

Abstract
Lysocardiolipin acyltransferase (LYCAT), a cardiolipin (CL)-remodeling enzyme, is crucial for maintaining normal mitochondrial function and vascular development. Despite the well-characterized role for LYCAT in the regulation of mitochondrial dynamics, its involvement in lung cancer, if any, remains incompletely understood. In this study, in silico analysis of TCGA lung cancer data sets revealed a significant increase in LYCAT expression, which was later corroborated in human lung cancer tissues and immortalized lung cancer cell lines via indirect immunofluorescence and immunoblotting, respectively. Stable knockdown of LYCAT in NSCLC cell lines not only reduced CL and increased monolyso-CL levels but also reduced in vivo tumor growth, as determined by xenograft studies in athymic nude mice. Furthermore, blocking LYCAT activity using a LYCAT mimetic peptide attenuated cell migration, suggesting a novel role for LYCAT activity in promoting NSCLC. Mechanistically, the pro-proliferative effects of LYCAT were mediated by an increase in mitochondrial fusion and a G1/S cell cycle transition, both of which are linked to increased cell proliferation. Taken together, these results demonstrate a novel role for LYCAT in promoting NSCLC and suggest that targeting LYCAT expression or activity in NSCLC may provide new avenues for the therapeutic treatment of lung cancer.
Keywords: non-small-cell lung cancer, lysocardiolipin acyltransferase, cardiolipin, cell motility, cell proliferation, cell cycle, cell migration, LYCAT, NSCLC
Lung cancer is the leading cause of cancer-related deaths in the United States, and non-small-cell lung cancer (NSCLC) accounts for ∼85% of all lung cancers with a 5-year survival of only ∼16% (1, 2). Overall, the toll of lung cancer deaths in the United States exceeds that of the next four major cancers combined (1, 2). Therefore, there is an urgent need to develop new technologies and biomarkers for early detection as well as to identify new targets for therapy. Emerging evidence suggests a key role of mitochondrial dysfunction in the development and progression of lung cancer (3, 4); therefore, targeting mitochondrial function will facilitate not only a better understanding of the molecular basis of lung cancer development but also the development of novel therapeutics.
Cardiolipin (CL), or 1,3-diphosphatidyl-sn-glycerol, is an anionic phospholipid that is synthesized exclusively in the mitochondria and predominantly localized in the inner mitochondrial membrane (5). CL plays an important role for optimal mitochondrial function, including electron transport, mitochondrial dynamics, mitochondrial biogenesis, apoptotic machinery, and vascular development. Deficiencies in CL content and modification in linoleic acid molecular species are associated with dysfunctional heart in diabetes, heart failure and ischemia-reperfusion injury, and Barth syndrome (6, 7). Abnormalities in CL molecular species correlate with prostate cancer cell proliferation (8), thyroid oncocytic tumors (9), and mouse brain tumors (10). However, very little is known about the role of CL and its modifying enzyme, LYCAT, in lung cancer.
In this study, mining the Cancer Genome Atlas (TCGA) gene expression data sets revealed a significant up-regulation of LYCAT in human non-small-cell lung cancers, which was later corroborated in human lung tumor tissues by immunohistochemistry. Furthermore, knockdown of LYCAT expression attenuated NSCLC proliferation and cell migration in vitro and subcutaneous tumor growth in vivo. Transcriptional profiling of LYCAT knockdown cells revealed several genes that regulate the G1/S cell cycle transition. Moreover, LYCAT knockdown resulted in a significant decrease in mitochondrial fragmentation. Collectively, our findings reveal a novel role for LYCAT in promoting lung tumor growth via altered mitochondrial dynamics. Therefore, targeting LYCAT expression and/or activity might represent a novel therapeutic strategy to treat lung cancer.
Results
LYCAT expression is up-regulated in human lung tumors
As a first step to evaluate a role for LYCAT in lung cancer, we examined the expression levels of LYCAT in TCGA lung cancer data sets. LYCAT expression was significantly up-regulated in the lung tumors when compared with normal lungs in both lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) data sets (Fig. 1A). Furthermore, to determine the correlation of LYCAT expression with overall patient survival, we performed Kaplan–Meier survival analysis by using KM Plotter (11). For these studies, 274 low- and 186 high-LYCAT–expressing patient samples from TCGA, which are separated by their median values, were employed. Increased expression of LYCAT was correlated with poor overall patient survival in LUAD, but not in LUSC, data sets (Fig. 1B). We next investigated whether LYCAT gene expression correlates with known drivers of lung cancer by using cBIOPortal for Cancer Genomics (RRID:SCR_014555). Of the seven frequently observed somatic gene alterations (EGFR (epidermal growth factor receptor), KRAS, ERBB2, ALK, TP53, BRAF, and RET (rearranged during transfection)), increased expression of LYCAT positively correlated with KRAS, ERBB2, and BRAF and negatively correlated with ALK (Fig. 1C). To further examine LYCAT expression in human lung cancer, we assessed the expression of LYCAT in both formalin-fixed paraffin-embedded (FFPE) tissues samples and fresh-frozen lung tumor tissue samples. Lung tumors have a significant increase in LYCAT expression compared with adjacent uninvolved lung tissues as determined by immunohistochemical analysis (Fig. 1D) and immunoblotting (Fig. 1E). There was no significant staining with IgG-only controls, indicating the specificity of anti-LYCAT antibody (Fig. 1D). Moreover, increased LYCAT expression was also observed in the lung tumors of urethane-treated mice (Fig. 1F). Urethane is a chemical carcinogen and a KRAS mutagen that induces lung tumors (12–14). Finally, the expression of LYCAT was analyzed in cell lysates of human bronchial epithelial cells and a panel of NSCLC cell lines (Fig. 1G). In agreement with the lung cancer tissue data, LYCAT expression was significantly increased in 70% of NSCLC cell lines (Fig. 1G). Collectively, these data demonstrate an enhanced expression of LYCAT in human lung tumors, NSCLC cells, and in the lungs of urethane-treated mice compared with normal lungs.
Figure 1.
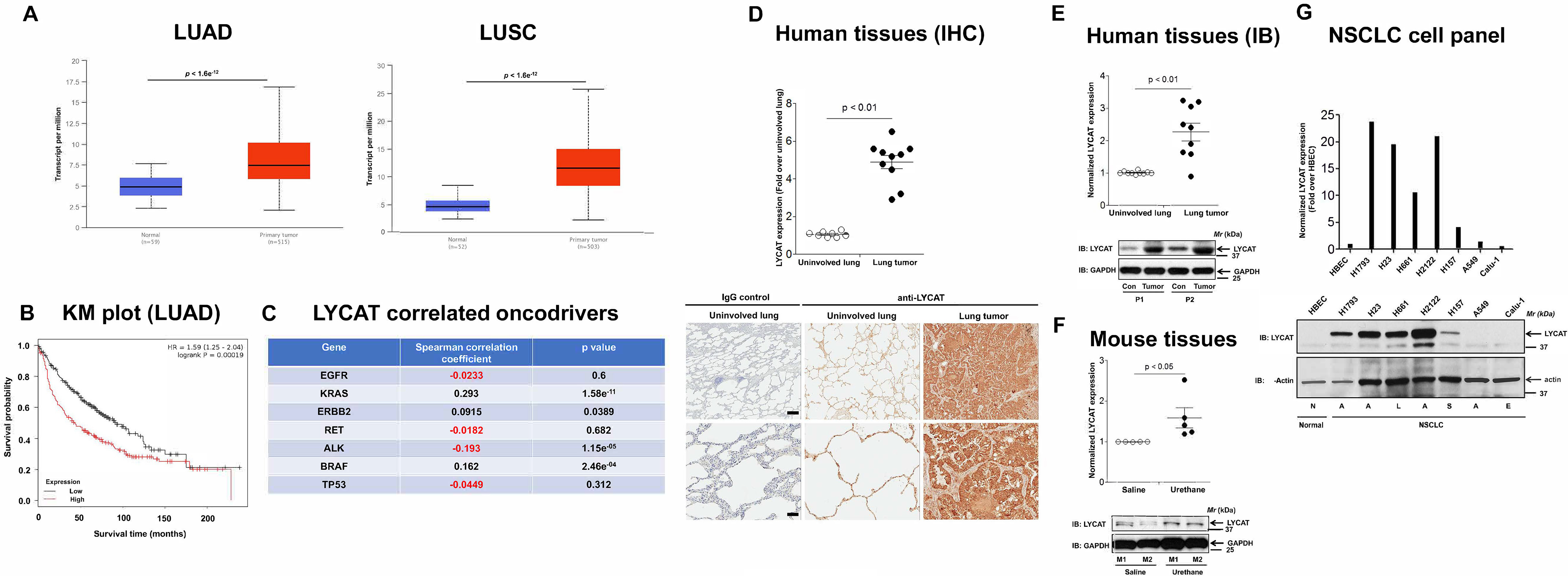
Expression of LYCAT is increased in lung cancer. A, bioinformatics analysis of TCGA lung cancer data sets showed increased LYCAT expression in LUAD and LUSC. B, Kaplan–Meier survival curves for patients with high and low LYCAT expression were determined using an online tool (KM Plotter). p values were calculated by log-rank test. For these studies, 274 low- and 186 high-LYCAT–expressing patient samples from TCGA, which are separated by their median values, were employed. C, Spearman correlation studies to determine the LYCAT-correlated oncodrivers in lung cancer by using cBioPortal. D, immunohistochemical (IHC) staining of FFPE sections of human lung tumors and adjacent uninvolved lung tissues from the same patients with LYCAT antibodies. Top, staining intensities; bottom, representative pictures. **, p < 0.01. Scale bar, 500 μm. E, expression of LYCAT in fresh-frozen human lung cancer tissues and adjacent uninvolved lung tissues as detected by immunoblotting (IB) with anti-LYCAT and anti-GAPDH antibodies. F, expression of LYCAT in chemical carcinogen (urethane)-induced mouse lung tumors was determined by immunoblotting (IB) with anti-LYCAT antibodies. G, expression of LYCAT in human nontransformed bronchial epithelial cells (HBEC) and a panel of NSCLC cell lines. N, normal; A, adenocarcinoma; S, squamous carcinoma; L, large-cell carcinoma; E, epidermoid carcinoma.
LYCAT knockdown inhibits NSCLC cell proliferation
Having determined an increased expression of LYCAT in lung cancer, we next sought to determine the role of LYCAT in driving cell proliferation. For these studies, we designed small hairpin RNAs (shRNAs) against LYCAT. Because LYCAT protein expression is up-regulated in lung cancer, we chose to evaluate the effects of LYCAT knockdown on cell proliferation in NSCLC cells (e.g. H2122 and H23 cells that express high amounts of LYCAT) (Fig. 1G). We generated and tested multiple stable clones with stable expression of nontargeting control shRNA and LYCAT targeting shRNA. H2122 clone (Fig. 2A) and H23 (Fig. 2D) clone with stable expression of LYCAT shRNA showed significant knockdown of LYCAT. Knockdown of LYCAT in two NSCLC cell lines resulted in a significant reduction in cell proliferation as determined by MTT (Fig. 2, B and E) and clonogenic (Fig. 2, C and F) cell proliferation assays. To exclude the possible off-target effects of LYCAT shRNA, we performed rescue experiments. For these studies, we employed adenoviruses that encode mouse LYCAT cDNAs. mLycat adenoviral transduction of H23 clones with stable expression of LYCAT shRNAs resulted in an increase in cell proliferation when compared with control adenovirus-treated H23 clone with stable LYCAT knockdown (Fig. 2G, compare lanes 1, 3, and 4). To determine the effects of LYCAT knockdown on NSCLC proliferation, we also employed CRISPR-Cas9–mediated gene editing of LYCAT in NSCLC cell lines: H2122 (Fig. 2, H–J) and H23 (Fig. 2, K–M). For generating LYCAT knockdown cell lines, H2122 and H23 cells were transfected with a pool of guide RNAs (sgRNAs) targeting LYCAT followed by clonal isolation via cell sorting. H2122 and H23 LYCAT knockout clones displayed significant reduction in LYCAT protein expression (Fig. 2, H and K) and cell proliferation compared with their respective parental cells, as evaluated by cell growth curves (Fig. 2, I and L) and clonogenic cell proliferation assays (Fig. 2, J and M). We speculate that the incomplete LYCAT gene knockout in H2122 and H23 cells might be due to the cell ploidy and the efficiency of LYCAT sgRNAs to knock out all the copies of LYCAT in NSCLC cells. Taken together, these data establish an important role for LYCAT in driving cell proliferation.
Figure 2.

LYCAT knockdown reduces NSCLC cell proliferation. A and D, H2122 and H23 NSCLC cells were transfected with LYCAT shRNA, followed by a single-cell isolation technique to develop LYCAT knockdown H2122 and H23 clones. Lysates of H2122 (A) and H23 (D) LYCAT knockdown clones and their parental cells were immunoblotted with anti-LYCAT antibodies. The proliferation rates of H2122 and H23 LYCAT knockdown clones were determined by MTT (B and E) and clonogenic (C and F) cell proliferation assays as described under “Experimental procedures.” Data represent mean ± S.E. (error bars) from three independent highly reproducible experiments. *, p < 0.05 versus parental control. G, Restoration of LYCAT expression in H23 LYCAT knockdown clones rescues the effect of LYCAT knockdown, as determined by hemocytometer cell count. *, p < 0.05 versus parental control. H–M, NSCLC cells were transfected with LYCAT sgRNAs, followed by cell sorting to develop LYCAT knockout H2122 and H23 clones. Lysates of H2122 (H) and H23 (K) LYCAT knockout clones and their controls were immunoblotted with anti-LYCAT antibodies. The proliferation rates of H2122 and H23 LYCAT knockout clones were determined by hemocytometer cell count (I and L) and clonogenic (J and M) cell proliferation assays as described under “Experimental procedures.” Data represent mean ± S.E. from three independent highly reproducible experiments. **, p < 0.01; *, p < 0.05 versus parental control.
Knockdown of LYCAT inhibits NSCLC cell migration in vitro and tumor growth in vivo
To test the effects of LYCAT knockdown on cell migration, we performed wound-healing assays (scratch assay). In these assays, a 3-mm scrape wound was created on confluent cultures of H2122 (Fig. 3A) and H23 (Fig. 3C) clones stably expressing LYCAT shRNA. After 24 h, the abilities of the cells to heal the wound were recorded in the presence of a mitosis blocker, Mitomycin C. H2122 and H23 clones with LYCAT knockdown showed reduced cell motility when compared with H2122 and H23 clones with control shRNA (Fig. 3, A and C). The migratory potential of H2122 and H23 LYCAT knockdown clones was also assessed by using transwell assays (Fig. 3, B and D). In these assays, cells were seeded into transwell inserts and allowed to migrate toward 10% serum-containing medium through a permeable membrane (8 μm). The migrated cells on the bottom of the insert were later fixed, stained with crystal violet, and counted, and they are represented in the graphs (Fig. 3, B and D). LYCAT knockdown resulted in reduced cell migration in both H2122 (Fig. 3B) and H23 (Fig. 3D) clones. Consistent with shRNA-mediated LYCAT knockdown, CRISPR-Cas9–mediated gene editing of LYCAT also resulted in reduced cell migration in both H2122 (Fig. 3E) and H23 (Fig. 3F) clones. We have also evaluated the invasive capabilities of LYCAT knockdown clones by employing transwell inserts coated with Matrigel (BD Biosciences). The cells that invaded Matrigel and migrated through the 8-μm pores were later stained and counted. When compared with parental cells, LYCAT knockdown showed reduced invasive capabilities in both H2122 (Fig. 3G) and H23 (Fig. 3H) cells. We also evaluated the effects of LYCAT knockdown on apoptosis. Annexin V–FITC staining of parental and LYCAT gene–edited cells showed no significant changes in the percentages of annexin V–FITC/propidium iodide–positive cells in both H2122 and H23 cell lines (Fig. 3I).
Figure 3.
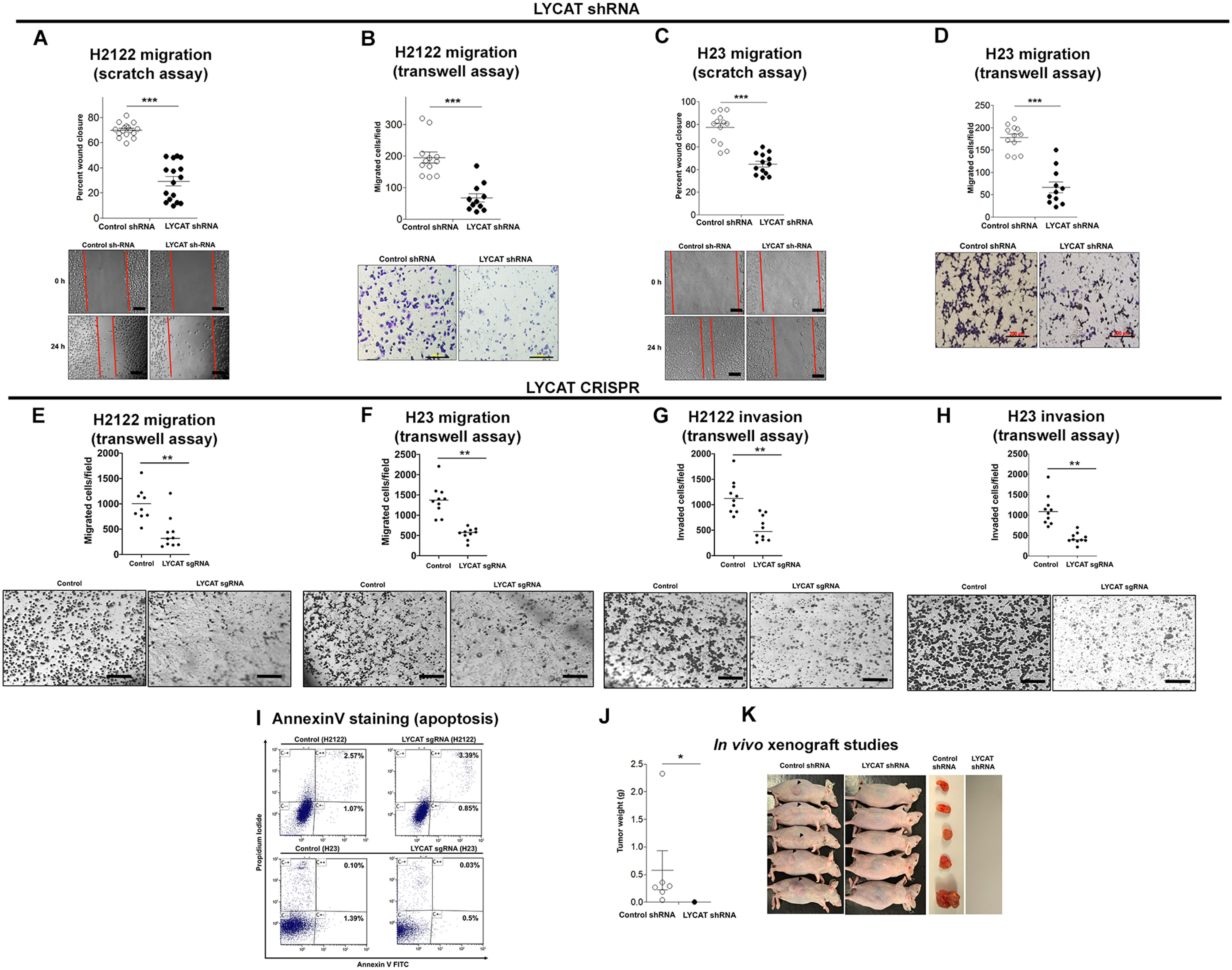
LYCAT knockdown inhibits NSCLC cell migration in vitro and oncogenic potential in vivo. A and C, a 3-mm scrape wound was created in confluent cultures of H2122 (A) and H23 (C) NSCLC cells stably expressing LYCAT shRNAs. Later, migration of the cells toward the scrape wound was recorded at 0 and 24 h. Scale bar, 100 μm. B and D, cell migration rates of H2122 (B) and H23 (D) LYCAT knockdown clones were assayed in transwell inserts as described under “Experimental procedures.” Scale bar, 200 μm. ***, p < 0.05 versus parental control. E and F, cell migration of CRISPR-Cas9 LYCAT gene–edited H2122 (E) and H23 (F) clones was assayed in transwell inserts as described under “Experimental procedures.” **, p < 0.01 versus parental control. Scale bar, 200 μm. G and H, cell invasion of CRISPR-Cas9 LYCAT gene–edited H2122 (G) and H23 (H) clones was assayed in transwell inserts coated with Matrigel as described under “Experimental procedures.” **, p < 0.01 versus parental control. Scale bar, 200 μm. I, the extent of apoptosis in CRISPR-Cas9 LYCAT gene–edited H2122 and H23 clones was assayed by annexin V–FITC staining as described under “Experimental procedures.” J and K, a single clone of H2122 LYCAT knockdown was subcutaneously injected into athymic nude mice. Tumor weights (J) and tumor-bearing mice (K) are displayed. *, p < 0.05 versus control. Error bars, S.E.
To evaluate the effects of LYCAT knockdown on tumor growth in vivo, we subcutaneously injected H2122 clones stably expressing LYCAT shRNA into the lateral flanks of athymic nude mice. After 8 weeks, all the mice injected with H2122 cells stably expressing control shRNA developed tumors at the injection site (Fig. 3, J and K), whereas mice injected with H2122 clones stably expressing LYCAT shRNA failed to develop tumors (Fig. 3, J and K). Taken together, these findings demonstrate that increased LYCAT expression is an important contributor of lung tumor development.
LYCAT knockdown decreases cardiolipin and increases monolysocardiolipin levels in NSCLC cells
Because LYCAT is involved in the dynamic remodeling of CL in mitochondria and membranes of mammalian cells (15, 16), we evaluated the effects of depletion of LYCAT on the molecular species of CL in H23 cells stably expressing LYCAT shRNA. LC–MS analysis of the molecular species of CL from control and shRNA LYCAT H23 cells revealed a major CL cluster between m/z 1349 and 1499 (Fig. 4A). A significant decrease in 11 molecular species of CL was observed in H23 cells stably expressing LYCAT shRNA (Fig. 4, B and C). Fragmentation analysis further revealed a decrease in 18:1/18:1/18:2/18:0 species in H23 cells stably expressing LYCAT shRNA (Fig. 4D). Hydrolysis of CLs by phospholipases (e.g. iPLA2 localized in the mitochondria or cPLA2) results in the generation of monolysocardiolipin (MCL; Fig. 4E). Interestingly, 50:4 and 44:3 molecular species of MCL were significantly increased in H23 cells stably expressing LYCAT shRNA when compared with control shRNA cells (Fig. 4, F and G). In total, depletion of LYCAT in NSCLC cells resulted in a decrease in CL levels and a corresponding increase in MCL levels.
Figure 4.
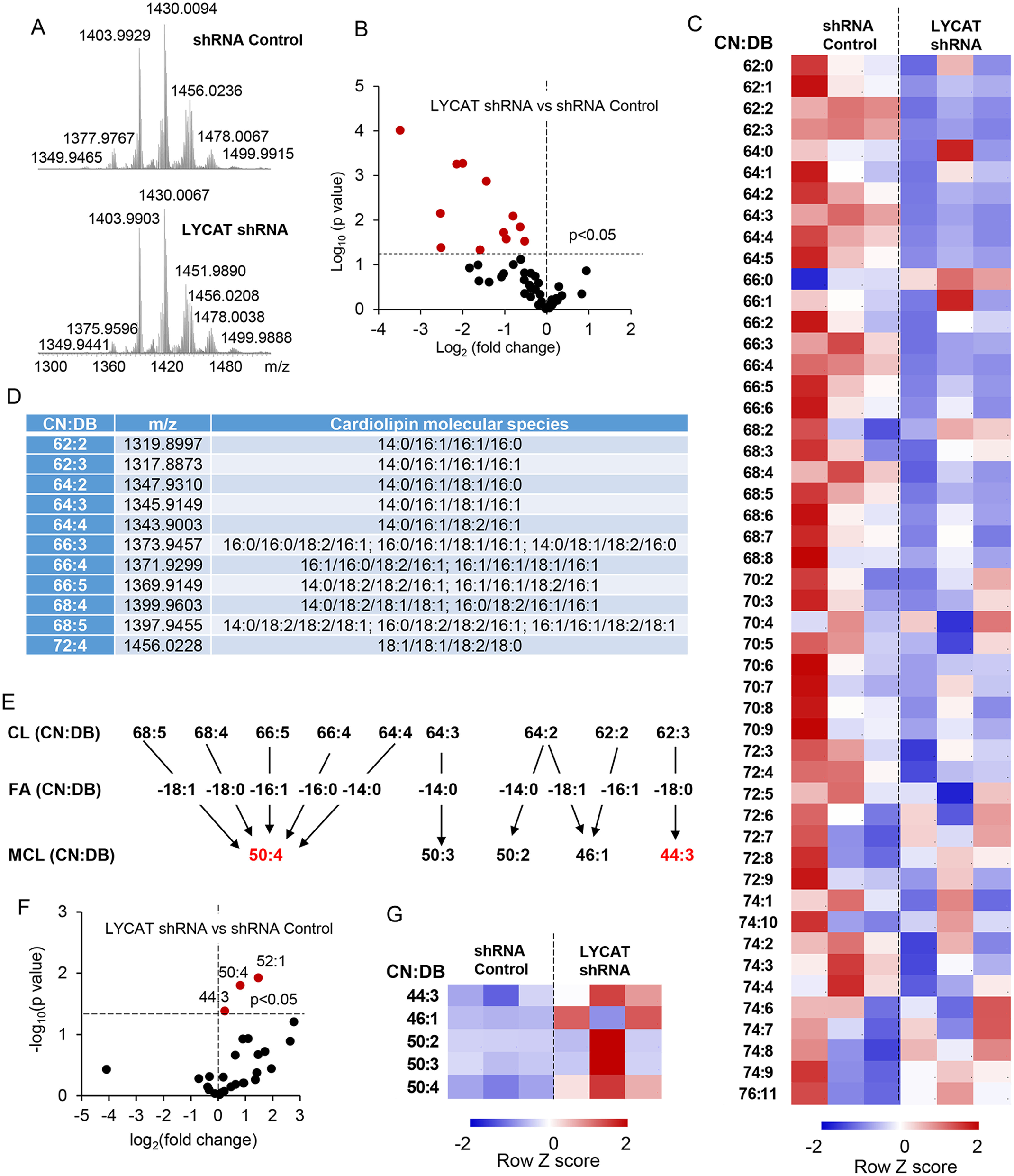
LYCAT knockdown–induced changes in CL and MCL levels. A, typical spectra of CL obtained from H23 cells. B, volcano plot of LYCAT shRNA–induced changes in CL (log2(-fold change)) versus significance (log10 p value). C, the contents of CL in H23 cells. The data are presented as heatmaps, autoscaled to z-scores, and coded blue (low values) to red (high values). D, CL molecular species that were significantly changed in LYCAT shRNA–treated cells. E, schema illustrating hydrolysis of parent CLs to yield detected MCLs. F, volcano plot of LYCAT shRNA–induced changes in MCL (log2(-fold change)) versus significance (log10 p value). G, the contents of MCL in H23 cells. The data are presented as heatmaps, autoscaled to z-scores, and coded blue (low values) to red (high values). CN, carbon number; DB, double bond.
Blocking LYCAT activity attenuates NSCLC cell migration
It is known that LYCAT activity is required for the dynamic remodeling of CL in mitochondria and membranes of mammalian cells (15, 16). To determine whether the biological effects of LYCAT on NSCLC were dependent on its activity, we developed peptide mimetics of LYCAT based on a unique sequence within LYCAT, which can bind to MCL (Fig. 5A). We believe that the peptide when added to cells competes with LYCAT for MCL, thereby preventing the formation of cardiolipin (Fig. 5A). For these studies, both the scrambled control peptide (VTSISADTHK) and peptide mimetic of 10 amino acids were tagged to HIV TAT sequence (YGRKKRRQRRR) for cell penetration. Later, these peptides were employed in in vitro LYCAT activity assays to evaluate the effects of LYCAT mimetic peptide in blocking LYCAT activity. In these assays, H2122 cells were preincubated either with control peptide or LYCAT mimetic peptide (30 μm) for 3 h, followed by incubation of cell lysates (200 µg of protein) with or without MCL (5 μm) and 25 μm 16-E8 NBD-16:0 fatty acyl-CoA, as described under “Experimental procedures.” Following incubation, the lipids were extracted under acidic conditions and dried under a nitrogen stream, and the fluorescence-labeled CL formed from exogenously added MCL and NBD-16:0 fatty acyl-CoA was separated by TLC and quantified by a GE ImageQuant LAS4000 analyzer and quantified by ImageJ (11). Preincubation of cells with the LYCAT mimetic peptide reduced the ability of cell lysates to generate fluorescence-labeled CL, showing the ability of the mimetic peptide to penetrate inside the cell and inhibit LYCAT activity (Fig. 5B).
Figure 5.
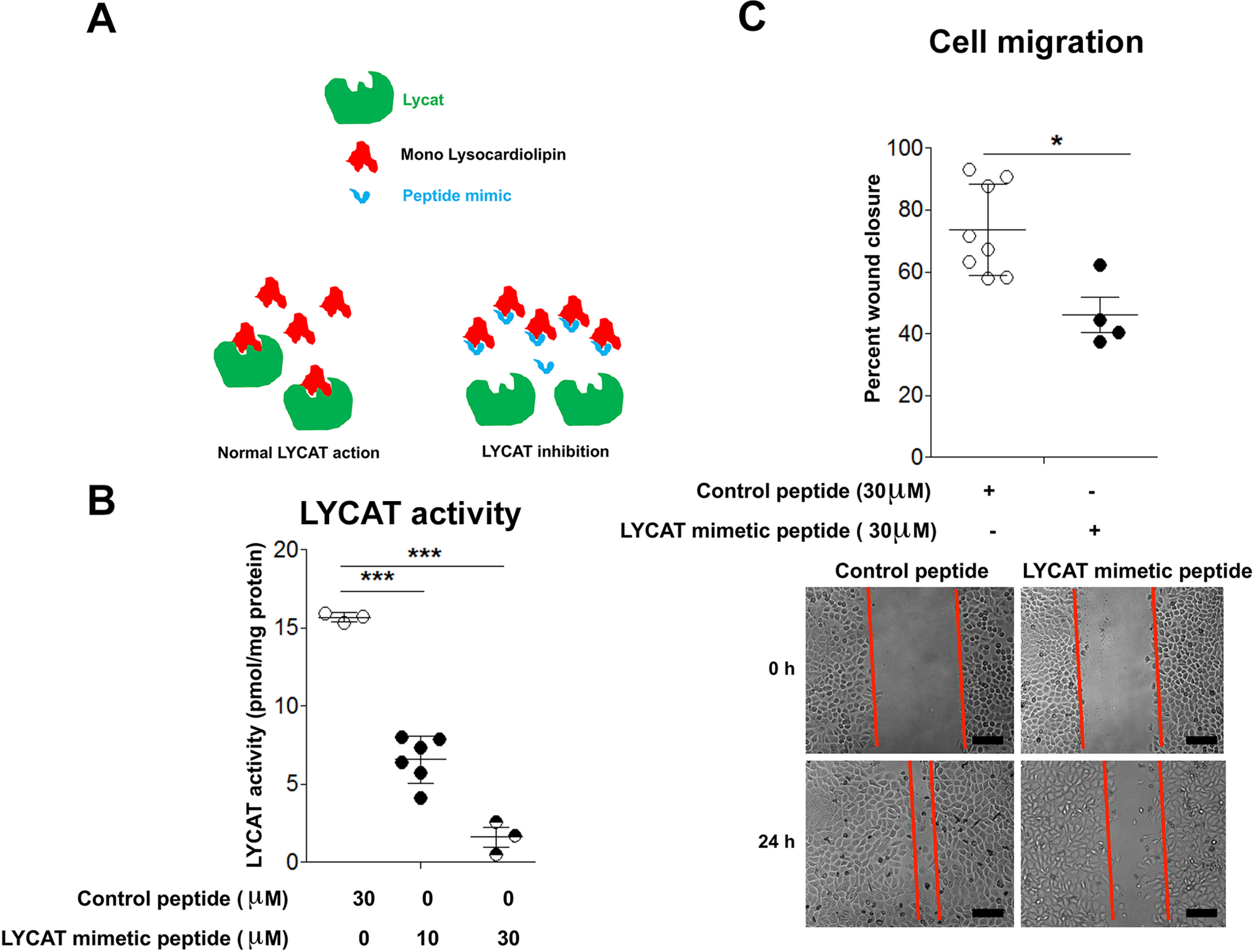
Inhibition of LYCAT activity reduces NSCLC cell migration. A, cartoon depicting the mechanism of action of LYCAT mimetic peptide. B, preincubation of H2122 cell lysates with LYCAT mimetic peptide (30 μm, 30 min) inhibits LYCAT activity. ***, p < 0.005 versus parental control peptide (n = 5). C, treatment of H2122 cells with LYCAT mimetic peptide (30 μm) inhibits cell migration as determined by using the scratch assay. *, p < 0.05 versus control peptide (n = 5). Scale bar, 100 μm. Error bars, S.E.
To determine the biological effects of LYCAT activity inhibition, we assessed the effects of the LYCAT peptide mimetic on cell culture assays that evaluate cell migration. For these assays, we selected H2122 NSCLC cell lines, as they exhibited high LYCAT expression. Treatment of H2122 cells with the LYCAT peptide mimetic (30 μm) resulted in a significant reduction in cell motility (Fig. 5C). These results suggest that LYCAT activity is essential for the serum-induced cell migration of NSCLC cells in vitro.
Transcriptional profiling of LYCAT-dependent genes
To determine the mechanism by which LYCAT contributes to lung tumor development, we analyzed the mRNA expression profiles of H2122 cells stably expressing either control or LYCAT shRNA. For these studies, RNA was isolated from a single clone at three different passages (n = 3, biological replicates). LYCAT knockdown in these cells was shown previously (Fig. 2A). The RNA-Seq analysis identified several differentially expressed (DE) genes. Further gene ontology and KEGG pathway analyses of the DE gene list using the DAVID database (17, 18) categorized the DE genes into 11 gene ontology categories (e.g. genes related to prostate cancer, ErbB signaling, bladder cancer, focal adhesion, mitogen-activated protein kinase signaling, extracellular matrix–receptor interaction, pathways in cancer, cytokine-cytokine receptor interaction, amoebiasis, axon guidance, and p53 signaling) (Fig. 6A). Of the 11 categories, it was interesting to note that the most significant pathway of DE genes was the p53 signaling pathway (Fig. 6A). Fifteen key genes were classified under the p53 signaling pathway, of which the top down-regulated (CCND2) and up-regulated (CDKN1A) genes upon LYCAT knockdown were selected for further validation (Fig. 6B). Probing the cell lysates of H2122 NSCLC cells stably expressing either control shRNA or LYCAT shRNA showed a significant up-regulation of CDKN1A and a significant down-regulation of CCND2, validating the RNA-Seq data (Fig. 6C). Furthermore, because CDKN1A and CCND2 are critical regulators of the cell cycle (19), we also evaluated whether knockdown of LYCAT impacts cell cycle distribution. For these studies, H2122 and H23 clones stably expressing control or LYCAT shRNA were utilized in flow cytometric analysis of cell cycle distribution with propidium iodide DNA staining dye. LYCAT shRNA knockdown in H2122 and H23 resulted in a decrease in the percentage of cells in S phase (Fig. 6, D and E) and a corresponding increase in G2/M phase (Fig. 6, D and E), indicating that LYCAT knockdown arrests NSCLC cells in G2/M. In total, these results demonstrate that LYCAT may affect G1/S transition during lung tumorigenesis.
Figure 6.
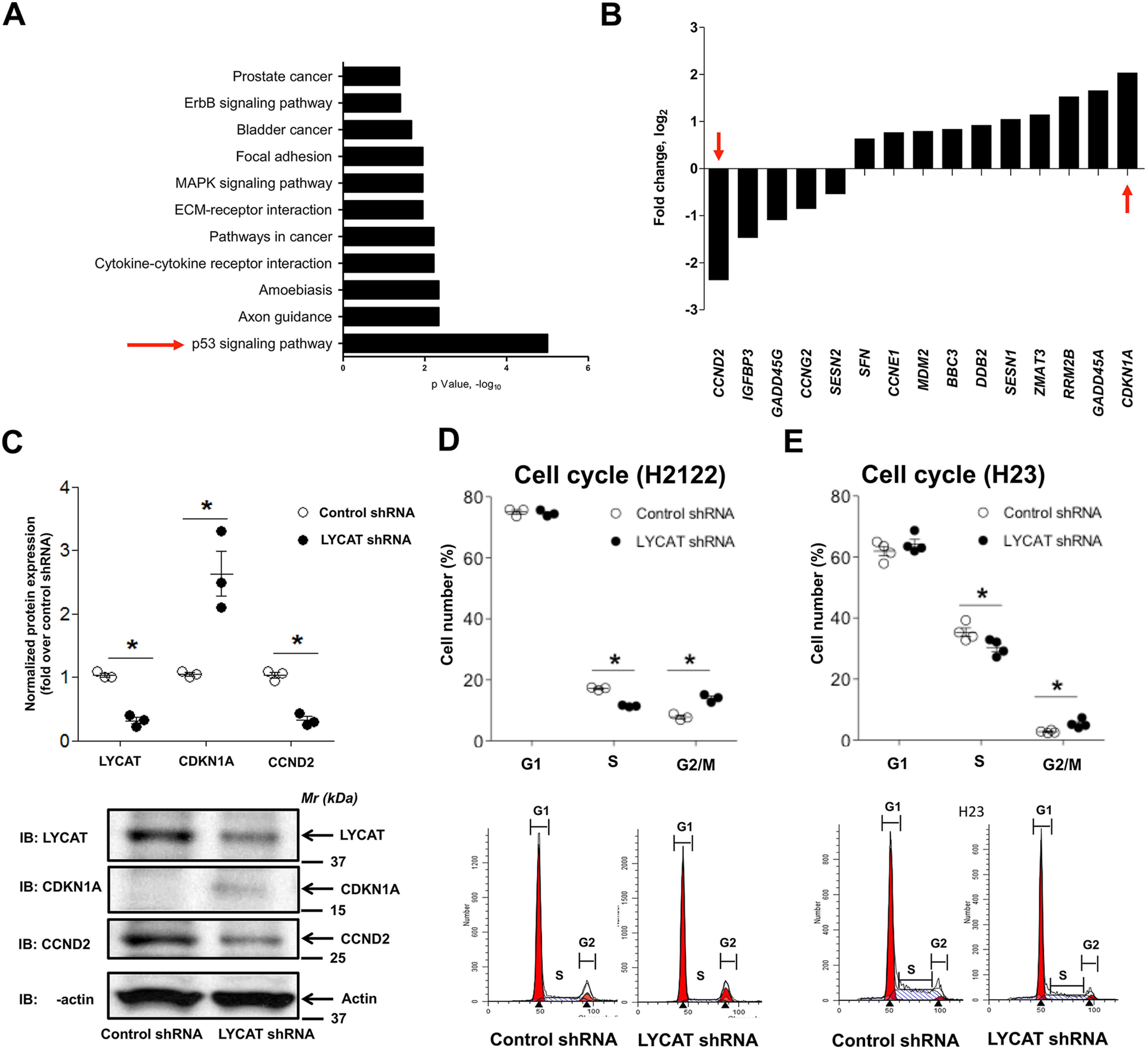
Inhibition of LYCAT results in altered cell cycle distribution. A, RNA-Seq analysis of the differentially expressed genes upon LYCAT knockdown. Top 10 gene ontology terms that are enriched in LYCAT-expressing NSCLC cells were shown in the figure. B, top 10 up- and down-regulated genes from the p53 signaling pathway. C, expression of top down-regulated (CCND2) and top up-regulated (CDKN1A) genes from the p53 signaling pathway in the lysates of H2122 cells stably expressing LYCAT shRNAs. Top, quantification of Western blots; bottom, representative images. *, p < 0.05 versus parental control. The β-actin blots in C and Fig. 7D are identical because they were from the same experiment and run at the same time, when β-actin immunostaining was performed. D, cell cycle distribution in H2122 (D) and H23 (E) cells stably expressing either control or LYCAT shRNAs. *, p < 0.05 versus parental control. Error bars, S.E.
LYCAT knockdown attenuates mitochondrial fusion in NSCLC cells
Mitochondria quickly adapt to the increasing energy demands by interchanging their architecture (mitochondrial dynamics) between a highly fused, tubular state (i.e. fusion) and a more fragmented perinuclear form (i.e. fission) (20). Because LYCAT is one of the two major cardiolipin-remodeling enzymes that is critical for the normal function of the mitochondria (16), and also because a role for the mitochondrial network during the G1/S phase of the cell cycle was highlighted in a recent study (21), we explored next the role of LYCAT in regulating mitochondrial dynamics in NSCLC. For these studies, H2122 (Fig. 7A) and H23 (Fig. 7B) cells with stable expression of either control or LYCAT shRNA were stained with MitoTracker Red, followed by confocal microscopy. LYCAT knockdown resulted in a significant decrease in mitochondrial fragmentation in both H2122 (Fig. 7A) and H23 (Fig. 7B) cells when compared with their corresponding control shRNA–transfected cells. Restoration of LYCAT expression in H23 cells stably expressing LYCAT shRNAs by way of adenoviral infection reversed the effects of LYCAT knockdown on mitochondrial fragmentation (Fig. 7C). In strong agreement with the MitoTracker studies, probing the cell lysates with dynamin-related protein 1 (DRP1), which drives mitochondrial fission (22), showed an increase in DRP1 expression upon LYCAT knockdown (Fig. 7D). Furthermore, an increase in mitofusin1 (MFN1) and mitofusin2 (MFN2), which control mitochondrial outer membrane fusion (22), was also observed upon LYCAT knockdown (Fig. 7D). Consistent with the shRNA-mediated LYCAT knockdown (Fig. 7, A–C), CRISPR-Cas9–mediated LYCAT gene editing also resulted in increased mitochondrial fusion (Fig. 7, E and F). Complementary studies employing LYCAT overexpression in Beas2B cells showed reduced mitochondrial fusion, as determined by MitoTracker staining (Fig. 7G) and markers of mitochondrial fusion/fission (Fig. 7H). These results demonstrate an important role for LYCAT in regulating mitochondrial dynamics in NSCLC.
Figure 7.
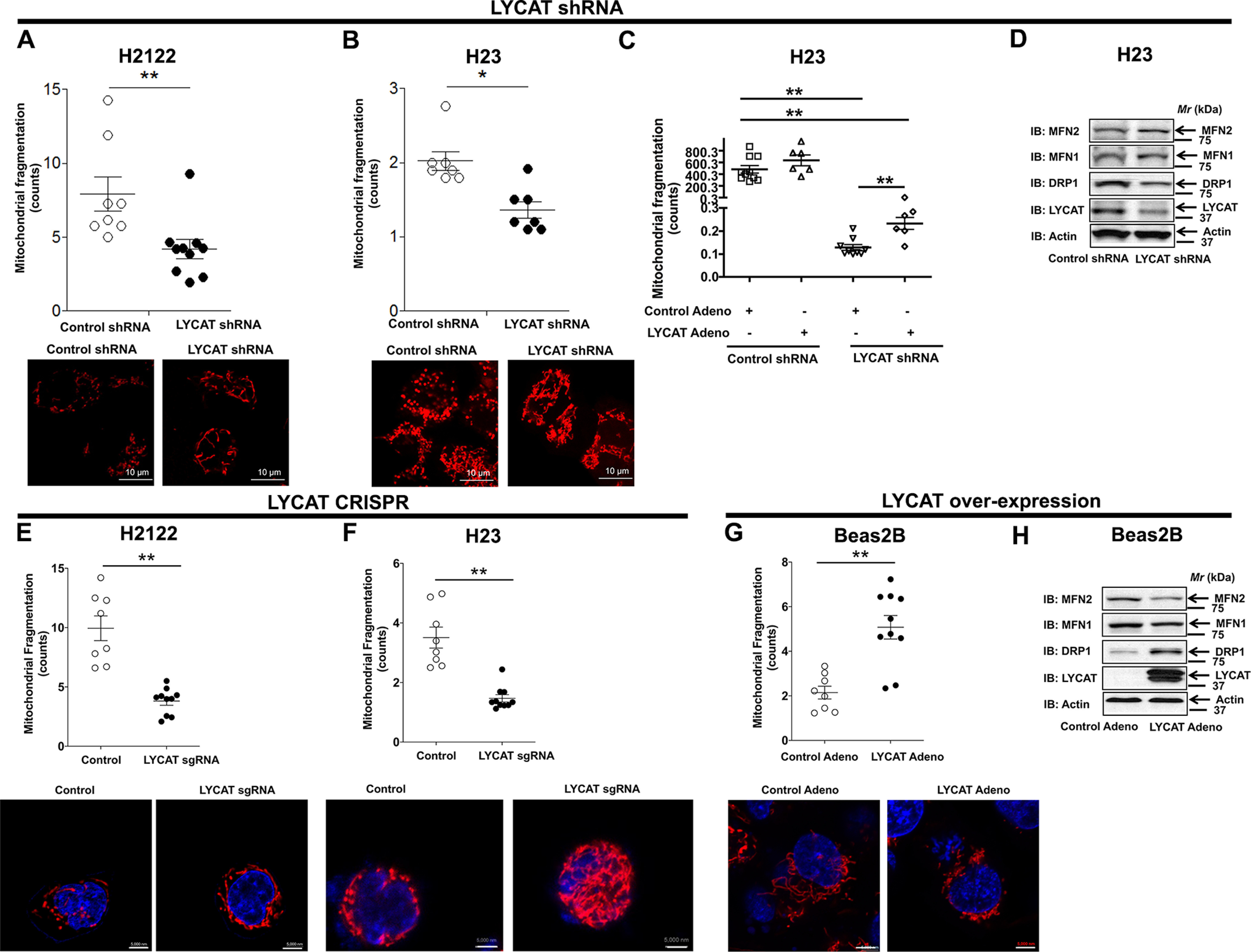
LYCAT knockdown inhibits mitochondrial fragmentation. A and B, H2122 (A) and H23 (B) cells stably expressing either control or LYCAT shRNAs were stained with MitoTracker Red fluorescence dye, and mitochondrial fragmentation was recorded by confocal microscopy. Scale bar, 10 μm. *, p < 0.05; **, p< 0.01 versus parental control. C, restoration of LYCAT expression in H23 LYCAT knockdown clones rescues the effect of LYCAT knockdown on mitochondrial fragmentation, as determined by MitoTracker staining and confocal microscopy. **, p < 0.01 versus parental control. D, expression of DRP1 that drives mitochondrial fission and MFN1/2, which control mitochondrial outer membrane fusion in CRISPR-Cas9 LYCAT–edited H23 cells. The β-actin blots in D and Fig. 6C are identical because they were from the same experiment and run at the same time, when β-actin immunostaining was performed. E and F, LYCAT–edited H2122 (E) and H23 (F) knockout cells were stained with MitoTracker Red fluorescence dye, and mitochondrial fragmentation was recorded by confocal microscopy. Scale bar, 5000 nm. *, p< 0.05; **, p < 0.01 versus parental control. G, LYCAT-overexpressing Beas2B cells were stained with MitoTracker Red fluorescence dye, and mitochondrial fragmentation was recorded by confocal microscopy. **, p < 0.01 versus control. H, cell lysates of LYCAT-overexpressing Beas2B cells were probed for the expression of the indicated proteins via immunoblotting (IB). Error bars, S.E.
Discussion
CL, or 1,3-diphosphatidyl-sn-glycerol, is an anionic phospholipid that is biosynthesized exclusively in the mitochondria and predominantly localized in the inner mitochondrial membrane (5). CL has four long-chain fatty acids, and the fatty acyl chain composition depends on the cell type. In heart mitochondria, the predominant molecular species of CL are tetralinoleoyl and trilinoleoyl-oleoyl species; however, the newly synthesized immature CL has more monounsaturated and saturated fatty acids. The fatty acyl chains of CL undergo rapid remodeling in the mitochondria to generate a linoleic acid–rich molecular species, which is catalyzed by tafazzin and LYCAT. Tafazzin, encoded by the TAZ gene, is highly expressed in cardiac and skeletal muscle (6, 7) and functions as a transacylase. Tafazzin catalyzes the direct transfer of linoleic acid from the sn-2 position of phosphatidylcholine to MCL, and mutations in the TAZ gene are responsible for Barth syndrome (6, 7). ALCAT (acyl-CoA:lysocardiolipin acyltransferase) catalyzes reacylation of MCL and dilyso-CL in the mitochondria and endoplasmic reticulum; however, it acts on other lysophospholipids, including lysophosphatidylinositol (23, 24). CL plays an important role in optimal mitochondrial function, including electron transport (10), mitochondrial dynamics (25), mitochondrial biogenesis (26), apoptotic machinery (27), and vascular development (28). Deficiencies in CL content and modification in linoleic acid molecular species are associated with dysfunctional heart in diabetes (29), heart failure (30), and ischemia-reperfusion injury (31). In this study, for the first time we have identified increased expression of LYCAT and CL levels in lung cancer (Figs. 1 and 4), suggesting that both LYCAT expression and CL levels might represent novel biomarkers for lung cancer detection. Therefore, future studies should focus on evaluating the diagnostic/prognostic utilities of LYCAT and CL levels in lung cancer. Furthermore, because cigarette smoking is closely associated with lung cancer, the impact of cigarette smoking on LYCAT expression and/or CL levels is also of great interest, and we have recently shown elevated LYCAT expression in lung tissues from smokers and bronchial epithelial cells exposed to cigarette smoke extract (31).
For the first time, we show that knockdown of LYCAT in NSCLC cells leads to reduced cell proliferation, cell migration, and cell invasion, both in vitro and in vivo (Figs. 2 and 3). Our studies revealed that LYCAT-mediated effects on NSCLC biology were independent of apoptosis, as determined by annexin V–FITC staining (Fig. 3I). However, for absolute determination of the role of apoptosis in LYCAT-mediated NSCLC biology, future studies should adopt other techniques to evaluate apoptosis (e.g. immunoblotting for cleaved caspase-3 or its substrate PARP (poly(ADP-ribose) polymerase) and multiplex assays that determine both cell viability and apoptosis (ApoLive glow, Promega Corp.) to name a few).
Interestingly, in contrast to tafazzin, no mutation(s) in LYCAT gene has been identified that is specifically associated with any human pathology. In another study, we have recently shown that LYCAT expression was significantly altered in PBMCs and lung tissues from idiopathic pulmonary fibrosis patients and in lung tissues from murine models of bleomycin- and radiation-induced pulmonary fibrosis (16). Further, in both bleomycin and radiation models, overexpression of hLYCAT in the lung reduced lung fibrosis, whereas down-regulation of native LYCAT expression increased fibrogenesis (16). Importantly, bleomycin challenge decreased CL levels in lung tissue ∼50% and mol % of C18:1 and C18:2 fatty acid, and overexpression of hLYCAT restored the CL and fatty acid profiles similar to control animals without bleomycin challenge, suggesting a protective role for enhanced LYCAT expression in ameliorating lung fibrosis (16). On the contrary, increased expression of LYCAT and CL levels was shown to have deleterious effects in lung cancer (Figs. 1–5), suggesting that blocking LYCAT activity might be beneficial in lung cancer patients. Currently, there are no small-molecule inhibitors developed to block LYCAT activity. In this study, we have identified a way to block the LYCAT activity by designing LYCAT mimetic peptides and went on to show that the treatment of NSCLC cells with LYCAT mimetic peptide could block LYCAT activity and LYCAT-mediated cell migration (Fig. 5). However, the specificity of the LYCAT peptide mimetic and the in vivo efficacy of LYCAT mimetic peptide(s) in blocking or reducing tumor size and development remain to be assessed.
Abnormalities in CL can impair mitochondrial electron transport chain activities and mitochondrial function. Alterations in CL molecular species correlated with prostate cancer cell proliferation (8), and alterations in CL content and composition of mitochondria occurred in thyroid oncocytic tumors (9). Abnormal CL content and deficiencies in polyunsaturated mature molecular species in mouse brain tumor mitochondria were associated with significant reduction in both individual and linked electron transport activities (10). There is evidence for the involvement of mitochondria in the development and progression of lung cancer; however, the role of LYCAT-mediated mitochondrial dynamics in lung tumorigenesis is unclear. In a recent study, the therapeutic utility of leflunomide in blocking pancreatic ductal adenocarcinoma growth via attenuating mitochondrial fragmentation was highlighted (32). Similarly, mitochondrial fusion can be induced by genetic or pharmacologic inhibition of DRP1 or through forced expression of MFN2. In this study, we have identified that loss of LYCAT expression results in decreased DRP1 expression (data not shown) and increased mitochondrial fusion (Fig. 7). Further, ecto-expression of mLycat in lung epithelial BEAS-2B cells stimulated DRP1 expression compared with control nontransfected cells, suggesting a direct or indirect effect of LYCAT expression in transcriptional regulation of genes that regulate mitochondrial dynamics. In fact, knockdown of LYCAT in H23 NSCLC cells regulated differential expression of 15 key genes under the p53 signaling pathway, two of which were the top down-regulated (CCND2) and up-regulated (CDKN1A) genes involved in the cell cycle. Moreover, it would be interesting to determine the effects of the LYCAT mimetic peptide (Fig. 5) on DRP1/MFN1/2 expression and mitochondrial fusion/fission in lung cancer.
In this study, we have shown an increased expression of LYCAT in lung cancer specimens and NSCLC cell lines; however, LYCAT is expressed in most of the mammalian cells and is essential to maintain normal mitochondrial function (10, 25, 26). Thus, knockdown of LYCAT in normal lung epithelial cells could affect mitochondrial electron transport, mitochondrial dynamics, and mitochondrial reactive oxygen species, which needs to be addressed. Our current observation that knockdown of LYCAT in NSCLC cells regulates differential expression of 11 gene ontology and KEGG pathways is supported by the findings of the mLycat gene in hemangioblast development in embryonic stem cell differentiation in vitro (33). In this study, Lycat siRNA reduced expression of endothelial (Flk1 and Cd31) and hematopoietic (Gata1, gata2, Runx1, and Scl) genes, suggesting a role for the acyltransferase in regulating development of hematopoietic and endothelial lineages during embryonic stem cell differentiation.
In conclusion, we have identified a novel tumor-promoting role for LYCAT in lung cancer. We also demonstrate that LYCAT promotes lung cancer via modulating CL levels. Furthermore, tumor-promoting effects of LYCAT were also determined to be via altered mitochondrial dynamics and cell cycle distribution. The nature of signaling intermediates between LYCAT-mediated CL levels and mitochondrial dynamics/cell cycle distribution awaits further study.
Experimental procedures
Reagents
Protease inhibitor mixture tablets (EDTA-free Complete) were from Roche Applied Science. TMRM dye, rabbit anti-LYCAT, and mouse anti-β-actin antibodies were from Sigma–Aldrich. Cell lysis buffer and rabbit anti-CDKN1A, CCND2, MFN1, MFN2, and DRP1 were from Cell Signaling Technology (Danvers, MA, USA). Anti-GAPDH antibodies were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). The Cell Cycle Assay Kit was ordered from Abcam Inc. (Cambridge, MA, USA). Horseradish peroxidase–linked anti-mouse IgG and anti-rabbit IgG antibodies were obtained from Bio-Rad.
Cell culture
Human nontransformed bronchial epithelial cell line (Beas2B) and NSCLC cell lines (A549, H2122, 1793, H23, 669, H157, Calu-1) were obtained from the tissue culture core of the University of Colorado, Anschutz Medical Campus. Beas2B, A549, H2122, 1793, H23, 669, H157, and Calu-1 were cultured in RPMI medium supplemented with 10% FBS in a humidified 5% CO2 incubator at 37 °C. All the cell lines were cultured biweekly, and stocks of cell lines were passaged no more than 10 times for use in experiments.
Knockdown protocol
Control shRNA and human LYCAT shRNA (HSH067542) were ordered from Genecopoeia (Rockville, MD, USA). NSCLC cells were transfected with 3 µg of control or LYCAT shRNA for 48 h by using Lipofectamine 2000 reagent according to the manufacturer's protocol. For generating H23 and H2122 clones with stable knockdown of nontargeting shRNAs and LYCAT shRNAs, H23 and H2122 cells were transfected with either PLKO1-control shRNA or PLKO1-LYCAT shRNA vectors followed by 1 μg/ml puromycin selection, respectively. The extent of LYCAT knockdown was confirmed by immunoblotting. Multiple clones were screened, and the best-performing clone was employed in the studies.
Gene editing of LYCAT
Guide RNAs targeting LYCAT were purchased from Santa Cruz Biotechnology (SC415182). H2122 and H23 cells were transfected with the guide plasmids using Lipofectamine reagent (Life Technologies) according to the manufacturer's recommendations. Twenty-four hours after transfection, GFP-positive cells were sorted under sterile conditions into 96-well plates. Multiple clones were isolated and assayed for LYCAT knockdown by using anti-LYCAT antibodies.
Cell proliferation studies
MTS cell growth assays were performed in triplicates by seeding 500 cells/well in a 96-well culture plate, followed by incubation at 37 °C in a 5% CO2 incubator. Cell proliferation was measured at 24, 48, and 72 h by adding 20 μl of MTS reagent (Cell Titer 96® Aqueous One Solution, G3582, Promega, Madison, WI, USA) to each well, followed by incubation at 37 °C. After 1 h, the absorbance of the formazan product was measured at 490 nm using a plate reader. Normalized absorbance values (sample readings − readings of medium-only blank) are represented in the graphs. In some experiments, cell proliferation was quantified using hemocytometer counting of trypsinized cells loaded with trypan blue dye.
Clonogenic assays were performed in triplicates by seeding 1000 cells/well in a 12-well culture plate, followed by incubation at 37 °C in a 5% CO2 incubator. After 5–7 days, colonies were stained using a staining solution (0.5% crystal violet, 12% glutaraldehyde, 87.5% H2O) for 1 h at room temperature. After destaining in water and drying, colonies were quantified using the Bio-Rad Chemidoc Imaging System and Quantity One Software. Cloning efficiency represents the mean number of colonies formed per well.
Cell migration assays
Wound-healing assay
Cells (100% confluence) in complete cell culture medium were applied for the scratch assay. A 2-mm scrape wound was created across the diameter with a pipette tip, followed by washout of the dead and floating cells. The cells were cultured in growth medium with mitomycin C (1 μg/ml). The wound was captured by an inverted microscope equipped with a digital camera at 0, 12, 24, and 48 h, and the healing of the wound was analyzed by determining the distance between the cells on either side of the scratch overtime and was represented in the figure as percentage of scratch closure.
Transwell assays
In total, 30,000 cells in serum-free medium were seeded into the transwell inserts (Corning) containing 8-μm permeable pores and allowed to migrate toward 10% FBS-containing medium. Later, the cells in the transwell inserts were removed, and the inserts were washed in PBS three times. The migrated cells on the bottom of the insert were fixed with 2% glutaraldehyde solution followed by crystal violet (1%) staining. After washing the inserts three times with PBS, the inserts were allowed to air-dry, and pictures were taken using an inverted microscope. Ten independent fields were counted for each transwell, and the average number of cells/field is represented in the graphs. For assessing cell invasion, 30,000 cells in serum-free medium were seeded in the Matrigel-coated transwell inserts (BD Biosciences). The cells were later processed in a manner similar to the cell migration assay.
Inhibitor studies
To check the effect of inhibitors on LYCAT activity or cell migration, H2122 cells were used. Confluent cells (∼90%) were pretreated with cell-permeable vehicle control peptide or LYCAT mimetic peptide of 21 amino acids, of which the first 11 amino acids constitute a leader sequence of YGRKKRRQRRR for cell permeation. These peptides were synthesized by Genicbio Ltd. (Shanghai, China).
MS analysis of phospholipids
Lipids were extracted by using the Folch procedure (34), and lipid phosphorus was determined by a micromethod (35). MS analysis of phospholipids was performed on a Q-Exactive hybrid-quadrupole-orbitrap mass spectrometer (Thermo Fisher Scientific) as described previously (36). Phospholipids were separated on a normal phase column (Silica Luna, 3 μm, 100A, 150 × 2 mm (Phenomenex, Torrance, CA) at 35° C using gradient solvents containing 5 mm CH3COONH4 (A: n-hexane/2-propanol/water, 43:57:1 (v/v/v); B: n-hexane/2-propanol/water, 43:57:8 (v/v/v)). The gradient conditions (all linear) were as follows: 0–23 min (10% B to 32% B); 23–32 min (32% B to 65% B); 32–35 min (65% B to 100% B); 35–62 min (hold at 100% B); 62–64 min (100% B to 10% B); 64–80 min (10% B). The flow rate was maintained at 200 μl/min except for the 35–62 min time frame, where the flow rate was increased to 225 μl/min. MS analysis was performed in negative ion mode at a resolution of 140,000 for the full MS scan in a data-dependent mode. The scan range for MS analysis was 400–1800 m/z with a maximum injection time of 128 ms using 1 microscan. An isolation window of 1.0 Da was set for the MS and MS2 scans. Capillary spray voltage was set at 3.5 kV, and capillary temperature was 320 °C. The S-lens Rf level was set to 60. Analysis of LC–MS data was performed using the software package Compound DiscovererTM (Thermo Fisher Scientific) with an in-house generated analysis workflow and oxidized phospholipid database.
Western blotting analysis
Cell lysates or tissue homogenates were prepared in lysis buffer containing EDTA-free complete protease inhibitors, and after centrifugation at 10,000 × g for 10 min, the lysate samples were boiled with Laemmli sample buffer for 5 min. 20 µg of protein were separated on 10% SDS-PAGE, transferred to polyvinylidene difluoride membranes, and blocked with TBST containing 5% BSA prior to incubation with primary antibodies (1:1000 dilution) overnight and secondary antibodies (1:2000 dilution) for 2 h at room temperature. Blots were developed using the ECL chemiluminescence kit, and integrated density of pixels in each membrane was quantified using ImageQuant 5.2 software (Molecular Dynamics, Sunnyvale, CA, USA).
LYCAT activity assay
The reaction mixture contained 50 mm Tris/HCl (pH 7.0), 25 μm 16-E8 NBD-16:0 CoA, monolysocardiolipin (5 μm) in 0.1% fatty acid–free BSA and cell lysate (200 µg of protein) in a total volume of 200 μl and was incubated at room temperature for 30 min (14, 15). The reaction was terminated by the addition of 1 ml of methanol/HCl (100:1, v/v), following which the lipids were extracted by adding 1 ml of chloroform and 0.7 ml of 1 n HCl (37). The lipid extract was vortexed and centrifuged at 5000 × g for 10 min, and the lower chloroform phase was dried under a nitrogen stream and separated by TLC on pre-made silica gel H plates containing 1% potassium oxalate with chloroform/methanol/ammonium hydroxide/water (65:25:4:1, v/v/v/v). Fluorescence-labeled cardiolipin and monolysocardiolipin, separated on the TLC plates, were analyzed by a GE ImageQuant LAS4000 analyzer and quantified by ImageJ. The specific activity of LYCAT was calculated and normalized to incubations of total cell lysates without monolysocardiolipin as substrate. The data are expressed as mean ± S.E.
RNA-Seq data collection and analysis
Total RNA from H2122 clones stably expressing either control shRNA or LYCAT shRNA was extracted with the Qiagen RNeasy Mini Kit (catalog no. 74104) according to the manufacturer's instructions. For these studies, RNA was isolated from a single clone at three different passages (biological replicates). The concentrations and purity of RNA were determined on a NanoDrop 1000 (Invitrogen), and RNA integrity was determined on the 2200 TapeStation system using RNA ScreenTape (Agilent, catalog no. 5067-5576). Samples with RNA integrity values between 7.0 and 8.4 were chosen for library preparation. The levels of remaining DNA were checked on a subset of samples using a Qubit fluorometer (Invitrogen). DNA amounts did not exceed 10% of the total amount of nucleic acid.
RNA-Seq library preparation
Libraries were prepared with the 3′ QuantSeq mRNA-Seq Library Prep Kit REV for Illumina (Lexogen), according to the manufacturer's instructions. In brief, 10–500 µg of total RNA was used to make each library. Library generation was initiated by oligo(dT) priming followed by first-strand cDNA synthesis, removal of RNA, and second-strand cDNA synthesis using random priming and DNA polymerase. During these steps, Illumina linker sequences and external barcodes were incorporated. Next, the libraries were subject to the final 20 cycles of PCR amplification.
RNA-Seq library validation and quantification
The quality of the libraries was checked on the 2200 TapeStation system using D1000 ScreenTape (Agilent, catalog no. 5067-5582), and as expected, peaks ranged from 264 to 294 bp. Libraries were quantified on the Qubit 2.0 fluorometer with the Qubit dsDNA HS assay kit (Life Technologies, Inc., catalog no. P7581). Individual libraries were pooled in equimolar amounts, and concentration of the final pool was determined by the PCR quantification method using the KAPA Library Quantification Kit (KAPA Biosystems). Sequencing was carried out on NextSeq 500 (Illumina), 1 × 75-nt reads, high output, to achieve approximately 20 × 106 clusters/sample.
RNA-Seq analysis
Raw reads were aligned to reference genome hg19 using bowtie2 (38). Gene expression was quantified using FeatureCounts (39). Normalized and differential expression statistics were computed using edgeR (40, 41), and p values were adjusted for multiple testing using the false discovery rate correction of Benjamini and Hochberg (42).
Cancer Genome Atlas analysis
For determining the expression of LYCAT in lung cancer, lung cancer data sets were downloaded from the TCGA data portal and analyzed using the “R” program for statistical computing. For analysis, 46 matched normal-tumor lung cancer data sets were utilized, and the LYCAT gene expression values (RNA sequence values) were represented in a graph.
Animal studies
Animal experiments were conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH). The animals were housed in the Biologics Research laboratory vivarium (University of Illinois, Chicago, IL, USA). All the animal experiments were approved by the Institutional Animal Care and Use Committee, under the title “the role of LYCAT in lung cancer” (16-203).
Tumor xenograft studies
H2212 clone (2 × 106 cells were resuspended in 100 μl of PBS) with stable expression of control shRNA or LYCAT shRNA were injected subcutaneously into the left and right flanks of 6-week-old female nude mice (Charles River Breeding Laboratories). 8 weeks postinjection, the mice were sacrificed, and the tumors from mice were isolated and weighted.
Mitochondrial dynamics, network imaging, and quantification
Cells were stained with MitoTracker Red (50 nm; Thermo Fisher Scientific, Waltham, MA, USA) in culture medium at 37 °C for 20 min and imaged with the Zeiss 510 META confocal laser-scanning microscope (excitation at 581 nm and emission recorded at 644 nm; Carl Zeiss MicroImaging, Thornwood, NY, USA). The analysis and quantification of mitochondrial fragmentation count (MFC) for each imaged cell were approached as described before (43). MFC = number of particles × 10,000/total mitochondrial pixels.
Annexin V/propidium iodide staining
To evaluate the extent of apoptosis, LYCAT gene–edited H2122 and H23 knockout clones were stained with the annexin V–FITC apoptosis detection kit (556547, BD Pharmingen) as per the manufacturer's recommendations. Flow cytometry was performed on a Beckman Gallios flow cytometer at the Research Resources Center Flow Cytometry Core Facility at the University of Illinois (Chicago, IL, USA).
Statistical analysis
Data are expressed as means ± S.E. from at least three independent experiments, and the results were subjected to statistical analysis by using two-way analysis of variance or a two-tailed Student's t test. Values of p < 0.05 were considered significant.
Data availability
Data have been deposited in the Gene Expression Omnibus under accession number GSE149054. All data presented and discussed are contained within the article.
Acknowledgments
We thank Dr. Balaji Ganesh (Director, Flow Cytometry Core Facility, Research Resources Center, University of Illinois, Chicago) for help in capturing and analyzing annexin V staining. We also thank Drs. Zarema Arbieva (Director, Core Genomics Facility) and Mark Maienschein-Cline (Director, Research Informatics Core) for RNA-Seq analysis and bioinformatics analysis. Bioinformatics analysis in the project described was performed by the UIC Research Informatics Core, supported in part by NCATS through Grant UL1TR002003.
Author contributions—L. S. H., S. R. K., A. P., P. S. Y., X. Z., S. P. R., A. H., R. R., R. K. B., and V. N. conceptualization; L. S. H., S. R. K., Y. Y. T., V. E. K., R. K. B., and V. N. data curation; L. S. H., S. R. K., Y. Y. T., V. E. K., T. S., P.-K. P.-K., and V. N. formal analysis; L. S. H., S. R. K., and S. A. validation; L. S. H., S. A., M. V., and V. N. investigation; L. S. H., S. R. K., S. A., M. V., P. S. Y., M. B., Y. Y. T., and V. E. K. methodology; L. S. H., R. K. B., and V. N. writing-original draft; R. A. W. and V. N. supervision; R. A. W., A. P., R. S., S. P. R., R. K. B., and V. N. writing-review and editing; A. P., R. S., X. Z., S. P. R., and R. K. B. resources; V. N. funding acquisition; V. N. project administration.
Funding and additional information—This work was supported in part by NHLBI, National Institutes of Health, Grants P01HL126609, RO1HL127342, and P01HL060678 and UIC College of Medicine Research Funds (to V. N.) and NIH grants CA165065, CA243142, HL114453, AI145406 and GM113908 (to V. E. K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- NSCLC
- non-small-cell lung cancer
- ALK
- anaplastic lymphoma kinase
- CL
- cardiolipin
- DE
- differentially expressed
- DRP1
- dynamin-related protein 1
- FFPE
- formalin-fixed paraffin-embedded
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- KRAS
- Kristen rat sarcoma
- LYCAT
- lysocardiolipin acyltransferase
- LUAD
- lung adenocarcinoma
- LUSC
- lung squamous cell carcinoma
- MFN
- mitofusin
- iPLA2
- calcium-independent phospholipase A2
- cPLA2
- cytosolic PLA2
- TCGA
- the Cancer Genome Atlas
- shRNA
- short hairpin RNA
- MCL
- monolysocardiolipin
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
References
- 1. Herbst R. S., Heymach J. V., and Lippman S. M. (2008) Lung cancer. N. Engl. J. Med. 359, 1367–1380 10.1056/NEJMra0802714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jemal A., Siegel R., Ward E., Hao Y., Xu J., Murray T., and Thun M. J. (2008) Cancer statistics, 2008. CA Cancer J. Clin. 58, 71–96 10.3322/CA.2007.0010 [DOI] [PubMed] [Google Scholar]
- 3. Santucci R., Sinibaldi F., Polticelli F., and Fiorucci L. (2014) Role of cardiolipin in mitochondrial diseases and apoptosis. Curr. Med. Chem. 21, 2702–2714 10.2174/0929867321666140414112156 [DOI] [PubMed] [Google Scholar]
- 4. Lennon F. E., and Salgia R. (2014) Mitochondrial dynamics: biology and therapy in lung cancer. Expert Opin. Investig. Drugs 23, 675–692 10.1517/13543784.2014.899350 [DOI] [PubMed] [Google Scholar]
- 5. Buckland A. G., Kinkaid A. R., and Wilton D. C. (1998) Cardiolipin hydrolysis by human phospholipases A2: the multiple enzymatic activities of human cytosolic phospholipase A2. Biochim. Biophys. Acta 1390, 65–72 10.1016/S0005-2760(97)00170-7 [DOI] [PubMed] [Google Scholar]
- 6. Houtkooper R. H., Turkenburg M., Poll-The B. T., Karall D., Pérez-Cerdá C., Morrone A., Malvagia S., Wanders R. J., Kulik W., and Vaz F. M. (2009) The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta 1788, 2003–2014 10.1016/j.bbamem.2009.07.009 [DOI] [PubMed] [Google Scholar]
- 7. Gonzalvez F., D'Aurelio M., Boutant M., Moustapha A., Puech J. P., Landes T., Arnauné-Pelloquin L., Vial G., Taleux N., Slomianny C., Wanders R. J., Houtkooper R. H., Bellenguer P., Møller I. M., Gottlieb E., et al. (2013) Barth syndrome: cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim. Biophys. Acta 1832, 1194–1206 10.1016/j.bbadis.2013.03.005 [DOI] [PubMed] [Google Scholar]
- 8. Sapandowski A., Stope M., Evert K., Evert M., Zimmermann U., Peter D., Päge I., Burchardt M., and Schild L. (2015) Cardiolipin composition correlates with prostate cancer cell proliferation. Mol. Cell Biochem. 410, 175–185 10.1007/s11010-015-2549-1 [DOI] [PubMed] [Google Scholar]
- 9. Zhang J., Yu W., Ryu S. W., Lin J., Buentello G., Tibshirani R., Suliburk J., and Eberlin L. S. (2016) Cardiolipins are biomarkers of mitochondria-rich thyroid oncocytic tumors. Cancer Res. 76, 6588–6597 10.1158/0008-5472.CAN-16-1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kiebish M. A., Han X., Cheng H., Chuang J. H., and Seyfried T. N. (2008) Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: lipidomic evidence supporting the Warburg theory of cancer. J. Lipid Res. 49, 2545–2556 10.1194/jlr.M800319-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Keith R. L., Karoor V., Mozer A. B., Hudish T. M., Le M., and Miller Y. E. (2010) Chemoprevention of murine lung cancer by gefitinib in combination with prostacyclin synthase overexpression. Lung Cancer 70, 37–42 10.1016/j.lungcan.2010.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. To M. D., Rosario R. D., Westcott P. M., Banta K. L., and Balmain A. (2013) Interactions between wild-type and mutant Ras genes in lung and skin carcinogenesis. Oncogene 32, 4028–4033 10.1038/onc.2012.404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Westcott P. M., Halliwill K. D., To M. D., Rashid M., Rust A. G., Keane T. M., Delrosario R., Jen K. Y., Gurley K. E., Kemp C. J., Fredlund E., Quigley D. A., Adams D. J., and Balmain A. (2015) The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 517, 489–492 10.1038/nature13898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang L. S., Jiang P., Feghali-Bostwick C., Reddy S. P., Garcia J. G. N., and Natarajan V. (2017) Lysocardiolipin acyltransferase regulates TGF-β mediated lung fibroblast differentiation. Free Radic. Biol. Med. 112, 162–173 10.1016/j.freeradbiomed.2017.07.023 [DOI] [PubMed] [Google Scholar]
- 16. Huang L. S., Mathew B., Li H., Zhao Y., Ma S. F., Noth I., Reddy S. P., Harijith A., Usatyuk P. V., Berdyshev E. V., Kaminski N., Zhou T., Zhang W., Zhang Y., Rehman J., et al. (2014) The mitochondrial cardiolipin remodeling enzyme lysocardiolipin acyltransferase is a novel target in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 189, 1402–1415 10.1164/rccm.201310-1917OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang D. W., Sherman B. T., Tan Q., Collins J. R., Alvord W. G., Roayaei J., Stephens R., Baseler M. W., Lane H. C., and Lempicki R. A. (2007) The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 8, R183 10.1186/gb-2007-8-9-r183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang D. W., Sherman B. T., Tan Q., Kir J., Liu D., Bryant D., Guo Y., Stephens R., Baseler M. W., Lane H. C., and Lempicki R. A. (2007) DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res 35, W169–W175 10.1093/nar/gkm415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Badia R., Pujantell M., Riveira-Muñoz E., Puig T., Torres-Torronteras J., Martí R., Clotet B., Ampudia R. M., Vives-Pi M., Este J. A., and Ballana E. (2016) The G1/S specific cyclin D2 is a regulator of HIV-1 restriction in non-proliferating cells. PLoS Pathog. 12, e1005829 10.1371/journal.ppat.1005829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tilokani L., Nagashima S., Paupe V., and Prudent J. (2018) Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 62, 341–360 10.1042/EBC20170104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horbay R., and Bilyy R. (2016) Mitochondrial dynamics during cell cycling. Apoptosis 21, 1327–1335 10.1007/s10495-016-1295-5 [DOI] [PubMed] [Google Scholar]
- 22. Song M., Mihara K., Chen Y., Scorrano L., and Dorn G. W. 2nd (2015) Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 21, 273–286 10.1016/j.cmet.2014.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cao J., Liu Y., Lockwood J., Burn P., and Shi Y. (2004) A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J. Biol. Chem. 279, 31727–31734 10.1074/jbc.M402930200 [DOI] [PubMed] [Google Scholar]
- 24. Cao J., Shen W., Chang Z., and Shi Y. (2009) ALCAT1 is a polyglycerophospholipid acyltransferase potently regulated by adenine nucleotide and thyroid status. Am. J. Physiol. Endocrinol. Metab. 296, E647–E653 10.1152/ajpendo.90761.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kameoka S., Adachi Y., Okamoto K., Iijima M., and Sesaki H. (2018) Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol. 28, 67–76 10.1016/j.tcb.2017.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schlame M., Rua D., and Greenberg M. L. (2000) The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 39, 257–288 10.1016/S0163-7827(00)00005-9 [DOI] [PubMed] [Google Scholar]
- 27. McMillin J. B., and Dowhan W. (2002) Cardiolipin and apoptosis. Biochim. Biophys. Acta 1585, 97–107 10.1016/S1388-1981(02)00329-3 [DOI] [PubMed] [Google Scholar]
- 28. Shen Z., Ye C., McCain K., and Greenberg M. L. (2015) The role of cardiolipin in cardiovascular health. Biomed Res. Int. 2015, 891707 10.1155/2015/891707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shi Y. (2010) Emerging roles of cardiolipin remodeling in mitochondrial dysfunction associated with diabetes, obesity, and cardiovascular diseases. J. Biomed. Res. 24, 6–15 10.1016/S1674-8301(10)60003-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mejia E. M., Cole L. K., and Hatch G. M. (2014) Cardiolipin metabolism and the role it plays in heart failure and mitochondrial supercomplex formation. Cardiovasc. Hematol. Disord. Drug Targets 14, 98–106 10.2174/1871529x14666140505123753 [DOI] [PubMed] [Google Scholar]
- 31. Bandela M., Bikkavilli R. K., Fu P., Suryadevara V., Dhavamani S., Papasani S., Singla S., Reddy S. P., Dudek S. M., and Natarajan V. (2019) Role of lysocardiolipin acyltransferase in lung epithelial cell apoptosis induced by cigarette smoke extract. Am. J. Respir. Crit. Care Med. 199, A5358 10.1164/ajrccm-conference.2019.199.1_MeetingAbstracts.A5358 [DOI] [Google Scholar]
- 32. Yu M., Nguyen N. D., Huang Y., Lin D., Fujimoto T. N., Molkentine J. M., Deorukhkar A., Kang Y., San Lucas F. A., Fernandes C. J., Koay E. J., Gupta S., Ying H., Koong A. C., Herman J. M., et al. (2019) Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 4, e126915 10.1172/jci.insight.126915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang C., Faloon P. W., Tan Z., Lv Y., Zhang P., Ge Y., Deng H., and Xiong J. W. (2007) Mouse lysocardiolipin acyltransferase controls the development of hematopoietic and endothelial lineages during in vitro embryonic stem-cell differentiation. Blood 110, 3601–3609 10.1182/blood-2007-04-086827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Folch J., Lees M., Sloane G. H., and Stanley G. H. (1957) A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509 [PubMed] [Google Scholar]
- 35. Fiske C. H., and Subbarow Y. (1929) Phosphorus compounds of muscle and liver. Science 70, 381–382 10.1126/science.70.1816.381-a [DOI] [PubMed] [Google Scholar]
- 36. Barroso B., and Bischoff R. (2005) LC-MS analysis of phospholipids and lysophospholipids in human bronchoalveolar lavage fluid. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 814, 21–28 10.1016/j.jchromb.2004.07.044 [DOI] [PubMed] [Google Scholar]
- 37. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 10.1139/y59-099 [DOI] [PubMed] [Google Scholar]
- 38. Langmead B., and Salzberg S. L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liao Y., Smyth G. K., and Shi W. (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- 40. McCarthy D. J., Chen Y., and Smyth G. K. (2012) Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 10.1093/nar/gks042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Robinson M. D., McCarthy D. J., and Smyth G. K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Benjamini Y., Drai D., Elmer G., Kafkafi N., and Golani I. (2001) Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 125, 279–284 10.1016/S0166-4328(01)00297-2 [DOI] [PubMed] [Google Scholar]
- 43. Rehman J., Zhang H. J., Toth P. T., Zhang Y., Marsboom G., Hong Z., Salgia R., Husain A. N., Wietholt C., and Archer S. L. (2012) Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 26, 2175–2186 10.1096/fj.11-196543 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data have been deposited in the Gene Expression Omnibus under accession number GSE149054. All data presented and discussed are contained within the article.