

Abstract
Leber's hereditary optic neuropathy (LHON) is a maternal inheritance of eye disease because of the mitochondrial DNA (mtDNA) mutations. We previously discovered a 3866T>C mutation within the gene for the ND1 subunit of complex I as possibly amplifying disease progression for patients bearing the disease-causing 11778G>A mutation within the gene for the ND4 subunit of complex I. However, whether and how the ND1 mutation exacerbates the ND4 mutation were unknown. In this report, we showed that four Chinese families bearing both m.3866T>C and m.11778G>A mutations exhibited higher penetrances of LHON than 6 Chinese pedigrees carrying only the m.3866T>C mutation or families harboring only the m.11778G>A mutation. The protein structure analysis revealed that the m.3866T>C (I187T) and m.11778G>A (R340H) mutations destabilized the specific interactions with other residues of ND1 and ND4, thereby altering the structure and function of complex I. Cellular data obtained using cybrids, constructed by transferring mitochondria from the Chinese families into mtDNA-less (ρ°) cells, demonstrated that the mutations perturbed the stability, assembly, and activity of complex I, leading to changes in mitochondrial ATP levels and membrane potential and increasing the production of reactive oxygen species. These mitochondrial dysfunctions promoted the apoptotic sensitivity of cells and decreased mitophagy. Cybrids bearing only the m.3866T>C mutation displayed mild mitochondrial dysfunctions, whereas those harboring both m.3866T>C and m.11778G>A mutations exhibited greater mitochondrial dysfunctions. These suggested that the m.3866T>C mutation acted in synergy with the m.11778G>A mutation, aggravating mitochondrial dysfunctions and contributing to higher penetrance of LHON in these families carrying both mtDNA mutations.
Keywords: Leber's hereditary optic neuropathy, mitochondrial DNA, mutation, ubiquinone oxidoreductase, pathophysiology, mitochondrial disease, mitochondrial respiratory chain complex, mitophagy, vision, pathogenesis, organelle, oxygen radicals, human genetics, apoptosis, molecular modeling, modifier, NADH:ubiquinone oxidoreductase, penetrance
Leber's hereditary optic neuropathy (LHON) is the most common maternally transmitted eye disorder, characterized by the degeneration of retinal ganglion cells (RGC) and loss of central vision (1–6). A number of mitochondrial DNA (mtDNA) mutations have been linked to the pathogenesis of LHON with various degrees of contribution (7–10). These mtDNA mutations included the primary mutations, which can each cause LHON, and the secondary mutations, which may interact with the primary mutation to increase the probability of clinical expression (1, 8). In the majority of cases worldwide, LHON was caused by three primary mtDNA point mutations, ND4 11778G>A, ND6 14484T>C, and ND1 3460G>A, affecting subunits of NADH:ubiquinone oxidoreductase (complex I) (7–15). These LHON-associated mtDNA mutations often occur near homoplasmy or at homoplasmy. These LHON-associated mtDNA mutations conferred mild mitochondrial dysfunctions, especially the reduced activity of complex I (16–19). Notably, families carrying the mtDNA mutation(s) exhibited gender bias and incomplete penetrance of optic neuropathy (20, 21). These indicated that nuclear and mitochondrial genetic modifiers are required for the phenotypic manifestation of these LHON-associated mtDNA mutations. In a recent study, we identified two nuclear modifiers (c.572G>T, p.Gly191Val in YARS2; and c.157C>T, p.Arg53Trp in PRICKLE3) necessary for the phenotypic expression of the m.11778G>A mutation (22, 23). Several mtDNA variants, including tRNAMet 4435A>G, tRNAThr 15951A>G, ND1 3394T>C, and ND6 14502T>C mutations, may act as mitochondrial genetic modifiers to increase the phenotypic expression of the m.11778G>A mutation (24–28). Furthermore, our previous investigations identified the m.3866T>C mutation in 7 probands (4 bearing only the m.3866T>C mutation and 3 carrying m.3866T>C and m.11778G>A mutations) in a large cohort of 1281 Chinese probands with LHON (15, 29). These Chinese pedigrees carrying only the m.3866T>C mutation exhibited extremely low penetrance of LHON, whereas the penetrances of LHON among those pedigrees bearing the m.3866T>C mutation together with m.11778G>A mutations were higher than those in families carrying only single mtDNA mutation (15, 29). These data suggested the role of m.3866T>C mutation in the pathogenesis of LHON. However, the pathogenic mechanisms underlying these LHON-associated mutations remain elusive.
The m.3866T>C mutation resulted in the change of highly conserved isoleucine at position 187 with threonine (I187T) in ND1, the core component of 45 subunits of complex I (29–31). Thus, the m.3866T>C mutation may perturb both the structure and function of complex I, thereby causing mitochondrial dysfunction. The higher penetrance of LHON in the families carrying both m.11778G>A and m.3866T>C mutations implied that biochemical consequences caused by the m.3866T>C mutation deteriorate the mitochondrial dysfunction associated with m.11778G>A mutation, thereby increasing the penetrance and risk of LHON. The structural consequences of m.11778G>A and m.3866T>C mutations were evaluated by the tertiary structure analysis of complex I from Homo sapiens and Mus musculus (31–33). Functional consequences of m.3866T>C mutation was further investigated through the cybrid cell lines constructed by transferring mitochondria from lymphoblastoid cell lines derived from an affected matrilineal relative carrying the m.11778G>A and m.3866T>C mutation, individuals bearing only m.3866T>C or m.11778G>A mutation, and a control subject belonging to the same mtDNA haplogroup, all into human mtDNA-less (ρ°) cells (19, 34). Using Western blotting and blue native PAGE (BN-PAGE) analyses, we examined if the m.3866T>C and m.11778G>A mutations exerted an effect on the stability of ND1 and ND4 as well as assembly of complex I. These cell lines were then assessed for effects of the mtDNA mutations on the enzymatic activities of respiratory chain complexes, the rate of O2 consumption, mitochondrial ATP production, mitochondrial membrane potential, and generation of reactive oxygen species (ROS). These cell lines were further evaluated for the effect of these mtDNA mutations on apoptotic state and mitophagy.
Results
Clinical and genetic evaluation of 10 Chinese families carrying the m.3866T>C mutation
Ten Han Chinese pedigrees bearing the m.3866T>C mutation (6 bearing only the m.3866T>C mutation, 4 carrying m.3866T>C mutation together with m.11778G>A mutation) were identified in a large cohort of 1793 Chinese probands with LHON (15). This translated to an incidence of 0.6% in this cohort. All available members of 10 pedigrees underwent comprehensive physical and ophthalmologic examinations to identify personal or family medical histories of visual impairments and other clinical abnormalities. As shown in Table S1 and Fig. S1, matrilineal relatives exhibited variable penetrance and expressivity of optic neuropathy among and within families. In particular, the severity of visual loss ranged from profound visual loss to normal vision. The age at onset of optic neuropathy of 6 pedigrees bearing only m.3866T>C mutation ranged from 5 to 39 years, with an average of 24.8 years, whereas the average age at onset of visual impairment of 4 pedigrees harboring both m.3866T>C and m.11778G>A mutations were 22.2 years, respectively. Strikingly, 6 pedigrees harboring only the m.3866T>C mutation exhibited extremely low penetrance of optic neuropathy, ranging from 11.1% to 29.2%, with an average of 17.0%. In contrast, the average penetrances of optic neuropathy of 4 pedigrees harboring both m.3866T>C and m.11778G>A mutations were 41.9%.
Entire sequence analysis of mtDNAs among these Chinese pedigrees revealed that these probands showed the presence of m.3866T>C or m.11778G>A mutations and distinct sets of mtDNA polymorphisms, including 129 known variants, as shown in Table S2. As shown in Table S1, the mtDNAs from 10 pedigrees resided at mtDNA haplogroups C4a (1), D4 (2), G2 (1), M 10 (4), H2 (1), and R (1), respectively (35). These mtDNA variants included 35 in the D-loop region, 4 in the 12S rRNA gene, 3 in the 16S rRNA gene, 5 in the tRNA gene, and 53 silent and 29 missense variants in the genes encoding polypeptides (36). These variants in RNAs and polypeptides were evaluated by phylogenetic analysis of these variants and sequences from 17 vertebrates, as shown in Table S2. These variants were further evaluated for the presence of 485 control subjects and potential structural and functional alterations. Of these, these variants may not have potential structural and functional alterations. These findings indicated that these mtDNA variants do not play an important role in the phenotypic manifestation of m.3866T>C mutations.
The effect of m.3866T>C and m.11778G>A mutations on stability of ND1 and ND4
To test the effect of m.3866T>C (I187T) and m.11778G>A (R340H) mutations on the structure and function of ND1 and ND4, we performed the tertiary structure analysis of complex I from H. sapiens and M. musculus. Based on the Cryo-EM structure of mammalian complex I (PDB entry 5XTD and 6G2J) (32, 33), the hydrophobic side chain of I187 forms the hydrophobic interaction with phosphatidylcholine in ND1 (Fig. 1A), whereas the side chain guanidino group of R340 forms specific electrostatic interactions with the backbone hydroxyl group of V221 in ND4, which is located at the matrix side and water-membrane interface (Fig. 1B). Hence, the replacement of hydrophobic isoleucine at position 187 with the hydrophilic threonine by m.3866T>C mutation in ND1 or arginine at position 340 with histidine by m.11778G>A mutation in ND4 may destabilize these interactions inside ND1 or ND4, thereby perturbing the structure and stability of ND1 and ND4 proteins, respectively.
Figure 1.
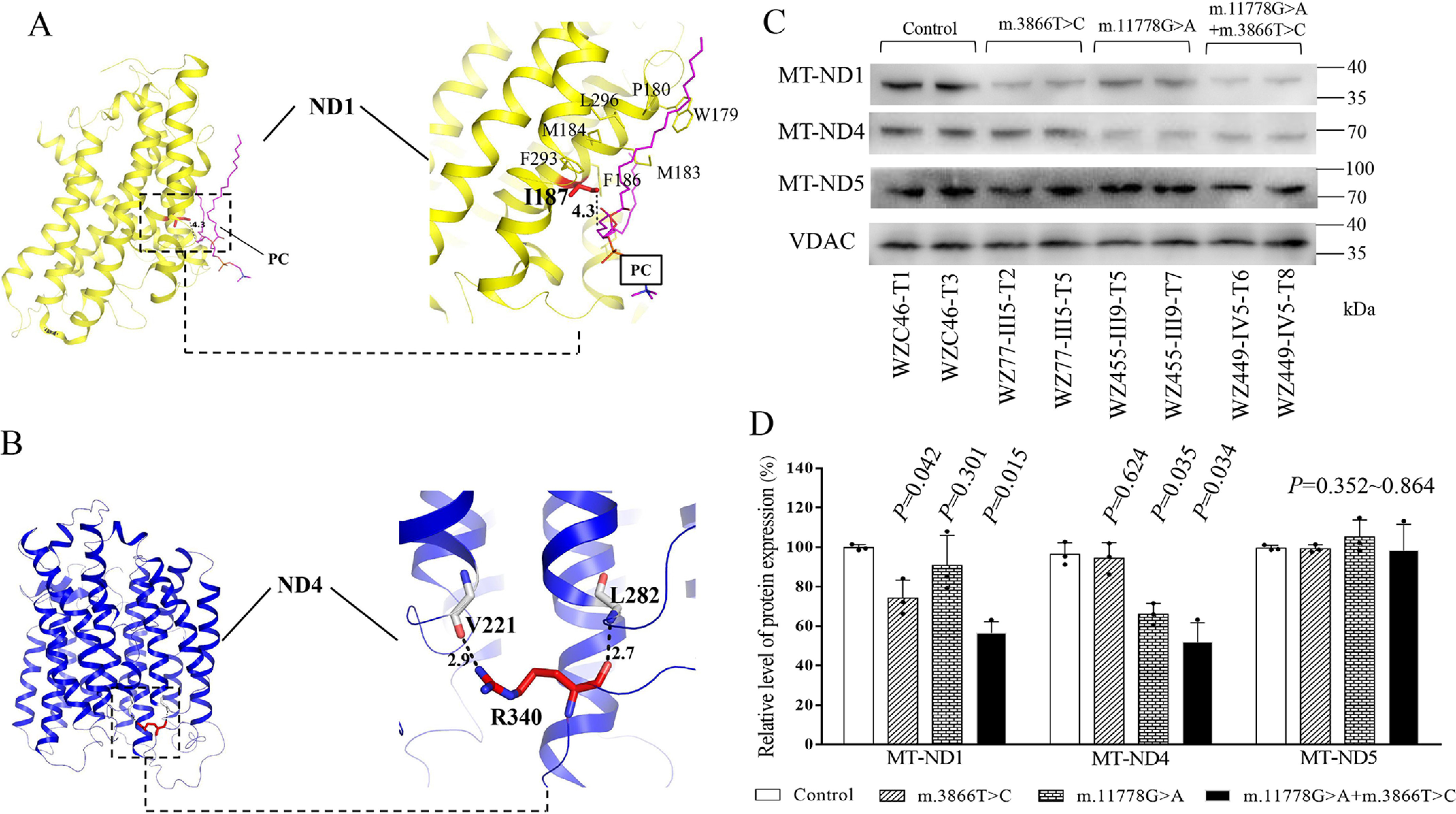
Alterations in the structure and stability of ND1 and ND4. A, interaction between I187 and phosphatidylcholine (PC) in MT-ND1 (shown in yellow). Based on the cryo-EM structure of complex I from H. sapiens (PDB entry 5XTD), the hydrophobic residue I187 interacts with a PC in ND1. I187 and PC are highlighted in red and magenta sticks, respectively. The other hydrophobic residues around I187 are shown in a yellow line. The distance between the side chain of I187 and the fatty acyl tail of PC is shown in a black number (in angstrom). B, the electrostatic interactions in R340 of ND4 (shown in blue). Based on the cryo-EM structure of complex I from M. musculus (PDB entry 6G2J), the side chain guanidino group of R340 forms specific electrostatic interactions with the backbone hydroxyl group of V221 in MT-ND4. R340 and its partner are highlighted in red and white sticks, respectively. Hydrogen bonds are shown as black-dashed lines, with the distance shown in black numbers (in angstroms). C, Western blot analysis. 5 µg of mitochondrial proteins from various cell lines was electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with ND1, N4, and ND6 antibodies, respectively, with VDAC as a loading control. D, quantification of ND1, ND4, and ND6 in mutant and control cell lines. The calculations were based on three independent determinations in each cell line. The error bars indicate two standard errors of the means. p indicates the significance, according to the t test, of the differences between mutant and control cell lines.
To experimentally test this hypothesis, we examined the levels of ND1 and ND4 proteins by Western blotting in these mutant cell lines carrying only m.3866T>C mutation, only m.11778G>A mutation, or both m.3866T>C and m.11778G>A mutations, as well as control cell lines lacking these mutations. As shown in Fig. 1, C and D, the levels of ND1 in mutant cell lines carrying only m.3866T>C, m.11778G>A, or both m.3866T>C and m.11778G>A mutations were 74.1%, 90.3%, and 56.4% relative to the average values of control cell lines. Furthermore, the levels of ND4 in mutant cell lines carrying only m.3866T>C, m.11778G>A, and both m.3866T>C and m.11778G>A mutations were 94.5%, 66.0%, and 51.9%, respectively, relative to the average control values.
The lack of effect of m.3866T>C and m.11778G>A mutations on expression of other subunits of complex I
The complex I of human and mice is composed of 45 subunits, including 7 subunits encoded by mtDNA and 38 subunits encoded by nuclear genes. These subunits are organized into six modules, Q, ND1, ND2, ND4, ND5, and N, with the help of specific assembly factors that begin to assemble separately and ultimately combine to form the complex I holocomplex in the inner membrane of mitochondria (37). As shown in Fig. 2A, ND1 interacts with NDUFA1, NDUFA3, NDUFA8, and NDUFA13, whereas ND4 interacts with ND5, NDUFS2, NDUFB1, NDUFB4, NDUFB5, NDUFB8, and NDUFB11 (32, 33). To examine whether the m.3866T>C or m.11778G>A mutation affected the expression of other subunits of complex I, we measured the levels of NDUFA3, NDUFA8, NDUFA13, ND5, NDUFS2, NDUFB4, NDUFB5, NDUFB8, and NDUFB11 by Western blot analysis among mutant and control cell lines, respectively. As shown in Fig. 1, C and D, and 2, B–D, the levels of these subunits in mutant cell lines were comparable with those in control cell lines.
Figure 2.
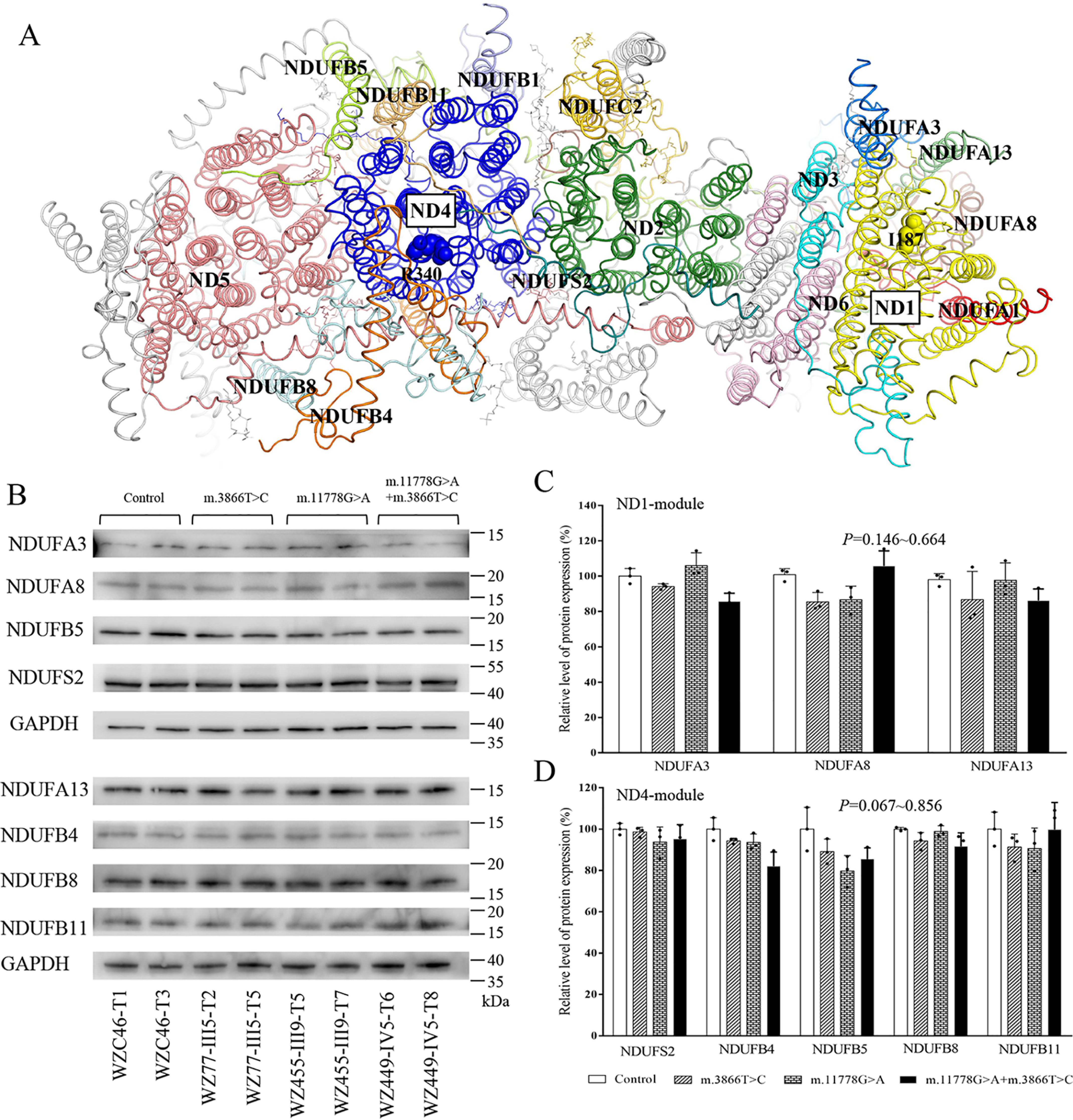
The m.3866T>C and m.11778G>A mutations did not affect the expression of subunits of complex I. A, structure of membrane domains of the human complex I (PDB entry 5XTD). The hydrophilic domains of complex I were removed. The subunits surrounding ND1 and ND4 are shown in different colors. In contrast, other subunits are represented in gray. B, Western blot analysis. 10 µg of total cellular proteins from various cell lines was electrophoresed through a denaturing polyacrylamide gel, electroblotted, and hybridized with NDUFA3, NDUFA8, NDUFA13, NDUFS2, NDUFB4, NDUFB5, NDUFB8, and NDUFB11 antibodies with GAPDH as a loading control. D, quantification of ND1, ND4, and ND6 in mutant and control cell lines. The calculations were based on three independent determinations. Graph details and symbols are explained in the legend to Fig. 1.
Altered assembly and activity of complex I
We analyzed the consequence of m.3866T>C and m.11778G>A mutations on the stability and activity of complex I using the in-gel activity assay. Mitochondrial membrane proteins isolated from mutant and control cell lines were separated by BN-PAGE and stained with specific substrates of complexes I, II, and IV (38–40). As illustrated in Fig. 3A, mutant cell lines carrying only m.3866T>C, only m.11778G>A, or both m.3866T>C and m.11778G>A mutations exhibited altered assembly of intact supercomplexes and complex I. As shown in Fig. 3B, the in-gel activities of complex I in mutant cell lines carrying only m.3866T>C, only m.11778G>A, and both m.3866T>C and m.11778G>A mutations were 72.7%, 66.7%, and 51.5%, respectively, relative to the average values of control cell lines. In contrast, the average in-gel activities of complexes II and IV in mutant cell lines were comparable with those of the control cell lines.
Figure 3.
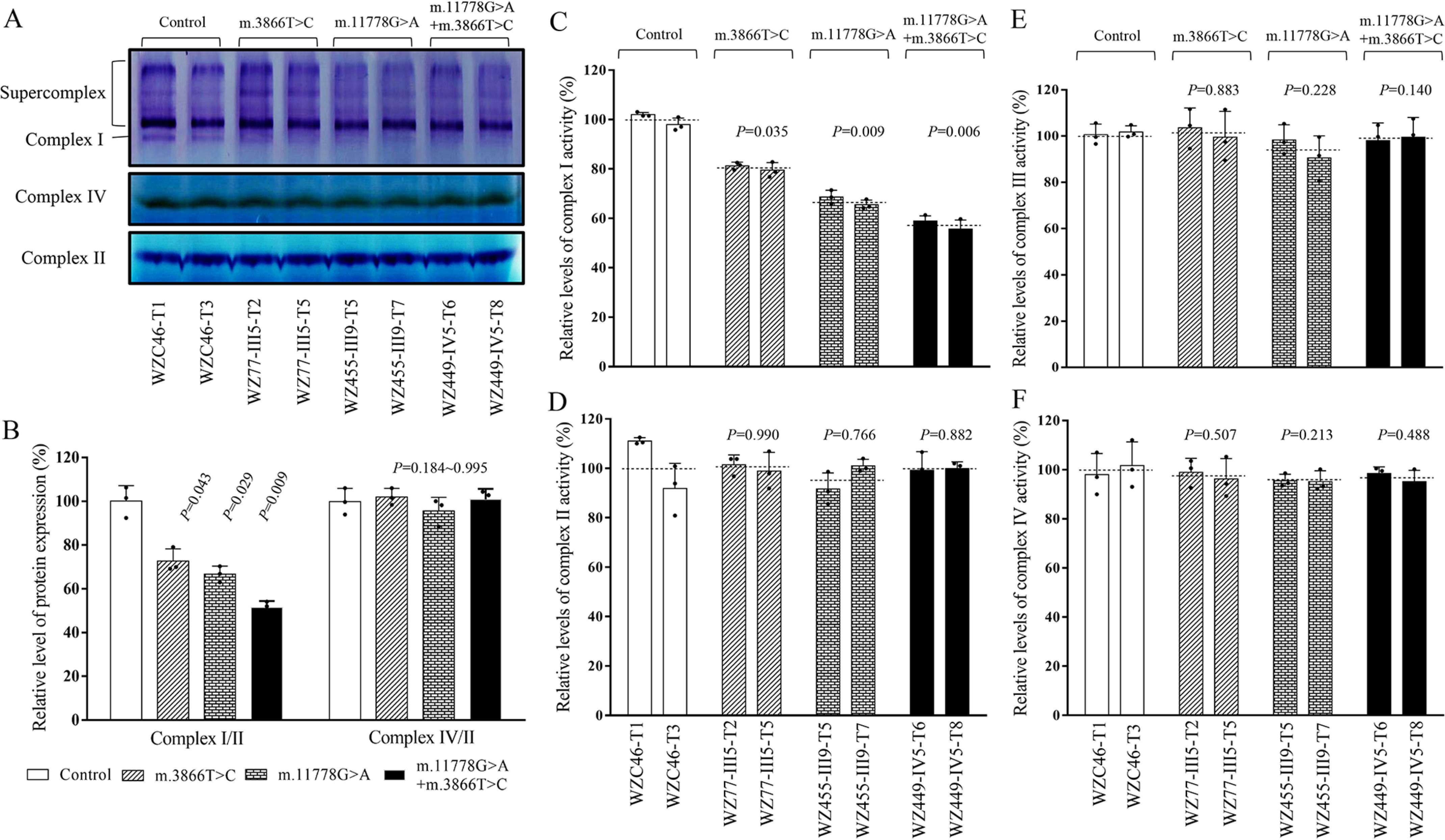
Defective stability and activity of complex I. A, in-gel activity of complexes I, II, and IV. Twenty micrograms of mitochondrial proteins from various mutant and control cell lines was used for BN-PAGE, and the activities of complexes were measured in the presence of specific substrates (NADH and NTB for complex I, sodium succinate, phenazine methosulfate, and NTB for complex II, and DAB and cytochrome c for complex IV). B, quantification of in-gel activities of complexes I, II, and IV. The calculations were based on three independent determinations in each cell line. C–F, Enzymatic activity of respiratory chain complexes. The activities of respiratory complexes I, II, III, and IV were investigated by enzymatic assay on complex I (C), II (D), III (E), and IV (F) in mitochondria isolated from various cell lines. The calculations were based on three independent determinations. Graph details and symbols are explained in the legend to Fig. 1.
We then assessed the consequences of this m.3866T>C mutation on complex I activity using isolated mitochondrial from our mutant and control cybrid cell lines (40–42). As shown in Fig. 3C, the activities of complex I in mutant cell lines carrying only m.3866T>C, only m.11778G>A, and both m.3866T>C and m.11778G>A mutations were 76.7%, 59.8%, and 53.4%, respectively, relative to the average values of control cell lines. As shown in Fig. 3, D–F, the average activities of complexes II, III, and IV in mutant cell lines were comparable with those of the control cell lines.
Respiration deficiency
To further examine whether the m.3866T>C mutation affected cellular bioenergetics, we examined the oxygen consumption rates (OCR) of various mutant and control cybrid cell lines with a Seahorse Bioscience XF-96 extracellular flux analyzer (Seahorse Bioscience) (43, 44). As shown in Fig. 4, the basal OCR in the mutant cybrids carrying only m.3866T>C, only m.11778G>A, or both m.3866T>C and m.11778G>A mutations were 82.9%, 79.1%, and 61.0%, respectively, relative to the mean value measured in the control cybrids. To assess which of the enzyme complexes of the respiratory chain was perturbed in the mutant cell lines, OCR was measured after the sequential addition of oligomycin (inhibit the ATP synthase), carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP; to uncouple the mitochondrial inner membrane and allow for maximum electron flux through the electron transport chain), rotenone (to inhibit complex I), and antimycin (to inhibit complex III). The difference between the basal OCR and the drug-insensitive OCR yielded an amount of ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and nonmitochondrial OCR. As shown in Fig. 4, the ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and nonmitochondrial OCR in mutant cybrids carrying both m.3866T>C and m.11778G>A mutations were 53.2%, 151.2%, 41.4%, 13.4%, and 73.5%, respectively, those in mutant cybrids carrying only the m.3866T>C mutation were 77.1%, 151.2%, 82.7%, 82.3%, and 96.5%, respectively, and those in mutant cybrids carrying only the m.11778G>A mutation were 69.8%, 186.9%, 61.0%, 35.1%, and 86.1%, respectively, relative to the mean values measured in the control cybrids.
Figure 4.

Respiration assays. A, an analysis of O2 consumption in the various cell lines using different inhibitors. The rates of O2 (OCR) were first measured on 2 × 104 cells of each cell line under basal conditions and then sequentially added to oligomycin (1.5 mm), carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) (0.5 mm), rotenone (1 mm), and antimycin A (1 mm) at the indicated times to determine different parameters of mitochondrial functions. B, graphs present the ATP-linked OCR, proton leak OCR, maximal OCR, reserve capacity, and nonmitochondrial OCR in mutant and control cell lines. Nonmitochondrial OCR was determined as the OCR after rotenone/antimycin A treatment. Basal OCR was determined as OCR before oligomycin minus OCR after rotenone/antimycin A. ATP-lined OCR was determined as OCR before oligomycin minus OCR after oligomycin. Proton leak was determined as basal OCR minus ATP-linked OCR. Maximal OCR was determined as the OCR after FCCP minus nonmitochondrial OCR. Reserve capacity was defined as the difference between maximal OCR after FCCP minus basal OCR. The average values of 4 determinations for each cell line are shown, and the horizontal dashed lines represent the average value for each group. Graph details and symbols are explained in the legend to Fig. 1.
Reductions in mitochondrial ATP production
To examine the capacity of oxidative phosphorylation, we measured the levels of cellular and mitochondrial ATP production using a luciferin/luciferase assay (44). Populations of cells were incubated in the media in the presence of glucose and 2-deoxy-d-glucose with pyruvate to inhibit the glycolysis. As shown in Fig. 5, the levels of mitochondrial ATP production (pyruvate and 2-deoxy-d-glucose) in the mutant cybrids bearing only m.3866T>C, only m.11778G>A, and both m.3866T>C and m.11778G>A mutations were 82.3%, 67.2%, and 43.2%, respectively, of average values of control cybrids, whereas the levels of cellular ATP production (glucose) in mutant cybrids were comparable with those measured in the control cybrids.
Figure 5.
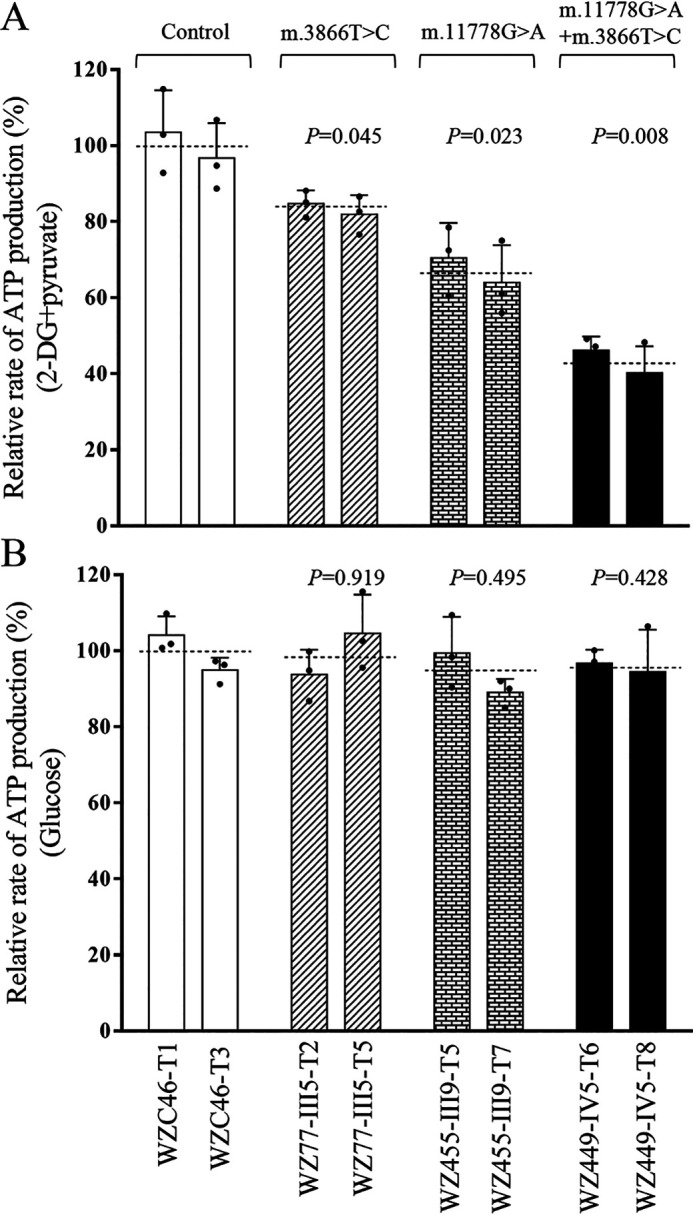
Measurement of cellular and mitochondrial ATP levels. ATP levels from mutant and control cell lines were measure using a luciferin/luciferase assay. Mutant and control cell lines were incubated with 10 mm glucose or 5 mm 2-deoxy-d-glucose plus 5 mm pyruvate to determine ATP generation under mitochondrial ATP synthesis. Average rates of ATP level per cell line in mitochondria are shown. A, ATP level in mitochondria. B, ATP level in total cells. Five determinations were made for each cell line. The average values of 5 determinations for each cell line are shown. Graph details and symbols are explained in the legend to Fig. 1.
Alterations in mitochondrial membrane potential
The mitochondrial membrane potential (ΔΨm) generated by proton pumps (complexes I, III, and IV) is an essential component in the process of energy storage during oxidative phosphorylation (45, 46). We examined the levels of mitochondrial membrane potential (ΔΨm) in the mutant and control cybrids using a fluorescence probe JC-10 assay system. As shown in Fig. 6, the levels of the ΔΨm in the mutant cybrids carrying only the m.3866T>C mutation, only the m.11778G>A mutation, or both the m.3866T>C and m.11778G>A mutations were 79.9%, 75.7%, and 53.7% of the mean values measured in the control cell lines. Conversely, the levels of ΔΨm in mutant cell lines in the presence of FCCP were comparable with those measured in the control cell lines.
Figure 6.

Mitochondrial membrane potential analysis. The mitochondrial membrane potential (ΔΨm) was measured in mutant and control cybrid cell lines using a fluorescence probe JC-10 assay system. The ratios of fluorescence intensities between excitation/emission of 490/590 nm and 490/530 nm (FL590/FL530) were recorded to delineate the ΔΨm level of each sample. The geometric mean relative ratios of FL590/FL530 between mutant and control cell lines were calculated to reflect the level of ΔΨm. A and C, flow cytometry images of cybrids WZC46 lacking the mtDNA mutation, WZ77-III5 carrying only the m.3866T>C mutation, WZ455-III9 carrying only the m.11778G>A mutation, and WZ449-IV5 bearing both m.3866T>C and m.11778G>A mutations without (A) and with (C) FCCP. B and D, relative ratios of JC-10 fluorescence intensities at excitation/emission of 490/530 nm and 490/590 nm in the absence and presence of FCCP. The average of 3–5 determinations for each cell line is shown. Graph details and symbols are explained in the legend to Fig. 1.
The increase of ROS production
Mitochondrial ROS play a critical role in physiological consequences (47). We next assessed ROS production in mutant cybrid cell lines via flow cytometry, comparing baseline staining intensity for each cell line to that upon oxidative stress to obtain a ratio corresponding to ROS generation (48, 49). Geometric mean intensity was recorded to measure the rate of mitoROS of each sample. The relative levels of geometric mean intensity in each cell line were calculated to delineate the levels of mitoROS in mutant and control cells. As shown in Fig. 7, A and B, the levels of ROS generation in the mutant cybrids carrying only the m.3866T>C mutation, only the m.11778G>A mutation, and both the m.11778G>A and m.3866T>C mutations were 119.3%, 122.1%, and 141.2% of the mean values measured in the control cell lines. Furthermore, we examined the levels of catalase and superoxide dismutase proteins (SOD1 and SOD2) in mutant and control cell lines by Western blot analysis (47, 50). As showed in Fig. 7, C and D, the marked increasing levels of these proteins were observed in the mutant cybrids. In particular, the levels of catalase, SOD1, and SOD2 in mutant cybrids carrying only the m.3866T>C mutation were 124.1%, 144.4%, and 177.6%, carrying only the m.11778G>A mutation were 156.2%, 170.6%, and 237.8%, and carrying both m.3866T>C and m.11778G>A mutations were 170.7%, 200.9%, and 272.2%, respectively, relative to the mean values measured in the control cell lines.
Figure 7.

Measurement of mitochondrial ROS. The rates of ROS generation by mitochondria in living cells from mutant and control cell lines were analyzed by a Novocyte flow cytometer (ACEA Biosciences) using the mitochondrial superoxide indicator MitoSOX-Red (5 mm). A, flow cytometry histogram showing MitoSOX-Red fluorescence of various cell lines. B, relative ratios of MitoSOX-Red fluorescence intensity. The average of 4 determinations for each cell line is shown. C, Western blot analysis of three antioxidative enzymes. Twenty micrograms of total proteins from various cell lines was electrophoresed, electroblotted, and hybridized with catalase, SOD1, and SOD2 antibodies and with GAPDH as a loading control. D, quantification of Sod2, Sod1, and catalase. Average relative values of Sod2, Sod1, and catalase were normalized to the average values of GAPDH in various cell lines. The values for the latter are expressed as percentages of the average values for the control cell lines. The average of three independent determinations for each cell line is shown. Graph details and symbols are explained in the legend to Fig. 1.
Promoting apoptosis
Deficient activities of oxidative phosphorylation have been linked to protection against certain apoptotic stimuli (19, 51). To evaluate if the m.3866T>C and m.11778G>A mutations affected the apoptotic processes, we examined the apoptotic state of mutant and control cybrid cell lines by using Annexin V/PI-based flow cytometry and Western blot analyses. As shown in Fig. 8A, the average ratio of Annexin V-positive cells in the mutant cybrids carrying only the m.3866T>C mutation, only the m.11778G>A mutation, and both m.11778G>A and m.3866T>C mutations were 223.9%, 276.6%, and 330.4% of the mean values measured in control cell lines. Furthermore, we examined the levels of three apoptosis-activated proteins (caspase 9, caspase 3, and poly-ADP-ribose polymerase [PARP]) in mutant and control cell lines by Western blot analysis (52). As shown in Fig. 8B, the markedly increasing levels of these proteins were observed in the mutant cybrids. In particular, the levels of cytochrome c, caspase 9, caspase 3, and PARP in mutant cybrids carrying both m.3866T>C and m.11778G>A mutations were 218.2%, 166.1%, 135.7%, and 193.4%, those in cybrids carrying only the m.3866T>C mutation were 125.5%, 143.5%, 122.4%, and 134.5%, and those in cybrids carrying only the m.11778G>A mutation were 185.7%, 154.5%, 127.9%, and 174.5% relative to the mean values measured in the control cybrids, respectively (Fig. 8, C and D).
Figure 8.

Analysis of apoptosis. A, Annexin V/PI apoptosis assay by flow cytometry. Cells were harvested and stained with Annexin V and 1 μl of propidium iodide. The percentage of Annexin V-positive cells then was assessed. B, relative Annexin V-positive cells from various cell lines. Three independent determinations were done in each cell line. C, Western blot analysis of four apoptosis-activated proteins. Twenty micrograms of total proteins from various cell lines was electrophoresed, electroblotted, and hybridized with caspases 9 and 3 and PARP antibodies, with GAPDH as a loading control. D, quantification of four apoptosis-activated proteins. Three independent determinations were done in each cell line. Graph details and symbols are explained in the legend to Fig. 1.
Alteration in mitophagy
The alterations in oxidative phosphorylation (OXPHOS) and mitochondrial membrane potential affected the mitophagic removal of damaged mitochondria (19, 53, 54). To investigate if the m.3866T>C and m.11778G>A mutations regulated the autophagy, the autophagic states of various mutant and control cybrids were analyzed using both fluorescence-based cytometry and Western blotting assays. First, CYTO-ID® autophagy detection kits were used with flow cytometry to examine the degree of autophagy of mutant and control cell lines. As shown in Fig. 9, A and B, significant shifts in the fluorescence peak to high intensity occurred in the mutant cybrids compared with those in control cybrids. The levels of autophagy in the mutant cybrids harboring only the m.3866T>C mutation, only the m.11778G>A mutation, and both m.11778G>A and m.3866T>C mutations were 82.3%, 65.5%, and 55.4% of the mean values measured in the control cybrids. To further evaluate the effect of mtDNA mutations on mitophagy, we performed a Western blot analysis using two markers: microtubule-associated protein 1A/1B light chain 3B (LC3) and sequestosome 1 (SQSTM1/p62) in various cell lines. LC3-II is recleaved by cysteine protease (Atg4B) following completion of the autophagosome and recycled, whereas SQSTM1/p62, one of the best-known autophagic substrates, interacts with LC3 to ensure the selective delivery of these proteins into the autophagosome (55). As shown in Fig. 9, C and D, the average levels of LC3-II/I+II in the mutant cybrids carrying only the m.3866T>C mutation, only the m.11778G>A mutation, and both m.11778G>A and m.3866T>C mutations were 90.3%, 81.1%, and 79.6% of the mean values measured in the control cybrids. Furthermore, the average levels of p62 in the mutant cell lines carrying only the m.3866T>C mutation, only the m.11778G>A mutation, and both m.11778G>A and m.3866T>C mutations were 118.4%, 152.8%, and 180.1% of the mean values measured in control cybrids.
Figure 9.

Analysis of mitophagy. A, flow cytometry histograms showing fluorescence of 6 mutant and 2 control cybrids using a CYTOID® autophagy detection kit. Cells were incubated with DMEM in the absence and presence of rapamycin (inducers of autophagy) and chloroquine (lysosomal inhibitor) at 37 °C for 18 h, added to CYTO-ID®-Green dye, and analyzed using a Novocyte flow cytometer (ACEA Biosciences). B, relative fluorescence intensity from various cell lines. Three independent determinations were done in each cell line. C, Western blot analysis for mitophagic response proteins LC3-I/(I+II) and P62. Twenty micrograms of total cellular proteins from various cell lines was electrophoresed, electroblotted, and hybridized with LC3 and p62, with GAPDH as a loading control. D, quantification of autophagy markers LC3 I/II and p62 in mutant and control cell lines, determined as described elsewhere (19). Four independent determinations were done in each cell line. Graph details and symbols are explained in the legend to Fig. 1.
Discussion
The pathogenicity of ND1 3866T>C mutation
In this present study, we further investigated the molecular mechanism of LHON-associated ND1 3866T>C mutation. The m.3866T>C mutation has been associated with LHON, either together with the m.11778G>A mutation or in isolation among Chinese families (15, 29). The incidences of the m.3866T>C mutation were 0.6% in the cohorts of 1793 Chinese probands with LHON. The occurrence of the m.3866T>C mutation in these genetically unrelated pedigrees affected by LHON and differing considerably in their mtDNA haplotypes (C4a, D4, G2, M10, H2, and R) strongly indicated that this mutation is involved in the pathogenesis of LHON (15, 29). However, 5 pedigrees carrying only m.3866T>C did not have a family history of optic neuropathy. The extremely lower penetrance of LHON in these families carrying only the m.3866T>C mutation was comparable with those in Chinese families bearing only one LHON-associated mtDNA mutation(s), including m.3394T>C, m.14502T>C, m.12338T>C, m.11778G>A, or m.14484T>C (26, 27, 56–59).
The m.3866T>C (I187T) mutation changed a conserved tyrosine at position 187 with isoleucine in ND1, which is an essential subunit of complex I (29, 60). Complex I is composed of 45 subunits, including 7 subunits encoded by mtDNA and 38 subunits encoded by nuclear genes. These subunits are organized into six modules, Q, ND1, ND2, ND4, ND5, and N, with the help of specific assembly factors that begin to assemble separately and ultimately combine to form the complex I holocomplex in the inner membrane of mitochondria (30–32). Based on the cryo-EM structure of mammalian complex I, the hydrophobic side chain of I187 forms the hydrophobic interaction with lipids in ND1. The substitution of hydrophobic isoleucine at position 187 for the hydrophilic threonine by the m.3866T>C mutation in ND1 may destabilize these interactions, thereby altering the structure and function of complex I. In this investigation, the instability of mutated ND1 was evidenced by the reduced levels of ND1 observed in cybrid cell lines bearing only the m.3866T>C mutation and both m.3866T>C and m.11778G>A mutations. However, the m.3866T>C mutation did not affect the expression of other subunits of complex I, such as NDUFA3, NDUFA8, and NDUFA13, which interacted with ND1. The mutated ND1 indeed perturbed the assembly of complex I, as in the case of m.14502T>C or m.3394T>C together with m.11778G>A mutations (26, 27). Both instability of ND1 and altered assembly of complex I were responsible for 20% reductions in the activity of complex I observed in cybrids carrying only the m.3866T>C mutation. Furthermore, the m.3866T>C mutation yielded reduced rates in the basal OCR and ATP-linked OCR in mutant cybrid cell lines. These respiratory deficiencies would lead to an impaired ATP synthesis, alterations in the mitochondrial membrane potentials, and increasing production of oxidative reactive species (61, 62). In the present study, there was a very significant correlation between the reduced levels of complex I activity, mitochondrial ATP production, mitochondrial membrane potentials, and the increasing rates of ROS production (p < 0.01) in the control and mutant cybrids harboring the m.3866T>C mutation. This correlation is clearly consistent with the important role that the defective activity of complex I in the mutant cell lines carrying m.3866T>C mutation plays in producing mitochondrial dysfunction for the development of LHON.
Mitochondrial dysfunctions and overproduction of ROS by LHON-associated mtDNA mutations often induced mitochondrion-dependent apoptotic death and defective mitophagy (19, 62, 63). In this study, the m.3866T>C mutation promoted the apoptotic process, evidenced by elevated release of cytochrome c into cytosol and increased levels of apoptosis-activated proteins: caspase 9, caspase 3, and poly-ADP-ribose polymerase in the cybrid cell lines carrying the m.3866T>C mutation. Moreover, we also documented the involvement of m.3866T>C mutation in decreased mitophagy, as shown by reduced levels of autophagy protein LC3 and accumulation of autophagic substrate p62, as in the cases of other LHON-linked mtDNA mutations (19, 53, 54). These data demonstrated the direct link between mitochondrial dysfunction caused by complex I mutation and apoptosis or mitophagy. However, the extremely low penetrance of LHON and mild mitochondrial dysfunction strongly suggested that the m.3866T>C mutation is the primary factor underlying the development of LHON but is insufficient to produce a clinical phenotype. The additional hereditary or environmental factors are necessary for the phenotypic expression of m.3866T>C mutation.
The synergy effect of m.3866T>C mutation on the phenotypic expression of the LHON-associated m.11778G>A mutation
Mitochondrial genetic backgrounds, such as specific mtDNA variants, may act as genetic modifiers to increase the phenotypic expression of the m.11778G>A mutation (24–28, 64). The average penetrances of optic neuropathy of 4 pedigrees harboring both m.3866T>C and m.11778G>A mutations exhibited levels significantly higher than those in 6 families carrying only the m.3866T>C mutation and other pedigrees harboring only the m.11778G>A mutation. (14, 15, 29, 58). These observations strongly suggested that the m.3866T>C mutation increases the expression of optic neuropathy in these families carrying the m.11778G>A mutation, as in the cases of m.4216T>C and m.13708G>A mutations in European families and m.4435A>G, m.3394T>C, m.15951A>G, and m.14502T>C mutations in other Chinese pedigrees (11, 24–27, 64).
These cybrid cell models, with identical nuclear background and mtDNA haplogroup, allow us to evaluate the biochemical consequences because of specific mtDNA mutations. In fact, m.11778G>A only altered the stability of ND4 but not other subunits, such as ND5, NDUFS2, NDUFB4, NDUFB5, NDUFB8, and NDUFB11, that interacted with ND4. As a result, m.11778G>A mutation conferred a mild reduction in the activity of complex I (16–18, 22, 23). In the present study, cybrid cell lines bearing both m.3866T>C and m.11778G>A mutations exhibited more severe mitochondrial dysfunctions than those in the cells carrying only the m.11778G>A or m.3866T>C mutation. In particular, the cell lines carrying both m.3866T>C and m.11778G>A mutations exhibited more unstable complex I than those bearing only the m.3866T>C or m.11778G>A mutation. As a result, the m.3866T>C mutation aggravated the biochemical defects caused by the m.11778G>A mutation, including the defective activity of complex I, decreases of mitochondrial ATP production and mitochondrial membrane potentials, and increasing production of ROS. In particular, the 57% decreases in the mitochondrial ATP production observed in the cybrids bearing both m.3866T>C and m.11778G>A mutations were below a proposed threshold level, which is 50% of control levels in the mitochondrial ATP, to develop a clinical phenotype (23, 65, 66). In particular, RGCs require the highest-ATP-demand cells in the retina (67, 68). Therefore, the retinal ganglion cells carrying both m.3866T>C and m.11778G>A mutations may be preferentially involved, because they are high-ATP-demand cells and somehow are exquisitely sensitive to subtle imbalance in cellular redox state or increased level of free radicals (1, 5, 6, 69–71). Mitochondrial dysfunction affected the apoptotic sensitivity and mitophagy of cells carrying the LHON-associated mtDNA mutations (19, 53, 71). In the present investigation, the cell lines carrying both m.3866T>C and m.11778G>A mutations exhibited more apoptotic susceptibility than those carrying only the m.3866T>C or m.11778G>A mutation. In particular, cell lines carrying both m.3866T>C and m.11778G>A mutations exhibited an increase of apoptosis and higher levels of apoptosis-activated proteins cytochrome c, caspases 3 and 9, and PARP than those in cell lines carrying only the m.3866T>C or m.11778G>A mutation. These data demonstrated that m.3866T>C mutation elevated the apoptosis induced by the m.11778G>A mutation. Mitophagy disposes of damaged mitochondria and maintains the healthy mitochondrial population in the cells. The impairment of OXPHOS and alteration of mitochondrial membrane potential affected the mitophagic removal of damaged cells (72, 73). In this study, the cell lines carrying both m.3866T>C and m.11778G>A mutations exhibited more increased levels of p62 than cells carrying only m.3866T>C or m.11778G>A mutation, which indicated the preferential accumulation of autophagic substrates, such as misfolded proteins, leading to deleterious effects. Furthermore, more severely decreased levels of LC3 were observed in cybrid cell lines carrying both m.3866T>C and m.11778G>A mutations than those in cell lines carrying only m.3866T>C or m.11778G>A mutation, which suggested more decreases in the capacity of the double mutant cells to generate autophagosomes, thereby perturbing the autophagic degradation of ubiquitinated proteins. These data indicated that m.3866T>C mutation worsened the defective mitophagy caused by the m.11778G>A mutation. The significant correlation between severe biochemical phenotypes in these mutants cybrids and higher penetrance of LHON in these pedigrees harboring both m.11778G>A and m.3866T>C mutation demonstrated that the m.3866T>C mutation enhanced the penetrance and occurrence of optic neuropathy in the Chinese families carrying both m.3866T>C and m.11778G>A mutations. Therefore, our findings highlighted the critical role of m.3866T>C mutation in the pathogenesis of LHON, manifested by sole mtDNA mutation or its synergy with the m.11778G>A mutation.
Experimental procedures
Families and subjects
A total of 1793 genetically unrelated Han Chinese subjects with LHON were recruited from eye clinics across China, as described previously (14, 15, 26). This study was in compliance with the Declaration of Helsinki. Informed consent, blood samples, and clinical evaluations were obtained from all participating family members under protocols approved by the Ethic Committees of Zhejiang University and Wenzhou Medical University. A comprehensive history and physical examination for these participating subjects were performed at length to identify both personal or family medical histories of visual impairment and other clinical abnormalities. The ophthalmic examinations of probands and other members of these families were conducted as detailed previously (24, 29). A total of 485 control DNA samples were obtained from adult Han Chinese from the same area.
Mitochondrial DNA analysis
The mutational screening of the m.3866T>C mutation was performed as detailed previously (15, 29). The entire mitochondrial genomes of 6 probands carrying only the m.3866T>C mutation and 4 individuals harboring both the m.3866T>C and m.11778G>A mutations were determined as detailed elsewhere (74). The resulting sequence data were compared with the updated consensus Cambridge sequence (GenBank accession number NC_012920) (36). The entire mtDNA sequences of 11 pedigrees and 1 control subject were assigned to the mitochondrial haplogroups using the nomenclature described previously (35). An analysis for the presence and levels of m.11778G>A and m.3866T>C mutations in mutant and control cell lines were carried out as described previously (23, 29). The quantification of mtDNA copy numbers from different cell lines was carried out as detailed elsewhere (73).
Cell lines and culture conditions
Lymphoblastoid cell lines derived from three probands (WZ77-III5 harboring only m.3866T>C mutation, WZ455-III9 bearing only m.11778G>A mutation, and WZ449-IV5 carrying both m.11778G>A and m.3866T>C mutations) and one control individual, WZC46, were immortalized by transformation with the Epstein-Barr virus, as described elsewhere (75). Lymphoblastoid cell lines were grown in RPMI 1640 medium (Corning) supplemented with 10% fetal bovine serum. The 143B.TK– cell line was grown in DMEM (containing 4.5 mg of glucose and 0.11 mg pyruvate per ml), supplemented with 100 µg of BrdU per ml and 5% FBS. The mtDNA-less ρ°206 cell line, derived from 143B.TK–, was grown under the same conditions as the parental line, except for the addition of 50 µg of uridine/ml (34). Transformation by cytoplasts of mtDNA-less ρ°206 cells was performed by using four immortalized lymphoblastoid cell lines, as detailed previously (34, 76). The cybrids derived from each donor cell line were analyzed for the presence and level of the m.3866T>C or m.11778G>A mutation and mtDNA copy numbers as detailed elsewhere (24, 29, 76). Two cybrids derived from each donor cell line with homoplasmy of mtDNA mutations and similar mtDNA copy numbers were used for the following biochemical characterization. All cybrid cell lines were maintained in the same medium as the 143B.TK– cell line.
Western blot analysis
Mitochondria were isolated from mutant and control cell lines by following the protocol described elsewhere (77). Western blot analysis was carried out by using total mitochondrial proteins isolated from mutant and control cell lines, as detailed elsewhere (44, 46). The first antibodies used for this investigation were from Proteintech Group (anti-VDAC1/2 [10866-1-AP], ND1 [19703-1-AP], NDUFA3 [17257-1-AP], NDUFB1 [16902-1-AP], NDUFB8 [14794-1-AP], NDUFB11 [16720-1-AP], SOD1 [10269-1-AP], SOD2 [24127-1-AP], and GAPDH [60004-1-Ig]), Abcam Biotechnology (ND4 [ab219822], ND5 [ab138136], catalase [ab52477], cytochrome c [ab133504], P62 [ab56416]), ABclonal Technology (NDUFA8 [A12118], NDUFA13 [A5412], NDUFB4 [A13820], NDUFB5 [A8327], NDUFS2 [A12858]), Cell Signaling Technology (PARP [9542], caspase 3 [14420], caspase 9[9508], and LC3I/II [12741]), and the secondary antibodies were from Beyotime Biotechnology (peroxidase AffiniPure goat anti-rabbit IgG [A0208] and goat anti-mouse IgG [A0216]). The protein signals were detected using the ECL system (Millipore). Quantification of density in each band was performed as detailed previously (22, 46).
BN gel electrophoresis
BN gel electrophoresis and in-gel activity assay were performed by using mitochondrial proteins, as detailed elsewhere (27, 38, 39).
Enzymatic assays
The enzymatic activities of complexes I, II, III, and IV in isolated mitochondria were assessed by using Synergy H1 (Biotek, Winooski, VT), as detailed previously (40–42, 78).
Measurements of oxygen consumption
The rates of oxygen consumption in cybrid cell lines were measured with a Seahorse Bioscience XF-96 extracellular flux analyzer (Seahorse Bioscience), as described elsewhere (43).
ATP measurements
The cellular and mitochondrial ATP levels were analyzed by the Cell Titer-Glo luminescent cell viability assay kit (Promega) according to the modified procedures of the manufacturer (44).
Assessment of mitochondrial membrane potential
Basal level of mitochondrial membrane potential was examined with JC-10 assay kit-flow cytometry (Abcam) according to the manufacturer's general recommendations, with some modifications, as detailed elsewhere (45, 46).
ROS measurements
The MitoSOX red mitochondrial superoxide indicator (Thermo Fisher) was used for ROS measurements by following the manufacturer's instructions, as detailed previously (48, 49).
Annexin V/PI apoptosis assay by flow cytometry
For discrimination of apoptotic and nonapoptotic cells by Annexin V/PI staining, cells were harvested and stained with Annexin V and 1 μl of propidium iodide (PI) (V13242, ThermoFisher Scientific) according to the manufacturer's instructions. Each sample was detected by NovoCyte (ACEA Biosciences) and analyzed using NovoExpress software (79).
Analysis of autophagy
The fluorescence-based cytometry to analyze the level of mitophagy was performed using the CYTO-ID® autophagy detection kit (Enzo) as detailed elsewhere (80, 81).
Statistical analysis
All statistical analyses were performed using the unpaired, two-tailed Student's t test contained in the GraphPad Prism 5 program (GraphPad Software) and Microsoft Excel (version 2016). A p value of <0.05 was considered statistically significant.
Data availability
Representative experiments are shown in the figures and supporting information. Supplemental data include two tables and one figure. For any additional information, please contact the corresponding author.
Supplementary Material
Acknowledgments
We are grateful to patients and their family members for their participation.
This article contains supporting information.
Author contributions—Y. J., J. Z., Y. L., Q. Y., M. C., S. X., X. M., Y. X., F. M., M. Z., and M.-X. G. data curation; Y. J., Q. Y., and M.-X. G. formal analysis; Y. J., J. Z., and S. X. validation; Y. J. and Y. L. methodology; Y. J. writing-original draft; J. Z. and M.-X. G. investigation; J. Z. visualization; S. X., M. Z., and M.-X. G. resources; R. Y. and M.-X. G. supervision; R. Y. and M.-X. G. funding acquisition; M.-X. G. conceptualization; M.-X. G. project administration; M.-X. G. writing-review and editing.
Funding and additional information—This work was supported by and the National Key R&D Program of China, grant 2018YFC1002700 and 2018YFC1004802, from the Ministry of Science and Technology of China, and grants 31970557, 31471191, and 81400434 from the Natural Science Foundation of China.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- BN-PAGE
- blue native PAGE
- OXPHOS
- oxidative phosphorylation
- PI
- propidium iodide
- ΔΨm
- mitochondrial membrane potential
- ROS
- reactive oxygen species
- PARP
- poly-ADP-ribose polymerase
- OCR
- oxygen consumption rate
- FCCP
- carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone
- mtDNA
- mitochondrial DNA
- RGC
- retinal ganglion cells.
References
- 1. Wallace D. C., and Lott M. T. (2017) Leber hereditary optic neuropathy: exemplar of an mtDNA disease. Handb. Exp. Pharmacol. 240, 339–376 10.1007/164_2017_2 [DOI] [PubMed] [Google Scholar]
- 2. Newman N. J. (1993) Leber's hereditary optic neuropathy. Ophthalmol. Clin. N. Am. 50, 540–548 10.1001/archneur.1993.00540050082021 [DOI] [PubMed] [Google Scholar]
- 3. Nikoskelainen E. K. (1994) Clinical picture of LHON. Clin. Neurosci. 2, 115–120 [Google Scholar]
- 4. Sadun A. A., La Morgia C., and Carelli V. (2011) Leber's hereditary optic neuropathy. Curr. Treat Options Neurol. 13, 109–117 10.1007/s11940-010-0100-y [DOI] [PubMed] [Google Scholar]
- 5. Carelli V., La Morgia C., Valentino M. L., Barboni P., Ross-Cisneros F. N., and Sadun A. A. (2009) Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim. Biophys. Acta 1787, 518–528 10.1016/j.bbabio.2009.02.024 [DOI] [PubMed] [Google Scholar]
- 6. Bahr T., Welburn K., Donnelly J., and Bai Y. (2020) Emerging model systems and treatment approaches for Leber's hereditary optic neuropathy: challenges and opportunities. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165743 10.1016/j.bbadis.2020.165743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wallace D. C., Singh G., Lott M. T., Hodge J. A., Schurr T. G., Lezza A. M., Elsas L. J., and Nikoskelainen E. K. (1988) Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science 242, 1427–1430 10.1126/science.3201231 [DOI] [PubMed] [Google Scholar]
- 8. Yu-Wai-Man P., Griffiths P. G., Hudson G., and Chinnery P. F. (2009) Inherited mitochondrial optic neuropathies. J. Med. Genet. 46, 145–158 10.1136/jmg.2007.054270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maresca A., Caporali L., Strobbe D., Zanna C., Malavolta D., La Morgia C., Valentino M. L., and Carelli V. (2014) Genetic basis of mitochondrial optic neuropathies. Curr. Mol. Med. 14, 985–992 10.2174/1566524014666141010132627 [DOI] [PubMed] [Google Scholar]
- 10. Ruiz-Pesini E., Lott M. T., Procaccio V., Poole J. C., Brandon M. C., Mishmar D., Yi C., Kreuziger J., Baldi P., and Wallace D. C. (2007) An enhanced mitomap with a global mtDNA mutational phylogeny. Nucleic Acids Res. 35, D823–D828 10.1093/nar/gkl927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brown M. D., Torroni A., Reckord C. L., and Wallace D. C. (1995) Phylogenetic analysis of Leber's hereditary optic neuropathy mitochondrial DNA's indicates multiple independent occurrences of the common mutations. Hum. Mutat. 6, 311–325 10.1002/humu.1380060405 [DOI] [PubMed] [Google Scholar]
- 12. Mackey D. A., Oostra R. J., Rosenberg T., Nikoskelainen E., Bronte-Stewart J., Poulton J., Harding A. E., Govan G., Bolhuis P. A., and Norby S. (1996) Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy. Am. J. Hum. Genet. 59, 481–485 [PMC free article] [PubMed] [Google Scholar]
- 13. Liang M., Jiang P., Li F., Zhang J., Ji Y., He Y., Xu M., Zhu J., Meng X., Zhao F., Tong Y., Liu X., Sun Y., Zhou X., Mo J. Q., et al. (2014) Frequency and spectrum of mitochondrial ND6 mutations in 1218 Han Chinese subjects with Leber's hereditary optic neuropathy. Invest. Ophthalmol. Vis. Sci. 55, 1321–1331 10.1167/iovs.13-13011 [DOI] [PubMed] [Google Scholar]
- 14. Jiang P., Liang M., Zhang J., Gao Y., He Z., Yu H., Zhao F., Ji Y., Liu X., Zhang M., Fu Q., Tong Y., Sun Y., Zhou X., Huang T., et al. (2015) Prevalence of mitochondrial ND4 mutations in 1281 Han Chinese subjects with Leber's hereditary optic neuropathy. Invest. Ophthalmol. Vis. Sci. 56, 4778–4788 10.1167/iovs.14-16158 [DOI] [PubMed] [Google Scholar]
- 15. Ji Y., Liang M., Zhang J., Zhu L., Zhang Z., Fu R., Liu X., Zhang M., Fu Q., Zhao F., Tong Y., Sun Y., Jiang P., and Guan M.-X. (2016) Mitochondrial ND1 variants in 1281 Chinese subjects with Leber's hereditary optic neuropathy. Invest. Ophthalmol. Vis. Sci. 57, 2377–2389 10.1167/iovs.16-19243 [DOI] [PubMed] [Google Scholar]
- 16. Brown M. D., Trounce I. A., Jun A. S., Allen J. C., and Wallace D.,C. (2000) Functional analysis of lymphoblast and cybrid mitochondria containing the 3460, 11778, or 14484 Leber's hereditary optic neuropathy mitochondrial DNA mutation. J. Biol. Chem. 275, 39831–39836 10.1074/jbc.M006476200 [DOI] [PubMed] [Google Scholar]
- 17. Hofhaus G., Johns D. R., Hurko O., Attardi G., and Chomyn A. (1996) Respiration and growth defects in transmitochondrial cell lines carrying the 11778 mutation associated with Leber's hereditary optic neuropathy. J. Biol. Chem. 271, 13155–13161 10.1074/jbc.271.22.13155 [DOI] [PubMed] [Google Scholar]
- 18. Qian Y., Zhou X., Liang M., Qu J., and Guan M. X. (2011) The altered activity of complex III may contribute to the high penetrance of Leber's hereditary optic neuropathy in a Chinese family carrying the ND4 G11778A mutation. Mitochondrion 11, 871–877 10.1016/j.mito.2011.06.006 [DOI] [PubMed] [Google Scholar]
- 19. Zhang J., Ji Y., Lu Y., Fu R., Xu M., Liu X., and Guan M. X. (2018) Leber's hereditary optic neuropathy (LHON)-associated ND5 12338T > C mutation altered the assembly and function of complex I, apoptosis and mitophagy. Hum. Mol. Genet. 27, 1999–2011 10.1093/hmg/ddy107 [DOI] [PubMed] [Google Scholar]
- 20. Riordan-Eva P., Sanders M. D., Govan G. G., Sweeney M. G., Da Costa J., and Harding A. E. (1995) The clinical features of Leber's hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain 118, 319–337 10.1093/brain/118.2.319 [DOI] [PubMed] [Google Scholar]
- 21. Newman N. J., Lott M. T., and Wallace D. C. (1991) The clinical characteristics of pedigrees of Leber's hereditary optic neuropathy with the 11778 mutation. Am. J. Ophthalmol. 111, 750–762 10.1016/S0002-9394(14)76784-4 [DOI] [PubMed] [Google Scholar]
- 22. Jiang P., Jin X., Peng Y., Wang M., Liu H., Liu X., Zhang Z., Ji Y., Zhang J., Liang M., Zhao F., Sun Y.-H., Zhang M., Zhou X., Chen Y., et al. (2016) The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber's hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum. Mol. Genet. 25, 584–596 10.1093/hmg/ddv498 [DOI] [PubMed] [Google Scholar]
- 23. Yu J., Liang X., Ji Y., Ai C., Liu J., Zhu L., Nie Z., Jin X., Wang C., Zhang J., et al. (2020) PRICKLE3 linked to ATPase biogenesis manifested Leber's hereditary optic neuropathy. J. Clin. Invest. 130, 134965 10.1172/JCI134965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qu J., Li R., Zhou X., Tong Y., Lu F., Qian Y., Hu Y., Mo J. Q., West C. E., and Guan M. X. (2006) The novel A4435G mutation in the mitochondrial tRNAMet may modulate the phenotypic expression of the LHON-associated ND4 G11778A mutation in a Chinese family. Invest. Ophthalmol. Vis. Sci. 47, 475–483 10.1167/iovs.05-0665 [DOI] [PubMed] [Google Scholar]
- 25. Li R., Qu J., Zhou X., Tong Y., Hu Y., Qian Y., Lu F., Mo J. Q., West C. E., and Guan M. X. (2006) The mitochondrial tRNAThr A15951G mutation may influence the phenotypic expression of the LHON-associated ND4 G11778A mutation in a Chinese family. Gene 376, 79–86 10.1016/j.gene.2006.02.014 [DOI] [PubMed] [Google Scholar]
- 26. Jiang P., Liang M., Zhang C., Zhao X., He Q., Cui L., Liu X., Sun Y.-H., Fu Q., Ji Y., Bai Y., Huang T., and Guan M.-X. (2016) Biochemical evidence for a mitochondrial genetic modifier in the phenotypic manifestation of Leber's hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum. Mol. Genet. 25, 3613–3625 10.1093/hmg/ddw199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ji Y., Zhang J., Yu J., Wang Y., Lu Y., Liang M., Li Q., Jin X., Wei Y., Meng F., Gao Y., Cang X., Tong Y., Liu X., Zhang M., et al. (2019) Contribution of mitochondrial ND1 3394T>C mutation to the phenotypic manifestation of Leber's hereditary optic neuropathy. Hum. Mol. Genet. 28, 1515–1529 10.1093/hmg/ddy450 [DOI] [PubMed] [Google Scholar]
- 28. Pello R., Martín M. A., Carelli V., Nijtmans L. G., Achilli A., Pala M., Torroni A., Gómez-Durán A., Ruiz-Pesini E., Martinuzzi A., Smeitink J. A., Arenas J., and Ugalde C. (2008) Mitochondrial DNA background modulates the assembly kinetics of OXPHOS complexes in a cellular model of mitochondrial disease. Hum. Mol. Genet. 17, 4001–4011 10.1093/hmg/ddn303 [DOI] [PubMed] [Google Scholar]
- 29. Zhou X., Qian Y., Zhang J., Tong Y., Jiang P., Liang M., Dai X., Zhou H., Zhao F., Ji Y., Mo J. Q., Qu J., and Guan M.-X. (2012) Leber's hereditary optic neuropathy is associated with the T3866C mutation in mitochondrial ND1 gene in three Han Chinese families. Invest. Ophthalmol. Vis. Sci. 53, 4586–4594 10.1167/iovs.11-9109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scheffler I. E. (2015) Mitochondrial disease associated with complex I (NADH-CoQ oxidoreductase) deficiency. J. Inherit. Metab. Dis. 38, 405–415 10.1007/s10545-014-9768-6 [DOI] [PubMed] [Google Scholar]
- 31. Zhu J., Vinothkumar K. R., and Hirst J. (2016) Structure of mammalian respiratory complex I. Nature 536, 354–358 10.1038/nature19095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo R., Zong S., Wu M., Gu J., and Yang M. (2017) Architecture of human mitochondrial respiratory megacomplex I2III2IV2. Cell 170, 1247–1257 10.1016/j.cell.2017.07.050 [DOI] [PubMed] [Google Scholar]
- 33. Agip A.-N. A., Blaza J. N., Bridges H. R., Viscomi C., Rawson S., Muench S. P., and Hirst J. (2018) Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 25, 548–556 10.1038/s41594-018-0073-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. King M. P., and Attadi G. (1996) Mitochondria-mediated transformation of human rhoo cells. Methods Enzymol. 264, 313–334 [DOI] [PubMed] [Google Scholar]
- 35. Kong Q.-P., Bandelt H.-J., Sun C., Yao Y.-G., Salas A., Achilli A., Wang C.-Y., Zhong L., Zhu C.-L., Wu S.-F., Torroni A., and Zhang Y.-P. (2006) Updating the East Asian mtDNA phylogeny: a prerequisite for the identification of pathogenic mutations. Hum. Mol. Genet. 15, 2076–2086 10.1093/hmg/ddl130 [DOI] [PubMed] [Google Scholar]
- 36. Andrews R. M., Kubacka I., Chinnery P. F., Lightowlers R. N., Turnbull D. M., and Howell N. (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 23, 147 10.1038/13779 [DOI] [PubMed] [Google Scholar]
- 37. Guerrero-Castillo S., Baertling F., Kownatzki D., Wessels H. J., Arnold S., Brandt U., and Nijtmans L. (2017) The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab. 25, 128–139 10.1016/j.cmet.2016.09.002 [DOI] [PubMed] [Google Scholar]
- 38. Jha P., Wang X., and Auwerx J. (2016) Analysis of mitochondrial respiratory chain super-complexes using blue native polyacrylamide gel electrophoresis (BN-PAGE). Curr. Protoc. Mouse Biol. 6, 1–14 10.1002/9780470942390.mo150182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wittig I., Braun H. P., and Schagger H. (2006) Blue native PAGE. Nat. Protoc. 1, 418–428 10.1038/nprot.2006.62 [DOI] [PubMed] [Google Scholar]
- 40. Li Y., D'Aurelio M., Deng J. H., Park J. S., Manfredi G., Hu P., Lu J., and Bai Y. (2007) An assembled complex IV maintains the stability and activity of complex I in mammalian mitochondria. J. Biol. Chem. 282, 17557–17562 10.1074/jbc.M701056200 [DOI] [PubMed] [Google Scholar]
- 41. Birch-Machin M. A., and Turnbull D. M. (2001) Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 65, 97–117 10.1016/s0091-679x(01)65006-4 [DOI] [PubMed] [Google Scholar]
- 42. Thorburn D. R., Chow C. W., and Kirby D. M. (2004) Respiratory chain enzyme analysis in muscle and liver. Mitochondrion 4, 363–375 10.1016/j.mito.2004.07.003 [DOI] [PubMed] [Google Scholar]
- 43. Dranka B. P., Benavides G. A., Diers A. R., Giordano S., Zelickson B. R., Reily C., Zou L., Chatham J. C., Hill B. G., Zhang J., Landar A., and Darley-Usmar V. M. (2011) Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic. Biol. Med. 51, 1621–1635 10.1016/j.freeradbiomed.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gong S., Peng Y., Jiang P., Wang M., Fan M., Wang X., Zhou H., Li H., Yan Q., Huang T., and Guan M.-X. (2014) A deafness-associated tRNAHis mutation alters the mitochondrial function, ROS production and membrane potential. Nucleic Acids Res. 42, 8039–8048 10.1093/nar/gku466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reers M., Smiley S. T., Mottola-Hartshorn C., Chen A., Lin M., and Chen L. B. (1995) Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 260, 406–417 10.1016/0076-6879(95)60154-6 [DOI] [PubMed] [Google Scholar]
- 46. Zhou M., Xue L., Chen Y., Li H., He Q., Wang B., Meng F., Wang M., and Guan M. X. (2018) A hypertension-associated mitochondrial DNA mutation introduces an m1G37 modification into tRNAMet, altering its structure and function. J. Biol. Chem. 293, 1425–1438 10.1074/jbc.RA117.000317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sena L. A., and Chandel N. S. (2012) Physiological roles of mitochondrial reactive oxygen species. Mol. Cell. 48, 158–167 10.1016/j.molcel.2012.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mahfouz R., Sharma R., Lackner J., Aziz N., and Agarwal A. (2009) Evaluation of chemiluminescence and flow cytometry as tools in assessing production of hydrogen peroxide and superoxide anion in human spermatozoa. Fertil. Steril. 92, 819–827 10.1016/j.fertnstert.2008.05.087 [DOI] [PubMed] [Google Scholar]
- 49. Jiang P., Wang M., Xue L., Xiao Y., Yu J., Wang H., Yao J., Liu H., Peng Y., Liu H., Li H., Chen Y., and Guan M.-X. (2016) A hypertension-associated tRNAAla mutation alters tRNA metabolism and mitochondrial function. Mol. Cell. Biol. 36, 1920–1930 10.1128/MCB.00199-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fan W., Zheng J., Kong W., Cui L., Aishanjiang M., Yi Q., Wang M., Cang X., Tang X., Chen Y., Mo J. Q., Sondheimer N., Ge W., and Guan M.-X. (2019) Contribution of a mitochondrial tyrosyl-tRNA synthetase mutation to the phenotypic expression of the deafness-associated tRNASer(UCN) 7511A>G mutation. J.Biol. Chem. 294, 19292–19305 10.1074/jbc.RA119.010598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jia Z., Zhang Y., Li Q., Ye Z., Liu Y., Fu C., Cang X., Wang M., and Guan M. X. (2019) A coronary artery disease-associated tRNAThr mutation altered mitochondrial function, apoptosis and angiogenesis. Nucleic Acids Res. 47, 2056–2074 10.1093/nar/gky1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Taylor R. C., Cullen S. P., and Martin S. J. (2008) Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 9, 231–241 10.1038/nrm2312 [DOI] [PubMed] [Google Scholar]
- 53. Sharma L. K., Tiwari M., Rai N. K., and Bai Y. (2019) Mitophagy activation repairs Leber's hereditary optic neuropathy-associated mitochondrial dysfunction and improves cell survival. Hum. Mol. Genet. 28, 422–433 10.1093/hmg/ddy354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Melser S., Lavie J., and Bénard G. (2015) Mitochondrial degradation and energy metabolism. Biochim. Biophys. Acta 1853, 2812–2821 10.1016/j.bbamcr.2015.05.010 [DOI] [PubMed] [Google Scholar]
- 55. Korolchuk V. I., Menzies F. M., and Rubinsztein D. C. (2009) A novel link between autophagy and the ubiquitin-proteasome system. Autophagy 5, 862–863 10.4161/auto.8840 [DOI] [PubMed] [Google Scholar]
- 56. Qu J., Zhou X., Zhao F., Liu X., Zhang M., Sun Y.-H., Liang M., Yuan M., Liu Q., Tong Y., Wei Q.-P., Yang L., and Guan M.-X. (2010) Low penetrance of Leber's hereditary optic neuropathy in ten Han Chinese families carrying the ND6 T11484C mutation. Biochim. Biophys. Acta 1800, 305–312 10.1016/j.bbagen.2009.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu X.-L., Zhou X., Zhou J., Zhao F., Zhang J., Li C., Ji Y., Zhang Y., Wei Q.-P., Sun Y.-H., Yang L., Lin B., Yuan Y., Li Y., Qu J., et al. (2011) Leber's hereditary optic neuropathy is associated with the T12338C mutation in mitochondrial ND5 gene in six Han Chinese families. Ophthalmology 118, 978–985 10.1016/j.ophtha.2010.09.003 [DOI] [PubMed] [Google Scholar]
- 58. Qu J., Zhou X., Zhao F., Zhang J., Sun Y. H., Tong Y., Wei Q. P., Cai W., Yang L., West C. E., and Guan M. X. (2009) Extremely low penetrance of Leber's hereditary optic neuropathy in 8 Han Chinese families carrying the ND4 G11778A mutation. Ophthalmology 116, 558–564 10.1016/j.ophtha.2008.10.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Caporali L., Iommarini L., La Morgia C., Olivieri A., Achilli A., Maresca A., Valentino M. L., Capristo M., Tagliavini F., Del Dotto V., Zanna C., Liguori R., Barboni P., Carbonelli M., Cocetta V., et al. (2018) Peculiar combinations of individually non-pathogenic missense mitochondrial DNA variants cause low penetrance Leber's hereditary optic neuropathy. PLoS Genet. 14, e1007210 10.1371/journal.pgen.1007210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zickermann V., Wirth C., Nasiri H., Siegmund K., Schwalbe H., Hunte C., and Brandt U. (2015) Structural biology. Mechanistic insight from the crystal structure of mitochondrial complex I. Science 347, 44–49 10.1126/science.1259859 [DOI] [PubMed] [Google Scholar]
- 61. Szczepanowska J., Malinska D., Wieckowski M. R., and Duszynski J. (2012) Effect of mtDNA point mutations on cellular bioenergetics. Biochim. Biophys. Acta 1817, 1740–1746 10.1016/j.bbabio.2012.02.028 [DOI] [PubMed] [Google Scholar]
- 62. Ghelli A., Zanna C., Porcelli A. M., Schapira A. H., Martinuzzi A., Carelli V., and Rugolo M. (2003) Leber's hereditary optic neuropathy (LHON) pathogenic mutations induce mitochondrial-dependent apoptotic death in transmitochondrial cells incubated with galactose medium. J. Biol. Chem. 278, 4145–4150 10.1074/jbc.M210285200 [DOI] [PubMed] [Google Scholar]
- 63. Danielson S. R., Wong A., Carelli V., Martinuzzi A., Schapira A. H., and Cortopassi G. A. (2002) Cells bearing mutations causing Leber's hereditary optic neuropathy are sensitized to Fas-induced apoptosis. J. Biol. Chem. 277, 5810–5815 10.1074/jbc.M110119200 [DOI] [PubMed] [Google Scholar]
- 64. Torroni A., Petrozzi M., D'Urbano L., Sellitto D., Zeviani M., Carrara F., Carducci C., Leuzzi V., Carelli V., Barboni P., et al. (1997) Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am. J. Hum. Genet. 60, 1107–1121 [PMC free article] [PubMed] [Google Scholar]
- 65. Houstek J., Klement P., Floryk D., Antonická H., Hermanská J., Kalous M., Hansíková H., Hout'ková H., Chowdhury S. K., Rosipal T., Kmoch S., Stratilová L., and Zeman J. (1999) A novel deficiency of mitochondrial ATPase of nuclear origin. Hum. Mol. Genet. 8, 1967–1994 10.1093/hmg/8.11.1967 [DOI] [PubMed] [Google Scholar]
- 66. Trounce I., Neill S., and Wallace D. C. (1994) Cytoplasmic transfer of the mtDNA nt 8993 T>G (ATP6) point mutation associated with Leigh syndrome into mtDNA-less cells demonstrates cosegregation with a decrease in state III respiration and ADP/O ratio. Proc. Natl. Acad. Sci. U.S.A. 91, 8334–8338 10.1073/pnas.91.18.8334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Country M. W. (2017) Retinal metabolism: a comparative look at energetics in the retina. Brain Res. 1672, 50–57 10.1016/j.brainres.2017.07.025 [DOI] [PubMed] [Google Scholar]
- 68. Wong-Riley M. T. (2010) Energy metabolism of the visual system. Eye Brain 2, 99–116 10.2147/EB.S9078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hayashi G., and Cortopassi G. (2015) Oxidative stress in inherited mitochondrial diseases. Free Radic. Biol. Med. 88, 10–17 10.1016/j.freeradbiomed.2015.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wong A., Cavelier L., Collins-Schramm H. E., Seldin M. F., McGrogan M., Savontaus M. L., and Cortopassi G. A. (2002) Differentiation-specific effects of LHON mutations introduced into neuronal NT2 cells. Hum. Mol. Genet. 11, 431–438 10.1093/hmg/11.4.431 [DOI] [PubMed] [Google Scholar]
- 71. Song L., Yu A., Murray K., and Cortopassi G. (2017) Bipolar cell reduction precedes retinal ganglion neuron loss in a complex 1 knockout mouse model. Brain Res. 1657, 232–244 10.1016/j.brainres.2016.12.019 [DOI] [PubMed] [Google Scholar]
- 72. Lee J., Giordano S., and Zhang J. (2012) Autophagy, mitochondria and oxidative stress: cross-talk and redox signaling. Biochem. J. 441, 523–540 10.1042/BJ20111451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang J., Liu X., Liang X., Lu Y., Zhu L., Fu R., Ji Y., Fan W., Chen J., Lin B., Yuan Y., Jiang P., Zhou X., and Guan M.-X. (2017) A novel ADOA-associated OPA1 mutation alters the mitochondrial function, membrane potential, ROS production and apoptosis. Sci. Rep. 7, 5704 10.1038/s41598-017-05571-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rieder M. J., Taylor S. L., Tobe V. O., and Nickerson D. A. (1998) Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res. 26, 967–973 10.1093/nar/26.4.967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Miller G., and Lipman M. (1973) Release of infectious Epstein-Barr virus by transformed marmoset leukocytes. Proc. Natl. Acad. Sci. U.S.A. 70, 190–194 10.1073/pnas.70.1.190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Guan M. X., Fischel-Ghodsian N., and Attardi G. (2001) Nuclear background determines biochemical phenotype in the deafness-associated mitochondrial 12S rRNA mutation. Hum. Mol. Genet. 10, 573–580 10.1093/hmg/10.6.573 [DOI] [PubMed] [Google Scholar]
- 77. King M. P., and Attardi G. (1993) Post-transcriptional regulation of the steady-state levels of mitochondrial tRNAs in HeLa cells. J. Biol. Chem. 268, 10228–10237 [PubMed] [Google Scholar]
- 78. Meng F., He Z., Tang X., Zheng J., Jin X., Zhu Y., Ren X., Zhou M., Wang M., Gong S., Mo J. Q., Shu Q., and Guan M.-X. (2018) Contribution of the tRNAIle 4317A→G mutation to the phenotypic manifestation of the deafness-associated mitochondrial 12S rRNA 1555A→G mutation. J. Biol. Chem. 293, 3321–3334 10.1074/jbc.RA117.000530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kabakov A. E., and Gabai V. L. (2018) Cell death and survival assays. Methods Mol. Biol. 1709, 107–127 10.1007/978-1-4939-7477-1_9 [DOI] [PubMed] [Google Scholar]
- 80. Arduíno D. M., Esteves A. R., Corte L., Silva D. F., Pate lB., Grazina M., Swerdlow R. H., Oliveira C. R., and Cardoso S. M. (2012) Mitochondrial metabolism in Parkinson's disease impairs quality control autophagy by hampering microtubule-dependent traffic. Hum. Mol. Genet. 21, 4680–4702 10.1093/hmg/dds309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ho T. T., Warr M. R., Adelman E. R., Lansinger O. M., Flach J., Verovskaya E. V., Figueroa M. E., and Passegué E. (2017) Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210 10.1038/nature21388 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Representative experiments are shown in the figures and supporting information. Supplemental data include two tables and one figure. For any additional information, please contact the corresponding author.