–12 wk (9, 21, 23). These data therefore supported our hypothesis that deletion of αv from B cells increased or accelerated autoimmunity in TLR7.tg mice.
FIGURE 1.
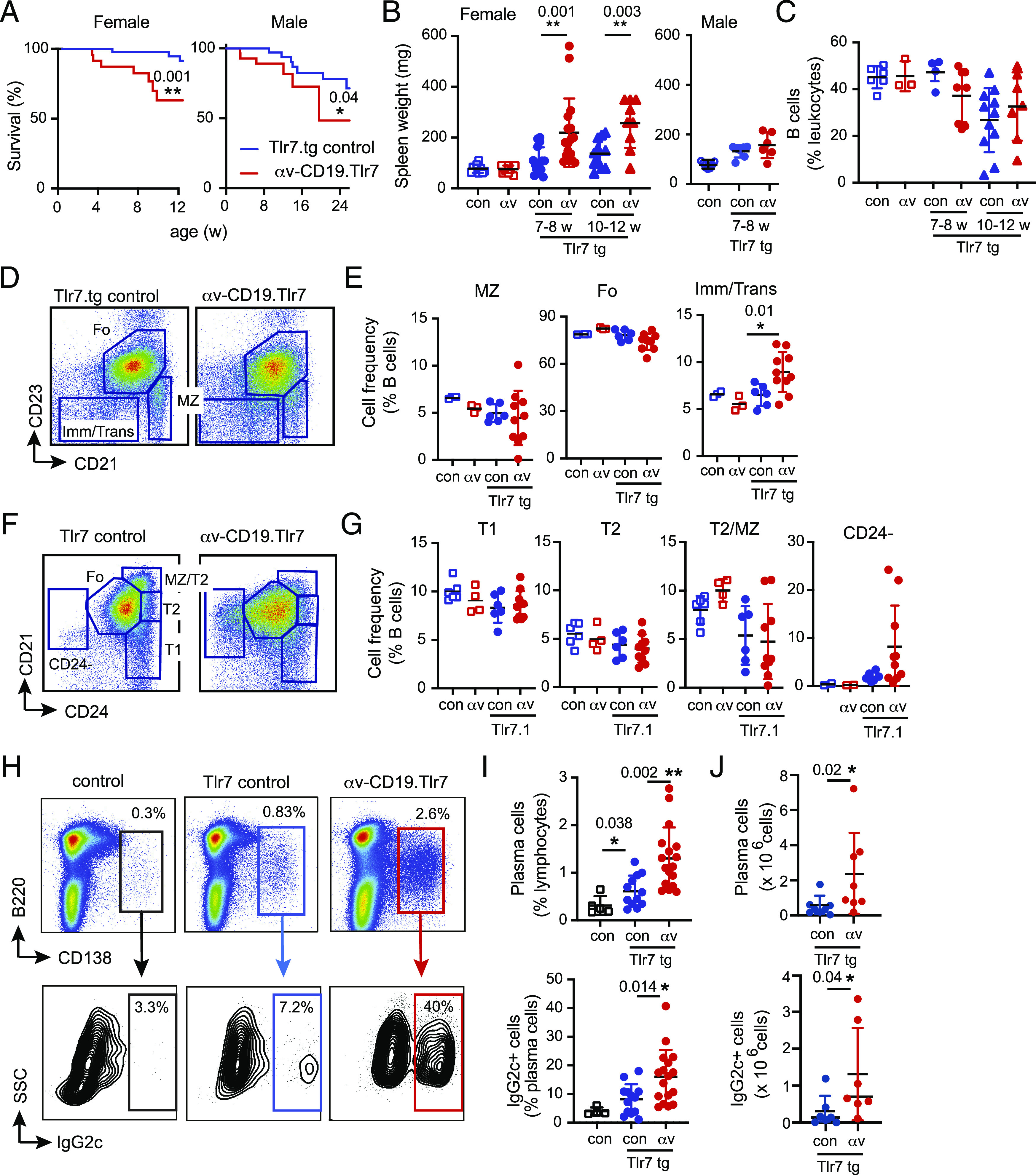
αv deletion promotes expansion of plasma cells. (A) Survival of female and male Tlr7.tg control and αv-CD19.Tlr7 mice (n ≥ 17 mice per group). (B) Spleen weight from control mice (con), αv-CD19 (αv), control Tlr7.tg (con-Tlr7.tg), and αv-CD19.Tlr7 mice (αv Tlr7.tg). Groups analyzed are 7–8-wk-old males (n ≥ 6 per group) and females (n ≥ 10 per group) and 10–12-wk-old females (n ≥ 7 mice per group). (C) Spleen B cell frequency in female mice at 7–8 and 10–12 wk of age (n ≥ 4 mice per group). (D–G) Splenocytes were gated on CD19+ cells, and the frequencies of immature/transitional (Imm/Trans), T1 and T2, MZ, Fo, and CD24-negative B cells determined by flow cytometry as shown. Analysis of non-tg control and αv-CD19 mice are included for comparison (n = 3–6 per group of non-tg and 6–10 per group of tg mice). (H–J) Spleen plasma cells were identified based on CD138 staining and analyzed for intracellular IgG2c and quantified by percentage of parent population (n = 5 for non-tg mice and n ≥ 13 per group for tg mice) (I) or total number of cells per spleen (n = 8 mice per group) (J). Data are presented as data points from individual mice. Shown are p values <0.05 for comparisons between control Tlr7.tg and αv-CD19.Tlr7 mice. *p < 0.05, **p < 0.01, calculated using log-rank test [survival curves (A)] or Mann–Whitney U test.
Expansion of extrafollicular plasma cells in αv-CD19.Tlr7 mice
To understand the role of αv-deficient B cells in early development of autoimmunity and to avoid possible confounding factors due to extensive immune dysregulation, early mortality, and sex, we focused our analysis of B cells on young (6–8 wk) female αv-CD19.Tlr7 mice. Tlr7.tg mice are reported to have a reduction in total spleen B cells, accompanied by an increase in the frequency of transitional cells and reduction in MZ B cells (9, 21). We therefore first analyzed spleen B cells in αv-CD19.Tlr7 and Tlr7.tg control mice, using CD21/CD23 and CD24 staining to identify transitional, MZ, and Fo B cell subsets. We did not observe any statistically significant changes in total B cell numbers in 7–8-wk-old Tlr7.tg mice compared with either non-tg controls or αv-CD19.Tlr7 mice (Fig. 1C). CD21high CD23low MZ B cell numbers were also not statistically significantly different between TLR7.tg strains and controls, although the relative proportions of MZ B cells were much more variable in αv-CD19.Tlr7 mice than in Tlr7.tg or non-tg controls, suggesting that they may be in the process of developing the reduction of MZ B cells previously reported for Tlr7.tg mice. Germinal center (GC) B cells are also reported to be expanded in Tlr7.tg mice, but they were not present at high numbers in these young mice, and their frequencies were similar between Tlr7.tg controls and αv-CD19.Tlr7 mice (Supplemental Fig. 1). We did observe significantly higher proportions of CD21midlow CD23low B cells in αv-CD19.Tlr7 mice compared with Tlr.tg and non-tg controls (Fig. 1D, 1E). This population includes immature and transitional B cells, which have been reported to expand in Tlr7.tg mice. We therefore used additional Ab panels to investigate changes in these and other B cell populations further. The percentage of immature B cells (based on expression of CD93) was not affected by αv deletion (Supplemental Fig. 1). The relative proportions of T1 (B220+ CD24high CD21low) or T2 (B220+ CD24high CD21mid) B cells also did not change significantly between Tlr7.tg and αv-CD19.Tlr7 mice except for a small increase in IgMhigh CD23mid T2 cells (Fig. 1F, 1G, Supplemental Fig. 1). Instead, the increase in CD23low cells seems to be driven by the emergence of a population of B220+ CD24− CD21mid cells. These cells are uniformly IgMlow and do not express high levels of CD11b or CD11c, which are seen on activated “age-associated B cells” associated with mouse autoimmunity models (24) (Supplemental Fig. 1). Based on these data, we concluded that many of the previously reported effects of Tlr7 overexpression on B cell populations are yet to develop in the young mice used for these experiments.
In contrast, we observed clear effects of Tlr7 overexpression and αv deletion on spleen plasma cells. The frequency of CD138+ plasma cells was increased in Tlr7.tg mice compared with non-tg controls, and this was even further enhanced in αv-CD19.Tlr7 mice (Fig. 1H–J). Furthermore, the proportion of plasma cells that had undergone class switching to IgG2c was markedly increased in αv-CD19.Tlr7 mice compared with Tlr7.tg controls (Fig. 1H–J). We also analyzed B cell phenotypes in older αv-CD19.Tlr7 mice and Tlr7.tg controls that had survived to 10–13 wk of age. At this age, we observed similar increases in immature/transitional, CD24−, and IgG2c+ plasma cells in both αv-CD19.Tlr7 mice and Tlr7.tg controls (Supplemental Fig. 1). These data are consistent with the earlier expansion of autoreactive B cells in the αv-CD19.Tlr7 mice and loss of the αv-CD19.Tlr7 mice with severe autoimmunity. Hence, taken together, these data indicated that deletion of αv did not directly affect the proportion of immature cells in the spleen but instead influenced their activation and/or differentiation into class-switched, Ab-producing cells during initiation of autoimmunity.
αv deletion has differential outcomes on activation of B cell subsets
Based on our previous studies, we hypothesized that deletion of αv caused increased B cell activation by increasing TLR7 signaling (19, 20). To test this, we measured activation of the transcription factor NF-κB upon TLR stimulation in purified B cell subsets from αv-CD19.Tlr7 and Tlr7.tg control mice. Treatment with the TLR7 ligand R848 induced stronger activation (nuclear localization) of NF-κB p65 in T1 cells from αv-CD19.Tlr7 mice compared with controls (Fig. 2A), confirming that loss of αv increases TLR signaling in these B cells, similar to our previous observations in MZ and GC B cells (19, 20). R848 also triggered NF-κB nuclear localization in Tlr7.tg Fo B cells, but this was not significantly increased by αv deletion. However, Fo cells from αv-CD19.Tlr7 mice showed high levels of NF-κB nuclear localization in the absence of exogenous TLR7 ligands, which was not seen in Tlr7.tg control Fo B cells (Fig. 2A). We attribute this to basal activation of autoreactive Fo B cells by endogenous ligands encountered during purification or from cellular debris in culture, and these data therefore support our model that deletion of αv increases TLR7 signaling. MZ B cells normally express high levels of TLR7 and respond robustly to TLR stimulation. Consistent with this, MZ B cells from both αv-CD19.Tlr7 and control Tlr.tg mice showed very high levels of TLR signaling even in the absence of exogenous ligands, with no clear difference between the genotypes (Fig. 2A). Thus, although we can see the effect of loss of αv in increasing TLR signaling in transitional and Fo B cells, this is harder to see in the MZ B cells because of massive increase in basal TLR7 signaling.
FIGURE 2.

Increased TLR signaling in αv-deficient Tlr7.1 tg B cells. (A) Sorted transitional (T1), MZ, and Fo B cells from Tlr7.tg control and αv-CD19.Tlr7 mice were cultured with 5 μg/ml TLR7 ligand R848 for 0–120 min. Nuclear NF-κB (p65) localization was measured by Western blot, and LSD1 is shown as nuclear loading control. (B and C) Cytokine (B) and IgG2c (C) levels in supernatants of cultured B cell subpopulations after stimulation with R848 for 24 h [cytokines (B)] or 4 d [IgG2c (C)]. Data shown are mean ± SD of replicate cultures from one experiment. The p values <0.05 (Student t test) for comparisons between control Tlr7.tg and αv-CD19.Tlr7 mice are shown (*p < 0.05). Similar results were seen in at least three independent experiments.
To understand how αv-mediated changes in TLR signaling affect B cell responses, we first measured cytokine production after Tlr7 stimulation in vitro. Fo and MZ B cells from αv-CD19.Tlr7 both showed significant increases in cytokine production after stimulation when compared with Tlr7.tg control or non-tg controls (IL-10 and IL-6 production by Fo B cells and IFN-β production by MZ B cells) (Fig. 2B). T1 B cells produced little or no detectable TNF-α, IL-6, or IFN-β after stimulation but did secrete low levels of IL-10, which was increased in Tlr7.tg T1 cells and further increased in αv-CD19.Tlr7 T1 cells (Fig. 2B). Tlr7.tg transitional B cells have been shown to respond to TLR7 ligands by differentiating into IgG2c-producing cells (21). In agreement with this previous report, transitional Tlr7.tg B cells differentiated into IgG2c-producing cells after stimulation with R848 (Fig. 2C), as seen by IgG2c production in supernatants. Under the same conditions, cultures of MZ and Fo B cells also differentiated into IgG2c-producing cells, although at considerably lower levels than in T1 cultures. αv deletion greatly increased IgG2c production in T1 cells, with cultures from αv-CD19.Tlr7 mice producing 2–3-fold higher levels of IgG2c Ab than the equivalent cells from Tlr7.tg controls (Fig. 2C). Together, these data demonstrate that αv normally regulates TLR7 signaling in transitional, MZ, and Fo B cells, and deletion of αv leads to increased Tlr7 responses in Tlr7.tg B cells. This has distinct functional consequences in B cell subsets such that loss of αv leads to increased cytokine production by MZ and Fo cells while it increases differentiation of transitional cells to IgG2c-producing cells.
αv-deficient B cells drive increased T cell activation
T cell activation by B cells is increasingly appreciated as an important contributor to autoimmune disease (16). We therefore determined whether the increased B cell TLR responses in αv-CD19.Tlr7 mice may affect T cells. Consistent with previous reports of T cell activation in Tlr7.tg mice, 7–8-wk-old Tlr7.tg control mice had reduced numbers of spleen T cells and a significant increase in the proportion of activated CD44+ CD62Llow T cells. The frequency of spleen T cells was similar in Tlr7.tg and αv-CD19.Tlr7 mice, but activation of both CD4 and CD8 subsets was significantly increased in αv-CD19.Tlr7 mice (Fig. 3A, 3B). To determine whether αv integrins affect direct T cell activation by B cells, B cells from αv-CD19.Tlr7 mice or Tlr7.tg controls were cocultured with naive CD4 T cells in the presence of anti-CD3 Abs and T cell activation and proliferation measured. T cells cultured with αv-knockout B cells had higher expression of the T cell activation markers CD69 and CD25 (Fig. 3C, 3D) and proliferated significantly more than T cells cultured with Tlr7.tg control B cells (Fig. 3E). T1, Fo, and MZ B cell subsets from αv-CD19.Tlr7 mice all shared this capacity for increased T cell activation, although this was most evident in MZ B cells (Fig. 3F). Hence, B cell–intrinsic effects of αv deletion promote increased activation of T cells in vivo, and our in vitro experiments support a direct role for αv-knockout B cells in T cell activation.
FIGURE 3.

Increased T cell activation in αv-CD19.Tlr7.1 tg mice. (A and B) Spleens from 7- to 8-wk-old wild-type control mice (con), Tlr7.tg controls (con-Tlr7.tg), and αv-CD19.Tlr7 mice (αv-Tlr7.tg) were analyzed for CD3+ CD4+ and CD3+ CD8+ T cells as a frequency of total splenocytes by FACS (A). CD4 and CD8 were further analyzed for expression of CD44 and CD62L cells as shown (B). Each data point represents a single mouse. The p values <0.05 (Mann–Whitney U test) for comparisons between control Tlr7.1 tg and αv-CD19.Tlr7.1 tg mice are shown (n = 6 non-tg and n ≥ 13 per group tg mice). *p < 0.05, **p < 0.01. (C–E) B cells from TLR7.tg controls and αv-CD19.Tlr7 mice were cultured with CD4+ T cells from control mice in the presence of anti-CD3. T cell activation (based on CD25 and CD69) (C and D) and cell proliferation measured by [3H]thymidine incorporation (E) were measured after 3 d. (F) Sorted transitional, MZ, and Fo B cells from control TLR7.1 tgs (con-Tlr7.1) and αv-CD19.Tlr7.1 tg mice (αv-Tlr7.1) were cultured with control CD4+ T cells in the presence of anti-CD3, and cell proliferation was measured after 48 h. All data shown are mean ± SD of replicate cultures from one experiment. Similar results were seen in at least three independent experiments. The p values <0.05 (Student t test) for comparisons between control Tlr7.1 tg and αv-CD19.Tlr7.1 tg mice are shown. *p < 0.05, **p < 0.01.
αv deletion increases autoantibody response
To determine whether αv-deficient B cells affected development of autoimmunity, we examined production of autoantibodies. Autoimmunity in Tlr7.tg mice is associated with high levels of autoantibodies to RNA and associated Ags (9). Serum titers of anti-ssRNA IgG Abs were increased in αv-CD19.Tlr7 mice compared with littermate Tlr7.tg controls (Fig. 4A). Titers of IgG2c anti-ssRNA were also increased (Fig. 4A), consistent with the expansion of IgG2cclass-switched plasma cells in αv-CD19.Tlr7 mice. However, IgM anti-ssRNA Abs, which were elevated in Tlr7.tg controls compared with non-tg controls, were not further affected by αv deletion (Fig. 4A). Autoantibodies against the RNA-associated protein snRNP were also increased in αv-CD19.Tlr7 mice compared with Tlr7.tg controls, and in this case, IgM, IgG, and IgG2c isotypes were all increased (Fig. 4B). Autoantibodies were also assessed using a fluorescent ANA assay. Both Tlr7.tg and αv-CD19.Tlr7 mice had ANA-positive serum but exhibited different patterns of staining. Although Tlr7.tg controls had nuclear or mixed nuclear/cytoplasmic staining patterns, most αv-CD19.Tlr7 mice showed staining that was concentrated around the nucleus or in the cytoplasm, indicating that deletion of αv may alter the range of autoantibody targets (Fig. 4C). To test this and better understand the effects of αv on autoantibody repertoire, we used an autoantigen array to profile serum Abs from αv-CD19.Tlr7 mice and controls. αv deletion led to increases in all autoantibodies present in Tlr7.tg mice, with increases in both IgM and IgG isotypes (Fig. 4D), indicating that αv deletion increased all TLR7-driven auto-Ab responses.
FIGURE 4.

αv deletion from B cells increases autoantibody production (A). (A and B) Serum titers of anti-RNA and anti-snRNP Abs from 7 to 8-wk-old wild-type control, control Tlr7.1 tg, and αv-CD19.Tlr7.1 tg mice (n ≥ 11 per group). The p values <0.05 (Student t test) for comparisons between control Tlr7.1 tg and αv-CD19.Tlr7.1 tg mice are shown (*p < 0.05, **p < 0.01). (C) HEp-2 cell staining patterns of serum Abs from 7- to 8-wk-old control Tlr7.1 tg and αv-CD19.Tlr7.1 tg mice. Two representative images are shown per genotype. Original magnification ×40. Pie charts show summary of patterns for 5–6 mice per genotype. (D) Serum IgM and IgG autoantibodies from 7- to 8-wk-old control Tlr7.1 tg (n = 4) and αv-CD19.Tlr7.1 tg (n = 3) mice measured using an autoantibody array chip containing 88 specific autoantigens. Data are shown as a heat map of z-scores. Autoantigen class is shown, along with control Abs (anti-KLH and anti-LPS).
αv deletion increases kidney Ab deposition and glomerulonephritis
To determine whether the increased serum autoantibodies in the αv-CD19.Tlr7 mice could accelerate autoimmune organ damage, we analyzed Ab and complement deposition in kidneys of the mice at 7–8 wk. αv-CD19.Tlr7 mice showed higher levels of IgG staining in glomeruli than the Tlr7.tg mice (Fig. 5A). αv-CD19.Tlr7 mice also showed prominent glomerular C3 deposition at much higher levels than in Tlr7.tg control mice (Fig. 5B), consistent with increase in IgG2c class-switched autoantibodies in these mice. Because our analysis was focused on younger mice, we did not expect to see severe glomerulonephritis in these mice. However, histological analysis of kidney sections indicated increased glomerular size in αv-CD19.Tlr7 mice compared with controls, supporting acceleration of autoimmune pathology in these mice (Fig. 5C). We therefore conclude that deletion of αv integrins from B cells significantly accelerated autoimmunity as measured by splenomegaly, autoantibody production, and Ab deposition in organs, and this results in increased pathology and mortality.
FIGURE 5.

αv deletion increases kidney Ab deposition and glomerulonephritis. (A and B) Glomerular immune complex deposits determined by immunofluorescence staining for IgG (A) and complement C3 (B). Left panels show representative images from 7- to 8-wk-old control Tlr7.1 tg and αv-CD19.Tlr7.1 tg mice. Original magnification ×60. Right panels show intensity and area of staining. (C) Histological analysis of H&E-stained representative sections. Original magnification ×40. Graph represents quantification of glomerular size. Data are mean ± SD for at least n = 3 mice per group. The p values <0.05 (Mann–Whitney U test) for comparisons between control Tlr7.1 tg and αv-CD19.Tlr7.1 tg mice are shown. *p < 0.05, **p < 0.01.
Discussion
Although TLR7 signaling has been shown to be critical for development of lupus-like autoimmunity in mice (6, 9, 16, 17), little is known about how TLR signaling is normally regulated and how loss of regulation in individual cell types contributes to autoimmune disease. Our results identify αv integrin as a cell-intrinsic regulator of pathogenic TLR signaling and show that loss of this regulatory pathway in B cells exacerbates TLR7-driven autoimmunity, complementing previous studies that have shown that TLR signaling in B cells is required for autoimmunity. Dysregulated TLR signaling has differential outcomes on B cell subsets, promoting plasma cell differentiation, IgG2c class switching, cytokine production, and T cell activation. Together, this results in increased autoantibody production and tissue damage in the Tlr7.tg model. These data highlight the central role of B cell TLR signaling in autoimmunity and show that increasing TLR signaling in B cells is enough to accelerate autoantibody production and class switching, kidney damage, and activation of other immune cells.
Based on our previous work, we propose that deletion of αv from B cells increases autoimmunity through cell-intrinsic dysregulation of B cell TLR signaling (19, 20). Our data support this model, showing that isolated αv-knockout Tlr7.tg B cells respond more strongly to TLR stimulation in culture. This was particularly clear in transitional B cells, in which αv deletion resulted in increased TLR7-driven NF-κB activation, similar to our previous observations in MZ and GC B cells (19, 20), and was also apparent in Fo B cells, in which αv-knockouts showed higher constitutive NF-κB activation. The high constitutive NF-κB activation in Tlr7.tg MZ B cells confounded our ability to show effects of αv on acute TLR signaling in this subset. However, the increase in T cell activation and cytokine secretion in αv-knockout MZ and Fo B cells confirmed that TLR signaling in these cell subsets is increased when αv is deleted. Further supporting our model, the effects of αv deletion on B cell TLR responses in culture matched the phenotypes of αv-CD19.Tlr7 mice, including increased IgG2c class switching and T cell activation. Notably, our data also reveal that dysregulation of TLR signaling had distinct effects on different B cell subsets.
The transitional B cell stage represents an early site of encounter of autoreactive B cells with self-antigens in the periphery and acts as a checkpoint for deletion of strongly autoreactive cells (25, 26). T1 B cells have been shown to be strongly affected by TLR7 overexpression, which leads to expansion of T1 B cells and drives their differentiation into autoreactive extrafollicular IgG2c-producing plasma cells in the red pulp (21). Supporting a regulatory role for αv in TLR signaling, αv deletion increased production of IgG2c by transitional cells in vitro and resulted in expansion of spleen IgG2c-producing plasma cells in vivo. Curiously, αv deletion did not appear to affect numbers of transitional cells in Tlr7.tg mice, although we have previously shown that transitional B cell numbers are increased in αv-CD19 mice (19), indicating that αv does regulate expansion of this B cell population in the absence of pathogenic TLR signaling. However, we did observe a previously unreported B220+ CD24− CD21mid population that develops in Tlr7.tg mice and is greatly expanded in αv-CD19.Tlr7 mice. This population appears to be distinct from the age-associated B cell populations seen in mouse autoimmunity models (24, 27, 28). The concurrent expansion of these cells and IgG2c-producing cells in young αv-CD19.Tlr7 mice and our observation that transitional cells produce more IgG2c in culture than other B cell subsets lead us to speculate that many of the IgG2c-producing cells we observe in αv-CD19.Tlr7 mice develop directly from an immature or transitional population as extrafollicular plasma cells (21). However, additional studies will be needed to confirm this. Recent studies of SLE patients have identified a new immature/transitional population of effector B cells that are poised to differentiate into autoantibody-producing plasma cells (29, 30) and develop through an extrafollicular pathway in response to TLR7 signaling. Thus, our data are in agreement with other studies of mouse models and SLE in highlighting the important role of TLR regulation at the immature stage of B cell development and suggest that activation of autoreactive immature cells and conversion to extrafollicular plasma cells is an important early step in autoimmunity.
Our data also point to important effects of αv-mediated regulation of TLR signaling on functions of other B cell subsets, including cytokine production and T cell activation, which were more pronounced in MZ and Fo B cells than transitional cells. T cell activation is an important feature of lupus-like autoimmunity and in mouse models and requires B cell TLR signaling (16). Our data show that dysregulated TLR signaling in B cells can further increase T cell activation. Whether this is due to increased cytokine production by B cells or changes in processing or presentation of Ags remains to be determined.
Regulation of TLR signaling by αvβ3 requires components of the autophagy pathway. Specifically, αvβ3 promotes recruitment of LC3 to TLR-containing endosomes, which is required for lysosomal fusion. Deletion of LC3b or a component of the LC3 conjugation machinery, Atg5, cause increased B cell responses to TLR ligands, reproducing the phenotype of αv-knockout cells (19). We would therefore expect that disruption of LC3 recruitment would also increase autoimmunity in the Tlr7.tg model. However, studies from the Huber group (31) report that deletion of Atg5 from B cells in the Tlr7.tg model has the opposite effect to αv deletion, reducing autoantibody production and autoimmune pathology. These seemingly contradictory findings reflect the involvement of LC3 in many distinct intracellular processes. Atg5 and LC3 are essential for classical autophagy (also referred to as macroautophagy), a process by which cells target, partition, and digest intracellular protein complexes, organelles, or pathogens via formation of a characteristic double-membrane autophagosome (32). Classical autophagy is essential for plasma cell homeostasis, and mice lacking Atg5 and other components of the LC3-lipidation pathway have fewer plasma cells and impaired Ab production (33, 34). Effects on plasma cells therefore explain the reduction in autoantibodies in Atg5-knockout mice. In contrast, αvβ3-mediated activation of Atg5 and recruitment of LC3 to Tlr endosomes occur via a noncanonical autophagy pathway, independent of macroautophagy. This pathway, which requires Rubicon and reactive oxygen species (19, 20), appears similar to the LC3-associated phagocytosis (LAP) pathway described by Green, Sanjuan, and Martinez (35–37). Disruption of LAP in mice results in SLE-like inflammatory responses (38), suggesting that noncanonical autophagy may also play an immune-regulatory role for noncanonical autophagy in myeloid cells, although it remains to be seen whether LAP involves αv and if it regulates TLR signaling in myeloid cells. These different and potentially opposing roles for core autophagy components in immune cells may explain the complex and poorly understood role of autophagy in SLE and other autoimmune diseases (32, 39–41).
We have previously shown that αvβ3 directs the intracellular trafficking of TLRs and their ligands to lysosomes, where they are degraded and signaling is terminated (19). Deletion of αv or β3 delays TLR trafficking, resulting in increased and prolonged signaling (19). Although previous studies have focused on the role of TLR trafficking in delivering TLRs to endosomes (42–45), regulating ligand engagement (46–52), or determining signaling through NF-κB and IRFs (53–57), our studies identified an additional and critical role for the rate of trafficking in regulating TLR signaling. Our finding that αv-CD19 mice develop spontaneous autoantibodies (19) and accelerate TLR7-driven autoimmunity suggest that this αv-mediated regulatory pathway is particularly important in preventing autoimmunity to self-derived TLR ligands, such as nuclear Ags. αv integrins have long been known to be involved in clearance of apoptotic cells and other cellular debris that serve as a rich source of autoantigens and nucleic acid TLR ligands. We propose that αvβ3 functions as a coreceptor for these self-derived TLR ligands and regulates TLR-ligand signaling to prevent autoimmunity. Supporting this model, deletion of MFG-E8 also promotes autoimmunity in mice (58) and directs the intracellular trafficking of apoptotic material in DCs (59). The recent findings that another integrin, αMβ2, also acts as a coreceptor for TLR ligands (60) and regulates TLR signaling in the context of autoimmunity (61, 62) suggest that this mechanism of TLR regulation may not be limited to αvβ3 integrins. Together, these data point to the importance of regulating the strength of TLR signaling in B cells for preventing pathogenic B cell activation by self-antigens. Moreover, these data highlight the underlying role of molecules such as integrins and autophagy proteins that regulate intracellular trafficking events in adjusting immune signaling and maintaining tolerance.
Supplementary Material
Acknowledgments
We thank all members of the Acharya, Stuart, Hamerman, and Lacy-Hulbert laboratories for assistance and advice on this project. We thank the vivarium staff and core facilities at Benaroya Research Institute for expert assistance. Graphical abstract was created using Biorender.com.
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (NIH) Grant DK093695 (to A.L.-H.) and National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH Grants AR076242 and AI150178 (both to J.A.H.).
The online version of this article contains supplemental material.
- ANA
- anti-nuclear Ab
- DC
- dendritic cell
- Fo
- follicular
- GC
- germinal center
- LAP
- LC3-associated phagocytosis
- MZ
- marginal zone
- pDC
- plasmacytoid dendritic cell
- SLE
- systemic lupus erythematosus
- snRNP
- small nuclear ribonucleoprotein
- T1
- transitional 1
- tg
- transgenic.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.García-Ortiz H., Velázquez-Cruz R., Espinosa-Rosales F., Jiménez-Morales S., Baca V., Orozco L. 2010. Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Ann. Rheum. Dis. 69: 1861–1865. [DOI] [PubMed] [Google Scholar]
- 2.Shen N., Fu Q., Deng Y., Qian X., Zhao J., Kaufman K. M., Wu Y. L., Yu C. Y., Tang Y., Chen J.-Y., et al. 2010. Sex-specific association of X-linked toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 107: 15838–15843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawasaki A., Furukawa H., Kondo Y., Ito S., Hayashi T., Kusaoi M., Matsumoto I., Tohma S., Takasaki Y., Hashimoto H., et al. 2011. TLR7 single-nucleotide polymorphisms in the 3′ untranslated region and intron 2 independently contribute to systemic lupus erythematosus in Japanese women: a case-control association study. Arthritis Res. Ther. 13: R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee Y. H., Lee H.-S., Choi S. J., Ji J. D., Song G. G. 2012. Associations between TLR polymorphisms and systemic lupus erythematosus: a systematic review and meta-analysis. Clin. Exp. Rheumatol. 30: 262–265. [PubMed] [Google Scholar]
- 5.Tian J., Ma Y., Li J., Cen H., Wang D.-G., Feng C.-C., Li R.-J., Leng R.-X., Pan H.-F., Ye D.-Q. 2012. The TLR7 7926A>G polymorphism is associated with susceptibility to systemic lupus erythematosus. Mol. Med. Rep. 6: 105–110. [DOI] [PubMed] [Google Scholar]
- 6.Pisitkun P., Deane J. A., Difilippantonio M. J., Tarasenko T., Satterthwaite A. B., Bolland S. 2006. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science 312: 1669–1672. [DOI] [PubMed] [Google Scholar]
- 7.Subramanian S., Tus K., Li Q.-Z., Wang A., Tian X.-H., Zhou J., Liang C., Bartov G., McDaniel L. D., Zhou X. J., et al. 2006. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl. Acad. Sci. USA 103: 9970–9975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolland S., Yim Y. S., Tus K., Wakeland E. K., Ravetch J. V. 2002. Genetic modifiers of systemic lupus erythematosus in FcgammaRIIB(-/-) mice. J. Exp. Med. 195: 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deane J. A., Pisitkun P., Barrett R. S., Feigenbaum L., Town T., Ward J. M., Flavell R. A., Bolland S. 2007. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 27: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kono D. H., Baccala R., Theofilopoulos A. N. 2013. TLRs and interferons: a central paradigm in autoimmunity. Curr. Opin. Immunol. 25: 720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Viglianti G. A., Lau C. M., Hanley T. M., Miko B. A., Shlomchik M. J., Marshak-Rothstein A. 2003. Activation of autoreactive B cells by CpG dsDNA. Immunity 19: 837–847. [DOI] [PubMed] [Google Scholar]
- 12.Lau C. M., Broughton C., Tabor A. S., Akira S., Flavell R. A., Mamula M. J., Christensen S. R., Shlomchik M. J., Viglianti G. A., Rifkin I. R., Marshak-Rothstein A. 2005. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/toll-like receptor 7 engagement. J. Exp. Med. 202: 1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leadbetter E. A., Rifkin I. R., Hohlbaum A. M., Beaudette B. C., Shlomchik M. J., Marshak-Rothstein A. 2002. Chromatin-IgG complexes activate B cells by dual engagement of IgM and toll-like receptors. Nature 416: 603–607. [DOI] [PubMed] [Google Scholar]
- 14.Koh Y. T., Scatizzi J. C., Gahan J. D., Lawson B. R., Baccala R., Pollard K. M., Beutler B. A., Theofilopoulos A. N., Kono D. H. 2013. Role of nucleic acid-sensing TLRs in diverse autoantibody specificities and anti-nuclear antibody-producing B cells. J. Immunol. 190: 4982–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan O., Shlomchik M. J. 1998. A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in MRL-lpr/lpr mice. J. Immunol. 160: 51–59. [PubMed] [Google Scholar]
- 16.Teichmann L. L., Schenten D., Medzhitov R., Kashgarian M., Shlomchik M. J. 2013. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity 38: 528–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christensen S. R., Shupe J., Nickerson K., Kashgarian M., Flavell R. A., Shlomchik M. J. 2006. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 25: 417–428. [DOI] [PubMed] [Google Scholar]
- 18.Jackson S. W., Scharping N. E., Kolhatkar N. S., Khim S., Schwartz M. A., Li Q.-Z., Hudkins K. L., Alpers C. E., Liggitt D., Rawlings D. J. 2014. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J. Immunol. 192: 4525–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acharya M., Sokolovska A., Tam J. M., Conway K. L., Stefani C., Raso F., Mukhopadhyay S., Feliu M., Paul E., Savill J., et al. 2016. αv Integrins combine with LC3 and atg5 to regulate Toll-like receptor signalling in B cells. Nat. Commun. 7: 10917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raso F., Sagadiev S., Du S., Gage E., Arkatkar T., Metzler G., Stuart L. M., Orr M. T., Rawlings D. J., Jackson S. W., et al. 2018. αv integrins regulate germinal center B cell responses through noncanonical autophagy. J. Clin. Invest. 128: 4163–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giltiay N. V., Chappell C. P., Sun X., Kolhatkar N., Teal T. H., Wiedeman A. E., Kim J., Tanaka L., Buechler M. B., Hamerman J. A., et al. 2013. Overexpression of TLR7 promotes cell-intrinsic expansion and autoantibody production by transitional T1 B cells. J. Exp. Med. 210: 2773–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akilesh H. M., Buechler M. B., Duggan J. M., Hahn W. O., Matta B., Sun X., Gessay G., Whalen E., Mason M., Presnell S. R., et al. 2019. Chronic TLR7 and TLR9 signaling drives anemia via differentiation of specialized hemophagocytes. Science 363: eaao5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buechler M. B., Teal T. H., Elkon K. B., Hamerman J. A. 2013. Cutting edge: type I IFN drives emergency myelopoiesis and peripheral myeloid expansion during chronic TLR7 signaling. J. Immunol. 190: 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubtsov A. V., Rubtsova K., Fischer A., Meehan R. T., Gillis J. Z., Kappler J. W., Marrack P. 2011. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood 118: 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.King L. B., Monroe J. G. 2000. Immunobiology of the immature B cell: plasticity in the B-cell antigen receptor-induced response fine tunes negative selection. Immunol. Rev. 176: 86–104. [DOI] [PubMed] [Google Scholar]
- 26.Su T. T., Guo B., Wei B., Braun J., Rawlings D. J. 2004. Signaling in transitional type 2 B cells is critical for peripheral B-cell development. Immunol. Rev. 197: 161–178. [DOI] [PubMed] [Google Scholar]
- 27.Manni M., Gupta S., Ricker E., Chinenov Y., Park S.-H., Shi M., Pannellini T., Jessberger R., Ivashkiv L. B., Pernis A. B. 2018. Regulation of age-associated B cells by IRF5 in systemic autoimmunity. Nat. Immunol. 19: 407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Myles A., Sanz I., Cancro M. P. 2019. T-bet+ B cells: a common denominator in protective and autoreactive antibody responses? Curr. Opin. Immunol. 57: 40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jenks S. A., Cashman K. S., Zumaquero E., Marigorta U. M., Patel A. V., Wang X., Tomar D., Woodruff M. C., Simon Z., Bugrovsky R., et al. 2018. Distinct effector B cells induced by unregulated toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. [Published erratum appears in 2020 Immunity 52: 203.] Immunity 49: 725–739.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tipton C. M., Fucile C. F., Darce J., Chida A., Ichikawa T., Gregoretti I., Schieferl S., Hom J., Jenks S., Feldman R. J., et al. 2015. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat. Immunol. 16: 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weindel C. G., Richey L. J., Bolland S., Mehta A. J., Kearney J. F., Huber B. T. 2015. B cell autophagy mediates TLR7-dependent autoimmunity and inflammation. Autophagy 11: 1010–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levine B., Kroemer G. 2019. Biological functions of autophagy genes: a disease perspective. Cell 176: 11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conway K. L., Kuballa P., Khor B., Zhang M., Shi H. N., Virgin H. W., Xavier R. J. 2013. ATG5 regulates plasma cell differentiation. Autophagy 9: 528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pengo N., Scolari M., Oliva L., Milan E., Mainoldi F., Raimondi A., Fagioli C., Merlini A., Mariani E., Pasqualetto E., et al. 2013. Plasma cells require autophagy for sustainable immunoglobulin production. Nat. Immunol. 14: 298–305. [DOI] [PubMed] [Google Scholar]
- 35.Sanjuan M. A., Dillon C. P., Tait S. W. G., Moshiach S., Dorsey F., Connell S., Komatsu M., Tanaka K., Cleveland J. L., Withoff S., Green D. R. 2007. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450: 1253–1257. [DOI] [PubMed] [Google Scholar]
- 36.Martinez J., Malireddi R. K. S., Lu Q., Cunha L. D., Pelletier S., Gingras S., Orchard R., Guan J.-L., Tan H., Peng J., et al. 2015. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 17: 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Martinez J., Almendinger J., Oberst A., Ness R., Dillon C. P., Fitzgerald P., Hengartner M. O., Green D. R. 2011. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. USA 108: 17396–17401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez J., Cunha L. D., Park S., Yang M., Lu Q., Orchard R., Li Q.-Z., Yan M., Janke L., Guy C., et al. 2016. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells [Published erratum appears in 2016 Nature 539: 124.]. Nature 533: 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Deretic V., Kimura T., Timmins G., Moseley P., Chauhan S., Mandell M. 2015. Immunologic manifestations of autophagy. J. Clin. Invest. 125: 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gros F., Arnold J., Page N., Décossas M., Korganow A.-S., Martin T., Muller S. 2012. Macroautophagy is deregulated in murine and human lupus T lymphocytes. Autophagy 8: 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pierdominici M., Vomero M., Barbati C., Colasanti T., Maselli A., Vacirca D., Giovannetti A., Malorni W., Ortona E. 2012. Role of autophagy in immunity and autoimmunity, with a special focus on systemic lupus erythematosus. FASEB J. 26: 1400–1412. [DOI] [PubMed] [Google Scholar]
- 42.Kim Y.-M., Brinkmann M. M., Paquet M.-E., Ploegh H. L. 2008. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 452: 234–238. [DOI] [PubMed] [Google Scholar]
- 43.Tabeta K., Hoebe K., Janssen E. M., Du X., Georgel P., Crozat K., Mudd S., Mann N., Sovath S., Goode J., et al. 2006. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via toll-like receptors 3, 7 and 9. Nat. Immunol. 7: 156–164. [DOI] [PubMed] [Google Scholar]
- 44.Lee B. L., Moon J. E., Shu J. H., Yuan L., Newman Z. R., Schekman R., Barton G. M. 2013. UNC93B1 mediates differential trafficking of endosomal TLRs. Elife 2: e00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiang C.-Y., Engel A., Opaluch A. M., Ramos I., Maestre A. M., Secundino I., De Jesus P. D., Nguyen Q. T., Welch G., Bonamy G. M. C., et al. 2012. Cofactors required for TLR7- and TLR9-dependent innate immune responses. Cell Host Microbe 11: 306–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Means T. K., Latz E., Hayashi F., Murali M. R., Golenbock D. T., Luster A. D. 2005. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J. Clin. Invest. 115: 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chaturvedi A., Dorward D., Pierce S. K. 2008. The B cell receptor governs the subcellular location of toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity 28: 799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lande R., Gregorio J., Facchinetti V., Chatterjee B., Wang Y.-H., Homey B., Cao W., Wang Y.-H., Su B., Nestle F. O., et al. 2007. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449: 564–569. [DOI] [PubMed] [Google Scholar]
- 49.Ivanov S., Dragoi A.-M., Wang X., Dallacosta C., Louten J., Musco G., Sitia G., Yap G. S., Wan Y., Biron C. A., et al. 2007. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 110: 1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tian J., Avalos A. M., Mao S.-Y., Chen B., Senthil K., Wu H., Parroche P., Drabic S., Golenbock D., Sirois C., et al. 2007. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE [Published erratum appears in 2007 Nat. Immunol. 8: 780.]. Nat. Immunol. 8: 487–496. [DOI] [PubMed] [Google Scholar]
- 51.Barton G. M., Kagan J. C., Medzhitov R. 2006. Intracellular localization of toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat. Immunol. 7: 49–56. [DOI] [PubMed] [Google Scholar]
- 52.Ewald S. E., Engel A., Lee J., Wang M., Bogyo M., Barton G. M. 2011. Nucleic acid recognition by toll-like receptors is coupled to stepwise processing by cathepsins and asparagine endopeptidase. J. Exp. Med. 208: 643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mantegazza A. R., Guttentag S. H., El-Benna J., Sasai M., Iwasaki A., Shen H., Laufer T. M., Marks M. S. 2012. Adaptor protein-3 in dendritic cells facilitates phagosomal toll-like receptor signaling and antigen presentation to CD4(+) T cells. Immunity 36: 782–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sasai M., Linehan M. M., Iwasaki A. 2010. Bifurcation of toll-like receptor 9 signaling by adaptor protein 3. Science 329: 1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kagan J. C., Su T., Horng T., Chow A., Akira S., Medzhitov R. 2008. TRAM couples endocytosis of toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 9: 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Honda K., Ohba Y., Yanai H., Negishi H., Mizutani T., Takaoka A., Taya C., Taniguchi T. 2005. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 434: 1035–1040. [DOI] [PubMed] [Google Scholar]
- 57.Barton G. M., Kagan J. C. 2009. A cell biological view of toll-like receptor function: regulation through compartmentalization. Nat. Rev. Immunol. 9: 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hanayama R., Tanaka M., Miyasaka K., Aozasa K., Koike M., Uchiyama Y., Nagata S. 2004. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 304: 1147–1150. [DOI] [PubMed] [Google Scholar]
- 59.Peng Y., Elkon K. B. 2011. Autoimmunity in MFG-E8-deficient mice is associated with altered trafficking and enhanced cross-presentation of apoptotic cell antigens. [Published erratum appears in 2012 J. Clin. Invest. 122: 782.] J. Clin. Invest. 121: 2221–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han C., Jin J., Xu S., Liu H., Li N., Cao X. 2010. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat. Immunol. 11: 734–742. [DOI] [PubMed] [Google Scholar]
- 61.Faridi M. H., Khan S. Q., Zhao W., Lee H. W., Altintas M. M., Zhang K., Kumar V., Armstrong A. R., Carmona-Rivera C., Dorschner J. M., et al. 2017. CD11b activation suppresses TLR-dependent inflammation and autoimmunity in systemic lupus erythematosus. J. Clin. Invest. 127: 1271–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reed J. H., Jain M., Lee K., Kandimalla E. R., Faridi M. H., Buyon J. P., Gupta V., Clancy R. M. 2013. Complement receptor 3 influences toll-like receptor 7/8-dependent inflammation: implications for autoimmune diseases characterized by antibody reactivity to ribonucleoproteins. J. Biol. Chem. 288: 9077–9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.