

Abstract
The angiotensin II (AngII) type 1 receptor (AT1R), a member of the G protein–coupled receptor (GPCR) family, signals through G proteins and β-arrestins, which act as adaptors to regulate AT1R internalization and mitogen-activated protein kinase (MAPK) ERK1/2 activation. β-arrestin–dependent ERK1/2 regulation is the subject of important studies because its spatiotemporal control remains poorly understood for many GPCRs, including AT1R. To study the link between β-arrestin–dependent trafficking and ERK1/2 signaling, we investigated three naturally occurring AT1R variants that show distinct receptor–β-arrestin interactions: A163T, T282M, and C289W. Using bioluminescence resonance energy transfer (BRET)–based and conformational fluorescein arsenical hairpin–BRET sensors coupled with high-resolution fluorescence microscopy, we show that all AT1R variants form complexes with β-arrestin2 at the plasma membrane and efficiently internalize into endosomes upon AngII stimulation. However, mutant receptors imposed distinct conformations in β-arrestin2 and differentially impacted endosomal trafficking and MAPK signaling. Notably, T282M accumulated in endosomes, but its ability to form stable complexes following internalization was reduced, markedly impairing its ability to co-traffic with β-arrestin2. We also found that despite β-arrestin2 overexpression, T282M's and C289W's residency with β-arrestin2 in endosomes was greatly reduced, leading to decreased β-arrestin–dependent ERK1/2 activation, faster recycling of receptors to the plasma membrane, and impaired AngII-mediated proliferation. Our findings reveal that naturally occurring AT1R variants alter the patterns of receptor/β-arrestin2 trafficking and suggest conformationally dependent β-arrestin–mediated MAPK activation as well as endosomal receptor–β-arrestin complex stabilization in the mitogenic response of AT1R.
Keywords: angiotensin II type 1 receptor (AT1R), arrestin, bioluminescence resonance energy transfer (BRET), endocytosis, ERK1/2, G protein-coupled receptor (GPCR), MAPK, naturally occurring variants, trafficking, angiotensin II, extracellular-signal-regulated kinase (ERK), mitogen-activated protein kinase
G protein–coupled receptors (GPCRs) transmit signals conveyed by extracellular stimuli such as proteins, hormones, small molecules, neurotransmitters, and ions by engaging G proteins and other signaling effectors inside cells (1). Their signaling is tightly regulated by a family of intracellular proteins known as arrestins (arrestin2 and arrestin3, also known as β-arrestin1 and β-arrestin2, respectively) through binding to agonist-activated GPCRs that have been phosphorylated by GPCR kinases, and terminate G protein-mediated signaling at the plasma membrane (PM) (for review, see Refs. 2–5). Receptor-bound β-arrestins also act as endocytic adaptors by interacting with components of the clathrin-coated vesicles (CCV), such as clathrin and AP-2, and initiate GPCR internalization from the PM (6, 7). Receptor-containing CCVs are pinched off from the PM by dynamin GTPases and fuse with larger endosomes, where receptors are either sorted for recycling to the PM or trafficked to other intracellular compartments for recycling or degradation (5, 8).
Engagement of β-arrestins to agonist-activated receptors at the PM is a general property of nearly all GPCRs, but the outcomes of such GPCR–β-arrestin complexes vary markedly among receptors. Depending on the strength of the interaction of β-arrestins with receptors, which influences their trafficking profiles, GPCRs can be divided into two distinct classes, colloquially referred to as Class A and B (9). Class A GPCRs, such as the β2-adrenergic receptor (β2AR), are characterized by their transient interaction with β-arrestins. Once internalized, β2AR–β-arrestin complexes rapidly dissociate at or near the PM before receptors traffic into endosomes (9). These receptors then rapidly recycle back to the PM where they regain their signaling functions. In contrast, class B GPCRs, such as the angiotensin II type 1 receptor (AT1R), the vasopressin V2 receptor (V2R), and bradykinin B2 receptor (B2R), are characterized by their ability to form long-lived complexes with β-arrestins and traffic into endosomes once internalized (9, 10). These stable complexes in endosomes slow the rate of receptor recycling, ultimately leading to a decline in receptor density and signaling at the PM.
In addition to modulating GPCR signaling through desensitization and internalization, β-arrestins also act as signaling scaffolds (3–5). They recruit components of the mitogen-activated protein kinase (MAPK) cascade, notably elements of the ERK1/2 pathway such as cRaf1, MEK1/2, and ERK1/2 to receptor–β-arrestin complexes for different GPCRs, including for AT1R (11–13). Binding of these MAPK components are sensitive to β-arrestin's conformation (13–15). β-arrestin subtypes have also been shown to play distinct functions in MAPK signaling (16). For example, depleting β-arrestin1 or β-arrestin2, respectively increase or decrease ERK1/2 activation by AT1R and B2R (10, 17). β-arrestins thus play dual and opposite functions on mitogenic signals, because on the one hand they desensitize G protein–mediated ERK1/2 signals, while on the other hand they act as signaling scaffolds for ERK1/2 signaling. The amplitude of MAPK signaling is therefore contingent on the relative contribution of the G protein– versus β-arrestin–dependent pathways, as well as the relative expression of the effectors of these pathways and their efficient engagement in cells (18). Subcellular compartmentalization and stabilization of GPCR–β-arrestin complexes with components of the MAPK pathway also play important and differential roles in ERK1/2 activation. For the B2R, β-arrestin–mediated ERK1/2 activation has been shown to occur both at the PM and in endosomes (10), whereas for V2R blocking receptor–β-arrestin targeting to CCV at the PM and the ensuing internalization of the complex is sufficient to inhibit ERK1/2 activation (19). The stable formation of V2R–β-arrestin complexes also contributes to the sustained ERK1/2 activation (20). For the β2AR, stabilization of receptor–β-arrestin complexes does not seem to be necessary for MAPK activation, because as recently reported, receptor primed, unbound β-arrestins in CCVs are still able to engage ERK1/2 at the PM (21). For AT1R, MAPK signaling required the trafficking of AT1R–β-arrestin complexes into endosomes (11, 17, 22, 23). However, the incidence of β-arrestin–mediated ERK1/2 signaling at the PM, as well as the need to stabilizing receptor–β-arrestin complexes into endosomes for sustained ERK1/2 activation, are unclear in the case of AT1R.
We investigated three naturally occurring variants of AT1R: A163T, T282M, and C289W (A4.60T, T7.33M, and C7.40W, as defined by Ballesteros-Weinstein numbering, respectively), which we previously showed to distinctly interact with β-arrestin (24), on β-arrestin–dependent receptor trafficking and ERK1/2 activation. These mutations are located in the transmembrane (TM) region of the AT1R (A163T in TM4, T282M and C289W in TM7), which we suspected would likely affect the stabilization of distinct receptor conformations once engaged by ligands and thus alter β-arrestin interactions. A163T (rs12721226, National Library of Medicine, NCBI database) is a variant discovered in humans that has normal physiological properties with the endogenous ligand AngII, but has significantly reduced affinity to the antagonist losartan (25, 26). T282M and C289W (rs104893677 and rs1064533, respectively) are rare mutations, where T282M has been linked to renal tubular dysgenesis and C289W has been shown to decrease AT1R's affinity to AngII (27, 28). Here, we demonstrate that these naturally occurring mutations convert, in some cases, AT1R from a class B to a class A GPCR in terms of their interactions and trafficking behavior with β-arrestin and negatively affected β-arrestin2–mediated ERK1/2 signaling in endosomes. Importantly, our findings indicate that sustained β-arrestin–mediated MAPK signaling is contingent on the ability of β-arrestin2 to form long-lived complexes with AT1R in endosomes and dependent on β-arrestin2's conformation, and that compartmentalization of mitogenic signal in endosomes is critical for the cell proliferative response mediated by AT1R.
Results
Different internalization and trafficking profiles of AT1R variants
We evaluated the ability of WT-AT1R and mutants to undergo agonist-mediated internalization and receptor and β-arrestin trafficking into endosomes. First, WT-AT1R, A163T, T282M, and C289W were tagged with the Renilla luciferase (RlucII) donor and individually expressed with the PM-anchored rGFP-CAAX acceptor in HEK293 cells to track their kinetics of PM removal following AngII stimulation through bioluminescence resonance energy transfer (BRET), as described previously (29, 30) (Fig. 1A). We previously observed that T282M displayed similar expression levels as compared with WT, whereas A163T and C289W expressed ∼40% greater and lesser than WT, respectively. The affinities of A163T for AngII was similar to that of WT (∼0.5 nm) but reduced for C289W and T282M (∼5 nm and ∼100 nm, respectively) (24). Therefore, to ensure maximal activation, we stimulated cells with 1 μm AngII to reach maximum occupancy on all receptors (>90%). Following AngII stimulation, all receptors internalized with similar kinetics, although C289W showed a significantly greater extent of receptor removal from the PM as compared with WT after 60 min of agonist stimulation, whereas T282M displayed a lower level of internalized receptors (Fig. 1A, left panel). We tracked the trafficking of receptors into endosomes using RlucII-tagged receptors and the rGFP-FYVE acceptor anchored in endosomes (29). All receptors initially displayed similar kinetics of endosomal accumulation (up to 10 min), although A163T accumulated to a greater extent than WT into endosomes after 60 min of AngII stimulation, whereas T282M displayed reduced accumulation (Fig. 1B, left panel). We monitored the trafficking of β-arrestin into endosomes following AngII stimulation by expressing receptors with β-arrestin2–RlucII and rGFP-FYVE (29). β-arrestin2 accumulated in endosomes with similar kinetics and magnitude as WT for both A163T and C289W, although its trafficking was significantly impaired for T282M (representing only <30% of that of WT) (Fig. 1C, left panel). The potency of AngII-promoted receptor internalization and trafficking to endosomes for A163T was similar to WT, but reduced for T282M and C289W, consistent with the lower AngII affinities of these mutants (Fig. 1, A and B, right panels) (24). Similarly, C289W and T282M displayed a reduced potency of AngII-promoted trafficking of β-arrestin into endosomes as compared with WT (8 and 27 nm, respectively, as compared with 1.7 nm for WT) (Fig. 1C, right panel). However, the trafficking efficacy of β-arrestin2 into endosome was greatly reduced for T282M even at saturating AngII concentrations that mediated maximal receptor internalization, suggesting intrinsic changes in the mutant's trafficking behavior with this β-arrestin.
Figure 1.

AngII-mediated internalization and trafficking profiles of WT and mutant AT1R with β-arrestin. A and B, HEK293 cells were transfected with RlucII-tagged WT or mutant AT1R and PM-anchored rGFP-CAAX (A) or endosomally anchored rGFP-FYVE (B). C, HEK293 cells were transfected with WT or mutant AT1R, β-arrestin2-RlucII, and rGFP-FYVE. Cells were stimulated with 1 μm AngII for up to 1 h in time course experiments (left panels) or stimulated with indicated AngII concentrations for 1 h in concentration-response experiments (right panels). For A, the percentage change in basal BRET is reported, whereas for B and C, the BRET change was normalized and reported as percentage of WT maximal response. Data are represented as mean ± S.D. of triplicates from three to six independent experiments, and for left panels, two-way ANOVA followed by Dunnett's multiple comparisons tests were performed for the last time point where *** = p < 0.001, ** = p < 0.01, * = p < 0.05, n = 3–6.
We next visualized the differential co-trafficking of WT and mutant receptors with β-arrestin in HEK293 cells using confocal microscopy with YFP-tagged receptors and β-arrestin2–mCherry. Prior to AngII stimulation (0 min), YFP-tagged receptors (green channel) were predominately expressed at the PM, whereas β-arrestin2–mCherry (red channel) was homogenously distributed in the cytosol, where neither protein co-localized. Following AngII stimulation at 37°C for 40 min, WT, A163T, and C289W strongly co-localized into endosomes with β-arrestin (Fig. 2A). Consistent with the BRET data, agonist-mediated activation of T282M led to receptors accumulating in endosomes with very little β-arrestin co-localizing in the same compartment (Fig. 2A, as depicted by the white arrows). Quantification of signal intensities across whole-field high-resolution structured illumination microscopy images (Fig. 2B) revealed strong co-localization of WT, A163T, and C289W with β-arrestin after AngII stimulation (r = 0.69850, 0.80893, and 0.75642, respectively), whereas T282M showed weak co-localization with β-arrestin (r = 0.35360). Cross-sectional analysis of fluorescence intensity through multiple endosomes also confirmed the high incidence of WT, A163T, and C289W with β-arrestin, and low co-localization for T282M (Fig. 2C). Together, these findings indicate that although T282M internalizes efficiently from the PM and traffic into endosomes to comparable levels as WT and other mutant receptors, its ability to co-traffic with β-arrestin2 into endosomes is greatly impaired.
Figure 2.
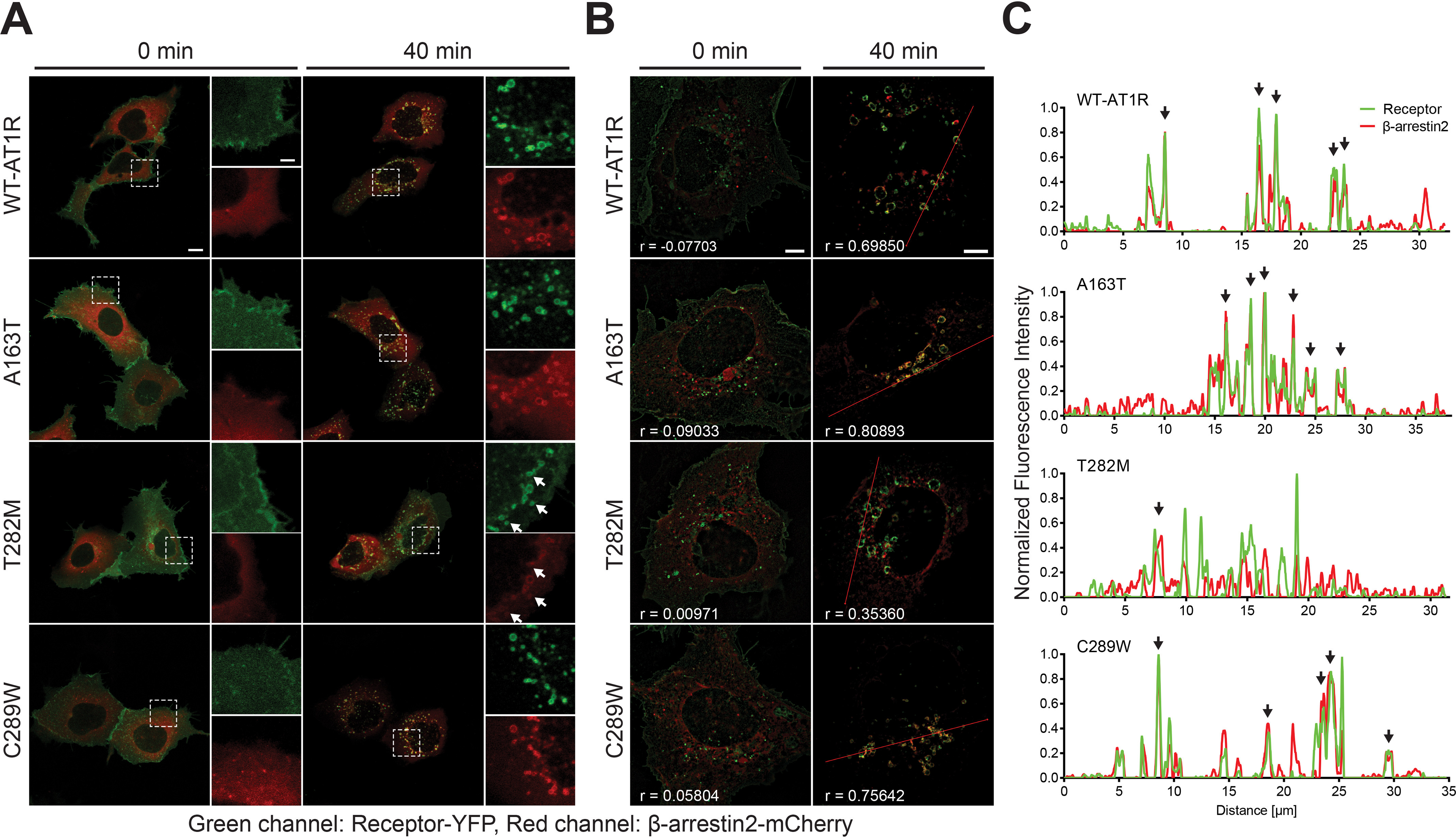
WT and mutant AT1R trafficking with β-arrestin as revealed by fluorescence microscopy. A and B, representative confocal (A) and structured illumination (B) microscopy images of HEK293 cells transfected with YFP-tagged WT or mutant AT1R (green) and β-arrestin2–mCherry (red). A, individual cells were tracked and imaged before and after 40-min stimulation with 1 μm AngII. Images represent overlay fluorescence signals, and dashed boxes represent blown-up section of cells shown in insets with individual fluorescence signals separated. White arrows depict endosomes containing receptors with minimal β-arrestin2. Scale bars: 10 μm; in insets, 2 μm. B, cells were stimulated with vehicle or 1 μm AngII for 40 min and then fixed with 4% PFA. Images represent overlay fluorescence signals. Pearson's correlation coefficients of receptor-YFP and β-arrestin2–mCherry intensities of the representative images were calculated and reported as r. Scale bars: 5 μm. C, line scan analysis from red lines shown in B, where black arrows indicate overlaps in fluorescence intensity peaks of receptor-YFP and β-arrestin2-mCherry.
Different receptor–β-arrestin2 complex stability of T282M during internalization
The impaired co-trafficking of T282M with β-arrestin into endosomes suggests a weakening of receptor–β-arrestin2 complexes during internalization. Therefore, we investigated the lability of these complexes at different stages of internalization: Engagement of β-arrestins to receptors at the PM and targeting of receptor–β-arrestin complexes into CCVs versus the removal of receptor–β-arrestin complexes from the PM into intracellular compartments. AT1R internalization is temperature sensitive because it requires the efficient activation of dynamin GTPase to pinch off the receptor–β-arrestin complexes in CCVs from the PM (29) (Fig. S1). We measured the efficiency of AngII to promote the formation of receptor–β-arrestin complexes through BRET by expressing YFP-tagged WT and mutant receptors and β-arrestin2–RlucII and stimulated cells with AngII for 30 min at 10°C, which restricts the formation of complexes at the PM. We compared those conditions to ones where cells were stimulated at 37°C with or without the expression of dynamin K44A (DynK44A), which respectively restricts the formation of receptor–β-arrestin complexes at the PM or allows their targeting to endosomes. Either lowering the temperature below physiological conditions or preventing the scission of CCVs with DynK44A greatly impaired AT1R internalization from the PM (Fig. S1) (29). Inhibiting endocytosis either by lowering the temperature of cells or by expressing DynK44A conserved the same rank order of potency for AngII-mediated receptor–β-arrestin2 complex formation between receptors as compared with that of AngII's action at 37°C (compare Fig. 3A, left and right panels to middle panel). Efficacy for AngII-mediated receptor–β-arrestin complex formation was similar between WT and receptor mutants when complexes were restricted to the PM, although we observed an increase in maximal complex formation for A163T, consistent with the higher expression of receptors (Fig. 3, A and B). On the other hand, a significant reduction in the extent of complex formation was observed for T282M when endocytosis was allowed. We reasoned that if β-arrestin's interaction with T282M was more labile than with WT, while the complex internalizes, it would be easier to disrupt during such process. We tested the effects of increasing β-arrestin2 expression on AngII-mediated BRET responses between β-arrestin2–RlucII and YFP-tagged WT and T282M at different stages of internalization (Fig. 3C). Experiments were conducted in CRISPR/Cas9 β-arrestin1/2–deleted HEK293 (DKO) cells (18) to eliminate any confounding effects of endogenous β-arrestin1 and 2 competing with β-arrestin2–RlucII binding to YFP-tagged receptors. Increasing β-arrestin2 expression had similar dose-dependent inhibitory effects on β-arrestin's interaction with both receptors when complexes were restricted at the PM (e.g. at 10°C or in conditions expressing DynK44A) (Fig. 3C, left and right panels, respectively). Indeed, the levels of β-arrestin2 co-transfection required to inhibit 50% of BRET signals from receptor-YFP–β-arrestin2-RlucII complexes (IC50) were similar for both WT and T282M (Fig. 3, C and D). In contrast, when complexes were allowed to internalize at 37°C, around three times less β-arrestin2 co-transfection was required to disrupt the T282M-YFP–β-arrestin2–RlucII complexes compared with WT. These results confirm that β-arrestin's interaction with T282M becomes more labile when complexes internalize from the PM.
Figure 3.
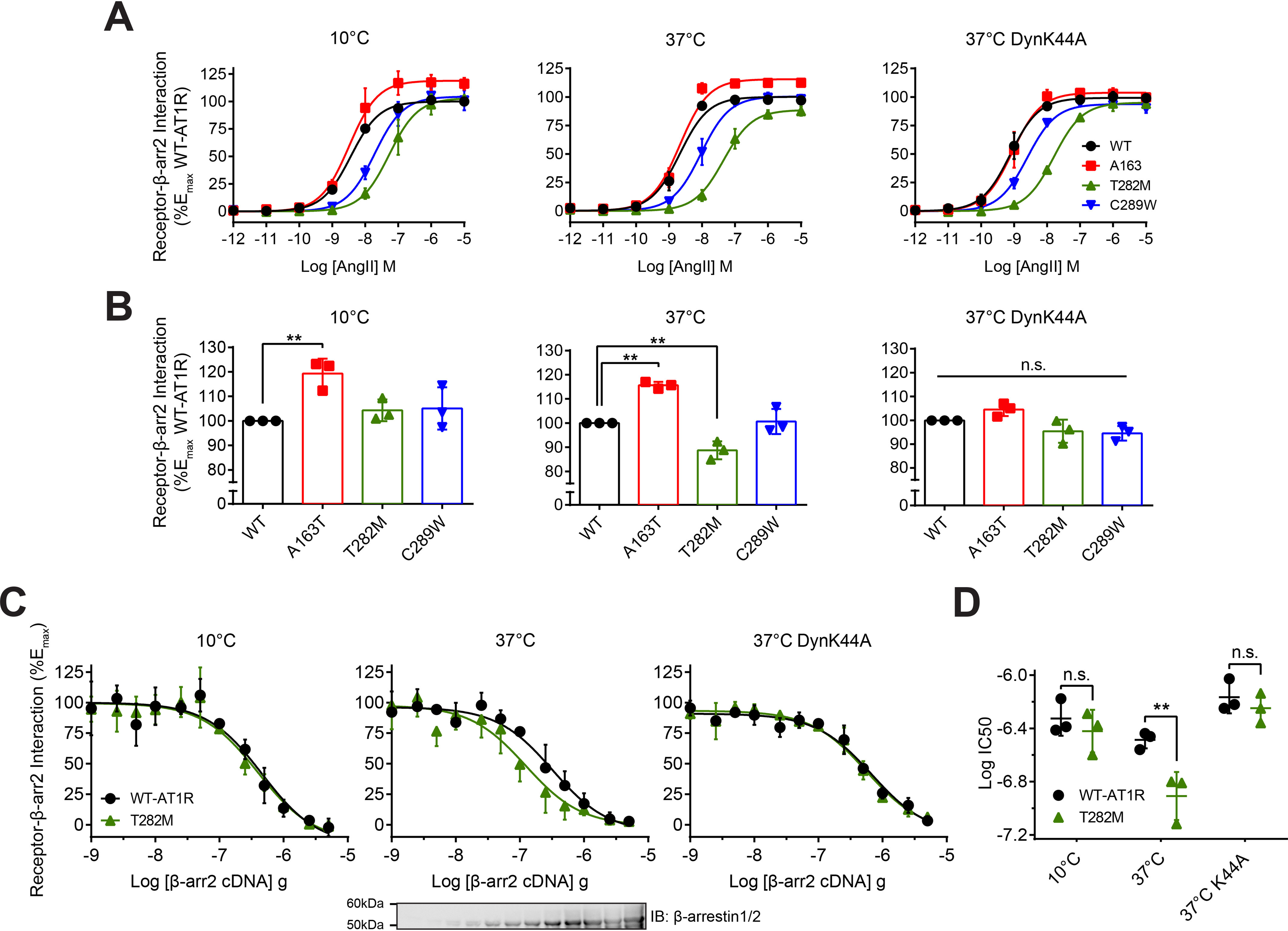
Stability of WT and mutant AT1R–β-arrestin complexes in different cellular compartments. A and B, HEK293 cells were transfected with YFP-tagged WT or mutant AT1R, β-arrestin2–RlucII, and with or without dominant-negative inhibitor of dynamin-mediated internalization, DynK44A. Cells were stimulated with AngII for 30 min at 10°C or 37°C in concentration-response experiments. For A, the BRET change was normalized and reported as percentage of WT maximal response. For B, the Emax of the concentration-response curves from A is represented as a scatter plot. Data are represented as mean ± S.D. of triplicates from three independent experiments, and for B, one-way ANOVA followed by Dunnett's multiple comparison test was performed where ** = p < 0.01, n = 3. C and D, HEK293/DKO cells were transfected with YFP-tagged WT or T282M, β-arrestin2–RlucII, and with or without DynK44A, along with increasing amounts of β-arrestin2. For C, the BRET change was normalized and reported as percentage of the maximal response for each receptor. Cells were also lysed and analyzed by Western blotting with antibodies specific for β-arrestin1/2. For D, the EC50 of the concentration-response curves from C is represented as a scatter plot. Data are represented as mean ± S.D. of triplicates from three independent experiments, and two-way ANOVA followed by Bonferroni-corrected t tests were performed where ** = p < 0.01, n = 3.
Binding of β-arrestin2 to mutant receptors promotes distinct conformations
The weakened T282M–β-arrestin2 complex, as determined by the ease at which the complexes were disrupted during internalization, as well as the differential trafficking into endosomes of mutant receptors with β-arrestin suggest distinct complex arrangements, which should be echoed in changes in β-arrestin2's conformation when binding to different receptors. To assess this possibility, we expressed WT and mutant receptors with a series of β-arrestin2–fluorescein arsenical hairpin (FlAsH)–BRET sensors (Rluc–β-arrestin2–FlAsH; F1–F6) (Fig. 4A), which capture changes in intramolecular BRET signals at each position following agonist-mediated engagement of β-arrestin with receptors (31). All mutant receptors were confirmed to recruit each FlAsH reporter comparably to that of WT (Fig. S2), whereas the F3 reporter was poorly recruited to all receptors and thus acted as the negative control (Fig. 4) (31). For A163T, the β-arrestin2 conformational signature was largely similar to that of WT, although we observed a slightly lower signal at the F6 position. On the other hand, C289W displayed significantly lower BRET changes at both F2 and F6, as compared with that of WT. T282M induced the most striking difference in the conformational signature of the β-arrestin2 sensors, as illustrated in the heat map of the responses at each reporter position (see Fig. 6B); it induced a significantly lower BRET change at F4, as well as a negative BRET change at F6, as compared with AT1R.
Figure 4.
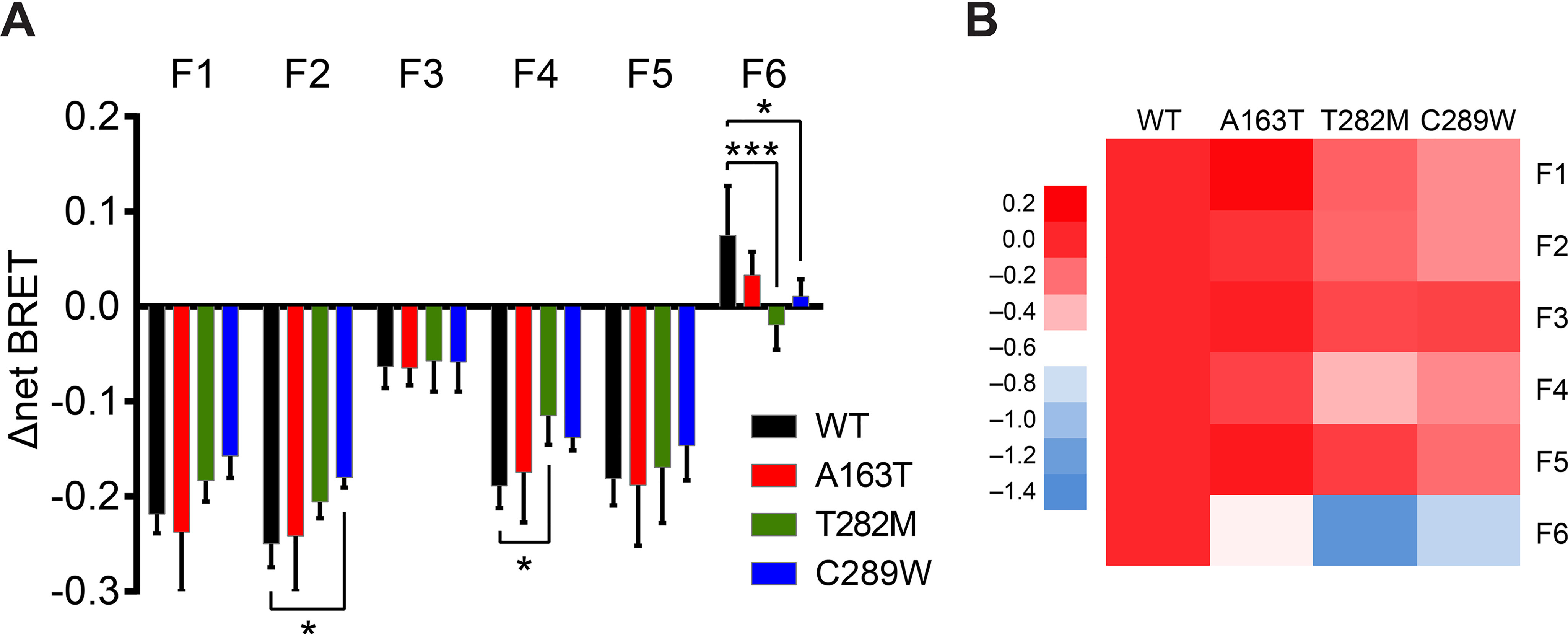
AngII-induced conformational signatures of β-arrestin2 binding to WT and mutant AT1R. A and B, HEK293 cells were transfected with WT or mutant AT1R and β-arrestin2-FlAsH reporters (F1–F6). Cells were stimulated with 1 μm AngII for 10 min, with six consecutive BRET measurements taken every minute after 5-min stimulation. For A, change in net BRET is reported and data are represented as mean ± S.D. of triplicates from four or five independent experiments. Two-way ANOVA followed by Dunnett's multiple comparison tests were performed where *** = p < 0.001, * = p < 0.05, n = 4–5. For B, the change in net BRET from A was normalized to those of WT at each reporter position and represented as a heat map to highlight relative differences in conformational signatures of β-arrestin2 binding between WT and mutant receptors.
Figure 6.
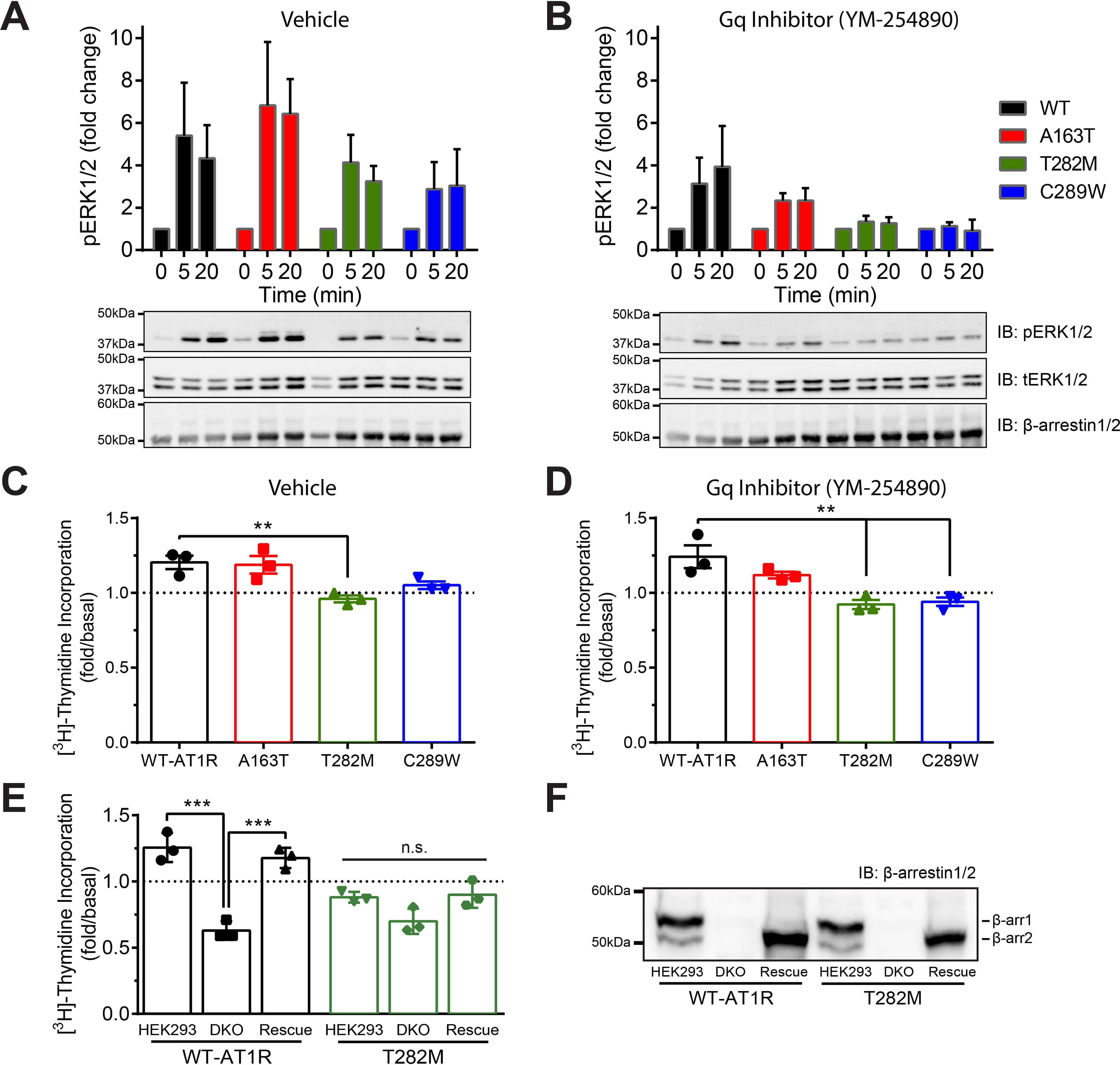
Role of Gαq/11 and β-arrestin in ERK1/2 activation and proliferation mediated by WT and mutant AT1R. A and B, HEK293 cells were transfected with WT or mutant AT1R and β-arrestin2 and pretreated with vehicle (A) or selective Gαq/11 inhibitor YM-254890 (B) to dissect G protein– versus β-arrestin–mediated ERK1/2 activation. Cells were stimulated with 1 μm AngII for 0, 5, or 20 min and then lysed and analyzed via Western blotting with antibodies specific for phospho-ERK, total-ERK, and β-arrestin1/2. Quantifications are represented as mean ± S.D. from four to six independent experiments. C and D, HEK293 cells were transfected with WT or mutant AT1R and β-arrestin2 and pretreated with vehicle (C) or YM-254890 (D). Cells were stimulated with or without 1 μm AngII for 24 h in presence of [3H]thymidine and then lysed to measure incorporated radioactivity. Data are represented as mean ± S.D. of duplicates from three independent experiments, and one-way ANOVA followed by Dunnett's multiple comparison tests were performed where ** = p < 0.01, n = 3. E and F, HEK293 and HEK293/DKO cells were transfected with WT or mutant AT1R, along with additional β-arrestin2 (rescue). Cells were stimulated with vehicle or 1 μm AngII for 24 h in presence of [3H]thymidine and then lysed to measure incorporated radioactivity. For F, cells from E were lysed and analyzed via Western blotting with antibodies specific for β-arrestin1/2. Data are represented as mean ± S.D. of duplicates from three independent experiments, and one-way ANOVA followed by Tukey's multiple comparison tests were performed for each receptor where *** = p < 0.001, n = 3.
Mutant receptors transit through endosomes and recycle at different rates
Given differences in receptor–β-arrestin2 complex formations, β-arrestin conformations and its trafficking in endosomes, we suspected that there would also be consequences on the subsequent recycling back to the PM of receptors following agonist removal. Therefore, we expressed WT or mutant receptors and stimulated cells with AngII for 30 min at 37°C to induce accumulation of receptor–β-arrestin complexes into endosomes, which was detected through BRET using rGFP-FYVE and either RlucII-tagged β-arrestin2 or RlucII-tagged receptors (Fig. 5, A and B, respectively). AngII was then washed off of cells to halt further internalization and allow the exiting of receptors and/or β-arrestin2 from the endosomal compartments. Following AngII stimulation, as expected, T282M induced significantly less accumulation of β-arrestin in endosomes (∼30% that of WT), whereas A163T and C289W displayed similar levels of β-arrestin accumulation in endosomes as compared with WT (Fig. 5A). The level of β-arrestin in endosomes decreased to basal levels immediately following AngII washout for T282M, whereas β-arrestin continued to accumulate into endosomes for WT and A163T. Surprisingly, despite C289W showing comparable levels of β-arrestin accumulation in endosomes as WT and A163T, β-arrestin exited rapidly from endosomes following AngII removal, similar to what was seen with T282M. Consistent with what was previously observed (Figs. 1 and 2), all receptors accumulated into endosomes to similar extents as WT following AngII stimulation (Fig. 5B). However, removal of AngII resulted in the exiting of T282M and C289W from endosomes, although at a slower rate than β-arrestin's removal from the same compartment (compare Fig. 5B to Fig. 5A), whereas WT and A163T continued to accumulate in endosomes up to 45 min post agonist washout. Finally, we assessed receptor recycling to the PM by expressing RlucII-tagged receptors with rGFP-CAAX (Fig. 5C). Following receptor internalization and agonist removal, T282M was rapidly recovered at the PM compared with WT and A163T and reached initial receptor density after 45 min. Interestingly, the initial rate of recycling for C289W was also faster than that of WT and A163T, and comparable with T282M. Altogether, these findings reveal that some mutants affect not only the accumulation, but also the removal of β-arrestin in endosomes and suggest that β-arrestin's removal from endosomes precedes AT1R's efflux from the same compartment, ultimately governing the rate of receptor recycling to the PM following receptor internalization.
Figure 5.

Recycling profiles of WT and mutant AT1R. A, HEK293 cells were transfected with WT or mutant AT1R, β-arrestin2–RlucII, and endosomally anchored rGFP-FYVE. B and C, HEK293 cells were transfected with RlucII-tagged WT or mutant AT1R and rGFP-FYVE (B) or PM-anchored rGFP-CAAX (C). To monitor the kinetics of receptor recycling, cells were stimulated with 100 nm AngII for 30 min and then washed with ice-cold acid and Tyrode's buffer to remove AngII from receptors on the PM. A–C, the percentage change in basal BRET is reported. Data are represented as mean ± S.D. of triplicates from three or four independent experiments. Two-way ANOVA followed by Dunnett's multiple comparison tests were performed where *** = p < 0.001, ** = p < 0.01, * = p < 0.05, n = 3–4.
β-arrestin2–dependent MAPK signaling regulates AngII-mediated cell proliferation
Because AT1R engages in both G protein– and β-arrestin–dependent MAPK signaling, we next evaluated β-arrestin2's role in such responses for WT and mutant receptors. We first confirmed that Gαq activation was similar between WT, T282M, and C289W, whereas A163T displayed a higher level of agonist-mediated Gαq activation (Fig. S3). Next, we treated cells with or without a selective Gαq/11 inhibitor (YM-254890) (24) prior to AngII stimulation to differentiate the G protein– and β-arrestin–mediated ERK signaling components. All receptors produced robust MAPK signaling (around 15- to 25-fold), although C289W displayed slightly lower ERK1/2 activation (Fig. S4A). When Gαq/11 signaling was inhibited, substantially lower agonist-mediated MAPK signaling was observed (Fig. S4B), suggesting that the response was primarily mediated by Gαq/11 and that the levels of endogenous β-arrestins as compared with receptors expressed was limited to desensitize receptors and to efficiently engage them in β-arrestin–mediated ERK1/2 activation. We therefore overexpressed β-arrestin2 with the receptors in HEK293 cells, because in addition to favoring β-arrestin–mediated ERK1/2 activation, it would reduce the contribution of G protein–dependent signaling to MAPK (Fig. 6, A and B) (18). Consistent with the role of β-arrestin2 in desensitizing G protein signaling, ERK1/2 activation only reached maximum levels of around 4- to 6-fold at 5 min of AngII stimulation and persisted at 20 min for all receptors. All receptors produced robust MAPK signal, although we observed a small reduction in maximum ERK1/2 activation for T282M and C289W, as compared with WT (Fig. 6A). When β-arrestin2 was overexpressed and Gαq/11 was inhibited, we observed a partial reduction in AngII-mediated ERK1/2 activation by WT (around 50 and 15% at 5 and 20 min, respectively), suggesting that β-arrestin–mediated MAPK signaling outweighed most of the loss of G protein–dependent ERK1/2 signaling for WT-AT1R, notably at later time of agonist stimulation where receptor–β-arrestin complexes are internalized. However, in similar conditions, substantially more decreases in MAPK signaling of A163T (around 70% for both time points) and of T282M and C289W were observed (around 90% for both time points) (Fig. 6B). Taken together, these results suggest that while T282M- and C289W-mediated ERK1/2 activation is primarily through Gαq/11 engagement, such response for WT and A163T involves both Gαq/11 and β-arrestin, albeit to different extents.
Finally, we assessed the ability of the receptors to promote cell proliferation, a process reported to be mediated through β-arrestin–dependent activation of AT1R in vascular smooth cells (Fig. 6, C–F) (32). We found that AngII stimulation of WT and A163T receptors promoted cell proliferation in HEK293 cells, which was sustained even when Gαq/11 was blocked (Fig. 6, C and D). On the other hand, AngII stimulation of T282M and C289W when Gαq/11 was inhibited or not, both failed to promote cell proliferation. We validated β-arrestin2's involvement in AT1R-mediated cell proliferation by using β-arrestin1/2–deleted DKO cells, complemented with or without β-arrestin2, and compared their proliferative response to parental HEK293 cells (Fig. 6, E and F). In HEK293 cells, AngII stimulation of WT led to a ∼25% increase over basal cell proliferation rate. On the other hand, we observed a ∼40% decrease in the rate of proliferation following AngII stimulation of DKO cells expressing WT, whereas the proliferative response was rescued by expressing β-arrestin2. Interestingly, AngII stimulation of T282M expressed in either HEK293 or DKO cells had no significant effects on cell proliferation. Altogether, these results indicate that cell proliferation through AT1R activation depends on β-arrestin2 and suggests that this proliferative response depends on the β-arrestin–mediated MAPK signaling of AT1R.
Discussion
Numerous studies have reported the critical role of β-arrestin–dependent ERK1/2 activation in GPCR signaling and function (2–5), although the spatiotemporal control of such signaling scaffolds remains largely understudied for many receptors, including AT1R. Our findings with natural variants of AT1R demonstrating the contribution of endosomal receptor–β-arrestin2 complexes toward ERK1/2 signaling are contemporary, considering that such a role has been questioned and the subject of recent studies (18, 33, 34).
We show that AT1R engages both in Gαq/11- and β-arrestin2–mediated ERK1/2 signaling, and that the contribution of each effectors/pathways in MAPK signaling varies between WT and mutant receptors. This was suggested when assessing receptor signaling in conditions of β-arrestin2 overexpression versus only in presence of endogenous β-arrestins. In the latter condition, MAPK signaling by AT1R and mutant receptors was found to be primarily mediated through Gαq/11 activation. Overexpressing β-arrestin2, however, favored more β-arrestin– over Gαq/11-mediated ERK1/2 activation, but only for WT-AT1R. It failed to have similar effects on T282M and C289W, suggesting a defect in β-arrestin–dependent MAPK for these mutant receptors. These observations also imply that the extent of the MAPK response by WT versus mutant receptors, and the signaling pathways utilized by these receptors will most likely be dependent on the relative complement of G proteins versus β-arrestins found in cells/tissues where these receptors are expressed.
We show that β-arrestin–dependent MAPK signaling correlated with the ability of this β-arrestin subtype to form long-lived complexes with receptors in endosomes, rather than at the PM. This was clearly demonstrated with T282M, which readily interacted with β-arrestin2 at the PM (e.g. in CCVs) but remained poorly associated into endosomes with β-arrestin2 and for which signaling to ERK1/2 was mainly driven by Gαq/11 activation, even in conditions of β-arrestin2 overexpression. Similarly, C289W, which was also rapidly removed from the endosomes with β-arrestin, failed to produce efficient β-arrestin2–dependent ERK1/2 signals.
The ability of AT1R mutants to traffic into endosomes, form long-lived complexes, and signal to ERK1/2 was also linked to β-arrestin2's conformation, as demonstrated here through the use of β-arrestin2–FlAsH probes (31). A163T, which was well expressed and efficiently internalized into endosomes with β-arrestin2, showed more Gαq/11-dependent ERK1/2 activation at the expense of reduced β-arrestin–mediated ERK1/2 signals. A163T also exhibited a different overall β-arrestin2 conformational signature than what was observed with WT, which was clearly revealed by a consistent reduction in β-arrestin's positive sensor signal at position F6. Similarly, we observed a reduction in negative signals at positions F2 and F4, which was readily significant for C289W and T282M, respectively; both mutants also showed significant signal reductions at position F6. Such changes in β-arrestin's conformational pattern, in particular at F4 and F6 positions, are consistent with long-lived receptor–β-arrestin complexes in endosomes reported for other GPCRs and for MAPK activation by different β-arrestin–biased ligands for AT1R, as well as the predictive trafficking behavior of class A versus B GPCRs (31, 35), which was also most evident for T282M.
Our data also reveal that mutations, as exemplified by T282M and to a lesser extent C289W, converted AT1R from a class B to a class A GPCR in terms of their trafficking behavior. The differences in trafficking behaviors observed between WT and some mutant receptors, as well as between receptor–β-arrestin complexes, may have been accentuated by the overexpression of β-arrestin2. However, even when β-arrestin2 was overexpressed, T282M was still unable to form long-lived complexes with β-arrestin in endosomes. Classically, the nature of the receptor's C-terminal domain and its ability to be phosphorylated at clusters of serine/threonine upon ligand binding to receptors determine the stability of GPCR–β-arrestin complexes and their trafficking to endosomes (9). Although the effects of T282M on the AT1R's ability to be phosphorylated was not assessed, our findings nonetheless imply that mutations induced specific AngII–AT1R conformations, which also impacted β-arrestin's conformations and the stability of its interaction with agonist-bound receptors. This is also supported by our recent observations that AT1R bearing the T282M mutation showed differences in contact interactions of the receptor's intracellular domains with β-arrestin, as compared with that of AngII-bound WT (35). Why then receptor–β-arrestin interactions seem more labile for certain mutants in endosomes than at the PM, is unknown. It may be because such complexes form stronger interactions at the PM, because they are stabilized by other binding partners like the interaction of β-arrestin with clathrin and AP-2 in CCVs (6, 7). It also remains to be determined the extent to which conformations and the stability of receptor–β-arrestin complexes in endosomes affect the binding of β-arrestin2 to the different MAPK components, such as cRaf1, MEK1/2, and ERK1/2.
β-arrestin–mediated ERK1/2 activation has been involved in numerous cell and physiological responses promoted by GPCRs, including cytoskeletal reorganization and cell migration, hypertrophy, cell survival and proliferation (3, 5). Such responses are cell context–dependent and differ among GPCRs and β-arrestin subtypes. For AT1R, we previously reported in vascular smooth muscles cells that although both β-arrestin-1 and 2 are involved in MAPK signaling, only β-arrestin2 contributed to ligand-mediated cell migration and proliferation (32). Here, our findings also support the role of β-arrestin2 in HEK293 cell proliferation because not only was such response decreased below basal rates upon agonist stimulation in β-arrestin1/2 DKO cells as compared with the parental cells, but also it was rescued when β-arrestin2 was reintroduced. Interestingly, our findings also suggest that β-arrestin2 likely promoted cell proliferation through a mechanism that requires the trafficking and signaling of receptors in endosomes, because blocking Gαq/11-mediated ERK1/2 signaling for WT and A163T did not significantly impede the cell proliferative response, whereas T282M and C289W, which signal to ERK1/2 mainly through Gαq/11 and have reduced residency in endosomes with β-arrestin2, were not able to promote significant proliferative responses in parental (for T282M and C289W) and in DKO (for T282M) cells. Our findings not only support a divergent role of β-arrestin2–dependent versus G protein–mediated ERK1/2 activation by GPCRs, but highlight the importance of discriminating between these compartmentalized mitogenic signals, as measuring global MAPK activation may be unpredictive of the cell response (20, 22, 23).
By characterizing β-arrestin2–mediated trafficking and signaling of three naturally occurring AT1R variants, we not only showed that mutations alter β-arrestin's conformation, its interaction with receptors, and the trafficking of receptor–β-arrestin complexes in cells, but more importantly demonstrate that such endosomal complexes, as well as β-arrestin's conformation, contribute to AT1R-mediated MAPK signaling and cell proliferation.
Experimental Procedures
Reagents
Human angiotensin II (Asp1-Arg2-Val3-Tyr4-Ile5-His6-Pro7-Phe8), poly-ornithine, poly-l-lysine, 1,2-ethanedithiol (EDT), 2,3-dimercapto-1-propanol, and horseradish peroxidase–conjugated rabbit secondary antibody were purchased from Sigma-Aldrich. Minimal essential media (MEM), FBS, and other cell culture additives were purchased from Gibco, Life Technologies. Lipofectamine 2000 was purchased from Invitrogen. FlAsH-EDT2 was synthesized at McGill University. YM-254890 was purchased from Wako Pure Chemical Industries (Osaka, Japan). Coelenterazine 400a and Coelenterazine H were purchased from Nanolight Technology (Pinetop, AZ). Chemiluminescence reagents and [3H]thymidine were purchased from PerkinElmer Life Sciences. Anti–phospho-ERK1/2 and anti-ERK1/2 antibody were purchased from Cell Signaling Technology, and the β-arrestin A1CT antibody was received as a gift from Dr. Robert Lefkowitz.
DNA constructs
FLAG-tagged human β-arrestin2 and WT and mutant FLAG-tagged human AT1R constructs were described previously (7, 24). β-arrestin2-RlucII, β-arrestin2-mCherry, rGFP-FYVE, rGFP-CAAX, AT1R-YFP, and Rluc-β-arrestin2-FlAsH BRET sensors were described previously (7, 19, 30–32).
Cell culture and transfection
HEK293 and HEK293/DKO cells were cultured in MEM supplemented with 10% FBS, 5 mm l-glutamine, and 20 μg/ml gentamicin. Cells were grown at 37°C in 5% CO2 and 90% humidity. Cells were seeded at a density of 7.5 × 105 cells per 100 mm dish, 1.5 × 105 cells per 35-mm glass bottom dish, or 1.5 × 105 cells per well in 6-well plate and were transiently transfected the next day with either WT or mutant AT1R variants and BRET sensor construct using lipofectamine or conventional calcium phosphate co-precipitation method. 18 h post transfection, the media were replaced and cells were divided for subsequent experiments. All assays were performed 48 h post transfection.
BRET measurements
One day post transfection, cells were detached and seeded in poly-ornithine–coated white 96-well plates at a density of 2.5 × 104 cells per well in media. The next day, cells were washed and incubated for 1 h with Tyrode's buffer (140 mm NaCl, 2.7 mm KCl, 1 mm CaCl2, 12 mm NaHCO3, 5.6 mm d-glucose, 0.5 mm MgCl2, 0.37 mm NaH2PO4, 25 mm HEPES, pH 7.4) at room temperature or 37°C. For concentration-response and time course experiments, cells were stimulated with various concentrations of AngII in Tyrode's for 5–60 min, or a single concentration of AngII for various durations, respectively. For recycling experiments, cells were stimulated with AngII for 30 min at 37°C and then washed three times with ice-cold acid (50 mm sodium citrate, pH 4.0), followed by two additional washes with ice-cold Tyrode's buffer. Cells were then incubated with Tyrode's buffer at 37°C for 45 min with BRET measurements taken at set intervals. For β-arrestin2–FlAsH BRET experiments, FlAsH labeling was performed as described previously (31). Briefly, 1.75 μl of FlAsH-EDT2 stock reagent was mixed with 3.5 μl of 25 mm EDT solution in DMSO and left for 10 min at room temperature. 100 μl of Tyrode's buffer was then added to the mix and left for 5 min at room temperature. The volume was then adjusted to 5 ml with Tyrode's buffer to complete the labeling solution. Cells were washed with Tyrode's buffer and incubated with 60 μl of labeling solution per well for 1 h at 37°C. Cells were then washed twice with 2,3-dimercapto-1-propanol wash buffer followed by another wash with Tyrode's buffer. Next, 90 μl of Tyrode's buffer was added per well and incubated for 1 h at 37°C. Cells were stimulated with ligand for 10 min, with six consecutive BRET measurements taken every minute after 5-min stimulation. For all BRET experiments, cell-permeable substrates, coelenterazine H (final concentration of 2 μm) for BRET1 and coelenterazine 400a (final concentration of 5 μm) for BRET2, was added 3–5 min prior to BRET measurements, with triplicates for each condition. BRET measurements were performed using either a Victor X (PerkinElmer) plate reader with a filter set (center wavelength/bandwidth) of 460/25 nm (donor) and 535/25 nm (acceptor) for BRET1, or Synergy2 (BioTek) plate reader with filter set 410/80 nm (donor) and 515/30 nm (acceptor) for BRET2. BRET ratios were determined by dividing the intensity of light emitted by the acceptor over the intensity of light emitted by the donor.
Fluorescence microscopy
For live cell confocal fluorescence microscopy, HEK293 cells were seeded in 35-mm glass bottom dishes and transfected with either YFP-tagged WT or mutant AT1R variants and β-arrestin2–mCherry. 18 h post transfection, the media was replaced and 48 h post transfection, cells were serum starved for 1 h before the experiment was performed. Individual cells were tracked and imaged at 0 and 40 min following AngII stimulation using a Zeiss LSM-510 Meta laser scanning microscope with a 63×/1.4 oil objective lens using excitation/emission filter sets: 543 nm/560 nm (long pass) for mCherry, and 514 nm/530–600 nm (band pass) for YFP. For structured illumination microscopy, cells were seeded in 6-well plates with poly-l-lysine–coated coverslips and transfected with either YFP-tagged WT or mutant AT1R variants and β-arrestin2–mCherry. 18 h post transfection, the media were replaced and 48 h post transfection, cells were serum starved for 1 h before the experiment was performed. Cells were stimulated with vehicle or AngII for 40 min and immediately washed with PBS and then fixed with PBS-containing 4% paraformaldehyde. After fixation, the coverslips were washed with PBS and mounted onto microscope slides. The slides were imaged the next day using Zeiss LSM-880 laser scanning microscope with a 100×/1.46 oil objective lens using excitation/emission filter sets: 561 nm/570 nm (long pass) for mCherry and 488 nm/495-590 nm (band pass) for YFP. Image processing and colocalization analysis were performed using Zen software from Zeiss.
Western blotting
One day post transfection, cells were detached and seeded in poly-l-lysine–coated 12-well plates at a density of 1.0 × 105 cells per well in media. The next day, cells were washed and serum starved for 1 h at 37°C and stimulated with AngII for 5–20 min. Immediately following stimulation, cells were rinsed once with ice-cold PBS and lysed in Laemmli buffer (2% SDS, 10% glycerol, 60 mm Tris, 0.02% bromphenol blue, 5% β-mercaptoethanol, pH 6.8). Samples were heated, sonicated, and then analyzed via Western blotting following SDS-PAGE. An anti-p44/42 antibody was used to probe for phospho-ERK (1:1000 dilution). The A1CT primary antibody was used to probe for β-arrestin1/2 (1:2000 dilution). A secondary anti-rabbit antibody conjugated to horseradish peroxidase was used to visualize the bands by chemiluminescence (1:10,000). Membranes probed with an anti-ERK1/2 antibody to control for loading. Chemiluminescence was detected using Chemidoc Touch Imaging System (Bio-Rad), and densitometry analysis of immunoblots was performed on Image LabTM 6.0 software (Bio-Rad). To assess MAPK activation, bands were normalized by dividing the intensity of phospho-ERK1/2 by ERK1/2 and expressed as relative ratios to determine the -fold response of AngII stimulation compared with vehicle.
[3H]thymidine incorporation
One day post transfection, cells were detached and seeded in poly-l-lysine–coated 24-well plates at a density of 2.0 × 104 cells per well in media. The next day, the media were changed to MEM supplemented with 0.5% FBS. 6 h later, cells were stimulated with either vehicle or AngII for 24 h, and 0.5 μCi of [3H]thymidine was added to each well during the last 6 h of stimulation. After 24-h stimulation, cells were washed two times with ice-cold PBS and incubated with 5% TCA for 15 min on ice. Finally, 0.2 M NaOH was used to lyse the cells before scraping. Incorporated radioactivity was measured using PerkinElmer Tri-Carb 28000TR liquid scintillation analyzer.
Data analysis and statistics
Statistical analyses were performed using GraphPad Prism 6 software (GraphPad Software Inc.; La Jolla, CA) using Student's t tests, one- or two-way analysis of variance (ANOVA), Bonferroni, Dunnett's, or Tukey's post hoc multiple comparisons test, when appropriate. Curves presented represent the best fits and were generated using GraphPad Prism software. P values < 0.05 were considered significant.
Data Availability
All data are contained within the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Min Fu and Shi Bo Feng from the Molecular Imaging Platform at the Research Institute of the McGill University Health Center for assistance in obtaining structured illumination microscopy images, and Dr. Robert. J. Lefkowitz for the generous gift of the β-arrestin A1CT antibody.
This article contains supporting information.
Author contributions—Y. C. and S. A. L. conceptualization; Y. C., S. K., and Y. N. data curation; Y. C., Y. N., A. C., and S. A. L. formal analysis; Y. C. and L. G. validation; Y. C., S. K., L. G., and S. A. L. investigation; Y. C. visualization; Y. C. methodology; Y. C., Y. N., and S. A. L. writing-original draft; Y. C. and S. A. L. writing-review and editing; S. A. L. supervision; S. A. L. funding acquisition; S. A. L. project administration.
Funding and additional information—This work was supported by Canadian Institutes of Health Research (CIHR) Grant MOP-74603 to (S.A.L.) and Fonds de recherche santé Québec (FRQS) doctoral training scholarship (to Y. C.) and master's training scholarship (to L. G.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G protein-coupled receptor
- CCV
- clathrin-coated vesicles
- PM
- plasma membrane
- β2AR
- β2-adrenergic receptor
- AT1R
- angiotensin II type 1 receptor
- V2R
- vasopressin V2 receptor
- B2R
- bradykinin B2 receptor
- MAPK
- mitogen-activated protein kinase
- ERK
- extracellular signal-regulated kinase
- BRET
- bioluminescence resonance energy transfer
- TM
- transmembrane
- Rluc
- Renilla luciferase
- DKO
- β-arrestin1/2–deleted HEK293
- FlAsH
- fluorescein arsenical hairpin
- EDT
- 1,2-ethanedithiol
- MEM
- minimum essential media
- ANOVA
- analysis of variance.
References
- 1. Wettschureck N., and Offermanns S. (2005) Mammalian G proteins and their cell type specific functions. Physiol. Rev. 85, 1159–1204 10.1152/physrev.00003.2005 [DOI] [PubMed] [Google Scholar]
- 2. Pierce K. L., and Lefkowitz R. J. (2001) Classical and new roles of β-arrestins in the regulation of G-protein-coupled receptors. Nat. Rev. Neurosci. 2, 727–733 10.1038/35094577 [DOI] [PubMed] [Google Scholar]
- 3. Peterson Y. K., and Luttrell L. M. (2017) The diverse roles of arrestin scaffolds in G protein-coupled receptor signaling. Pharmacol. Rev. 69, 256–297 10.1124/pr.116.013367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gurevich V. V., and Gurevich E. V. (2019) GPCR signaling regulation: The role of GRKs and arrestins. Front. Pharmacol. 10, 125 10.3389/fphar.2019.00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Laporte S. A., and Scott M. G. H. (2019) β-Arrestins: Multitask scaffolds orchestrating the where and when in cell signalling. Methods Mol. Biol. 1957, 9–55 10.1007/978-1-4939-9158-7_2 [DOI] [PubMed] [Google Scholar]
- 6. Goodman O. B. Jr., Krupnick J. G., Santini F., Gurevich V. V., Penn R. B., Gagnon A. W., Keen J. H., and Benovic J. L. (1996) β-Arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature 383, 447–450 10.1038/383447a0 [DOI] [PubMed] [Google Scholar]
- 7. Laporte S. A., Oakley R. H., Zhang J., Holt J. A., Ferguson S. S., Caron M. G., and Barak L. S. (1999) The β2-adrenergic receptor/βarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc. Natl. Acad. Sci. U. S. A. 96, 3712–3717 10.1073/pnas.96.7.3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sorkin A., and von Zastrow M. (2009) Endocytosis and signalling: Intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 10, 609–622 10.1038/nrm2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oakley R. H., Laporte S. A., Holt J. A., Caron M. G., and Barak L. S. (2000) Differential affinities of visual arrestin, βarrestin1, and βarrestin2 for G protein-coupled receptors delineate two major classes of receptors. J. Biol. Chem. 275, 17201–17210 10.1074/jbc.M910348199 [DOI] [PubMed] [Google Scholar]
- 10. Zimmerman B., Simaan M., Akoume M. Y., Houri N., Chevallier S., Séguéla P., and Laporte S. A. (2011) Role of βarrestins in bradykinin B2 receptor-mediated signalling. Cell. Signal. 23, 648–659 10.1016/j.cellsig.2010.11.016 [DOI] [PubMed] [Google Scholar]
- 11. Luttrell L. M., Roudabush F. L., Choy E. W., Miller W. E., Field M. E., Pierce K. L., and Lefkowitz R. J. (2001) Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Natl. Acad. Sci. U. S. A. 98, 2449–2454 10.1073/pnas.041604898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meng D., Lynch M. J., Huston E., Beyermann M., Eichhorst J., Adams D. R., Klussmann E., Klusmann E., Houslay M. D., and Baillie G. S. (2009) MEK1 binds directly to βarrestin1, influencing both its phosphorylation by ERK and the timing of its isoprenaline-stimulated internalization. J. Biol. Chem. 284, 11425–11435 10.1074/jbc.M806395200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Song X., Coffa S., Fu H., and Gurevich V. V. (2009) How does arrestin assemble MAPKs into a signaling complex? J. Biol. Chem. 284, 685–695 10.1074/jbc.M806124200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coffa S., Breitman M., Hanson S. M., Callaway K., Kook S., Dalby K. N., and Gurevich V. V. (2011) The effect of arrestin conformation on the recruitment of c-Raf1, MEK1, and ERK1/2 activation. PLoS One 6, e28723 10.1371/journal.pone.0028723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gurevich V. V., and Gurevich E. V. (2019) The structural basis of the arrestin binding to GPCRs. Mol. Cell. Endocrinol. 484, 34–41 10.1016/j.mce.2019.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Srivastava A., Gupta B., Gupta C., and Shukla A. K. (2015) Emerging functional divergence of β-arrestin isoforms in GPCR function. Trends Endocrinol. Metab. 26, 628–642 10.1016/j.tem.2015.09.001 [DOI] [PubMed] [Google Scholar]
- 17. Ahn S., Wei H., Garrison T. R., and Lefkowitz R. J. (2004) Reciprocal regulation of angiotensin receptor-activated extracellular signal-regulated kinases by β-arrestins 1 and 2. J. Biol. Chem. 279, 7807–7811 10.1074/jbc.C300443200 [DOI] [PubMed] [Google Scholar]
- 18. Luttrell L. M., Wang J., Plouffe B., Smith J. S., Yamani L., Kaur S., Jean-Charles P. Y., Gauthier C., Lee M. H., Pani B., Kim J., Ahn S., Rajagopal S., Reiter E., Bouvier M., et al. (2018) Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal. 11, eaat7650 10.1126/scisignal.aat7650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beautrait A., Paradis J. S., Zimmerman B., Giubilaro J., Nikolajev L., Armando S., Kobayashi H., Yamani L., Namkung Y., Heydenreich F. M., Khoury E., Audet M., Roux P. P., Veprintsev D. B., Laporte S. A., et al. (2017) A new inhibitor of the β-arrestin/AP2 endocytic complex reveals interplay between GPCR internalization and signalling. Nat. Commun. 8, 15054 10.1038/ncomms15054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tohgo A., Choy E. W., Gesty-Palmer D., Pierce K. L., Laporte S., Oakley R. H., Caron M. G., Lefkowitz R. J., and Luttrell L. M. (2003) The stability of the G protein-coupled receptor-β-arrestin interaction determines the mechanism and functional consequence of ERK activation. J. Biol. Chem. 278, 6258–6267 10.1074/jbc.M212231200 [DOI] [PubMed] [Google Scholar]
- 21. Eichel K., Jullié D., Barsi-Rhyne B., Latorraca N. R., Masureel M., Sibarita J. B., Dror R. O., and von Zastrow M. (2018) Catalytic activation of β-arrestin by GPCRs. Nature 557, 381–386 10.1038/s41586-018-0079-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tohgo A., Pierce K. L., Choy E. W., Lefkowitz R. J., and Luttrell L. M. (2002) β-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J. Biol. Chem. 277, 9429–9436 10.1074/jbc.M106457200 [DOI] [PubMed] [Google Scholar]
- 23. Ahn S., Shenoy S. K., Wei H., and Lefkowitz R. J. (2004) Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J. Biol. Chem. 279, 35518–35525 10.1074/jbc.M405878200 [DOI] [PubMed] [Google Scholar]
- 24. Namkung Y., LeGouill C., Kumar S., Cao Y., Teixeira L. B., Lukasheva V., Giubilaro J., Simões S. C., Longpré J. M., Devost D., Hébert T. E., Piñeyro G., Leduc R., Costa-Neto C. M., Bouvier M., et al. (2018) Functional selectivity profiling of the angiotensin II type 1 receptor using pathway-wide BRET signaling sensors. Sci. Signal. 11, eaat1631 10.1126/scisignal.aat1631 [DOI] [PubMed] [Google Scholar]
- 25. Arsenault J., Lehoux J., Lanthier L., Cabana J., Guillemette G., Lavigne P., Leduc R., and Escher E. (2010) A single-nucleotide polymorphism of alanine to threonine at position 163 of the human angiotensin II type 1 receptor impairs Losartan affinity. Pharmacogenet. Genomics 20, 377–388 10.1097/FPC.0b013e32833a6d4a [DOI] [PubMed] [Google Scholar]
- 26. Yoshida T., Kato K., Fujimaki T., Yokoi K., Oguri M., Watanabe S., Metoki N., Yoshida H., Satoh K., Aoyagi Y., Nishigaki Y., Tanaka M., Nozawa Y., Kimura G., and Yamada Y. (2009) Association of genetic variants with chronic kidney disease in Japanese individuals. Clin. J. Am. Soc. Nephrol. 4, 883–890 10.2215/CJN.04350808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gribouval O., Gonzales M., Neuhaus T., Aziza J., Bieth E., Laurent N., Bouton J. M., Feuillet F., Makni S., Ben Amar H., Laube G., Delezoide A. L., Bouvier R., Dijoud F., Ollagnon-Roman E., et al. (2005) Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat. Genet. 37, 964–968 10.1038/ng1623 [DOI] [PubMed] [Google Scholar]
- 28. Hansen J. L., Haunsø S., Brann M. R., Sheikh S. P., and Weiner D. M. (2004) Loss-of-function polymorphic variants of the human angiotensin II type 1 receptor. Mol. Pharmacol. 65, 770–777 10.1124/mol.65.3.770 [DOI] [PubMed] [Google Scholar]
- 29. Namkung Y., Le Gouill C., Lukashova V., Kobayashi H., Hogue M., Khoury E., Song M., Bouvier M., and Laporte S. A. (2016) Monitoring G protein-coupled receptor and β-arrestin trafficking in live cells using enhanced bystander BRET. Nat. Commun. 7, 12178 10.1038/ncomms12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cao Y., Namkung Y., and Laporte S. A. (2019) Methods to monitor the trafficking of β-arrestin/G protein-coupled receptor complexes using enhanced bystander BRET. Methods Mol. Biol. 1957, 59–68 10.1007/978-1-4939-9158-7_3 [DOI] [PubMed] [Google Scholar]
- 31. Lee M. H., Appleton K. M., Strungs E. G., Kwon J. Y., Morinelli T. A., Peterson Y. K., Laporte S. A., and Luttrell L. M. (2016) The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature 531, 665–668 10.1038/nature17154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zimmerman B., Beautrait A., Aguila B., Charles R., Escher E., Claing A., Bouvier M., and Laporte S. A. (2012) Differential β-arrestin-dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci. Signal. 5, ra33 10.1126/scisignal.2002522 [DOI] [PubMed] [Google Scholar]
- 33. Grundmann M., Merten N., Malfacini D., Inoue A., Preis P., Simon K., Rüttiger N., Ziegler N., Benkel T., Schmitt N. K., Ishida S., Müller I., Reher R., Kawakami K., Inoue A., et al. (2018) Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun. 9, 341 10.1038/s41467-017-02661-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. O'Hayre M., Eichel K., Avino S., Zhao X., Steffen D. J., Feng X., Kawakami K., Aoki J., Messer K., Sunahara R., Inoue A., von Zastrow M., and Gutkind J. S. (2017) Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signaling to ERK. Sci. Signal. 10, eaal3395 10.1126/scisignal.aal3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gagnon L., Cao Y., Cho A., Sedki D., Huber T., Sakmar T. P., and Laporte S. A. (2019) Genetic code expansion and photocross-linking identify different β-arrestin binding modes to the angiotensin II type 1 receptor. J. Biol. Chem. 294, 17409–17420 10.1074/jbc.RA119.010324 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript.