

Key Points
Clec9A-targeted Ag induces humoral responses.
cDC1 directly present native Ag to B cells for B cell activation.
Clec9A maintains native Ag on the cDC1 surface but DEC205 does not.
Visual Abstract
Abstract
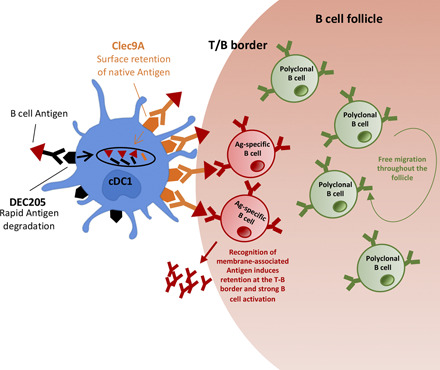
Follicular dendritic cells and macrophages have been strongly implicated in presentation of native Ag to B cells. This property has also occasionally been attributed to conventional dendritic cells (cDC) but is generally masked by their essential role in T cell priming. cDC can be divided into two main subsets, cDC1 and cDC2, with recent evidence suggesting that cDC2 are primarily responsible for initiating B cell and T follicular helper responses. This conclusion is, however, at odds with evidence that targeting Ag to Clec9A (DNGR1), expressed by cDC1, induces strong humoral responses. In this study, we reveal that murine cDC1 interact extensively with B cells at the border of B cell follicles and, when Ag is targeted to Clec9A, can display native Ag for B cell activation. This leads to efficient induction of humoral immunity. Our findings indicate that surface display of native Ag on cDC with access to both T and B cells is key to efficient humoral vaccination.
Introduction
T-dependent humoral immunity requires a complex cellular interplay to facilitate both T and B cell activation and differentiation. T cell activation generally depends on Ag presentation by dendritic cells (DC), which provide antigenic as well as costimulatory signals. B cell activation is also dependent on antigenic signals, but in this case, native unprocessed Ags are required, generally in a form capable of cross-linking the BCR. This is often in the context of membranes on host cells, such as macrophages, follicular DC (FDC), and even conventional DC (cDC) (1). This context also enables B cells to efficiently capture Ag for presentation to Th cells (2). Display of native Ag by host cells is often facilitated by receptors that bind complement or Abs or by surface receptors that bind pathogen carbohydrates (3).
Although FDC are crucial for presentation of Ags to B cells during germinal center (GC) reactions (3), they can also participate in initial B cell activation (4), as can the CD169+ macrophage (5). These macrophages line the marginal sinus in the lymph nodes (LN) and the marginal zone in the spleen and capture various forms of Ag (6–11), providing them to B cells (6–8). Ag encounter by cognate B cells leads to their activation and migration to the T–B border in the pursuit of T cell help.
Several early studies also implicated cDC in presentation of native Ags to B cells for induction of humoral immunity (12–18). These studies were followed by a seminal report using adoptive transfer of Ag-pulsed cDC, which visualized cDC presenting native Ag to B cells in vivo (19). These cDC migrated to regions in the LN surrounding the high endothelial venules (HEV), where they presented native Ag to B cells entering the LN as they began their migration to the follicles. Such presentation by Ag-bearing cDC led to efficient Ag-uptake by B cells and to the subsequent activation and migration of these B cells to the T–B border for the acquisition of T cell help.
Although these earlier studies focused on cDC in general, subsequent studies revealed that cDC consist of functionally distinct subsets that express an array of surface molecules. In the murine spleen, cDC can be broadly divided into two subsets: XCR1+ cDC and CD11b+ cDC, now referred to as cDC1 and cDC2 (20–22). cDC1 differentially express various molecules, including XCR1, CD8α, and the C-type lectin-like molecules Clec9A (also termed DNGR1) and DEC205 (23–27). These cDC are very efficient at processing Ags for delivery into the MHC class I pathway (28–31). The other main cDC subset, cDC2, is identified by its differential expression of molecules, such as CD11b, Sirpα, and the C-type lectin-like molecule DCIR2 (26, 30, 32). cDC2 preferentially process exogenous Ags for delivery into the MHC class II (MHC II) presentation pathway (30).
Although adoptive transfer studies revealed a capacity for cDC to present native Ag to B cells, it was unclear which subsets, cDC1 versus cDC2, participated in responses to in vivo–administered Ags. One study used Ag targeting to the DCIR2 receptor on cDC2 to examine this subset’s role in B cell activation (33). Injection of DCIR2-targeted Ag but not untargeted Ag led to T-independent B cell activation within 24 hours, suggesting that targeted Ag was captured by cDC2 and directly presented to B cells. A more recent study, examining humoral immunity to influenza virus, implicated cDC2 in B cell responses (34). This study introduced virus into the lung, then examined transport to the draining LN and induction of T follicular helper (Tfh) cells and Ab responses. Although both migratory cDC1 and cDC2 were shown to transport Ag to the LN, impairment of cDC2 migration in DOCK8-deficient mice abrogated humoral immunity, despite the normal migration of Ag-bearing cDC1. The requirement for cDC2 was explained by differential localization, with cDC1 migrating to the deep T cell zone and cDC2 migrating to the T–B border.
Consistent with this notion, Ags delivered to cDC2 via various receptors induce strong Ag-specific Ab responses, often in the absence of adjuvant, whereas Ags delivered to cDC1 via targeting DEC205 generally induce poor responses (33, 35, 36). However, not all targeting strategies align with this view. Our studies have shown that Ags delivered to cDC1 via Clec9A induce potent humoral responses associated with strong GC Tfh responses and memory (24, 37–39). These findings imply that under the right circumstances cDC1 can participate in humoral immunity. They also raise the possibility that properties of the targeted surface molecule itself (DEC205 versus Clec9A) may influence immune outcome.
Clec9A is a C-type lectin-like molecule (23–25) selectively expressed by DC and their precursors, with high levels on mouse cDC1 and low levels on plasmacytoid DC (24, 25). Clec9A is a type II membrane protein with a single extracellular C-type lectin-like domain and a cytoplasmic YXXL HemITAM that can signal through Syk (23–25), although its activation in cDC has not been demonstrated (40). Our studies examining immunity in which Ag is targeted to Clec9A have shown induction of strong T-dependent humoral responses that are independent of Fc receptor signaling, even in the absence of adjuvant (24, 38). This finding extends from mice to nonhuman primates (37), suggesting it is a general property of targeting this receptor. The strong humoral response induced by Ags targeted to Clec9A is also associated with formation of both effector and memory Tfh cells (38, 39). Together, these findings suggest that targeting Clec9A induces what is essentially conventional T-dependent humoral immunity. Because engagement of Clec9A on cDC1 does not lead to obvious signaling or direct cDC1 activation or to induction of cDC1 gene expression (24, 25, 40), the mechanistic basis for such effective priming has, until now, been somewhat obscure. It is evident that persistence within the circulation is a property of Clec9A-targeted Ag, which may, in part, explain its potency (38). What is not clear, however, is whether cDC1 can directly present Ag to B cells and whether such a process contributes to the efficient induction of humoral immunity.
In this study, we show that Clec9A-targeted Ag generates strong humoral immunity through efficient B cell activation as a consequence of direct recognition of Ag displayed on the surface of targeted cDC1. Such recognition is not essential for Ag capture but is important for B cell activation, which initiates relocalization of Ag-specific B cells to the T–B border. Importantly, B cells residing in follicles have direct access to cDC1-presented Ags within border regions of the follicle, suggesting that, like macrophages of the subcapsular sinus, cDC1 are able to directly present Ag to cognate follicular B cells. The efficient and prolonged display of Ag on the surface of cDC1 after Clec9A targeting contrasts with the transient and inefficient display after DEC205 targeting, explaining relatively poor humoral responses when engaging this receptor. Our studies show an important role for Ag display by cDC1 in Clec9A-targeted immunity and highlight the importance of this property in humoral responses by this cDC subset.
Materials and Methods
Mice and immunization
All procedures were performed in strict accordance with the recommendations of the Australian code of practice for the care and use of animals for scientific purposes. The protocols were approved by the Melbourne Health Research Animal Ethics Committee, The University of Melbourne.
C57BL/6 (B6), B6.SJLPtprcaPep3b/BoyJ (CD45.1+), Clec9a−/− (41), uGFP, DsRed, B6.129S2-H2dlAb1-Ea (MHC II−/−), μMT, B1-8hi, B1-8hi.uGFP, Xcr1Venus/+, OB1.Rag−/−, OB1.Rag−/−.uGFP and OT-IICD45.1+ mice were used between 6 and 12 wk of age.
DC-targeting Ab constructs and vaccination
Whole chicken OVA protein or the OVAFGD construct (see later) and the enterovirus 71 peptide were genetically fused to the H chain C-terminal regions of anti-mouse Clec9A (clone 24/04-10B4), anti-DEC205 (clone NLDC145), and rat IgG2a isotype control (clone GL117) via an alanine linker using a published protocol (38). The amino acid sequence of the OVAFGD construct is as follows: AAAALESIINFEKLTELKISQAVHAAHAEINEAGREVKLPGFGDSIE. The underlined amino acid sequences correspond to linear epitopes for OT-I, OT-II, and OB1 in that order. DC-targeting Ab constructs were expressed using Freestyle 293-F cells and then purified using protein G as previously described (38, 42). 4-hydroxy-3-nitrophenylacetyl (NP) was chemically conjugated to various mAbs and mAb–OVA fusion proteins, as indicated in the figure legends, using a published protocol (42). NP to Ig conjugation ratios ranged from seven to eight as determined by spectrophotometry. DC-targeting mAb constructs (0.1–5 μg) were prepared in PBS and injected via the tail vein in the absence of adjuvant.
Adoptive transfer of B cells and CD4+ T cells
OB1 B cells were prepared from the spleen of OB1.Rag−/− or OB1.Rag−/−.uGFP mice. Splenocytes from these mice yielded ∼90% pure Ag-specific CD19+ B cells upon removal of erythrocytes. B cells from B1-8hi, B1-8hi.uGFP, B6, uGFP, or DsRed mice were negatively enriched from the spleens depleted of erythrocytes using anti-mouse CD43 (Ly-48) MACS MicroBeads (Miltenyi Biotec) as per manufacturer’s protocol. In some experiments, non-B cells were first stained with anti-CD43 PE (S7; BD Biosciences) and then removed using Anti-PE MicroBeads (Miltenyi Biotec) as per manufacturer’s protocol.
CD4+ T cells were negatively enriched from cutaneous LNs and the spleen from OT-II mice using a previously published protocol (39). Briefly, single-cell suspensions depleted of erythrocytes were prepared and stained with anti–Mac-1 (M1/70), anti-F4/80 (F4/80), anti-erythrocytes (anti-Ter119), anti–Gr-1 (RB6-8C5), anti–I-A/E (M5/114), and anti-CD8 (53.6-7). Non-CD4 T cells were magnetically removed using BioMag Goat Anti-Rat IgG beads (QIAGEN).
In some experiments, purified B cells and T cells were labeled with various inorganic dyes for imaging or to assess cell division. Cells prepared in 0.1% BSA/PBS were labeled with 2.5–5 μM CFSE or 5 μM CellTrace Violet dye (CTV) for 9.5 min at 37°C. For CellTracker Deep Red dye (CTDR), cells prepared in PBS were labeled with 500 nM of the dye for 15 min at 37°C.
All cells were injected via the tail vein (i.v.) using 29-gauge BD Ultra-Fine Insulin Syringes in 200 μl of HBSS. A total of 5 × 104–106 T cells and/or 105–106 B cells were transferred for FACS experiments 1–2 d before immunization as indicated in figure legends. For imaging experiments, 1–2 × 106 T/B cells were transferred 1–5 d before immunization.
ELISA
Nunc-Immuno MicroWell 96-Well ELISA Plates (300 μl, round bottom) were coated overnight at 4°C with 50 μl of PBS containing 5 μg/ml OVA-NP16–17 (in-house). For anti-SP70 IgG ELISA. Round-bottom 96-well ELISA Plates (Costar) were coated with 3 μg/ml OVA-conjugated SP70 peptide (Mimotopes). Unbound protein was washed away (PBS, 0.05% Tween 20). Serially diluted plasma samples (PBS, 5% skim milk powder) were plated and incubated overnight at 4°C. Bound mouse IgG Abs were detected using donkey anti-mouse IgG-HRP (catalog number AP192P; 1:20,000; Millipore) and visualized using 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (Sigma-Aldrich). OD at 405 nm was subtracted by background OD at 492 or 490 nm. The end-point titers were calculated by using cut-off values determined as twice (or five times for SP70) the OD of control wells without plasma.
Flow cytometry
All Abs were purchased from BioLegend, BD Biosciences, or eBioscience/Thermo Fisher Scientific. Cells were surface labeled with mAbs specific for B220 (RA3-6B2), CCR7 (4B12), CD3ε (145-2C11), CD4 (GK1.5, RM4.5), CD8α (53-6.7), CD11b (M1/70), CD11c (N418), CD19 (1D3), CD45.1 (A20), CD45.2 (104), CD69 (H1.2F3), CD86 (GL1), CD95 (Jo2), Ig L chain κ (RMK-45), Ig L chain λ (RML-42), MHC II I-A/I-E (M5/114), NK1.1 (PK136), PD1 (RMP1-40), TCR Vα2 (B20.1), and/or XCR1 (ZET). NP-binding B cells were detected using NP-allophycocyanin generated in-house. Fc-mediated binding was blocked with purified anti-CD16/32 (2.4G2) and 1% normal mouse/rat serum (Jackson ImmunoResearch). Biotinylated Abs were detected using streptavidin PECy7/Brilliant Violet 605/allophycocyanin (BioLegend). Dead cells were excluded by propidium iodide (Sigma-Aldrich) or LIVE/DEAD Fixable Near-IR or Violet Dead Cell Stain Kits (Thermo Fisher Scientific). Analysis was restricted to single cells, which were identified using forward scatter height and forward scatter area. All surface staining was performed at 4°C for 30 min with the exception of Abs against CCR7 and CXCR5, which were incubated at room temperature (RT) for 45 min. To stain Bcl6, cells were first surface stained, washed, and fixed/permeabilized using eBioscience Intracellular Fixation and Permeabilization Buffer Set as per manufacturer’s protocol and then stained with anti-Bcl-6 (K112-91) PE (BD Biosciences) at RT for 45 min. Data were acquired on a FACSFortessa (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Immunofluorescence and confocal microscopy
LNs and spleens were embedded in OCT (Sakura) and snap-frozen using liquid nitrogen. Tissue sections were cut at a 12-μm thickness with a Cryostat (CM3050S; Leica) and air-dried before being fixed in acetone for 5 min. Sections were blocked with Protein Block Serum-Free (X0909; DAKO) for 10 min at RT. Sections were then stained with anti-IgD–Alexa Fluor 647 (A647) (11-26c.2a; BioLegend), anti-CD4–Alexa Fluor 488 (RM4.5; BioLegend), and anti-laminin (rabbit polyclonal; AbD Serotec) prepared in 2% normal donkey serum/PBS for 1.5–2 h at RT. Anti-laminin was detected by staining with polyclonal donkey anti-rabbit IgG–Alexa Fluor 568 (Thermo Fisher Scientific) for 30 min at RT. Slides were mounted using ProLong Gold Antifade Mountant (Thermo Fisher Scientific). Z-stacked tile images were acquired with an LSM710 confocal microscope (Carl Zeiss) and analyzed using Fiji/ImageJ (National Institutes of Health).
For quantification of OB1 cell distribution in the spleen, white pulp was defined using laminin staining as a guide. Within white pulp, IgD+ CD4− B zone and IgD− CD4+ T zone were detected, and the T–B border was manually drawn using the Freehand selection tool within Fiji/ImageJ software. The x, y coordinates of points along the border line at one-pixel intervals were determined using a custom Fiji/ImageJ plug-in. Binary mask images of the T zone and B zone were generated based on the white pulp, and the T–B border was determined as above. OB1 cells in the T zone and B zone were visualized using the Image Calculator “AND” function, and subsequently, these cells were detected and the x, y coordinates were determined semiautomatically using the Analyze Particle function (10–50 μm2 particle size). Minimum distance of individual cells from the T–B border was then calculated using R spatstat.
Imaging of thick spleen sections and cleared LNs
CTV-labeled polyclonal B cells and CTDR-labeled T cells were adoptively transferred into Xcr1Venus mice. Twenty-four hours later, skin-draining LNs and spleen were harvested and fixed in 2% periodate-lysine-paraformaldehyde (pH 7.4) at 4°C for 2 h and overnight, respectively. Using a vibrating microtome (Leica Biosystems), spleens were sectioned to 150 μm. Samples were washed in PBS prior to immersion in special wash buffer (0.2% BSA, 10% 10× PBS, 0.2% Tween 20, 0.2% Triton X-100, 0.2% 10% SDS; Baxter) at 4°C overnight. For immunolabeling, samples were incubated with B220–Pacific Blue, CD8–A647, and GFP polyclonal Abs diluted in wash buffer (0.2% BSA, 10% 10× PBS, 0.1% Tween 20, 0.2% 10% SDS; Baxter) for 3 d at 4°C. After incubation, samples were washed thoroughly with wash buffer twice and immersed in FUnGI clearing agent overnight and then transferred into 85% glycerol in PBS before imaging (43). Thick spleen sections were imaged using an LSM 710 confocal microscope (Carl Zeiss), and cleared LNs were imaged with an upright FVMPERS two-photon microscope (Olympus). Fluorescence excitation was provided by Mai Tai (690–1040 nm) and InSight (690–1300 nm) pulsed infrared lasers (Spectra-Physics), and fluorescence emission was detected using external non-descanned photomultiplier tubes. CTV, Pacific Blue, and Venus were excited at 820 nm, and CTDR and A647 at were excited at 1150 nm.
Intravital two-photon microscopy
Surgically exposed left inguinal LNs were prepared for intravital imaging via a modified version of a published protocol (19, 44) using an upright LSM 710 NLO multiphoton microscope (Carl Zeiss) or FVMPERS multiphoton microscope (Olympus) with a 20×/1.0 numerical aperture water immersion objective enclosed in an environmental chamber maintained at 37°C with heated air. Fluorescence excitation was provided by a Chameleon Vision II Ti:Sapphire laser (Coherent) with dispersion correction and fluorescence emission detected using external non-descanned photomultiplier tubes. EGFP, DsRed, and Venus were excited at 900–920 nm, and CTV and CTDR were excited at 800–820 nm when the LSM 710 microscope was used. For four-dimensional datasets, three-dimensional stacks were captured every 60 s for 30–90 min. For images acquired on the FVMPERS microscope, fluorescence excitation was provided by a Mai Tai (690–1040 nm) and InSight (690–1300 nm) pulsed infrared lasers (Spectra-Physics), and fluorescence emission was detected using external non-descanned photomultiplier tubes. CTV, Pacific Blue, and Venus were excited at 820 nm, and CTDR and A647 were excited at 1150 nm. For four-dimensional datasets, three-dimensional stacks were captured every 30 s for 17–40 min.
Raw imaging data were processed with Imaris 8.1.2+ (Bitplane). Autofluorescence was removed via the channel arithmetic function from Imaris XT (Bitplane). Cellular motion was tracked semiautomatically via a built-in spot tracking function aided by manual correction. Only the cells tracked for at least 5 min were included in analyses. To facilitate comparison across different movies, OB1 B cell track velocities were normalized against the median of polyclonal B cell track velocities.
To determine the motility of B cells that come in contact with XCR1-Venus+ cDC1 in Fig. 8E, the distance between the centroid of the B cell spot objects and the rendered surface of XCR1-Venus+ cDC1 was calculated using Imaris XT. B cells were considered in contact with cDC1 if they were within a 5-μm distance and not in contact if they were at least 15 μm away.
FIGURE 8.

Ag-specific B cells that encounter cDC1 have reduced migration velocity after Clec9A-targeted immunization. (A–D) OB1GFP+ and either DsRed+ or CTV+ polyclonal B cells were cotransferred into WT or Clec9a−/− mice 1–4 d before intravital two-photon imaging of inguinal LN was performed using the LSM 710 microscope. (A) Paired analyses of polyclonal B cells and OB1 median velocities in unimmunized WT mice. (B) OB1 median velocity in (A) normalized to polyclonal B cells median velocity. (C and D) B cell motility 1.5–3 h after immunization with indicated doses of αClec9A–OVAFGD. Analysis was performed as described in (A) and (B), respectively, for (C) and (D). (A–D) Each symbol represents a mouse. Blue, red, and green symbols indicate cells were CTV+, DsRed+, and GFP+, respectively. Pooled data from five (A and B), one [(C and D), 5 μg dose] or two [(C and D), 0.5 and 2 μg doses] adoptive transfer experiments. Statistical analysis was performed using paired Student t test (A and C) or unpaired Student t test (D). (E and F) CTV+ OB1 and CTDR+ polyclonal B cells were cotransferred into lethally irradiated B6 chimeric mice reconstituted with bone marrow cells from Xcr1Venus/+ mice. Intravital two-photon imaging of inguinal LN was performed 1.5–3 h postimmunization with 0.5 of μg αClec9A–OVAFGD i.v. using the LSM 710 microscope. (E) A single-frame three-dimensional-perspective image from a time-lapse video shows 10-min tracks of OB1 and polyclonal B cells. A surface object was created from Venus+ cDC1 in the paracortical region, and cell tracks were grouped into those that came into close proximity (<5-μm distance) with the Venus+ cDC surface (white tracks) or those that remained distant (>15-μm distance) from the surface (blue or red) during an ∼40-min imaging session. Scale bars, 50 μm. (F) Median velocities of cell tracks classified as in (E). Each symbol represents a mouse. Pooled data from two adoptive transfer experiments (n = 4 mice). ns = p ≥ 0.05, *p < 0.05, **p < 0.01 by one-way ANOVA, followed by Tukey test.
To determine contact between B cells and XCR1-Venus+ cDC1 in Fig. 7B, in which the generation of multiple cDC1 surfaces were required, surfaces were generated for cDC1 and spots were generated for B cells; both were tracked using custom Python scripts (with the use of the additional libraries; NumPy and scikit-image) interfaced with Imaris XT. Contact threshold was determined when the distance between the centroid of the B cell spot objects and the rendered surface of XCR1-Venus+ cDC1 was ≤5 μm. Movies were generated in Imaris and composed in After Effects (Adobe).
FIGURE 7.

B cells in the border regions of follicles are in close contact with cDC1. (A and B) A total of 5 × 106 CTDR+ polyclonal B cells (red) and 5 × 106 CTV+ polyclonal T cells (blue) were adoptively transferred into Xcr1Venus/+ mice (green) 1 d before intravital two-photon imaging of an inguinal LN using an FVMPERS microscope. One hour before imaging anti-mouse CD62L (MEL14 mAb) was administered i.v. to prevent new entry of lymphocytes into the LN. (A) Single frames from three mice were imaged. See also Supplemental Video 2. (B) B cell tracks for frames displayed in (A) were highlighted in red when B cells came in close proximity to cDC1 (≤5-μm distance) or blue when at greater distances (>5-μm distance). Scale bar, 50 μm. (C and D) 24 × 106 CTV+ polyclonal B cells (red) and 5 × 106 CTDR+ polyclonal T cells (blue) were adoptively transferred into Xcr1Venus/+ mice (green) 1 d before sacrifice to demarcate B cell follicles and T cell zones for imaging of whole cleared LN and 200–250 μm thick spleen vibratome sections. Cleared LN and thick spleen sections were labeled with anti-B220 (red) and anti-CD8 (blue) before imaging. (C) Representative cleared LN section and (D) representative thick spleen section. Scale bar, 100 μm. Representative image from one experiment (n = 3 mice) is shown. See also Supplemental Movies 3 and 4.
Mathematical modeling of B cell migration in follicles
Average follicle volumes were assessed in three dimensions in either LN or spleen. For LNs, inguinal and brachial LNs were fixed in 4% paraformaldehyde, labeled with B220-PB (BioLegend) and mounted in 85% glycerol for three-dimensional imaging on an Olympus FVMPERS multiphoton microscope. The images were analyzed in Imaris (Bitplane), and the follicle volumes were measured based on the external boundaries of the B cell areas. For the spleen, excised spleens were fixed in 4% paraformaldehyde, frozen in OCT, and thick spleen sections (40 μM) were cut on a Cryostat. The sections were labeled with B220-PB, imaged on a Zeiss LSM 700 confocal microscope, and the follicle diameters measured based on the diameter of the B cell area. The follicle volumes were then calculated assuming a spherical shape with the formula . In total, volumes for five LN follicles and 31 spleen follicles were measured, and the average follicle volume was determined to be 1.86 ×107 μm3.
The B cells are assumed to move with Brownian motion, so their location at time t in minutes is given by , where D is the diffusion coefficient in micrometers-squared per minute and Wt is standard Brownian motion in three dimensions. The cells were observed at 1-min intervals, moving at an average speed of 6.5 μm/min. Thus, the expected displacement after 1 min is . Each component of Wt is normally distributed with mean zero and variance t min, so the Euclidean distance of Wt from the origin is distributed according to the Chi distribution with three degrees of freedom. This gives and [(45) page 237]. Equating the observed and theoretical displacement when t = 1 min gives .
The Brownian motion of the B cells can be approximated as a discrete random walk in three dimensions, taking steps of length d micrometer every τ minutes. Let Sn be a random walk in three dimensions with step size d, then the mean-squared displacement after n steps (i.e., after nτ minutes) is . The mean-squared displacement of Brownian motion after nτ minute is given by [(46) page 56]. Equating these gives min. Then, if and , Sn converges to Brownian motion [(47) page 50]. Experiments showed that a step size of d = 2.5 μm is sufficiently small to approximate Brownian motion. Smaller step sizes produced essentially the same results. In this case, min.
The B cell follicles are assumed to be spherical. Experiments showed that deformations to a spherical follicle (keeping volume constant) produced a higher proportion of cells contacting the border in a 24-h period, so a spherical follicle represents a “worst-case” scenario. The cells’ starting positions within the follicle were chosen uniformly at random. The movement of N cells over a 24-h period was simulated in follicles of different volumes, and the proportion of cells contacting the border was recorded. As N becomes large, this proportion approaches the probability that a cell will contact the follicle border. N was chosen to be 10,000 because much larger choices for N produced essentially the same results.
In addition, in simulations in which cells start at the center of the follicle, >97, 83, and 79% of cells contact the border of follicles of sizes 1.86, 4.20, and 5.03 × 107 μm3, respectively. This is a worst-case scenario for the cells’ starting positions.
Statistical analysis
Figures were generated using GraphPad Prism 5 (GraphPad Software). Statistical analyses were performed with GraphPad Prism 5. The statistical test used and the p values are indicated in each figure legend. A p value < 0.05 was considered to indicate statistical significance.
Data availability
All data are available from the authors upon request.
Results
Clec9A targeting of the B cell epitope is required for efficient humoral immunity
Ags targeted to Clec9A on cDC1 induce efficient humoral immunity, underscored by generation of Tfh cells and GC reactions (24, 38, 39, 48). Because cDC are well known for their capacity to prime T cells, targeting Ag to cDC was expected to improve T cell responses, but whether this also facilitated cDC–B cell interactions that were important in humoral responses was unknown. To address this issue, we determined whether cDC–B cell collaboration contributed to the efficacy of Clec9A targeting by examining the effect of targeting the B cell epitope to cDC1 (as opposed to targeting the T cell epitope). NP, a well-characterized B cell hapten, was conjugated to a rat anti-mouse Clec9A mAb (αClec9A-NP) or to a rat IgG2a isotype control mAb (isotype-NP) (Fig. 1A). In B6 mice, the foreign rat Abs act as a source of Th cell epitopes (37), whereas NP acts as a potent B cell epitope. Using this model, we devised an immunization strategy that separated the contributions provided by targeting the Th epitopes versus the B cell epitopes to cDC1. This was achieved by comparing NP-specific IgG responses after vaccination with the following three alternative Ab combinations: group I, αClec9A-NP (which targets both T and B cell epitopes to Clec9A on cDC); group II, a mix of unmodified αClec9A (which targets the rat Th cell epitope alone to Clec9A on cDC) together with isotype-NP (which does not target the B cell epitope to Clec9A); or group III, isotype-NP alone (in which neither Th nor B cell epitopes are targeted to Clec9A). For group II, NP-specific B cells capturing isotype-NP should be able to acquire cognate help from rat Ig-specific T cells induced by αClec9A because both αClec9A and isotype-NP share rat Ig Th epitopes (Fig. 1A). This analysis showed that targeting both the T and B cell epitopes using αClec9A-NP (group I) induced a robust NP-specific IgG response, whereas untargeted T and B cell epitopes in the form of isotype-NP (group III) failed to induce any detectable response (Fig. 1B). Importantly, group II revealed that, whereas targeting the Th epitopes using αClec9A improved the response to the untargeted B cell epitopes (isotype-NP) (Fig. 1B; compare group II and III), the magnitude of this response was significantly less than that achieved when both the T and B cell epitopes were targeted to Clec9A on cDC1 (compare group I and II). This was the case over a wide range of Ag doses and more evident when the vaccine dose was limiting (Fig. 1B, 1C). Thus, there was a clear benefit to targeting the B cell epitope to Clec9A expressed by cDC1, implicating potential cDC1–B cell collaboration.
FIGURE 1.

Clec9A-mediated DC–B cell interactions promote Ab responses. (A) Diagrammatic representation of αClec9A-NP, αClec9A, and isotype-NP constructs. All three Abs are rat IgG2a and share common Th epitopes depicted as black bands. NP molecules coupled to αClec9A-NP and isotype-NP are shown as white hexagons. αClec9A-NP targets both NP and rat Ig Th epitopes to Clec9A. αClec9A only targets the rat Ig Th epitopes to cDC1. Isotype-NP does not target either NP or rat Ig Th epitopes to cDC1. (B) B6 mice were immunized i.v. with the following mixtures of Ag: group (I), 10 μg of αClec9A-NP; group (II), 10 μg of isotype-NP plus 10 μg of αClec9A; or group (III), 10 μg of isotype-NP, or (C) with lower doses of these Ag (as indicated). Anti-NP IgG titers were measured by ELISA 14 d postimmunization. Each symbol represents an individual mouse, and horizontal lines indicate the geometric mean (B and C). Pooled data from four independent experiments (n = 18–20 mice per group) (B); one to three independent experiments (n = 5–15 per group) [(C), 0.1 and 0.5 μg doses] or two to four independent experiments (n = 10–20 per group) [(C), 2 μg dose]. Statistical analysis was performed on log-transformed data sets by one-way ANOVA, followed by a Tukey test (B and C). See also Supplemental Fig. 1A for SP 70 Ag. ns = p ≥ 0.05, *p < 0.05, ***p < 0.001, ****p < 0.0001.
It was important to check whether the same benefit of targeting the B cell epitope to Clec9A applied when this epitope was a peptide sequence of a protein rather than a hapten. We tested this using the 15 aa SP70 peptide from the hand, foot, and mouth disease enterovirus 71, which contains a B cell–neutralizing epitope (and no T cell epitopes). We have previously demonstrated that by linking this otherwise weak immunogen to the rat anti-mouse Clec9A, thus providing both T cell epitopes and effective Clec9A targeting, an Ab response to SP70 is obtained, even in the absence of adjuvant (42). Experiments analogous to those in Fig. 1 were accordingly performed, substituting SP70 for NP. At lower doses of the SP70–Ig construct, a consistently significant anti-SP70 Ab titer was obtained only when both the T and the B cell epitopes were targeted to Clec9A (Supplemental Fig. 1A). This indicated that when this protein Ag was limiting, targeting both the B and T cell epitopes to Clec9A was important for an effective Ab response to ensue.
Ag capture by B cells is not Clec9A dependent
The above findings indicated that humoral responses were more effective when the B cell epitope was targeted to Clec9A rather than when left untargeted and soluble. This increased effectiveness most likely related to Clec9A targeting resulting in membrane-associated Ag, which enhanced B cell responses by allowing more effective Ag capture by specific B cells (via concentrating the Ag) and/or by inducing more efficient B cell activation (via receptor cross-linking). To assess these and other possibilities, we developed an approach that enabled monitoring of specific B cells by using the OVA-specific BCR transnuclear line, OB1, which produces B cells that recognize a linear epitope of OVA (i.e., an OVA peptide sequence centered on the FGD motif) (49). To examine the response of OB1 B cells after Clec9A targeting, we generated a construct termed αClec9A–OVAFGD that contained T cell (OT-I and OT-II) and B cell (OB1) epitopes of OVA genetically fused to the 10B4 αClec9A mAb (Fig. 2A). When OT-II cells and OB1 cells were cotransferred into congenic CD45.1+ mice, both populations proliferated more extensively in response to αClec9A–OVAFGD than to isotype–OVAFGD (Fig. 2B–E), validating the antigenicity of this construct for both T and B populations. Using this model, we could now explore the nature of cDC1–B cell collaboration engaged by Clec9A targeting.
FIGURE 2.

αClec9A–OVAFGD preferentially induces proliferation of OB1 and OT-II cells in vivo. (A) The amino acid sequence of OVAFGD. The sequence was cloned in-frame with the C-terminal region of the H chains of mAbs via alanine linkers. The amino acid sequences corresponding to OT-I, OT-II, and OB1 epitopes are underlined. (B–D) A total of 1 × 106 CTV+ OB1 cells and 1 × 106 CTV+ OT-II cells expressing GFP (OT-IIGFP) were cotransferred into CD45.1+ recipient mice 1 d before i.v. immunization with 2 μg of isotype–OVAFGD (Iso–OVAFGD) or αClec9A–OVAFGD. (B) Representative histograms showing proliferation of OB1 cells [left, gated on CD45.2+ CD19+ CD4− GFP− live cells as shown in (E)] and OT-II [right, gated on CD45.2+ CD19− CD4+ GFP+ live cells as shown in (E)] from the spleen on day 3. The gate identifies CTVlo-divided cells, and the number indicates the mean percentage of divided cells ± SEM. (C and D) CTVlo-divided OB1 cells (C) and OT-II cells (D) in the spleen were enumerated on day 3 postimmunization. Each symbol represents a mouse, and horizontal lines indicate the mean. Pooled data from three experiments with n = 5–10 mice. ns = p ≥ 0.05, ***p < 0.001 by one-way ANOVA, followed by a Tukey test.
As suggested above, one potential benefit of targeting the B cell epitope to Clec9A is that it may allow B cells to more efficiently acquire Ag from cDC1 (19). To test the efficacy of B cell acquisition of Ag in this setting, we labeled αClec9A Abs with A647. B6 mice were then adoptively transferred with OB1 B cells and immunized with either αClec9A–OVAFGD-A647 or isotype–OVAFGD–A647; then spleens were harvested at 1, 3, and 6 h postimmunization to assess OB1 cells for their acquisition of Ag by measuring A647 fluorescence (Fig. 3). The flow cytometry gating strategy for OB1 cells is shown in Supplemental Fig. 1B. This experiment showed efficient capture of Ag by OB1 B cells whether or not this Ag was targeted to Clec9A. Similar findings were apparent at 24 h (Supplemental Fig. 1C, 1D). Thus, efficient humoral responses induced by Clec9A-targeting did not appear to be linked to improved Ag acquisition by B cells.
FIGURE 3.

Clec9A targeting of Ag does not enhance its capture by B cells relative to untargeted Ag. A total of 1 × 106 OB1GFP+ cells were transferred into B6 mice 2 d before i.v. immunization with 0.5 μg of mAb–OVAFGD–A647 or isotype–OVAFGD (iso–OVA)–647, or with nothing (none). (A) The uptake of A647 by OB1 cells in the spleen was assessed at 1, 3, and 6 h postimmunization, and representative histograms are presented. (B) The geometric mean fluorescence intensity (gMFI) A647 fluorescence of OB1 cells from individual mice from (A). Similar analyses were performed at 24 h (Supplemental Fig. 1B–D). Each symbol represents a mouse, and horizontal lines indicate the mean. Data were pooled from two experiments with n = 6–7 mice. ****p < 0.0001 by one-way ANOVA, followed by Tukey test.
DC presentation of Ag to B cells leads to efficient B cell activation
Ag acquisition by OB1 B cells was clearly very efficient whether or not it was delivered to Clec9A, most likely because the antigenic component of both constructs was efficiently captured by the BCR of OB1 cells. Because Ag capture was not improved yet Ab responses were strongly enhanced by Ag targeting to Clec9A, we asked whether targeting instead improved initial B cell activation. To address this point, we measured activation markers on OB1 B cells in the spleen 24 h after vaccination with αClec9A–OVAFGD at a time point prior to cognate pre-Tfh cell expansion. This showed that Clec9A-targeting induced downregulation of CXCR5 and upregulation of CCR7 on OB1 B cells (Fig. 4A, 4B), both important chemokine receptors that control localization within secondary lymphoid organs. Such vaccination also led to upregulation of the MHC II and CD86 (Fig. 4A, 4B). Activation of OB1 B cells was strongly promoted by Clec9A targeting, as limited activation was seen in Clec9a−/− mice, in which no targeting would occur. The low level of activation seen in Clec9a−/− mice may have occurred because of small aggregates of Ag and/or contributions from Fc receptor–mediated cross-linking on cells such as marginal zone macrophages. Activation was also Ag-specific because nonspecific endogenous B cells were not activated by αClec9A–OVAFGD in wild-type (WT) or Clec9a−/− mice (Supplemental Fig. 2A). The initial activation of Ag-specific B cells was also independent of T cell priming, as similar activation was seen when OB1 cells were exposed to αClec9A–OVAFGD in MHC II–deficient mice, which lack CD4+ T cells (Fig. 4C). Of note, use the Clec9A-deficient mice as a control for Ag targeting in experiments, such as shown in Fig. 4A, is preferred over an isotype control mAb because Ag is then identical in the targeted and nontargeted conditions. Isotype control Abs always differ slightly from targeted mAb because of sequence variation within the Ag-binding domains. Although the use of Clec9A-deficient mice might affect Ag processing under some circumstances, use of these mice for examining initial steps in B cell activation, which is T-independent (Fig. 4C), should otherwise mimic the use of an isotype control mAb.
FIGURE 4.

Targeting Ag to Clec9A enhances early B cell activation, which occurs independent of T cell help. To examine B cell activation, 1 × 106 OB1GFP+ cells were adoptively transferred into B6 or Clec9a−/− mice 1 d before vaccination with 0.5 μg of αClec9A-OVAFGD. (A) Activation of OB1 cells in the spleen (GFP+CD19+CD3−PI−) was assessed 24 h postimmunization based on the expression levels of CXCR5, CCR7, CD86, and MHC II. (B) geometric mean fluorescence intensity (gMFI) values plotted are normalized against cells from naive mice. The activation phenotype of endogenous B cells (GFP−CD19+CD3−PI−) is shown in Supplemental Fig. 2A and was assessed as in (A). (C) Early Clec9A-mediated B cell activation is independent of T cells. A total of 1 × 106 CTV+ OB1 cells were adoptively transferred into B6 or MHC II−/− mice 1 d before vaccination with 2 μg of αClec9A–OVAFGD. Activation of OB1 cells (CTV+CD19+CD3−PI−) in the spleen was assessed based on the expression levels of CXCR5 and CCR7. gMFI values plotted are normalized against cells from naive mice. (D) Rapid activation of B cells is a general feature of Clec9A-targeted vaccination. Targeting the B cell hapten NP to Clec9A also enhances early activation to NP-specific B cells. A total of 1 × 106 GFP+ or CTV-labeled B1-8hi cells were adoptively transferred into B6 or Clec9a−/− mice 1 d before vaccination with 0.1 μg of αClec9A-NP. Activation of B1-8hi cells (GFP+/CTV+ IgLκ− NP-binding CD19+CD3−PI−). Similar experiments were performed using alternative αClec9A mAbs labeled with NP (Supplemental Fig. 2B). Each symbol represents a mouse, and horizontal lines indicate the mean. (B–D) Pooled data from two experiments with n = 6–8 mice (B), two experiments with n = 2–5 mice (C), or three independent experiments with n = 8–12 mice (D). ns = p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way ANOVA, followed by Tukey test (B–D).
To test whether these findings extended to B cells of a different specificity, we also examined activation of NP-specific B1-8hi IgH knock-in B cells (50) 24 h after vaccination with the NP-conjugated 10B4 clone of αClec9A (Fig. 4D). This showed that Ag-specific B cells were activated upon targeting NP to Clec9A, and such activation was absent in Clec9A-deficient mice. Furthermore, injection of two other αClec9A clones conjugated to NP, 7H11 and 1F6, caused similar activation of B1-8hi B cells (Supplemental Fig. 2B). Targeting Ag to Clec9A thus enables cDC1 to directly activate Ag-specific B cells independent of its effect on Tfh priming or the Ag used.
Clec9A-mediated activation of Ag-specific B cells leads to their migration toward T–B border for cognate pre-Tfh help
To investigate whether the observed changes in the surface receptors, suggestive of B cell activation, resulted in functional consequences, such as migration to the T–B border, we examined B cell localization after Clec9A targeting. Histological examination was undertaken of spleens from mice that received OB1 B cells and were then primed with αClec9A–OVAFGD (Fig. 5). This showed that OB1 B cells began to migrate to the T–B border by 6 h postimmunization and continued to accumulate in this region 12–24 h postimmunization. This migration depended on Clec9A targeting, as it was not evident in B6 mice primed with untargeted isotype–OVAFDG.
FIGURE 5.

Clec9A-mediated activation of B cells facilitates their migration toward the T–B border. A total of 1 × 106 CTV+ OB1 cells were adoptively transferred into B6 mice 1 d before vaccination with 0.5 μg of αClec9A–OVAFGD or isotype–OVAFGD. The spleens were harvested at 6, 12, and 24 h postimmunization for histological analysis. (A) Representative immunofluorescence images of splenic white pulp showing changes in the distribution of CTV+ OB1 cells (white) over time. Frozen spleen sections were stained for IgD (blue), CD4 (red), and laminin (data not shown) to highlight the B cell follicles and the T cell zone. Scale bar, 100 μm. (B) Bar graphs showing relative distribution of OB1 cells in the white pulp over time. OB1 cells in the B cell follicles (blue) and T zone (red) were identified and subdivided based on the minimum distance from the T–B border. Cells within 5 μm of the T–B border were considered to be on the “border” region (black). Bars represent the mean and error bars indicate the SEM. The average frequency ± SEM of OB1 in the B cell follicles and T zone are indicated in each graph. Pooled data from two independent experiments are presented with n = 3 mice per time point for each treatment.
To monitor changes in localization of B cells more closely, we examined the LN of vaccinated mice using intravital two-photon microscopy. These studies examined responses in the inguinal LN rather than the spleen because of technical limitations of imaging at the depth of the splenic white pulp. Furthermore, robust Tfh cell and GC B cell responses occur in both the spleen and LN following i.v. immunization with Clec9A-targeted Ags in the absence of adjuvant (Supplemental Fig. 3). Similar to spleen, OB1 B cells downregulated CXCR5 and upregulated CCR7 in the LN upon i.v. injection of αClec9A–OVAFGD in WT hosts (Fig. 6A), and these cells then accumulated at the T–B border by 20–28 h postimmunization in WT hosts (Fig. 6B, Supplemental Video 1). At this time point, OB1 B cells had somewhat higher migration speeds compared with polyclonal B cells (Fig. 6C, 6D), consistent with the behavior of B cells seeking cognate help at the T–B border (51). Control OB1 B cells primed in Clec9a−/− mice did not change CXCR5/CCR7 expression (Fig. 6A), although they did tend to migrate somewhat faster and closer to the T–B border compared with polyclonal B cells, suggesting a minor level of activation by untargeted Ag (Fig. 6B–D, Supplemental Video 1). Immunization with Clec9A-targeted Ag thus preferentially activated B cells early, which in turn led to their migration to the T–B border in the spleen and LN.
FIGURE 6.

Ag-specific B cells are activated upon Clec9A-targeted vaccination and relocate to the T–B border by a process that is not dependent on the new entry of B cells into the LN. (A) Ag-specific B cells in inguinal LN are rapidly activated upon Clec9A-targeted vaccination. A total of 1 × 106 OB1GFP+ were adoptively transferred into B6 or Clec9a−/− mice 1 d before vaccination with 0.5 μg of αClec9AOVAFGD. B cell activation in the inguinal LN was assessed 24 h postimmunization based on the expression levels of CXCR5 [(A), upper panel] and CCR7 [(A), lower panel]. OB1 cells were identified as GFP+CD19+CD3−PI−, and geometric mean fluorescence intensity (gMFI; normalized against OB1 from naive mice) is plotted. (B–D) Accumulation of Ag-specific B cells at the T–B border in inguinal LN after Clec9A-targeting. OB1GFP+ and DsRed+ polyclonal B cells were cotransferred into B6 or Clec9a−/− mice. Intravital two-photon imaging of inguinal LN was performed between 20 and 28 h postimmunization with 0.5 μg of αClec9A–OVAFGD using an LSM 710 microscope. (B) Single-frame, three-dimensional perspective images from time-lapse movies of B6 (upper left) and Clec9a−/− (lower left) inguinal LN and 30 min cell tracks illustrating distinct patterns of OB1 (green) and polyclonal B cell (red) coverage (right). Scale bars, 50 μm. See also Supplemental Video 1. (C) Median velocities of both B cell types in individual mice. (D) Median OB1 velocities normalized against median polyclonal B cell velocities in individual mice. See also Supplemental Fig. 3 for Clec9A-targeted Ag induction of Tfh and GC responses in the LN. (E–J) Blocking entry into LNs does not prevent B cell relocation to the T–B border. OB1 B cell localization in inguinal LN 24 h postvaccination with 0.5 μg anti–Clec9A–OVAFGD. B6 (E, F, H, and I) or Clec9a−/− (G and J) mice were injected with 1 × 106 CTV+ OB1 B cells and either treated with anti-CD62L (E and H) or rat IgG2a isotype control (F and I) 15 min before vaccination or left untreated (G and J) to assess the effect of blocking further entry of OB1 B cells into LN via HEV. (E–G) Representative images showing OB1 B cell localization patterns. CTV+ OB1 (white), IgD (blue), CD4 (red). Scale bar, 300 μm. (H–J) Histograms show distribution patterns of OB1 B cells in conditions illustrated in (E and F). B zone (blue), T zone (red), and the T–B border (±5 μm of the border; black), mean ± SEM. Number indicates the percentage of OB1 B cells in the B zone or T zone, mean ± SEM. Each symbol (A, C, and D) represents a mouse, and horizontal lines (A and D) indicate the mean. Representative data from three independent experiments [n = 8–13 mice per group (A), n = 4–5 mice per group (B–D)]. ns = p ≥ 0.05, ***p < 0.001, ****p < 0.0001 (A) by one-way ANOVA followed by Tukey test (A), paired Student t test (C) or unpaired Student t test (D). (E–J) Data pooled from two independent experiments. (H) n = 5 mice. (I) n = 4 mice. (J) n = 7 mice. One LN section was analyzed per mouse. Evidence for functional blocking of OB1 LN entry by anti-CD62L is shown in Supplemental Fig. 4A–C.
B cells do not require migration from HEV to recognize Clec9A-targeted Ag on cDC1
Evidence that Clec9A-targeted Ag but not untargeted Ag was able to rapidly activate specific B cells and cause their relocation to the T–B border within a few hours of Ag administration suggested that cDC1 directly presented targeted Ag to B cells. However, where this Ag-recognition event took place was unclear. As cDC1 are thought to be primarily located in the paracortex of the LN, two main but not mutually exclusive possibilities existed: either B cells encountered Ag-bearing cDC1 within the paracortex as they migrated from the HEV to follicles, as previously reported for adoptively transferred cDC (19), or they were able to access Ag-bearing cDC1 at the T–B border regions as they migrated within the follicles. To test whether Clec9A-targeted activation of OB1 cells required their migration from HEV to the follicles (through the T cell zone rich in cDC1), we examined OB1 cell responses in mice treated with anti-CD62L to prevent new entry of circulating B cells into the LN from the blood and compared this treatment to mice given an isotype control mAb (Fig. 6E–J, Supplemental Fig. 4A–C). Although blockade of CD62L prior to injection of αClec9A–OVAFGD prevented new entry of OB1 cells into LN (Supplemental Fig. 4A–C), this treatment did not prevent their accumulation at the T–B border after administration of Clec9A-targeted Ag (Fig. 6E, 6H), suggesting that OB1 cells already present within follicles prior to CD62L blockade could efficiently respond to this form of Ag. As expected, OB1 cells did not accumulate at the T–B border in Clec9A-deficient mice (Fig. 6G, 6J). These observations suggested that Ag-specific B cells that were resident within the B cell follicles at the time of priming were able to access Ag presented on cDC1 (presumably at the border of the follicles) and become sufficiently activated to alter their migration patterns and localize at the T–B border. This finding did not exclude B cell–cDC1 interactions occurring during migration from HEV to the follicles, but CD62L-blocking clearly showed that such interactions were not essential.
B cells interact with cDC1 at the border of B cell follicles
As Clec9A-targeted Ag does not activate cDC1 (24, 38), it is envisaged that in the immediate period after Ag administration that these cDC, as well as cDC2 and FDC will remain largely in their steady-state positions. Although FDC are found in B cell follicles and cDC2 reside largely in the bridging channels in the spleen and the interfollicular regions and outer T cell zones in LNs, cDC1 are located in the T cell zones of both tissues, as well as in the red pulp of the spleen. To support the notion that the location of cDC1 in the T cell zone includes areas directly in contact with B cells, we used two-photon microscopy to examine B cell–cDC1 colocation within LNs. Nontransgenic polyclonal B cells and T cells were separately labeled with fluorescent dyes and then transferred into transgenic mice expressing the YFP fluorescent protein Venus in cDC1 under the Xcr1 promoter. This combination of fluorescently labeled cells enabled visualization of T cells, B cells, and cDC1 and delineation of the T cell zone and B cell follicles. The day after cell transfer, mice were injected with anti-CD62L mAb 1 h before imaging to prevent new entry of B cells into the LNs. Two-photon microscopy showed T cell regions rich in cDC1 and B cell follicles with extensive bordering regions containing cDC1 (Fig. 7A, 7B, Supplemental Video 2). B cells within these border regions showed ample contacts with cDC1 (mean of 29 ± 7% of B cell tracks in close proximity to cCD1), intermingling extensively with this population. CD62L blockade prior to imaging, favored the view that cDC1 were able to interact with B cells when associated with follicles. Using tissue clearing and two-photon microscopy to image large volumes of the fixed LN tissue provided further evidence that B cell follicles were in close contact with bordering cDC1 (Fig. 7C, Supplemental Video 3). Finally, confocal imaging of thick slices from the spleen extended this analysis, showing close association of B cells with cDC1 in this organ, the latter of which were not only within the T–B border regions but also appeared to encroach within the follicles to some extent (Fig. 7D, Supplemental Video 4). Together, these data provide compelling evidence that B cells within follicles have ample opportunity to interact with cDC1 bordering these follicles.
Mathematical modeling supports the view that B cells within follicles can be activated by Clec9A-targeted Ag on cDC1
Given that CD62L blockade did not impair B cell activation to Clec9A-targeted Ag (Fig. 6E–J) and that ample interactions between B cells and cDC1 could be observed in the steady state (Fig. 7), we proposed that most B cells within follicles would be able to contact Ag-bearing cDC1 surrounding these follicles within the timeframe of <24 h required for most OB1 cells to be activated by Clec9A-targeted Ag (see Fig. 4). This view was supported by mathematical modeling of B cell migration, which showed that within an average-sized follicle of 1.86 ×107 μm3, >99% of B cells could contact the border regions of the follicle at least once within a 24 h timeframe. Similarly, in a 4.20 × 107 μm3 follicle, which is larger than 97% of follicles measured, >95% of B cells were able to contact the border regions in the same period; in the largest follicle measured, with a volume of 5.03 × 107 μm3, >93% of B cells were able to contact the border region in the same period. Thus, mathematical modeling of B cell migration within follicles supported the view that efficient activation could be induced by Clec9A-targeted Ag through contact of B cells with bordering cDC1 bearing targeted Ag.
Follicle-associated B cells are activated by Clec9A-targeted Ag on cDC1
To assess whether Ag-specific B cells interacted with Ag-bearing cDC1 at the periphery of follicles after Clec9A-targeted vaccination, we visualized B cells (in mice that did not contain fluorescent cDC1) a few hours after immunization to determine whether Ag-specific B cells slowed down relative to polyclonal B cells, indicative of Ag recognition (51) (Fig. 8, Supplemental Video 5). GFP+ OB1 B cells and either DsRed+ or CTV-labeled polyclonal B cells were cotransferred into B6 mice prior to i.v. immunization with various doses of Clec9A-targeted Ag. As expected, OB1 and polyclonal B cells displayed similar migration velocities in the absence of Ag (Fig. 8A, 8B). By contrast, injection of low doses (0.5–2 μg) of αClec9A–OVAFGD caused a moderate but consistent, Clec9A-dependent reduction in OB1 velocity at 1.5–3 h postvaccination (Fig. 8C, 8D). Increasing the Ag dose to 5 μg caused a more pronounced reduction in velocity, but at this high dose, effects on migration velocity were independent of Clec9A targeting. These experiments suggested that Ags targeted to Clec9A stimulated B cells rapidly and more efficiently than untargeted Ags.
To address whether reduced migration velocity was a consequence of Ag recognition on cDC1, we measured the velocity of OB1 B cells in Xcr1Venus mice after Clec9A-targeted vaccination. These mice received fluorescently labeled OB1 and polyclonal B cells and were then immunized with 0.5 μg of αClec9A–OVAFGD, the dose at which the Clec9A-dependent reduction of OB1 migration velocities was most evident (Fig. 8C, 8D). Analysis of OB1 cell velocity showed that those OB1 cells in close proximity to cDC1 had reduced median migration velocities compared with OB1 that remained in the central B cell follicle (Fig. 8E, 8F, Supplemental Video 6). Polyclonal B cells migrated at similar velocities regardless of their proximity to cDC1, indicating that recognition of cognate Ag on paracortical cDC1 promoted an initial migration arrest of Ag-specific OB1 B cells.
Together, these findings support a model in which cDC1 present Clec9A-targeted native Ag directly to B cells at the border of the follicles (and potentially also during their migration from HEV to the follicles), leading to B cell activation and preferential location to the T–B border. In this model, untargeted Ag is ineffective because it fails to cause B cell activation (despite being captured by B cells), most likely because it is not presented in a membrane-associated form capable of efficient BCR cross-linking.
DC surface expression of the B cell epitope is a feature of Clec9A priming
Targeting the B epitope to Clec9A on cDC1 led to efficient B cell–DC interactions and B cell activation, implying that cDC1 displayed native Clec9A-targeted Ag on their surface. To formally test this possibility, mice were injected i.v. with Clec9A–OVAFDG, and 2 h later, cDC1 were harvested from the spleen and examined for surface expression of the FDG epitope recognized by OB1 B cells (Fig. 9A, 9B). The flow cytometry gating strategy for cDC1 is shown in Supplemental Fig. 4D. This showed that the OB1 epitope was present on the surface of cDC1, supporting the idea that cDC1 can present this Ag to OB1 B cells after Clec9A targeting.
FIGURE 9.

cDC1 surface expression of the B cell epitope is a feature of Clec9A-targeted vaccination. (A and B) B6 mice were vaccinated with 2 μg of isotype–OVAFGD, αClec9A–OVAFGD, or αDEC–OVAFGD. Low-density cells were enriched from the spleen at 2 h postvaccination and surface stained for the OVAFGD epitope using biotinylated anti-OVAFGD Ab (generated from serum of OB1 mice), followed by streptavidin–allophycocyanin. (A) Representative histograms show staining for OVAFGD detectable on the surface of cDC1 (CD11chiCD8α+CD11bloB220−CD3−CD19−PI− low-density single cells) from vaccination mice (black lines) and unvaccinated mice (gray shading). (B) The geometric mean fluorescence intensity (gMFI) of the surface OVAFGD levels from vaccinated mice normalized against cDC1 from naive mice (n = 4 mice per group); mean ± SEM. (C and D) cDC1 efficiently capture Ag targeted via Clec9A or DEC205 in vivo. B6 mice were vaccinated with 2 μg of isotype–OVAFGD (Iso–OVAFGD)–A647, αClec9A–OVAFGD–A647, or αDEC–OVAFGD–A647. Low-density cells enriched from the spleen at 30 min or 3 h postvaccination were examined by flow cytometry. (C) Representative histograms show the level of A647 on cDC1 (CD11chiXCR1+CD11blo B220−CD3−CD19−PI− low-density cells). (D) gMFI of A647 in cDC1 from vaccinated mice normalized against cDC1 from naive mice (n = 6 mice per group for each time point); mean ± SEM. (E) Targeting Ag to DEC205 does not enhance early B cell activation. A total of 1 × 106 CTV+ OB1 cells were adoptively transferred into CD45.1+ mice 1 d before vaccination with 2 μg of Iso–OVAFGD, αClec9A–OVAFGD, or αDEC–OVAFGD. Activation of OB1 (CTV+CD19+CD3−CD45.1−PI−) was assessed 24 h postimmunization based on the expression of CXCR5, CCR7, CD86, and MHC II. Each symbol represents a mouse, and horizontal lines indicate the mean (n = 8) mice per group. Pooled data from two (A–D) or three (E) independent experiments. ns = p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way ANOVA followed by Tukey test (B, D, and E).
As mentioned earlier, targeting DEC205, in contrast to targeting Clec9A, induces comparatively poor humoral responses in the absence of adjuvant (24, 38), despite high expression of DEC205 by cDC1. One difference between these two receptors is that the DEC205–αDEC205 mAb adducts traffic to the late endosomal compartment (52), whereas Clec9A complexes are restricted mostly to the recycling endosomes (40). To test whether this difference in receptor trafficking may affect display of Ag, we compared surface expression after targeting either Clec9A or DEC205. In contrast to Clec9A-targeted Ag, DEC205-targeted Ag was almost undetectable on the surface of cDC1 2 hr after vaccination (Fig. 9A, 9B). This was despite the fact that cDC1 targeted by αDEC205–OVAFGD–A647 efficiently captured Ag (Fig. 9C, 9D). As expected, lack of surface display of the FGD Ag after DEC205 targeting led to poor activation of OB1 B cells, as measured by minimal changes to various activation markers (Fig. 9E). This contrasted with the efficient activation of B cells by Clec9A-targeted Ag. Surface display of the B cell epitope on cDC1 after targeting is therefore highly beneficial for B cell activation and the subsequent priming of T-dependent humoral responses.
Discussion
Although DC are important for the generation of Ab responses, this has generally been considered a secondary consequence of their role in priming Tfh cells (53). Nevertheless, DC have been implicated in having a more direct role in promoting Ab production (12, 15, 18, 19, 54–56). Whereas adoptive transferred cDC have clearly been shown to directly present Ag to B cells (19), knowledge of which cDC subsets participate in this function is somewhat limited. Ag-targeting studies have implicated cDC2 in this process (33, 35, 36) but have given somewhat disparate findings for cDC1 (24, 33, 36). The recent identification of cDC2 as critical for Tfh cell responses and humoral immunity to influenza virus administered to the lung further argue for a role for cDC2 but not cDC1 in Ag presentation to B cells (34).
In this study, we targeted molecularly defined B/T epitopes to Clec9A, a receptor predominantly expressed on cDC1. Whereas cDC1 are normally considered specialized for cross-presentation and the subsequent activation of cytotoxic T cells, we show that Ags taken up by Clec9A are retained in a form suitable for recognition by B cells. B cells appear to recognize this Ag on cDC1 at the T–B border of the follicles (or as they migrate from the HEV to the follicles) and are then rapidly activated, modulating their chemokine receptors to facilitate localization at this border. A previous study erroneously concluded that cDC1 lack the capacity to directly present Ags to B cells, based on the observation that Ags captured through DEC205 poorly activated B cells (33). Like DEC205, Clec9A is an endocytic receptor, and Ags captured through this receptor are processed through MHC class I/II to prime T cells, including Tfh (38). Nonetheless, we found that substantial amounts of native B cell epitopes also remained intact on the surface of cDC1 upon targeting to Clec9A, and this Ag directly induced B cell activation. This contrasted with Ags targeted to DEC205, which were not retained on the surface of DC and failed to activate B cells. Interestingly, Clec9A has been shown to preferentially shuttle exogenous Ag through early/recycling endosomes (40), whereas DEC205 preferentially delivers them to more degradative lysosomes (52), perhaps explaining the differential capacity to retain Ag for display. Similarly, a previous study examining culture-generated DC showed internalization of immune complexes through FcγRIIB led to Ag recycling to the cell surface and efficient priming of T-independent B cell responses, whereas internalization through activating FcRs (e.g., FcγRI and FcγRIII), which access lysosomes, did not (12). Although we have uncovered the capacity of Clec9A to enable surface Ag display through artificially targeting these Ags to Clec9A using mAbs, it is possible that many Ags naturally become associated with this receptor. The recognition of exposed F-actin by Clec9A (57–59) may result in the binding of dead cell–derived Ags linked to or trapped by actin filaments. These Ags may be self- or pathogen-derived, potentially leading to autoreactive or anti-pathogen Ab responses. To this point, however, we have been unable to identify a physiological role for Clec9A in humoral immunity to infections such as HSV and malaria. This likely relates to the highly redundant nature of mechanisms available to provide membrane-linked Ags to B cells.
A study using Ags, such as OVA and influenza virus, implicated migratory cDC2 as the primary drivers of T-dependent Ab responses, at least after intranasal immunization (34). Our findings suggest that the nature of the Ag tested deserves careful consideration because effective priming by a particular cDC subset may be dictated by the receptor capturing the Ag for display. Both OVA and influenza virus are heavily glycosylated Ags, and therefore carbohydrate recognition receptors differentially expressed by cDC2 may be required to capture these types of Ags for effective B cell activation and subsequent induction of Tfh cells and humoral immunity. SIGN-R1–expressing cDC participate in priming humoral immunity to influenza virus, supporting receptor dependence for this response (55).
In their early interaction with B cells, cDC provide more than just efficient delivery of Ag because Ag-specific B cells were shown to have good access to untargeted soluble Ag. Clec9A-bound Ag on cDC1 was more potent at activating B cells than soluble Ag and effectively modulated the expression of chemokine receptors and upregulated MHC II and the costimulatory molecule CD86. We attribute this to the fact that Ags tethered on the cell membrane are able to cross-link the BCR far more efficiently than soluble Ags (2). Although untargeted soluble Ags might eventually become deposited on FDCs upon complement-mediated opsonization, this process is likely to be inefficient and slow. Thus, naive B cells may not have the opportunity to ever recognize epitopes on untargeted Ags in a form that is membrane tethered if these Ags are in low abundance and/or prone to degradation. We cannot exclude the possibility that cytokines and costimulatory molecules produced by cDC1 also contribute to B cell activation. Chappell et al. (33) show that steady-state cDC2 directly interacted with Ag-specific B cells to promote early activation upon targeting Ag through DCIR2. However, in the absence of adjuvant, such targeting to cDC2 exclusively elicits low-affinity Ab responses through the induction of short-lived extrafollicular Ab-secreting cells without initiating GC responses (33). It is plausible that steady-state cDC1 and cDC2 provide qualitatively different signals to responding B cells rather than simply acting as platforms for Ag display. Alternatively, localization of Ag-loaded DC at the T–B border may be critical. In the steady state, cDC2 are largely restricted to the splenic marginal zone bridging channels and only migrate to the T–B border upon activation by adjuvant (33, 60, 61). In contrast, cDC1 reside largely within the T cell zone but, as we have shown by imaging, this includes the T–B border, where access to both B cells and T cells is evident. The differential requirement by cDC1 and cDC2 for adjuvant activation after targeting may thus relate to their location. That said, although Clec9A-mediated B cell activation and subsequent Ab production occur in the absence of adjuvants and cDC1 activation (24, 38), the Ab response to weak Ags is further enhanced by coadministration of adjuvants with the targeted Ags (42). Precisely how changes in cDC1 localization, Ag processing capacity, and the expression of costimulatory molecules/cytokines induced by adjuvants influence B cells requires further study. Similarly, those chemokine signals that influence cDC1 location, even in the steady state, particularly those that place cDC1 adjacent to B cell follicles will be interesting to unravel.
There is a need for rationally designed vaccines that can predictably and reliably induce strong immune responses. Essentially all successful vaccines for infectious disease developed to date rely primarily on their ability to elicit strong humoral responses, although CTL responses may contribute to clearing infections. In this respect, targeting cDC1 via Clec9A is a promising strategy because this DC subset and this receptor are conserved in mice, nonhuman primates, and humans. Previous results showed that such targeting markedly enhanced Ab responses in mice and primates, even in the absence of adjuvants (24, 37). Furthermore, in the presence of adjuvants that activate DC, cytotoxic T cells can also be induced and Ab production is further enhanced (37, 38, 62), stimulating both arms of the adaptive immune system. Our findings provide a rational basis for targeting cDC surface receptors, such as Clec9A, which not only provide efficient platforms for T cell activation but also maintain surface display of native Ag for efficient B cell activation and subsequent humoral immunity.
Supplementary Material
Acknowledgments
We thank Dr. Caetano Reis e Sousa for the kind gift of Ab from the αClec9A clones 7H11 and 1F6. We thank the Biological Research Facility and the Advance Cytometry Facility at the Peter Doherty Institute for Infection and Immunity for technical support. We also thank Ming Li and Melanie Damtsis for technical assistance.
This work was supported by the Australian Research Council (CE140100011) and the National Health and Medical Research Council (1113293, 1124706, 1154457).
The online version of this article contains supplemental material.
- A647
- Alexa Fluor 647
- cDC
- conventional DC
- αClec9A
- rat anti-mouse Clec9A mAb
- CTDR
- CellTracker Deep Red dye
- CTV
- CellTrace Violet dye
- DC
- dendritic cell
- FDC
- follicular DC
- GC
- germinal center
- HEV
- high endothelial venule
- LN
- lymph node
- MHC II
- MHC class II
- NP
- 4-hydroxy-3-nitrophenylacetyl
- RT
- room temperature
- Tfh
- T follicular helper
- WT
- wild-type.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Cyster J. G. 2010. B cell follicles and antigen encounters of the third kind. Nat. Immunol. 11: 989–996. [DOI] [PubMed] [Google Scholar]
- 2.Batista F. D., Iber D., Neuberger M. S. 2001. B cells acquire antigen from target cells after synapse formation. Nature 411: 489–494. [DOI] [PubMed] [Google Scholar]
- 3.Heesters B. A., van der Poel C. E., Das A., Carroll M. C. 2016. Antigen presentation to B cells. Trends Immunol. 37: 844–854. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki K., Grigorova I., Phan T. G., Kelly L. M., Cyster J. G. 2009. Visualizing B cell capture of cognate antigen from follicular dendritic cells. J. Exp. Med. 206: 1485–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.den Haan J. M. M., Martinez-Pomares L. 2013. Macrophage heterogeneity in lymphoid tissues. Semin. Immunopathol. 35: 541–552. [DOI] [PubMed] [Google Scholar]
- 6.Phan T. G., Grigorova I., Okada T., Cyster J. G. 2007. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat. Immunol. 8: 992–1000. [DOI] [PubMed] [Google Scholar]
- 7.Carrasco Y. R., Batista F. D. 2007. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity 27: 160–171. [DOI] [PubMed] [Google Scholar]
- 8.Junt T., Moseman E. A., Iannacone M., Massberg S., Lang P. A., Boes M., Fink K., Henrickson S. E., Shayakhmetov D. M., Di Paolo N. C., et al. 2007. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature 450: 110–114. [DOI] [PubMed] [Google Scholar]
- 9.Delemarre F. G., Kors N., van Rooijen N. 1990. Elimination of spleen and of lymph node macrophages and its difference in the effect on the immune response to particulate antigens. Immunobiology 182: 70–78. [DOI] [PubMed] [Google Scholar]
- 10.Buiting A. M. J., De Rover Z., Kraal G., Van Rooijen N. 1996. Humoral immune responses against particulate bacterial antigens are dependent on marginal metallophilic macrophages in the spleen. Scand. J. Immunol. 43: 398–405. [DOI] [PubMed] [Google Scholar]
- 11.Veninga H., Borg E. G. F., Vreeman K., Taylor P. R., Kalay H., van Kooyk Y., Kraal G., Martinez-Pomares L., den Haan J. M. M. 2015. Antigen targeting reveals splenic CD169+ macrophages as promoters of germinal center B-cell responses. [Published erratum appears in 2016 Eur. J. Immunol. 46: 493.] Eur. J. Immunol. 45: 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergtold A., Desai D. D., Gavhane A., Clynes R. 2005. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity 23: 503–514. [DOI] [PubMed] [Google Scholar]
- 13.MacPherson G., Kushnir N., Wykes M. 1999. Dendritic cells, B cells and the regulation of antibody synthesis. Immunol. Rev. 172: 325–334. [DOI] [PubMed] [Google Scholar]
- 14.Sornasse T., Flamand V., De Becker G., Bazin H., Tielemans F., Thielemans K., Urbain J., Leo O., Moser M. 1992. Antigen-pulsed dendritic cells can efficiently induce an antibody response in vivo. J. Exp. Med. 175: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wykes M., Pombo A., Jenkins C., MacPherson G. G. 1998. Dendritic cells interact directly with naive B lymphocytes to transfer antigen and initiate class switching in a primary T-dependent response. J. Immunol. 161: 1313–1319. [PubMed] [Google Scholar]
- 16.Ludewig B., Maloy K. J., López-Macías C., Odermatt B., Hengartner H., Zinkernagel R. M. 2000. Induction of optimal anti-viral neutralizing B cell responses by dendritic cells requires transport and release of virus particles in secondary lymphoid organs. Eur. J. Immunol. 30: 185–196. [DOI] [PubMed] [Google Scholar]
- 17.Colino J., Shen Y., Snapper C. M. 2002. Dendritic cells pulsed with intact Streptococcus pneumoniae elicit both protein- and polysaccharide-specific immunoglobulin isotype responses in vivo through distinct mechanisms. J. Exp. Med. 195: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balázs M., Martin F., Zhou T., Kearney J. 2002. Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity 17: 341–352. [DOI] [PubMed] [Google Scholar]
- 19.Qi H., Egen J. G., Huang A. Y. C., Germain R. N. 2006. Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science 312: 1672–1676. [DOI] [PubMed] [Google Scholar]
- 20.Guilliams M., Dutertre C.-A., Scott C. L., McGovern N., Sichien D., Chakarov S., Van Gassen S., Chen J., Poidinger M., De Prijck S., et al. 2016. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity 45: 669–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guilliams M., Ginhoux F., Jakubzick C., Naik S. H., Onai N., Schraml B. U., Segura E., Tussiwand R., Yona S. 2014. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat. Rev. Immunol. 14: 571–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durai V., Murphy K. M. 2016. Functions of murine dendritic cells. Immunity 45: 719–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huysamen C., Willment J. A., Dennehy K. M., Brown G. D. 2008. CLEC9A is a novel activation C-type lectin-like receptor expressed on BDCA3+ dendritic cells and a subset of monocytes. J. Biol. Chem. 283: 16693–16701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caminschi I., Proietto A. I., Ahmet F., Kitsoulis S., Shin Teh J., Lo J. C. Y., Rizzitelli A., Wu L., Vremec D., van Dommelen S. L., et al. 2008. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood 112: 3264–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sancho D., Mourão-Sá D., Joffre O. P., Schulz O., Rogers N. C., Pennington D. J., Carlyle J. R., Reis e Sousa C. 2008. Tumor therapy in mice via antigen targeting to a novel, DC-restricted C-type lectin. J. Clin. Invest. 118: 2098–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vremec D., Shortman K. 1997. Dendritic cell subtypes in mouse lymphoid organs: cross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J. Immunol. 159: 565–573. [PubMed] [Google Scholar]
- 27.Dorner B. G., Dorner M. B., Zhou X., Opitz C., Mora A., Güttler S., Hutloff A., Mages H. W., Ranke K., Schaefer M., et al. 2009. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity 31: 823–833. [DOI] [PubMed] [Google Scholar]
- 28.den Haan J. M., Lehar S. M., Bevan M. J. 2000. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 192: 1685–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pooley J. L., Heath W. R., Shortman K. 2001. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8- dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 166: 5327–5330. [DOI] [PubMed] [Google Scholar]
- 30.Dudziak D., Kamphorst A. O., Heidkamp G. F., Buchholz V. R., Trumpfheller C., Yamazaki S., Cheong C., Liu K., Lee H.-W., Park C. G., et al. 2007. Differential antigen processing by dendritic cell subsets in vivo. Science 315: 107–111. [DOI] [PubMed] [Google Scholar]
- 31.Smith C. M., Belz G. T., Wilson N. S., Villadangos J. A., Shortman K., Carbone F. R., Heath W. R. 2003. Cutting edge: conventional CD8 alpha+ dendritic cells are preferentially involved in CTL priming after footpad infection with herpes simplex virus-1. J. Immunol. 170: 4437–4440. [DOI] [PubMed] [Google Scholar]
- 32.Lahoud M. H., Proietto A. I., Gartlan K. H., Kitsoulis S., Curtis J., Wettenhall J., Sofi M., Daunt C., O’keeffe M., Caminschi I., et al. 2006. Signal regulatory protein molecules are differentially expressed by CD8- dendritic cells. J. Immunol. 177: 372–382. [DOI] [PubMed] [Google Scholar]
- 33.Chappell C. P., Draves K. E., Giltiay N. V., Clark E. A. 2012. Extrafollicular B cell activation by marginal zone dendritic cells drives T cell-dependent antibody responses. J. Exp. Med. 209: 1825–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krishnaswamy J. K., Gowthaman U., Zhang B., Mattsson J., Szeponik L., Liu D., Wu R., White T., Calabro S., Xu L., et al. 2017. Migratory CD11b + conventional dendritic cells induce T follicular helper cell-dependent antibody responses. Sci. Immunol. 2: eaam9169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corbett A. J., Caminschi I., McKenzie B. S., Brady J. L., Wright M. D., Mottram P. L., Hogarth P. M., Hodder A. N., Zhan Y., Tarlinton D. M., et al. 2005. Antigen delivery via two molecules on the CD8- dendritic cell subset induces humoral immunity in the absence of conventional “danger”. Eur. J. Immunol. 35: 2815–2825. [DOI] [PubMed] [Google Scholar]
- 36.Shin C., Han J.-A., Koh H., Choi B., Cho Y., Jeong H., Ra J.-S., Sung P. S., Shin E.-C., Ryu S., Do Y. 2015. CD8α(-) dendritic cells induce antigen-specific T follicular helper cells generating efficient humoral immune responses. Cell Rep. 11: 1929–1940. [DOI] [PubMed] [Google Scholar]
- 37.Li J., Ahmet F., Sullivan L. C., Brooks A. G., Kent S. J., De Rose R., Salazar A. M., Reis e Sousa C., Shortman K., Lahoud M. H., et al. 2015. Antibodies targeting Clec9A promote strong humoral immunity without adjuvant in mice and non-human primates. Eur. J. Immunol. 45: 854–864. [DOI] [PubMed] [Google Scholar]
- 38.Lahoud M. H., Ahmet F., Kitsoulis S., Wan S. S., Vremec D., Lee C.-N., Phipson B., Shi W., Smyth G. K., Lew A. M., et al. 2011. Targeting antigen to mouse dendritic cells via Clec9A induces potent CD4 T cell responses biased toward a follicular helper phenotype. J. Immunol. 187: 842–850. [DOI] [PubMed] [Google Scholar]
- 39.Kato Y., Zaid A., Davey G. M., Mueller S. N., Nutt S. L., Zotos D., Tarlinton D. M., Shortman K., Lahoud M. H., Heath W. R., Caminschi I. 2015. Targeting antigen to Clec9A primes follicular Th cell memory responses capable of robust recall. J. Immunol. 195: 1006–1014. [DOI] [PubMed] [Google Scholar]
- 40.Zelenay S., Keller A. M., Whitney P. G., Schraml B. U., Deddouche S., Rogers N. C., Schulz O., Sancho D., Reis e Sousa C. 2012. The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J. Clin. Invest. 122: 1615–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caminschi I., Vremec D., Ahmet F., Lahoud M. H., Villadangos J. A., Murphy K. M., Heath W. R., Shortman K. 2012. Antibody responses initiated by Clec9A-bearing dendritic cells in normal and Batf3(-/-) mice. Mol. Immunol. 50: 9–17. [DOI] [PubMed] [Google Scholar]
- 42.Park H.-Y., Tan P. S., Kavishna R., Ker A., Lu J., Chan C. E. Z., Hanson B. J., MacAry P. A., Caminschi I., Shortman K., et al. 2017. Enhancing vaccine antibody responses by targeting Clec9A on dendritic cells. NPJ Vaccines 2: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rios A. C., Capaldo B. D., Vaillant F., Pal B., van Ineveld R., Dawson C. A., Chen Y., Nolan E., Fu N. Y., Jackling F. C., et al. 3DTCLSM Group 2019. Intraclonal plasticity in mammary tumors revealed through large-scale single-cell resolution 3D imaging. [Published erratum appears in 2019 Cancer Cell 35: 953.] Cancer Cell 35: 618–632.e6. [DOI] [PubMed] [Google Scholar]
- 44.Miller M. J., Wei S. H., Cahalan M. D., Parker I. 2003. Autonomous T cell trafficking examined in vivo with intravital two-photon microscopy. Proc. Natl. Acad. Sci. USA 100: 2604–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abell M. L. L., Braselton J. P., Rafter J. A., Rafter J. A. 1999. Statistics With Mathematica. Academic Press, Cambridge, MA. [Google Scholar]
- 46.Mazo R. M. 2009. Brownian Motion: Fluctuations, Dynamics, and Applications. Oxford University Press, Oxford, U.K. [Google Scholar]
- 47.Lawler G. F. 2010. Random Walk and the Heat Equation. American Mathematical Society, Providence, RI. [Google Scholar]
- 48.Park H.-Y., Light A., Lahoud M. H., Caminschi I., Tarlinton D. M., Shortman K. 2013. Evolution of B cell responses to Clec9A-targeted antigen. J. Immunol. 191: 4919–4925. [DOI] [PubMed] [Google Scholar]
- 49.Dougan S. K., Ogata S., Hu C. C. A., Grotenbreg G. M., Guillen E., Jaenisch R., Ploegh H. L. 2012. IgG1+ ovalbumin-specific B-cell transnuclear mice show class switch recombination in rare allelically included B cells. Proc. Natl. Acad. Sci. USA 109: 13739–13744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shih T.-A. Y., Roederer M., Nussenzweig M. C. 2002. Role of antigen receptor affinity in T cell-independent antibody responses in vivo. Nat. Immunol. 3: 399–406. [DOI] [PubMed] [Google Scholar]
- 51.Okada T., Miller M. J., Parker I., Krummel M. F., Neighbors M., Hartley S. B., O’Garra A., Cahalan M. D., Cyster J. G. 2005. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PLoS Biol. 3: e150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mahnke K., Guo M., Lee S., Sepulveda H., Swain S. L., Nussenzweig M., Steinman R. M. 2000. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J. Cell Biol. 151: 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi Y. S., Kageyama R., Eto D., Escobar T. C., Johnston R. J., Monticelli L., Lao C., Crotty S. 2011. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 34: 932–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dubois B., Vanbervliet B., Fayette J., Massacrier C., Van Kooten C., Brière F., Banchereau J., Caux C. 1997. Dendritic cells enhance growth and differentiation of CD40-activated B lymphocytes. J. Exp. Med. 185: 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gonzalez S. F., Lukacs-Kornek V., Kuligowski M. P., Pitcher L. A., Degn S. E., Kim Y.-A., Cloninger M. J., Martinez-Pomares L., Gordon S., Turley S. J., Carroll M. C. 2010. Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat. Immunol. 11: 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Macpherson A. J., Uhr T. 2004. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 303: 1662–1665. [DOI] [PubMed] [Google Scholar]
- 57.Zhang J.-G., Czabotar P. E., Policheni A. N., Caminschi I., Wan S. S., Kitsoulis S., Tullett K. M., Robin A. Y., Brammananth R., van Delft M. F., et al. 2012. The dendritic cell receptor Clec9A binds damaged cells via exposed actin filaments. Immunity 36: 646–657. [DOI] [PubMed] [Google Scholar]
- 58.Ahrens S., Zelenay S., Sancho D., Hanč P., Kjær S., Feest C., Fletcher G., Durkin C., Postigo A., Skehel M., et al. 2012. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 36: 635–645. [DOI] [PubMed] [Google Scholar]
- 59.Hanč P., Fujii T., Iborra S., Yamada Y., Huotari J., Schulz O., Ahrens S., Kjær S., Way M., Sancho D., et al. 2015. Structure of the complex of F-actin and DNGR-1, a C-type lectin receptor involved in dendritic cell cross-presentation of dead cell-associated antigens. Immunity 42: 839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yi T., Cyster J. G. 2013. EBI2-mediated bridging channel positioning supports splenic dendritic cell homeostasis and particulate antigen capture. eLife 2: e00757.23682316 [Google Scholar]
- 61.Gatto D., Wood K., Caminschi I., Murphy-Durland D., Schofield P., Christ D., Karupiah G., Brink R. 2013. The chemotactic receptor EBI2 regulates the homeostasis, localization and immunological function of splenic dendritic cells. [Published erratum appears in 2013 Nat. Immunol. 14: 876.] Nat. Immunol. 14: 446–453. [DOI] [PubMed] [Google Scholar]
- 62.Idoyaga J., Lubkin A., Fiorese C., Lahoud M. H., Caminschi I., Huang Y., Rodriguez A., Clausen B. E., Park C. G., Trumpfheller C., Steinman R. M. 2011. Comparable T helper 1 (Th1) and CD8 T-cell immunity by targeting HIV gag p24 to CD8 dendritic cells within antibodies to Langerin, DEC205, and Clec9A. Proc. Natl. Acad. Sci. USA 108: 2384–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the authors upon request.